

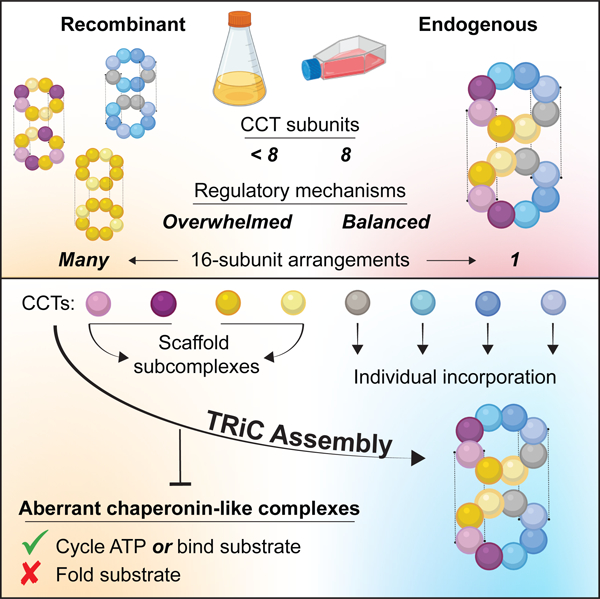
Summary
How the essential eukaryotic chaperonin TRiC/CCT assembles from eight distinct subunits into a unique double-ring architecture remains undefined. We show TRiC assembly involves a hierarchical pathway that segregates subunits with distinct functional properties until holocomplex completion. A stable, likely early intermediate arises from small oligomers containing CCT2, CCT4, CCT5, and CCT7, contiguous subunits that constitute the negatively charged hemisphere of the TRiC chamber, which has weak affinity for unfolded actin. Remaining subunits CCT8, CCT1, CCT3 and CCT6, which comprise the positively charged chamber hemisphere that binds unfolded actin more strongly, join the ring individually. Unincorporated late-assembling subunits are highly labile in vivo, which prevents their accumulation and premature substrate binding. Recapitulation of assembly in a recombinant system demonstrates the subunits in each hemisphere readily form stable noncanonical TRiC-like holocomplexes with aberrant functional properties. Thus regulation of TRiC assembly along a biochemical axis disfavors formation of stable alternative chaperonin complexes.
Keywords: TRiC/CCT, chaperonin, complex assembly, protein folding, actin folding
Graphical Abstract
eTOC:
Moreira, Collier, et al. show that TRiC/CCT chaperonin assembly involves a hierarchical pathway of intermediates segregating subunit properties along a biochemical axis with subunits CCT2, CCT4, CCT5, and CCT7 nucleating assembly while CCT8, CCT1, CCT3 and CCT6 join the ring individually. Unassembled oligomers and subunits are degraded by proteasomal and lysosomal pathways, preventing assembly of noncanonical chaperonin arrangements with aberrant functions.
Introduction
Many essential cellular processes rely on large hetero-oligomeric complexes that must assemble in a unique stoichiometry and topological arrangement in order to function properly. As machines evolve increasing complexity, their spontaneous assembly becomes less efficient 1–3. Deviations from the correct assembly can have deleterious effects, due to both loss-of-function of the complex and gain-of-function of the unassembled subunits or the incorrectly assembled complex. In addition, unassembled subunits are often unstable and their buildup may lead to proteotoxicity 4,5. Accordingly, cells develop mechanisms to ensure these machines adopt specific arrangements. Decades of research have shed light on the elaborate pathways that build various oligomeric complexes 6–14.
Assembly of the 1 MDa eukaryotic chaperonin TRiC/CCT (TCP1-ring complex, or cytosolic chaperonin containing TCP1), which functions as an important node in eukaryotic protein folding and proteostasis machinery, has not been defined. TRiC is an essential protein-folding machine that consists of eight paralogous subunits (CCT1-CCT8) which assemble into a double-ring structure with defined subunit order. Each ring contains one of each paralog, CCT1–8 (or CCTα, β, γ, δ, ε, ζ, η, θ), which share only ~30% sequence identity but high structural similarity 15,16. TRiC helps maintain cellular proteostasis 17 by assisting the nascent chain transition to folded and functional protein for an estimated 10% of the proteome 18,19,20–23. Within TRiC, CCT1–8 adopt a precise and unique arrangement that, to current knowledge, is conserved across all eukaryotes 19,24,25. The subunits assemble in an invariant order within each ring and stack along a two-fold symmetrical axis (Figure 1A). This arrangement spatially separates the properties of individual subunits to create an asymmetric chaperonin complex. The folding chamber is segregated into hemispheres by net charge 24,26 as well as ATP binding and hydrolysis capacities 20,27,28. Thus, the unique arrangement of 16 subunits in TRiC partitions the biochemistry and allosteric structural transitions underlying its ability to process substrates 15,20,27,29,30.
Figure 1. TRiC assembles hierarchically from defined sets of CCT subunits.
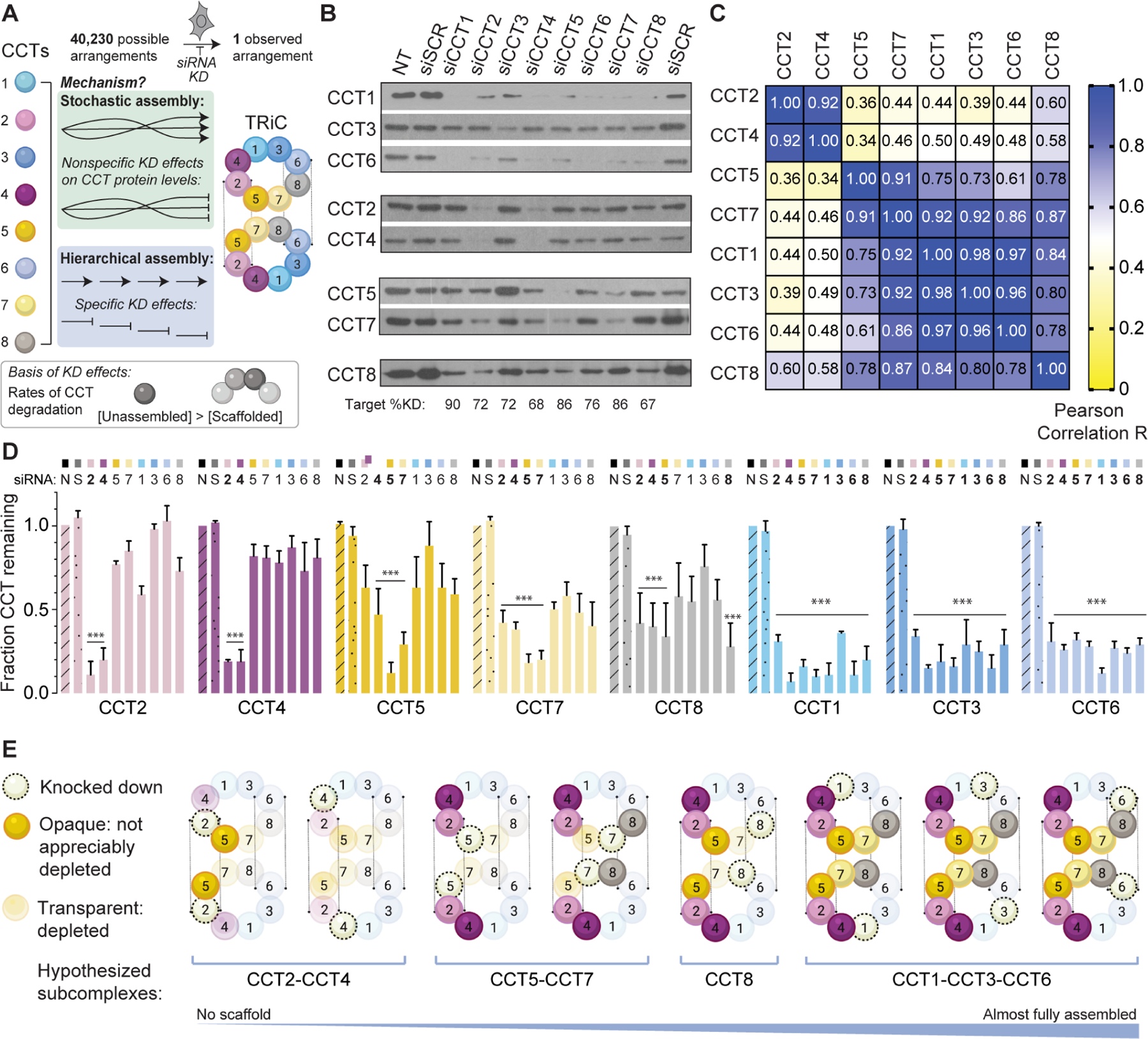
A. Unique arrangement of eight CCTs within the TRiC double ring complex and schematic of the knockdown experiment to probe assembly. B. SDS-PAGE immunoblots for each TRiC subunit in HeLa cell lysate following siRNA treatments. NT = non-transfected; siSCR = scrambled siRNA control. Subunits with similar depletion patterns are grouped together. Bottom text indicates knockdown efficiencies. C. Heat map of Pearson correlation coefficients between CCT pairs after knockdowns. D. Fraction of CCT protein remaining after each knockdown (top) normalized to NT control (n=3, errors bars represent SEM, *** = p < 0.0001 using two-way ANOVA and Dunnett’s multiple comparisons test). E. Summary of the TRiC subunit depletion patterns incorporated into a proposed assembly order. See also Figure S1.
How TRiC assembles to such a well-defined structure given the degeneracy of the subunit-subunit interfaces is unclear. In principle, individual CCTs could form two stacked identical 8-subunit rings in over 40,000 distinct configurations; if subunit repetition is allowed, millions of arrangements are possible. Recombinant expression efforts have demonstrated that CCT subunits can indeed adopt some of these alternative arrangements. For instance, human CCT5 self-assembles into complete 16-mers 31 and yeast CCT1, CCT2, and CCT6 are also reported to form homo-oligomeric double rings 32. In addition, human CCT4 and CCT5 can promiscuously interact with CCTs that are not their cognate neighbors in native TRiC 33,34. While roles have been ascribed to some CCTs outside of their function within TRiC 35,36, noncanonical chaperonin-like assemblies of the types formed recombinantly have not been observed physiologically.
To reconcile the ability of individual subunits to form alternative oligomers with the unique arrangement in vivo, most TRiC assembly models postulate that CCT oligomers constitute intermediates in a pathway. For example, CCT5 homo-oligomers were proposed to precede successive subunit exchange reactions leading to the final arrangement 34. However, this model predicts a larger stoichiometric imbalance than typically observed and does not explain the lack of detectable homo-oligomers in cells. In addition, yeast CCT5 only self-assembles if its N-terminus is truncated 32. Thus a pathway initiated by homo-oligomers would differ between human and yeast, despite strong likelihood of a conserved assembly mechanism across eukaryotes 37. Structural analysis has also led to the suggestion that TRiC assembles both rings simultaneously 38, contrasting a prior proposal that subunits integrate into pre-existing TRiC during cycles of disassembly into labile single rings 39. Intermediates supporting or disproving these models have yet to be isolated or confirmed.
Here we show TRiC assembly in mammalian cells involves a hierarchical pathway of intermediates that are separated temporally by biochemical properties and rapidly degraded, preventing their accumulation. Recombinant production of CCT oligomers in insect cells reveals the extent of CCT subunit capacity to form alternative noncanonical TRiC-like architectures without the full complement of all eight CCTs and with aberrant substrate binding and folding abilities. Our data demonstrate that the ordered assembly of CCT subunits into TRiC is not dictated predominantly by intrinsic thermodynamic or structural properties but is regulated to prevent improper assembly of noncanonical chaperonin arrangements with aberrant functions.
Results
TRiC assembles hierarchically through defined sets of CCT subunits
We first reasoned that if TRiC assembly is ordered, limiting the availability of individual subunits should block the process at distinct stages (Figure 1A). Any remaining unassembled subunits would likely be cleared 4. Because complete knockout of any CCT gene is lethal, we used siRNA to deplete individual subunits in mammalian cells and then examined the effects on levels of remaining subunits by Western blotting (Figure 1A). qPRC analyses indicated the siRNAs depleted only the mRNA of the targeted CCT subunit (Figure S1A), corresponding to 67–90% reductions in target protein (Figure 1B). Notably, we observed that depletion of individual CCT subunits lead to subunit-specific reductions in the levels of non-targeted subunits (Figure 1B). Pearson correlations revealed four CCT subsets of subunits similarly affected by knockdown of certain paralogs, namely CCT2 & CCT4; CCT5 & CCT7; CCT1, CCT3, & CCT6; and CCT8 alone (Figure 1C). Of note, CCT2 & CCT4 were only destabilized by siRNA of each other, while knockdown of CCT5 & CCT7 destabilized all subunits except CCT2 & CCT4 (Figure 1C). Interestingly, each subset comprises subunits that are directly adjacent within a correctly assembled TRiC ring, leading us to interpret the destabilization of a given subunit as loss of its scaffold during TRiC assembly (Figure 1D). The observed patterns of destabilization suggest a hierarchical model of TRiC assembly in which a scaffold forms first from CCT2 and CCT4, followed by incorporation of CCT5 and CCT7; then CCT8; and lastly, CCT1, CCT3, and CCT6 (Figure 1E). This empirical order of incorporation requires no rearrangement or subunit displacement. Since CCT2 forms homotypic contacts across the rings, the proposed early intermediate also sets into place one of the homotypic inter-ring contacts, suggesting both rings could assemble concomitantly.
The hierarchy of TRiC assembly is not thermodynamically predetermined
Given the similarity in fold and interface topology between all CCTs, we next asked what determines the preferential formation of subcomplexes. For some hetero-oligomeric complexes, the assembly pathway is driven by the hierarchy of binding free energies 37,40. Accordingly, we asked whether the TRiC assembly pathway deduced from in vivo experiments simply reflects the stabilities of native intersubunit interfaces. We used the PISA algorithm (Proteins, Interfaces, Structures, and Assemblies, EMBL) 41 to estimate the free energy of formation ΔG for each CCT-CCT surface based on an open-state model of human TRiC (average resolution 3.8 Å) derived by cryo-electron microscopy 42. We inspected the smaller inter-ring interfaces and larger intra-ring interfaces (Figure S1D–E). Intra-ring stability predictions varied widely, and there was no relationship to the order of assembly. The lowest inter-ring ΔG did not correspond to the first such contact predicted by our model, CCT2-CCT2, but instead to CCT3-CCT8, a pair that is not observed in vivo. These analyses suggest the assembly pathway of TRiC does not reflect the predicted free energy hierarchy of either intra-ring or inter-ring subunit-specific interfaces, pointing to external regulation guiding this pathway.
Labile CCT assembly intermediates accumulate in cells prior to degradation
Our proposed model hypothesizes that TRiC assembly involves formation of physical subcomplexes amongst CCTs similarly impacted by siRNA knockdown. Formation of these subcomplexes would stabilize them against degradation, whereas unassembled subunits would be more easily degraded. To test this, we performed native gel electrophoresis and immunoblotting analysis of HeLa lysates (Figure 2 and S2). All CCT-targeting siRNA led to a decrease in fully assembled TRiC and an increase in faster-migrating CCT species (Figure 2A; Figure S2A). We only observed detectable monomer-size bands (herein CCTM) for CCT8 (Figure 2A). Other bands indicated intermediate-sized subcomplexes (herein CCTSC), which contained CCT2, CCT5, and CCT7 (Figure 2A; Figure S2A). In contrast, neither CCT1, CCT3, nor CCT6 were detected in subcomplexes; instead these subunits migrated only with fully assembled TRiC. Our CCT4 antibodies were too weak to conclude from native gels whether or not this subunit was present in subcomplexes. Thus, blocking TRiC assembly causes subunits CCT2, CCT5 and CCT7 at minimum to accumulate in multimeric subcomplexes.
Figure 2. Unintegrated CCTs form selective subcomplexes in cells.
