

Abstract
There is an urgent clinical need to develop nerve-blocking agents capable of inducing long duration sensory block without muscle weakness or paralysis to treat post-operative and chronic pain conditions. Here, we report a galacturonic acid-capsaicin (GalA-CAP) prodrug as an effective nociceptive-selective axon blocking agent. Capsaicin selectively acts on nociceptive signaling without motor nerve blockade or disruption of proprioception and touch sensation, and the galacturonic acid moiety enhance prodrug permeability across the restrictive peripheral nerve barriers (PNBs) via carrier-mediated transport by the facilitative glucose transporters (GLUTs). In addition, following prodrug transport across PNBs, the inactive prodrug is converted to active capsaicin through linker hydrolysis, leading to sustained drug release. A single injection of GalA-CAP prodrug at the sciatic nerves of rats led to nociceptive-selective nerve blockade lasting for 234 ± 37 hours, which is a sufficient duration to address the most intense period of postsurgical pain. Furthermore, the prodrug markedly mitigated capsaicin-associated side effects, leading to a notable decrease in systemic toxicity, benign local tissue reactions, and diminished burning and irritant effects.
Keywords: nociceptive-selective nerve blockade, prodrug, capsaicin, galactose, Glucose Transporters
1. Introduction
Injection of conventional amino-amide and amino-ester local anesthetics around nerves can effectively inhibit peripheral nerve signal transduction and relieve pain[1]. However, clinically available local anesthetics affects all nerve fiber types upon local application, impeding action potential propagation in nociceptive, sensory, and motor neurons and inducing unnecessary motor paralysis in the affected region[2–4]. As a consequence, patients have to deal with varying degrees of muscle weakness for adequate pain relief. This is particularly problematic for postoperative pain management that typically lasts for 5–7 days and long-term chronic pain management for as long as 12 weeks[5]. Therefore, there is a clinical need to develop a nerve blocking agent that can produce nociceptive-selective nerve blockade (i.e., a selective nerve blockade that blocks nociceptive signaling without motor nerve blockade and disruption of proprioception and touch sensation) lasting 7–14 days with a single perineural injection with minimal local or systemic side effects.
The transient receptor potential vanilloid-1 (TRPV1) receptor is a key nociceptive channel expressed in neurons of various sensory ganglia and is pivotal. It plays a vital role in transmitting sensory information from the periphery to the somatosensory cortex[6]. It is activated by various stimuli including high temperatures, low pH, and a range of natural products (resiniferatoxin, capsaicin, gingerol, etc.), as well as various venoms[7–8].. Recent studies have focused on the structure of TRPV1 and its potential for developing innovative therapeutics for diseases in which TRPV1 is involved, such as pain, cancer, and neurodegenerative diseases[9]. Capsaicin, the active component of chili peppers, is an agonist for the TRPV1 receptor and can bind to residue Tyr511 to open the TRPV1 receptors on primary afferent C-fibers. Capsaicin and other TRPV1 agonists can help relieve pain because prolonged or repeated activation of TRPV1 can lead to receptor desensitization and insensitivity to subsequent stimuli and lead to the depletion of certain neurotransmitters, such as substance P, which are involved in transmitting pain signals[10]. Since TRPV1 is expressed primarily on the central and peripheral terminals of nociceptive neurons in axons and the dorsal root ganglion cells (DRGs)[11], capsaicin selectively acts on nociceptive signaling without motor nerve blockade or disruption of proprioception and touch sensation. As a consequence, capsaicin is a potential nociceptive-selective blocking agent for nociceptive signals. The use of capsaicin to inhibit nociceptive signaling without impairing motor function has recently been reported[12]. Perineural injection of capsaicin with QX-314, a quaternary lidocaine derivative with obligate positive charges, produces a nociceptive-selective axon blockade lasting 2 hours in rats. However, 2 hours is relatively short, making this formulation clinically inadequate for post-operative pain and chronic pain syndromes lasting days to weeks.
Applying capsaicin for prolonged duration nociceptive-selective axon blockade poses two major challenges: i) Insufficient drug permeation due to peripheral nerve barriers (PNBs). In order to act on nociceptive sensory neurons, the perineurally injected capsaicin must first cross the tight junction forming restrictive perineurium composed of multiple concentric layers of specialized epithelioid myofibroblasts[13]. It is well established that a very small percentage (<1%) of local anesthetics injected subcutaneously penetrate the perineurium to subsequently modulate axonal signal transduction[3]. These drugs are significantly adsorbed into adjacent tissues and/or taken up into the systemic circulation. ii) Dose limitation due to the potential local and systemic drug toxicity. Local toxic effects include irritation, redness, and burning sensation at the injection site, while systemic adverse effects can lead to dizziness, nausea, vomiting, hypotension, tachycardia, seizures, and respiratory depression[14]. Moreover, capsaicin toxicity can cause myotoxicity and neurotoxicity, leading to muscle weakness, poor stamina, and lack of muscle control, particularly with prolonged use or high doses[15].
Efforts have been made to increase the injection dose of capsaicin while reducing its toxicity. These efforts include the development of capsaicin prodrugs, such as Vocapsaicin[16–17], and drug delivery systems[18–21] that can achieve sustained release of capsaicin to improve capsaicin’s pharmacokinetic profile. However, there is currently no research reported on improving capsaicin permeation through the PNBs.
Peripheral nerves are metabolically active. Sugar molecules, such as glucose, galactose, mannose, can gain access into peripheral nerve endoneurium from the blood circulation or interstitial fluid across restrictive cellular barriers[22]. Perineurial cells and endothelial cells that form the perineurium and blood-nerve barrier (BNB) express multiple glucose transporters (GLUTs), with glucose transporter protein type 1 (GLUT1) and glucose transporter protein type 3 (GLUT3) being the most prevalent[23–24]. GLUT1 is predominantly found in endoneurial capillaries and the perineurium, while GLUT3 is primarily located in myelinated fibers, endoneurial capillaries, and the perineurium[25]. GLUTs facilitate the transport of sugar molecules across these PNBs by carrier-mediated mechanisms[26–30].
In this work, we develop sugar-capsaicin prodrugs, in which capsaicin is covalently linked to a sugar molecule through hydrolysable bonds. Our hypothesis is that the transport of sugar-capsaicin prodrugs across PNBs is mediated by facilitative GLUTs, with resultant increased prodrug permeability into peripheral nerves (Scheme 1). Following PNB transport into the endoneurium, hydrolysis of a specific sugar-capsaicin prodrug bond occurs. This process converts the inactive prodrug to active capsaicin, thereby inducing a nociceptive-selective axon blockade. Of note, the prodrug bond hydrolysis is a slow process that results in sustained release of active capsaicin over time.
Scheme 1.
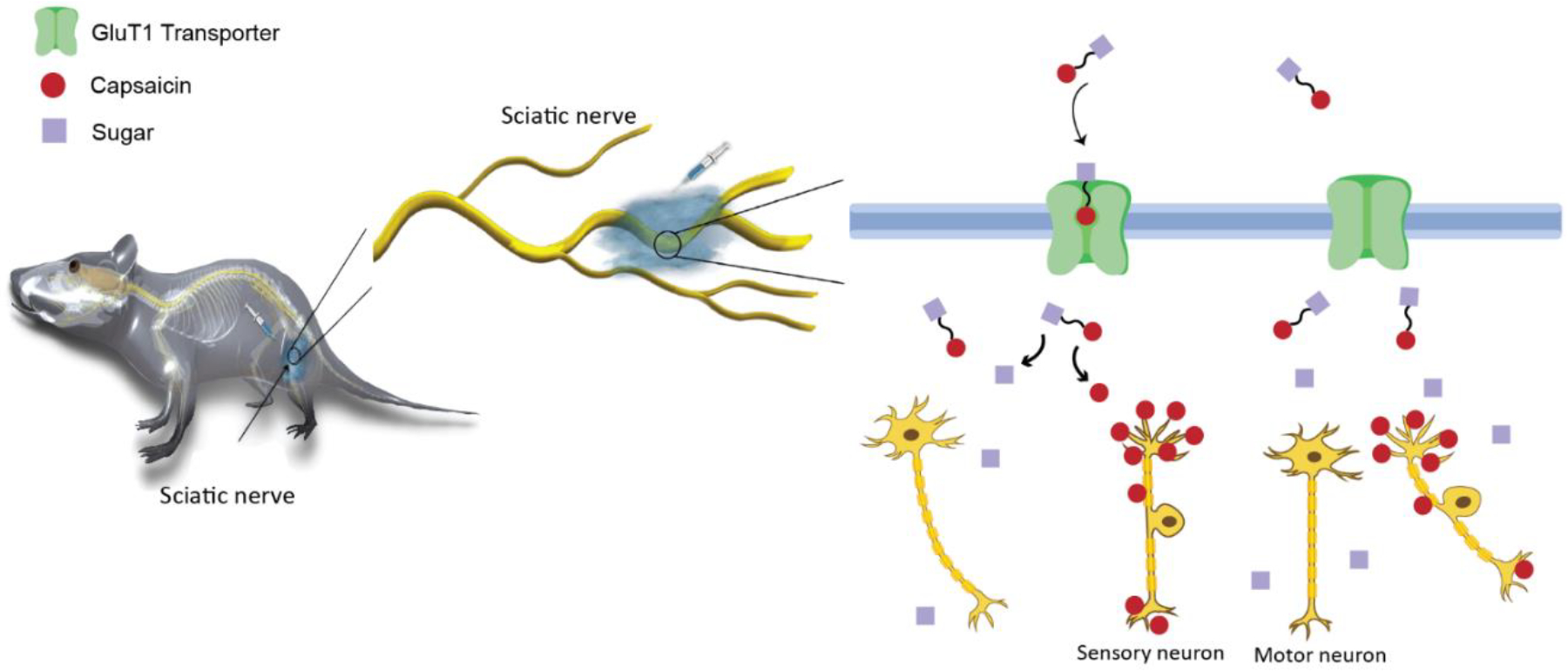
Schematic illustration of the sugar-capsaicin prodrug for sensory selective nerve blockade. Sugar-capsaicin prodrugs penetrate the peripheral nerve barrier, mediated by the GLUT transporters. After crossing the peripheral nerve barriers, the inactive prodrug is converted to active capsaicin by hydrolysis of the ester bond. Capsaicin acts selectively on sensory but not motor neurons.
This approach ensures a consistent supply of drug levels necessary for maintaining a prolonged nociceptive-selective axon blockade. Conversely, it prevents the initial release of a local drug bolus, mitigating the potential for drug-related local and systemic adverse effects. With this hypothesis, sugar-capsaicin prodrugs are expected to facilitate safer injection of higher capsaicin doses to produce prolonged duration nociceptive-selective axon blockade with limited side effects.
2. Results
2.1. Synthesis and characterization of the galacturonic acid-capsaicin prodrug
We designed and synthesized a prodrug by conjugating capsaicin (2) with galacturonic acid (i.e., the oxidized form of galactose) with a hydrolysable carboxylic acid ester bond. Galactose was selected as the sugar moiety due to its high binding affinities to GLUT1/3 (Km=17 mM for GLUT1, 8.5 mM for GLUT3). Galactose has been used as the active ingredient in prodrugs to enhance drug uptake by the brain and cancer cells through binding to the highly expressed GLUT1 in the blood brain barrier[31–32] and tumors[33]. The hydroxyl group at the carbon 6’‒position of galactose (Figure 1a) is the most promising functional group for conjugation to drug molecules because this conjugation generally does not affect the affinity of galactose to GLUT transporters[34]. In our design, the carboxylic acid group at the C-1’ position of galacturonic acid (Figure 1a, corresponding to the C-6’ position of galactose) was used to react with the hydroxyl group of capsaicin to form the carboxylic acid ester linkage.
Figure 1.
