

ABSTRACT
CsrA is an RNA-binding protein that regulates processes critical for growth and survival, including central carbon metabolism, motility, biofilm formation, stress responses, and expression of virulence factors in pathogens. Transcriptomics studies in Escherichia coli suggested that CsrA repressed genes involved in surviving extremely acidic conditions. Here, we examine the effects of disrupting CsrA-dependent regulation on the expression of genes and circuitry for acid stress survival and demonstrate CsrA-mediated repression at multiple levels. We show that this repression is critical for managing the trade-off between growth and survival; overexpression of acid stress genes caused by csrA disruption enhances survival under extreme acidity but is detrimental for growth under mildly acidic conditions. In vitro studies confirmed that CsrA binds specifically to mRNAs of structural and regulatory genes for acid stress survival, causing translational repression. We also found that translation of the top-tier acid stress regulator, evgA, is coupled to that of a small leader peptide, evgL, which is repressed by CsrA. Unlike dedicated acid stress response genes, csrA and its sRNA antagonists, csrB and csrC, did not exhibit a substantial response to acid shock. Furthermore, disruption of CsrA regulation of acid stress genes impacted host-microbe interactions in Caenorhabditis elegans, alleviating GABA deficiencies. This study expands the known regulon of CsrA to genes of the extreme acid stress response of E. coli and highlights a new facet of the global role played by CsrA in balancing the opposing physiological demands of stress resistance with the capacity for growth and modulating host interactions.
IMPORTANCE
To colonize/infect the mammalian intestinal tract, bacteria must survive exposure to the extreme acidity of the stomach. E. coli does this by expressing proteins that neutralize cytoplasmic acidity and cope with molecular damage caused by low pH. Because of the metabolic cost of these processes, genes for surviving acid stress are tightly regulated. Here, we show that CsrA negatively regulates the cascade of expression responsible for the acid stress response. Increased expression of acid response genes due to csrA disruption improved survival at extremely low pH but inhibited growth under mildly acidic conditions. Our findings define a new layer of regulation in the acid stress response of E. coli and a novel physiological function for CsrA.
KEYWORDS: CsrA, acid stress, posttranscriptional regulation, translation regulation, protein-RNA interaction
INTRODUCTION
Bacteria have sophisticated regulatory systems that detect and respond to environmental changes. An important example is the carbon storage regulatory (Csr) system, which is present in E. coli and other species. The Csr system plays critical regulatory roles in biofilm formation, central carbon metabolism, stress response systems, motility, quorum sensing, and virulence factor expression in pathogens (1–7). The central component of the Csr system is CsrA, a homodimeric protein that recognizes and binds to specific RNA sequences (1, 7). CsrA generally represses stress responses and systems associated with the stationary phase of growth while activating the expression of genes associated with exponential growth (7, 8). CsrA-mediated regulation involves binding to sites containing a critical GGA motif, which is often found in the single-stranded loop of an RNA hairpin (9, 10). These binding sites are typically located in the 5′ leader or early mRNA coding regions. CsrA binding can regulate translation initiation, RNA stability, riboswitch activity, or transcription elongation (5, 7, 11–18).
The expression of csrA is tightly regulated, both transcriptionally and posttranscriptionally (19). In addition, CsrA activity is extensively regulated by other components of the Csr system. In E. coli, two small RNA (sRNA) antagonists, CsrB and CsrC, contain multiple high-affinity CsrA binding sites that act to sequester CsrA from other regulatory targets (20, 21). csrB/C transcription is activated in response to environmental stresses, the accumulation of metabolic end products such as acetate, and quorum sensing (22, 23). CsrB/C levels are also regulated by CsrD via RNase E-dependent turnover, which is activated by glucose in E. coli (24–27). These findings imply that elevated CsrB/C levels cause decreased CsrA activity under environmental stresses and upon metabolic end product accumulation but increased activity in the presence of preferred carbon sources.
Recent high-throughput studies identified CsrA as a likely regulator of acid stress resistance genes in E. coli (Fig. 1) (3, 9, 28–30). Despite being characterized as a neutrophile, E. coli can survive extremely acidic conditions for extended periods of time (31). Tolerance for extremely acidic environments is thought to protect against exposure to gastric acidity (32) and contribute to the low oral infectious dose of certain E. coli pathogens (33, 34).
Fig 1.
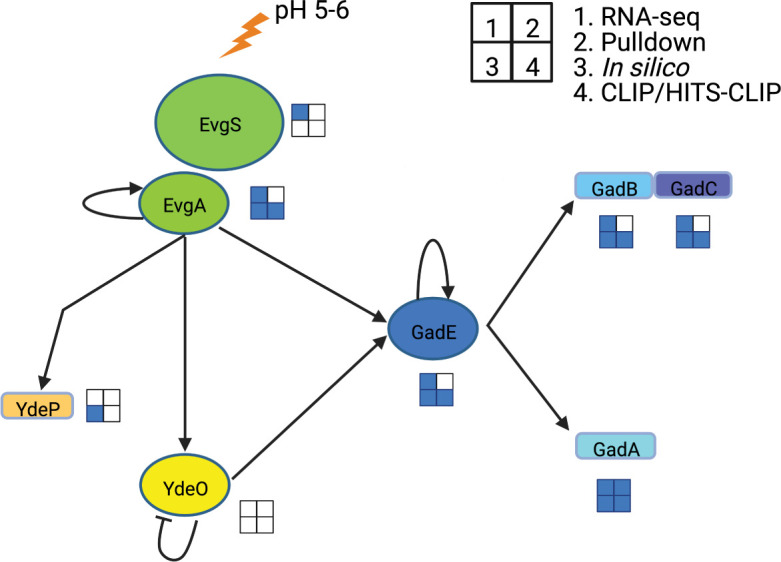
Hypothetical targets of CsrA-dependent regulation in the EvgA-YdeO-GadE circuit. RNA-seq studies include Log2 fold changes > 1 (3, 24, 28). Pulldown refers to CsrA-bound RNA targets (29). In silico predictions refer to the computational identification of putative CsrA-binding sites (30). CLIP-seq (3) and HITS-CLIP-seq (28) refer to CsrA-binding targets identified by these methods. Blue squares indicate that the gene was identified as a possible regulatory target of CsrA in the respective study.
Extremely acidic conditions harm cells by damaging lipids, DNA and RNA, denaturing proteins, and disrupting metabolism (31–33). E. coli utilizes several strategies to survive and grow under acidic conditions, including systems that leverage metabolic reactions to neutralize the internal pH (31–33, 35–37). The most critical system for survival in extremely acidic conditions is the glutamate-dependent acid resistance (GDAR) system (31, 38, 39). Three proteins constitute GDAR, including GadA and GadB, which convert glutamate to CO2 and γ-aminobutyric acid (GABA), and GadC, which imports glutamate and exports GABA (Fig. 1) (40, 41). The GDAR system is activated by acidic conditions of ~pH 5.5. When the environmental pH drops, GadB and presumably GadA associate with GadC and adopt an active conformation (40, 42, 43). GABA is an important neurotransmitter and has been suggested to be a critical interkingdom signaling molecule, playing roles in the gut-brain axis, in part through organisms carrying the GDAR system (44–46). Another protein critical for acid resistance is YdeP, which can support survival during exponential growth in the absence of exogenous amino acids through an unknown mechanism (Fig. 1) (39, 47–49).
Transcriptional regulation of GDAR and ydeP is complex and involves several circuits such as EvgA-YdeO-GadE that activate the expression of gadA, gadBC, and ydeP during the exponential phase (Fig. 1) (39, 50). The EvgA-YdeO-GadE circuit responds to mildly acidic pH when the sensor kinase EvgS phosphorylates the response regulator EvgA (Fig. 1) (51, 52). EvgA in turn activates the transcription of ydeP and overexpression of evgA leads to increased expression of gadA and gadB (53). EvgA also activates the transcription of ydeO and gadE. YdeO is a transcription factor that activates gadE transcription, represses its expression, and indirectly represses ydeP expression (39, 50, 54). GadE is a transcription factor that is critical for the expression of the GDAR system and for activating its expression. GadE is under complex transcriptional and posttranscriptional regulation involving over a dozen regulators (55–57).