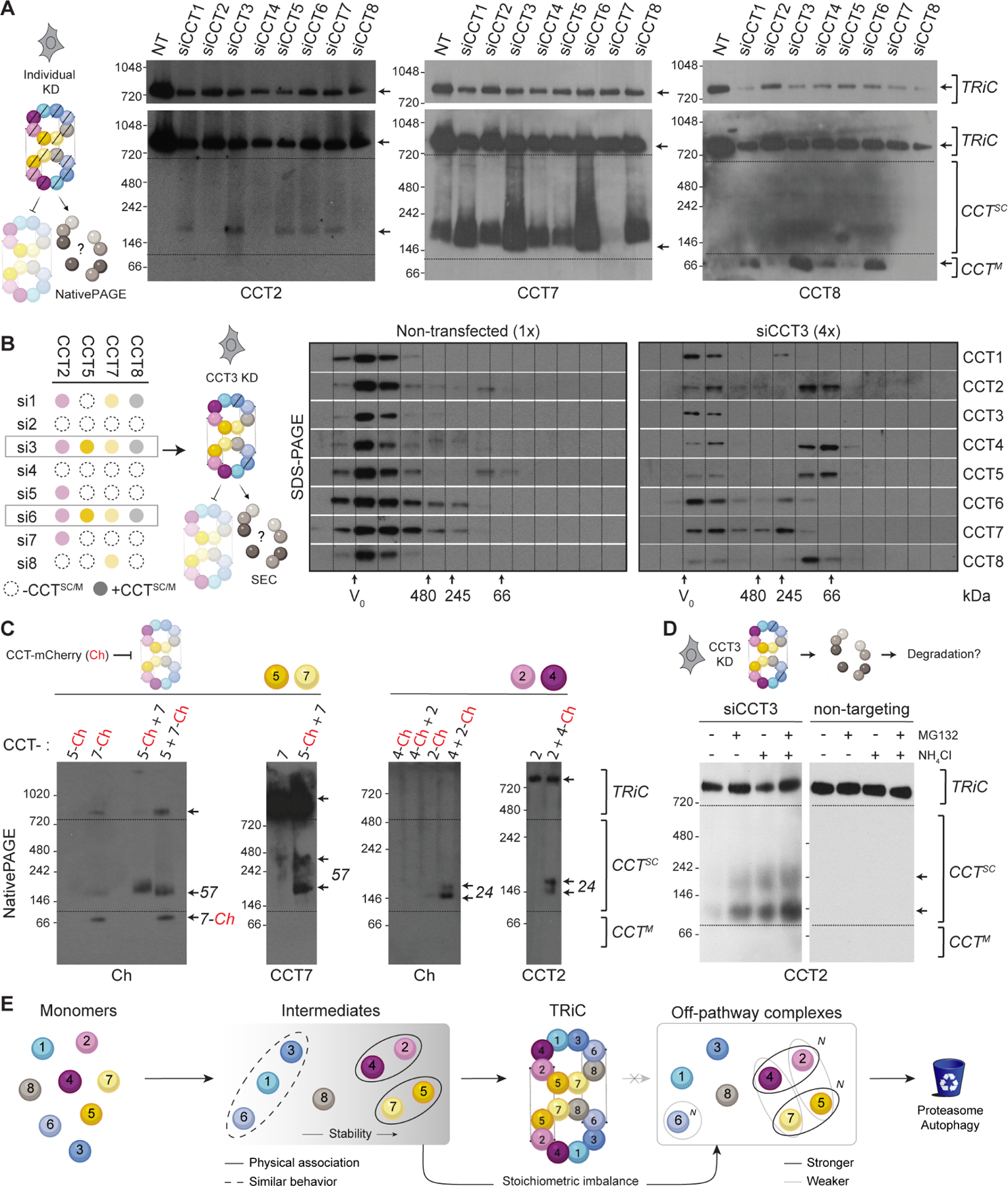
A. NativePAGE immunoblots of HeLa cell lysates after individual CCT knockdowns. Low exposure blots (top) show TRiC depletion; higher exposure blots (bottom) highlight subcomplexes (CCTsc) or monomers (CCTM). Representative blots from each CCT subset with detectable species smaller than TRiC are shown; Figure S2 contains blots for remaining subunits. NT = non-transfected. B. Left, summary of results from A informing right, immunoblots of HeLa cell lysate depleted of CCT3 compared to control lysate after fractionation by size exclusion chromatography. TRiC elutes near the void volume V0; subcomplexes elute in later fractions. Due to clearance mechanisms and signal dispersion, 4-fold higher amounts of CCT3 knockdown lysates compared to controls were loaded to detect CCT subcomplexes. C. Subcomplexes between assembly partners CCT5-CCT7 and CCT2-CCT4 detected by NativePAGE after co-overexpression in HeLa cells. The mCherry tag hinders higher-order assembly. D. NativePAGE immunoblots for CCT2 in HeLa cells treated with siRNA targeting CCT3 or scrambled control after treatment with proteasome inhibitor MG132 and/or autophagy inhibitor NH4Cl. E. Summary of in vivo CCT subcomplex capacities based on knockdown and overexpression experiments. N indicates higher-order oligomerization. See also Figure S2.
Formation of CCT subcomplexes that are putative assembly intermediates was further investigated using size exclusion chromatography. We fractionated cell lysates after stimulating subcomplex accumulation by siRNA knockdown of CCT3 (Figure 2B, left). All non-targeted CCTs eluted in fractions corresponding to species smaller than TRiC (Figure 2B). CCT2, CCT4, CCT5, and CCT8 displayed the largest size shifts, with the majority of each subunit eluting below ~200 kDa (Figure 2B). These approaches together confirm that disrupting TRiC assembly leads to accumulation of CCT subcomplexes.
Because these subcomplexes were not abundant, we next overexpressed sets of CCTs to examine their ability to form subcomplexes. We used C-terminal mCherry (Ch) tags, reasoning that because these termini protrude into the central chamber of TRiC, the tag would sterically hinder assembly and promote accumulation of intermediates. As expected, Ch-tagged subunits accumulated in subcomplexes more readily than untagged subunits (Figure 2C). Substantiating the formation of CCT2-CCT4 and CCT5-CCT7 subcomplexes, joint overexpression of CCT2 with CCT4 and of CCT5 with CCT7 increased the yield of their respective subcomplexes compared to individual overexpression (Figures 2C and S2B–C, 57 and 24 arrows). When we co-overexpressed CCT2 or CCT4 with CCT7 or CCT5, pairings that were not significantly correlated in our siRNA experiments, the resulting subcomplexes were much less abundant; pairing CCTs with YFP produced no subcomplexes (Figure S2C). In contrast to CCT2, CCT4, CCT5, and CCT7, overexpression of subunits CCT3, CCT6, CCT1, and CCT8 alone or in any combination did not result in hetero-oligomeric subcomplexes (Figure S2D–E). Individual overexpression of CCT6 led to a smear of higher-order CCT6-positive species (Figure S2D–E), which could reflect a capacity to form homo-oligomers. Thus, whereas unassembled CCT8 is detectable as a monomer, CCT1, CCT3, and CCT6 do not appear to persist as stable monomers or subcomplexes even when overexpressed together.
The low levels of subcomplexes observed in cells, even upon overexpression, suggests that unassembled CCTs are subject to rapid clearance. We next asked which degradation pathways process orphaned CCTs. We generated TRiC assembly intermediates via siRNA knockdown of CCT3 and treated cells with either proteasome inhibitor MG132 or lysosome-autophagy inhibitor ammonium chloride. Each inhibitor increased the levels of two differently sized CCT2-containing subcomplexes, with an additive effect observed in the presence of both (Figure 2D; Figure S2F). It thus appears that both proteasomal and lysosomal degradation contribute to quality control of unassembled CCTs.
These analyses extend our model for the TRiC assembly pathway (Figure 2E). We posit that CCT5, CCT7, CCT2, and CCT4 form assembly intermediate subcomplexes, with preference for CCT2-CCT4 and CCT5-CCT7 pairs of cognate neighbors. CCT8 is relatively stable as a monomer whereas CCT1, CCT3 and CCT6 are not, being rapidly degraded without forming detectable subcomplexes unless incorporated into TRiC. While our experiments do not delve into the temporal sequence of events leading to assembled TRiC, it is reasonable to propose that the CCT2-CCT4 and CCT5-CCT7 subcomplexes template incorporation of the highly labile subunits in the other hemisphere of the TRiC ring. Unassembled subunits and intermediates are degraded via both autophagic and proteasomal pathways.
CCTs can form subcomplexes in insect cells that do not re-assemble into TRiC
Given the low abundance of CCT subcomplexes in mammalian cells, we tried generating CCT assembly intermediates from an insect cell expression system that produces human TRiC (Figure 3 and S3) 43. We first expressed pairs of contiguous subunits, namely CCT2-CCT4, CCT5-CCT7, CCT3-CCT6, and CCT1-CCT8 (Figure 3A). Subcomplex formation was assessed by NativePAGE, both through total protein staining in lysates and by radiography after pulse-labeling (Figure 3B). CCT5-CCT7 and CCT2-CCT4 formed similar subcomplexes to those observed in mammalian cells (Figure 3B; see also Figure S4A). Unassembled CCT8 again migrated as a monomer. However, unlike mammalian cells, we observed larger subcomplexes for the other subunits in insect cell lysates. Several subunit pairs yielded CCT complexes migrating as a 1 MDa complex with similar mobility to TRiC (Figure 3B). Immunoblots confirmed the presence of multiple exogenous CCTs in these 1 MDa-sized complexes, in addition to intermediate-size oligomers containing CCT1, CCT3 and CCT6 (Figure 3C), which are not observed in mammalian cells. Of these subunits, only CCT6 could form larger complexes when overexpressed in mammalian cells. This suggests the high expression in the insect system overwhelms the PQC capacity of endogenous clearance pathways, allowing non-physiological or otherwise labile complexes to be obtained.
Figure 3. Recombinant CCTs assemble into various complexes via preferentially but not exclusively canonical contacts.
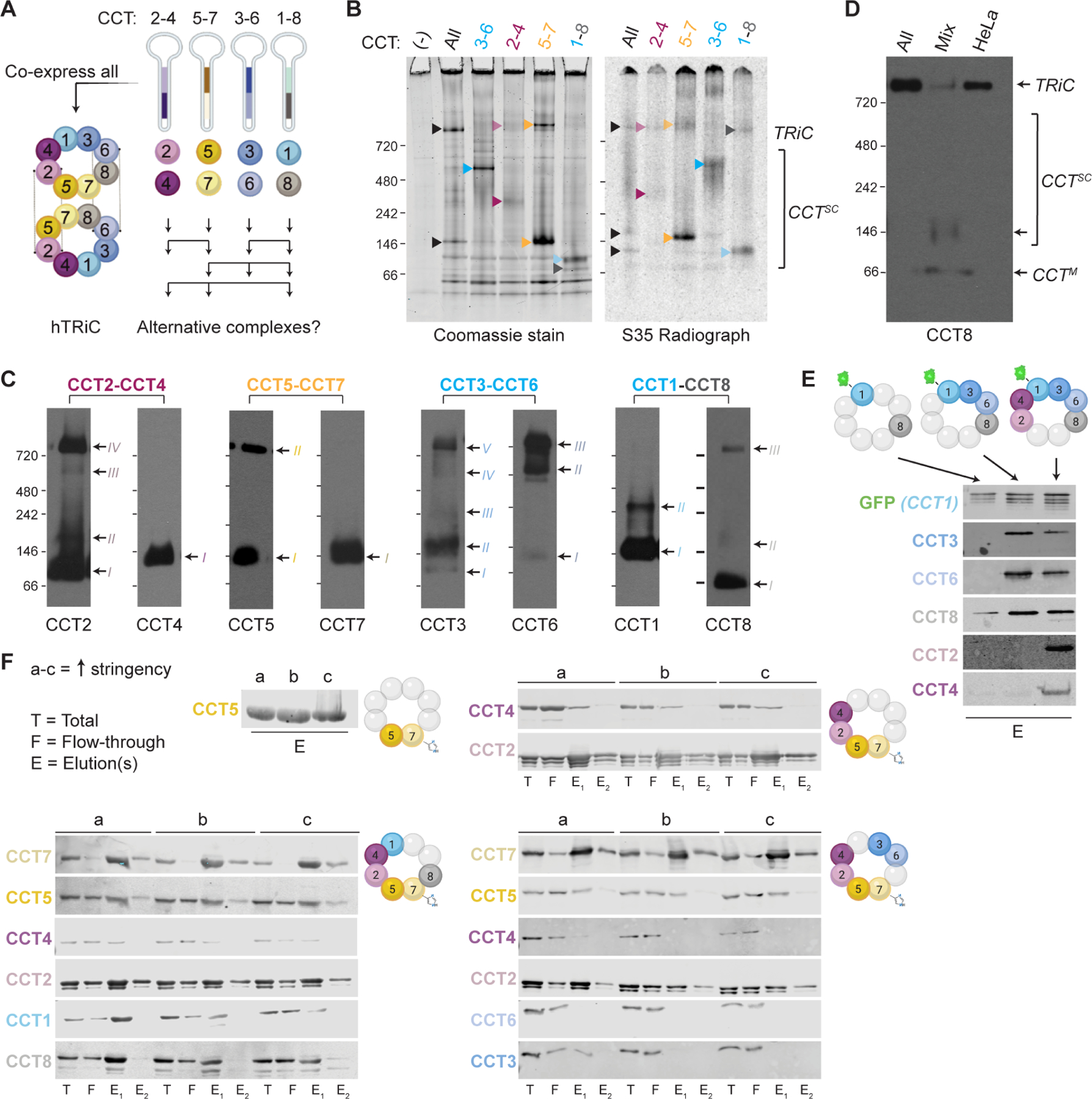
A. Schematic approach to express recombinant TRiC or alternative CCT combinations in an insect cell (Hi5) system using dual cassette baculovirus vectors. B. Total protein stain (left) and 35S radiograph (right) of Hi5 cell lysates expressing all CCT subunits (TRiC) or pairs. Only the radiograph samples were metabolically labeled with 35S-methionine. Arrows indicate CCT complexes. C. NativePAGE immunoblots for all recombinant subunits in Hi5 cells expressing each CCT pair. Arrows and Roman numerals indicate distinct complexes containing each subunit. The non-adjacent pair CCT1-CCT8 show the least co-migration. D. NativePAGE immunoblot for CTT8 in Hi5 cells expressing all TRiC subunits or pairs mixed post-lysis. TRiC does not assemble in the mixture. HeLa cell lane shows endogenous TRiC. E. SDS-PAGE immunoblots for CCT subunits after GFP immunoprecipitation from Hi5 cells co-expressing the colored subunits above each lane. F. SDS-PAGE immunoblots for CCT subunits after affinity purification of CCT7His from Hi5 cells. Colored subunits to the right of each set of blots were co-expressed and beads were washed with stringencies of 100 mM NaCl, 500 mM NaCl, and 500 mM NaCl with 0.5% NP-40 (a, b, c). Broad CCT recovery secondary to the subunit pulled down indicates that complexes form in the absence of complete TRiC, with canonical ring contacts mediating recovery more than noncanonical contacts. See also Figure S3.
Since all eight recombinant CCTs formed detectable complexes in insect cells, we next attempted to reconstitute TRiC from these subcomplexes. We co-incubated lysates containing all eight subunits and tested whether they could reassemble into TRiC. To account for the potential requirement of nucleotide and/or endogenous factors, we incubated lysates at varying temperatures and in the presence or absence of supplemental ATP-Mg and HeLa cell lysate. None of the CCT combinations or conditions yielded correctly assembled TRiC (Figures 3D; S3A–B).
Because these results imply specific requirements of co-expression for correct assembly, we next examined CCT assembly in relation to which subunits were co-expressed. To facilitate analysis, we tagged two subunits in opposing TRiC hemispheres: CCT1 with GFP (CCT1GFP) and CCT7 with hexahistidine (CCT7His). Both tags were introduced in an accessible surface loop where they do not affect assembly of recombinant TRiC 33,43. First, we sequentially expressed the contiguous subunits forming the CCT7His-containing hemisphere and captured the resulting complexes by His-tag affinity pulldowns under varying stringency (Figure S3C). CCT5 was readily recovered with CCT7His under all conditions (Figure 3F). CCT2 and CCT4 were also recovered in CCT7 pulldowns when additionally co-expressed, indicating these four contiguous subunits form a complex (Figure 3F). CCT2 was more resistant to stringent CCT7His isolation conditions than CCT4 (Figure 3F), consistent with formation of a canonical CCT7His-5-2-4 subcomplex. Of note, additionally co-expressing non-contiguous subunits CCT3 and CCT6 in this setting did not lead to their detectable incorporation into a CCT7His-5-2-4 complex (Figure 3F). In contrast, additional co-expression of CCT1 and CCT8, which respectively flank CCT4 and CCT7 in TRiC, enabled CCT3 and CCT6 association with the CCT7His-5-2-4 complex (Figure 3F). These experiments suggest the preferential assembly of subunits contiguous in the ring.
Next, we used the GFP tag in CCT1GFP to similarly examine the assembly specificity of the other TRiC hemisphere. CCT8, which is not adjacent to CCT1 in TRiC, was weakly recovered in the pulldown (Figure 3E). However, CCT1-adjacent subunits CCT3 and CCT6 readily co-eluted with CCT1GFP; furthermore, inclusion of these subunits bridged the association with CCT8, leading it to be pulled down more strongly (Figure 3E). Continuing with expression of contiguous subunits around the ring, additional co-expression of CCT2 and CCT4 led to co-elution of all six subunits (Figure 3E). In sum, these co-expression experiments recapitulated the formation of the CCT7-5-2-4 hemisphere inferred to form from stable subcomplexes in mammalian cells; but also revealed the ability to form a CCT1-3-6-8 complex in the absence of the CCT7-5-2-4 scaffold, even though these four subunits are highly labile and do not form any detectable subcomplexes when over-expressed in mammalian cells. These results indicate that contiguous CCTs oligomerize during or closely following biogenesis in a process that does not require a canonical scaffold; and that, under physiological conditions, this oligomerization is somehow inhibited for CCT1, CCT3, CCT6, and CCT8.