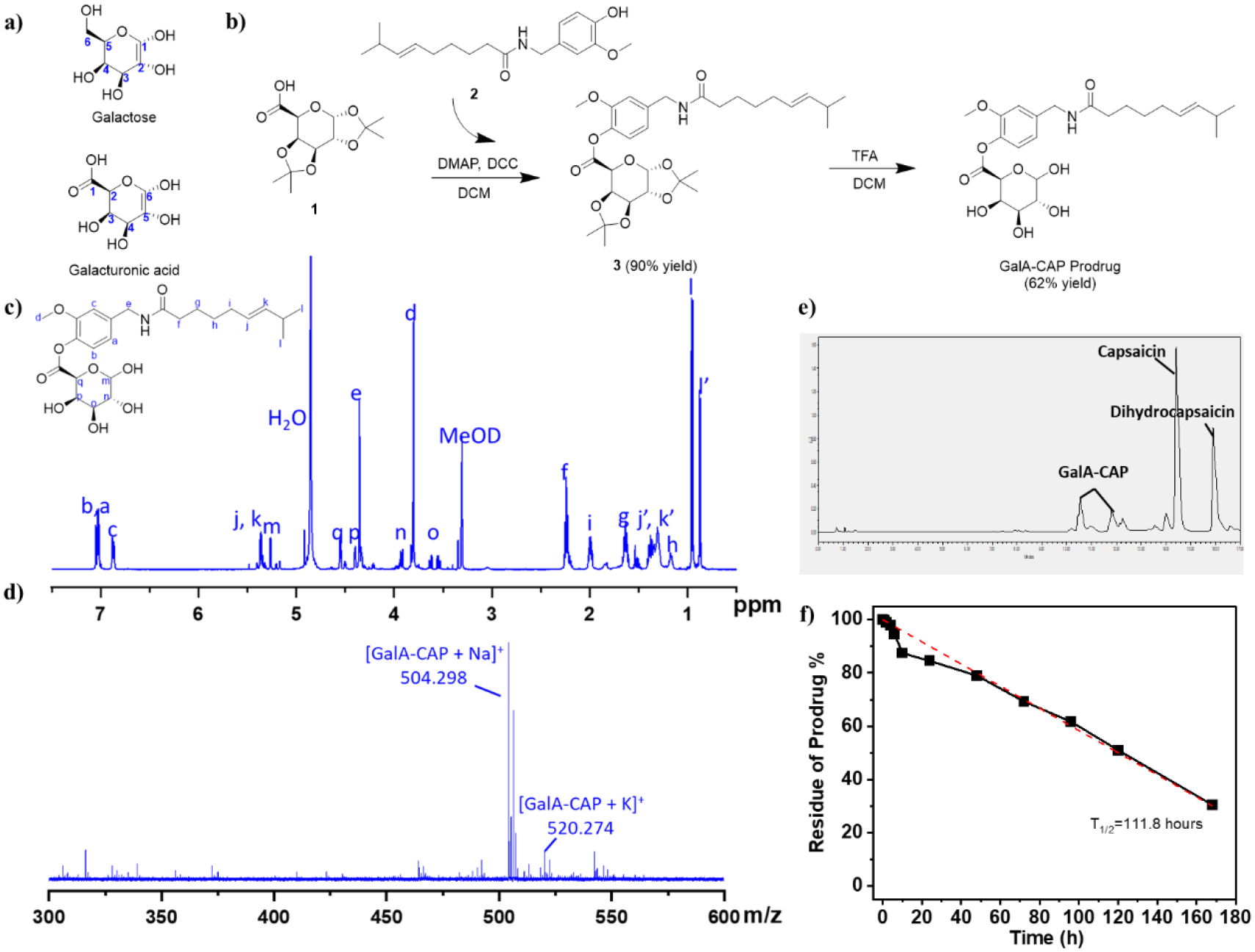
Synthesis route and characterization of the GalA-CAP prodrug. (a) Structure of galactose and galacturonic acid. (b) Synthesis route a of the GalA-CAP prodrug. (c) 1H NMR (500 MHz) spectrum of GalA-CAP prodrug in methanol-d4. j’, k’, l’ are assigned to dihydrocapsaicin. (d) Mass spectrum of GalA-CAP prodrug. (e) UPLC chromatography illustrated the GalA-CAP prodrug degradation to capsaicin after 7 days. (f) Hydrolysis kinetics of GalA-CAP prodrug in DI water at 37 °C determined by UPLC. The concentration of GalA-CAP prodrug is 1 mg/mL. The solution was stirred at 200 rpm and kept at 37°C.
Specifically, the galacturonic acid-capsaicin prodrug (GalA-CAP) was synthesized through a two-step reaction. The first step is a condensation reaction between capsaicin (2) and 1,2,3,4-di-o-isopropylidene-α-d-galacturonic acid (DIGA, 1) to synthesize a DIGA-capsaicin ester (3, Figure 1b, Figures S1–S3). Dicyclohexylcarbodiimide (DCC) was used as a condensing agent and 4-dimethylaminopyridine (DMAP) as a catalyst in this reaction. The second step involves removal of the ketal protection of hydroxyl groups in the DIGA-capsaicin ester using a trifluoroacetic acid solution, resulting in the formation of the GalA-CAP prodrug (Figure 1b).
The GalA-CAP prodrug was characterized by NMR (Figures 1c, S4) and MALDI-TOF mass spectrometry (Figure 1d). In the 1H NMR spectrum of the GalA-CAP prodrug (Figures 1c), all peaks can be assigned to each proton of the prodrug molecule. In particular, the peak at 4.36 ppm is assigned to the N’‒methylene (-CH2) proton of the capsaicin moiety. After conjugation with the galacturonic acid, it shifted from 4.26 ppm of free capsaicin to 4.36 ppm of the prodrug. The peaks at 3.5‒4.6 ppm are assigned to the methylene and methine proton of the conjugated galacturonic acid. The MALDI-TOF/MS spectrum in the positive ion mode displayed a predominant peak at m/z 504.298, which is assigned to the GalA-CAP ([M + Na]+). The purity of prodrug determined from ultra-performance liquid chromatography (UPLC) is 96.23 %, while the detected impurities are capsaicin and dihydrocapsaicin (3.76 %, Figure S5). These results demonstrated the successful synthesis of the designed prodrug.
Hydrolysis kinetics of GalA-CAP was evaluated in deionized (DI) water at 37 °C. 10 mg of prodrug was suspended in 10 mL DI water and the sample was kept in a constant temperature bath at 37 °C. At predetermined time intervals, the component of sample was analyzed and quantified via UPLC. Results demonstrated that capsaicin/dihydrocapsaicin was the decomposed product, indicating the prodrug converted to active capsaicin through the hydrolysis of the carboxylic acid ester bond (Figure 1e). The hydrolysis was found to follow pseudo-first-order kinetics. The pseudo-first-order half-life (t1/2) was calculated from the slope of linear plots of the logarithm of remaining prodrug against time. The t1/2 was determined to be 111.8 hours (Figure 1f). The presence of esterase significantly accelerated the prodrug hydrolysis. Specifically, when there were 60 units/mL and 10 units/mL of esterase in 1 mL of PBS buffer, 91.1% and 87.3% of the prodrug were converted to capsaicin within 10 minutes, respectively (Table S1).
2.2. Preparation of injectable prodrug formulations
By adding the sugar moiety, the hydrophilicity of the GalA-CAP prodrug (Log P = 1.53) increased compared to capsaicin (Log P = 3.66). However, it is still poorly water soluble. In order to make the injectable prodrug formulation, Tween 20 was used as s surfactant. Specifically, the GalA-CAP prodrug and capsaicin were dissolved in methanol with Tween 20 at room temperature. The mixture was then subjected to a vacuum process overnight at room temperature to remove the methanol. Subsequently, the mixture was suspended in PBS buffer to achieve the desired concentrations of the drug and Tween 20.
Dynamic light scattering (DLS) analysis revealed that the prodrug formed aggregates in the prepared formulation. The size of aggregates was found to be linearly correlated with the prodrug concentration. Specifically, the aggregate size increased from 842 ± 293 nm to 4167 ± 350 nm when the concentrations increased from 1.67 mg/mL to 10 mg/mL (equivalent to 0.5 to 3 mg in 0.3 mL PBS buffer for injection with 2.5 % (w/v) Tween 20, Figure S6).
2.3. Nociceptive axon blockade and systemic toxicity
The efficacy and safety for nociceptive-selective nerve blockade of the synthesized GalA-CAP prodrug was evaluated using a rat sciatic nerve block model. Male Sprague–Dawley rats (250–350 g) were injected at the left sciatic nerve with 0.3 mL of 1×PBS containing 7.5‒12 mg Tween 20 and 0.5‒3 mg GalA-CAP prodrug or capsaicin. Neurobehavioral tests were performed to determine the duration of functional deficits, including nociceptive (measure paw withdrawal latency using a hotplate test) and motor axon blockade (measure the maximum weight a rat could bear using a weight-bearing test), in both hindpaws. The duration of deficits on the injected (left, ipsilateral) side reflected the duration of nerve block. Deficits on the uninjected (right, contralateral) side and other possible side effects (respiratory distress or seizure) reflected undesired systemic distribution of the injected formulation.
Groups of rats receiving sciatic nerve injections of both the GalA-CAP prodrug and free capsaicin showed dose-dependent increases in the frequency of successful nerve blocks and in the average duration of nerve block. Specifically, both groups achieved a 100% successful analgesic block (i.e. all rats tested showed thermal paw withdrawal latencies > 7 seconds) at injection doses above 1 mg (Figure 2a). In addition, the injection did not produce any motor deficits (i.e., from motor fascicular blockade) in the injected hind limb. We successfully induced a reversible nociceptive axon blockade following a single prodrug injection of 2 and 3 mg lasting for 150.0 ± 76.8 and 234 ± 37 hours, respectively (Figure 2b). Importantly, the injection did not cause any capsaicin-related side effects, such as spontaneous pain behavior (e.g. flinching, licking), contralateral nociceptive axon blockade in the uninjected right hind limb, irreversible nociceptive axon blockade, seizure, or acute respiratory distress (Figure 2c–f). Full thermal stimulus sensitivity of injected rats was restored after 13 days, as evidenced by a return to baseline thermal latency (Figures 3a–h). No motor deficits were observed, as demonstrated by the consistent maximum bearing weight across both hind paws (Figures 3i–p). However, increasing the dosage of GalA-CAP prodrug to 4 mg led to systemic toxicity and adverse effects, such as seizures observed in 2 out of 4 rats. The side effects are likely due to the higher systemic distribution of the metabolically converted capsaicin.
Figure 2.
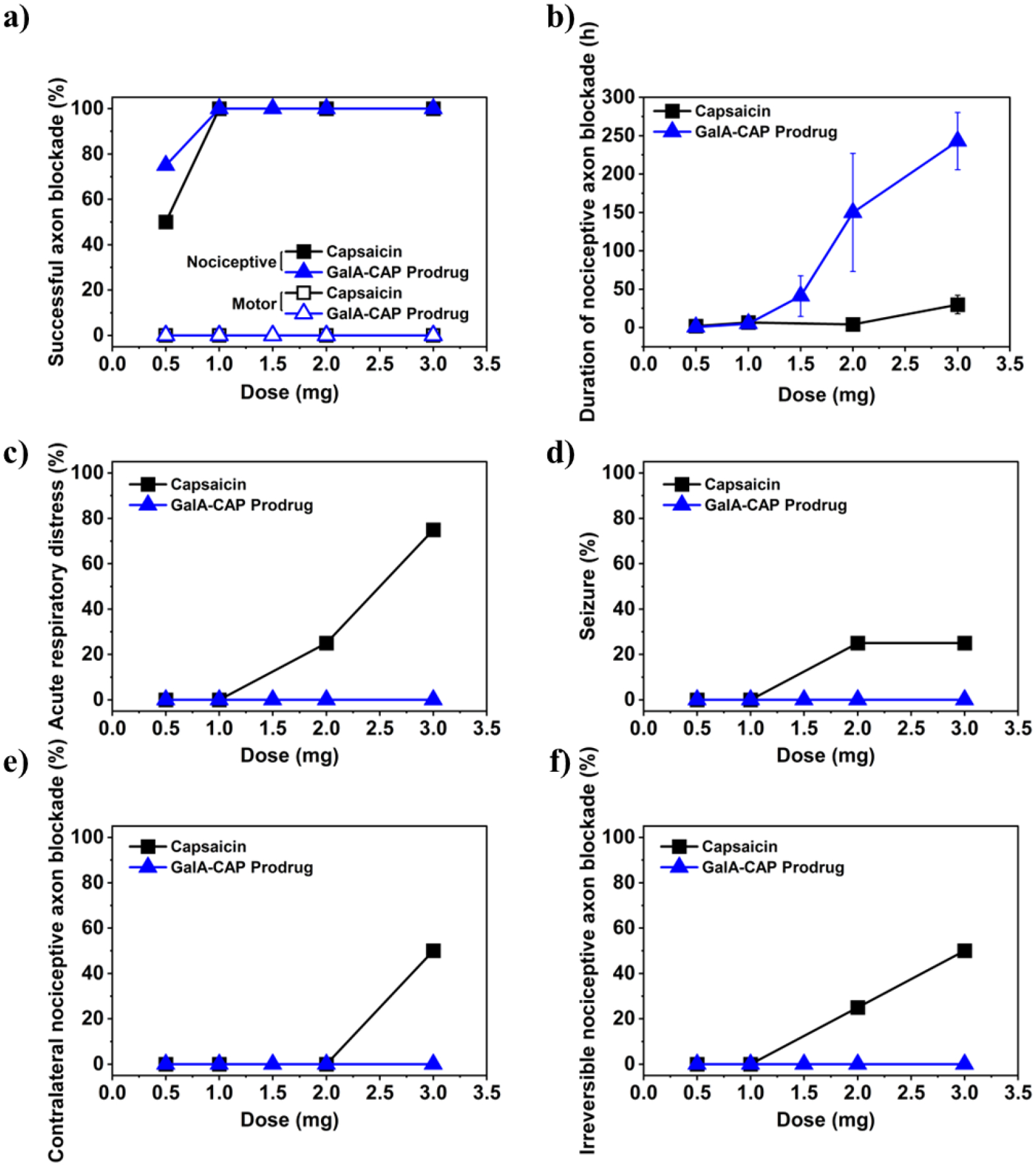
Dose effects of capsaicin and GalA-CAP prodrug on (a) frequency of successful sensory nerve blockade, (b) duration of nociceptive axon blockade, (c) frequency of acute respiratory distress, (d) frequency of severe seizure, (e) frequency of contralateral nociceptive axon blockade, and (f) frequency of irreversible nociceptive axon blockade. n = 4 for all groups except for n = 8 for 3 mg GalA-CAP prodrug in (b). Data are means ± SD.
Figure 3.
