

SUMMARY
Chromatin structure is regulated through posttranslational modifications of histone variants that modulate transcription. Although highly homologous, histone variants display unique amino acid sequences associated with specific functions. Abnormal incorporation of histone variants contributes to cancer initiation, therapy resistance, and metastasis. This study reports that, among its biologic functions, histone H3.1 serves as a chromatin redox sensor that is engaged by mitochondrial H2O2. In breast cancer cells, the oxidation of H3.1Cys96 promotes its eviction and replacement by H3.3 in specific promoters. We also report that this process facilitates the opening of silenced chromatin domains and transcriptional activation of epithelial-to-mesenchymal genes associated with cell plasticity. Scavenging nuclear H2O2 or amino acid substitution of H3.1(C96S) suppresses plasticity, restores sensitivity to chemotherapy, and induces remission of metastatic lesions. Hence, it appears that increased levels of H2O2 produced by mitochondria of breast cancer cells directly promote redox-regulated H3.1-dependent chromatin remodeling involved in chemoresistance and metastasis.
In brief
Palma et al. report that H3.1Cys96 oxidation is a functional posttranslational modification involved in the regulation of gene expression. In breast cancer, H3.1Cys96 oxidation promotes resistance to first-line chemotherapeutic drugs and stimulates metastasis. They also show that suppressing H3.1Cys96 oxidation inhibits metastasis while resensitizing cancer cells to chemotherapy.
Graphical Abstract
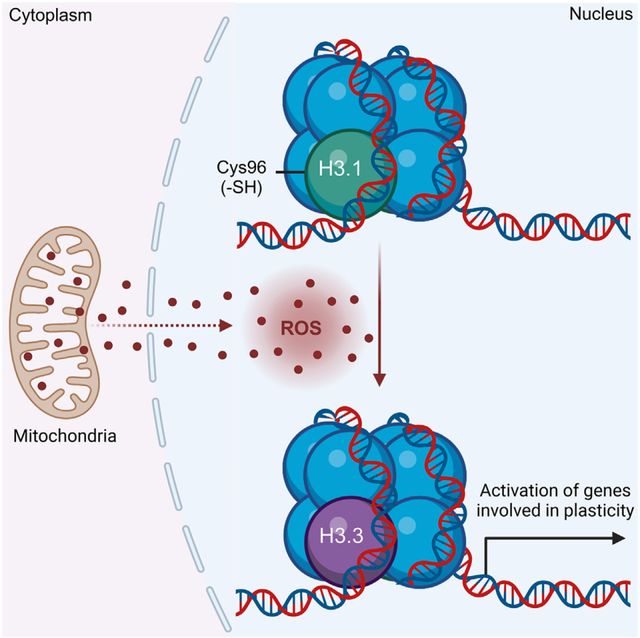
INTRODUCTION
Most cells are confined to a narrow range of differentiated phenotypes that maintain tissue function and preserve organismal viability. Differentiated phenotypes are, in large part, conserved by restrictions imposed on chromatin structure that prevent transcriptome drifts toward poorly differentiated states. At its most fundamental level, chromatin is organized by nucleosomes, which, in turn, are made up of DNA wrapped around histone proteins. Histone variant composition and posttranslational modifications shape protein-DNA interactions to promote chromatin remodeling in response to environmental stimuli. This either further reinforces cellular identity and function or relaxes structural constraints to enable phenotypic plasticity during stress conditions. Maladaptive chromatin remodeling occurs in cancer cells and promotes tumor evolution toward more malignant, untreatable disease.1,2
Posttranslational modifications of histones include well-known methylations, acetylations, and phosphorylations, among other modifications, that are positively or negatively associated with cancer initiation and progression.3,4 In addition to histone modifications, histone protein variants, which differ slightly in amino acid sequence, have been shown to affect chromatin architecture and influence transcription through distinct variant protein features and structural functions.5–7 For example, the human histone H3 variant H3.3 is enriched in nucleosomes at sites of active transcription,6 whereas the canonical H3.1 and H3.2 variants often delineate silent chromatin.8 Accumulating evidence implicates genomic distributions of H3.1, H3.2, and H3.3 as a key determinant of cellular gene expression, identity, and function, although the mechanisms that control nucleosome histone variant composition and exchange are not completely understood.5,9,10 Modest sequence changes observed among histone proteins have permitted phylogenetic analysis that identified H3.3 as the ancestor for all H3 variants in animals and fungi.11 In contrast, the H3.1, one of the canonical variants, is believed to occur only in mammals,11 and is distinguished from H3.2 and H3.3 variants by a unique cysteine residue at position 96 (Cys96). The proximity of Cys96 to acidic residues (e.g., Glu97) also suggests that it is likely to be susceptible to oxidation. In support of this idea, previous studies indicated that, in vitro, H3.1 is readily oxidized12 or glutathyolated13 by H2O2 and oxidized gluathione (GSSG), respectively. In light of these findings, we hypothesized that in cells, H3.1 Cys96 serves as a chromatin-embedded redox sensor. This is important because several disease processes, including cancer initiation and progression, involve the progressive dysregulation of mitochondrial oxidative metabolism leading to increased generation of reactive oxygen species (ROS).14–17 Hence, we used a combination of recently developed biosensors and chemogenic systems to generate H2O2 in situ to investigate whether H3.1 histone is sensitive to H2O2. We also investigated the functional consequences of H3.1Cys96 oxidation to chromatin structure, transcriptional outcomes, and the progression of breast cancer cells to more malignant phenotypes. In summary, the results discussed below support the hypothesis that the local redox environment of the nucleus can shape epigenetic control (posttranslational modifications of histones [e.g., cysteine oxidation]) of cellular identity via depleting the histone H3.1 variant. Depletion of H3.1 promotes its exchange by the H3.3 to activate plasticity gene transcription. Based on these findings, we propose that altered redox metabolism in cancer cells exploits the substitution of the H3.1 variant by H3.3 to enable adaptation to the harsh microenvironment of tumors, thereby allowing the acquisition of more aggressive and plastic phenotypes. Also, targeting this mechanism could be helpful to improve therapeutic outcomes.
RESULTS
Nucleus localization signal-D-amino acid oxidase (NLS-DAO) and NLS-Orp1-roGFP2 comprise a sensitive, inducible system to elevate and measure nuclear ROS (nROS) without causing oxidative DNA damage
To enable functional studies of altered nROS accumulation, a two-part system was created to allow (1) the reproducible generation of controlled amounts of H2O2 exclusively in the nucleus using NLS-DAO, an enzyme that generates H2O2 from d-amino acids and molecular oxygen (Figure S1A),18 and (2) ratiometrically measure levels of H2O2 generated in the nucleus, in real time, with NLS-tagged Orp1-roGFP2 reporter (NLS-Orp1-roGFP2), a redox bionsensor that quantifies redox potential as a ratio of excitation maxima for the reduced form (λex = 488 nm) against the oxidized form (λex = 405 nm) (Figure S1A).19
When this system was expressed in nontransformed MCF10AER/vSrc cells, NLS-Orp1-roGFP2 and NLS-DAO responded to 10 nM d-alanine (d-Ala) by increasing nuclear H2O2 (nH2O2), which was detected by relative changes in NLS-Orp1roGFP2 fluorescence (Figures S1B and S1C). Furthermore, comparisons with bolus H2O2 treatment indicated that NLS-Orp1roGFP2 expressed in MCF10AER/vSrc cells had both dynamic range and a rapid and reversible response to small variations in nH2O2 levels (Figures S1D and S1E). Levels of d-Ala sufficient to produce changes in NLS-Orp1roGFP2 fluorescence were titrated down to 10 nM (Figures S1F, top, and S1G). Used at this concentration, d-Ala was able to produce localized levels of H2O2 that were not sufficient to cause significant direct oxidative DNA damage as detected by 8-Oxo-2′-deoxyguanosine (8-oxo-dG) (Figures S1F, bottom, and S1G) or H2AX phosphorylation (Figure S1H) at peak activity (0–4 h) or well after substrate exhaustion (24 h) compared to the positive control (500 μM H2O2). d-Ala 10 nM was also insufficient to trigger ATM phosphorylation, another sensitive marker of DNA damage response activation (Figure S1H). In addition, despite elevating nH2O2 levels (Figure S1G, top), 10 nM d-Ala did not significantly increase DNA double-strand breaks assessed by comet assay (Figures S1I–S1K). Finally, our results indicated that there was no measurable increase in amplex red oxidation, a highly sensitive extracellular probe for H2O2, when d-Ala was used at levels below 10 μM (Figure S1L). These results support the conclusion that NLS-DAO and NLS-Orp1-roGFP2 can be used in combination as a sensitive, inducible system to simultaneously generate and measure nROS without causing the accumulation of oxidative DNA damage.
Steady-state nROS levels correlate with aggressive breast cancer cell phenotypes
Using a selection of breast cancer cell lines, we found that immortalized but not transformed mammary epithelial cells (MCF10AER/vSrc; Figure 1A) had the lowest levels of nH2O2; however, fully transformed MCF10AER/vSrc cells showed rapid and sustained increase in nH2O2 (Figures 1A–1C). MCF10AER/vSrc transformation was induced by the activation, with 4-hydroxytamoxifen (4-OHT), of an estrogen receptor (ER)-driven construct stably transfected into these cells that controls the expression of the v-Src oncogene.20,21 Other fully transformed human breast cancer cells lines (MCF7, BT474, BT20, and MB231) showed a trend toward higher nH2O2 in the more aggressive cell lines (BT20 and MB231), compared with less aggressive cells (MCF7 and BT474) (Figures 1B and 1C). These data suggest a link between higher levels of nROS and more aggressive, less differentiated breast cancer cell phenotypes.
Figure 1. Steady-state levels of nuclear H2O2 (nH2O2) are higher in transformed breast cancer cells and promote plasticity gene expression.
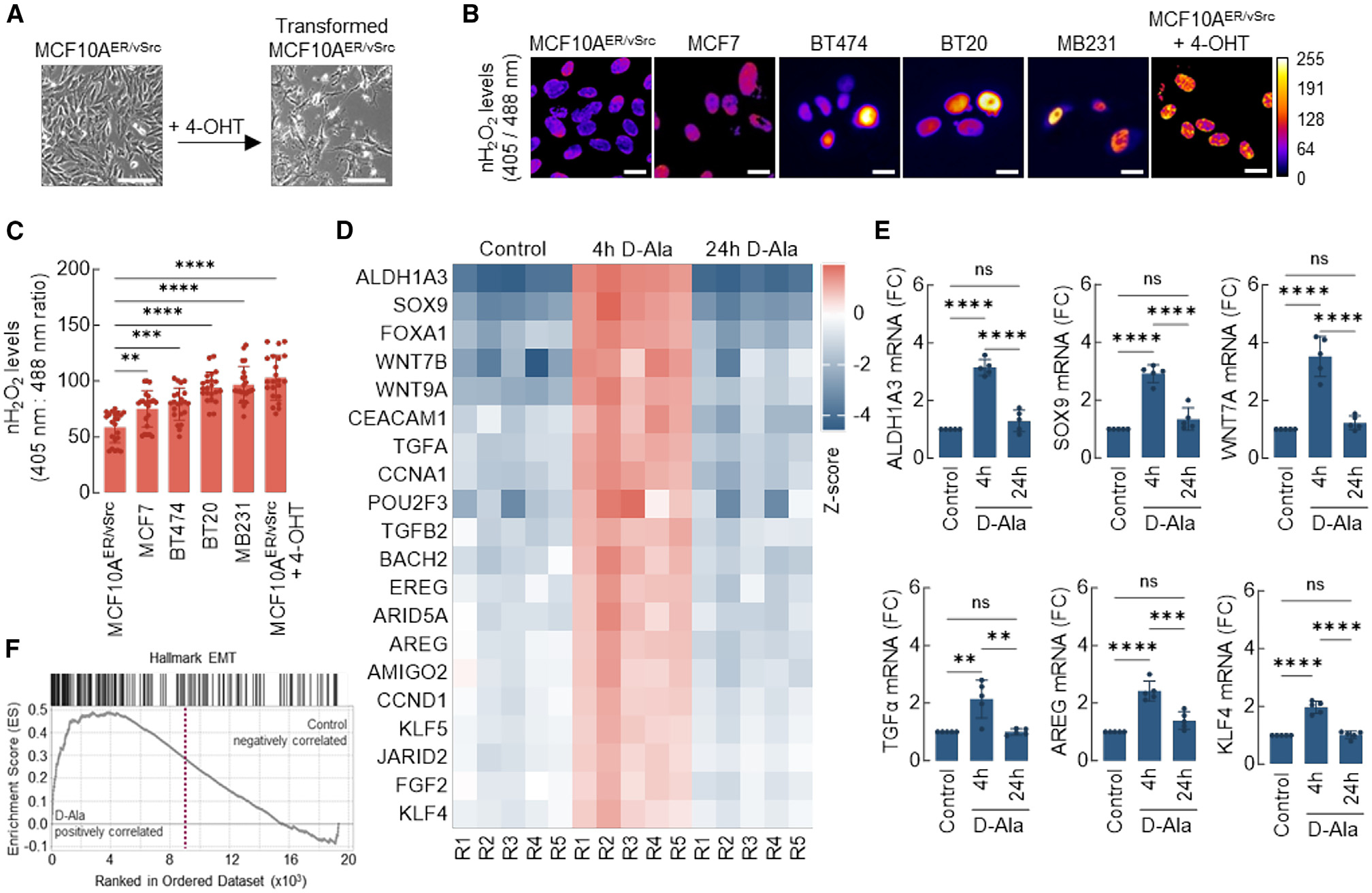
(A) Nontumorigenic epithelial MCF10AER/vSrc cells were transformed to a mesenchymal/tumorigenic phenotype via stimulation with 4-OHT. Scale bars, 200 μm.
(B) Expression of nuclear-targeted Orp1-roGFP2 in different cell lines. The levels of H2O2 in the nucleus of normal epithelial (MCF10AER/vSrc) and cancer cell lines (MCF7, BT474, BT20, MB231, and transformed MCF10AER/vSrc) were determined using confocal microscopy. Oxidized (λex = 405 nm) and reduced (λex = 488 nm) roGFP2 signals were acquired, and oxidized/reduced ratios were calculated using ImageJ (shown as a heatmap). Scale bars, 20 μm. On the side scale, 0 (colder colors) represents low ROS levels, whereas warmer colors toward the 255 mark represent higher ROS levels.
(C) Quantification of oxidized:reduced ratio of nuclear Orp1-roGFP2 in (B). Statistical significance was determined by 1-way ANOVA with post hoc Tukey test. Bars represent mean ± SEM. **p < 0.01; ***p < 0.001; ****p < 0.0001.