CCT subunits encode the capacity to form stable non-canonical 1 MDa chaperonins
Having gained insight into the formation of CCT subcomplexes through insect cell co-expression, we next examined their assembly state and stoichiometry. NativePAGE after affinity purification revealed co-expressed contiguous CCT subunits formed sets of distinct subcomplexes, ranging from 1 MDa to smaller oligomers (Figure S3D). To profile their composition, we excised various bands from native gels and analyzed them by mass spectrometry (MS). Since untagged TRiC exhibits some affinity for nickel resin, the 1 MDa complexes obtained by simple CCT7-His-tag pulldowns were contaminated with insect TRiC (tCCTs). Mass spectrometry analysis of the two distinct 1 MDa bands from His-pulldowns following CCT57His expression showed one corresponded to insect TRiC and the other to a predominantly CCT5-containing 1 MDa oligomer (Figure S3D). All co-expressions including CCT5 also yielded a dimer population predominantly containing CCT5. Combinations containing CCT1GFP-3-6-8 formed a 1 MDa complex as well as smaller and less abundant heteromeric subcomplexes. Consistent with our other data, CCT8 was only detected in either 1 MDa complexes or as a monomer. CCT1GFP-containing subcomplexes smaller than 250 kDa contained Hsp70s and to a lesser extent Hsp90, suggesting a possible role for the chaperones in stabilization or clearance of these species.
In summary, expressing human CCTs in insect cells recapitulates the stepwise formation of TRiC assembly intermediates along canonical interfaces. However, it also reveals that 1 MDa complexes readily form even without the presence of a full complement of CCT subunits. While these 1 MDa CCT complexes preferentially utilize canonical interfaces when available at the point of biogenesis, they can use non-canonical interfaces to complete formation of stable 1 MDa TRiC-sized complexes. These results indicate that absent the PQC pathways that would otherwise block or remove certain CCT species during assembly, noncanonical contacts can become stabilized by forming a 1 MDa complex. We hypothesize that the high expression levels in insect cells overwhelm these PQC pathways when components of the canonical scaffold are absent. These findings indicate active regulation of TRiC assembly mediates formation of its unique canonical arrangement under physiological conditions.
CCT subsets assemble in TRiC-like stable noncanonical holocomplexes
To understand how an incomplete set of CCT subunits can give rise to a 1 MDa complex, we examined these recombinant TRiC-sized complexes (hereafter ‘holocomplexes’ or ‘HC’, as opposed to smaller ‘subcomplexes’ or ‘SC’) in more detail. We focused on three HCs comprising different contiguous segments of the TRiC ring: (1) HCCCT4257His containing only subunits from the negatively charged hemisphere of the TRiC chamber; (2) HCCCT1CBP368, containing subunits from the positively charged hemisphere (GFP was exchanged for a CBP tag within CCT1 to facilitate purification); and (3) a six-CCT HCCCT57His8631, lacking CCT2-CCT4, which was the most stable and highest yield HC in our experiments. (For clarity, we omit the tags from CCT set names hereafter.) We also attempted to purify the two-subunit HCs formed by co-expressing CCT2-CCT4 or CCT5-CCT7, but these HCs were less stable; we could only purify stable SCs with sizes corresponding to the different CCT2-CCT4 and CCT5-CCT7 intermediates observed in mammalian cells (Figure S4A).
All HCs were separated from SCs by size exclusion chromatography and analyzed by a suite of structural techniques (Figure 4A). HCs share the approximate molecular weight of TRiC by NativePAGE (Figure 4B). Immunoblotting and mass spectrometry validated the presence of all expressed subunits in each purified HC, and confirmed the absence of unexpressed subunits (Figure S4C–D). Hsp70 was detected by mass spectrometry in solution with some of the HCs (Figure S4D), possibly indicating a dynamic association of this chaperone with larger CCT species.
Figure 4. Heterogeneous TRiC-like complexes assemble from CCT subsets.
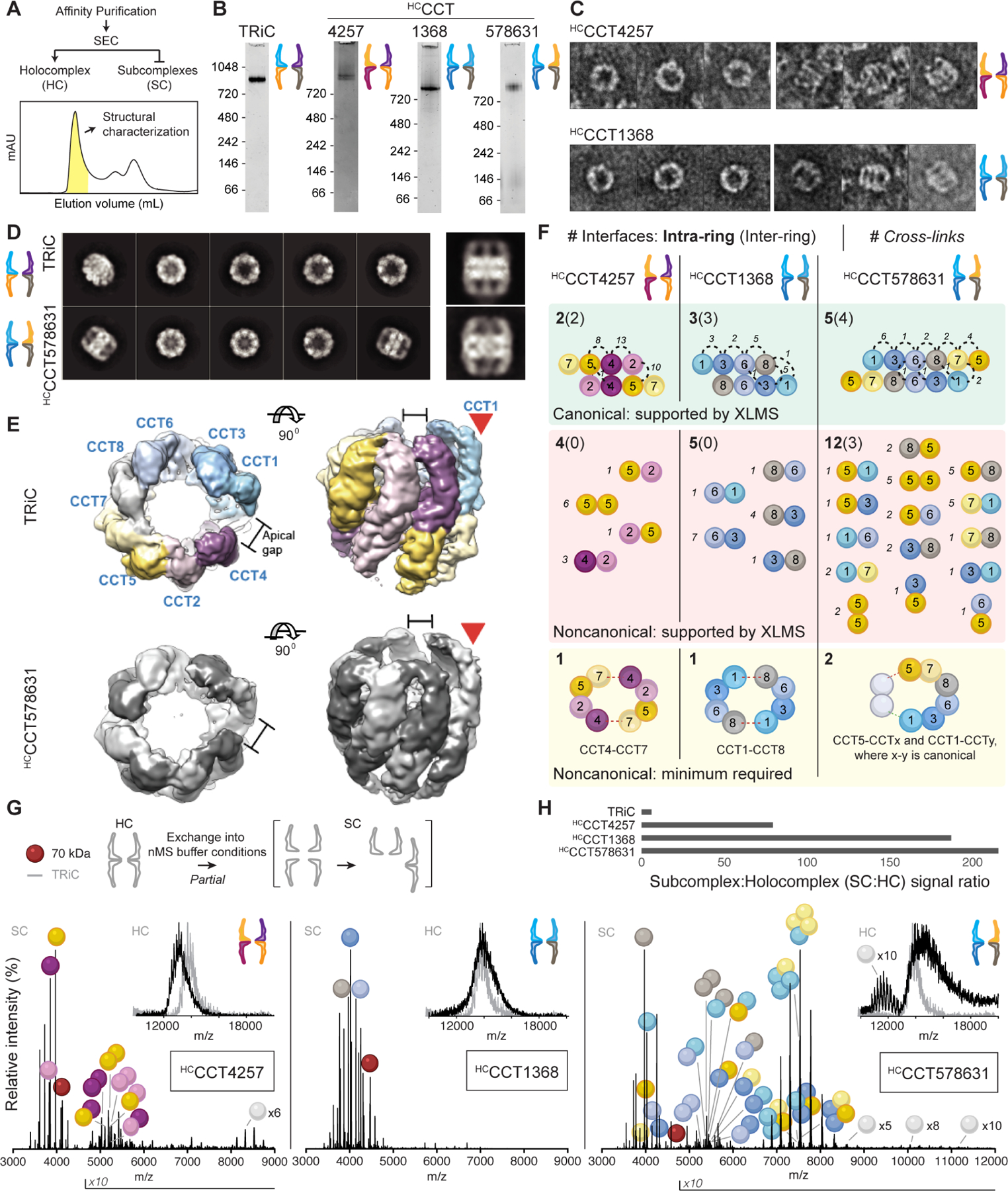
A. After enrichment by affinity purification from Hi5 cells, holocomplexes (HCs) are purified by size exclusion chromatography. The representative trace shown is from the purification of HCCCT4257. B. Coomassie-stained NativePAGE of HCs containing four or six CCTs reveal similar sizes to TRiC and monodispersity. C. Negative stain electron micrographs of HCCCT4257 and HCCCT1368 showing top (left) and side (right) views of double ring topologies. D. 2D class averages of electron micrographs of TRiC and HCCCT578631 with representative side views on the right. E. Reconstructed 3D maps of TRiC and HCCCT578631 reveal shared asymmetric structural features (red arrows). Subunits within HCCCT578631 are unassigned. F. Summary of cross-linking mass spectrometry of HCs. Cross-links are mapped to canonical, noncanonical, intra-ring, and inter-ring CCT-CCT interfaces. Subunit orientations in cartoons connote distinct interfaces; e.g. 4–5 and 5–4 represent two interfaces where CCT4 is positioned on either side of CCT5. Cross-links were inconsistent with a parsimonious model of holocomplex assembly via minimal noncanonical contacts, and instead indicated that multiple arrangements form. G. Native mass spectra of holocomplexes (insets) and their dissociation products. The most abundant charge state of each assigned peak series is annotated with the colors of CCT monomers, dimers, and so on. CCTxN designates charge series closely matching CCT oligomers with ambiguous sub-stoichiometry. Red spheres denote charge series with masses close to 70 kDa, likely Hsp70 isoforms based on proteomic analysis. Intensities are normalized to 100%; absolute maximum signal intensities are 4, 1.7, and 8.3 × 105 from left to right, meaning differences in oligomerization could be partly concentration-dependent. H. Concentration-independent ratios of native MS signal demonstrate that holocomplexes are less stable than TRiC. See also Figure S4.
Negative stain electron microscopy (EM) indicated that each of the three HCs exclusively formed TRiC-like double ring structures (Figure 4C–4D), without discernible spiraling or other departures from regular chaperonin symmetry previously reported for homo-oligomeric CCT HCs 32. The six-CCT HCCCT578631 yielded EM data of sufficient quality for image classification and determination of a structure with average resolution 7.6 Å (Figure S4E–G). Despite the absence of CCT2 and CCT4, this structure was very similar to that of native TRiC, containing two 8-subunit rings stacked back to back (Figure 4D). Although we could not assign the subunits in the map at this resolution, the TRiC and HCCCT578631 HC complexes presented intriguing differences and similarities. For instance, the apical domain of CCT2 within TRiC is highly dynamic, creating a flexible region (Figure S4H). This CCT2-specific feature is missing in HCCCT578631, which instead contains a relatively ordered set of apical domains. However, TRiC and HCCCT578631 share an asymmetric feature where a subunit protrudes (Figure 4E, S4H). In TRiC, this protrusion creates a gap between CCT1 and CCT4. Because CCT4 is not present in HCCCT578631, the molecular architecture surrounding this feature is necessarily different, though the feature itself is reproduced. Thus, EM confirms that the double ring chaperonins containing a noncanonical complement of CCT subunits bear a striking resemblance to TRiC, and further indicates that the presence of specific subunits in the arrangements confer unique structural characteristics to the complex.
Our analyses raise the question of what contacts constitute the arrangements within the noncanonical chaperonin rings. First, we examined inter-subunit contacts using cross-linking mass spectrometry (XLMS). Each HC was treated with the amine-reactive cross-linker DSS (Figure S4I). Each link that satisfied the distance constraint (11.4 Å) was assigned by structural modeling to support a specific CCT-CCT interface: canonical or noncanonical, and lateral (intra-ring) or equatorial (intra-ring) (Figure 4F; Table S2). While previous cross-linking analyses of TRiC supported canonical intersubunit contacts 24,33, the HCs examined here yielded a mix of canonical and noncanonical interfaces. We considered whether HC rings could be composed of enjoined TRiC hemispheres, which would require only one or two noncanonical interfaces in the case of the 4-subunit CCT and 6-subunit CCT oligomers, respectively (Figure 4F, yellow). However, we observed multiple distinct noncanonical contacts for all HCs (Figure 4F, red). This suggests an alternative pathway for HC assembly, conceivably initiated through formation of canonical interface contacts followed by multiple off-pathway routes to complete the 16-mer.
The intersubunit contacts within the HCs were also analyzed by native (non-denaturing) mass spectrometry (nMS), an orthogonal approach to XLMS that does not rely on lysine availability and can provide higher order stoichiometric information. We previously found that nMS of TRiC can yield well-resolved CCT monomers derived from disassembly of a subpopulation of TRiC during the experiment 33. Here, ionized HCs yielded lower order oligomers in addition to monomers (Figure 4G). We used subunit masses to assign CCT oligomers. Mass spectra of HCCCT4257, containing the subunits from the negatively charged hemisphere, featured various dimers (Figure 4G). In contrast, no intermediate assemblies were detected for HCCCT1368, consisting of the subunits in the positively charged hemisphere. Strikingly, when CCT5 and CCT7 were present alongside the positive subunits to make the 6-CCT subunit HCCCT578631, a range of dimers and tetramers were readily detected (Figure 4G). The dimers and tetramers implied both canonical and noncanonical CCT interfaces. We cannot distinguish whether oligomers measured by nMS derive directly from dissociated holocomplexes or from re-associating subunits. However, we have shown previously that generating a monomer pool sufficient to re-form dimers from native TRiC requires the addition of denaturing solvent which was absent on these experiments 33. Notably, the greater extents of HC dissociation relative to TRiC as measured by signal ratios indicates that noncanonical HCs are less stable than TRiC (Figure 4H).
These structural analyses indicate CCT subsets can form stable double ring chaperonin holocomplexes with alternative arrangements within each 8-mer ring that are topologically similar to TRiC. The diversity of interfaces revealed by XLMS of alternative complexes and the breadths of signals measured by nMS indicate that HCs can adopt an array of compositions rather than a single arrangement. The alternative chaperonin arrangements appear to begin forming along a canonical scaffold (Figure 3E–F and Figure 4F) but fill empty spaces in degenerate arrangements to complete a stable double-ring architecture. This is further supported by the stoichiometries suggested by the nearly equimolar CCT abundances in label-free quantitative MS, with no differences larger than a factor of two (Figure S4D). Thus, without sufficient regulation, the complete double ring architecture appears to be the favored assembly state of CCTs even when native TRiC cannot form. Our finding that subunits can in principle substitute for each other in assembly pathways orthogonal to the one that forms TRiC resonates with our PISA analysis (Figure S1D–E), which suggests that TRiC assembly into its native arrangement is not driven by the intrinsic thermodynamic stability of the native intersubunit interfaces. Rather, the biological assembly pathway must disfavor non-canonical interactions.
Noncanonical CCT holocomplexes segregate biochemical TRiC functions
Given their structural similarity to TRiC, we next asked to what extent each HC can function like a chaperonin. HCs necessarily differ from TRiC and from each other in their biochemical properties, including the net charge on the inner surface of the folding chamber and the ability to hydrolyze ATP (Figure 5A). We used functional assays to correlate these biochemical properties with activity of the double ring HCs, independently of shared topology.
Figure 5. CCT holocomplexes segregate TRiC functions.
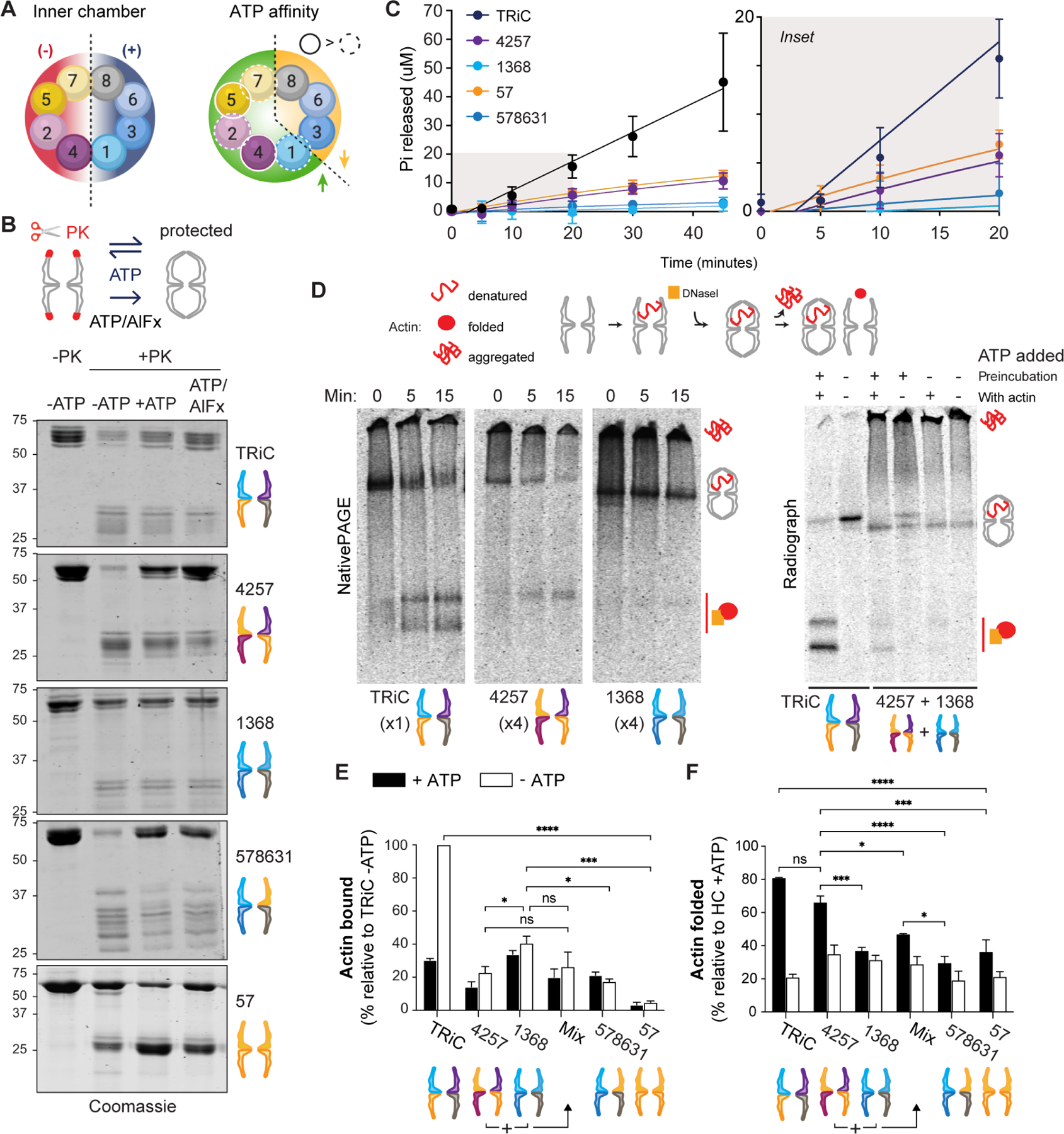
A. Segregation of net charge on the inner chamber surface (left) and of ATP affinity (right) within each TRiC ring, and thus between HCs. B. Conformational cycling assay read out by SDS-PAGE. ATP triggers chamber opening (unprotected) and closure (protected). More protection is observed for HCs containing high-ATP-affinity subunits. The transition state analog ATP/AlFx stabilizes the PK-protected state. C. ATPase activity of HCs by quinaldine red assay measuring Pi release over time. Error bars represent the SD. Right, magnified view of early time points. D. Radioactive labeled actin binding and folding assay. Representative radiographs after NativePAGE are shown; additional in Figure S6. Left, 15 minute time course in presence of ATP; right, the same assay using HCCCT4257 and HCCCT1368 incubated together for 30 minutes at 30°C (+/− ATP during preincubation) prior to introducing actin (+/− ATP). E. Actin bound by HCs relative to actin bound by TRiC (n = 3). F. Actin folded as a percentage of total actin present with each HC (n = 3). Error bars represent SEM. ns = not significant if p> 0.05, * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. All statistical comparisons to TRiC not indicated reached significance p < 0.01 (**). Two-way ANOVA, Tukey’s multiple comparisons test. See also Figure S5.