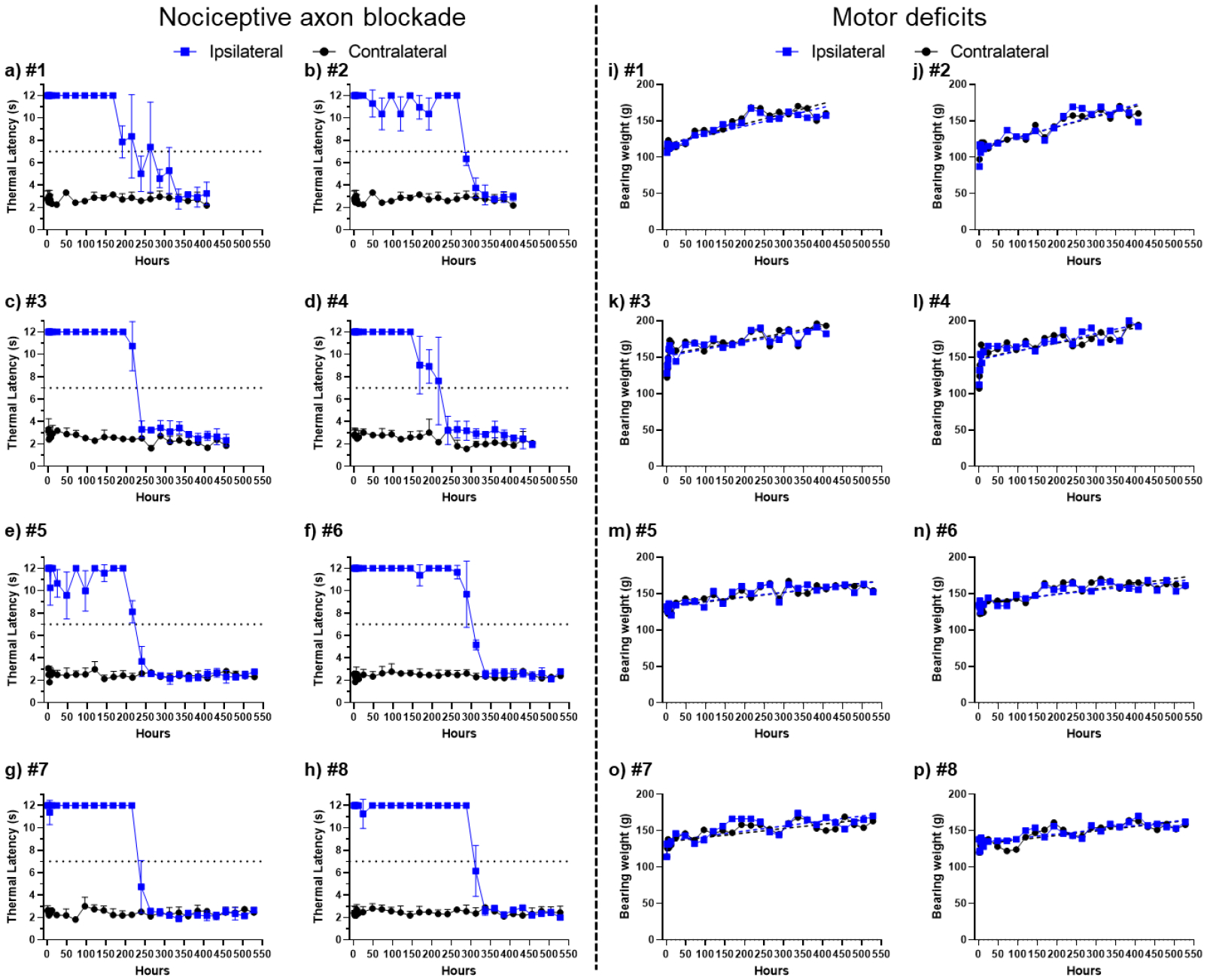
Assessment of nociceptive axon blockade (a–h) and motor function impairment (i–p) in rats post-administration with 3 mg of GalA-CAP prodrug formulated with 2.5% (w/v) Tween 20 in 0.3 mL PBS buffer. Data in panels (a–h) are means ± SD.
In contrast, injection of 2 and 3 mg of pure capsaicin produced a nociceptive axon block lasting for 4 ± 2 and 30 ± 12 hours, respectively. We observed that all rats injected with 3 mg of pure capsaicin exhibited at least one systemic adverse effect (Table S2). Specifically, 75% developed acute respiratory distress, 25% experienced a seizure, 50% developed contralateral nociceptive axon block. Injection with 2 mg and 3 mg of capsaicin caused irreversible nociceptive axon blockade in 25% and 75% of animals in the injected hind limb, respectively, indicating capsaicin-induced nerve damage (Figures 2f, S7, S8). The observation of irreversible nociceptive axon blockade aligns with previous reports of an initial transient thermal latency spike succeeded by a prolonged latency period that was never recovered[21].
2.4. Tissue Reaction
Conventional local anesthetics, particularly with prolonged duration, can be associated with myotoxicity and inflammation[35].. In order to study the local effects of prodrug and pure capsaicin injections, we euthanized treated rats 4 days (n = 3), 14 days (n = 4) and 28 days (n = 4) after injection. We harvested, sectioned, and stained the sciatic nerves and their surrounding tissues for histologic evaluation. We processed and stained hamstring muscles with hematoxylin–eosin (H&E). We generated epon-embedded semi-thin sciatic nerve sections and stained them with toluidine blue (gold-standard for peripheral nerve morphology).
We observed that the surrounding tissues in rats injected with 2–3 mg of capsaicin appeared reddish and swollen (Figure S9). Microscopic examination revealed a trace of perifascicular internalization of the nucleus (myotoxicity score = 1) in one rat (out of 3) injected with 2 mg of capsaicin after 4 days (Figure 4a). The cause of myotoxicity can be attributed to capsaicin, because previous studies using the same rat model have shown that injections of PBS buffer and Tween 20 with the tested concentration do not cause myotoxicity and neurotoxicity[36–38].. Mild inflammation was observed in tissues harvested from rats after 4 days of injection of 2 mg of capsaicin, with a median score of 3 (range [1–4]). In contrast, the surrounding tissues in rats injected with 2–3 mg of the GalA-CAP prodrug did not appear edematous or discolored, and there were no obvious signs of tissue injury. Microscopic examination did not reveal myotoxicity (median 0 for all groups, Table S3). Mild inflammation was observed in tissue harvested from rats after 4 days of injection of 2 mg of the GalA-CAP prodrug, with a median score of 1 (range [1–3]). There are no signs of inflammation in the deeper layers of the muscle. The trace of inflammation faded over time, and the score was lowered to 1 after 28 days of injecting 3 mg of the GalA-CAP prodrug or free capsaicin.
Figure 4.
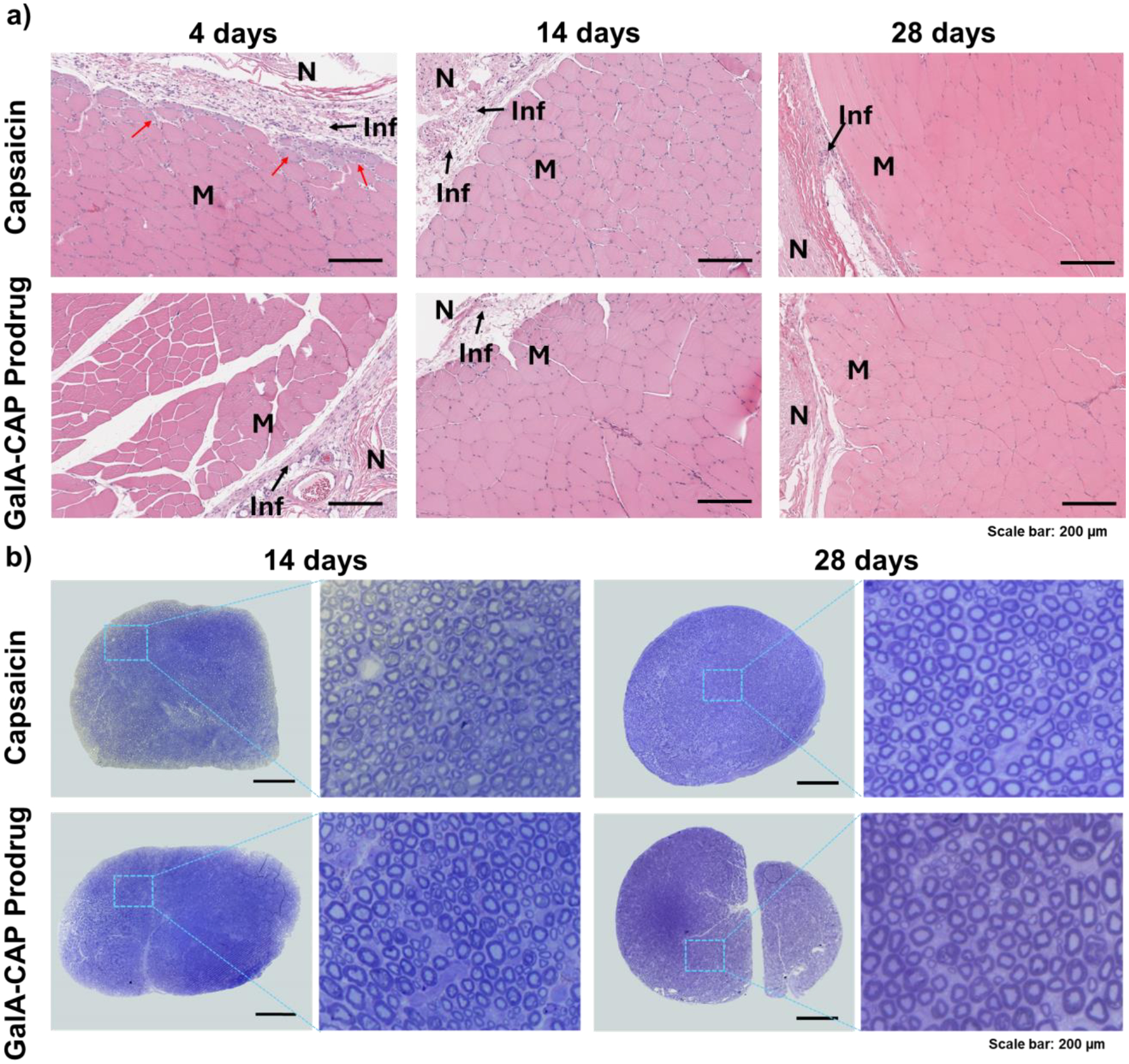
Representative histology of dissected sciatic nerves and surrounding muscle tissue. H&E-stained sections of muscle tissue (a) and toluidine blue-stained sections of sciatic nerves (b). Tissues harvested at 4- and 14-days post-injection were injected 2 mg capsaicin and GalA-CAP prodrug, whereas those harvested at 28 days post-injection were injected 3 mg capsaicin and GalA-CAP prodrug. Red arrow denotes perifascicular internalization of nucleus. M: Muscle; N: Nerve region; Inf: Inflammation. Scale bar: 200 μm.
We also observed myelinated axons in toluidine blue-stained sections (Figure 4b). The myelinated nerves appeared normal, showing no signs of nerve degradation, with representative images provided. The sciatic nerves harvested from rats 28 days post-injection of 3 mg capsaicin and GalA-CAP prodrug were then examined by transmission electron microscopy (TEM) to better visualize the unmyelinated C-fibers, where TRPV1 is mostly expressed and targeted by the capsaicin. “No injection” controls were selected to better represent the original condition of the C-fibers and to avoid any potential confounding effects caused by the injection process. The hypertrophy of C-fibers was observed in nerves harvested from rats administered with 3 mg capsaicin (Figure 5). The observed lesions and degeneration in the unmyelinated sensory axons align with the irreversible nociceptive axon blockade induced by the 3 mg capsaicin injection (Figures S8, S10). The damage to nociceptive axons caused by capsaicin is well-documented. Topical application of a high dosage of capsaicin could cause nearly complete degeneration of epidermal nerve fibers and the subepidermal neural plexus in treated skin[39]. Such deterioration of nerves could potentially be attributed to osmotic swelling of C-fibers stemming from unsustainable intracellular calcium concentrations[40]. However, the diameters of unmyelinated axons in rats injected with 3 mg of GalA-CAP prodrug were not significantly increased compared to those of the control group without injection, which could be attributed to the mild release of capsaicin by the prodrug. These findings, which align with the results from animal behavioral tests, highlight the impact of histological changes and underscore the importance of reversible nerve blockade in analgesic treatments.
Figure 5.