(D) Heatmap of core-enriched genes 4 or 24 h after 10 nM d-Ala treatment. Z scores were calculated based on the expression of each gene.
(E) qRT-PCR analysis of a subset of genes differentially expressed in (D). Statistical significance was determined by 1-way ANOVA with post hoc Tukey test. Bars represent mean ± SEM. **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant.
(F) GSEA enrichment plot of Hallmark genes associated with EMT and upregulated in cells 4 h after 10 nM d-Ala treatment.
See also Figures S1 and S2.
Elevated nROS activate transcription of genes associated with epithelial-to-mesenchymal transition (EMT) and cell plasticity acquisition
To determine the impact of nROS on the transcriptome, samples from the MCF10A/NLS-DAO/NLS-Orp1-roGFP2 system with activated nH2O2 production (10 nM d-Ala) were collected for RNA sequencing (RNA-seq) at 4 and 24 h after treatment. Comparative whole transcriptome analysis between control cells and cells induced with d-Ala to produce nH2O2 indicated the upregulation of numerous genes associated with lineage plasticity at the 4-h time point in d-Ala-treated cells, including ALDH1A3, SOX9, Wnt family transcription factors (Wnt7a and Wnt9b), stem cell transcription factors (POU2F3 and KLF4), and EMT-associated cytokines (transforming growth factor [TGF]-α, TGF-β, and amphiregulin [AREG]) (Figures 1D and S2A). Upregulated transcription was verified for a subset of these genes by qRT-PCR (Figure 1E). Consistently, gene set enrichment analysis (GSEA) at the same time point indicated a robust induction of EMT transcriptional activation by nH2O2 (Figure 1F), whereas Gene Ontology analysis indicated the activation of pathways associated with tumorigenesis and cell plasticity, such as cell migration, adhesion, proliferation, and differentiation (Figure S2B). At the 24-h time point (several hours after exhaustion of d-Ala substrate), the transcriptomic changes detected in d-Ala-treated cells at 4 h had largely reversed back to baseline levels (Figures S2C and S2D). Taken together, these results suggest that a flow of H2O2 into the nucleus triggers the transcription of genes associated with aggressive, mesenchymal phenotypes in immortalized mammary epithelial cells. Also, that such transcriptional changes may be at least temporarily dependent on the continued presence of H2O2 in the nucleus.
nROS promote nucleosome histone H3.1 exchange for histone H3.3 and chromatin decompaction
Recent studies (by Gomes et al.)5 showed that the exchange of H3 histone variants, H3.1 for H3.3, precedes cancer cell metastasis and is required for TGF-β and tumor necrosis factor α-induced EMT-driven acquisition of aggressive behavior. Intriguingly, H3.1 is unique among H3-type histones in harboring an oxidizable cysteine residue at position 96. This led to our hypothesis that H3.1 oxidation at Cys96 is involved in its replacement by H3.3 and, subsequently, the activation of EMT gene expression driven by nH2O2. Chromatin immunoprecipitation sequencing (ChIP-seq) experiments with antibodies specific for H3.1/H3.2 (canonical) and H3.3 histones performed in our MCF10A NLS-DAO model for nH2O2 generation demonstrated the significant loss of H3.1/H3.2 histones near transcription start sites (TSSs) at 4 h after nH2O2 induction, as compared to chromatin in control cells (Figure 2A). At the same time point, decreased histone H3.1 promoter occupancy mirrored an increased histone H3.3 deposition at promoters in nH2O2-treated cells (Figure 2A). Histone H3.3 differs from H3.1/H3.2 in that its chromatin incorporation is cell-cycle independent.22,23 H3.3 incorporation is also known to correlate with chromatin decompaction and transcriptional activation.23,24 Consistently, transmission electron microscopy (TEM) assessment of chromatin structure in nH2O2-treated cells showed that control MCF10A displayed a typical pattern of condensed heterochromatin adjacent to the inner membrane of the nuclear envelope (Figure 2B, top, and 2C). The elevation of nH2O2 reduced heterochromatin content by 4 h post-induction (Figure 2B, bottom, and 2C). Heterochromatin dissolution accompanied nuclear enlargement (Figures 2B and 2D), consistent with chromatin decompaction. In addition, western blotting indicated an overall net loss of H3.1 upon increased nH2O2, with an increase in the H3.3/H3.1-H3.2 ratio (Figures 2E and 2F). In Figure 1B, v-Src induced epithelial cell transformation in MCF10AER/vSrc20,21 was shown to follow an increase in nH2O2 levels. Furthermore, the activation of v-Src led to a time-dependent loss of H3.1-H3.2 and a relative increase in H3.3 (Figures 2G and 2H), co-occurring with the acquisition of spindle-shape morphology indicative of increased malignancy (Figure 1A).20,21 To test the functional requirement for nH2O2 to promote H3.1-H3.2/H3.3 exchange, EMT, and malignant transformation, we performed additional experiments in MCF10AER/vSrc cells that stably expressed NLS-catalase, an efficient scavenger of nH2O2. NLS-catalase prevented the accumulation of nH2O2 in MCF10AER/vSrc cells during v-Src-induced transformation and also after 4 h of NLS-DAO activation by 10 nM d-Ala treatment (Figures 2I and 2J). Interestingly, the expression of NLS-catalase preserved H3.1-H3.2 levels in both contexts (Figures 2K–2N) as well as net H3.3:H3.1-H3.2 ratios, consistent with a requirement for increased nH2O2 to deplete the canonical H3.1 variant. In addition, NLS-catalase expression prevented EMT-associated transcriptional activation of mesenchymal markers, SOX9 and fibronectin, in response to NLS-DAO/d-Ala stimulation (Figures 2O and 2P). Finally, because previous studies have shown that H3.1-H3.2 replacement by H3.3 is required for TGF-β-induced EMT,5 we investigated whether increased nH2O2 is also an essential step during TGF-β-induced EMT. MCF10A control cells and those expressing NLS-catalase were treated with TGF-β (10 ng/mL) for 14 days (Figure 2Q). EMT was verified by the increased expression of fibronectin and decreased E-cadherin. Control cells responded to TGF-β, and the expression of NLS-catalase blocked the transition to mesenchymal phenotypes, suggesting that nH2O2 necessarily participates in TGF-β-induced, H3 variant-dependent activation of EMT gene expression. Taken together, the results presented in Figure 2 support a model wherein nH2O2 elevation mediates nucleosomal H3 variant exchange to facilitate EMT transcriptional reprogramming.
Figure 2. nH2O2 promotes H3.1/H3.2 and H3.3 remodeling associated with EMT activation.
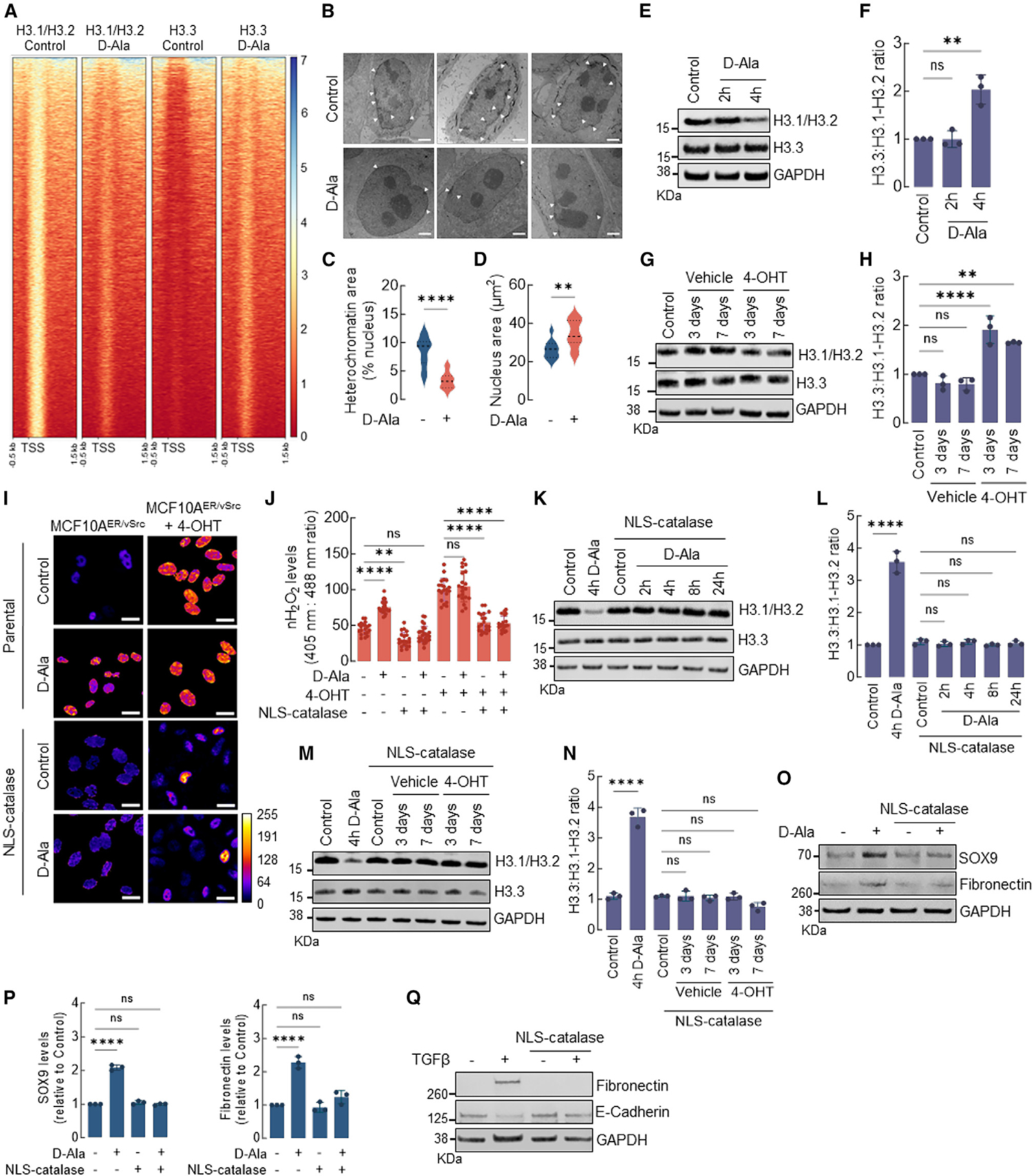
(A) Heatmap of H3.1/H3.2 and H3.3 ChIP-seq peaks across TSS (−0.5 kb, +1.5 kb) in MCF10AER/vSrc cells 4 h after 10 nM d-Ala treatment. On the side scale, 0 represents low occupancy, whereas higher numbers represent increased occupancy levels.
(B) Representative images of heterochromatin distribution in cells 4 h after 10 nM d-Ala treatment. Heterochromatin was assessed by TEM. White arrows indicate heterochromatin. Scale bars, 2 μm.
(C) Quantification of perinuclear heterochromatin in (B) using Trainable Weka Segmentation plug-in in Fiji ImageJ. Statistical significance was determined by t test. ****p < 0.0001.
(D) Area of the nucleus of cells in (B). Statistical significance was determined by t test. **p < 0.01.
(E) Western blot analysis of H3.1-H3.2 and H3.3 in MCF10AER/vSrc cells 2 or 4 h after 10 nM d-Ala treatment.
(F) H3.3:H3.1-H3.2 ratio quantification of (E). Statistical significance was determined by 1-way ANOVA with post hoc Tukey test. Bars represent mean ± SEM. **p < 0.01; ns, not significant.
(G) Western blot analysis of H3.1-H3.2 and H3.3 in MCF10AER/vSrc transformed cells (3 and 7 days after treatment with 4-OHT).
(H) H3.3:H3.1-H3.2 ratio quantification of (G). Statistical significance was determined by 1-way ANOVA with post hoc Tukey test. Bars represent mean ± SEM. **p < 0.01; ****p < 0.0001; ns, not significant.
(I) The levels of H2O2 in the nucleus of MCF10AER/vSrc cells expressing NLS-catalase and treated with 10 nM d-Ala or transformed with 4-OHT were determined using confocal microscopy. Oxidized (λex = 405 nm) and reduced (λex = 488 nm) roGFP2 signals were acquired, and the oxidized:reduced ratio was calculated using ImageJ (shown as a heatmap). White bars represent 20 μm. On the heatmap scale, 0 represents low ROS levels, whereas warmer colors toward the 255 mark represent relatively higher ROS levels.
(J) Quantification of oxidized:reduced ratio of nuclear Orp1-roGFP2 in (I). Statistical significance was determined by 1-way ANOVA with post hoc Tukey test. Bars represent mean ± SEM. **p < 0.01; ****p < 0.0001; ns, not significant.
(K) Western blot analysis of H3.1-H3.2 and H3.3 in MCF10AER/vSrc cells expressing NLS-catalase. Indicated times represent points when cells were harvested relative to t = 0 h (10 nM d-Ala addition to the media).
(L) H3.3:H3.1-H3.2 ratio quantification in (K). Statistical significance was determined by 1-way ANOVA with post hoc Tukey test. Bars represent mean ± SEM. ****p < 0.0001; ns, not significant.
(M) Western blot analysis of H3.1-H3.2 and H3.3 in MCF10AER/vSrc transformed cells (3 and 7 days after treatment with 4-OHT) expressing NLS-catalase.
(N) H3.3:H3.1-H3.2 ratio quantification in (M). Statistical significance was determined by 1-way ANOVA with post hoc Tukey test. Bars represent mean ± SEM. ****p < 0.0001; ns, not significant.
(O) Western blot analysis of SOX9 and fibronectin (EMT markers) in MCF10AER/vSrc cells expressing NLS-catalase 8 h after 10 nM d-Ala treatment.
(P) Quantification of SOX9 and fibronectin protein levels in (O). Statistical significance was determined by 1-way ANOVA with post hoc Tukey test. Bars represent mean ± SEM. ****p < 0.0001; ns, not significant.
(Q) EMT induction in MCF10A cells treated with TGF-β. Parental and NLS-catalase-expressing cells were treated with 10 ng/mL TGF-β for 14 days. The levels of the mesenchymal marker fibronectin and the epithelial marker E-cadherin were determined by western blot.