First, we asked if the lid changes conformation upon ATP hydrolysis using proteolytic protection assays. In the open apo-state, the TRiC lid segments protruding from the apical domains of CCT subunits are flexible and susceptible to proteinase K (PK) digestion, causing the appearance of a new band pattern between 25 and 35 kDa by SDS-PAGE (Figure 5B). The addition of ATP triggers conformational changes that include chamber closure, a state in which the structured lid segments are protected from digestion. Addition of the transition state analog ATP/AlFx further stabilizes the closed conformation, protecting the lid from PK (Figure 5B). Similar to TRiC, the lid segments of HCCCT4257 were susceptible to digestion in the apo-state and more protected in the presence of ATP. This is evidence of conformational cycling of HCCCT4257, possibly due to the CCTs with relatively high ATP affinity 27. However, ATP addition conferred greater PK protection to HCCCT4257 relative to TRiC, suggesting it cycles more slowly (Figure 5B). In contrast, ATP did not appreciably affect the PK sensitivity of HCCCT1368, which remained primarily in a PK-protected conformation. This HC contains subunits with lower ATP affinity, likely compromising its ATP cycling. Strikingly, inclusion of CCT5 and CCT7 to form HCCCT578631 restored the ATP-dependent change in PK sensitivity. In addition, HCCCT578631 yielded a unique digestion pattern with larger fragments, perhaps due to its reduced flexibility compared to TRiC revealed by cryo-EM (Figure S4H). We also examined the highly labile purified HCCCT57, finding it to be more protected than TRiC in the absence of ATP but still capable of conformational transitions (Figure 5B, bottom panel). These experiments indicate that HCs containing high ATP affinity subunits can conformationally cycle in response to ATP while the primarily low ATP affinity subunit HCs are less responsive.
Next we examined the ATPase activity of HCs using an inorganic phosphate (Pi) release assay 44. All HCs exhibited lower ATPase activity than TRiC (Figure 5C). HCCCT4257 and HCCCT57 hydrolyzed ATP at a faster rate than HCCCT1368, consistent with subunits’ relative affinities for ATP. HCCCT578631 exhibited a similar hydrolysis rate to HCCCT1368. Together, these assays demonstrate that, despite lacking a full complement of CCTs, non-canonical HCs retain varying abilities to hydrolyze ATP and engage in chamber opening and closure. Importantly, all noncanonical HC chaperonins exhibited aberrant cycling, likely due to disrupted intersubunit communication.
We next examined chaperonin activity of the HCs. We used a native gel assay to examine binding and folding of the obligate TRiC substrate actin in the presence and absence of ATP (Figure 5D). In this assay, when unfolded 35S-actin binds to TRiC or a HC, it migrates at a characteristic 1 MDa band whereas folded 35S-actin is detected as a faster-migrating complex with DNase I 45. 35S-actin that fails to bind chaperonin aggregates and does not enter the gel 20. Compared to TRiC, both HCCCT4257 and HCCCT1368 exhibited reduced binding to 35S-actin following a 15 minute incubation (Figure 5D; note higher molar ratios of HC). Extending incubation to 60 min offered no improvement (Figure 5E–F, S5A–B). However, we observed striking differences between these two HCs (Figure 5E). HCCCT1368 bound 35S-actin much more efficiently than HCCCT4257 (Figure 5D), reaching approximately 40% of binding relative to TRiC (Figure 5E). However, upon ATP addition, little to no HCCCT1368-bound actin was folded. In contrast, the smaller amount of actin that bound HCCCT4257 was efficiently folded upon ATP addition (Figure 5F). These experiments suggest a clear functional segregation among the subunits in the two hemispheres of TRiC that is remarkably retained when the hemispheres are assembled separately into HCs. We also confirmed that subcomplexes SCCCT24 and SCCCT57 did not demonstrate any activity toward the substrate (Figure S5A).
Lastly, we considered whether incubating HC species together to achieve a full complement of CCTs, prior to or alongside denatured actin, may improve binding or folding activity. This was not the case (Figure 5D–F; Figure S5A). Inclusion of subunits CCT5 and CCT7 to form the 6-CCT subunit HCCCT578631 did not augment folding ability relative to HCCCT1368 (Figure 5F). Altogether, these results indicate that only the TRiC holocomplex can mediate efficient actin binding and folding. Additionally, HCs consisting of the early-assembling subunits CCT2, CCT4, CCT5 and CCT7 do not efficiently bind denatured actin but are important for actin folding and for chaperonin cycling in response to ATP. In contrast, assemblies of subunits CCT6, CCT3, CCT8 and CCT1, which do not accumulate to any significant extent during normal biogenesis in mammalian cells, can bind actin effectively but cannot fold it. Taken together these analyses suggest a functional logic for the hierarchy of TRiC assembly in the physiological pathway observed in mammalian cells, which is to prevent accumulation of noncanonical chaperonin species with potential dominant negative activities toward substrate.
Discussion
This study provides insight into the longstanding question of how TRiC assembles to form the unique arrangement of this hetero-oligomeric complex conserved across eukaryotic cells. Assembly appears to proceed along a hierarchical pathway involving formation of CCT subcomplexes comprising the contiguous hemisphere containing CCT7, CCT5, CCT2, and CCT4, which templates individual incorporation of the remaining subunits as labile monomers that are otherwise rapidly degraded (Figure 6A). Surprisingly, CCT subunits have the potential to engage in noncanonical oligomerization to form stable TRiC-like complexes with aberrant chaperone activities. Thus, TRiC assembly is not driven by energetics alone but instead regulated to both promote a unique arrangement and to disfavor numerous alternative arrangements that are potentially harmful to the cell.
Figure 6. Model of TRiC hierarchical assembly and consequences of its dysregulation.
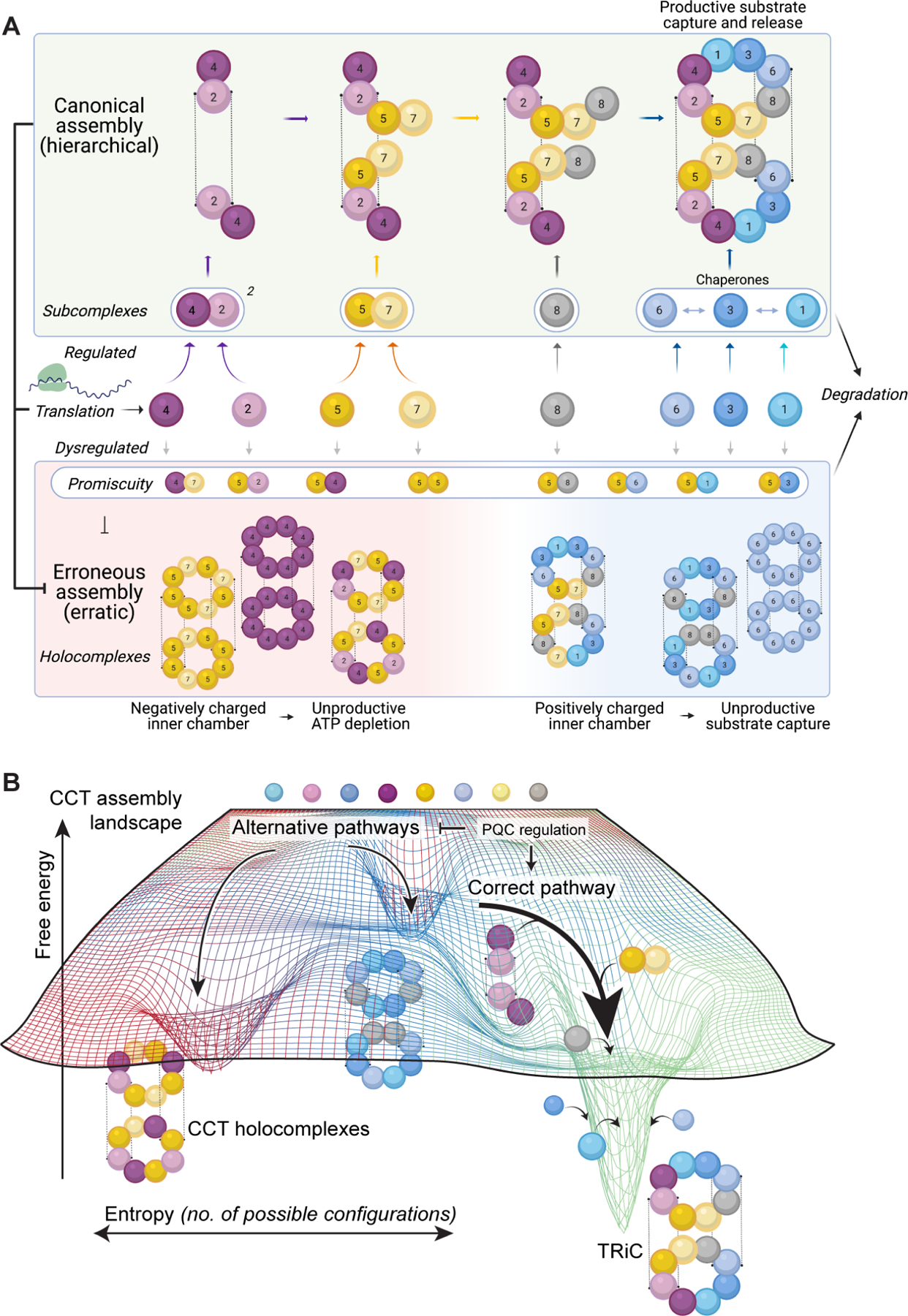
Co- or immediately post-translationally, CCT proteins from the negatively charged hemisphere assemble into subcomplexes and form a scaffold. CCT8 joins as a monomer, followed lastly by CCT1, CCT3, and CCT6, which are deterred from subcomplex formation amongst each other. Orphaned subunits and intermediates are rapidly degraded, and can form transient lower-order off-pathway associations with each other. This keeps the free energy landscape (B) broad, kinetically separating different holocomplexes and ensuring the cell can funnel subunits toward the proper pathway. Without ordered assembly, noncanonical holocomplexes can form with improper biochemical symmetry and aberrant function.
A hierarchical pathway assembles TRiC via subunit-specific CCT subcomplexes
Our data suggest that CCT2 and CCT4, adjacent in TRiC, form the initial scaffold. When CCT2 and CCT4 are co-overexpressed in mammalian cells, or in insect cells without other human CCTs, a single subcomplex containing both subunits forms with a mass between 200 and 400 kDa. Because CCT2 makes homotypic contacts across the stacked TRiC rings, its scaffold with CCT4 could adopt a topology similar to these subunits’ arrangement in the final complex, CCT4-CCT2:CCT2-CCT4 (where a hyphen represents a lateral intra-ring interface and the colon an equatorial inter-ring interface). Since other CCTs readily form recombinant TRiC-like structures without CCT2 or CCT4, the role of CCT2-CCT4 nucleating assembly in the cell appears to be regulatory rather than a structural requirement.
The next step in the hypothesized assembly pathway is the addition of CCT5 and CCT7 to the CCT4-CCT2 scaffold. We detected both CCT5 and CCT7 at dimeric sizes after disrupting assembly and after their co-overexpression in mammalian cells. Because CCT5 and CCT7 are cognate neighbors in each TRiC ring but do not appreciably contact their counterparts in the opposing ring, their incorporation as dimers is logical. However, these subunits can exceed the dimer state when in excess. In insect cells, CCT5 and CCT7 also formed a 1 MDa holocomplex. Recombinant CCT5-CCT7 complexes were chaperone-inert toward denatured actin despite a moderate rate of ATP hydrolysis. Interestingly, the inclusion of CCT2 and CCT4 conferred weak actin binding; however, the complex (HCCCT4257) folded very little of the actin it bound. This accords with prior work implicating subunits CCT2 and CCT4 in actin folding 20.
Our model posits that CCT8 joins the complex independently. When TRiC assembly was disrupted or CCT8 overexpressed, a population of monomeric CCT8 was always observed. Recombinantly, CCT8 was the only subunit appreciably detected in monomer-sized bands by mass spectrometry. As a monomer, CCT8 would diffuse and locate the proto-complex more rapidly than a multimer. PISA analysis revealed that CCT8 inter- and intra-ring interfaces rely on more electrostatic contacts than other interfaces in open-state TRiC, which would confer solubility as well as a relatively fast association rate 46,47. We propose that association of CCT8 with the CCT4-CCT2-CCT5-CCT7 intermediate would prevent further assembly into the noncanonical TRiC-like 1 MDa holocomplex observed in the recombinant system, driving assembly toward the correct TRiC end product. CCT8 overexpression alone is reported to stimulate TRiC assembly in tissues and extend organismal lifespan under proteotoxic stress 48.
Finally, CCT1, CCT3, and CCT6 join the complex to complete formation of TRiC. Our experiments cannot resolve the order in which these labile intermediates assemble, perhaps via redundant pathways of incorporation. Unlike the proposed early-assembling subunits, we found no evidence that CCT1, CCT3, and CCT6 are stable as monomers nor that they engage in lower-order oligomerization in mammalian cells. Nonetheless, when overexpressed in insect cells and absent early-assembling subunits, recombinant CCT1, CCT3, and CCT6 readily co-assemble with CCT8 into a stable TRiC-like HCCCT1368 HC that binds denatured actin relatively efficiently but is unable to fold it and is not responsive to ATP. Thus, assembly of even low amounts of these noncanonical complexes would be deleterious to the cell. Our results accord with previous ATP-binding studies 27 and with reported roles for individual subunits facilitating TRiC substrate recognition, not only of actin 20,26,49 but also VHL 50, tubulin 42,49,51, and AML1-ETO 49. The data suggest that, once a canonical core has formed, a double ring architecture can be reached to complete formation of noncanonical 16-mers in multiple ways. We propose that because there is no a priori requirement for a separate scaffold in order for CCT1, CCT3, and CCT6 to oligomerize into a double-ring architecture, their premature assembly is disfavored in vivo. This would serve to preclude the formation of substrate-binding surfaces and assembly with monomeric cellular CCT8 to yield detrimental dead-end chaperonin complexes.
Energetic and kinetic rationale for a hierarchical TRiC assembly pathway
While our data supports a hierarchy of events leading to the formation of TRiC, it leaves open possible ambiguity within the timeline of these events. For example, co-overexpressed CCT2 formed low abundance subcomplexes with CCT5 in addition to those formed with CCT4 in mammalian cells, which could indicate redundancy in TRiC nucleation. In addition, as discussed, CCT1, CCT3, and CCT6 could incorporate in parallel routes to reach the same end product. Our proposed hierarchy thus represents an average assembly pathway that may not rely entirely on obligate and rigid transitions but may be better conceived as a landscape that funnels the complex toward an energy minimum (Figure 6B). This is analogous to the multiple paths of ordered assembly proposed and later confirmed to form the 30S ribosome 9,52. This mode of assembly would confer robustness under conditions where subunit availability becomes moderately unbalanced. In remarkable agreement with our experimental observations, simulations have postulated that addition of individual subunits to preassembled clusters facilitates economical chaperonin formation 53,54. A process of early subcomplex formation followed by integration of smaller units can not only underlie the productive assembly of TRiC but also the formation of aberrant chaperonins in the recombinant system.