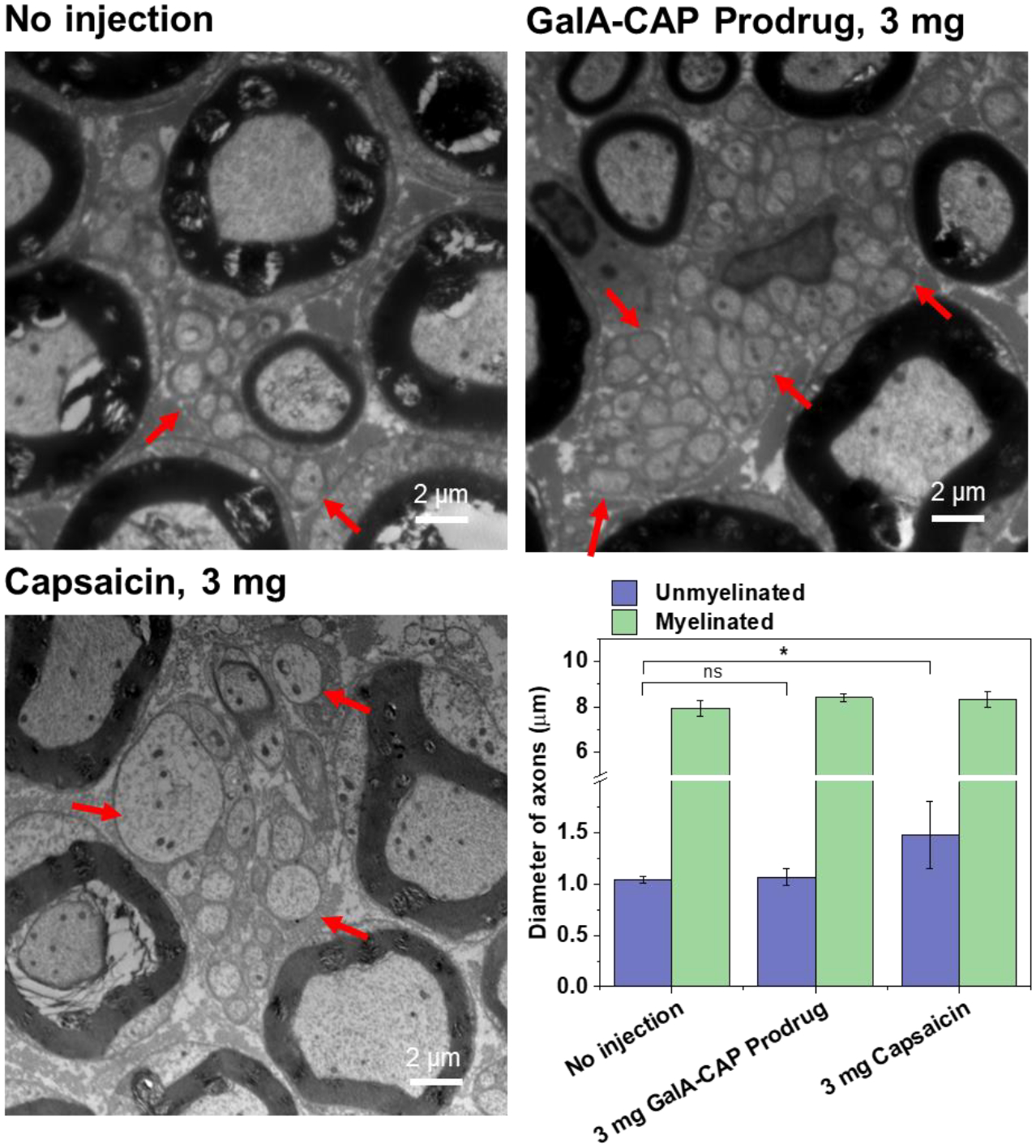
Representative TEM images of dissected sciatic nerves and the diameter of axons. Red arrows denote unmyelinated fibers. The nerve tissues were harvested at 28 days post-injection, and the nerve from rats without injections were harvested at the same time. Scale bar: 2 μm. n = 200 fibers for diameter measurement. Data shown mean ± SD. *P < 0.05, ns‒not significant, analysis of variance (ANOVA), n = 4.
2.5. Enhanced efficacy and reduced side effects due to facilitated GLUTs transport.
We hypothesize that the enhanced efficacy and reduced side effects of the prodrug in nociceptive-selective nerve blockade are due to that the galacturonic acid moiety enhances prodrug permeability across the restrictive PNBs via carrier-mediated transport by the facilitative GLUTs. To confirm this hypothesis, the prodrug was co-injected at the rat sciatic nerve with the glucose and the GLUT inhibitor (KL-11743), respectively. Glucose has a comparable binding affinity for GLUTs (Km=17 mM for GLUT1, 11 mM for GLUT3) to galactose[34], and was previously reported to reduce the GLUT1-mediated uptake of the prodrugs by the brain when co-administrated with glucosyl ketoprofen and indomethacin prodrugs[32].. KL-11743 is a commercially available small-molecule inhibitor that impedes the function of class I glucose transporters (GLUT 1–4) both in vitro and in vivo[41].. Here they are used as competitive substrates for the prodrug because the preferential binding between glucose/KL-11743 and GLUTs will reduce the possibility of the prodrug binding to GLUT, and thus hindering GLUT transporter-mediated transport of prodrugs through the PNBs permeation.
2 mg of the GalA-CAP prodrug was co-dissolved with varying doses of glucose or KL-11743 in 0.3 mL of PBS buffer (with 2.5% (w/v) Tween 20). The prepared formulation was administered at the sciatic nerve of rat. The duration of the thermal nociception block was reassessed. As expected, the duration of nerve block was significantly reduced with increasing glucose dose (Figure 6a, c). Specifically, when 2 mg of the GalA-CAP prodrug was injected together with 14.8 mM (0.8 mg) glucose, the nociceptive axon blockade duration significantly decreased to 26.25 ± 31.88 hours, compared to 150 ± 76.84 hours in the absence of glucose, and this duration further reduced to 2.50 ± 2.38 hours with the co-application of 74.1 mM (4 mg) glucose.
Figure 6.
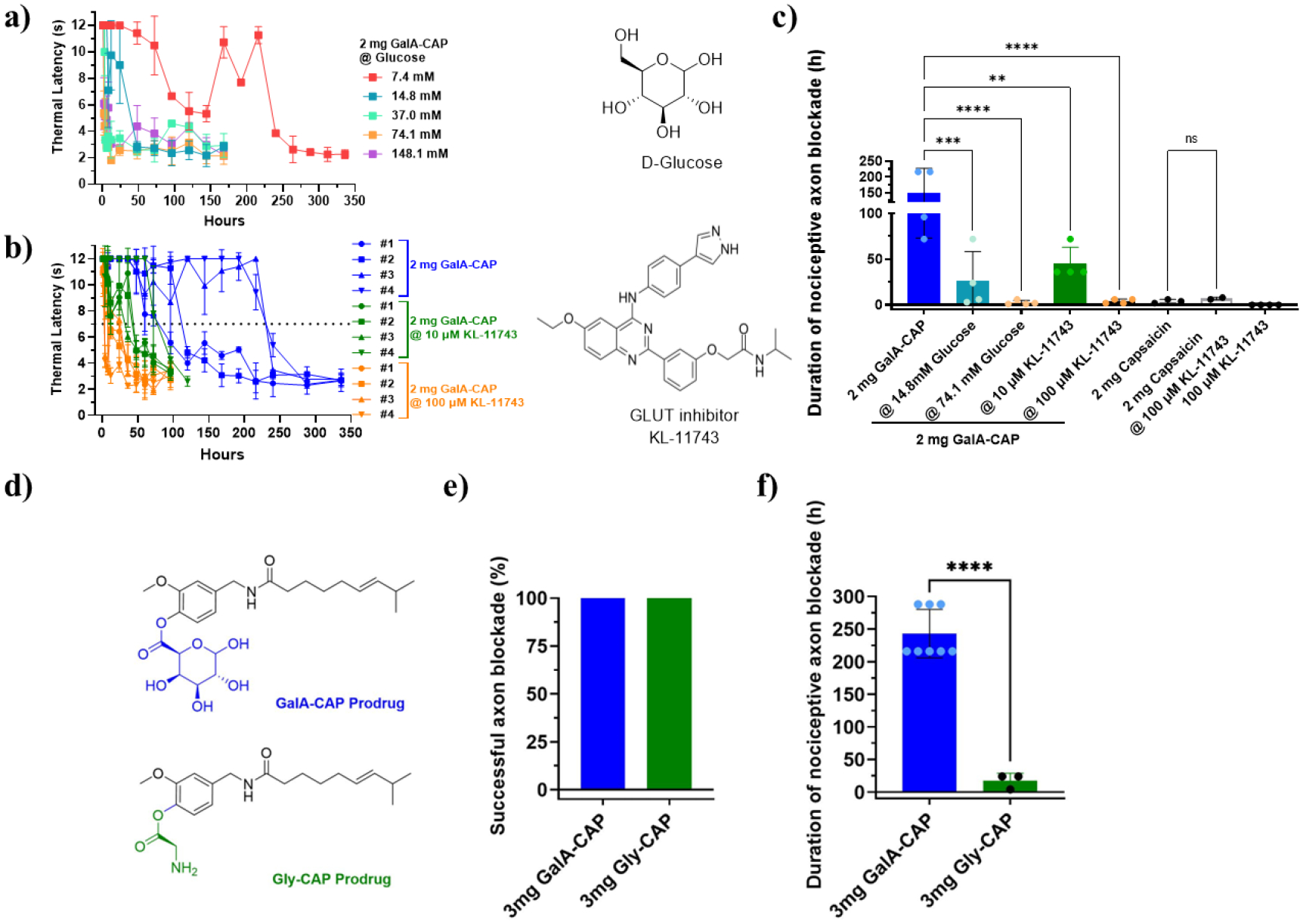
Inhibition of the prodrug uptake by glucose and KL-11743. (a) Representative time courses of thermal latency in rats co-injected with 2 mg GalA-CAP prodrug and different doses of glucose. (b) Time courses of thermal latency of the ipsilateral side of rats co-injected with 2 mg GalA-CAP prodrug and different doses of KL-11743. (c) The average duration of nociceptive axon blockade in rats co-injected with 2 mg GalA-CAP prodrug or capsaicin with different doses of inhibitors. (d) Structures of GalA-CAP prodrug and Gly-CAP prodrug. Comparison on frequency of successful axon blockade (e) and duration of nociceptive axon blockade (f) between 3 mg GalA-CAP prodrug and 3 mg Gly-CAP prodrug. Data shown are mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns: not significant, analysis of variance (ANOVA).
A similar trend of dose-dependent inhibition was observed when KL-11743 was co-administered with the prodrug (Figure 6b, c). The nociceptive axon blockade durations were shortened to 45 ± 18 hours and 4 ± 2.31 hours when co-administered with 10 μM (1.57 μg) and 100 μM (15.7 μg) of KL-11743, respectively. These inhibition experimental results demonstrated that GLUT facilitates the transport of the GalA-CAP prodrug across the nerve barriers. Notably, the KL-11743 didn’t cause any nerve blockade when injected solely and didn’t affect the efficacy of capsaicin (Figure S11, S12).
To further confirm the enhanced efficacy and reduced side effects of the GlaA-CAP prodrug due to facilitated GLUTs transport, we synthesized the glycine-capsaicin prodrug (Gly-CAP). In Gly-CAP, glycine was linked to capsaicin through an ester bond (Figure 6d, Scheme S1, Figures S13–S15). The Gly-CAP prodrug exhibited a similar hydrolysis profile in both water and enzyme environments as the GalA-CAP prodrug (Figure S16, Table S1). However, Gly-CAP does not have a sugar moiety and is not expected to be actively transported across the perineurium through the GLUT1 transporter. 3 mg of the Gly-CAP prodrug in 0.3 mL PBS (with 2.5% w/v Tween 20) was injected at the sciatic nerve of rats. As expected, the injection of 3 mg of the Gly-CAP prodrug demonstrated similar in vivo effects to pure capsaicin. It achieved a 100% successful nociceptive axon blockade (Figure 6e). The average duration of nociceptive axon blockade in rats injected with 3 mg of Gly-CAP prodrug lasted for 17.33 ± 11.55 hours, with one rat exhibiting irreversible nerve blockade (Figures 6f, S17). Additionally, 1 in 4 rats experienced a seizure, and 2 rats developed contralateral nociceptive axon block.
2.6. Increased uptake of glycosyl fluorescein by nerve.
The enhanced prodrug permeation across the PNBs by GLUTs was also confirmed by the increased uptake of glycosyl fluorescein by nerves. We compared the nerve permeation of galactosamine-fluorescein (Gal-FL, Mn = 568, Log P = 1.46), glucose-UDP-fluorescein (Glc-UDP-FL, Mn = 1283, Log P = 1.33, Figure S18), fluorescein isothiocyanate (FITC, Mn = 389, Log P = 3.77), and poly (ethylene glycol)750-fluorescein (PEG750-FL, Mn = 1138, cLog P = −0.18). We synthesized Gal-FL by conjugating the primary amine at the C-2’ position of galactosamine to FITC (Figure S19). Gal-FL has the same galactose sugar moiety as GalA-CAP prodrug (Mn = 481, Log P = 1.53) and a similar molecular weight and log P value, so it could mimic the physicochemical properties and permeation behavior across the PNBs as the GalA-CAP prodrug. In order to confirm the role of GLUTs in facilitating the prodrug crossing PNBs, we need to demonstrate that prodrugs with different sugar moieties exhibit similar behavior. Therefore, Glc-UDP-FL, a commercially available compound, was selected due to its glucose sugar moiety. FITC was used as a negative control group. In addition to using FITC as a control group, PEG750-FL was synthesized (Figure S20) and employed as an additional control because it has a comparable molecular weight and hydrophilicity to Glc-UDP-FL, which helps minimize the impact of molecular weight and hydrophilicity on the permeation behavior of compounds across the PNBs.
0.3 mL of Gal-FL or Glc-UDP-FL or FITC or PEG750-FL (15.6 μM) in PBS buffer was injected at the sciatic nerve. Four hours later, animals were euthanized, and the nerve and surrounding tissue were harvested. Frozen sections of the tissues were produced. Confocal microscopy imaging was used to track the location of fluorescein molecules.
In rats administered with free FITC, fluorescence was predominantly external to the nerve, accruing in the epineurium and perineurium. In contrast, the fluorescence signal of Gal-FL and Glc-UDP-FL penetrated deep into the nerve (Figure 7a, S21). However, when Gal-FL and Glc-UDP-FL were co-administered with 100 μM KL-11743, a much lower fluorescence signal was observed inside the nerves. This indicates that KL-11743 inhibited the function of GLUTs and significantly impeded the permeation of Gal-FL and Glc-UDP-FL into the nerve. In addition, a much lower fluorescence signal was observed outside the nerves as well. This is because Gal-FL and Glc-UDP-FL are more hydrophilic than FITC, so they migrated more rapidly away from the injection site compared to FITC.