The goal of our study was to investigate CsrA-dependent regulation of the acid stress response in E. coli. We demonstrate that CsrA represses acid stress circuitry at multiple levels. Disruption of CsrA causes overexpression of this circuitry, conferring a survival advantage under extremely acidic conditions but a growth defect under mildly acidic conditions. Our findings highlight a new and important role played by CsrA in managing the trade-off between bacterial growth and stress survival.
RESULTS
Disruption of csrA causes a pH-dependent growth defect
Recent studies identified several likely mRNA targets of CsrA-dependent regulation involved in growth and survival in acidic conditions including evgA and gadA (Fig. 1; Table S1) (3, 9, 28–30). To assess the physiological implications of CsrA-dependent regulation of acid stress systems, wild-type (WT) E. coli strain MG1655 and its isogenic csrA::kan mutant were grown under different pH conditions ranging from 7.5 to 5 (Fig. 2A; Table S1). At pH 6 or below, the csrA mutant displayed a strong growth defect that intensified as the pH decreased further. While the growth rate of the WT strain also decreased under acidic conditions, the effect was substantially weaker. Complementing the csrA mutation with a plasmid-borne wild-type csrA gene restored WT growth at pH 5.5, confirming the role of CsrA in this phenotype (Fig. S2). These results suggest that CsrA represses genes detrimental for the growth under acidic conditions, activates genes beneficial for growth under acidic conditions, or both. Interestingly, pH 6 is the upper limit of activation for the EvgS sensor kinase and increased activity and expression of the EvgAS TCS are associated with a pH-dependent growth defect, suggesting that CsrA may negatively regulate EvgAS function or downstream regulatory targets of EvgAS (47, 49).
Fig 2.

The csrA mutant exhibits pH-dependent growth and survival phenotypes. pBR322 is a control plasmid and p2VR112 overexpresses csrA. Stippling indicates that the strain carries a plasmid. (A) Relative growth rates of E. coli MG1655 (WT) and its isogenic csrA::kan mutant are shown. Error bars represent the standard deviation (sd) from four independent experiments. (B) Cells grown exponentially under mildly acidic conditions and then challenged by simulated stomach acid (pH 1.6) for 10 min. (C) Cells grown exponentially under neutral conditions and then challenged by extremely acidic conditions without glutamate (0 mM glutamate) for 2 h. No survival of any strain was seen under these conditions. (D) Cells grown exponentially under neutral conditions and then challenged by extremely acidic conditions with 1.5 mM glutamate for 2 h. (E) Cells grown exponentially under mildly acidic conditions and then challenged by extremely acidic conditions without glutamate for 2 h. (F) Cells grown exponentially under mildly acidic conditions and then challenged by extremely acidic conditions with 1.5 mM glutamate for 2 h. Error bars represent standard deviation (sd) from at least three independent experiments. Statistical significance was determined using unpaired t-tests and is denoted as follows: *P < 0.05; **P < 0.01; ***P < 0.001.
CsrA regulates survival under extremely acidic conditions
When E. coli cells were grown in neutral media (pH 7) and then shocked by exposure to extremely acidic conditions (pH 2.5) in the absence of exogenous glutamate, little to no survival was observed for WT or csrA-mutant strains (Fig. 2C). However, when shocked under extremely acidic conditions in the presence of glutamate, mean survival of the csrA mutant was ~12 fold higher than WT, suggesting that the GDAR system is more highly expressed or more active in the csrA mutant (Fig. 2D; Table S1). Complementation of the csrA mutant with plasmid-borne csrA reversed this survival phenotype, confirming the role of CsrA in the acid stress response.
When cells were grown in mildly acidic media (pH 5.5), preadapting them before challenge with extreme acidity, the csrA mutant exhibited a much higher survival rate than WT in all conditions (Fig. 2B, E, and F). Without added glutamate, the csrA mutant survived ~100-fold better than WT, and with added glutamate ~5-fold better than WT (Fig. 2E and F). Complementation of the csrA mutation reversed these phenotypes (Fig. 2).
The effect of CsrA on survival also extended to a condition designed to simulate fasted human stomach contents after drinking a glass of water (Biorelevant Media). After a 10-min challenge, the csrA mutant survived ~55-fold better than WT. Complementation of csrA abolished this survival phenotype (Fig. 2B). These results suggest that CsrA likely impacts survival in stomach acidity during host colonization. Survival under extremely acidic conditions in the absence of external amino acids has been associated with evgA and ydeP overexpression (49), offering a possible explanation for this phenotype.
Acid stress phenotypes of the csrA mutant depend on the EvgA-YdeO-GadE circuitry
We used epistasis analysis to identify genes responsible for the pH-dependent physiology of the csrA mutant (Fig. 1; Table S1). Deletion of evgS, gadB, or gadC did not suppress the growth defect of the csrA mutant (Fig. 3A, G, and H). However, deletion of several other genes in the EvgA-YdeO-GadE circuit suppressed the pH-dependent growth defect of the csrA mutant grown under mildly acidic conditions, including evgA, ydeO, ydeP, gadE, and gadA (Fig. 3B through F; Fig. S3). Complementing these genes in mutant strains restored the growth defect of the csrA mutant. Previous studies indicated that overexpression of evgA and ydeP impaired the growth of an otherwise WT strain (49), similar to the growth defective phenotype observed in the csrA mutant. Our results suggest that the EvgA-YdeO-GadE circuit participates in the pH-dependent growth defect of the csrA mutant and imply that CsrA may regulate the EvgA-YdeO-GadE circuit. Deletion of evgS, gadB, or gadC did not suppress the growth defect of the csrA mutant (Fig. 3A, G, and H).
Fig 3.

Suppressors of the pH-dependent growth defect of the csrA mutant. (A through H) Growth rates (μ) of exponential phase E. coli strains in mildly acidic (pH 5.5) M9 media normalized to WT. Stippling indicates complemented strains. Gene products of possible suppressors are indicated in the heading of each panel. Error bars represent the standard deviation (sd) from six independent experiments. Statistical significance was determined using an unpaired t-test and is denoted as follows: *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. The absence of a comparison bar indicates a comparison to WT.
We next tested whether the deletion of genes in the EvgA-YdeO-GadE circuit affected the survival of WT and csrA mutant strains that were preadapted by growth under mildly acidic conditions (pH 5.5) and then subjected to extremely acidic conditions (pH 2.5). Deleting evgS, evgA, ydeO, ydeP, or gadA abolished the survival of WT and csrA mutant strains in the absence of exogenous glutamate (Fig. 4A and C). Deleting gadE reduced the survival of WT and the csrA mutant under extremely acidic conditions with or without added glutamate (Fig. 4C and D). Interestingly, deleting gadB did not affect survival with or without glutamate, whereas deleting gadA resulted in a survival defect when grown without glutamate but not in its presence (Fig. 4C and D).
Fig 4.

Suppression of the csrA mutant survival phenotype in preadapted cells. Exponentially growing cells (OD = 0.5) were preadapted at pH 5.5 and then challenged for 2 h at pH 2.5. (A) Knockouts of genes relevant to the EvgA-YdeO regulon were challenged under extremely acidic conditions without glutamate or (B) extremely acidic conditions with 1.5 mM glutamate. (C) Knockouts of genes relevant to the GDAR system were challenged under extremely acidic conditions without glutamate or (D) extremely acidic conditions with 1.5 mM glutamate. Error bars represent the standard deviation (sd) from six independent experiments. Statistical significance was determined using unpaired t-tests and is denoted as follows: *P < 0.05; **P < 0.01; ***P < 0.001. The absence of a comparison bar indicates a comparison to WT.