Our model is also consistent with physical constraints conferred by inter-subunit interface free energies. If intra-ring affinities are unsuitably high, polymerization will out-compete assembly; however, if these affinities are too low, the correct arrangement will become unstable 54. Consistent with this analysis, we find that CCTs do not interact especially tightly in vitro. Our model also avoids the slow process of two large, multiparticle single rings needing to meet in a specific orientation, which would constitute a substantial kinetic trap, particularly in the crowded cellular environment. A pathway of register-offset assembly proceeding along each ring increases specificity by requiring subunits to match both lateral and equatorial interfaces to incorporate. In addition, it implies slower off-rates when both interfaces must dissociate to detach, rendering the remainder of assembly kinetically accessible.
Evolutionary considerations of a hierarchical TRiC assembly pathway
While the hierarchical pathway assembling TRiC proceeds along an interaction landscape that ultimately disfavors noncanonical assemblies (Figure 6A), noncanonical interactions are not particularly unfavorable and CCT interfaces can form promiscuously. Evolutionary analyses may illuminate this paradox. TRiC evolved from multiple duplications of an ancestral homo-oligomeric chaperonin gene 25,55. Accordingly, promiscuous CCT interactions are likely evolutionary remnants where each gene product retained residual affinities while evolving enough contacts to entrench a new position within the ring 33,34,56. This promiscuity could be intertwined with regulation of assembly, ensuring noncanonical oligomers are transient and small in vivo (Figure 6A). Evolutionarily divergent from CCT8 but no longer considered CCTs, another family known as the BBS (Bardet-Biedl syndrome) proteins reportedly interact with six CCTs to form a complex of unknown topology specific to cilia 57–59. Future phylogenetic work will shed light on these evolutionary processes.
Therapeutic implications of TRiC assembly
Increases in CCT levels occur in many cancers 60–69 while proteostasis declines in aging and neurodegenerative diseases 70–72. Individual CCT subunits and even domains can ameliorate phenotypes linked to neurodegenerative diseases 73–75. Thus, better understanding of TRiC assembly could be leveraged in the rational design of TRiC activators and inhibitors with therapeutic applications in cancers and proteinopathies.
Limitations of the study
Limitations of the study include: (i) it does not provide insight into detailed mechanistic aspects; this will be an area for future research. (ii) It does not clarify if TRiC assembly requires dedicated factors or is mediated by generalist chaperone machinery. We did not detect assembly factors in association with various recombinant CCT oligomers by mass spectrometry other than chaperone Hsp70. Hsp70 associates with CCT nascent chains 76, supporting a role for this chaperone in their cotranslational folding. Additional assembly factors interacting dynamically with TRiC assembly intermediates may have dissociated prior to proteomic analyses. (iii) Coupling of folding and degradation by proteasomal and lysosomal pathways remain to be determined. CCT subunits can be extensively ubiquitylated 77,78, and a proteasome-targeting pathway for orphan CCT subunits was recently reported,79; however, we also find evidence of autophagic degradation of CCTs, with an unusual cumulative effect upon inhibition of both pathways. It is possible that different populations of CCT subcomplexes are degraded via distinct mechanisms. (iv) The study does not reveal how specific contacts between CCT2, CCT4, CCT5 and CCT7 mediate their ability to form stable subcomplexes, nor (v) how temporal and spatial control of translation, central to assembly of many protein complexes80, might mediate TRiC assembly.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Judith Frydman (jfrydman@stanford.edu).
Materials Availability
Plasmids generated in this study can be requested directly to lead contact.
Data and Code Availability
All unprocessed western blot, unprocessed negative stain images and XL-MS data have been deposited at Mendeley Data. Cryo-EM Map of HCCCT578631 has been deposited at EMDB, Electron Microscopy Data Bank. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD043532. All data are publicly available as of the date of publication. Accession numbers and DOI are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CCT1 | Santa Cruz Biotechnology | Cat # sc-374088 RRID:AB_10918262 |
| CCT1 | Abcam | Cat# ab240903 RRID:AB_2920800 |
| CCT2 | Abcam | Cat # ab92746 RRID:AB_10565196 |
| CCT2 (rabbit) | 81 | N/A |
| CCT3 | Abcam | Cat # ab106932 RRID:AB_10865346 |
| CCT3 | Santa Cruz Biotechnology | Cat # sc-271336 RRID:AB_10610107 |
| CCT4 | Santa Cruz Biotechnology | Cat # sc-137092 RRID:AB_2073768 |
| CCT5 | Abcam | Cat # ab129016 RRID:AB_11154964 |
| CCT5 (rabbit) | 81 | N/A |
| CCT6 | Santa Cruz Biotechnology | Cat # sc-514466 RRID:AB_11154964 |
| CCT7 | Santa Cruz Biotechnology | Cat # sc-271951 RRID:AB_10709576 |
| CCT8 | Santa Cruz Biotechnology | Cat # sc-377261 RRID:AB_2917991 |
| IRDye 680RD Donkey anti-rabbit IgG | LI-COR Biosciences | Cat# 926-68073 RRID:AB_10954442 |
| IRDye 800CW Donkey anti-mouse IgG | LI-COR Biosciences | Cat# 926-32212 RRID:AB_621847 |
| IRDye 800CW Donkey anti-Goat IgG (H + L) | LI-COR Biosciences | Cat# 926-32214 RRID:AB_621846 |
| IRDye 800CW Goat anti-Rat IgG (H + L) | LI-COR Biosciences | Cat# 926-32219 RRID:AB_1850025 |
| Peroxidase AffiniPure Donkey Anti-Rabbit IgG (H+L) | Jackson Immuno Research | Cat# 711-035-152 RRID:AB_10015282 |
| Peroxidase AffiniPure Donkey Anti-Mouse IgG (H+L) | Jackson Immuno Research | Cat# 715-035-151 RRID:AB_2340771 |
| Peroxidase AffiniPure Donkey Anti-Goat IgG (H+L) | Jackson Immuno Research | Cat# 705-035-003 RRID:AB_2340390 |
| Anti-GFP-MBP nanobody | Modified by MPB addition, this study | N/A |
| Bacterial and virus strains | ||
| E. coli strain DH10Bac | Thermo Fisher | Cat# 10361012 |
| Chemicals, peptides, and recombinant proteins | ||
| HCCCT578631 | This study | N/A |
| HCCCT1368 | This study | N/A |
| HCCCT4257 | This study | N/A |
| HCCCT57 | This study | N/A |
| TRIC | Collier et al., 2021 | N/A |
| DMEM supplemented with glucose | Thermo Fisher Scientific | Cat# 10566024 |
| Fetal Bovine Serum | Thermo Fisher Scientific | Cat# 26400044 |
| DharmaFECT #4 transfection reagent | DharmaFECT | Cat# T-2004-03 |
| DharmaFECT Cell Culture Reagent (DCCR) | Thermo Fisher Scientific | Cat# B-004500-100 |
| Complete, EDTA-free Protease Inhibitor Cocktail | Roche | Cat# 4693132001 |
| Lipofectamine™ 2000 | Thermo Fisher Scientific | Cat# 11668019 |
| Z-Leu-Leu-Leu-al | Sigma-Aldrich | Cat# C2211-5MG |
| Ammonium Chloride | Sigma-Aldrich | Cat# A9434 |
| Benzonase® Nuclease | Sigma-Aldrich | Cat# E1014-5KU |
| Ni Sepharose® High Performance | Sigma-Aldrich | Cat# GE17-5268-02 |
| Calmodulin Sepharose™ 4B | VWR | Cat# 95016-874 |
| Amylose Resin | New England Biolabs | Cat# E8021S |
| DSS-H12/D12 | Creatie Molecules Inc. | Cat# 001S |
| Adenosine 5’-triphosphate disodium salt hydrate | Sigma-Adlrich | Cat# A7699 |
| DNaseI | Worthington Biochemical | Cat# LS006333 |
| DNaseI | Sigma-Aldrich | Cat# D5025 |
| Critical commercial assays | ||
| Quick-RNA miniprep Kit | Zymo Research | Cat# R1054 |
| iScript cDNA Synthesis Kit | Bio-Rad | Cat# 1708891 |
| Itaq™ Universal SYBR assay | Bio-Rad | Cat# 1708891 |
| [35S] EasyTag™ express protein labeling mix | Perkin Elmer | Cat# NEG772002MC |
| Deposited data | ||
| Unprocessed western blot deposited in Mendeley Data | This paper | DOI: 10.17632/ymrbcv2wrt.1 |
| Unprocessed images of negative stain | This paper | DOI: 10.17632/ymrbcv2wrt.1 |
| XL-MS raw data in Mendeley Data | This paper | DOI: 10.17632/ymrbcv2wrt.1 |
| Map of HCCCT578631 | This paper | EMD-32393 |
| Coordinates of the TRiC in open conformation | Gestaut et al. 2022 | PDB:7WU7 |
| Experimental models: Cell lines | ||
| Hela cells (CCL-2) | ATCC | CCL-2 |
| SF9 cells | Thermo Fisher Scientific | 12659-017 |
| High Five cells (Hi5) | Invitrogen | B85502 |
| Oligonucleotides | ||
| Primers used in this study are listed in Table S1 | This paper | N/A |
| Recombinant DNA | ||
| pFB-Duet CCT7His, CCT5 | Gestaut et al., 2019 | pDG463 |
| pFB-Duet CCT2, CCT4, | Gestaut et al., 2019 | pDG443 |
| pFB-Duet CCT6, CCT3 | Gestaut et al., 2019 | pDG445 |
| pFB-Duet CCT8, CCT1 | Gestaut et al., 2019 | pDG446 |
| pFB-Duet CCT1CBP, CCT8 | Gestaut et al., 2019 | pDG518 |
| pFB-Duet CCT1GFP, CCT8 | Gestaut et al., 2019 | pDG517 |
| pFB-Duet HIS-ACTB | This study | pYC26 |
| pcDNA3.1 CCT2-Ch C-terminal mCherry | (Zhao et al., 2016) | pJF1919 |
| pcDNA3.1 CCT4-Ch C-terminal mCherry | (Zhao et al., 2016) | pJF1921 |
| pcDNA3.1 CCT5-Ch C-terminal mCherry | (Zhao et al., 2016) | pJF1922 |
| pCMV6 CCT1 | Eric Jonasch | N/A |
| pCMV6 CCT2 | Eric Jonasch | N/A |
| pCMV6 CCT3 | Eric Jonasch | N/A |
| pCMV6 CCT4 | Eric Jonasch | N/A |
| pCMV6 CCT5 | Eric Jonasch | N/A |
| pCMV6 CCT6 | Eric Jonasch | N/A |
| pCMV6 CCT7 | Eric Jonasch | N/A |
| pcDNA YFP | Addgene | #13033 |
| siGENOME Human TCP1 (6950) siRNA - SMARTpool | Thermo Scientific | M-012749-01-0005 |
| siGENOME Human CCT2 (10576) siRNA - SMARTpool | Thermo Scientific | M-020107-02-0005 |
| siGENOME Human CCT3 (7203) siRNA - SMARTpool | Thermo Scientific | M-018339-01-0005 |
| siGENOME Human CCT4 (10575) siRNA - SMARTpool | Thermo Scientific | M-020147-00-0005 |
| siGENOME Human CCT5 (22948) siRNA - SMARTpool | Thermo Scientific | M-012797-02-0005 |
| siGENOME Human CCT6A (908) siRNA - SMARTpool | Thermo Scientific | M-016559-01-0005 |
| siGENOME Human CCT7 (10574) siRNA - SMARTpool | Thermo Scientific | M-020115-01-0005 |
| siGENOME Human CCT8 (10694) siRNA - SMARTpool | Thermo Scientific | M-008566-02-0005 |
| siGENOME Non-Targeting siRNA Pool #2 |
Thermo Scientific | D-001206-14-05 |
| Software and algorithms | ||
| ImageJ | Schneider et al. 2021 | https://imagej.nih.gov/ij/index.html |
| GraphPad Prism 7.04 & 9.1.2 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| CFX Maestro Manager | BioRad | https://www.bio-rad.com/en-us/product/cfx-maestro-software-for-cfx-real-time-pcr-instruments?ID=OKZP7E15 |
| Image Studio Lite | Li-COR Biosciences | https://www.licor.com/bio/image-studio-lite/ |
| Pymol 1.8.6.2 | Schrӧdinger | https://pymol.org/2/ |
| UniDec v3.1.0 | Marty et al., 2015 | https://github.com/michaelmarty/UniDec |
| MaCSED v0.3 | Justin Benesch | benesch.chem.ox.ac.uk/resources.html |
| MaxQuant v1.6.14 | Tyanova et al., 2016a | https://maxquant.net/download_asset/perseus/latest |
| Perseus v1.6.5 | Tyanova et al., 2016b | https://maxquant.net/download_asset/perseus/latest |
| cryosparc2 | Punjani et al., 2017 | https://cryosparc.com/ |
| RStudio Versi on 1.2.1335 | The R foundation | https://www.r-project.org/ |
| xQuest | Walzthoeni et al., 2012 | https://bioinformaticshome.com/tools/proteomics/descriptions/xQuest.html |
| UCSF ChimeraX | Pettersen et al., 2021 | https://www.cgl.ucsf.edu/chimerax/ |
Experimental Model and Study Participant Details
Cell culture and growth conditions
HeLa cells were maintained at 37°C in 5% CO2 and grown in DMEM supplemented with glucose (ThermoFisher #10566024) containing 10% FBS (ThermoFisher #26400044) and 100 U ml−1 of penicillin/streptomycin (ThermoFisher #15140163) and culture was split 1:10 every four days. SF9 cells were maintained in suspension at 27°C with shaking at 130 rpm and grown in SF900-III SFM (Gibco #12658027) medium supplemented with 2% FBS, 100 U ml−1 of penicillin/streptomycin. Cells are grown to 5×106 cells ml−1, diluted every 3 days to 0.5×106 cells ml−1.
High Five cells were maintained in High Five™ Cells in Express Five™ Medium (Gibco #B85502) supplemented with 20 mM Glutamine and 100 U ml−1 of penicillin/streptomycin. Cells are grown to 5×106 cells ml−1, diluted every 3 days to 0.5×106 cells ml−1.
METHOD DETAILS
Cell treatments and transfection
Cells were transfected with siRNA per manufacturer’s protocol (DharmaFECT). In short, HeLa cells were seeded at 3.5×105 cells per well in antibiotic-free complete media and grown to confluency. 25 µM siRNA solution and 2 uL DharmaFECT formulation #4 were diluted into DharmaFECT Cell Culture Reagent (DCCR) and incubated 30 minutes before being added to cells. Treated cells were incubated at 37°C in 5% CO2 for 72 hours before harvesting. Cells were lysed in 25 mM HEPES pH 7.5, 150 mM KCl, 1.5 mM MgCl2, 0.5% NP-40, 10% glycerol, 1 mM DTT, complete protease inhibitor cocktail (Roche). Lysates were incubated at 4°C for 10 minutes and clarified at 19,000×g for 10 minutes. 10–20 µg lysate was used for NativePAGE and immunoblotting.
CCT overexpression was done by transfection with Lipofectamine 2000 (Invitrogen) per manufacturer’s protocol. In short, 1 × 106 cells per well were seeded in 6-well plates; 2 µg each plasmid was used per 5 ul lipofectamine 2000 reagent; and cells were harvested after 24 hours and lysed as described above. For protein clearance assays, cells were treated for 2 hours with 20 µM MG132 (Z-Leu-Leu-Leu-al, Sigma) alone or in combination with 10 mM NH4Cl2 (Sigma) 72 hours post-siRNA transfection and harvested as described above.
Molecular Cloning
Human b-tubulin was amplified using primers listed in Table S1, from human cDNA library and fused with N-terminal 6x-HIS tag. The gene fragment was cloned into pFastbac duo-vector (Thermo Fisher Scientific) via XhoI and KpnI sites under p10 promoter for insect cell expression. The ligated product was transformed into DH5a strain. Plasmid was extracted from DH5a using QIAprep Spin miniprep kit (Qiagen) and sequencing verified before transformed into MAX efficiency DH10Bac competent cells (Thermo Fisher Scientific) according to manufacturer’s instructions. Recombinant Bacmid was isolated using Qiagen mini-prep and used to transfect SF9-II cells (Thermo Fisher Scientific) for baculovirus production.