Figure 7.
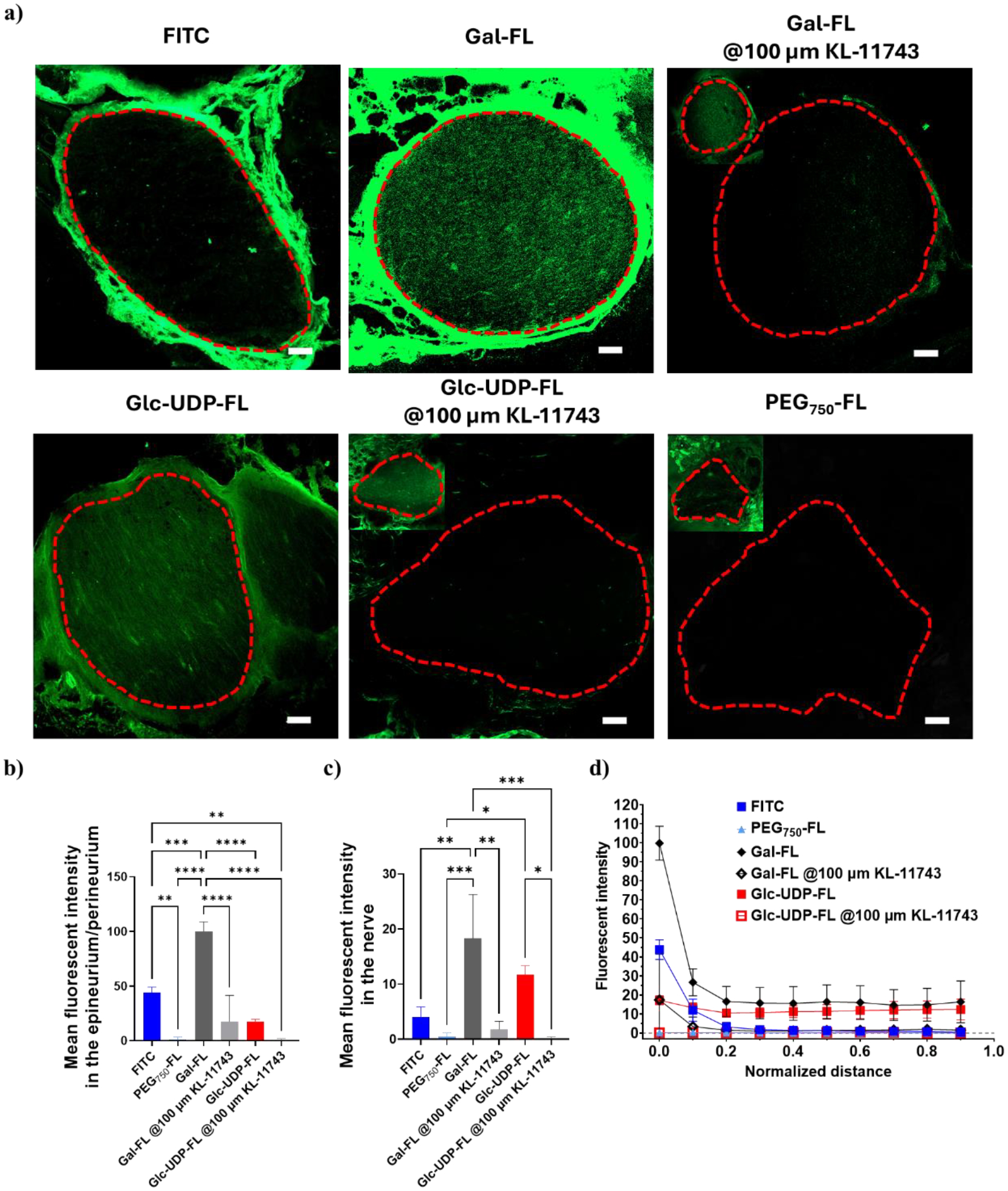
In vivo nerve penetration of FITC, Gal-FL, Glc-UDP-FL, and PEG750-FL. (a) Representative confocal images of sciatic nerves and surrounding tissues 4 h after injecting FITC, Gal-FL, Gal-FL@ 100 μM KL-11743, Glc-UDP-FL, Glc-UDP-FL@ 100 μM KL-11743, and PEG750-FL. The image taken from higher laser intensity were inserted on the left top for groups of Gal-FL@ 100 μM KL-11743, Glc-UDP-FL@ 100 μM KL-11743, and PEG750-FL, due to poor signals collected using the same camera settings. The contour of nerves was circled in red dash lines. Scale bar: 100 μm. (b) Mean fluorescent intensity in epineurium and perineurium. (c) Mean fluorescent intensity in nerves. (d) Relationship between mean fluorescent intensity and normalized distance from the epineurium/perineurium of the nerve. n = 3, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Data are means ± SD.
In rats administered with PEG750-FL, weak fluorescent signals were observed within both the epineurium and endoneurium. This finding excluded the possibility that Gal-FL and Glc-UDP-FL facilitate the nerve permeation of FITC due to the increased molecular weight and hydrophilicity of FITC. Similar to the Glc-UDP-FL group, the limited accumulation of PEG750-FL in the tissue around nerves could be attributed to its higher hydrophilicity, which causes it to quickly move away from the injection site after being administered.
Quantitative analysis showed that the mean fluorescent intensity in epineurium/perineurium and inside the nerve was significantly higher in animals injected with Gal-FL or Glc-UDP-FL than those injected with free FITC or PEG750-FL. However, this increase disappeared when the Gal-FL and Glc-UDP-FL were administered along with a GLUT inhibitor (Figure 7b, c). At a normalized distance of 0.1 (Figure S22), Gal-FL group displayed the spike fluorescent intensity surrounding the nerve (range 0) and evenly distributed fluorescent signals within the nerve (ranges 0.1–0.9) (Figures 7d), suggesting that the perineurium still remains the primary rate-limiting barrier for the diffusion of Gal-FL molecules.
Overall, these results indicate that sugar conjugation augments the trans-perineurial transport of fluorescein molecules through GLUT.
2.7. Enhanced efficacy and reduced side effects due to sustained release of capsaicin from the less active prodrug.
We hypothesize that the GalA-CAP prodrug is an inactive or less active form. Following prodrug transport across PNBs, the inactive or less active prodrug is gradually converted to active capsaicin through linker hydrolysis, leading to sustained drug release. This sustained drug release also contributes to enhanced efficacy and reduced side effects of the prodrug in nociceptive-selective nerve blockade. The in vitro prodrug hydrolysis assessment has confirmed the sustained drug release. Thus, here we need to confirm that the GalA-CAP prodrug is not as active as capsaicin in nerve blocks. We hypothesize that the GalA-CAP prodrug is an inactive or less active form.
We synthesized the galactose-capsaicin prodrug (Gal-CAP), in which the galactose was linked to capsaicin through an ether bond that is not hydrolysable (Scheme S2, Figures S23–S28). This modification preserves the physicochemical properties of the GalA-CAP prodrug while preventing esterase-mediated hydrolysis and subsequent conversion to capsaicin (Figure 8a). The Gal-CAP prodrug was injected at the sciatic nerve of rats. Results showed that the Gal-CAP prodrug demonstrated less effectiveness compared to capsaicin, with only 37.5% (3 out of 8) of the rats injected with 3 mg of Gal-CAP prodrug showing successful nociceptive axon blockade (Figure 8b). The average duration of nociceptive axon blockade in rats injected with 3 mg of prodrug only lasted for 0.88 ± 1.46 hours (Figures 8c, S29).
Figure 8.
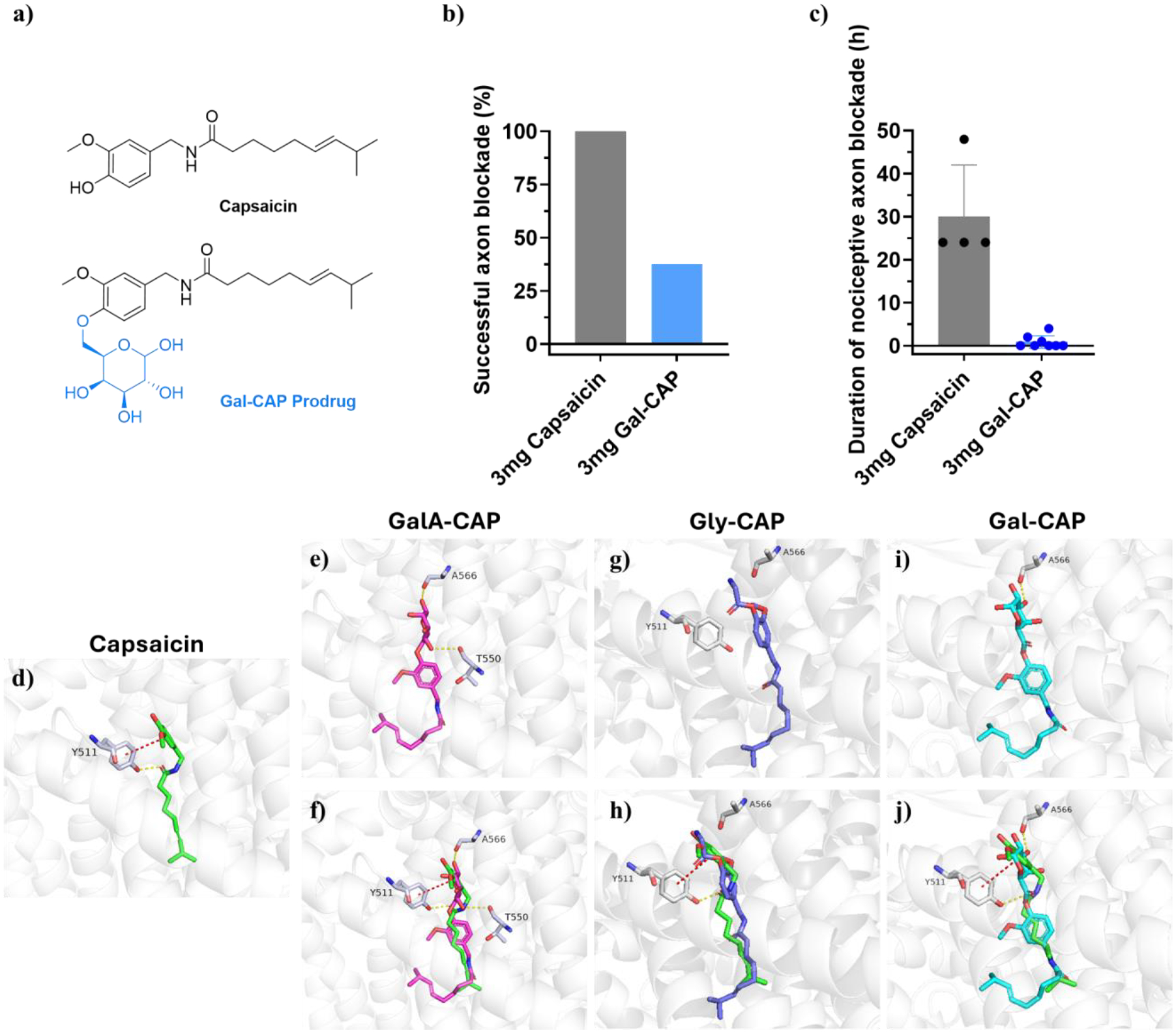
Enhanced efficacy and reduced side effects due to sustained release of capsaicin from the less active prodrug. (a) Structures of Gal-CAP prodrug and capsaicin. Comparison on frequency of successful axon blockade (b) and duration of nociceptive axon blockade (c) between 3 mg GalA-CAP prodrug, 3 mg Gly-CAP prodrug, 3 mg Gal-CAP prodrug, and 3 mg capsaicin. Data shown are mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, analysis of variance (ANOVA). (d-j) Molecular docking of capsaicin and prodrugs. Molecular docking of capsaicin and prodrugs. (d) Published binding mode of capsaicin with TRPV1 (generated from PDB code 7LPE). The atoms of capsaicin are colored as follows: carbon, green; oxygen, red; nitrogen, blue. The cartoon shows the protein and key residues are colored as follows: carbon, grey; oxygen, red; nitrogen, blue. Yellow dotted lines indicate H-bonds, and the red dotted lines represent π-π stacking interactions. (e, g, i) Proposed binding modes of GalA-CAP, Gly-CAP, and Gal-CAP prodrug with TRPV1. (f, h, j) Superposed docking poses of capsaicin and GalA-CAP, Gly-CAP, or Gal-CAP prodrug. The atoms of prodrugs are colored as follows. GalA-CAP: carbon, cyan; oxygen, red; nitrogen, blue. Gly-CAP: carbon, purple; oxygen, red; nitrogen, blue. Gal-CAP: carbon, pink; oxygen, red; nitrogen, blue.
The results from molecular docking studies also confirmed these experimental findings. Instead of π-π stacking interactions with residue Tyr511 of the TRPV1 receptor by capsaicin (Figure 8d), the GalA-CAP and Gal-CAP prodrugs, as well as the Gly-CAP prodrug, reveal unique and different binding modes with TRPV1 (Figure 8e–j). These results suggest that the GalA-CAP and Gly-CAP prodrug require hydrolysis into active capsaicin via ester bond cleavage to exert their functional effects and should follow similar cascade mechanisms as capsaicin.