A redox-sensitive cysteine residue in histone H3.1 is engaged by nH2O2 to promote exchange for H3.3 and activate EMT gene expression
Canonical histones H3.1-H3.2 share 97% homology with the H3.3 variant, differing in sequence by only five amino acids. In the case of H3.1, this includes a distinctive cysteine (Cys96) present in H3.1 alone. H3.1Cys96 is a redox-sensitive residue known to undergo stress-induced oxidation associated with nucleosome instability.25 To test the hypothesis that nH2O2-dependent H3.1 modification of Cys96 regulates H3.1 stability in the nucleus, MCF10AER/vSrc cells expressing mutant H3.1(C96S)-FLAG and NLS-DAO were developed. Upon stimulation with 10 nM d-Ala, unlike the wild-type (WT) H3.1 protein, the levels of oxidation-resistant H3.1(C96S) mutant protein remained unaltered (Figures 3A and 3B). This indicated that H3.1 regulation by nH2O2 requires Cys96. The idea that Cys96 is oxidized to promote H3.1 exchange was further tested using DCP-Bio1, a dimedone derivative that labels oxidized Cys-SOH sulfenic acid residues (Figure S3A). DCP-Bio1 is also conjugated to biotin to facilitate isolation/visualization with streptavidin-conjugated reagents. In Figure 3C, results indicate that the incubation of recombinant H3.1 with H2O2 produced DCP-Bio1 adducts that were detected by Alexa Fluor-labeled streptavidin. Results also indicated that neither H3.2 nor H3.3 produced adducts with DCP-Bio1 when treated with H2O2 at the same molar ratio, indicating that these variants are more resistant to oxidation when compared to H3.1. To test the significance of Cys96 for promoter occupancy and the activation of EMT-associated genes, ChIP-qPCR for H3.1(WT)-FLAG and H3.3(WT)-HA (hemagglutinin) variants as well as the H3.1(C96S)-FLAG expressed in MCF10AER/vSrc cells was performed (Figure 3D). After treatment with d-Ala for 8 h, we found a decreased association of H3.1(WT)-FLAG with promoter regions of key EMT genes such as SOX9, fibronectin, and ZEB1. In cells expressing the mutant H3.1(C96S)-FLAG, d-Ala treatment had a dramatically different effect—slightly increasing instead of decreasing H3.1(C96S)-FLAG association with EMT gene promoter regions. In the same experiments, an nH2O2-dependent increase in H3.3(WT)-HA occupancy at the SOX9, ZEB1, and fibronectin promoter regions was observed in control cells, but not in those transfected with the H3.1(C96S)-FLAG mutant. We also confirmed that artificial upregulation of H3.3 using an inducible H3.3(WT)-HA construct did not trigger by itself the expression of EMT genes (Figures 3E and S3B), nor did it interfere with endogenous levels of H3.1-H3.2 (Figures 3E and 3F). Similarly, overexpressing H3.1(C96S) by itself did not significantly change the levels of endogenous H3.3 (Figures S3C and S3D). Because increased accessibility is a direct outcome of the replacement of H3.1-H3.2 by H3.3, we next examined whether nH2O2-driven replacement of H3.1 by H3.3 increased EMT gene promoter accessibility. As expected, increased promoter accessibility was observed in cells expressing the H3.1(WT) variant 4 h after d-Ala challenge. In cells expressing the H3.1(C96S) mutant, d-Ala treatment had no impact on the accessibility of the same EMT gene sets (Figure 3G). Finally, increased accessibility led the upregulation of SOX9 and ZEB1 at the protein level in cells expressing H3.1(WT) (Figure 3H, left, and S3E). However, in cells expressing H3.1(C96S), levels of these EMT markers remained unchanged (Figure 3H, right, and S3E). Expanded analysis of canonical (H3.1) and replacement (H3.3) histone variants binding to promoter regions (Figure S4A), promoter accessibility (Figure S4B), and mRNA transcript levels (Figure S4C) confirmed a strong positive correlation between H2O2-driven replacement of H3.1 by H3.3 and increased chromatin accessibility and gene expression for all of the tested EMT genes, particularly 4 h after d-Ala stimulation. At 24 h, binding to H3.1/H3.2 remained lower than at baseline for the promoters of all of the tested EMT genes as well as AREG (Figure S4A), consistent with the persistent downregulation of H3.1-H3.2 at the protein level detected in Figure 3A (left). Interestingly, binding to H3.3, which denotes sites of active transcription, was also significantly lower at 24 h compared with 4 h. In fact, at 24 h, binding of H3.3 to EMT gene promoters had mostly returned to baseline levels. This potentially explains the RNA-seq results shown in Figure 1D that indicated that EMT genes were transiently upregulated at 4 h, although H3.1-H3.2 downregulation persisted for at least 24 h.
Figure 3. H3.1 histone is distinctively sensitive and is evicted from chromatin by nH2O2.
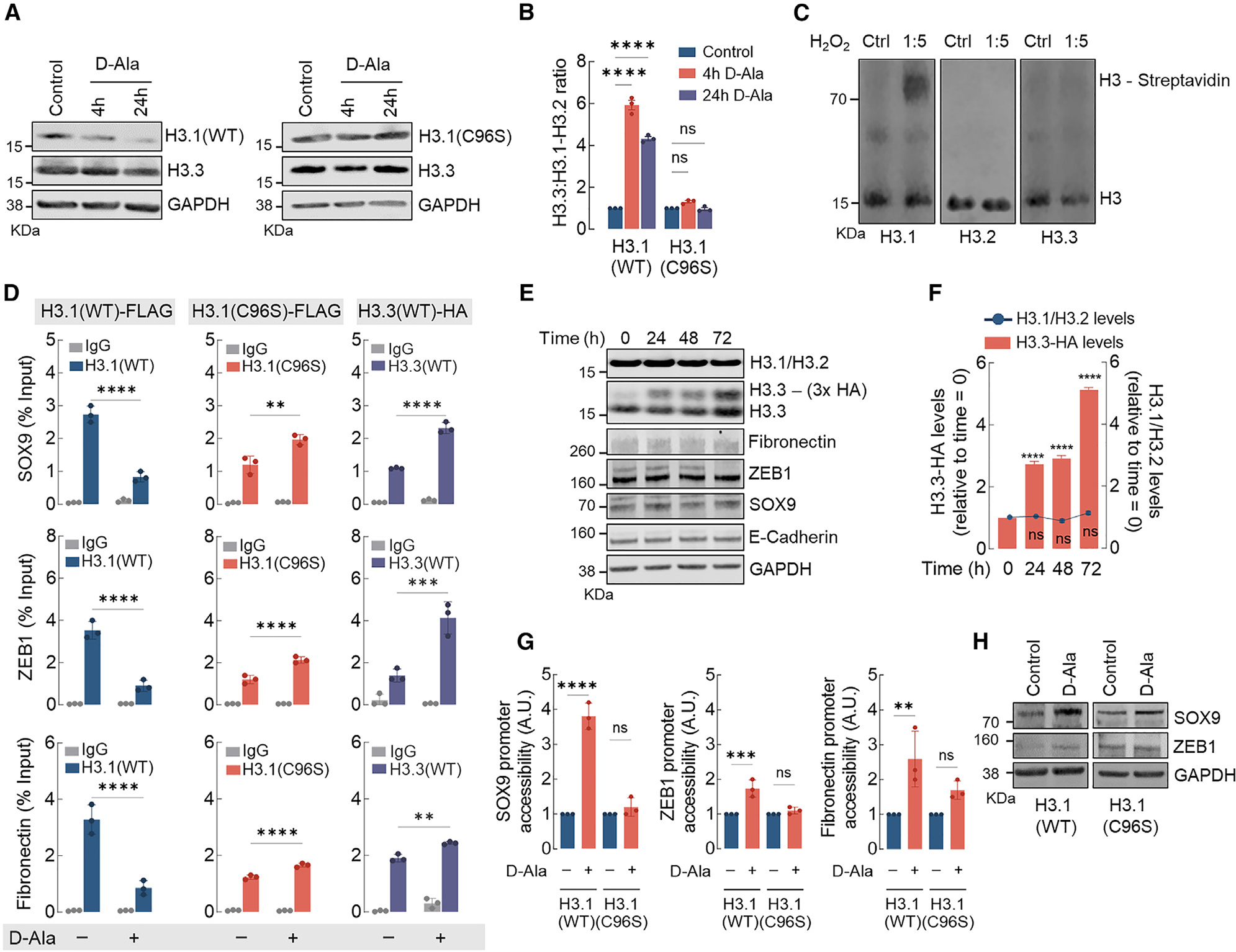
(A) Western blot analysis of H3.1 (WT and C96S) and H3.3 (endogenous) in MCF10AER/vSrc cells 4 and 24 h after 10 nM d-Ala treatment.
(B) H3.3/H3.1-H3.2 ratio quantification of (A). Statistical significance was determined by 2-way ANOVA with post hoc Bonferroni test. Bars represent mean ± SEM. ****p < 0.0001; ns, not significant.
(C) H3 variants labeling with DCP-Bio1. Recombinant H3.1, H3.2, and H3.3 were treated with H2O2 and incubated with biotin-conjugated DCP-Bio1, a dimedone derivative that labels oxidized Cys-SOH sulfenic acid residues. Biotin-conjugated DCP-Bio1 adducts were determined by western blot after incubation with Alexa Fluor-labeled streptavidin.
(D) H3.1(WT)-FLAG, H3.1(C96S)-FLAG, and H3.3-HA enrichment (ChIP-qPCR) in promoter regions of SOX9, ZEB1, and fibronectin in MCF10AER/vSrc cells 8 h after 10 nM d-Ala treatment. Statistical significance was determined by 2-way ANOVA with post hoc Bonferroni test. Bars represent mean ± SEM. **p < 0.01; ***p < 0.001; ****p < 0.0001.
(E) H3.3 expression does not affect the expression of H3.1-H3.2 or EMT genes. Overexpressing inducible H3.3-HA did not interfere with levels of endogenous H3.1-H3.2 or mesenchymal markers (fibronectin, ZEB1, and SOX9) and epithelial marker (E-cadherin).
(F) H3.1-H3.2 levels in response to H3.3-HA overexpression in (E). Statistical significance was determined by 1-way ANOVA with post hoc Tukey test. Bars represent mean ± SEM. ****p < 0.0001; ns, not significant.
(G) SOX9, ZEB1, and fibronectin gene accessibility in MCF10AER/vSrc cells expressing H3.1(WT) or H3.1(C96S) 4 h after 10 nM d-Ala treatment. Chromatin accessibility was determined in the promoter region of SOX9, ZEB1, and fibronectin. Statistical significance was determined by 1-way ANOVA with post hoc Tukey test. Bars represent mean ± SEM. **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant.
(H) Western blot analysis of SOX9 and ZEB1 in MCF10AER/vSrc cells expressing H3.1(WT) or H3.1(C96S) 24 h after 10 nM d-Ala treatment.
See also Figures S3 and S4.
Mitochondria-generated nH2O2 promotes H3.1 depletion in the nucleus
Mitochondria is a major source of H2O2 involved in the activation of oncogenic signaling in actively evolving tumors.26 In addition, recent reports showed that mitochondrial ROS may be trafficked to the nucleus through specialized structures,27 suggesting that mitochondria can potentially regulate chromatin remodeling via H2O2-driven H3.1 oxidation. To test this idea, we used an established model of malignant transformation based on the activation of the v-Src oncogene in MCF10A nontumorigenic cells.28 The activation of v-Src was induced in MCF10AER/vSrc cells stably transfected with the Orp1roGFP2 reporter targeted to either the nucleus (NLS-Orp1roGFP2) or the mitochondria (mito-Orp1roGFP2). v-Src-induced transformation significantly increased H2O2 in both the mitochondria and the nucleus (Figures 4A–4D) that was suppressed by the expression of mitochondria-targeted catalase. Consistent with mitochondrial H2O2 mediating changes in chromatin structure, mito-catalase expression suppressed H3.1 histone depletion in v-Src-transformed cells (Figures 4E and 4F) and phenocopied the effects of NLS-catalase in suppressing TGF-β-induced EMT (Figure 4G). Finally, mito-catalase resulted in lower expression levels of nH2O2-responsive genes in v-Src transformed cells, including SOX9, ZEB1, and fibronectin (Figures 4H and 4I). The results in Figure 4 are consistent with mitochondrial H2O2 regulating chromatin remodeling, tumor cell acquisition of lineage plasticity, and activation of EMT via interaction with H3.1 Cys96.
Figure 4. Mitochondria-generated H2O2 promotes H3.1 depletion in the nucleus and activates EMT.
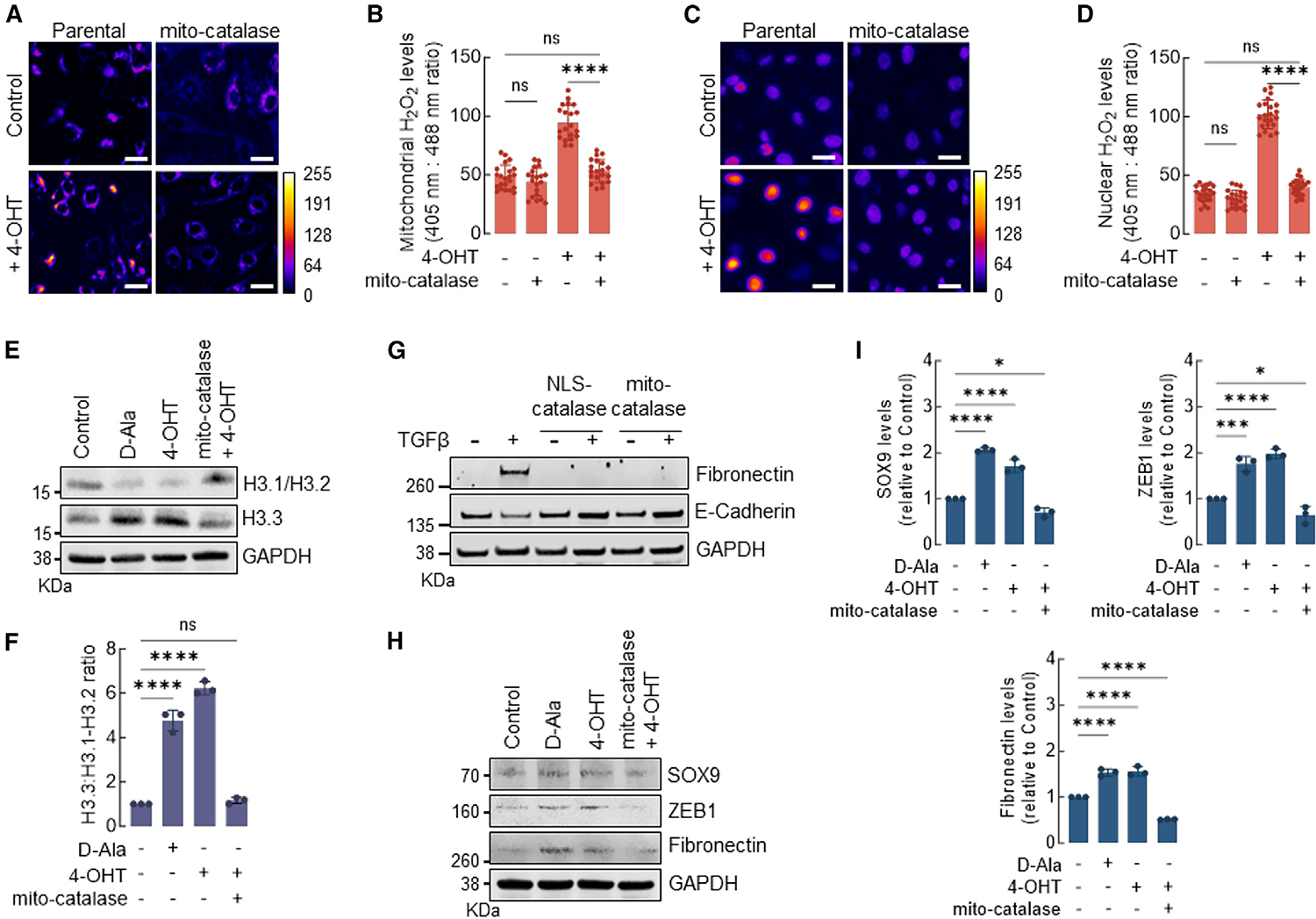
(A) The levels of H2O2 in the mitochondria of MCF10AER/vSrc cells expressing mito-catalase and/or transformed with 4-OHT were determined using fluorescence microscopy. Oxidized (λex = 405 nm) and reduced (λex = 488 nm) roGFP2 signals were acquired, and the oxidized:reduced ratio was calculated using ImageJ (shown as a heatmap). White bars represent 20 μm. On the heatmap scale, 0 represents low ROS levels, whereas warmer colors toward the 255 mark represent relatively higher ROS levels.