Western blot analysis
SDS-PAGE gels were 10% acrylamide Tris-Glycine (cast in-house). Blue and clear NativePAGE gels (4–16% Bis-Tris) were run per manufacturer recommendations (Invitrogen), with 1 mg ml−1 cysteine and 1 mM DTT added to the running buffer. Western blotting was carried out via transfer to a nitrocellulose membrane and immunoblotting against each subunit. Santa Cruz antibodies were used for: CCT1 (sc-374088), CCT3 (sc-271336) CCT4 (sc-137092), CCT6 (sc-514466), CCT7 (sc-271951), and CCT8 (sc-377261); Abcam antibodies were used for: CCT1 (ab240903), CCT2 (ab92746), CCT3 (ab106932), and CCT5 (ab129016). Rabbit polyclonal antibodies were used for CCT2 and CCT5 81. Blots used for quantification were detected by chemiluminescence using HRP conjugated secondary antibodies and blotted against x-ray film. Secondary HRP conjugated antibodies from Jackson ImmunoResearch are: Donkey anti-Rabbit (#711-035-152), Donkey anti-Mouse (#715-035-151), Donkey anti-Goat (#705-035-003). HeLA lysate dilutions were blotted using fluorescently conjugated antibodies to capture the linear range of detection for the primary antibodies used in this study (Figure S1B–C). Fluorescent antibodies from LI-COR Biosciences used were: Donkey anti-rabbit (#926-68073), Donkey anti-mouse (#926-32212), Donkey anti-goat (#926-32214). Only film exposures within a linear range and comparable to fluorescence signals were used for quantification. For quantification of HeLA lysate after siRNA treatment, three blots from three independent experiments were quantified by densitometry. The average fraction remaining after siRNA treatment compared to NT control was plotted after background subtraction and normalized to GAPDH.
For size exclusion chromatography of cell lysate, untreated and siRNA-treated HeLa cells were lysed as described above. 2 mg total protein was separated using a Superdex 200 Increase 10/300 GL column in MQA buffer (50 mM NaCl, 20 mM HEPES pH 7.4, 5 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 10% Glycerol) and collected in 1 ml fractions. 15 µl each fraction in the untreated sample was used for immunoblotting each CCT subunit, and 60 µl of each fraction was used for the siRNA treated samples.
RNA isolation and quantitative reverse transcription PCR (qRT-PCR)
For each biological replicate, cells were washed with PBS and total RNA was extracted using Quick-RNA miniprep (Zymo Research) according to manufacturer’s instructions including a DNAseI treatment step. RNA was subject to Bioanalyzer to determine concentration, 100 ng RNA were used to synthesize cDNA using iScript reverse transcriptase kit (BioRad) following manufacturer’s protocol. RT-qPCR was performed on a CFX-96 thermocycler using Itaq™ Universal SYBR Green Supermix (BioRad) in a 96-well plate format. All measurements were performed in a CFX Connect real-time PCR cycler (BioRad). Ct values were normalized by GAPDH mRNA and analyzed by dCT method. Results for three biological replicates were averaged, mean and SD are plotted as −Log2. Primer sequences for all genes analyzed are listed in Table S1.
Protein expression and purification
Multi-step purification of subcomplexes and holocomplexes
Complexes were expressed as described previously 43 using the Bac-to-Bac baculovirus expression system (Invitrogen). P3 virus stocks were prepared as described using plasmids pDG463, pDG443, pDG445 or pDG446, each encoding a CCT pair. To generate CCT subcomplexes and holocomplexes, 1 liter of Hi5 cells at density 1 × 106 cells x ml−1 was infected with 5 ml of each desired virus. For example, for dual CCT expression experiments a single virus was used; to purify HCCCT578631, three viruses were used. Infected cells were harvested after 72 hours and lysed using an Emulsiflex (Avestin) in buffer consisting of 50 mM HEPES pH 7.4, 100 mM NaCl, 4 mM imidazole, 0.05% NP-40, 10% glycerol, 1 mM DTT, 2 mM PMSF, complete protease inhibitor cocktail (Roche), and benzonase 5 U ml−1 (Sigma-Aldrich). Lysate was clarified at 50,000 × g for 1 hour. Holocomplexes and subcomplexes (Figures 4 and 5) were purified by gravity flow using Ni Sepharose HP (Cytiva) or Calmodulin Sepharose 4B (Cytiva) per manufacturer’s instructions and then separated by SEC (Superdex 200 26/60, Cytiva) in MQA buffer.
Single step affinity purification of subcomplexes and holocomplexes
For single step separations (Figure 3, Figure S3C–D), lysates expressing CCT7His were incubated with Ni Sepharose HP (Cytiva) 1 hour at 4°C with rotation, washed 2 times with lysis buffer (varying stringencies as described in the text) and eluted in 200 mM imidazole for 10 minutes at 4°C.
Immunoprecipitation of GFP-tagged subcomplexes and holocomplexes
Lysates expressing CCT1GFP were incubated with anti-GFP-MBP nanobody 82 (modified by MBP addition and purified in-house) for 30 minutes at 4°C with rotation, then incubated with amylose resin (NEB) for 1 hour at 4°C with rotation, washed 2 times with lysis buffer and eluted in 10 mM maltose for 20 minutes at 4°C. Fractions containing complexes of interest were identified by SDS-PAGE, concentrated to 300 μl in an Amicon Ultra-4 centrifugal filter 100 X kDa MWCO (Millipore), aliquoted and snap frozen. Concentrations were measured by BSA assay.
Metabolic labeling of recombinant CCTs
After 72 hours expression, 50 ml aliquots of infected Hi5 cells expressing CCT combinations as described above were starved for 30 minutes in Sf-900 II™ serum-free media (Invitrogen) lacking methionine and cysteine, then labeled with 20 µCi ml−1 [35S] EasyTag™ express protein labeling mix (Perkin Elmer) for 30 minutes, harvested and snap frozen. Detection was performed by exposing a storage phosphor screen (GE), scanned on a GE Amersham Molecular Dynamics Typhoon 9410 (GE) and analyzed using Image J.
Metabolic labeling of actin
For 35S-HIS-ACTB expression, 50 ml Hi5 cells were infected with 5 ml P3 virus (pYC26) and allowed to express for 2 days. Cells were harvested and starved for 15 minutes in Sf-900 II™ serum-free media (Invitrogen cat #21012026) lacking methionine and cysteine, supplemented with 2% dialyzed FBS. The culture was incubated for 19 hours with 0.02 µCi ml−1 [35S] EasyTag™ express protein labeling mix (Perkin Elmer), supplemented with 0.01% Express Five™ serum-free media. Cells were harvested and lysed in 1 M Tris-HCl pH 8.0, 0.5 mM MgCl2, 0.5 mM ATP, 4% Triton X-100, 1 mg ml−1 tween-20, 1 mM DTT, containing complete protease inhibitor cocktail (Roche) and 5 U ml−1 benzonase. Lysates were clarified at 20,000×g for 20 minutes, diluted into 5.5 M GnHCl with 20 mM imidazole, and incubated at room temperature for 30 minutes. β-actin was captured by gravity flow using Ni Sepharose HP (Cytiva), buffer exchanged using an amicon ultra concentrator with MWCO 10 kDa (Millipore) into 50 mM Tris pH 8.0, 6 M GnHCl, 50 mM DTT, concentrated to 40 ul, aliquoted and snap frozen.
TRiC reconstitution experiments
Five mL of Hi5 cell cultures, each infected with a single virus expressing a different CCT pair, or equivalent volume of cells expressing all 8 CCTs were harvested and lysed individually in 1.5 ml lysis buffer (50 mM HEPES, 50 mM KCl, 1.5 mM MgCl2, 0.5% NP-40, 10% glycerol, 2 mM PMSF, complete protease inhibitor cocktail (Roche), and benzonase 5 U ml−1 (Sigma-Aldrich)). Lysates expressing pairs were mixed in the presence or absence of additional 6 mM ATP or 25 mM MgCl2, and ATP regeneration system (0.2 mg ml−1 creatine kinase and 20 mM creatine phosphate kinase) as described in the text, incubated for 30 minutes at 4°C or 23°C where indicated, and centrifuged at 19,000×g for 20 minutes. For other listed formulations, 5 μg HCCCT2457, 4 μg TRiC or 3.4 μg HeLa cell lysate were used. 10 µl each sample was analyzed by NativePAGE (Invitrogen). Gels were transferred to a PVDF membrane (Biorad) and immunoblotted against CCT8 or, when 35S labeled, exposed to a storage phosphor screen (GE), scanned on a GE Amersham Molecular Dynamics Typhoon 9410 (GE) and analyzed using Image J.
Negative staining and electron microscopy
Complexes were diluted to 0.05 mg ml−1 in 20 mM HEPES, 50 mM NaCl, 5mM MgCl2, 1 mM EDTA, 1 mM DTT. 5 μl sample was placed for 1 minute on a formvar/carbon coated 200 holey mesh Ultra Thin copper grid (FCF200-Cu-UA, Electron Microscopy Sciences). The grid was blotted on filter paper, floated over a drop of stain for 20 seconds in 0.75% uranyl formate made fresh from powder (Electron Microscopy Sciences) and blotted again, the stain and blotting repeated 2 more times, then air dried. Grids were imaged using a JEOL JEM1400 transmission electron microscope.
Cryo-EM grid preparation and data collection
For cryo-EM specimen preparation and imaging, 3 μl aliquots of 0.5 mg ml−1 HCCCT578631 were applied to glow-discharged holey-carbon Quantifoil R1.2/1.3 grids, blotted for 2–3 seconds, and then plunge-frozen in liquid ethane using a EM GP plunge freezer (LEICA). Grids were transferred into cartridges and loaded into a 300 KeV JEM 3200FSC (JEOL) electron microscope with an in-column Ω filter (25 eV energy slit). We recorded images at 0.7–2.5 μm underfocus on a K2 summit direct electron detector (Gatan) at the magnification corresponding to a sampling of 1.43 Å pixel−1.
Cryo-EM map reconstruction
We imported 401 movies from analysis of HCCCT578631 and all image processing was performed in cryosparc2 83. Briefly, we performed patch-based correction for micrographs and performed contrast transfer function parameters estimation using Gctf 84. Manually picked particles were averaged and served as templates for automatic particle picking. We then performed 2D reference-free averaging and converged particles were subjected to ab initio map reconstruction to generate an initial model. We applied C1 symmetry for final reconstruction and global non-uniform refinement resulted in maps at 7.4 Å resolution based on the gold-standard Fourier shell correlation (FSC) at 0.143. The TRiC structure used herein is in preparation as part of separate work and will be cited at the time of publication. The TRiC map was low-pass filtered at 7.4 Å for direct comparison with HCCCT578631.
Native mass spectrometry
Samples were buffer exchanged into 500 mM ammonium acetate using a minimum of five resuspensions in Amicon centrifugal filters with 50 kDa molecular weight cutoff (Millipore). A Q-Exactive Plus UHMR (Ultra-High Mass Range) orbitrap mass spectrometer (ThermoFisher Scientific) was used with parameters described previously 33. Raw data were summed in XCalibur for visualization and imported into UniDec v3.1.0 85 for initial assignments of charge series. Clusters of charge series (e.g. monomers, dimers, tetramers, and higher-order species) were processed separately to improve fitting by grouping peaks with similar intensities. Charge series identified in UniDec were then input into the Mass and Charge State Evaluation and Determination tool (MaCSED v0.3, available online at benesch.chem.ox.ac.uk/resources.html) for manual mass assignment and extraction of the error associated with each assignment. Monomers and oligomers were assigned using subunit masses from samples prepared in the same system 33. Where multiple CCT combinations were possible within the standard deviation of a measurement, the oligomer was left unassigned.
LCMS
Samples derived from NativePAGE bands were prepared and analyzed as described previously 33. Briefly, bands were excised and protein was digested with trypsin in-gel before destaining, reduction, and alkylation. Gel pieces were dehydrated and dried completely, and peptides were extracted in 50% acetonitrile, 5% formic acid. Peptides were cleaned using C-18 resin and analyzed on a Velos Pro Elite Orbitrap Mass Spectrometer (Thermo Fisher Scientific) coupled with a NanoAcquity UPLC system (Waters). Data were processed using MaxQuant v1.6.14 86 and Perseus v1.6.5 87 with parameters and filtering as described 33. Searches were carried out against the T. ni proteome (with partial tCCT sequences replaced by complete NCBI sequences) plus all human CCTs expressed in the given sample. Relative intensity-based absolute quantification (iBAQ) values were calculated as fractions of the summed intensity 88, and proteins present at greater than 1% of the total protein content by iBAQ estimation were plotted in pie charts using R.
Purified holocomplexes were prepared for LCMS and analyzed using StageTips as described previously 89.
Cross-linking mass spectrometry
Samples were cross-linked and analyzed as described previously 43 using cross-linking reagent DSS-H12/D12 (Creative Molecules Inc). In brief, 50 μg of each purified holocomplex was incubated with 1 mM DSS for 30 minutes at 37°C, then the reaction was quenched with 50 mM Tris pH 7.4 for 15 minutes. Cross-linked samples were digested with endoproteinase Lys-C and trypsin, purified by solid-phase extraction and fractionated by size exclusion chromatography. Fractions were analyzed in duplicate by LC-MS/MS on an Orbitrap Elite mass spectrometer (ThermoFisher Scientific). MS data were searched using xQuest 90 and results were filtered at a 5% false discovery rate at the non-redundant peptide pair level. Distances were extracted using PDB (Protein Data Bank) entry 6NRA as a structural model by measuring intra-ring and inter-ring distances of the subunit pairs first to determine canonical contacts. If the distance constraint of 30 Å was exceeded, then distance was modeled to the analogous residue of the closest inter-ring or intra-ring neighboring subunit and designated to be a noncanonical contact. Results summarized in Figure 4F are detailed in Table S2.
PK protection assay
Holocomplexes were diluted to 250 nM in 14 μl ATPase buffer (20 mM Tris HCl pH 7.4, 100 mM KCl, 5 mM MgCl2, 10% glycerol, 1 mM DTT) and heated to 37°C. 6 μl either H2O (open chaperonin condition) or 2 μl 10 mM ATP, 2 μl 10mM AlNO3 and 2 μl 60 mM NaF in that order (closed chaperonin condition) was added bringing total reaction volumes to 20 μl. Samples were incubated for 10 minutes before a 5 minute digestion with 20 ng μl−1 Proteinase K (diluted into each sample from 20X stock) (Sigma-Aldrich, P2308) at room temperature. Reactions were stopped by adding 1 μl 200 mM PMSF and put on ice. Samples were analyzed by 15% SDS-PAGE and stained with Coomassie blue.
ATPase assay
The assay was done as previously described with modifications as noted 44. In short, stock solutions of 0.05% w/v quinaldine red (QR), 2.32% w/v polyvinyl alcohol, 5.72% w/v ammonium heptamolybdate tetrahydrate in 6M HCl, and water were mixed in a 2:1:1:2 ratio to prepare the QR reagent fresh prior to each experiment. 100 nM TRiC or HC was diluted in ATPase buffer (25 μl total reaction volume), preheated to 37°C and added to 2.5 μl water or 10 mM ATP to start the reaction, then incubated for the indicated time points. Reactions were stopped by addition of 5 μl 60 mM EDTA in a Corning 96-well opaque non-sterile polystyrene plate (Sigma-Aldrich, #CLS3992) on ice. After all time points were collected, reactions were developed by adding 80 μl QR reagent for 15 minutes then quenched by adding 10 μl 32% (w/v) sodium citrate. Fluorescence intensity was measured (excitation 430 nm, emission 530 nm) using a CLARIOstar plate reader (BMG Labtech). Analysis was performed by fitting a phosphate standard curve with a one-phase decay function and derived the parameters used to calculate the amount of phosphate released from CCT complexes.
Actin binding and folding assay
TRiC was diluted to 0.25 μM and holocomplexes to 1 μM in ATPase buffer. 35S-actin was diluted 1:100 (1 μM final) with TRiC or holocomplexes and allowed to bind for 30 minutes on ice. Samples were spun for 15 minutes at 19,000×g at 4°C to remove aggregated actin. Supernatants were transferred to fresh tubes containing H2O or final 1 mM ATP and incubated for 1 hour at 37°C. 5 μg DNaseI (Worthington Biochemical #LS006333 and Sigma-Aldrich #D5025) was added to each sample to bind folded actin. Samples were incubated for 10 minutes at 4°C and spun at 19,000×g at 4°C to remove large aggregates. All of each sample was run on 4–12% NativePAGE (Invitrogen) gels which were then dried at 80°C for 2 hours, exposed to a storage phosphor screen (GE), scanned on a GE Amersham Molecular Dynamics Typhoon 9410 (GE) and analyzed using Image J.
Actin binding (Figure 5E) was quantified as follows, where each value is a signal intensity measurement:
Percent actin folded by holocomplexes relative to total actin engaged (bound or folded) by TRiC (Figure S5C) was quantified as follows:
Percent actin folded by holocomplexes relative to individually limiting amounts of actin engaged (bound or folded) by each holocomplex (Figure 5F) was quantified as follows:
QUANTIFICATION AND STATISTICAL ANALYSIS
Data visualization
Structural and experimental schematics in Figures 1A, 1E, 2A–E, 3A, 3F, 4F, 5A, and 6A were generated using BioRender.com. Structures in Figure 4E, S2, S4F were visualized in UCSF ChimeraX 91.