2.8. Reduced irritancy of GalA-CAP and Gal-CAP prodrug
One limiting factor of capsaicin in its clinical use is the uncomfortable burning sensation post-application. As a result, pretreatment or co-administration with other local anesthetics is necessary to alleviate such uncomfortable burning sensations[42–43].. We assessed the relative irritancy of prodrugs using a previously established animal behavioral test[43]. We measured the frequency and duration of licking behavior during the first 5 minutes after the intraplantar administration of 0.1% (w/v) capsaicin, 0.05% (w/v) capsaicin, 0.1% (w/v) GalA-CAP prodrug (containing 0.063% (w/v) active capsaicin), 0.1% (w/v) Gal-CAP prodrug (containing 0.065% (w/v) active capsaicin), and 1×PBS buffer. The frequency of licking within 5 minutes after the injection of 0.1% GalA-CAP prodrug, 0.1% Gal-CAP prodrug decreased to 1 ± 0 times (1, 1, 1, and 1 time for 4 rats, respectively) and 1 ± 1.41 times (0, 0, 1, and 3 times for 4 rats, respectively). In contrast, the frequency of licking within 5 minutes after injection of 0.1% capsaicin and 0.05% capsaicin was 9.25 ± 4.79 times (5, 7, 9, and 16 times for 4 rats, respectively) and 4.75 ± 1.26 times (3, 5, 5, and 6 for 4 rats, respectively) (Figure 9). Likewise, the total duration of licking behavior was significantly reduced to 4.28 ± 5.81 seconds and 4.55 ± 5.77 seconds after the injection of 0.1% GalA-CAP prodrug, 0.1% Gal-CAP prodrug, respectively, compared to 48.77 ± 19.23 seconds after the injection of 0.1% capsaicin and 31.00 ± 14.03 seconds after the injection of 0.05% capsaicin. These results suggest that the conjugation with galacturonic acid or galactose substantially reduced the irritant properties of capsaicin.
Figure 9.
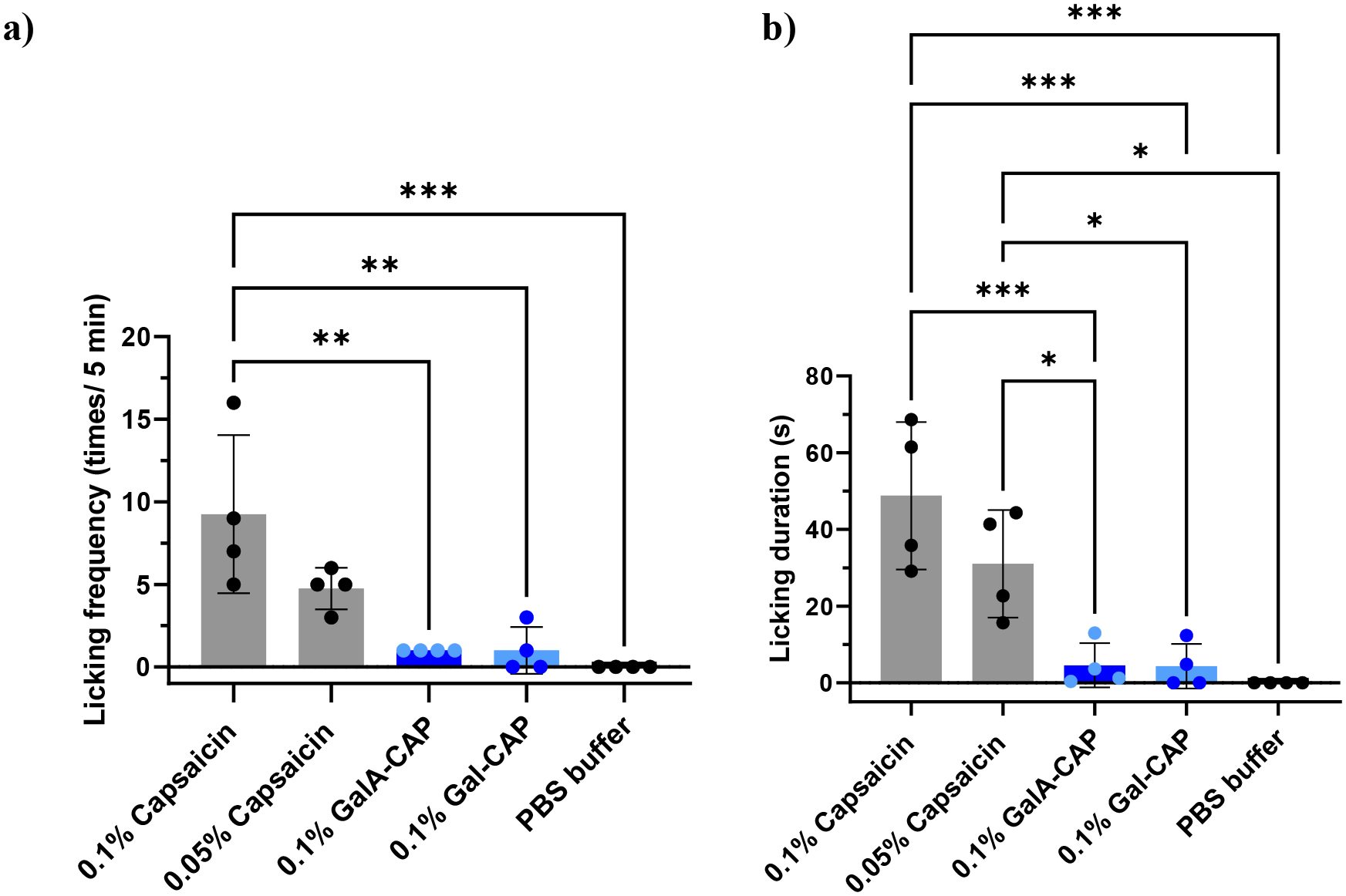
Licking frequency (a) and licking duration (b) during the first 5 min after intraplantar application of 0.1% (w/v) capsaicin, 0.05% (w/v) capsaicin, 0.1% (w/v) GalA-CAP prodrug, 0.1% (w/v) Gal-CAP prodrug and 1×PBS buffer. *P < 0.05, ***P < 0.001 analysis of variance (ANOVA), n = 4.
3. Discussion
There are no clinically available nociceptive-selective axon blocking drugs. Conventional amino-amide and amino-ester local anesthetics produce motor axonal signaling inhibition of approximately the same duration as in sensory axons, making these drugs clinically inadequate for post-operative or chronic pain management in weight-bearing limbs. The galacturonic acid-capsaicin prodrug has the potential to become the first clinically applicable long-duration nociceptive-selective local analgesic drug to treat chronic pain through a single injection.
This study presents the first evidence of GLUT-mediated peripheral nerve drug uptake. It is widely accepted that sugar-based prodrugs can bind to GLUTs and therefore enhance drug uptake by cells or permeation through biological barriers. For example, GLUTs facilitate the transport of sugar-based prodrugs from the blood circulation across the blood-brain barrier (BBB) into the brain[30, 44–45].. Dalpiaz et al. observed that glucose-conjugated dopamine interacted with GLUT1 and inhibited the transport of 3-O-methylglucose by human retinal pigment epithelium (HRPE) cells[31].. Fernández et al. found that the dopamine derivative, substituted at position C-6 of glucose, showed high binding affinity to GLUT1 in human erythrocytes[46]. Halmos et al. found that a glucose-chlorambucil derivative inhibited the uptake of [14C] D-glucose by the GLUT1 transporter in human erythrocytes[47]. Similarly, in cancer treatment, Lin et al. reported that glycan-based paclitaxel prodrugs enhanced delivery to cancer cells (NPC-TW01) through GLUTs[48]. However, it remains unclear whether GLUTs actively transport peripherally injected sugar-based prodrugs across PNBs into peripheral nerve endoneurium. We show that PNB-expressing GLUTs mediate sugar-based prodrug transport into peripheral nerve perineurium. As a result, there is increased uptake of peripherally injected drugs to inhibit axonal signal conduction, with longer duration of effect and reduced local and systemic adverse effects.
Enhancing local anesthetic permeability through PNBs has important clinical and scientific implications. Previous reports indicate that less than 1% of the local anesthetic injected perineurally penetrate the perineurium and ultimately act on the peripheral nerve axons[3]. Co-administration of local anesthetics with chemical permeation enhancers (CPEs) has been the most effective way to increase local anesthetic peripheral nerve bioavailability and thereby enhance its analgesic effect[49–50].. CPEs can interact with the intercellular lipids through physical processes including extraction, fluidization, increased disorder, and phase separation, thereby increasing the flux of local anesthetics into peripheral nerves[24]. However, CPEs are generally corrosive and excessive amounts can cause nerve damage[51–52].. In contrast, the galacturonic acid-capsaicin prodrug uses constitutionally expressed endogenous GLUTs to enhance local anesthetic peripheral nerve permeability. This strategy supports enhanced local anesthetic drug permeability and retention in peripheral nerves without causing CPE-related side effects.
The clinical application of this capsaicin-based prodrug extends beyond its primary use as a nerve-blocking agent for pain management. Due to capsaicin’s various biological effects, including antioxidant[53], antimicrobial[54], anti-inflammatory[55], anticancer[56] and anti-obesity properties[57–58], the prodrug could potentially be customized for a wide range of therapeutic interventions.
This prodrug approach is not limited to GLUTs but can also be applied to various other transporters expressed in PNBs, such as monocarboxylate transporters[59–60]. The prodrug strategy is not only applicable to capsaicin, but can be effectively applied to other local anesthetics like bupivacaine and lidocaine, as well as many anticancer drugs, antibiotic drugs, etc.
4. Conclusion
A nociceptive-selective axon blocking agent was developed that provided days of nociceptive-selective axon blockade with minimal local and systemic toxicity. This innovative design incorporates three crucial components: a capsaicin that selectively inhibits nociceptive sensory nerve signaling without compromising other sensory perceptions or motor function; a sugar moiety that enhances prodrug flux into nerves; and a degradable linker that ensures a controlled and sustained release of active capsaicin at a safe rate.
5. Experimental Section
Materials.
All chemicals and solvents of the highest purity available were used as purchased. 1,2,3,4-di-o-isopropylidene-α-d-galacturonic acid was purchased from Combi-Blocks, Inc. Capsaicin, methoxypolyethylene glycol amine (Mn = 750), and 1,2,3,4-di-o-isopropylidene-α-d-galactopyranose were supplied by Sigma-Aldrich (St. Louis, MO). DMAP, DCC, silica gels, PBS buffer (pH 7.4), methanol-d4 (100%, 99.96 atom% D), FITC, Tissue-Tek O.C.T. Compound, Glc-UDP-FL, and other organic solvents were purchased from VWR International Ltd (Radnor, PA).
Synthesis and characterization of GalA-CAP prodrug.
Dicyclohexylcarbodiimide (DCC, 376 mg, 1.82 mmol) was added to a stirring solution of capsaicin (2, 555 mg, 1.82 mmol), 4-Dimethylaminopyridine (DMAP, 278 mg, 2.27 mmol) and 1,2,3,4-di-o-isopropylidene-α-d-galacturonic acid (1, 500 mg, 1.82 mmol) in anhydrous dichloromethane (DCM). The reaction mixture was stirred at room temperature overnight. The solvent was removed under vacuum and the crude product was partitioned between ethyl acetate and water. The organic phase was dried on Na2SO4, filtered and then concentrated in vacuum. The crude product was purified using flash chromatography on silica gel eluting with ethyl acetate, obtaining 1,2,3,4-di-o-isopropylidene-α-d-galacturonic acid-capsaicin ester (3) as a yellow oil (920 mg, 90% yield).
1H-NMR (500 MHz, methanol-d4) δ (ppm) 0.89 (d, 6H, l’–H); 0.99 (d, 6H, l–H); 1.20 (m, 2H, h–H); 1.31/1.40 (d, 12H, r–H); 1.66 (m, 2H, g–H); 2.02 (m, 2H, i–H); 2.26 (m, 2H, f–H); 3.83 (s, 3H, d–H); 4.38 (s, 2H, e–H); 4.52 (q, H, o–H); 4.72 (d, H, n–H); 4.75 – 4.81 (m, 2H, p, q–H); 5.34 – 5.42 (m, 2H, j, k–H); 5.64 (d, H, m–H); 6.90 (d, H, c–H); 7.02 – 7.05 (m, 2H, a, b–H); 13C-NMR (125 MHz, methanol-d4) δ (ppm) 21.64 (C17’, dihydrocapsaicin); 21.74 (C17); 23.58 (C24); 25.2 (C12); 25.71 (C11’, dihydrocapsaicin); 27 (C14’, dihydrocapsaicin); 27.74 (C16’, dihydrocapsaicin); 35.60 (C10); 35.73 (C10’, dihydrocapsaicin); 38.76 (C15’, dihydrocapsaicin); 42.41 (C8); 54.99 (C7); 68.52 (C21); 70.35 (C19); 70.82 (C20); 72.04 (C22); 96.62 (C18); 109.05 (C26); 109.89 (C25); 111.93 (C5); 119.51 (C2); 112.28 (C3); 126.53 (C4); 137.53 (C15); 138.34 (C1); 151.14 (C6); 166.82 (C23); 174.7 (C9); MS (m/z) calculated for C30H43NO9 [M] = 561.29; found 562.312 [M + H]+; 584.209 [M + Na]+; 600.267 [M + K]+.