(B) Quantification of oxidized:reduced ratio of mitochondrial Orp1-roGFP2 in (A). Statistical significance was determined by 1-way ANOVA with post hoc Tukey test. Bars represent mean ± SEM. ****p < 0.0001; ns, not significant.
(C) The levels of H2O2 in the nucleus of MCF10AER/vSrc cells expressing NLS-catalase and/or transformed with 4-OHT were determined using fluorescence microscopy. Oxidized (λex = 405 nm) and reduced (λex = 488 nm) roGFP2 signals were acquired, and the oxidized:reduced ratio was calculated using ImageJ (shown as a heatmap). White bars represent 20 μm. On the heatmap scale, 0 represents low ROS levels, whereas warmer colors toward the 255 mark represent relatively higher ROS levels.
(D) Quantification of oxidized:reduced ratio of nuclear Orp1-roGFP2 in (C). Statistical significance was determined by 1-way ANOVA with post hoc Tukey test. Bars represent mean ± SEM. ****p < 0.0001; ns, not significant.
(E) Western blot analysis of H3.1-H3.2 and H3.3 in MCF10AER/vSrc cells expressing mito-catalase and/or transformed with 4-OHT (7 days). Parental cells were harvested 4 h after 10 nM d-Ala treatment and were used as positive controls.
(F) H3.3/H3.1-H3.2 ratio quantification in (E). Statistical significance was determined by 1-way ANOVA with post hoc Tukey test. Bars represent mean ± SEM. ****p < 0.0001; ns, not significant.
(G) EMT induction of MCF10A cells with TGF-β. Cells expressing NLS-catalase or mito-catalase were treated with 10 ng/mL TGF-β for 14 days. The levels of fibronectin (mesenchymal marker) and E-cadherin (epithelial marker) were determined by western blot.
(H) Western blot analysis of SOX9, ZEB1, and fibronectin in MCF10AER/vSrc cells expressing mito-catalase and/or transformed with 4-OHT (7 days).
(I) Quantification of SOX9, ZEB1, and fibronectin protein levels in (H). Statistical significance was determined by 1-way ANOVA with post hoc Tukey test. Bars represent mean ± SEM. *p < 0.05; ***p < 0.001; ****p < 0.0001.
nH2O2 promotes breast cancer cell chemoresistance
The finding that nH2O2 originating from mitochondria drives chromatin remodeling and EMT in multiple models of breast cancer transformation led us to hypothesize that it may also be associated with the development of chemoresistance.29–31 To test this idea, we used two murine mammary tumor cell lines, Py230 and Py8119, both derived from aggressive MMTV.PyVT oncogene-driven mammary tumors (Figures 5A–5D). Importantly, Py230 represents more epithelial (E-cadherin+, cytokeratin-8+), whereas Py8119 displays a more mesenchymal phenotype (N-cadherin+, cytokeratin-14+, and vimentin+). Drug resistance in Py230NLS–Orp1roGFP2 cells expressing an inducible NLS-catalase construct controlled by the tetracycline (TetON) promoter was stimulated by treating the cells with doxorubicin (DXR) or paclitaxel (PTX) for 7 days. Figures 5A and 5B show that Py230 cells displayed low levels of nH2O2 before drug treatment. Treatment with either DXR (Figure 5A) or PTX (Figure 5B) rapidly killed most cells, leaving a population of drug-resistant clones characterized by sustained, high levels of nH2O2. Interestingly, after a brief latency period (between days 4 and 6), persistent cells began to proliferate robustly in the presence of DXR or PTX. To evaluate the significance of elevated nH2O2 during the development and/or maintenance of chemoresistance, NLS-catalase expression was induced while cells were actively proliferating. As shown by cell viability quantitation and fluorescence micrographs, NLS-catalase expression effectively suppressed nH2O2 while resensitizing cells to DXR and PTX. This indicates that nH2O2 accumulation promotes resistance against first-line chemotherapeutic drugs. Similar trends were observed with the more mesenchymal Py8119 cell line (Figures 5C and 5D), with a few notable differences. First, pre-treatment levels of nROS were considerably higher in Py8119 cells compared to Py230 cells at baseline. Also, a larger number of persistent cells survived initial treatment. In addition, persistent cells showed a significantly shorter latency time before robust proliferation resumed in the presence of DXR and PTX. Figures 5E and 5F show that levels of nROS and the development and subsiding of chemoresistance closely tracked and were inversely associated with H3.1 abundance. As shown in Figures 5E and 5F, Py230 cells displayed high levels of H3.1 at baseline. The declined in H3.1 protein abundance induced by DXR treatment corresponded to the increase in nROS levels as well as the emergence of a DXR-resistant population. Through the proliferation phase in the presence of DXR, H3.1 levels remained low, while nROS levels remained high. The activation of NLS-catalase expression suppressed both nROS and chemoresistance while rescuing basal levels of H3.1 histone. In addition to rescuing H3.1, NLS-catalase induction downregulated the canonical EMT transcription factor ZEB1 (Figures 5G and S5A) in both cell lines. To confirm that H3.1 depletion is a mechanistic driver of chemoresistance induced by H2O2 in the nucleus, additional experiments were performed in which the expression of the H2O2-resistant mutant H3.1(C96S) was induced instead of NLS-catalase in MCF10AER/vSrc cells. The results shown in Figures S5B and S5C indicate that the expression of H3.1(C96S) recapitulated the effects of NLS-catalase in suppressing the development or reversing acquired chemoresistance against DXR. These results confirm that H2O2-driven H3.1 depletion via Cys96 oxidation is involved in the evolution of resistance against multiple chemotherapeutic drugs.
Figure 5. nH2O2 promotes chemoresistance, tumorigenicity, and metastasis of mammary xenograft tumors.
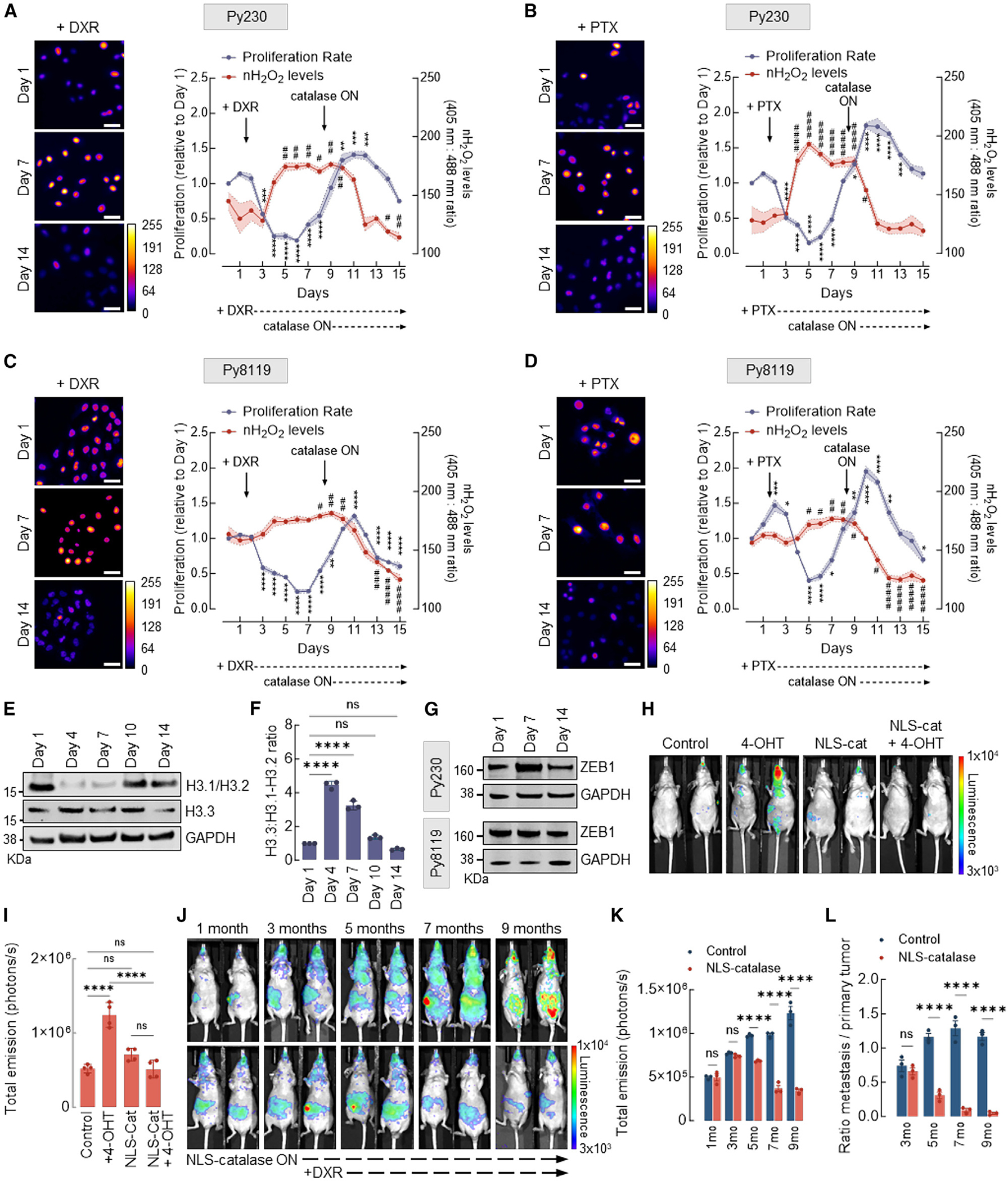
(A) Cell proliferation and levels of H2O2 in the nucleus of murine mammary carcinoma Py230 cells expressing inducible NLS-catalase and treated with 200 nM DXR. Oxidized (λex = 405 nm) and reduced (λex = 488 nm) roGFP2 signals were acquired, and the ratio oxidized:reduced was calculated using ImageJ (shown as a heatmap). Cell proliferation is relative to day 1. White bars represent 50 μm. Statistical significance was determined by 1-way ANOVA with post hoc Tukey test (proliferation and nH2O2 calculated separately). Lines and shadows represent mean ± SEM. **p < 0.01, ***p < 0.001, and ****p < 0.0001 (for proliferation) and #p < 0.05 and ##p < 0.01 (for nH2O2 levels). On the heatmap scale, 0 represents low ROS levels, whereas warmer colors toward the 255 mark represent relatively higher ROS levels.
(B) Cell proliferation and levels of H2O2 in the nucleus of Py230 cells expressing inducible NLS-catalase and treated with 10 nM PTX. Oxidized (λex = 405 nm) and reduced (λex = 488 nm) roGFP2 signals were acquired, and the ratio oxidized:reduced was calculated using ImageJ (shown as a heatmap). Cell proliferation is relative to day 1. White bars represent 50 μm. Statistical significance was determined by 1-way ANOVA with post hoc Tukey test (proliferation and nH2O2 calculated separately). Lines and shadows represent mean ± SEM. *p < 0.05, ***p < 0.001, and ****p < 0.0001 (for proliferation) and #p < 0.05 and ####p < 0.0001 (for nH2O2 levels). On the heatmap scale, 0 represents low ROS levels, whereas warmer colors toward the 255 mark represent relatively higher ROS levels.
(C) Cell proliferation and levels of H2O2 in the nucleus of Py8119 cells expressing inducible NLS-catalase and treated with 200 nM DXR. Oxidized (λex = 405 nm) and reduced (λex = 488 nm) roGFP2 signals were acquired, and the ratio oxidized:reduced was calculated using ImageJ (shown as a heatmap). Cell proliferation is relative to day 1. White bars represent 50 μm. Statistical significance was determined by 1-way ANOVA with post hoc Tukey test (proliferation and nH2O2 calculated separately). Lines and shadows represent mean ± SEM. **p < 0.01 and ****p < 0.0001 (for proliferation) and #p < 0.05, ##p < 0.01, ###p < 0.001, and ####p < 0.0001 (for nH2O2 levels). On the heatmap scale, 0 represents low ROS levels, whereas warmer colors toward the 255 mark represent relatively higher ROS levels.
(D) Cell proliferation and levels of H2O2 in the nucleus of Py8119 cells expressing inducible NLS-catalase and treated with 10 nM PTX. Oxidized (λex = 405 nm) and reduced (λex = 488 nm) roGFP2 signals were acquired, and the ratio oxidized:reduced was calculated using ImageJ (shown as a heatmap). Cell proliferation is relative to day 1. White bars represent 50 μm. Statistical significance was determined by 1-way ANOVA with post hoc Tukey test (proliferation and nH2O2 calculated separately). Lines and shadows represent mean ± SEM. *p < 0.05, **p < 0.01, and ****p < 0.0001 (for proliferation) and #p < 0.05, ##p < 0.01, and ####p < 0.0001 (for nH2O2 levels). On the heatmap scale, 0 represents low ROS levels, whereas warmer colors toward the 255 mark represent relatively higher ROS levels.