Statistical analysis
Gel and densitometry for bands in Figure 1B, and 5D were done with ImageJ. Data in Figures 1C, 1D, 5E, 5F, and S1B, were plotted using Prism (n=3). Data in Figure 1C represent the mean as a fraction of CCT protein remaining after knockdown normalized to NT control and SEM (p = 0.0001). Data in Figure 5C, S1A, and S1C represent the mean and SD. Data in Figure 5F, 5E represent the mean as a fraction of actin bound by TRiC and as a fraction of total actin present in each holocomplex, respectively. Error bars represent SEM (ns = not significant if p> 0.05, * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001). All statistical comparisons to TRiC not indicated reached significance p < 0.01 (**). Two-way ANOVA, Tukey’s multiple comparisons test. Data in Figure S6C represent the mean of actin folded by each complex relative to actin bound by TRiC and SEM (p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001, two-way ANOVA, Tukey’s multiple comparisons test). Scatter plots in Figure S2 were calculated and produced using R. Pie charts in Figure S4E, S5D were produced using R.
Supplementary Material
Table S2. Structural modeling of holocomplex cross-links, related to Figure 4.
Highlights.
TRiC assembles hierarchically through defined intermediates along a biochemical axis
Subcomplexes of adjacent subunits CCT2-4-5-7 nucleate concurrent double ring assembly
Monomeric CCT8 and labile CCT1, CCT3 and CCT6 join individually to form final complex
Hierarchical regulation disfavors alternate CCT arrangements with aberrant function
Acknowledgments
Supported by NIH grants GM074074 (J.F.), GM05643321 (J.F.), T32 AG0047126 (M.P.C.), National Research Foundation (NRF) 2021M3A9I4021220 and SUHF foundation (to S.-H.R). Resources provided by Stanford Research Computing Center, cryoEM data collected at NCMI at Baylor College of Medicine (P41GM103832 and S10OD021600), proteomic data collected at NCRR, UCSF (ARRA 1S10RR026780-01). We thank Pierre Rodriguez-Aliaga, Yanyan Zhao, Yu-Chan Chen, Al Burlingame and Wah Chiu for help, and members of the Frydman lab for discussions and advice.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interest Statement
JF is a member of the Molecular Cell advisory board. The authors declare no additional competing interests.
References
- 1.Gray MW, Lukes J, Archibald JM, Keeling PJ, and Doolittle WF (2010). Cell biology. Irremediable complexity? Science 330, 920–921. [DOI] [PubMed] [Google Scholar]
- 2.Stoltzfus A (2012). Constructive neutral evolution: exploring evolutionary theory’s curious disconnect. Biol. Direct 7, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mallik S, and Tawfik DS (2020). Determining the interaction status and evolutionary fate of duplicated homomeric proteins. PLoS Comput. Biol 16, e1008145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Juszkiewicz S, and Hegde RS (2018). Quality Control of Orphaned Proteins. Mol. Cell 71, 443–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brennan CM, Vaites LP, Wells JN, Santaguida S, Paulo JA, Storchova Z, Harper JW, Marsh JA, and Amon A (2019). Protein aggregation mediates stoichiometry of protein complexes in aneuploid cells. Genes Dev 33, 1031–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martinez-Salas E, Embarc-Buh A, and Francisco-Velilla R (2020). Emerging Roles of Gemin5: From snRNPs Assembly to Translation Control. Int. J. Mol. Sci 21. 10.3390/ijms21113868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marshall RS, and Vierstra RD (2019). Dynamic Regulation of the 26S Proteasome: From Synthesis to Degradation. Front Mol Biosci 6, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng J, Berninghausen O, and Beckmann R (2021). A distinct assembly pathway of the human 39S late pre-mitoribosome. Nat. Commun 12, 4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Talkington MWT, Siuzdak G, and Williamson JR (2005). An assembly landscape for the 30S ribosomal subunit. Nature 438, 628–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bubeck D (2014). The making of a macromolecular machine: assembly of the membrane attack complex. Biochemistry 53, 1908–1915. [DOI] [PubMed] [Google Scholar]
- 11.Singh S, Vanden Broeck A, Miller L, Chaker-Margot M, and Klinge S (2021). Nucleolar maturation of the human small subunit processome. Science 373. 10.1126/science.abj5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nikolay R, Hilal T, Schmidt S, Qin B, Schwefel D, Vieira-Vieira CH, Mielke T, Bürger J, Loerke J, Amikura K, et al. (2021). Snapshots of native pre-50S ribosomes reveal a biogenesis factor network and evolutionary specialization. Mol. Cell 81, 1200–1215.e9. [DOI] [PubMed] [Google Scholar]
- 13.Tomko RJ Jr, and Hochstrasser M (2013). Molecular architecture and assembly of the eukaryotic proteasome. Annu. Rev. Biochem 82, 415–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park S, Li X, Kim HM, Singh CR, Tian G, Hoyt MA, Lovell S, Battaile KP, Zolkiewski M, Coffino P, et al. (2013). Reconfiguration of the proteasome during chaperone-mediated assembly. Nature 497, 512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gestaut D, Limatola A, Joachimiak L, and Frydman J (2019). The ATP-powered gymnastics of TRiC/CCT: an asymmetric protein folding machine with a symmetric origin story. Curr. Opin. Struct. Biol 55, 50–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Skjærven L, Cuellar J, Martinez A, and Valpuesta JM (2015). Dynamics, flexibility, and allostery in molecular chaperonins. FEBS Lett 589, 2522–2532. [DOI] [PubMed] [Google Scholar]
- 17.Kampinga HH, Mayer MP, and Mogk A (2019). Protein quality control: from mechanism to disease : EMBO Workshop, Costa de la Calma (Mallorca), Spain, April 28 - May 03, 2019. Cell Stress Chaperones 24, 1013–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yokota SI, Yanagi H, Yura T, and Kubota H (2000). Upregulation of cytosolic chaperonin CCT subunits during recovery from chemical stress that causes accumulation of unfolded proteins. Eur. J. Biochem 267, 1658–1664. [DOI] [PubMed] [Google Scholar]
- 19.Lopez T, Dalton K, and Frydman J (2015). The Mechanism and Function of Group II Chaperonins. J. Mol. Biol 427, 2919–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Balchin D, Milicic G, Strauss M, Hayer-Hartl M, and Hartl FU (2018). Pathway of Actin Folding Directed by the Eukaryotic Chaperonin TRiC. Cell 174, 1507–1521 e16. [DOI] [PubMed] [Google Scholar]
- 21.Lewis SA, Tian G, and Cowan NJ (1997). The alpha- and beta-tubulin folding pathways. Trends Cell Biol 7, 479–484. [DOI] [PubMed] [Google Scholar]
- 22.Saegusa K, Sato M, Sato K, Nakajima-Shimada J, Harada A, and Sato K (2014). Caenorhabditis elegans chaperonin CCT/TRiC is required for actin and tubulin biogenesis and microvillus formation in intestinal epithelial cells. Mol. Biol. Cell 25, 3095–3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amit M, Weisberg SJ, Nadler-Holly M, McCormack EA, Feldmesser E, Kaganovich D, Willison KR, and Horovitz A (2010). Equivalent mutations in the eight subunits of the chaperonin CCT produce dramatically different cellular and gene expression phenotypes. J. Mol. Biol 401, 532–543. [DOI] [PubMed] [Google Scholar]
- 24.Leitner A, Joachimiak LA, Bracher A, Monkemeyer L, Walzthoeni T, Chen B, Pechmann S, Holmes S, Cong Y, Ma B, et al. (2012). The molecular architecture of the eukaryotic chaperonin TRiC/CCT. Structure 20, 814–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Archibald JM, Logsdon JM Jr., and Doolittle WF (2000). Origin and evolution of eukaryotic chaperonins: phylogenetic evidence for ancient duplications in CCT genes. Mol. Biol. Evol 17, 1456–1466. [DOI] [PubMed] [Google Scholar]
- 26.Kalisman N, Schröder GF, and Levitt M (2013). The crystal structures of the eukaryotic chaperonin CCT reveal its functional partitioning. Structure 21, 540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reissmann S, Joachimiak LA, Chen B, Meyer AS, Nguyen A, and Frydman J (2012). A gradient of ATP affinities generates an asymmetric power stroke driving the chaperonin TRIC/CCT folding cycle. Cell Rep 2, 866–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chagoyen M, Carrascosa JL, Pazos F, and Valpuesta JM (2014). Molecular determinants of the ATP hydrolysis asymmetry of the CCT chaperonin complex. Proteins 82, 703–707. [DOI] [PubMed] [Google Scholar]
- 29.Yamamoto YY, Uno Y, Sha E, Ikegami K, Ishii N, Dohmae N, Sekiguchi H, Sasaki YC, and Yohda M (2017). Asymmetry in the function and dynamics of the cytosolic group II chaperonin CCT/TRiC. PLoS One 12, e0176054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jin M, Han W, Liu C, Zang Y, Li J, Wang F, Wang Y, and Cong Y (2019). An ensemble of cryo-EM structures of TRiC reveal its conformational landscape and subunit specificity. Proc. Natl. Acad. Sci. U. S. A 116, 19513–19522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sergeeva OA, Chen B, Haase-Pettingell C, Ludtke SJ, Chiu W, and King JA (2013). Human CCT4 and CCT5 chaperonin subunits expressed in Escherichia coli form biologically active homo-oligomers. J. Biol. Chem 288, 17734–17744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu C, Wang H, Jin M, Han W, Wang S, Wang Y, Wang F, Su C, Hong X, Zhao Q, et al. (2021). Cryo-EM study on the homo-oligomeric ring formation of yeast TRiC/CCT subunits reveals TRiC ring assembly mechanism. Cold Spring Harbor Laboratory, 2021.02.24.432666. 10.1101/2021.02.24.432666. [DOI] [Google Scholar]
- 33.Collier MP, Moreira KB, Li KH, Chen Y-C, Itzhak D, Samant R, Leitner A, Burlingame A, and Frydman J (2021). Native mass spectrometry analyses of chaperonin complex TRiC/CCT reveal subunit N-terminal processing and re-association patterns. Sci. Rep 11, 13084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sergeeva OA, Haase-Pettingell C, and King JA (2019). Co-expression of CCT subunits hints at TRiC assembly. Cell Stress Chaperones 24, 1055–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spiess M, Echbarthi M, Svanström A, Karlsson R, and Grantham J (2015). Over-Expression Analysis of All Eight Subunits of the Molecular Chaperone CCT in Mammalian Cells Reveals a Novel Function for CCTdelta. J. Mol. Biol 427, 2757–2764. [DOI] [PubMed] [Google Scholar]
- 36.Elliott KL, Svanström A, Spiess M, Karlsson R, and Grantham J (2015). A novel function of the monomeric CCTε subunit connects the serum response factor pathway to chaperone-mediated actin folding. Mol. Biol. Cell 26, 2801–2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marsh JA, Hernandez H, Hall Z, Ahnert SE, Perica T, Robinson CV, and Teichmann SA (2013). Protein complexes are under evolutionary selection to assemble via ordered pathways. Cell 153, 461–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zang Y, Jin M, Wang H, Cui Z, Kong L, Liu C, and Cong Y (2016). Staggered ATP binding mechanism of eukaryotic chaperonin TRiC (CCT) revealed through high-resolution cryo-EM. Nat. Struct. Mol. Biol 23, 1083–1091. [DOI] [PubMed] [Google Scholar]
- 39.Liou AKF, McCormack EA, and Willison KR (1998). The Chaperonin Containing TCP-1 (CCT). Displays a Single-Ring Mediated Disassembly and Reassembly Cycle. Biological Chemistry 379. 10.1515/bchm.1998.379.3.311. [DOI] [PubMed] [Google Scholar]
- 40.Peterson LX, Togawa Y, Esquivel-Rodriguez J, Terashi G, Christoffer C, Roy A, Shin W-H, and Kihara D (2018). Modeling the assembly order of multimeric heteroprotein complexes. PLoS Comput. Biol 14, e1005937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krissinel E, and Henrick K (2007). Inference of macromolecular assemblies from crystalline state. J. Mol. Biol 372, 774–797. [DOI] [PubMed] [Google Scholar]
- 42.Gestaut D, Zhao Y, Park J, Ma B, Leitner A, Collier M, Pintilie G, Roh S-H, Chiu W, and Frydman J (2022). Structural visualization of the tubulin folding pathway directed by human chaperonin TRiC/CCT. Cell 185, 4770–4787.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gestaut D, Roh SH, Ma B, Pintilie G, Joachimiak LA, Leitner A, Walzthoeni T, Aebersold R, Chiu W, and Frydman J (2019). The Chaperonin TRiC/CCT Associates with Prefoldin through a Conserved Electrostatic Interface Essential for Cellular Proteostasis. Cell 177, 751–765 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miyata Y, Chang L, Bainor A, McQuade TJ, Walczak CP, Zhang Y, Larsen MJ, Kirchhoff P, and Gestwicki JE (2010). High-throughput screen for Escherichia coli heat shock protein 70 (Hsp70/DnaK): ATPase assay in low volume by exploiting energy transfer. J. Biomol. Screen 15, 1211–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meyer AS, Gillespie JR, Walther D, Millet IS, Doniach S, and Frydman J (2003). Closing the Folding Chamber of the Eukaryotic Chaperonin Requires the Transition State of ATP Hydrolysis. Cell 113, 369–381. 10.1016/s0092-8674(03)00307-6. [DOI] [PubMed] [Google Scholar]
- 46.Schreiber G, Haran G, and Zhou H-X (2009). Fundamental aspects of protein-protein association kinetics. Chem. Rev 109, 839–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Z, Witham S, and Alexov E (2011). On the role of electrostatics in protein-protein interactions. Phys. Biol 8, 035001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Noormohammadi A, Khodakarami A, Gutierrez-Garcia R, Lee HJ, Koyuncu S, König T, Schindler C, Saez I, Fatima A, Dieterich C, et al. (2016). Somatic increase of CCT8 mimics proteostasis of human pluripotent stem cells and extends C. elegans lifespan. Nat. Commun 7, 13649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roh S-H, Kasembeli M, Galaz-Montoya JG, Trnka M, Lau WC-Y, Burlingame A, Chiu W, and Tweardy DJ (2016). Chaperonin TRiC/CCT Modulates the Folding and Activity of Leukemogenic Fusion Oncoprotein AML1-ETO. J. Biol. Chem 291, 4732–4741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Spiess C, Miller EJ, McClellan AJ, and Frydman J (2006). Identification of the TRiC/CCT substrate binding sites uncovers the function of subunit diversity in eukaryotic chaperonins. Mol. Cell 24, 25–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Muñoz IG, Yébenes H, Zhou M, Mesa P, Serna M, Park AY, Bragado-Nilsson E, Beloso A, de Cárcer G, Malumbres M, et al. (2011). Crystal structure of the open conformation of the mammalian chaperonin CCT in complex with tubulin. Nat. Struct. Mol. Biol 18, 14–19. [DOI] [PubMed] [Google Scholar]
- 52.Mulder AM, Yoshioka C, Beck AH, Bunner AE, Milligan RA, Potter CS, Carragher B, and Williamson JR (2010). Visualizing ribosome biogenesis: parallel assembly pathways for the 30S subunit. Science 330, 673–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Whitelam S, and Geissler PL (2007). Avoiding unphysical kinetic traps in Monte Carlo simulations of strongly attractive particles. J. Chem. Phys 127, 154101. [DOI] [PubMed] [Google Scholar]
- 54.Whitelam S, Feng EH, Hagan MF, and Geissler PL (2009). The role of collective motion in examples of coarsening and self-assembly. Soft Matter 5, 1251–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fares MA, and Wolfe KH (2003). Positive selection and subfunctionalization of duplicated CCT chaperonin subunits. Mol. Biol. Evol 20, 1588–1597. [DOI] [PubMed] [Google Scholar]