Deleting evgS in a csrA mutant abolished the survival phenotype but it did not alter the growth defect of this strain (Fig. 3A and 4A). This paradox may be due to the influence of two factors: in the absence of EvgS, EvgA can be phosphorylated by acetyl-phosphate and EvgS is thought to be necessary for dephosphorylation of EvgA (58–60). Perhaps, sufficient phosphorylated EvgA accumulates in an evgS knockout to cause impaired growth of the csrA mutant but not enough to increase survival of the csrA mutant.
Glutamate supplementation eliminated survival defects of the evgS, evgA, ydeO, ydeP, and gadA deletions in WT and csrA mutant backgrounds, consistent with similar findings from prior studies (Fig. 4B and D); however, gadA was not tested during exponential growth in those studies (39, 48, 49). Deleting either gadA or gadB when supplemented with glutamate had little to no effect on the survival of the WT or csrA mutant strains. However, deleting both gadA and gadB in the WT and csrA mutant backgrounds abolished survival in the presence or absence of glutamate (Fig. 4C and D), highlighting the redundant nature of these genes. Deleting gadE impaired the survival of the WT and csrA mutant strains when supplemented with glutamate (Fig. 4D), similar to previous observations (61).
CsrA binds to 5′ leaders of mRNAs in the EvgA-YdeO-GadE circuit
While previous CLIP-Seq and RNA pulldown studies suggested that CsrA binds to evgA, gadE, gadA, and gadB mRNAs, these in vivo studies did not determine if CsrA binds independently of other factors or assess the binding affinity and specificity (3, 28, 29). Hence, gel shift assays were utilized to explore direct CsrA binding to the 5′ leader of transcripts in the EvgA-YdeO-GadE circuit independently of other factors (Fig. 5A).
Fig 5.
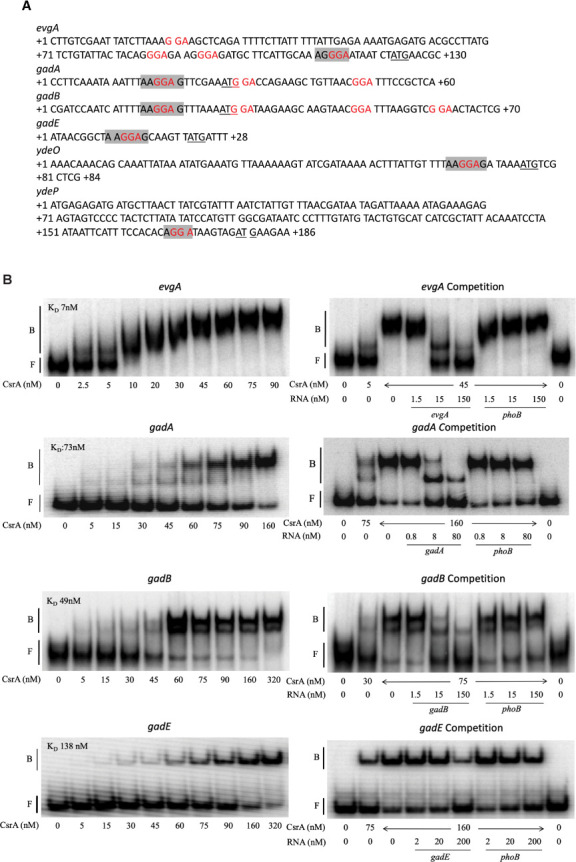
CsrA binding interactions with evgA, gadA, gadB, gadE, ydeO, and ydeP mRNA. (A) Sequences of probes used for gel shift assays. GGA motifs that may be components of CsrA binding sites are shown in red, start codons are underlined, and the Shine-Dalgarno (SD) sequence is shaded. (B) 5′-end-labeled transcripts were incubated with CsrA at the indicated concentrations. Competition reactions were performed in the presence of unlabeled specific (self) or unlabeled nonspecific (phoB) competitor RNAs at the concentrations shown. The positions of free (F) and bound (B) RNA are marked.
Previous studies demonstrated that CsrA-mediated regulation of complex genetic circuitry can occur via the top-tier regulator, as is the case for the regulation of motility via flhDC (4) or biofilm regulation via nhaR (62). EvgA appears to be the top-tier regulator of the acid stress response and is the first gene of the evgA-evgS operon. CsrA bound with high affinity to the 5′ leader of the evgA transcript (Fig. 5B), with an apparent KD of 7 nM. As the CsrA concentration was increased, two shifted species were observed, suggesting that more than one CsrA dimer can bind to the evgA 5′ leader transcript. This RNA contains 4 GGA motifs that might be components of CsrA-binding sites (Fig. 5A). Competition assays with unlabeled specific (evgA) and nonspecific (phoB) RNA established the specificity of this interaction.
CsrA also bound to the 5′ leaders of gadA, gadB, gadE, and ydeO with apparent KD values of 73 nM, 49 nM, 138 nM, and 84 nM, respectively (Fig. 5B). Two distinct shifts were observed for the gadA and gadB transcripts, suggesting that more than one CsrA dimer may bind to these transcripts. Given that the gadA RNA contains 3 GGAs and gadB contains 4 GGAs, this explanation is plausible. Competition assays indicated that CsrA binding to both gadA and gadB RNA was specific (Fig. 5B). On a native gel, the ydeO transcript adopted two forms (F1 and F2), and CsrA bound to both forms. Because this transcript produced a single band on a denaturing gel (data not shown), these forms represent alternative RNA conformations. The potential regulatory implications of these two RNA conformations are not known. CsrA did not bind to the 5′ leader of ydeP, suggesting that ydeP is not a direct target of CsrA-dependent regulation.
Together, these findings reveal that CsrA binds directly to the transcripts of the EvgA-YdeO-GadE circuit in the absence of other factors. Interestingly, CsrA bound most tightly to mRNA of the top-tier regulator EvgA and with weaker affinity to mRNAs of downstream genes within the circuit.
CsrA regulates evgA translation via translational coupling to a small leader peptide
Having identified the regulatory circuitry of interest for CsrA effects on acid stress resistance and the genes that CsrA likely regulates directly, we next assessed the effects of CsrA on gene expression using lacZ reporter fusions. First, evgA expression was examined under neutral and acidic conditions throughout the growth cycle in WT and csrA mutant backgrounds. Expression of an evgA’-‘lacZ translational fusion was higher in the csrA mutant under neutral and acidic conditions (Fig. 6A and B). In addition, a PlacUV5-evgA′-′lacZ leader fusion was tested in which the evgA promoter was replaced with the lacUV5 promoter. This promoter is unaffected by the csrA::kan mutation either directly or indirectly and was used to assess posttranscriptional regulation. The leader fusion was expressed at a higher level in the csrA mutant (Fig. 6C and D). Both the translational and leader fusions for evgA showed a similar level of higher expression (~2 fold) in the csrA mutant, suggesting that the difference of expression of evgA’-‘lacZ in the csrA mutant is likely a result of disrupted posttranscriptional regulation rather than transcriptional regulation. To determine whether CsrA directly represses evgA translation, a PT7-evgA’-’lacZ translational fusion expressed from a T7 promoter was used in the PURExpress system. The decrease in expression caused by increasing concentrations of CsrA indicated that CsrA directly represses evgA translation (Fig. 7B).
Fig 6.

Effects of CsrA on evgA expression in vivo. Translation fusion expression of evgA (A) at pH 7 and (B) at pH 5.5. Leader fusion expression of evgA (C) at pH 7 and (D) at pH 5.5. Translational fusions of evgL (E) at pH 7 and (F) at pH 5.5. (G) Translational fusion expression of evgA with the wild type evgL start codon mutated to a stop codon (A89T:T90A). Growth curves (OD600) are shown in the panel insets. Error bars represent the standard deviation (sd) from three independent experiments.
Fig 7.