(E) Western blot analysis of H3.1/H3.2 and H3.3 in Py230 cells treated with 200 nM DXR at the indicated time points.
(F) H3.3:H3.1-H3.2 ratio quantification in (E). Statistical significance was determined by 1-way ANOVA with post hoc Tukey test. Bars represent mean ± SEM. ****p < 0.0001; ns, not significant.
(G) Western blot analysis of ZEB1 in Py230 cells (top) and Py8119 cells (bottom) treated with 200 nM DXR at the indicated different time points.
(H) Assessment of tumors in NU/J mice injected with 4-OHT-transformed MCF10AER/vSRC cells and expressing NLS-catalase. The injection of MCF10AER/vSrc cells (±4-OHT, ±NLS-catalase expression) was performed subcutaneously in the mammary fat pad, and the bioluminescent signal of tumors in representative animals is shown (13 weeks after injection). Luciferase signal was detected using high exposure for 120 s.
(I) Quantification of total emission in (H). Statistical significance was determined by 1-way ANOVA with post hoc Tukey test. Bars represent mean ± SEM. ****p < 0.0001; ns, not significant.
(J) Assessment of tumors in NU/J mice injected with 4-OHT-transformed MCF10AER/vSRC cells and expressing inducible NLS-catalase. The injection of 4-OHT-transformed MCF10AER/vSrc cells was performed subcutaneously in the mammary fat pad, and tumor growth and metastasis were measured monthly. The induction of NLS- catalase occurred after the third month, and the treatment with DXR was performed on a weekly basis after the fifth month. Luciferase signal was detected using high exposure for 120 s.
(K) Quantification of total emission in (J). Statistical significance was determined by 2-way ANOVA with post hoc Bonferroni test. Bars represent mean ± SEM. ****p < 0.0001; ns, not significant.
(L) Ratio metastasis:primary tumor of (J). Statistical significance was determined by 2-way ANOVA with post hoc Bonferroni test. Bars represent mean ± SEM. ****p < 0.0001; ns, not significant.
See also Figure S5.
nH2O2 promotes mammary xenograft tumorigenicity and metastasis
We next sought to examine the impact of nH2O2 on tumor xenograft behavior in vivo using luciferase-labeled MCF10AER/vSrc cells with or without stable expression of NLS-catalase. Briefly, cells were suspended in Matrigel containing 10 μM 4-OHT to induce v-Src expression and implanted in the inguinal mammary fat pad of immunocompromised NU/J mice. Although tumors developed as expected in mouse recipients of transformed MCF10AER/vSrc cells, the expression of NLS-catalase in the same cell line abrogated tumor formation (Figures 5H and 5I), indicating that quenching nH2O2 was sufficient to suppress tumorigenicity in vivo.
Since quenching nH2O2 with NLS-catalase inhibited tumor formation, fully transformed MCF10AER/vSrc cells expressing luciferase were engineered to co-express NLS catalase under the control of an inducible TetON promoter. When xenografted into the inguinal mammary fat pad of female NU/J mice, rapidly proliferating MCF10AER/vSrc tumors developed in all of the recipients, followed by progression to metastatic disease within 3 months. After 3 months, half of the mice received doxycycline to activate NLS-catalase expression. In the group receiving doxycycline, metastatic lesions entered remission by 2 months after activation of NLS-catalase expression in most of the animals (Figures 5J–5L). Furthermore, NLS-catalase expression both significantly impeded primary tumor development (Figures 5H and 5I) and had a dramatic effect in reducing the number and progression of metastatic lesions (Figures 5J–5L), consistent with a potentially adaptive function for nH2O2 that may be required to sustain metastatic colonies growing in heterologous organ sites. To test, however, whether a combination of NLS-catalase induction and chemotherapy could also eliminate established primary xenograft tumors, however, treatments with DXR were initiated after the fifth month. Results shown in Figures 5J–5L indicate that combination with DXR significantly reduced established primary tumors while maintaining mice virtually metastasis-free. Therefore, the data in Figure 5 support a model in which nH2O2 has specific genome regulatory activities with clinical relevance to the adaptive transcriptomic and phenotypic reprogramming of tumor cells in vivo. In addition, suppressing nH2O2 in vivo may be an attainable strategy to treat chemoresistant metastatic disease.
DISCUSSION
In eukaryotes, the progressive accumulation of H3.3 over canonical H3.1-H3.2 represents an indelible hallmark of aging.32 The accumulation of H3.3 changes patterns of DNA methylation that have been used to accurately determine the chronologic age of an organism.33 Taken together, these observations suggest that H3.3 accumulation is mechanistically linked with the regulation of expression of aging-associated genes, some of which are directly involved in triggering or accelerating the progression of diseases more prevalent with aging (e.g., cancer).34–36
Another hallmark of aging is progressive metabolic/mitochondrial dysfunction associated with an increase in the production and/or a decrease in the detoxification of ROS.37 Both an increase in ROS and the exchange of histone variants have also been shown to occur in cancer and to be involved in its progression toward metastasis.5,26 In 2020, Desai et al.38 showed that mitochondria and the nucleus are directly connected and communicate, at least in part, via the transfer of mitochondria-derived ROS. Consistently, we found that transformed cancer cells displaying more aggressive behavior existed in states characterized by higher steady-state levels of nROS. To recapitulate this effect and understand its consequences for the regulation of gene expression, a system to directly deliver H2O2 to the nucleus was developed. Experiments using this engineered DAO system confirmed that ROS generated in the nucleus caused the loss of H3.1-H3.2 histones, changing the H3.1-H3.2/H3.3 ratio to ultimately promote chromatin decompaction and the expression of genes normally associated with heterochromatic domains, including EMT genes. H3.1 replacement by H3.3 preceded and was required to induce EMT by TGF-β or triggered directly via the oxidation of H3.1-Cys96 by H2O2 generated in the nucleus. Mutation of the redox-active Cys96 residue, which essentially converted H3.1 into H3.2, conferred resistance to ROS-induced as well as TGF-β-induced loss of canonical histones and EMT, confirming that Cys96 oxidation is a critical event in H3.1 destabilization and chromatin remodeling associated with cellular phenotypic plasticity acquisition. Consistently, expressing an H3.1 mutant lacking Cys96 or quenching ROS with catalase directed to the nucleus (NLS-catalase) suppressed H3 variant exchange induced by ROS as well as ROS-induced EMT gene expression. In vivo, suppressing nROS inhibited tumorigenesis and tumor growth and strongly suppressed metastasis. Surprisingly, suppression of nROS in established metastatic lesions drove them into remission, indicating that nROS are not only critical to enable the metastatic colonization but also to support the viability and growth of the metastatic colonies in heterotypic organs. Hence, by demonstrating that an increase in nROS structurally regulates EMT gene accessibility, likely via the oxidation of redox-sensitive H3.1, our results mechanistically connect several hallmark features common to aging, metabolic changes occurring with aging, and cancer, including (1) the enrichment of H3.3 over H3.1, (2) an increase in steady-state ROS, (3) epigenetic activation of EMT and stemness genes shown, here, to be induced by and dependent on mitochondrial H2O2 reaching the nucleus, and (4) increased cellular resistance to chemotherapeutics and metastatic potential that could be, at least partially, related to epigenetic reprogramming induced by changes in the redox state of the nucleus, as shown here and in other studies.39 Taken together, our results suggest that mitochondrial-ROS-driven chromatin remodeling is part of the survival response to acute metabolic stress that unlocks a larger range of transcriptional possibilities tumor cells can use to accelerate the progression to malignancy. They also suggest that the expansion of the cells’ transcriptional potential via oxidative chromatin remodeling spurs significant clonogenic diversity, a problem that is difficult to circumvent in clinical oncology. Therefore, there is significant therapeutic potential in the manipulation of mechanisms based on redox epigenetics for the development of combined modality approaches for cancer therapy.27
Limitations of the study
One limitation of our study relates to the difficulty of discerning between H3.1 and H3.2 variants using the available technologies. For instance, the anti-H3.1-H3.2 antibody used in this study is not capable of discerning between these variants that differ by only one atom. Hence, it was not possible to quantify residual H3.1 relative to H3.2 in studies relying on immune detection using antibodies. In addition, it is likely that nH2O2 affects H3-independent mechanisms of transcription, including the direct oxidation of DNA segments, transcription factors, and transcription coactivators/co-repressors, adding additional layers of complexity, not entertained here, to the redox regulation of gene expression.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Marcelo G. Bonini (marcelo.bonini@northwestern.edu).
Materials availability
Plasmids and cell lines generated in this study will be available upon request.
Data and code availability
Bulk RNAseq and ChIPseq have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| 8-oxo-dG | R&D Systems | Cat# 4354-MC-050; RRID: AB_1857195 |
| Alexa Fluor 647 | Invitrogen | Cat# A28181; RRID: AB_2536165 |
| ATM | Cell Signaling Technology | Cat# 92356; RRID: AB_2800184 |
| DYKDDDDK (FLAG Tag) | Cell Signaling Technology | Cat# 14793; RRID: AB_2572291 |
| E-Cadherin | Cell Signaling Technology | Cat# 3195; RRID: AB_2291471 |
| Fibronectin | Cell Signaling Technology | Cat# 26836; RRID: AB_2924220 |
| GAPDH | Cell Signaling Technology | Cat# 97166; RRID: AB_2756824 |
| HA-Tag | Cell Signaling Technology | Cat# 3724; RRID: AB_1549585 |
| Histone H2A.X | Cell Signaling Technology | Cat# 7631; RRID: AB_10860771 |
| Histone H3.1/H3.2 | Millipore | Cat# ABE154; RRID: AB_2811170 |
| Histone H3.3 (H3F3A) | Millipore | Cat# 09-838; RRID: AB_10845793 |
| IRDye Goat anti-Mouse | LI-COR Biosciences | Cat# 925-68070; RRID: AB_10953628 |
| IRDye Goat anti-Rabbit | LI-COR Biosciences | Cat# 92632211; RRID: AB_621843 |
| Normal Rabbit IgG | Cell Signaling Technology | Cat# 2729; RRID: AB_1031062 |
| phospho-ATM (Ser1981) | Cell Signaling Technology | Cat# 13050; RRID: AB_2798100 |
| phospho-Histone H2A.X (Ser139) | Cell Signaling Technology | Cat# 80312; RRID: AB_2799949 |
| SOX9 | Cell Signaling Technology | Cat# 82630; RRID: AB_2665492 |
| ZEB1 | Cell Signaling Technology | Cat# 3396; RRID: AB_1904164 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| DH5α competent cells | Invitrogen | 18265017 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| 4-hydroxytamoxifen (4-OHT) | Sigma Aldrich | H7904 |
| Ampicillin | Sigma Aldrich | A5354 |
| Blasticidin | Gibco | A1113903 |
| Bovine Serum Albumin (BSA) | Sigma Aldrich | A9648 |
| Catalase from bovine liver | Sigma Aldrich | C100 |
| Charcoal stripped FBS | Sigma Aldrich | F6764 |
| Cholera toxin | Sigma Aldrich | C8052 |
| D-Alanine | Sigma Aldrich | A7377 |
| DCP-Bio1 | Kerafast | EE0028 |
| DMEM/F12 medium (no phenol red) | Gibco | 21041025 |
| DPBS | Gibco | 14190144 |
| Doxorubicin hydrochloride | Sigma Aldrich | D1515 |
| Doxycycline hydrochloride | Sigma Aldrich | D3072 |
| EGF | Sigma Aldrich | SRP3027 |
| Epon resine | Electron Microscopy Sciences | 14910 |
| Ethanol | Sigma Aldrich | E7023 |
| F-12K | ATCC | 30-2004 |
| Formaldehyde | Sigma Aldrich | 28908 |
| FBS | Sigma Aldrich | F2442 |
| Geneticin | Gibco | 10131-027 |
| Glycine | Sigma Aldrich | G8898 |
| Gluteraldehyde | Sigma Aldrich | G6257 |
| Histone H3.1 human | New England Biolabs | M2503S |
| Histone H3.2 human | New England Biolabs | M2506S |
| Histone H3.3 human | New England Biolabs | M2507S |
| Hydrocortisone | Sigma Aldrich | H0135 |
| Hygromycin | Gibco | 10687010 |
| Insulin | Sigma Aldrich | I3536 |
| Intercept antibody diluent | LI-COR | 92765001 |
| Intercept blocking buffer | LI-COR | 92760001 |
| Lead citrate Reynold’s stain | Electron Microscopy Sciences | 22410-01 |
| Lipofectamine 3000 | Thermo Fisher Scientific | L3000015 |
| Magna ChIP A/G kit | Millipore | CS204457 |
| Matrigel matrix | Corning | 356237 |
| MEM medium | Gibco | 11090081 |
| MITO+ serum extender | Corning | 355006 |
| MSTP | Axon MedChem | 2876 |
| Nitrocellulose membrane | BIO-RAD | 1620112 |
| Novex MES buffer | Invitrogen | B0002 |
| NuPAGE 4–12% Bis-Tris gels | Invitrogen | NP0335 |
| NuPAGE LDS sample buffer | Invitrogen | NP0007 |
| Osmium tetroxide | Sigma Aldrich | 201030 |
| Paclitaxel | Sigma Aldrich | T7191 |
| PFA | Sigma Aldrich | F8775 |
| Penicillin/Streptomycin | Gibco | 15140122 |
| Protease inhibitor cocktail | Roche | 04693132001 |
| Puromycin | Gibco | A1113802 |
| RediJect D-Luciferin bioluminescent substrate | PerkinElmer | 770504 |
| RIPA buffer | Thermo Fisher Scientific | 89901 |
| Sodium cacodylate buffer | Electron Microscopy Sciences | 11650 |
| TGFβ | Sigma Aldrich | H8541 |
| Uranyl acetate | Electron Microscopy Sciences | 22400 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Comet Assay Kit | Trevigen | 4250-050-K |
| Amplex Red Assay Kit | Thermo Fisher Scientific | A22188 |
| RNeasy Kit | Qiagen | 74104 |
| High Capacity cDNA Reverse Transcription Kit | Applied Biosystem | 4368814 |
| Chromatin Accessibility Assay Kit | Abcam | ab185901 |
| Magna ChIP A/G Kit | Millipore | 1710085 |
| Fast SYBR Green Master Mix | Applied Biosystems | 4385612 |
|
| ||
| Deposited data | ||
|
| ||
| RNAseq | This paper | GEO: GSE255598 |
| ChIPseq | This paper | GEO: GSE255615 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| BT20 | ATCC | HTB-19 |
| BT474 | ATCC | HTB-20 |
| HEK293T/7 | ATCC | CRL-11268 |
| MCF7 | ATCC | HTB-22 |
| MCF10A | ATCC | CRL-10317 |
| MCF10AER/vSrc | Iliopoulos et al. | N/A |
| MDA-MB231 | ATCC | HTB-26 |
| Py230 | ATCC | CRL-3279 |
| Py8119 | ATCC | CRL-3278 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| NU/J mice | The Jackson Laboratory | Cat#: 002019; RRID: IMSR_JAX:002019 |
|
| ||
| Oligonucleotides | ||
|
| ||
| RT-qPCR primers | This paper | Table S1 |
| Chromatin accessibility primers | This paper | Table S1 |
| ChIP-qPCR primers | This paper | Table S1 |
|
| ||
| Recombinant DNA | ||
|
| ||
| pEIGW roGFP2-ORP1 | Addgene | Cat#: 64993; RRID: Addgene_64993 |
| pLenti CMV GFP Hygro | Addgene | Cat#: 17446; RRID: Addgene_17446 |
| pLVX-Tet3G blasticidin | Addgene | Cat#: 128061; RRID: Addgene_128061 |
| pLenti CMV GFP Blast | Addgene | Cat# 17445; RRID: Addgene_17445 |
| pLenti CMV GFP Zeo | Addgene | Cat#: 17449; RRID: Addgene_17449 |
| p8.91 | Addgene | Cat#: 187441 RRID: Addgene_187441 |
| pCMV VSV-G | Addgene | Cat#: 8454 RRID: Addgene_8454 |
| pLenti NLS-roGFP2-ORP1 | This paper | N/A |
| pLenti mito-roGFP2-ORP1 | This paper | N/A |
| pLenti NLS-DAO | This paper | N/A |
| pLenti NLS-catalase | This paper | N/A |
| pLenti Inducible TET-ON NLS-catalase | This paper | N/A |
| pLenti mito-catalase | This paper | N/A |
| pLenti H3.1(WT)-FLAG | This paper | N/A |
| pLenti H3.1(C96S)-FLAG | This paper | N/A |
| pLenti H3.3(WT)-HA | This paper | N/A |
| pLenti Inducible TET-ON H3.1(C96S)-FLAG | This paper | N/A |
| pLenti Inducible TET-ON H3.3(WT)-HA | This paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Aura | Spectral Instruments Imaging | https://spectralinvivo.com/software/ |
| Bowtie2 (2.2.9) | Langmead et al.40 | https://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Fiji | Schindelin et al.41 | https://imagej.net/downloads |
| GSEA | Subramanian et al.42 | http://software.broadinstitute.org/gsea/index.jsp |
| GraphPad Prism 10 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Homer (4.11) | Heinz et al.43 | http://homer.ucsd.edu/homer/index.html |
| Metascape | Zhou et al.44 | https://metascape.org/gp/index.html#/main/step1 |
| VolcaNoseR | Goedhart et al.45 | https://github.com/JoachimGoedhart/VolcaNoseR |
|
| ||
| Other | ||
|
| ||
| Eagle 4k HR 200kV CCD camera | FEI Company | N/A |
| IVIS in vivo imager | LAGO | N/A |
| Lionheart FX microscope | Biotek | N/A |
| Nikon A1R confocal microscope | Nikon | N/A |
| Novaseq 6000 | Illumina | N/A |
| Odyssey FC (LI-COR) | LI-COR | N/A |
| QuantStudio 6 Flex | Applied Biosystems | N/A |
| Tecnai Spirit G2 transmission electron microscope | FEI Company | N/A |
| Ultracut UC7 Ultramicrotome | Leica Microsystems | N/A |
| Eagle 4k HR 200kV CCD camera | FEI Company | N/A |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Animals
NU/J mice strains were purchased from The Jackson Laboratory. All animal protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of Northwestern University and followed the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. 6~8 weeks female mice were used in this study and were housed in specific pathogen-free facilities under a 12-h light/dark cycle, controlled temperature, and with a regular feeding and cage-cleaning schedule.