To a stirred solution of 3 (920 mg, 1.60 mmol) in DCM (10 mL), trifluoroacetic acid (5 mL) was added, and the mixture was stirred at room temperature for 48 h. After the reaction was completed, 10 mL of water was added. The organic layer was dried over sodium sulfate, filtered, concentrated, and submitted to a flash chromatography on silica gel, eluting with DCM/MeOH (90/10). After the evaporation of solvent, the GalA-CAP prodrug was obtained as a brown solid (490 mg, 62% yield).
1H-NMR (500 MHz, methanol-d4) δ (ppm) 0.87 (d, 6H, l’–H); 0.96 (d, 6H, l–H); 1.17 (m, 2H, h–H); 1.64 (m, 2H, g–H); 2.0 (m, 2H, i–H); 2.24 (m, 2H, f–H); 3.8 (s, 3H, d–H); 4.38 (s, 2H, e–H); 3.34 – 5.30 (m, 5H, m, n, o, p, q–H); 5.37 (m, 2H, j, k–H); 6.88 (d, H, c–H); 7.02 – 7.05 (m, 2H, a, b–H); 13C-NMR (125 MHz, methanol-d4) δ (ppm) 21.64 (C17’, dihydrocapsaicin); 21.74 (C17); 25.16 (C12); 25.71 (C11’, dihydrocapsaicin); 27 (C14’, dihydrocapsaicin); 27.74 (C16’, dihydrocapsaicin); 28.92 (C11); 29.31 (C13’, dihydrocapsaicin); 31.87 (C16); 35.60 (C10); 35.73 (C10’, dihydrocapsaicin); 38.76 (C15’, dihydrocapsaicin); 42.41 (C8); 54.99 (C7); 68.52 (C19); 70.37 (C21); 74.18 (C22); 93.18 (C18); 111.93 (C5); 119.51 (C2); 122.28 (C3); 126.53 (C4); 137.7 (C15); 138.34 (C1); 168.15 (C23); 174.78 (C9); MS (m/z) calculated for C24H35NO9 [M] = 481.23; found 504.298 [M + Na]+; 520.274 [M + K]+.
Measurement of chemical stability.
10 mg of the prodrugs was dissolved in 10 mL of Milli-Q water accordingly and cultured at 37 °C on the hotplate. 100 μL of the samples were withdrawn at each time point (0 h, 1 h, 2 h, 4 h, 6 h, 10 h, 24 h, 48 h, 72 h, 96 h, 120 h, and 168 h) and submitted to UPLC with a photodiode array detector and HSS T3 (1.8 μm, 2.1 mm × 100 mm) column (Acquity UPLC System; Waters, Milford, MA, USA) for quantification immediately. 10 μL of the collected sample were injected into the HSS T3 column for analysis. For esterase stability studies, 20 or 120 μL of an esterase solution (0.5 units/mL) was added to 480 or 380 μL of PBS buffer to make the esterase solution at the concentration of 20 units/mL or 120 units/mL, respectively. The esterase solution was then added to 500 μL of a stock solution of the prodrugs (2 mg/mL) in PBS buffer preincubated at 37 °C. 50 μL of the aliquots were removed after 10 minutes and immediately analyzed by UPLC as described above.
Synthesis and characterization of Gly-CAP prodrug.
To a solution of the capsaicin (555 mg, 1.82 mmol), Boc-Glycine-OH (397 mg, 2.27 mmol), and DMAP (278 mg, 2.27 mmol) in DCM (6 mL) at room temperature were added DCC (376 mg, 1.82 mmol). The reaction mixture was stirred at room temperature for 12 h. The solvent was removed under vacuum and the crude product was partitioned between ethyl acetate and water. The organic layer was dried over anhydrous Na2SO4 and concentrated in vacuum; the following purification by silica gel chromatography (Hexene/ethyl acetate, 50/50) led to the crude BOC-Gly-capsaicin as a yellow solid. Subsequently, to a stirred solution of BOC-Gly-capsaicin in ethyl acetate (3 mL), 4 M hydrochloric acid in 1,4-dioxane solution (10 mL) was added, and the mixture was stirred at room temperature for 12 h. After the reaction was completed, the organic layer was concentrated in vacuo; the following purification by recrystallization in ethyl acetate, obtaining Gly-CAP prodrug as white solid (524 mg, 72% yield).
1H-NMR (500 MHz, methanol-d4) δ (ppm) 0.81 (d, 6H, l’–H); 0.91 (d, 6H, l–H); 1.16 (m, 2H, h–H); 1.64 (m, 2H, g–H); 1.94 (m, 2H, i–H); 2.25 (m, 2H, f–H); 3.82 (s, 3H, d–H); 4.15 (s, 2H, m–H); 4.36 (s, 2H, e–H); 5.36 (m, 2H, j, k–H); 6.91 (d, H, a–H); 7.07 (d, 2H, b, c–H); 13C-NMR (125 MHz, methanol-d4) δ (ppm) 20.15 (C17’, dihydrocapsaicin); 20.24 (C17); 23.65 (C12); 24.17 (C11’, dihydrocapsaicin); 25.48 (C14’, dihydrocapsaicin); 26.22 (C16’, dihydrocapsaicin); 27.43 (C11); 27.80 (C13’, dihydrocapsaicin); 29.37 (C16); 30.36 (C10); 34.07 (C10’, dihydrocapsaicin); 37.24 (C15’, dihydrocapsaicin); 40.86 (C8); 53.58 (C7); 110.21 (C5); 117.82 (C2); 120.43 (C3); 124.97 (C14); 136.40 (C4); 137.42 (C15); 149.29 (C6); 164.07 (C18); 174.3 (C9); MS (m/z) calculated for C20H30N2O4 [M] = 362.22; found 363.196 [M + H]+; 385.174 [M + Na]+; 401.140 [M + K]+.
Synthesis and characterization of Gal-CAP prodrug.
A solution of p-toluenesulfonyl chloride (549 mg, 2.88 mmol) in anhydrous DCM (3 mL) was added to a cooled (0 °C) solution of 1,2,3,4-di-o-isopropylidene-α-d-galactopyranose (4, 200 mg, 0.77 mmol) and DMAP (179 mg, 1.47 mmol) in anhydrous pyridine (3 mL). The solution was warmed to room temperature and stirred for 24 h. The solvent was removed under reduced pressure and the residue dissolved in water and extracted with ethyl acetate. The organic layer was washed with 0.5 M HCl and saturated sodium bicarbonate, brine, then dried over sodium sulfate, filtered, and evaporated to give the product (5). The product (5) was then used directly in the next step without further purification.
Compound 5 (400 mg, 0.97 mmol) was added to a pressure glassware containing capsaicin (400 mg, 1.31 mmol), K2CO3 (500 mg, 3.62 mmol) and 10 mL DMSO, and stirred for 48 h at 95 °C. The solvent was removed under heating and vacuum, and the crude product was washed with water, extracted with ethyl acetate and submitted to a flash chromatography on silica gel (ethyl acetate/hexane = 50/50) to give the product (6, 20% yield).
1H-NMR (500 MHz, methanol-d4) δ (ppm) 0.87 (d, 6H, l’–H); 0.95 (d, 6H, l–H); 1.16 (m, 2H, h–H); 1.31/1.40 (d, 12H, r–H); 1.61 (m, 2H, g–H); 1.97 (m, 2H, i–H); 2.21 (m, 2H, f–H); 3.81 (s, 3H, d–H); 4.27 (s, 2H, e–H); 4.02/4.64 (m, 2H, s–H); 4.13 – 4.36 (m, 4H, p, q, n, o–H); 5.30 – 5.39 (m, 2H, j, k–H); 5.49 (d, H, m–H); 6.69 – 6.92 (m, 3H, a, b, c–H); 13C-NMR (125 MHz, methanol-d4) δ (ppm) 21.66 (C17’, dihydrocapsaicin); 21.74 (C17); 23.77 (C24); 25.2 (C12); 25.71 (C11’, dihydrocapsaicin); 27 (C14’, dihydrocapsaicin); 27.74 (C16’, dihydrocapsaicin); 28.92 (C11); 29.31 (C13’, dihydrocapsaicin); 31.87 (C16); 35.60 (C10); 35.73 (C10’, dihydrocapsaicin); 38.76 (C15’, dihydrocapsaicin); 42.41 (C8); 55.32 (C7); 66.13 (C23); 68.27 (C21); 70.66 (C19); 70.97 (C20); 96.39 (C18); 108.50 (C5); 109.08 (C2); 114.68 (C26); 115.26 (C25); 120.02 (C3); 126.48 (C4); 132.77 (C14); 137.73 (C15); 147.29 (C1); 150.01 (C6); 174.57 (C9); MS (m/z) calculated for C30H45NO8 [M] = 547.31; found 570.271 [M + Na]+; 586.258 [M + K]+.
To a stirred solution of 6 (100 mg, 0.13 mmol) in DCM (10 mL), trifluoroacetic acid (5 mL) was added, and the mixture was stirred at room temperature for 48 h. After the reaction, 10 mL of water was added. The organic layer was dried over sodium sulfate, filtered, concentrated, and submitted to a flash chromatography on silica gel (ethyl acetate/hexane = 50/50 to ethyl acetate). The product portions were collected and evaporated to give the product as a brown solid (82% yield).
1H-NMR (500 MHz, methanol-d4) δ (ppm) 0.84 (d, 6H, l’–H); 0.92 (d, 6H, l–H); 1.12 (m, 2H, h–H); 1.59 (m, 2H, g–H); 1.96 (m, 2H, i–H); 2.19 (m, 2H, f–H); 3.78 (s, 3H, d–H); 4.25 (s, 2H, e–H); 3.20 – 5.20 (m, 5H, m, n, o, p, q–H); 5.32 (m, 2H, j, k–H); 6.79 (d, H, c–H); 6.84 – 6.92 (m, 2H, a, b–H); 13C-NMR (125 MHz, methanol-d4) δ (ppm) 21.66 (C17’, dihydrocapsaicin); 21.74 (C17); 25.19 (C12); 25.71 (C11’, dihydrocapsaicin); 27 (C14’, dihydrocapsaicin); 27.74 (C16’, dihydrocapsaicin); 28.92 (C11); 29.31 (C13’, dihydrocapsaicin); 31.87 (C16); 35.60 (C10); 35.73 (C10’, dihydrocapsaicin); 38.76 (C15’, dihydrocapsaicin); 42.41 (C8); 55.13 (C7); 69.87 (C19); 72.95 (C21); 73.6 (C22); 97.37 (C18); 115.55 (C5); 117.91 (C2); 119.85 (C3); 126.49 (C4); 137.73 (C15); 174.70 (C9); MS (m/z) calculated for C24H37NO8 [M] = 467.25; found 490.176 [M + Na]+; 506.154 [M + K]+.
Animal studies.
Animal studies were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee of the University of Alabama (Protocol ID: 19-11-2992). Adult male Sprague-Dawley rats weighing 250–350 g (Charles River Laboratories, Wilmington, MA, USA) were group-housed under a 12 h/12 h light/dark cycle. Prior to injection, rats were briefly anesthetized with isoflurane through a facemask. Drug formulations were prepared by combining a Tween 20 solution with either free capsaicin or the GalA-CAP prodrug (both dissolved in methanol), followed by a vacuum process to evaporate the solvent. Prior to injection, the formulations were reconstituted in 0.3 mL of PBS buffer to create a 2.5% w/v Tween 20 solution containing a predetermined quantity of capsaicin or prodrug, achieved through vortex mixing. Solutions with 3 mg of capsaicin included 12 mg (4% w/v) of Tween 20 to enhance dissolution and avoid precipitation of capsaicin. The injection was conducted in the left leg by introducing a 23 G × 3/4” needle posteromedially into the greater trochanter, pointing in an anteromedial direction. Once in contact with bone, 0.3 mL of the test formulation was injected. Neurobehavioral testing was performed at predetermined intervals after injection. The right leg served as a control for systemic toxicity.
Sensory nerve blockade was assessed by a modified hotplate test. The time that rats left their hindpaws on a hot plate (Model 39D Hot Plate Analgesia Meter, IITC Inc., Woodland Hills, CA) at 56 °C was measured by a stopwatch. The time is referred to as thermal latency. The paw was removed from the hotplate after 12 seconds to avoid injury to the rat. All experiments were repeated 3 times at each time point, and the average was used for data analysis. Motor function was tested by measuring extensor postural thrust. Briefly, the rat was placed on top of a digital balance and allowed to bear its weight on one hindpaw at a time. The maximum weight that the rat can bear without its ankle touching the balance was measured.
The duration of sensory block is the time it takes for the thermal latency to return to 7 seconds (the baseline thermal latency is approximately 2 seconds; therefore, 7 seconds represents the midpoint between maximal latency, set as the 12-second cutoff, and the baseline). The duration of motor block was defined as the time to return 50% of weight bearing.
Molecular docking.
Selected protein crystal complex (PDB: 7LPE) was repaired, hydrogenated, decrystallized, and energy optimized using the Protein Preparation module of Meastro, and to define binding pockets. The prodrug molecules were then optimized for 3D conformation and energy minimization using the LigPre module. Finally, the small molecules were flexibly docked into the binding pocket using the XP docking module of Glide. Top poses were used for visualization analysis by Pymol 2.5.
In vivo distribution of FITC, PEG750-FL, Gal-FL and Glc-UDP-FL.
Gal-FL was synthesized by mixing FITC (12 mg, 0.031 mmol) with galactosamine hydrochloride (6.7 mg, 0.031 mmol) in 2 mL solvent (water/ethanol: 25/75). After the pH was adjusted to 7 using sodium bicarbonate, the mixture was stirred at room temperature for 24 hours. The mixture was used for further analysis and the preparation of the injection solution directly.