- 56.Emlaw JR, Tessier CJG, McCluskey GD, McNulty MS, Sheikh Y, Burkett KM, Musgaard M, and daCosta CJB (2021). A single historical substitution drives an increase in acetylcholine receptor complexity. Proc. Natl. Acad. Sci. U. S. A 118. 10.1073/pnas.2018731118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mukherjee K, Conway de Macario E, Macario AJL, and Brocchieri L (2010). Chaperonin genes on the rise: new divergent classes and intense duplication in human and other vertebrate genomes. BMC Evol. Biol 10, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mukherjee K, and Brocchieri L (2013). Ancient Origin of Chaperonin Gene Paralogs Involved in Ciliopathies. J Phylogenetics Evol Biol 1. 10.4172/2329-9002.1000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Seo S, Baye LM, Schulz NP, Beck JS, Zhang Q, Slusarski DC, and Sheffield VC (2010). BBS6, BBS10, and BBS12 form a complex with CCT/TRiC family chaperonins and mediate BBSome assembly. Proc. Natl. Acad. Sci. U. S. A 107, 1488–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li L, Wei Y, To C, Zhu C-Q, Tong J, Pham N-A, Taylor P, Ignatchenko V, Ignatchenko A, Zhang W, et al. (2014). Integrated Omic analysis of lung cancer reveals metabolism proteome signatures with prognostic impact. Nature Communications 5. 10.1038/ncomms6469. [DOI] [PubMed] [Google Scholar]
- 61.Huang X, Wang X, Cheng C, Cai J, He S, Wang H, Liu F, Zhu C, Ding Z, Huang X, et al. (2014). Chaperonin containing TCP1, subunit 8 (CCT8) is upregulated in hepatocellular carcinoma and promotes HCC proliferation. APMIS 122, 1070–1079. [DOI] [PubMed] [Google Scholar]
- 62.Qiu X, He X, Huang Q, Liu X, Sun G, Guo J, Yuan D, Yang L, Ban N, Fan S, et al. (2015). Overexpression of CCT8 and its significance for tumor cell proliferation, migration and invasion in glioma. Pathol. Res. Pract 211, 717–725. [DOI] [PubMed] [Google Scholar]
- 63.Meng Y, Yang L, Wei X, Luo H, Hu Y, Tao X, He J, Zheng X, Xu Q, Luo K, et al. (2021). CCT5 interacts with cyclin D1 promoting lung adenocarcinoma cell migration and invasion. Biochem. Biophys. Res. Commun 567, 222–229. [DOI] [PubMed] [Google Scholar]
- 64.Coghlin C, Carpenter B, Dundas SR, Lawrie LC, Telfer C, and Murray GI (2006). Characterization and over-expression of chaperonin t-complex proteins in colorectal cancer. J. Pathol 210, 351–357. [DOI] [PubMed] [Google Scholar]
- 65.Ma J, Yang L, Feng H, Zheng L, Meng H, and Li X (2021). CCT6A may act as a potential biomarker reflecting tumor size, lymphatic metastasis, FIGO stage, and prognosis in cervical cancer patients. J. Clin. Lab. Anal 35, e23793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Showalter AE, Martini AC, Nierenberg D, Hosang K, Fahmi NA, Gopalan P, Khaled AS, Zhang W, and Khaled AR (2020). Investigating Chaperonin-Containing TCP-1 subunit 2 as an essential component of the chaperonin complex for tumorigenesis. Sci. Rep 10, 798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Boutros PC, Lau SK, Pintilie M, Liu N, Shepherd FA, Der SD, Tsao M-S, Penn LZ, and Jurisica I (2009). Prognostic gene signatures for non-small-cell lung cancer. Proc. Natl. Acad. Sci. U. S. A 106, 2824–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Guest ST, Kratche ZR, Bollig-Fischer A, Haddad R, and Ethier SP (2015). Two members of the TRiC chaperonin complex, CCT2 and TCP1 are essential for survival of breast cancer cells and are linked to driving oncogenes. Exp. Cell Res 332, 223–235. [DOI] [PubMed] [Google Scholar]
- 69.Zhang Y, Wang Y, Wei Y, Wu J, Zhang P, Shen S, Saiyin H, Wumaier R, Yang X, Wang C, et al. (2016). Molecular chaperone CCT3 supports proper mitotic progression and cell proliferation in hepatocellular carcinoma cells. Cancer Lett 372, 101–109. [DOI] [PubMed] [Google Scholar]
- 70.Vilchez D, Saez I, and Dillin A (2014). The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat. Commun. 5, 5659. [DOI] [PubMed] [Google Scholar]
- 71.Koyuncu S, Loureiro R, Lee HJ, Wagle P, Krueger M, and Vilchez D (2021). Rewiring of the ubiquitinated proteome determines ageing in C. elegans. Nature 596, 285–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hipp MS, Kasturi P, and Hartl FU (2019). The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol 20, 421–435. [DOI] [PubMed] [Google Scholar]
- 73.Sontag EM, Joachimiak LA, Tan Z, Tomlinson A, Housman DE, Glabe CG, Potkin SG, Frydman J, and Thompson LM (2013). Exogenous delivery of chaperonin subunit fragment ApiCCT1 modulates mutant Huntingtin cellular phenotypes. Proc. Natl. Acad. Sci. U. S. A 110, 3077–3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kitamura A, Kubota H, Pack C-G, Matsumoto G, Hirayama S, Takahashi Y, Kimura H, Kinjo M, Morimoto RI, and Nagata K (2006). Cytosolic chaperonin prevents polyglutamine toxicity with altering the aggregation state. Nat. Cell Biol 8, 1163–1170. [DOI] [PubMed] [Google Scholar]
- 75.Tam S, Geller R, Spiess C, and Frydman J (2006). The chaperonin TRiC controls polyglutamine aggregation and toxicity through subunit-specific interactions. Nat. Cell Biol 8, 1155–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stein KC, Kriel A, and Frydman J (2019). Nascent Polypeptide Domain Topology and Elongation Rate Direct the Cotranslational Hierarchy of Hsp70 and TRiC/CCT. Mol. Cell 75, 1117–1130 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Steger M, Demichev V, Backman M, Ohmayer U, Ihmor P, Müller S, Ralser M, and Daub H (2021). Time-resolved in vivo ubiquitinome profiling by DIA-MS reveals USP7 targets on a proteome-wide scale. Nat. Commun 12, 5399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, Sowa ME, Rad R, Rush J, Comb MJ, et al. (2011). Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 44, 325–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yagita Y, Zavodszky E, Peak-Chew S-Y, and Hegde RS (2023). Mechanism of orphan subunit recognition during assembly quality control. Cell 10.1016/j.cell.2023.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Morales-Polanco F, Lee JH, Barbosa NM, and Frydman J (2022). Cotranslational Mechanisms of Protein Biogenesis and Complex Assembly in Eukaryotes. Annu Rev Biomed Data Sci 5, 67–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Thulasiraman V, Yang CF, and Frydman J (1999). In vivo newly translated polypeptides are sequestered in a protected folding environment. EMBO J. 18, 85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kirchhofer A, Helma J, Schmidthals K, Frauer C, Cui S, Karcher A, Pellis M, Muyldermans S, Casas-Delucchi CS, Cardoso MC, et al. (2010). Modulation of protein properties in living cells using nanobodies. Nat. Struct. Mol. Biol 17, 133–138. [DOI] [PubMed] [Google Scholar]
- 83.Punjani A, Rubinstein JL, Fleet DJ, and Brubaker MA (2017). cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296. [DOI] [PubMed] [Google Scholar]
- 84.Zhang K (2016). Gctf: Real-time CTF determination and correction. J. Struct. Biol 193, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Marty MT, Baldwin AJ, Marklund EG, Hochberg GK, Benesch JL, and Robinson CV (2015). Bayesian deconvolution of mass and ion mobility spectra: from binary interactions to polydisperse ensembles. Anal. Chem 87, 4370–4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tyanova S, Temu T, and Cox J (2016). The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc 11, 2301–2319. [DOI] [PubMed] [Google Scholar]
- 87.Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, and Cox J (2016). The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740. [DOI] [PubMed] [Google Scholar]
- 88.Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, and Selbach M (2011). Global quantification of mammalian gene expression control. Nature 473, 337–342. [DOI] [PubMed] [Google Scholar]
- 89.Rappsilber J, Mann M, and Ishihama Y (2007). Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc 2, 1896–1906. [DOI] [PubMed] [Google Scholar]
- 90.Walzthoeni T, Claassen M, Leitner A, Herzog F, Bohn S, Förster F, Beck M, and Aebersold R (2012). False discovery rate estimation for cross-linked peptides identified by mass spectrometry. Nat. Methods 9, 901–903. [DOI] [PubMed] [Google Scholar]
- 91.Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, Morris JH, and Ferrin TE (2021). UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci 30, 70–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2. Structural modeling of holocomplex cross-links, related to Figure 4.
Data Availability Statement
All unprocessed western blot, unprocessed negative stain images and XL-MS data have been deposited at Mendeley Data. Cryo-EM Map of HCCCT578631 has been deposited at EMDB, Electron Microscopy Data Bank. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD043532. All data are publicly available as of the date of publication. Accession numbers and DOI are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CCT1 | Santa Cruz Biotechnology | Cat # sc-374088 RRID:AB_10918262 |
| CCT1 | Abcam | Cat# ab240903 RRID:AB_2920800 |
| CCT2 | Abcam | Cat # ab92746 RRID:AB_10565196 |
| CCT2 (rabbit) | 81 | N/A |
| CCT3 | Abcam | Cat # ab106932 RRID:AB_10865346 |
| CCT3 | Santa Cruz Biotechnology | Cat # sc-271336 RRID:AB_10610107 |
| CCT4 | Santa Cruz Biotechnology | Cat # sc-137092 RRID:AB_2073768 |
| CCT5 | Abcam | Cat # ab129016 RRID:AB_11154964 |
| CCT5 (rabbit) | 81 | N/A |
| CCT6 | Santa Cruz Biotechnology | Cat # sc-514466 RRID:AB_11154964 |
| CCT7 | Santa Cruz Biotechnology | Cat # sc-271951 RRID:AB_10709576 |
| CCT8 | Santa Cruz Biotechnology | Cat # sc-377261 RRID:AB_2917991 |
| IRDye 680RD Donkey anti-rabbit IgG | LI-COR Biosciences | Cat# 926-68073 RRID:AB_10954442 |
| IRDye 800CW Donkey anti-mouse IgG | LI-COR Biosciences | Cat# 926-32212 RRID:AB_621847 |
| IRDye 800CW Donkey anti-Goat IgG (H + L) | LI-COR Biosciences | Cat# 926-32214 RRID:AB_621846 |
| IRDye 800CW Goat anti-Rat IgG (H + L) | LI-COR Biosciences | Cat# 926-32219 RRID:AB_1850025 |
| Peroxidase AffiniPure Donkey Anti-Rabbit IgG (H+L) | Jackson Immuno Research | Cat# 711-035-152 RRID:AB_10015282 |
| Peroxidase AffiniPure Donkey Anti-Mouse IgG (H+L) | Jackson Immuno Research | Cat# 715-035-151 RRID:AB_2340771 |
| Peroxidase AffiniPure Donkey Anti-Goat IgG (H+L) | Jackson Immuno Research | Cat# 705-035-003 RRID:AB_2340390 |
| Anti-GFP-MBP nanobody | Modified by MPB addition, this study | N/A |
| Bacterial and virus strains | ||
| E. coli strain DH10Bac | Thermo Fisher | Cat# 10361012 |
| Chemicals, peptides, and recombinant proteins | ||
| HCCCT578631 | This study | N/A |
| HCCCT1368 | This study | N/A |
| HCCCT4257 | This study | N/A |
| HCCCT57 | This study | N/A |
| TRIC | Collier et al., 2021 | N/A |
| DMEM supplemented with glucose | Thermo Fisher Scientific | Cat# 10566024 |
| Fetal Bovine Serum | Thermo Fisher Scientific | Cat# 26400044 |
| DharmaFECT #4 transfection reagent | DharmaFECT | Cat# T-2004-03 |
| DharmaFECT Cell Culture Reagent (DCCR) | Thermo Fisher Scientific | Cat# B-004500-100 |
| Complete, EDTA-free Protease Inhibitor Cocktail | Roche | Cat# 4693132001 |
| Lipofectamine™ 2000 | Thermo Fisher Scientific | Cat# 11668019 |
| Z-Leu-Leu-Leu-al | Sigma-Aldrich | Cat# C2211-5MG |
| Ammonium Chloride | Sigma-Aldrich | Cat# A9434 |
| Benzonase® Nuclease | Sigma-Aldrich | Cat# E1014-5KU |
| Ni Sepharose® High Performance | Sigma-Aldrich | Cat# GE17-5268-02 |
| Calmodulin Sepharose™ 4B | VWR | Cat# 95016-874 |
| Amylose Resin | New England Biolabs | Cat# E8021S |
| DSS-H12/D12 | Creatie Molecules Inc. | Cat# 001S |
| Adenosine 5’-triphosphate disodium salt hydrate | Sigma-Adlrich | Cat# A7699 |
| DNaseI | Worthington Biochemical | Cat# LS006333 |
| DNaseI | Sigma-Aldrich | Cat# D5025 |
| Critical commercial assays | ||
| Quick-RNA miniprep Kit | Zymo Research | Cat# R1054 |
| iScript cDNA Synthesis Kit | Bio-Rad | Cat# 1708891 |
| Itaq™ Universal SYBR assay | Bio-Rad | Cat# 1708891 |
| [35S] EasyTag™ express protein labeling mix | Perkin Elmer | Cat# NEG772002MC |
| Deposited data | ||
| Unprocessed western blot deposited in Mendeley Data | This paper | DOI: 10.17632/ymrbcv2wrt.1 |
| Unprocessed images of negative stain | This paper | DOI: 10.17632/ymrbcv2wrt.1 |
| XL-MS raw data in Mendeley Data | This paper | DOI: 10.17632/ymrbcv2wrt.1 |
| Map of HCCCT578631 | This paper | EMD-32393 |
| Coordinates of the TRiC in open conformation | Gestaut et al. 2022 | PDB:7WU7 |
| Experimental models: Cell lines | ||
| Hela cells (CCL-2) | ATCC | CCL-2 |
| SF9 cells | Thermo Fisher Scientific | 12659-017 |
| High Five cells (Hi5) | Invitrogen | B85502 |
| Oligonucleotides | ||
| Primers used in this study are listed in Table S1 | This paper | N/A |
| Recombinant DNA | ||
| pFB-Duet CCT7His, CCT5 | Gestaut et al., 2019 | pDG463 |
| pFB-Duet CCT2, CCT4, | Gestaut et al., 2019 | pDG443 |
| pFB-Duet CCT6, CCT3 | Gestaut et al., 2019 | pDG445 |
| pFB-Duet CCT8, CCT1 | Gestaut et al., 2019 | pDG446 |
| pFB-Duet CCT1CBP, CCT8 | Gestaut et al., 2019 | pDG518 |
| pFB-Duet CCT1GFP, CCT8 | Gestaut et al., 2019 | pDG517 |
| pFB-Duet HIS-ACTB | This study | pYC26 |
| pcDNA3.1 CCT2-Ch C-terminal mCherry | (Zhao et al., 2016) | pJF1919 |
| pcDNA3.1 CCT4-Ch C-terminal mCherry | (Zhao et al., 2016) | pJF1921 |
| pcDNA3.1 CCT5-Ch C-terminal mCherry | (Zhao et al., 2016) | pJF1922 |
| pCMV6 CCT1 | Eric Jonasch | N/A |
| pCMV6 CCT2 | Eric Jonasch | N/A |
| pCMV6 CCT3 | Eric Jonasch | N/A |
| pCMV6 CCT4 | Eric Jonasch | N/A |
| pCMV6 CCT5 | Eric Jonasch | N/A |
| pCMV6 CCT6 | Eric Jonasch | N/A |
| pCMV6 CCT7 | Eric Jonasch | N/A |
| pcDNA YFP | Addgene | #13033 |
| siGENOME Human TCP1 (6950) siRNA - SMARTpool | Thermo Scientific | M-012749-01-0005 |
| siGENOME Human CCT2 (10576) siRNA - SMARTpool | Thermo Scientific | M-020107-02-0005 |
| siGENOME Human CCT3 (7203) siRNA - SMARTpool | Thermo Scientific | M-018339-01-0005 |
| siGENOME Human CCT4 (10575) siRNA - SMARTpool | Thermo Scientific | M-020147-00-0005 |
| siGENOME Human CCT5 (22948) siRNA - SMARTpool | Thermo Scientific | M-012797-02-0005 |
| siGENOME Human CCT6A (908) siRNA - SMARTpool | Thermo Scientific | M-016559-01-0005 |
| siGENOME Human CCT7 (10574) siRNA - SMARTpool | Thermo Scientific | M-020115-01-0005 |
| siGENOME Human CCT8 (10694) siRNA - SMARTpool | Thermo Scientific | M-008566-02-0005 |
| siGENOME Non-Targeting siRNA Pool #2 |
Thermo Scientific | D-001206-14-05 |
| Software and algorithms | ||
| ImageJ | Schneider et al. 2021 | https://imagej.nih.gov/ij/index.html |
| GraphPad Prism 7.04 & 9.1.2 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| CFX Maestro Manager | BioRad | https://www.bio-rad.com/en-us/product/cfx-maestro-software-for-cfx-real-time-pcr-instruments?ID=OKZP7E15 |
| Image Studio Lite | Li-COR Biosciences | https://www.licor.com/bio/image-studio-lite/ |
| Pymol 1.8.6.2 | Schrӧdinger | https://pymol.org/2/ |
| UniDec v3.1.0 | Marty et al., 2015 | https://github.com/michaelmarty/UniDec |
| MaCSED v0.3 | Justin Benesch | benesch.chem.ox.ac.uk/resources.html |
| MaxQuant v1.6.14 | Tyanova et al., 2016a | https://maxquant.net/download_asset/perseus/latest |
| Perseus v1.6.5 | Tyanova et al., 2016b | https://maxquant.net/download_asset/perseus/latest |
| cryosparc2 | Punjani et al., 2017 | https://cryosparc.com/ |
| RStudio Versi on 1.2.1335 | The R foundation | https://www.r-project.org/ |
| xQuest | Walzthoeni et al., 2012 | https://bioinformaticshome.com/tools/proteomics/descriptions/xQuest.html |
| UCSF ChimeraX | Pettersen et al., 2021 | https://www.cgl.ucsf.edu/chimerax/ |