CsrA binding represses translation of evgA in vitro. (A) evgA leader sequence. GGA motifs are shown in red, the start codon of evgL in teal, the start codon of evgA in blue, and toeprint positions from AMV are in yellow and SSIII is in brown. The evgL and evgA SD sequences and the evgL stop codon are underlined. (B) Effects of CsrA on in vitro translation of evgA'-'lacZ and pnp'-'lacZ (control) translational fusions driven by a T7 RNAP promoter. Relative β-galactosidase activity depicts the mean and standard deviation of activity relative to reaction mixtures without CsrA. (C) CsrA-dependent toeprints on evgA RNA using AMV reverse transcriptase. Nucleotides in red indicate a GGA motif, blue indicates the evgA start codon, and teal the evgL start codon. Arrows (+) indicate bands upon the addition of CsrA and (–) indicate the loss of a band upon the addition of CsrA. (D) Toeprinting of the evgA transcript using SSIII reverse transcriptase. Color coding and arrows are the same as in (C). (E) CsrA-evgA RNA footprint. 5′-end-labeled evgA RNA was treated with RNase T1 ±CsrA, as shown. RNA exposed to partial alkaline hydrolysis (OH), RNase T1 digestion of denatured RNA (T1), and untreated control RNA (C) are also shown. Red vertical lines correspond to CsrA binding sites that contain the GGA motifs.
Recent studies identified a leader peptide, EvgL, in the evgA leader mRNA such that the evgL stop codon overlaps with the start codon of evgA, suggestive of translational coupling (9, 63). We investigated the possibility that evgA translation is coupled to that of evgL by determining if evgL is translated and then by examining the impact of evgL translation on evgA translation. Hence, we constructed an evgL’-‘lacZ translational fusion and found that expression was regulated by CsrA under both neutral and acidic conditions in a pattern similar to the expression of evgA (Fig. 6E and F). To assess the effects of evgL translation on evgA translation, we changed the start codon of evgL to a stop codon in the context of an evgA’-‘lacZ translational fusion. When evgL translation was disrupted by the stop codon, evgA’-‘lacZ expression was eliminated, which is indicative of translational coupling (Fig. 6G). Similar CsrA-mediated regulation via translational coupling of iraD to a short leader peptide was observed previously (64).
RNA footprint and toeprint analyses of CsrA binding suggested that CsrA-mediated regulation of evgA expression involves a more complex mechanism. Footprinting with RNase T1 showed CsrA-dependent protection of GGA4, which overlaps the evgA SD sequence. Footprints were not observed for the other GGA motifs (Fig. 7E). However, toeprinting assays, which identify the 3′ boundary of a bound protein or stable RNA secondary structure (65), suggested that CsrA has four binding sites in the evgA 5′ leader. The use of Avian Myeloblastosis Virus reverse transcriptase (AMV) indicated that CsrA interacts with the two GGAs in the 5′ leader that overlap the evgL SD sequence (GGA2) and just upstream of its start codon (GGA3), as well as the GGA overlapping the evgA SD sequence (GGA4), while toeprinting with SuperScript III reverse transcriptase (SSIII) suggested that CsrA interacts with GGA1 in the 5′ leader (Fig. 7C and D). These results imply that CsrA regulates the translation of evgA directly and via translational coupling with evgL.
CsrA regulates the expression of the EvgA-GadE-YdeO circuit at multiple levels
We next assessed the effects of CsrA on the expression of other acid stress resistance genes using lacZ translational fusions for mid-level regulators, ydeO and gadE, and for genes under the control of EvgA (ydeP) or GadE (gadA and gadB). The ydeO’-‘lacZ fusion was expressed at a higher level in the csrA mutant strain under neutral (pH 7) and mildly acidic conditions (pH 5.5) (Fig. 8A and B). Expression of the gadE’-‘lacZ translational fusion was similar in WT and csrA mutant strains under neutral conditions during exponential growth, but the expression in the csrA mutant was higher in the stationary phase (Fig. 8C). By contrast, under mildly acidic conditions, gadE’-‘lacZ expression was higher in the csrA mutant strain throughout growth (Fig. 8D).
Fig 8.

Effects of the csrA mutation on expression of ydeO, gadE, gadA, gadB, and ydeP under neutral and mildly acidic conditions. Expression of lacZ translational fusions in WT and csrA mutant strains. (A) ydeO at pH 7, (B) ydeO at pH 5.5, (C) gadE at pH 7, (D) gadE at pH 5.5, (E) gadA at pH 7, (F) gadA at pH 5.5, (G) gadB at pH 7, (H) gadB at pH 5.5, (I) ydeP at pH 7, and (J) ydeP at pH 5.5. Expressions in the WT and csrA mutant strains are shown in black and red, respectively. Growth is shown in panel insets. Error bars represent the standard deviation (sd) from three independent experiments.
Expression of both gadA’-‘lacZ and gadB’-‘lacZ translational fusions exhibited the effects of CsrA (Fig. 8E through H), the details of which helped to explain their differential effects on physiology, where GadA played an important role in the growth defect of the csrA mutant but GadB did not (Fig. 3). Under mildly acidic conditions, both fusions were expressed at higher levels in the csrA mutant during the exponential phase of growth. At maximal expression in the csrA mutant (~4 h), the gadA’-‘lacZ fusion was expressed at a level that was ~2.5-fold higher than the gadB’-‘lacZ fusion. In addition, complementing the gadA knockout with gadB on a plasmid reintroduced the growth defect of the csrA mutant, suggesting that the level of expression, and not the particular isozyme, is responsible for the effects on growth (Fig. S4). Under neutral conditions, gadB’-‘lacZ expression showed little to no effect of CsrA during the exponential growth but expression was higher in the csrA mutant in the stationary phase, while expression of the gadA’-‘lacZ fusion was comparable in the WT and csrA mutant strains (Fig. 8E and G).
Interestingly, the ydeP’-‘lacZ translational fusion was expressed at a higher level throughout growth in the csrA mutant strain under both neutral and mildly acidic conditions (Fig. 8I and J). Since CsrA did not bind to the ydeP 5′ leader (Fig. 5B), these results imply that CsrA-dependent regulation is mediated indirectly, likely via direct effects of CsrA on evgA expression.
The translational fusion assays showed that the disruption of CsrA-dependent regulation results in increased expression of genes in the EvgA-YdeO-GadE circuit under acidic conditions, highlighting the importance of CsrA-dependent regulation of the acid stress response. As described above, we observed increased expression of evgA in the csrA mutant (Fig. 6A and B). While evgA did not respond to acidic conditions in the WT background, this was not the case in the csrA mutant in which expression increased earlier during growth at ~3 h (Fig. 6A and B). This lack of an evgA response to mildly acidic conditions was observed in previous studies using transcriptional fusions, despite the fact that EvgA is reported to be autoregulatory (50, 52, 66). To investigate whether CsrA regulated the circuitry downstream of GadA primarily via evgA, we examined the effects of knocking out evgA on the expression of a gadE'-'lacZ translational fusion. Deletion of evgA did not eliminate the effects of CsrA on gadE expression (Fig. S5). This result is consistent with the gel shift data indicating that CsrA binds directly to the gadE transcript (Fig. 5B) and confirms the multi-tier nature of CsrA-dependent regulation of this circuit. However, gadE is subject to several transcription activation and repression mechanisms and is itself positively autoregulatory, which may complicate the interpretation of CsrA effects on gadE expression in an evgA mutant (50, 67).
CsrA directly regulates gadA and gadB
We next examined the molecular mechanism of CsrA-dependent regulation of gadA and gadB expression. To determine whether CsrA repressed translation of gadA and gadB, we constructed plasmids with PT7-gadA’-’lacZ and PT7-gadB’-’lacZ translational fusions in which the 5′ leaders were fused to a T7 promoter and assessed their expression in the PURExpress system. In both cases, CsrA repressed gadA and gadB, indicative of direct translational repression (Fig. 9B).
Fig 9.