Cell lines
Human-derived cell lines BT20, BT474, MCF7, MCF10A, MDA-MB231 and HEK293T/17 were obtained from ATCC. Human-derived cell line MCF10AER/vSrc was generously provided by Dr. Kevin Struhl from Harvard University, Cambridge, MA, Mouse-derived cell lines Py230 and Py8119 were obtained from ATCC.
MCF7, BT474, BT20, MDA-MB231, and HEK293T/17 cells were cultured in MEM medium (Gibco) supplemented with 10% FBS (Sigma Aldrich) and 1% penicillin-streptomycin (Gibco). MCF10A cells were cultured in DMEM/F12 (no phenol red) medium (Gibco) supplemented with 10% FBS (Sigma Aldrich), 20 ng/mL EGF (Sigma Aldrich), 10 μg/mL insulin (Sigma Aldrich), 0.5 μg/mL hydrocortisone (Sigma Aldrich), 0.1 μg/mL cholera toxin (Sigma Aldrich), and 1% penicillin-streptomycin (Sigma Aldrich). MCF10AER/vSrc cells were cultured in DMEM/F12 medium (Gibco) supplemented with 5% charcoal stripped (Sigma Aldrich), 20 ng/mL EGF (Sigma Aldrich), 10 μg/mL insulin (Sigma Aldrich), 0.5 μg/mL hydrocortisone (Sigma Aldrich), 0.1 μg/mL cholera toxin (Sigma Aldrich), and 1% penicillin-streptomycin (Sigma Aldrich). Py230 cells were cultured in F-12K medium supplemented with 5% FBS (Sigma Aldrich), 0.1% MITO+ serum extender (Corning), and 1% penicillin-streptomycin (Gibco). Py8119 cells were cultured in F-12K medium supplemented with 5% FBS (Sigma Aldrich) and 1% penicillin-streptomycin (Gibco). All cell lines were maintained in a humidified incubator at 37°C with 5% CO2 and atmospheric oxygen. Cells were regularly tested for mycoplasma.
METHOD DETAILS
Plasmids, lentivirus and viral transduction
NLS-roGFP2-Orp1 and mito-roGFP2-Orp1 (puromycin as selection mark) were obtained from cloning the c-myc sequence or an MTS sequence respectively into the pEIGW roGFP2-ORP1 vector (Addgene). NLS-DAO plasmid (geneticin as selection mark) was obtained from cloning the c-myc sequence into the DAAO vector (a generous gift from Thomas Michel, Harvard University). NLS-catalase and mito-catalase (both hygromycin as selection mark) were obtained from cloning catalase and c-myc or MTS sequences respectively into pLenti CMV GFP Hygro vector (Addgene). Inducible TET-ON NLS-catalase was obtained from cloning catalase and c-myc, and hygromycin selection marker sequences into pLVX-Tet3G blasticidin vector (Addgene) and removing blasticidin resistance sequence. H3.1(WT)-FLAG and H3.1(C96S)-FLAG were obtained from cloning H3.1(WT) or H3.1(C96S) and FLAG tag sequences into pLenti CMV GFP Blast vector (Addgene). H3.3(WT)-HA was obtained from cloning H3.3(WT) and HA-tag sequences into pLenti CMV GFP Zeo vector (Addgene). Inducible TET-ON H3.1(C96S)-FLAG was obtained from cloning H3.1(WT) or H3.1(C96S) and FLAG tag sequences into pLVX-Tet3G blasticidin vector (Addgene). Inducible TET-ON H3.3(WT)-HA was obtained from cloning H3.3(WT) and HA-Tag sequences into pLVX-Tet3G blasticidin vector (Addgene).
For lentivirus production, HEK293T/17 cells in phenol red free OPTI-MEM (Gibco) were co-transfected with a construct of interest and packaging plasmids Gag-Pol 8.91 (Addgene) and VSV-G (Addgene) using Lipofectamine 3000 (Thermo Fisher Scientific). Cells were incubated at 37°C and after 24h and 48h lentivirus-rich medium was collected, centrifuged (500 × g, 5 min). Supernatant was collected, filtered (0.45 μm) and used to transduction. Transduced cells were cultured in the appropriate medium for 48h and selected with antibiotics.
MCF10AER/vSrc cells malignant transformation
MCF10AER/vSrc cells contain a ER/v-Src construct, which consists of a derivative of the v-Src oncogene fused to the ligand-binding domain of the estrogen receptor (ER).46 To prevent the activation of v-Src by ER ligands, cells were grown in media containing charcoal stripped FBS devoid of phenol red. Activation of v-Src expression downstream of ER was achieved by incubating cells with 1 μM 4-OHT for 7 days.
D-Ala treatment
D-Ala treatment was performed in different experiments and consisted in replacing the medium with new, fresh medium containing the indicated concentration of D-Ala. Control cells received fresh medium only.
Oxidized and reduced roGFP2-Orp1 detection and signal processing
After specific treatments, cells were imaged in Nikon A1R confocal microscope (Nikon) or in Lionheart FX microscope (Biotek). roGFP2-Orp1 was excited sequentially at 405 nm and 488 nm and the emission was recorded at 525 nm. The generated images were analyzed using FIJI ImageJ. Each channel was converted to 8-bit and Median (radius = 2 pixels) and Gaussian blur (radius = 2 pixels) were applied. The background was subtracted and a threshold was set to avoid artifacts. Only the nuclear or mitochondrial area were considered for the analysis. Oxidation levels were then determined on a pixel by pixel basis dividing the signal emitted for each channel (405 nm/488 nm) using Imaging Expressing Parser. Final ratiometric values were obtained using Analyzing particles (size: 10 μm2 - infinity; circularity: 0.50–1.00). Obtained values were plotted in GraphPad Prism 10 and a heatmap (Lookup table: Fire) was created for visualization.
8-Oxo-dG detection
MCF10AER/vSrc cells were plated in black bottom 96-well plates and treated with different concentrations of D-Ala or 500μM H2O2. 4h after treatment with D-Ala or 30 min after treatment with H2O2, cells were fixed with 4% PFA (Sigma Aldrich) for 10 min at room temperature and permeabilized with ethanol (Sigma Aldrich) for 3 min. Then, cells were washed 3x with DPBS pH 7.4 (Gibco), blocked with 5% (w/v) bovine serum albumin (BSA) (Sigma Aldrich) for 1h at room temperature. Samples were incubated with anti-8-oxo-dG overnight at 4°C (R&D Systems), followed by staining with secondary antibody Alexa Fluor 647 (Invitrogen). 8-oxo-dG fluorescence images and oxidized/reduced roGFP2-Orp1 signals were acquired in Lionheart FX microscope (Biotek).
Comet assay
Comet assay was performed using the Comet Assay Kit (Trevigen) according to the instructions provided by the manufacturer. Briefly, MCF10AER/vSrc cells were seeded in 6-well plates and 4h hours after treatment with 10 nM D-Ala or 30 min after treatment with 500 μM H2O2 for 30 min they were washed, detached and resuspended in DPBS (Gibco). Cell suspension was mixed with LM-agarose at a ratio of 1:10 (v/v), transferred onto slides, and lysed at 4°C. The slides were treated with an alkaline unwinding solution for 60 min and placed on a horizontal electrophoresis unit filled with fresh buffer alkaline unwinding solution. Electrophoresis was performed at 20V for 35 min at 4°C in the dark and subsequentially staining. Images were collected under automatic microscope Lionheart FX (Biotek). The tail moment and % DNA in tail were analyzed using the plug-in Open Comet in FIJI ImageJ.
Amplex red assay
Extracellularly-released H2O2 was measured using an Amplex Red Hydrogen Peroxide/Peroxidase Assay Kit (Thermo Fisher Scientific), according to the manufacturer protocol. Briefly, MCF10AER/vSrc cells were plated in black bottom 96-well plates and treated with different concentrations of D-Ala (10 nM, 100 nM, 10μM, 100μM and 1mM). 50 μL Amplex Red reagent with 0.2 units/mL horseradish peroxidase was added to samples and fluorescence was measured for 2h at 37°C (λex = 530 nm, λem = 590 nm). The mean reading was plotted on a simultaneously prepared H2O2 standard curve.
Western blot analysis
Cells were lysed in cold RIPA buffer (Thermo Fisher Scientific) supplemented with protease inhibitor cocktail (Roche) for 30 min at 4°C. Equal amounts of total protein (30–50 μg) were separated in 4–12% Bis-Tris polyacrylamide gels (Invitrogen) and transferred to a 0.22 μm nitrocellulose membrane (BIO-RAD). Membranes were blocked for 1h at room temperature with Intercept Blocking Buffer (LI-COR) and incubated with appropriate primary antibodies overnight at 4°C (1:1000 in Intercept Antibody Diluent (LI-COR)). Species-specific antibodies (IRDye Goat anti-Mouse or IRDye Goat anti-Rabbit - LI-COR) were used for labeling after primary antibody incubation (1:10000 in Intercept Antibody Diluent (LI-COR)). Signal was detected using an Odyssey FC (LI-COR) imaging station. Quantification was performed using FIJI ImageJ.
RNA extraction and RT-qPCR
Total RNA was extracted using RNeasy Kit (Qiagen) according to the manufacturer’s instructions. cDNA was synthesized using High Capacity cDNA Reverse Transcription kit (Applied Biosystem). Quantitative PCR was performed on an Applied Biosystems QuantStudio 6 Flex Real-Time PCR System using Fast SYBR Green Master Mix (Applied Biosystems). The relative gene expression was calculated using the 2−ΔΔCt method with GAPDH as endogenous control for normalization. Primers used for RT-qPCR are listed in the Table S1.
RNA-seq and data analysis
RNA from 5 biological replicates/condition was isolated as described above 4h or 24 h after treatment with 10 nM D-Ala. Sequencing libraries were generated using Illumina Novaseq platform. Sample quality, library complexity, and alignment statistics were checked. The sequencing reads were aligned against the reference human genome hg38. Differentially expressed genes (DEG) were identified using EdgeR with FC cut-off = 2 in expression compared to control, by pairwise approach. Heatmap of representative genes was generated using GraphPad Prism 10, after normalization of expression levels of DEGs, while volcano plots were generated using VolcaNoseR (https://huygens.science.uva.nl/VolcaNoseR/). GO analysis was performed using the list of DEGs uploaded to Metascape against reference species (Homo sapiens). GSEA (Hallmark genes analysis) was performed using the GSEA software.