PEG750-FL was synthesized by mixing FITC (10 mg, 25.7 μmol) with methoxy polyethylene glycol amine (Mn = 750, 190 mg, 253 μmol) at an elevated temperature (80 °C) for 24 hours. The mixture was used for further analysis and the preparation of the injection solution directly.
A previous established but modified method was used to study the distribution of fluorescence in the nerve[61].. Under isoflurane-oxygen anesthesia, 0.3 mL of test solution (either FITC, Gal-FL, Glc-UDP-FL, or PEG750-FL in PBS buffer) was injected beside the sciatic nerve. The absorption intensities were determined by a plate reader (SpectraMax i3x, Molecular Devices) to ensure that the same dose of four compounds were injected. After 4 hours, the sciatic nerves were harvested and embedded into OCT compound and frozen sections were prepared. A coverslip was placed, and the slides were imaged by Leica TCS SP2 confocal microscopy.
To measure the depth that the injected compounds penetrated the nerve. The quantitative analysis of the confocal fluorescence images was conducted by dividing the nerve structure into 10 concentric areas equally separated by distance based on the nerve shape (Figure S21). All the fluorescence intensities throughout the nerve were calculated by ImageJ.
Tissue harvesting and histology.
Rats were sacrificed 14 days after the injection unless there was still a sensory block, and the sciatic nerve was harvested together with surrounding tissues to characterize the possible chronic inflammation and nerve degeneration. Muscle samples were fixed in 10% neutral buffered formalin and processed for histology (hematoxylin–eosin stained slides) using standard techniques. Muscle samples were stained with hematoxylin and eosin, and then scored for inflammation (0–4 points) and myotoxicity (0–6 points)[62].
To evaluate the neurotoxicity, the sciatic nerve samples were processed and fixed in Karnovsky’s KII Solution (2.5% glutaraldehyde, 2.0% paraformaldehyde, 0.025% calcium chloride in 0.1 M cacodylate buffer, pH 7.4). Samples were treated with osmium tetroxide for post-fixation for 2 hours and were subsequently dehydrated in graded ethanol solutions for 10 min each (30%, 60%, 90%, 100%, 100%, 100%). Then, the nerves were infiltrated with ethanol / Epon resin mixtures (1:1, 1:2, 0:1). After being sectioned by a diamond knife through ultramicrotome, nerve sections of 800 nm were stained with toluidine blue, followed by light microscopy. Nerve sections of 100 nm were observed by transmission electron microscopy (TEM, Hitachi H-7650), and the diameter of the nerves was quantified by Image J software.
Statistical Analysis.
Data are presented as means ± SDs (n = 3 in distribution measurement study, n = 4 or 8 in neurobehavioral studies). The statistical differences between groups were tested by one-way analysis of variance (ANOVA) for multiple comparisons. Statistical analyses were performed with GraphPad Prism 9 software. Statistical significance was defined as a p < 0.05.
Supplementary Material
Acknowledgements:
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Numbers: R01GM144388, and by the National Institute of Neurological Disorders and Stroke under Award Number R61NS123196.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest:
The authors declare no conflict of interest.
References
- [1].Becker DE, Reed KL, Anesthesia progress 2012, 59, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Butterworth J. F. t., Reg Anesth Pain Med 2010, 35, 167. [DOI] [PubMed] [Google Scholar]
- [3].Lirk P, Hollmann MW, Strichartz G, Anesthesia & Analgesia 2018, 126, 1381. [DOI] [PubMed] [Google Scholar]
- [4].Li Q, Li X, Bury E, Lackey K, Koh A, Wesselmann U, Yaksh T, Zhao C, Advanced Functional Materials 2023, 33, 2301025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kehlet H, Jensen TS, Woolf CJ, Lancet 2006, 367, 1618. [DOI] [PubMed] [Google Scholar]
- [6].Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D, Nature 1997, 389, 816. [DOI] [PubMed] [Google Scholar]
- [7].Chávez AE, Chiu CQ, Castillo PE, Nat. Neurosci 2010, 13, 1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, Raumann BE, Basbaum AI, Julius D, Neuron 1998, 21, 531. [DOI] [PubMed] [Google Scholar]
- [9].Zeng J, Lu Y, Chu H, Lu L, Chen Y, Ji K, Lin Y, Li J, Wang S, Heliyon 2024. [DOI] [PMC free article] [PubMed]
- [10].Mohapatra DP, Nau C, Journal of Biological Chemistry 2003, 278, 50080. [DOI] [PubMed] [Google Scholar]
- [11].Haanpää M, Treede RD, European Neurology 2012, 68, 264. [DOI] [PubMed] [Google Scholar]
- [12].Binshtok AM, Bean BP, Woolf CJ, Nature 2007, 449, 607. [DOI] [PubMed] [Google Scholar]
- [13].Peltonen S, Alanne M, Peltonen J, Tissue Barriers 2013, 1, e24956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ilie MA, Caruntu C, Tampa M, Georgescu SR, Matei C, Negrei C, Ion RM, Constantin C, Neagu M, Boda D, Experimental and therapeutic medicine 2019, 18, 916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chard PS, Bleakman D, Savidge JR, Miller RJ, Neuroscience 1995, 65, 1099. [DOI] [PubMed] [Google Scholar]
- [16].Knotts T, Mease K, Sangameswaran L, Felx M, Kramer S, Donovan J, J Orthop Res 2022, 40, 2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Asiri YI, Zaheen Hassan M, Arabian Journal of Chemistry 2023, 16, 105180. [Google Scholar]
- [18].Lan Y, Sun Y, Yang T, Ma X, Cao M, Liu L, Yu S, Cao A, Liu Y, Molecular Pharmaceutics 2019, 16, 3430. [DOI] [PubMed] [Google Scholar]
- [19].Linderoth L, Peters GH, Madsen R, Andresen TL, Angew. Chem. Int. Ed 2009, 48, 1823. [DOI] [PubMed] [Google Scholar]
- [20].Feng Y, Zhu Y, Wan J, Yang X, Firempong CK, Yu J, Xu X, Journal of Functional Foods 2018, 44, 137. [Google Scholar]
- [21].Shomorony A, Santamaria CM, Zhao C, Rwei AY, Mehta M, Zurakowski D, Kohane DS, Anesthesia & Analgesia 2019, 129, 709. [DOI] [PubMed] [Google Scholar]
- [22].Cura AJ, Carruthers A, Compr Physiol 2012, 2, 863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Iwanaga T, Takahashi-Iwanaga H, Nio-Kobayashi J, Ebara S, Biomedical research (Tokyo, Japan) 2022, 43, 145. [DOI] [PubMed] [Google Scholar]
- [24].Moghadam SH, Saliaj E, Wettig SD, Dong C, Ivanova MV, Huzil JT, Foldvari M, Mol Pharm 2013, 10, 2248. [DOI] [PubMed] [Google Scholar]
- [25].Magnani P, Cherian PV, Gould GW, Greene DA, Sima AA, Brosius FC 3rd, Metabolism 1996, 45, 1466. [DOI] [PubMed] [Google Scholar]
- [26].Mueckler M, Thorens B, Molecular aspects of medicine 2013, 34, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Burant CF, Bell GI, Biochemistry 1992, 31, 10414. [DOI] [PubMed] [Google Scholar]
- [28].Rumsey SC, Kwon O, Xu GW, Burant CF, Simpson I, Levine M, J Biol Chem 1997, 272, 18982. [DOI] [PubMed] [Google Scholar]
- [29].Thorens B, Mueckler M, Am J Physiol Endocrinol Metab 2010, 298, E141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sun Y, Zabihi M, Li Q, Li X, Kim BJ, Ubogu EE, Raja SN, Wesselmann U, Zhao C, Advanced Therapeutics 2023, 6, 2200150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Dalpiaz A, Filosa R, de Caprariis P, Conte G, Bortolotti F, Biondi C, Scatturin A, Prasad PD, Pavan B, Int J Pharm 2007, 336, 133. [DOI] [PubMed] [Google Scholar]
- [32].Gynther M, Ropponen J, Laine K, Leppanen J, Haapakoski P, Peura L, Jarvinen T, Rautio J, J Med Chem 2009, 52, 3348. [DOI] [PubMed] [Google Scholar]
- [33].Mikuni K, Nakanishi K, Hara K, Hara K, Iwatani W, Amano T, Nakamura K, Tsuchiya Y, Okumoto H, Mandai T, Biol Pharm Bull 2008, 31, 1155. [DOI] [PubMed] [Google Scholar]
- [34].Melisi D, Curcio A, Luongo E, Morelli E, Rimoli MG, Curr. Top. Med. Chem 2011, 11, 2288. [DOI] [PubMed] [Google Scholar]
- [35].Rwei AY, Lee JJ, Zhan C, Liu Q, Ok MT, Shankarappa SA, Langer R, Kohane DS, Proc. Nat. Acad. Sci. U.S.A 2015, 112, 15719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Li Y, Ji T, Torre M, Shao R, Zheng Y, Wang D, Li X, Liu A, Zhang W, Deng X, Yan R, Kohane DS, Nat. Commun 2023, 14, 6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ji T, Li Y, Deng X, Rwei AY, Offen A, Hall S, Zhang W, Zhao C, Mehta M, Kohane DS, Nat Biomed Eng 2021, 5, 1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Simons EJ, Bellas E, Lawlor MW, Kohane DS, Molecular pharmaceutics 2009, 6, 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Simone DA, Nolano M, Johnson T, Wendelschafer-Crabb G, Kennedy WR, J. Neurosci 1998, 18, 8947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Fischer MJM, Ciotu CI, Szallasi A, Frontiers in Physiology 2020, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Olszewski K, Barsotti A, Feng XJ, Momcilovic M, Liu KG, Kim JI, Morris K, Lamarque C, Gaffney J, Yu X, Patel JP, Rabinowitz JD, Shackelford DB, Poyurovsky MV, Cell Chem Biol 2022, 29, 423. [DOI] [PubMed] [Google Scholar]
- [42].Christensen JD, Vecchio SL, Andersen HH, Elberling J, Arendt-Nielsen L, The Journal of Pain 2021, 22, 778. [DOI] [PubMed] [Google Scholar]
- [43].Binshtok AM, Gerner P, Oh SB, Puopolo M, Suzuki S, Roberson DP, Herbert T, Wang CF, Kim D, Chung G, Mitani AA, Wang GK, Bean BP, Woolf CJ, Anesthesiology 2009, 111, 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Botti G, Dalpiaz A, Pavan B, Pharmaceutics 2021, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Rautio J, Laine K, Gynther M, Savolainen J, The AAPS journal 2008, 10, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Fernández C, Nieto O, Fontenla JA, Rivas E, de Ceballos ML, Fernández-Mayoralas A, Org. Biomol. Chem 2003, 1, 767. [DOI] [PubMed] [Google Scholar]
- [47].Halmos T, Santarromana M, Antonakis K, Scherman D, Eur. J. Pharmacol 1996, 318, 477. [DOI] [PubMed] [Google Scholar]
- [48].Lin Y-S, Tungpradit R, Sinchaikul S, An F-M, Liu D-Z, Phutrakul S, Chen S-T, J. Med. Chem 2008, 51, 7428. [DOI] [PubMed] [Google Scholar]
- [49].Zhao C, Liu A, Santamaria CM, Shomorony A, Ji T, Wei T, Gordon A, Elofsson H, Mehta M, Yang R, Kohane DS, Nature Communications 2019, 10, 2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Mehta M, Zhao C, Liu A, Innocent C, Kohane DS, Translational Vision Science & Technology 2022, 11, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Santamaria CM, Zhan C, McAlvin JB, Zurakowski D, Kohane DS, Anesth Analg 2017, 124, 1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Li X, Li Q, Song S, Stevens AO, Broemmel Z, He Y, Wesselmann U, Yaksh T, Zhao C, Advanced Therapeutics 2023, 6, 2200199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Manjunatha H, Srinivasan K, Can. J. Physiol. Pharmacol 2007, 85, 588. [DOI] [PubMed] [Google Scholar]
- [54].Marini E, Magi G, Mingoia M, Pugnaloni A, Facinelli B, Frontiers in Microbiology 2015, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kim C-S, Kawada T, Kim B-S, Han I-S, Choe S-Y, Kurata T, Yu R, Cell. Signal 2003, 15, 299. [DOI] [PubMed] [Google Scholar]
- [56].Friedman JR, Nolan NA, Brown KC, Miles SL, Akers AT, Colclough KW, Seidler JM, Rimoldi JM, Valentovic MA, Dasgupta P, J. Pharmacol. Exp. Ther 2018, 364, 462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Zheng J, Zheng S, Feng Q, Zhang Q, Xiao X, Biosci. Rep 2017, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Lu M, Chen C, Lan Y, Xiao J, Li R, Huang J, Huang Q, Cao Y, Ho CT, Food Funct 2020, 11, 2848. [DOI] [PubMed] [Google Scholar]
- [59].Sun Y, Zhao D, Wang G, Jiang Q, Guo M, Kan Q, He Z, Sun J, Asian J Pharm Sci 2019, 14, 631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Rawat A, Morrison BM, Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutics 2021, 18, 2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Liu Q, Santamaria CM, Wei T, Zhao C, Ji T, Yang T, Shomorony A, Wang BY, Kohane DS, Nano Lett 2018, 18, 32. [DOI] [PubMed] [Google Scholar]
- [62].McAlvin JB, Padera RF, Shankarappa SA, Reznor G, Kwon AH, Chiang HH, Yang JS, Kohane DS, Biomaterials 2014, 35, 4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.