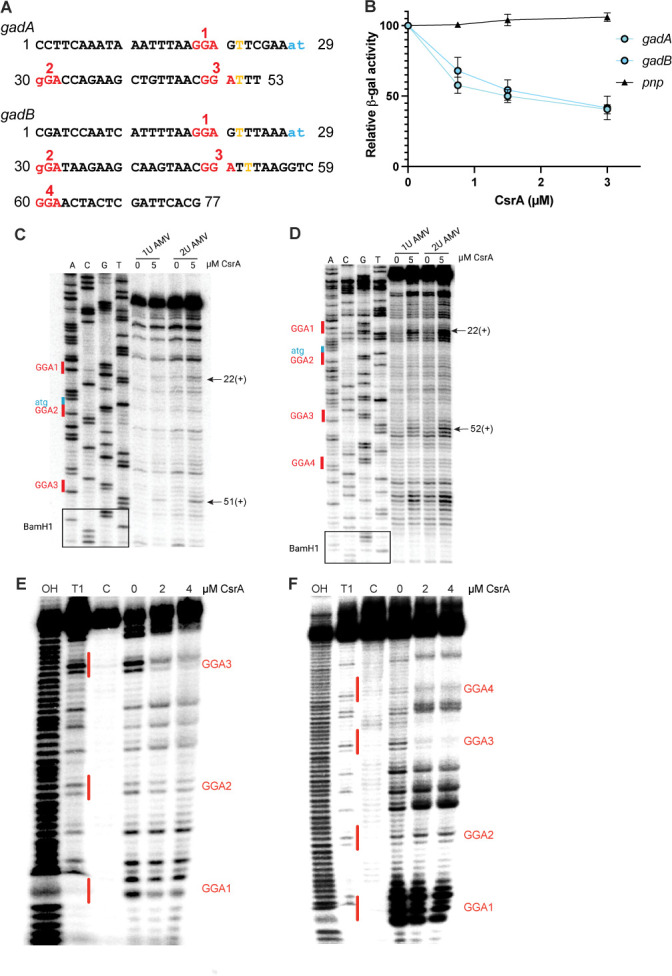
CsrA binding represses translation of gadA and gadB in vitro. (A) gadA and gadB leader sequences. The GGA motifs are shown in red and the start codons are in blue. Toeprint positions are indicated in yellow. (B) Effects of CsrA on in vitro translation of gadA'-'lacZ, gadB'-'lacZ and pnp'-'lacZ (control) translational fusions driven by a T7 RNAP promoter. Relative β-galactosidase activity depicts the mean and standard deviation relative to reaction mixtures lacking CsrA. (C) CsrA-gadA RNA toeprint using AMV reverse transcriptase. Toeprint positions are indicated on the right. (D) CsrA-gadB RNA toeprint using AMV reverse transcriptase. Toeprint positions are indicated on the right. (E) CsrA-gadA RNA footprint and (F) CsrA-gadB RNA footprint. 5′-end-labeled RNA was treated with RNase T1 ±CsrA as shown. Partial alkaline hydrolysis (OH) and RNase T1 digestion of denatured RNA (T1), and untreated control RNA are also shown.
We also performed RNA toeprinting and footprinting experiments to further investigate the regulatory mechanisms by which CsrA represses gadA and gadB translation. CsrA-dependent toeprints were observed just downstream of two GGA motifs in the gadA transcript. One GGA overlaps the gadA SD sequence (GGA1) and the second is in the early coding sequence (GGA3). A toeprint was not observed corresponding to GGA2 (Fig. 9A and C). In the case of gadB, CsrA-dependent toeprints were observed in regions that overlap the SD sequence (GGA1), and two in the early coding region (GGA3 and GGA4). A toeprint was not observed corresponding to GGA2 (Fig. 9A and D). To further examine the sites of CsrA binding, RNase T1 footprinting was performed. In both cases, this analysis suggested that the two most important CsrA interaction sites corresponded to GGA1, which overlaps the respective SD sequences, and GGA3, which is in the early coding sequences (Fig. 9E and F). These binding patterns are somewhat similar to CsrA-sdiA interaction, although CsrA binds exclusively within the coding region of sdiA (5).
Acid shock does not trigger the expression of csr genes
Previous studies on the Csr system demonstrated that it is often subject to feedback loops in which CsrA represses the expression of genes involved in activating transcription of CsrB and CsrC sRNAs (2, 5, 7). Furthermore, while acid stress does not appear to affect CsrA transcription (50), the protonated forms of formate or acetate stimulate transcription of csrB and csrC by binding to the sensor-kinase BarA of the BarA-UvrY TCS which phosphorylates the transcription factor UvrY, which, in turn, activates csrB/C transcription (22). We therefore explored the effects of a pH shift from 7 to 5.5 on the expression of a csrA’-‘lacZ translational fusion, as well as on the expression of csrB-lacZ and csrC-lacZ transcriptional fusions in exponentially growing cells. A ydeP’-‘lacZ translational fusion served as a positive control, as ydeP expression increases in response to a decrease in pH (50, 51). The ydeP’-‘lacZ fusion showed the expected response (Fig. S6A). Expression of the csrA’-‘lacZ fusion was unaffected by the decrease in pH, consistent with previous observations on its transcription (Fig. S6B) (50). Expression of the csrC-lacZ fusion was not affected by the decrease in pH, whereas the csrB-lacZ showed a small increase in expression, starting after 25 min of exposure to acidic conditions (Fig. S6C and D). These results reveal that the key genes of the Csr system do not respond or respond weakly to acid stress.
The csrA mutant alleviates GABA deficit in Caenorhabditis elegans
The csrA mutant overexpresses genes involved in the production of GABA, a neurotransmitter that regulates neuronal excitability in certain organisms including C. elegans. We investigated whether the csrA mutation would increase GABA production sufficiently to restore a GABA deficiency in C. elegans by assessing its effect on a GABA-dependent biological response. Nematodes harboring loss-of-function mutations in the unc-25 gene are deficient in endogenous GABA synthesis and exhibit pronounced convulsions upon exposure to pentylenetetrazole (PTZ), a competitive GABAA receptor antagonist that is used to chemically induce seizures to model epilepsy and other seizure disorders (68–71). Animals deficient in GABA lack the inhibitory signaling necessary to prevent the excitatory effects of PTZ on cholinergic signaling, thereby leading to PTZ-induced convulsions (71). Prior research demonstrated that exogenous GABA supplementation effectively restores normal phenotypes in unc-25 mutants (72), and dietary utilization of a GABA-producing E. coli strain has neuroprotective effects in nematodes (73).
After 15 min of PTZ exposure, the number of convulsing unc-25 nematodes fed the WT E. coli strains OP50 or MG1655 was approximately fivefold higher than nematodes fed the csrA mutant derivative of MG1655 or those exogenously supplemented with GABA (Fig. 10). We used epistasis analysis to identify the genes responsible for the restorative effects of the csrA mutant on the GABA-deficient phenotype and discovered that deletion of either of the GABA producing glutamate decarboxylases, gadA or gadB, dampened the effects of the csrA mutant on convulsions (Fig. 10). Interestingly, the gene encoding the glutamate-GABA antiporter gadC appeared to have limited involvement in the restorative phenotype afforded by the csrA mutant, perhaps due to the activity of additional GABA transporters, such as the GABA permease YhiM (74). Together, these findings underscore the potential therapeutic relevance of csrA and bacterial GABA production in host disorders where GABA plays a critical biological role.
Fig 10.