Chromatin immunoprecipitation
Approximately 5 × 106 cells were fixed with 1% methanol-free formaldehyde solution (Thermo Scientific) for 10 min at RT. Then, reaction was quenched with addition of 125 mM glycine (Sigma Aldrich) for 5 min. Chromatin isolation and immunoprecipitation were carried out using Magna ChIP A/G kit (Millipore). Chromatin was isolated from nuclei and sonicated in Diagenode Bioruptor Pico for 10 cycles of 30 s with intervals of 30 s at 4°C to generate DNA fragments with size range of 200–600 bp. Sheared chromatin equivalent to 2 × 106 cells was diluted 1:10 in ChIP Dilution Buffer and protease inhibitor cocktail and incubated for 16h with 20 μL Magnetic Protein A/G Beads (Millipore) and 5 μL antibodies. The antibodies used were H3.1-H3.2 (Millipore), H3.3 (Millipore), FLAG Tag (Cell Signaling), HA-TAG (Cell Signaling) or Normal Rabbit IgG (Cell Signaling). Immunoprecipitated DNA fragments were eluted and purified according to manufacturer’s instructions.
ChIP-seq and data analysis
One sample containing pooled DNA fragments of 3 biological replicates was tested for concentration, integrity and purity. Concentration, sample integrity and purity were checked. ChIP’s DNA was subjected to end-repair and then was 3′ adenylated. Adaptors were ligated to the ends of these 3′ adenylated fragments. DNA fragments were amplified with adaptors from previous step. PCR products were purified. The double stranded PCR products were heat denatured and circularized by the splint oligo sequence and the single strand circle DNA (ssCir DNA) were formatted as the final library. Library was qualified and amplified to make DNA nanoball (DNB). DNBs were loaded into the patterned nanoarray, and single end 50 bases reads were performed. The quality of reads was evaluated using FastQC. Reads were trimmed from the 3′ ends using cutadpat and aligned using Bowtie 2 (version 2.2.9) with default parameters to the human genome (hg38). Reads were then analyzed with Homer (version 4.11) to call H3.1-H3.2 and H3.3 binding regions and to identify regions of differential binding.
ChIP-qPCR
ChIP products were analyzed by qPCR using Fast SYBR Green (Applied Biosystems) on a Quant Studio 6 Flex PCR system (Applied Biosystems). Each ChIP sample was normalized by its respective input’s adjusted , where
To express ChIP enrichment in percentage of input, the formula used was
A list of primers used for ChIP-qPCR is shown in Table S1.
Transmission electron microscopy
MCF10AER/vSrc cells were treated with 10 nM D-Ala and after 4h they were fixed in mixture of 2.5% glutaraldehyde (Sigma Aldrich) and 2% PFA (Sigma Aldrich) in 0.1 M cacodylate buffer (Electron Microscopy Sciences) overnight at 4°C. After post-fixation in 1% osmium tetroxide (Sigma Aldrich) and 3% uranyl acetate (Electron Microscopy Sciences), cells were dehydrated in series of ethanol, embedded in Epon resin (Electron Microscopy Sciences) and polymerized for 48 h at 60°C. Ultrathin sections were made using Ultracut UC7 Ultramicrotome (Leica Microsystems) and contrasted with 3% uranyl acetate (Electron Microscopy Sciences) and Lead citrate Reynold’s stain (Electron Microscopy Sciences). Samples were imaged using a FEI Tecnai Spirit G2 transmission electron microscope (FEI Company) operated at 80 kV. Images were captured by Eagle 4k HR 200kV CCD camera (FEI Company).
EMT induction with TGFb
MCF10A cells with or without expression of NLS-catalase or mito-catalase were treated with 10 ng/mL TGFβ (Sigma) every 2 days for a total of 14 days. Cells were then prepared for western blot analysis with specific antibodies.
Cysteine oxidation detection using DCP-Bio1
Recombinant human histones H3.1, H3.2 and H3.3 (New England Biolabs) were incubated with H2O2 in a 1:5 (histone:H2O2) proportion for 30 min at 37°C. After H2O2 incubations, histones were incubated with DPBS (Gibco) containing 1 mM DCP-Bio1 (Kerafast), a biotin-conjugated dimedone-derivative that labels oxidized Cys-SOH sulfenic acid residues, 10 mM MSTP thiol blocker (Axon MedChem), and 200 u/mL catalase from bovine liver (Sigma Aldrich) on ice for 30 min. Samples were then incubated with Alexa Fluor 647-conjugated streptavidin overnight at 4°C. Recombinant histones and subsequently separated in polyacrylamide gels, transferred to nitrocellulose membranes and labeled with specific antibodies. Signal was detected using an Odyssey FC (LI-COR) imaging station.
Chromatin accessibility assay
Chromatin accessibility was analyzed using Chromatin Accessibility Assay Kit (Abcam) according to the manufacturer’s instructions. MCF10AER/vSrc cells treated with D-Ala in different time points were lysed and chromatin was extracted. Chromatin was thereafter digested using a nuclease mix and the purified DNA was analyzed by qPCR using specific primers (Table S1). Results were calculated by fold enrichment using a ratio of amplification efficiency of nuclease-treated samples over untreated nuclease-free control samples.
Drug resistance studies
Py230, Py8119 cells were expressing inducible TET-ON NLS-catalase were treated with 200 nM doxorubicin (DXR) (Sigma Aldrich) or 10 nM paclitaxel (PTX) (Sigma Aldrich). The expression of NLS-catalase occurred by treating the cells with 100 ng/mL doxycycline (Sigma Aldrich). The redox state of the nucleus and cell death/proliferation were measured daily using the Lionheart FX microscope (Biotek). Cells were collected in specific time points for western blot analysis of histone variants and the EMT marker ZEB1. Similar experiment was performed with MCF10AER/vSrc cells expressing inducible TET-ON H3.1(C96S).
Animal studies
All mouse experimentation was conducted in accordance with standard operating procedures approved by the Institutional Animal Care and Use Committee of our Institution. NU/J mice were acquired from The Jackson Laboratory. For subcutaneous xenograft, 25 μL medium containing 1 × 106 luciferase-expressing MCF10AER/vSrc cells (control or 4-OHT transformed, with or without NLS-catalase expression) were mixed with 75 μL Matrigel matrix (BD Bioscience) and injected into the inguinal mammary fat pad of the female mice at 6 to 8 weeks of age. Imaging of grafted tumors was performed using an IVIS in vivo imager (LAGO) after intraperitoneal injection of RediJect D-Luciferin Bioluminescent Substrate (PerkinElmer) (150 mg/kg) 13 weeks after cells injection. Luminescent intensity was acquired using high exposition for 120s and total emission (photons/s) was calculated. Similarly, NU/J animals were injected with transformed MCF10AER/vSrc cells expressing NLS-catalase under an inducible TET-ON promoter. The expression of NLS-catalase was induced using doxycycline. Animals were treated with 5 mg/kg Doxorubicin (DXR) after the fifth month of the injection. Imaging of grafted tumors was performed monthly using an IVIS in vivo imager (LAGO) after intraperitoneal injection of RediJect D-Luciferin Bioluminescent Substrate (PerkinElmer) (150 mg/kg).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed with GraphPad Prism 10 by using t test, One-way Anova with Tukey post hoc test or two-way ANOVA with Bonferroni post hoc test. Data with a normal distribution are expressed as the mean ± SEM. Experiments were replicated three to five times unless otherwise indicated. For western blots and imaging, representative data are shown. A value of p < 0.05 was considered statistically significant. The following annotation was employed: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, #p < 0.05, ##p < 0.01, ###p < 0.001, ####p < 0.0001.
Supplementary Material
Highlights.
Cysteine oxidation is a functional modification of the H3.1 histone variant
H3.1Cys96 oxidation promotes its exchange by H3.3
H3.1Cys96 oxidation promotes multidrug resistance in breast cancer
H3.1Cys96 modification by ROS promotes metastasis
ACKNOWLEDGMENTS
This project was supported by National Cancer Institute grants R01CA216882 (to M.G.B.), U54268084 and U54261694 (to V.B.), R01CA225002 (to V.B.); National Institute of Environmental Health Sciences grants R01ES035723 and R01ES028149 (both to M.G.B.); National Institute of Allergy and Infectious Diseases grant R01AI131267 (to M.G.B.); National Heart, Lung and Blood Institute grants R01HL163820 (to M.G.B.) and P01HL149620 (to S.R.); and National Institute of Diabetes and Digestive and Kidney Diseases grant R01DK134064 (to S.R.). We are also grateful for the financial support from the Lefkofsky Foundation Innovator Award (to M.G.B. and V.B.). We are also grateful to Northwestern University Center for Advanced Microscopy (CAM), Quantitative Data Sciences Core (QDSC) and NUSeq for technical assistance.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.113897.
REFERENCES
- 1.Morris SA (2019). The evolving concept of cell identity in the single cell era. Development 146. 10.1242/dev.169748. [DOI] [PubMed] [Google Scholar]
- 2.Bussotti G, Piel L, Pescher P, Domagalska MA, Rajan KS, Cohen-Chalamish S, Doniger T, Hiregange DG, Myler PJ, Unger R, et al. (2021). Genome instability drives epistatic adaptation in the human pathogen Leishmania. Proc. Natl. Acad. Sci. USA 118, e2113744118. 10.1073/pnas.2113744118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jain SU, Khazaei S, Marchione DM, Lundgren SM, Wang X, Weinberg DN, Deshmukh S, Juretic N, Lu C, Allis CD, et al. (2020). Histone H3.3 G34 mutations promote aberrant PRC2 activity and drive tumor progression. Proc. Natl. Acad. Sci. USA 117, 27354–27364. 10.1073/pnas.2006076117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao Y, and Garcia BA (2015). Comprehensive Catalog of Currently Documented Histone Modifications. Cold Spring Harbor Perspect. Biol. 7, a025064. 10.1101/cshperspect.a025064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gomes AP, Ilter D, Low V, Rosenzweig A, Shen ZJ, Schild T, Rivas MA, Er EE, McNally DR, Mutvei AP, et al. (2019). Dynamic Incorporation of Histone H3 Variants into Chromatin Is Essential for Acquisition of Aggressive Traits and Metastatic Colonization. Cancer Cell 36, 402–417.e13. 10.1016/j.ccell.2019.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McKittrick E, Gafken PR, Ahmad K, and Henikoff S (2004). Histone H3.3 is enriched in covalent modifications associated with active chromatin. Proc. Natl. Acad. Sci. USA 101, 1525–1530. 10.1073/pnas.0308092100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Venkatesh S, and Workman JL (2015). Histone exchange, chromatin structure and the regulation of transcription. Nat. Rev. Mol. Cell Biol. 16, 178–189. 10.1038/nrm3941. [DOI] [PubMed] [Google Scholar]
- 8.Stroud H, Otero S, Desvoyes B, Ramírez-Parra E, Jacobsen SE, and Gutierrez C (2012). Genome-wide analysis of histone H3.1 and H3.3 variants in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 109, 5370–5375. 10.1073/pnas.1203145109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin CJ, Conti M, and Ramalho-Santos M (2013). Histone variant H3.3 maintains a decondensed chromatin state essential for mouse preimplantation development. Development 140, 3624–3634. 10.1242/dev.095513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Song Y, Seol JH, Yang JH, Kim HJ, Han JW, Youn HD, and Cho EJ (2013). Dissecting the roles of the histone chaperones reveals the evolutionary conserved mechanism of transcription-coupled deposition of H3.3. Nucleic Acids Res. 41, 5199–5209. 10.1093/nar/gkt220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Postberg J, Forcob S, Chang WJ, and Lipps HJ (2010). The evolutionary history of histone H3 suggests a deep eukaryotic root of chromatin modifying mechanisms. BMC Evol. Biol. 10, 259. 10.1186/1471-2148-10-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Panyim S, Sommer KR, and Chalkley R (1971). Oxidation of the cysteine-containing histone F3. Detection of an evolutionary mutation in a conservative histone. Biochemistry 10, 3911–3917. 10.1021/bi00797a018. [DOI] [PubMed] [Google Scholar]
- 13.García-Giménez JL, Romá-Mateo C, Pérez-Machado G, Peiró-Chova L, and Pallardó FV (2017). Role of glutathione in the regulation of epigenetic mechanisms in disease. Free Radic. Biol. Med. 112, 36–48. 10.1016/j.freeradbiomed.2017.07.008. [DOI] [PubMed] [Google Scholar]
- 14.Spitz DR (2019). Manipulations of Redox Metabolism for Enhancing Radiation Therapy Responses: A Historical Perspective and Novel Hypothesis. Semin. Radiat. Oncol. 29, 1–5. 10.1016/j.semradonc.2018.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sena LA, and Chandel NS (2012). Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 48, 158–167. 10.1016/j.molcel.2012.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aykin-Burns N, Ahmad IM, Zhu Y, Oberley LW, and Spitz DR (2009). Increased levels of superoxide and H2O2 mediate the differential susceptibility of cancer cells versus normal cells to glucose deprivation. Biochem. J. 418, 29–37. 10.1042/BJ20081258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ekoue DN, He C, Diamond AM, and Bonini MG (2017). Manganese superoxide dismutase and glutathione peroxidase-1 contribute to the rise and fall of mitochondrial reactive oxygen species which drive oncogenesis. Biochim. Biophys. Acta Bioenerg. 1858, 628–632. 10.1016/j.bbabio.2017.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Halvey PJ, Hansen JM, Johnson JM, Go YM, Samali A, and Jones DP (2007). Selective oxidative stress in cell nuclei by nuclear-targeted D-amino acid oxidase. Antioxidants Redox Signal. 9, 807–816. 10.1089/ars.2007.1526. [DOI] [PubMed] [Google Scholar]