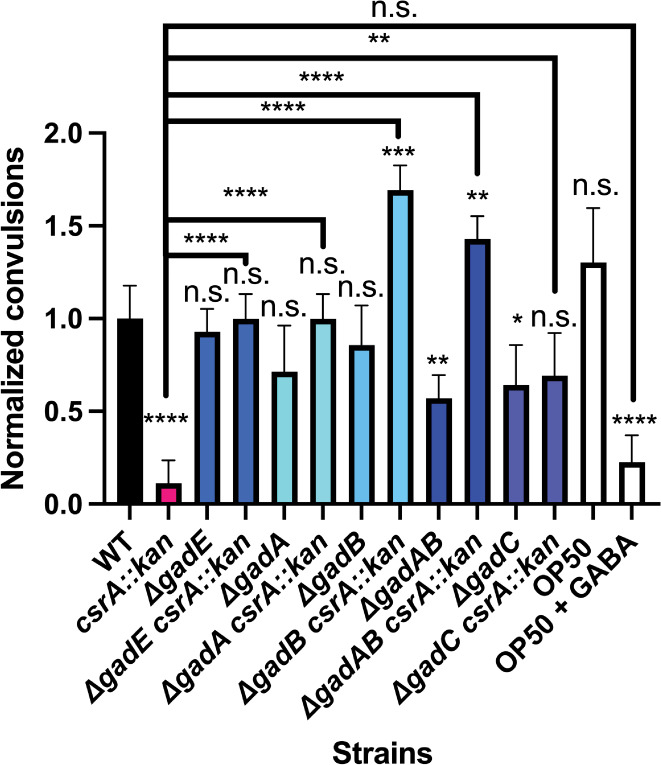
The effect of the csrA mutation on GABA-deficient C. elegans. Data are represented as the average percent of worms convulsing normalized to that in worms fed WT E. coli (MG1655) after 15 min of exposure to PTZ. Each data point is the average of a minimum of three independent experiments for a minimum of 60 worms. Error bars represent standard deviation (sd). Statistical significance was determined using unpaired t-tests and is denoted as follows: *P < 0.05; **P < 0.01; ***P < 0.001 ****P < 0.0001. The absence of a comparison bar indicates a comparison to WT.
DISCUSSION
Despite being a neutrophile, E. coli is capable of surviving extremely acidic conditions for extended periods of time through its expression of several acid resistance systems (31, 38). These systems are critical for survival in extremely acidic conditions but are detrimental to growth under mildly acidic conditions, presumably due to the metabolic drain that they exert (39, 48, 50, 75, 76). As a result, the acid stress systems are subject to complex and fine-tuned transcriptional regulatory circuits that are still not fully understood despite having been extensively studied (50, 55, 61, 77). In comparison, posttranscriptional regulation of the acid stress response has not been extensively studied, despite evidence suggesting that posttranscriptional regulation plays an important role in modulating the expression of many genes, which is the basis of CsrA-mediated regulation (3, 28, 36, 57, 78, 79).
We demonstrate that CsrA represses the EvgA-YdeO-GadE circuit of E. coli at multiple levels, from the top-tier regulator evgA to the structural genes gadA and gadB. Expression and activity of this circuitry are triggered by mildly acidic (pH 5–6) conditions, in anticipation of more extreme conditions (pH 1–3). CsrA is critical in preventing overexpression of the EvgA-YdeO-GadE circuit, and, in turn, preventing growth defects under mild acidity. CsrA deficiency results in severe growth defects under mildly acidic conditions and increased acid resistance in extremely acidic conditions, highlighting the critical role played by CsrA in balancing the trade-off between growth and survival.
Epistasis analysis showed that CsrA-dependent regulation of the EvgA-YdeO-GadE circuit was multitier in nature, and deleting evgA did not completely abolish the overexpression of gadE’-‘lacZ in a csrA mutant background (Fig. S5). Given that CsrA binds directly to gadE mRNA, the latter results were not surprising and demonstrate that CsrA represses gadE expression at more than one level, and not simply via effects on EvgA (Fig. 5B; Table S5).
High-affinity binding of CsrA to the evgA, gadE, ydeO, gadB, and gadA transcripts and in vivo lacZ reporter results also indicate that CsrA is involved in binding to and repressing multiple genes of the EvgA-YdeO-GadE circuit (Fig. 5, 7 and 9). The binding data are consistent with in vitro expression results, which demonstrated that CsrA directly repressed not only the top-tier regulator evgA but also the structural genes gadA and gadB. In the case of gadA and gadB, toeprinting showed that CsrA interacted with CsrA binding sites overlapping the SD sequence and the early coding sequences, which was supported by footprinting assays (Fig. 7 and 9). In the case of evgA, toeprinting and footprinting showed that CsrA interacted at multiple sites raising the possibility of a complex mechanism. This hypothesis is consistent with the results of lacZ reporter assays showing that translation of the leader peptide evgL is required for translation of evgA, implying that CsrA-dependent regulation of evgA involves translational coupling of the two genes, which is similar to CsrA-dependent regulation of iraD expression (64). However, unlike iraD where CsrA binds entirely upstream of the leader peptide coding sequence, CsrA binds both upstream and within the coding sequence of evgL. CsrA-dependent regulation of evgLA may require binding of a CsrA dimer to one site and then bridging to a second site in the segment preceding the evgL coding sequence as demonstrated previously (80). Doing so may enable another CsrA dimer to bind to a segment preceding evgL and to the site overlapping the evgA SD sequence, which is also within the coding region of evgL. This binding pattern would be consistent with gel shift results showing that CsrA binding to the 5′ leader of evgA results in at least two different shifts (Fig. 6). However, further analysis is required to fully elucidate the regulatory mechanism.
Interestingly, gadA but not gadB played an important role in the physiology of the csrA mutant under acidic conditions, affecting both growth and survival. These two isozymes were considered as interchangeable due to their high sequence similarity and similar phenotypes in the stationary phase (81). Complementing a gadA knockout with gadB restored the pH-dependent growth defect of the csrA mutant, suggesting that the combined level of GadA/B activity is more important for this growth phenotype than the particular isozyme (Fig. S4). This interpretation is consistent with the lacZ reporter results under acidic conditions in a csrA mutant background, where gadA’-‘lacZ was more highly expressed than gadB’-‘lacZ (Fig. 8). GadA also appears to play a role in survival under extremely acidic conditions without glutamate supplementation in exponentially growing cells. Previous work quantifying the amount of GadA and GadB produced in E. coli ATCC 11246, which is closely related to E. coli MG1655, showed that 80% of the isolated protein was GadA, explaining the disproportionate role of GadA in surviving extreme acidity (82, 83). While mutating gadA and gadB had differing effects on the physiology in a csrA mutant background, this pattern did not extend to the C. elegans studies. However, it is important to note that the latter studies were conducted on agar plates, where the cells were presumably in a stationary phase. Previous studies examining acid stress survival in the stationary phase noted no difference in the effects of gadA and gadB, suggesting that their distinct physiology may be limited to the exponential phase of growth (38, 84).
It is also interesting that reporter fusions for csrA, csrB, and csrC did not respond in a substantial way to acid stress induction, suggesting that expression of the Csr system itself is not strongly responsive to drops in pH. Despite this finding, we cannot definitively conclude that the Csr system is unresponsive to acid stress as the influence of CsrB/C RNA turnover was not examined in this study.
As CsrA is important for growth in E. coli and other species, it has become an appealing target for drug design (85–87). However, inhibiting CsrA causes increased biofilm formation and enhances the expression of virulence factors in some species, both of which are counterproductive for therapeutics (6, 88). The present study adds to the growing list of reasons why targeting CsrA for drug design may be counterproductive for antibiotic-based therapy of E. coli infections. Specifically, impairing CsrA-dependent regulation would greatly increase the expression of acid tolerance regulators of E. coli. This increase is relevant as acid tolerance has already been proposed to be involved in the low infectious dose of certain pathogenic E. coli (33). However, disrupting CsrA-dependent regulation of gadA/B may prove to be an appealing strategy for GABA production. GABA is a neurotransmitter that plays a key role in human mental health and microbiome research suggests that GABA-producing bacteria are inversely correlated with depression (44). Previous work explored engineering E. coli for batch production of GABA or even as a probiotic to deliver GABA directly; however, no previous studies have explored disrupting CsrA repression of gadA/B (89, 90). Our findings suggest that loss of repression of GDAR genes may be useful for increasing GABA levels in the host.
MATERIALS AND METHODS
Bacterial strains, plasmids, bacteriophage, culture conditions and oligonucleotides
All E. coli strains, plasmids, and bacteriophages used in this study are listed in Tables S2 to S4, respectively. Bacterial strains were grown and maintained in LB medium (0.5% yeast extract, 1% tryptone, and 1% NaCl, pH 7.4). Overnight cultures were inoculated in LB medium from frozen glycerol stocks of bacterial strains. These cultures were grown at 37°C or 30°C with shaking (250 rpm). Gene deletions were introduced by P1vir transduction from E. coli donor strains from the Keio library (91). To remove antibiotic resistance cassettes, pCP20 encoding the Flp recombinase was used (92). Plasmids to complement knockouts were sourced from the ASKA collection (93).
M9 supplemented medium (1 × M9 salts supplemented with 2 mM MgSO4, 0.1 mM CaCl2, 0.2% casamino acids, and 0.4% glucose) was used for assessing growth and gene expression. For media used in survival assays, M9 was only supplemented with 0.4% glucose, and the pH was adjusted to pH 2.5 using HCl. When studying GDAR activity, growth media were supplemented with 1.5 mM glutamate. The pH of the medium was adjusted to 2.5 using HCl. For simulated stomach acid (pH 1.6), Fasted State Simulated Gastric Fluid (FaSSGF) Biorelevant medium was used.
Where appropriate, media contained the following antibiotics: ampicillin (100 µg/mL), tetracycline (15 µg/mL), kanamycin (100 µg/mL), and chloramphenicol (25 µg/mL). Oligonucleotide primers used in this study were synthesized by Integrated DNA Technologies.
Deletion of csrA results in severe growth defects and genetic instability (94). To avoid this problem, experiments were performed with strains carrying a csrA truncation (csrA::kan) that retains ~10% of its RNA binding activity (95). The csrA::kan allele was moved into various strains via P1 transduction (1).
Construction of lacZ reporter fusions
Translational fusions to ‘lacZ were constructed in plasmid vector pLFT (29). Posttranscriptional fusions to ‘lacZ were constructed in plasmid vector placUV5 (29). All fusions were integrated into the chromosome using the CRIM system (96–98). Translational fusions were constructed as follows. About 500 nt of DNA upstream of the promoter region, the promoter region, and one or more codons downstream of the translation start site were amplified by PCR using the relevant primers (Table S3). Since gadE transcriptional regulation involves an extremely large upstream region, the translational fusion started ~750 nt upstream of the first promoter (67). The PCR products and pLFT were digested with PstI and BamHI and then ligated together. The products were transformed into DH5α λpir cells. The plasmids were isolated and fusion sequences were sequenced and verified before being integrated into the λatt site of strain MG1655 ΔlacZ using the helper plasmid pFINT as previously described (29). Single integrates were confirmed via PCR (96). Refer to Table S3 for the primer sequences.
The evgA leader fusion was constructed by PCR amplifying the 5′ leader of evgA (−114 to +7 relative to the first nucleotide of the start codon as +1) with primers that introduced restriction sites flanking the leader sequence. The resulting PCR product and pLacUV5 were digested with EcoRI and BamHI and then ligated together. To avoid the accumulation of spontaneous mutations that occurred when pLacUV5 plasmids were maintained in DH5α λpir cells, the ligated plasmids were integrated directly into the λatt site of MG1655 ΔlacZ using the Intλ expressing helper plasmid pFINT (29). The resulting single integrates were confirmed by PCR and sequencing.
β-galactosidase assay
Bacterial cultures containing lacZ fusions were grown in LB at 37°C to exponential phase (OD600 = 0.5), diluted to an optical density at 600 nm (OD600) of 0.02 in fresh M9 media supplemented with 0.4% glucose and 0.2% casamino acids. Cells were harvested at appropriate time points throughout growth during experiments. Acid induction experiments were based on a previously published protocol where a predetermined volume (1.6 mL) of 0.75 M HCl or water was added to actively growing cultures, HCl was added to decrease the pH from 7 to 5.5 and water was used as a negative control (50). β-Galactosidase activity was determined as described previously (29). Total cellular protein was measured following precipitation with 10% trichloroacetic acid, using the bicinchoninic acid (BCA) assay (Pierce Biotechnology) with bovine serum albumin as the protein standard.
Growth curves and kinetics
Bacterial cultures were grown in LB at 37°C overnight and diluted to an OD600 of 0.01 in fresh M9 media. OD600 was measured every h for 8 h and at 24 h. The growth rate constant (μ) was calculated from the exponential phase of growth: μ = 2.303(logOD2 − logOD1)/(t2 – t1).
Survival assays
Bacterial cultures were grown in LB at 37°C overnight and diluted to OD600 of 0.01 in fresh M9 media. Cells were grown to an OD600 of 0.5, pelleted by centrifugation, and then washed with a M9 medium containing 0.4% glucose. The wash medium was the same pH as the growth media and contained no added amino acids. Cells were then diluted 1:50 into pH 2.5 M9 medium containing 0.4% glucose with or without 1.5 mM glutamate and incubated for 2 h without shaking at 37°C. Aliquots were serially diluted in triplicate and were plated onto LB agar plates. Colonies were counted after being grown overnight at 37°C. Percent survival was calculated as follows: .
Gel shift assays
Binding of CsrA to the 5′ leader of gadA (60 nt; −27 to +33 relative to the first nucleotide of the start codon as +1), gadB (73 nt; −27 to +43 relative to the start codon), gadE (28 nt; −21 to +7 relative to the start codon), evgA (132 nt; −124 to +8 relative to the start codon), ydeO (84 nt; −74 to +10 relative to the start codon), and ydeP (188 nt; −178 to +10 relative to the start codon) was monitored using a gel shift assay. The transcripts were synthesized in vitro using the MEGAshortscript Kit (Invitrogen). Templates for in vitro transcription reactions were generated by PCR and subjected to PCR cleanup using Monarch PCR & DNA Clean up Kit (New England BioLabs, NEB) before using for in vitro transcription reactions. In vitro synthesized transcripts were gel purified on denaturing urea polyacrylamide gels, eluted overnight, extracted with phenol-chloroform, ethanol precipitated, resuspended in TE buffer, and quantified with a spectrophotometer. Twenty pmol of the RNA was dephosphorylated with Antarctic phosphatase (NEB) and 5′-end-labeled with γ-32P ATP and T4 polynucleotide kinase (NEB), gel purified, eluted overnight, phenol-chloroform extracted, ethanol precipitated, and resuspended in TE. The concentration of labeled RNA was determined using a standard curve constructed with γ-32P ATP. Binding reactions contained 0.08–0.2 nM end-labeled RNA, 10 mM MgCl2, 100 mM KCl, 32.5 ng SUPER-ase In (Ambion) with various concentrations of CsrA-H6 as indicated in the figures and incubated at 37°C for 30 min (99). Reaction mixtures were separated on a native polyacrylamide gel and imaged using a phosphorimager. The radioactive signals of free and shifted/bound RNA-protein complexes were quantified with Quantity One software and used for determining the apparent equilibrium dissociation constants (KD).
Coupled transcription-translation assays
Coupled transcription-translation assays were conducted in vitro using the PURExpress kit (NEB) following a published protocol (12). Plasmid pGadA-T7 contains a T7 promoter to drive transcription of the gadA translational fusion (nt +1 to +54 relative to the transcriptional start site). Plasmid pGadB-T7 contains a T7 promoter to drive transcription of the gadB translational fusion (nt +1 to +78 relative to the transcriptional start site). Plasmid pEvgA-T7 contains a T7 promoter to drive transcription of the evgA translational fusion (nt +1 to +122 relative to the transcriptional start site). The transcription start site of the σS promoter was chosen over the sigma σ70 promoter due to previous results suggesting it may be more active (66). A similar PT7-pnp'-'lacZ translational fusion plasmid was used as a negative control (65). These plasmids were used as templates for coupled transcription-translation reactions according to the manufacturer’s instructions. Each 6.7 µL reaction contained 250 ng of plasmid DNA and various concentrations of purified CsrA-His6 with 1 U of RNase inhibitor (Promega) and 2.5 mM dithiothreitol (DTT), 2.7 µL of solution A and 2 µL of solution B. The mixtures were incubated for 2 h at 37°C and β-galactosidase activity was determined. OD420 data without CsrA were normalized to 100%.