- 19.Matlashov ME, Belousov VV, and Enikolopov G (2014). How much H(2)O(2) is produced by recombinant D-amino acid oxidase in mammalian cells? Antioxidants Redox Signal. 20, 1039–1044. 10.1089/ars.2013.5618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Debnath J, Muthuswamy SK, and Brugge JS (2003). Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 30, 256–268. 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- 21.Iliopoulos D, Hirsch HA, and Struhl K (2009). An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell 139, 693–706. 10.1016/j.cell.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clément C, Orsi GA, Gatto A, Boyarchuk E, Forest A, Hajj B, Miné-Hattab J, Garnier M, Gurard-Levin ZA, Quivy JP, and Almouzni G (2018). High-resolution visualization of H3 variants during replication reveals their controlled recycling. Nat. Commun. 9, 3181. 10.1038/s41467-018-05697-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tagami H, Ray-Gallet D, Almouzni G, and Nakatani Y (2004). Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell 116, 51–61. 10.1016/s0092-8674(03)01064-x. [DOI] [PubMed] [Google Scholar]
- 24.Torné J, Ray-Gallet D, Boyarchuk E, Garnier M, Le Baccon P, Coulon A, Orsi GA, and Almouzni G (2020). Two HIRA-dependent pathways mediate H3.3 de novo deposition and recycling during transcription. Nat. Struct. Mol. Biol. 27, 1057–1068. 10.1038/s41594-020-0492-7. [DOI] [PubMed] [Google Scholar]
- 25.García-Giménez JL, Òlaso G, Hake SB, Bönisch C, Wiedemann SM, Markovic J, Dasí F, Gimeno A, Pérez-Quilis C, Palacios O, et al. (2013). Histone h3 glutathionylation in proliferating mammalian cells destabilizes nucleosomal structure. Antioxidants Redox Signal. 19, 1305–1320. 10.1089/ars.2012.5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DeBerardinis RJ, and Chandel NS (2016). Fundamentals of cancer metabolism. Sci. Adv. 2, e1600200. 10.1126/sciadv.1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luo M, Shang L, Brooks MD, Jiagge E, Zhu Y, Buschhaus JM, Conley S, Fath MA, Davis A, Gheordunescu E, et al. (2018). Targeting Breast Cancer Stem Cell State Equilibrium through Modulation of Redox Signaling. Cell Metab. 28, 69–86.e6, e66. 10.1016/j.cmet.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hirsch HA, Iliopoulos D, and Struhl K (2013). Metformin inhibits the inflammatory response associated with cellular transformation and cancer stem cell growth. Proc. Natl. Acad. Sci. USA 110, 972–977. 10.1073/pnas.1221055110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pastushenko I, and Blanpain C (2019). EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 29, 212–226. 10.1016/j.tcb.2018.12.001. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Y, and Weinberg RA (2018). Epithelial-to-mesenchymal transition in cancer: complexity and opportunities. Front. Med. 12, 361–373. 10.1007/s11684-018-0656-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krebs AM, Mitschke J, Lasierra Losada M, Schmalhofer O, Boerries M, Busch H, Boettcher M, Mougiakakos D, Reichardt W, Bronsert P, et al. (2017). The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 19, 518–529. 10.1038/ncb3513. [DOI] [PubMed] [Google Scholar]
- 32.Tvardovskiy A, Schwämmle V, Kempf SJ, Rogowska-Wrzesinska A, and Jensen ON (2017). Accumulation of histone variant H3.3 with age is associated with profound changes in the histone methylation landscape. Nucleic Acids Res. 45, 9272–9289. 10.1093/nar/gkx696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Field AE, Robertson NA, Wang T, Havas A, Ideker T, and Adams PD (2018). DNA Methylation Clocks in Aging: Categories, Causes, and Consequences. Mol. Cell 71, 882–895. 10.1016/j.molcel.2018.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu Y, Yan Y, Principe DR, Zou X, Vassilopoulos A, and Gius D (2014). SIRT3 and SIRT4 are mitochondrial tumor suppressor proteins that connect mitochondrial metabolism and carcinogenesis. Cancer Metabol. 2, 15. 10.1186/2049-3002-2-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.He C, Danes JM, Hart PC, Zhu Y, Huang Y, de Abreu AL, O’Brien J, Mathison AJ, Tang B, Frasor JM, et al. (2019). SOD2 acetylation on lysine 68 promotes stem cell reprogramming in breast cancer. Proc. Natl. Acad. Sci. USA 116, 23534–23541. 10.1073/pnas.1902308116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Campisi J, and Yaswen P (2009). Aging and cancer cell biology, 2009. Aging Cell 8, 221–225. 10.1111/j.1474-9726.2009.00475.x. [DOI] [PubMed] [Google Scholar]
- 37.Palma FR, Gantner BN, Sakiyama MJ, Kayzuka C, Shukla S, Lacchini R, Cunniff B, and Bonini MG (2024). ROS production by mitochondria: function or dysfunction? Oncogene 43, 295–303. 10.1038/s41388-023-02907-z. [DOI] [PubMed] [Google Scholar]
- 38.Desai R, East DA, Hardy L, Faccenda D, Rigon M, Crosby J, Alvarez MS, Singh A, Mainenti M, Hussey LK, et al. (2020). Mitochondria form contact sites with the nucleus to couple prosurvival retrograde response. Sci. Adv. 6, eabc9955. 10.1126/sciadv.abc9955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Coelho DR, Palma FR, Paviani V, He C, Danes JM, Huang Y, Calado JCP, Hart PC, Furdui CM, Poole LB, et al. (2022). Nuclear-localized, iron-bound superoxide dismutase-2 antagonizes epithelial lineage programs to promote stemness of breast cancer cells via a histone demethylase activity. Proc. Natl. Acad. Sci. USA 119, e2110348119. 10.1073/pnas.2110348119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, and Chanda SK (2019). Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10, 1523. 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goedhart J, and Luijsterburg MS (2020). VolcaNoseR is a web app for creating, exploring, labeling and sharing volcano plots. Sci. Rep. 10, 20560. 10.1038/s41598-020-76603-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aziz N, Cherwinski H, and McMahon M (1999). Complementation of defective colony-stimulating factor 1 receptor signaling and mitogenesis by Raf and v-Src. Mol. Cell Biol. 19, 1101–1115. 10.1128/MCB.19.2.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Bulk RNAseq and ChIPseq have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| 8-oxo-dG | R&D Systems | Cat# 4354-MC-050; RRID: AB_1857195 |
| Alexa Fluor 647 | Invitrogen | Cat# A28181; RRID: AB_2536165 |
| ATM | Cell Signaling Technology | Cat# 92356; RRID: AB_2800184 |
| DYKDDDDK (FLAG Tag) | Cell Signaling Technology | Cat# 14793; RRID: AB_2572291 |
| E-Cadherin | Cell Signaling Technology | Cat# 3195; RRID: AB_2291471 |
| Fibronectin | Cell Signaling Technology | Cat# 26836; RRID: AB_2924220 |
| GAPDH | Cell Signaling Technology | Cat# 97166; RRID: AB_2756824 |
| HA-Tag | Cell Signaling Technology | Cat# 3724; RRID: AB_1549585 |
| Histone H2A.X | Cell Signaling Technology | Cat# 7631; RRID: AB_10860771 |
| Histone H3.1/H3.2 | Millipore | Cat# ABE154; RRID: AB_2811170 |
| Histone H3.3 (H3F3A) | Millipore | Cat# 09-838; RRID: AB_10845793 |
| IRDye Goat anti-Mouse | LI-COR Biosciences | Cat# 925-68070; RRID: AB_10953628 |
| IRDye Goat anti-Rabbit | LI-COR Biosciences | Cat# 92632211; RRID: AB_621843 |
| Normal Rabbit IgG | Cell Signaling Technology | Cat# 2729; RRID: AB_1031062 |
| phospho-ATM (Ser1981) | Cell Signaling Technology | Cat# 13050; RRID: AB_2798100 |
| phospho-Histone H2A.X (Ser139) | Cell Signaling Technology | Cat# 80312; RRID: AB_2799949 |
| SOX9 | Cell Signaling Technology | Cat# 82630; RRID: AB_2665492 |
| ZEB1 | Cell Signaling Technology | Cat# 3396; RRID: AB_1904164 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| DH5α competent cells | Invitrogen | 18265017 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| 4-hydroxytamoxifen (4-OHT) | Sigma Aldrich | H7904 |
| Ampicillin | Sigma Aldrich | A5354 |
| Blasticidin | Gibco | A1113903 |
| Bovine Serum Albumin (BSA) | Sigma Aldrich | A9648 |
| Catalase from bovine liver | Sigma Aldrich | C100 |
| Charcoal stripped FBS | Sigma Aldrich | F6764 |
| Cholera toxin | Sigma Aldrich | C8052 |
| D-Alanine | Sigma Aldrich | A7377 |
| DCP-Bio1 | Kerafast | EE0028 |
| DMEM/F12 medium (no phenol red) | Gibco | 21041025 |
| DPBS | Gibco | 14190144 |
| Doxorubicin hydrochloride | Sigma Aldrich | D1515 |
| Doxycycline hydrochloride | Sigma Aldrich | D3072 |
| EGF | Sigma Aldrich | SRP3027 |
| Epon resine | Electron Microscopy Sciences | 14910 |
| Ethanol | Sigma Aldrich | E7023 |
| F-12K | ATCC | 30-2004 |
| Formaldehyde | Sigma Aldrich | 28908 |
| FBS | Sigma Aldrich | F2442 |
| Geneticin | Gibco | 10131-027 |
| Glycine | Sigma Aldrich | G8898 |
| Gluteraldehyde | Sigma Aldrich | G6257 |
| Histone H3.1 human | New England Biolabs | M2503S |
| Histone H3.2 human | New England Biolabs | M2506S |
| Histone H3.3 human | New England Biolabs | M2507S |
| Hydrocortisone | Sigma Aldrich | H0135 |
| Hygromycin | Gibco | 10687010 |
| Insulin | Sigma Aldrich | I3536 |
| Intercept antibody diluent | LI-COR | 92765001 |
| Intercept blocking buffer | LI-COR | 92760001 |
| Lead citrate Reynold’s stain | Electron Microscopy Sciences | 22410-01 |
| Lipofectamine 3000 | Thermo Fisher Scientific | L3000015 |
| Magna ChIP A/G kit | Millipore | CS204457 |
| Matrigel matrix | Corning | 356237 |
| MEM medium | Gibco | 11090081 |
| MITO+ serum extender | Corning | 355006 |
| MSTP | Axon MedChem | 2876 |
| Nitrocellulose membrane | BIO-RAD | 1620112 |
| Novex MES buffer | Invitrogen | B0002 |
| NuPAGE 4–12% Bis-Tris gels | Invitrogen | NP0335 |
| NuPAGE LDS sample buffer | Invitrogen | NP0007 |
| Osmium tetroxide | Sigma Aldrich | 201030 |
| Paclitaxel | Sigma Aldrich | T7191 |
| PFA | Sigma Aldrich | F8775 |
| Penicillin/Streptomycin | Gibco | 15140122 |
| Protease inhibitor cocktail | Roche | 04693132001 |
| Puromycin | Gibco | A1113802 |
| RediJect D-Luciferin bioluminescent substrate | PerkinElmer | 770504 |
| RIPA buffer | Thermo Fisher Scientific | 89901 |
| Sodium cacodylate buffer | Electron Microscopy Sciences | 11650 |
| TGFβ | Sigma Aldrich | H8541 |
| Uranyl acetate | Electron Microscopy Sciences | 22400 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Comet Assay Kit | Trevigen | 4250-050-K |
| Amplex Red Assay Kit | Thermo Fisher Scientific | A22188 |
| RNeasy Kit | Qiagen | 74104 |
| High Capacity cDNA Reverse Transcription Kit | Applied Biosystem | 4368814 |
| Chromatin Accessibility Assay Kit | Abcam | ab185901 |
| Magna ChIP A/G Kit | Millipore | 1710085 |
| Fast SYBR Green Master Mix | Applied Biosystems | 4385612 |
|
| ||
| Deposited data | ||
|
| ||
| RNAseq | This paper | GEO: GSE255598 |
| ChIPseq | This paper | GEO: GSE255615 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| BT20 | ATCC | HTB-19 |
| BT474 | ATCC | HTB-20 |
| HEK293T/7 | ATCC | CRL-11268 |
| MCF7 | ATCC | HTB-22 |
| MCF10A | ATCC | CRL-10317 |
| MCF10AER/vSrc | Iliopoulos et al. | N/A |
| MDA-MB231 | ATCC | HTB-26 |
| Py230 | ATCC | CRL-3279 |
| Py8119 | ATCC | CRL-3278 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| NU/J mice | The Jackson Laboratory | Cat#: 002019; RRID: IMSR_JAX:002019 |
|
| ||
| Oligonucleotides | ||
|
| ||
| RT-qPCR primers | This paper | Table S1 |
| Chromatin accessibility primers | This paper | Table S1 |
| ChIP-qPCR primers | This paper | Table S1 |
|
| ||
| Recombinant DNA | ||
|
| ||
| pEIGW roGFP2-ORP1 | Addgene | Cat#: 64993; RRID: Addgene_64993 |
| pLenti CMV GFP Hygro | Addgene | Cat#: 17446; RRID: Addgene_17446 |
| pLVX-Tet3G blasticidin | Addgene | Cat#: 128061; RRID: Addgene_128061 |
| pLenti CMV GFP Blast | Addgene | Cat# 17445; RRID: Addgene_17445 |
| pLenti CMV GFP Zeo | Addgene | Cat#: 17449; RRID: Addgene_17449 |
| p8.91 | Addgene | Cat#: 187441 RRID: Addgene_187441 |
| pCMV VSV-G | Addgene | Cat#: 8454 RRID: Addgene_8454 |
| pLenti NLS-roGFP2-ORP1 | This paper | N/A |
| pLenti mito-roGFP2-ORP1 | This paper | N/A |
| pLenti NLS-DAO | This paper | N/A |
| pLenti NLS-catalase | This paper | N/A |
| pLenti Inducible TET-ON NLS-catalase | This paper | N/A |
| pLenti mito-catalase | This paper | N/A |
| pLenti H3.1(WT)-FLAG | This paper | N/A |
| pLenti H3.1(C96S)-FLAG | This paper | N/A |
| pLenti H3.3(WT)-HA | This paper | N/A |
| pLenti Inducible TET-ON H3.1(C96S)-FLAG | This paper | N/A |
| pLenti Inducible TET-ON H3.3(WT)-HA | This paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Aura | Spectral Instruments Imaging | https://spectralinvivo.com/software/ |
| Bowtie2 (2.2.9) | Langmead et al.40 | https://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Fiji | Schindelin et al.41 | https://imagej.net/downloads |
| GSEA | Subramanian et al.42 | http://software.broadinstitute.org/gsea/index.jsp |
| GraphPad Prism 10 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Homer (4.11) | Heinz et al.43 | http://homer.ucsd.edu/homer/index.html |
| Metascape | Zhou et al.44 | https://metascape.org/gp/index.html#/main/step1 |
| VolcaNoseR | Goedhart et al.45 | https://github.com/JoachimGoedhart/VolcaNoseR |
|
| ||
| Other | ||
|
| ||
| Eagle 4k HR 200kV CCD camera | FEI Company | N/A |
| IVIS in vivo imager | LAGO | N/A |
| Lionheart FX microscope | Biotek | N/A |
| Nikon A1R confocal microscope | Nikon | N/A |
| Novaseq 6000 | Illumina | N/A |
| Odyssey FC (LI-COR) | LI-COR | N/A |
| QuantStudio 6 Flex | Applied Biosystems | N/A |
| Tecnai Spirit G2 transmission electron microscope | FEI Company | N/A |
| Ultracut UC7 Ultramicrotome | Leica Microsystems | N/A |
| Eagle 4k HR 200kV CCD camera | FEI Company | N/A |