Toeprint assays
CsrA-RNA toeprint assays followed a previously published procedure (19). gadA RNA (from nt −27 to +26 relative to the gadA translational start), gadB RNA (nt −27 to +50 relative to the gadB translational start), and evgA RNA (nt −114 to +8 relative to the evgA translational start) were synthesized with the RNAMaxx kit (Agilent technologies) using PCR-generated DNA templates. Each gel-purified RNA (150 nM) in TE buffer was hybridized to a 5′ end-labeled DNA oligonucleotide complementary to the 3′ end of the vector-derived 3′ extension by heating for 3 min at 80°C followed by slow cooling for 10 min at room temperature. Toeprint reaction mixtures (10 µL) contained 2 µL of the hybridization mixture (30 nM final concentration), 1 µM CsrA-H6, 375 µM each dNTP, 10 mM DTT and Superscript III (SSIII), or AMV reverse transcriptase buffer. Mixtures were incubated for 30 min at 37°C to allow CsrA-RNA complex formation. After the addition of 0.125–0.25 U SSIII (Invitrogen) or 0.5–2 U AMV reverse transcriptase (Sigma Aldrich) incubation was continued for 15 min at 37°C. Reactions were terminated by the addition of 10 µL of gel loading buffer (95% formamide, 0.025% SDS, 20 mM EDTA, 0.025% bromophenol blue, and 0.025% xylene cyanol). Samples were heated to 90°C for 5 min and fractionated through standard 6% polyacrylamide-8 M urea sequencing gels. Toeprint patterns were visualized with a phosphorimager.
Footprint assays
CsrA footprints were performed according to the published procedure (95). The gadA, gadB, and evgA RNAs described for the toeprint assay were used for footprinting. Gel-purified RNA was dephosphorylated and then 5′ end-labeled using T4 polynucleotide kinase (NEB) and [γ−32P]ATP (7,000 Ci/mmol). Labeled RNAs were renatured by heating for 1 min at 90°C followed by slow cooling for 10 min at room temperature. Binding reaction mixtures (10 µL) contained 2 nM labeled RNA, 10 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 100 mM KCl, 40 ng of yeast RNA, 7.5% glycerol, 0.1 mg/mL xylene cyanol, and various concentrations of purified CsrA-H6. After a 30-min incubation at 37°C to allow for CsrA-RNA complex formation, RNase T1 (0.016 U) was added, and the incubation was continued for 15 min at 37°C. The reactions were stopped by adding 10 µL of gel loading buffer. Samples were heated for 5 min at 90°C and fractionated through standard 6% polyacrylamide-8 M urea sequencing gels. Cleaved patterns were examined using a phosphorimager.
C. elegans studies
Nematodes were maintained as previously described (100, 101). The C. elegans strains used in this study, CB156 (unc-25) and N2 (Bristol), were obtained from the Caenorhabditis Genetics Center. Bacterial strains used in all C. elegans experiments were grown overnight in Lennox Broth (LB) at 37°C with shaking, seeded onto nematode growth media (NGM), and dried in a biosafety cabinet for 2–4 h. Worms were age-synchronized using the standard sodium hypochlorite method. Age-synchronized worms were plated onto NGM seeded with appropriate bacteria and were kept at 23°C for 2 days. Positive control plates were bathed in 30 mM GABA solution (102) for 3 h prior to starting the experiments. Worms were picked and placed on NGM plates supplemented with PTZ (7 mg/mL) for 15 min (68). Following the incubation, the number of worms displaying a convulsing (head bobbing) phenotype was counted. As a control, PTZ-treated N2 worms did not display any convulsions, consistent with previous reports (68).
Datamining methods
Hypothetical targets of CsrA-dependent regulation were selected from previously published studies. Evidence for CsrA affecting transcript levels was determined by examining RNA-seq data (3, 24, 28). Genes were considered hypothetical targets if the log2 fold change was >1 or < −1 and was statistically significant. Potential RNA targets derived from pulldown results (29), in silico prediction (30), CLIP-seq (3), and HITS-CLIP-seq (28) were obtained from previously published studies. The data were compiled and are represented in Table S1; Fig. 1.
ACKNOWLEDGMENTS
We are grateful to Drs. Ellington, Mosby-Haundrup, Trujillo-Rodriguez, and Schuster for providing support and supplies during the COVID-19 pandemic. The C. elegans strains were provided by the CGC, which is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440).
This project was funded by National Institutes of Health grants GM059969 to T.R. and P.B. and GM098399 to P.B.
The project was conceived by M.G.G., P.B., and T.R. The experiments were carried out and the data analyzed by M.G.G., A.P., C.P, H.Y., and A.C.W. under the supervisions of D.C., P.B., and T.R. The manuscript was drafted by M.G.G. and edited and revised by all authors.
Contributor Information
Paul Babitzke, Email: pxb28@psu.edu.
Michael Y. Galperin, NCBI, NLM, National Institutes of Health, Bethesda, Maryland, USA
DATA AVAILABILITY
The authors confirm all supporting data, code, and protocols have been provided within the article or through supplementary data files.
SUPPLEMENTAL MATERIAL
The following material is available online at https://doi.org/10.1128/jb.00354-23.
Tables S1 to S4 and Figures S1 to S6.
ASM does not own the copyrights to Supplemental Material that may be linked to, or accessed through, an article. The authors have granted ASM a non-exclusive, world-wide license to publish the Supplemental Material files. Please contact the corresponding author directly for reuse.
REFERENCES
- 1. Romeo T, Gong M, Liu MY, Brun-Zinkernagel AM. 1993. Identification and molecular characterization of csrA, a pleiotropic gene from Escherichia coli that affects glycogen biosynthesis, gluconeogenesis, cell size, and surface properties. J Bacteriol 175:4744–4755. doi: 10.1128/jb.175.15.4744-4755.1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pourciau C, Pannuri A, Potts A, Yakhnin H, Babitzke P, Romeo T. 2019. Regulation of iron storage by CsrA supports exponential growth of Escherichia coli. mBio 10:e01034-19. doi: 10.1128/mBio.01034-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Potts AH, Vakulskas CA, Pannuri A, Yakhnin H, Babitzke P, Romeo T. 2017. Global role of the bacterial post-transcriptional regulator CsrA revealed by integrated transcriptomics. Nat Commun 8:1596. doi: 10.1038/s41467-017-01613-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wei BL, Brun-Zinkernagel AM, Simecka JW, Prüss BM, Babitzke P, Romeo T. 2001. Positive regulation of motility and flhDC expression by the RNA-binding protein CsrA of Escherichia coli. Mol Microbiol 40:245–256. doi: 10.1046/j.1365-2958.2001.02380.x [DOI] [PubMed] [Google Scholar]
- 5. Yakhnin H, Baker CS, Berezin I, Evangelista MA, Rassin A, Romeo T, Babitzke P. 2011. CsrA represses translation of sdiA, which encodes the N-acylhomoserine-l-lactone receptor of Escherichia coli, by binding exclusively within the coding region of sdiA mRNA. J Bacteriol 193:6162–6170. doi: 10.1128/JB.05975-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Potts AH, Guo Y, Ahmer BMM, Romeo T. 2019. Role of CsrA in stress responses and metabolism important for Salmonella virulence revealed by integrated transcriptomics. PLOS ONE 14:e0211430. doi: 10.1371/journal.pone.0211430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pourciau C, Lai Y-J, Gorelik M, Babitzke P, Romeo T. 2020. Diverse mechanisms and circuitry for global regulation by the RNA-binding protein CsrA. Front Microbiol 11:601352. doi: 10.3389/fmicb.2020.601352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Romeo T, Vakulskas CA, Babitzke P. 2013. Post-transcriptional regulation on a global scale: form and function of Csr/Rsm systems: the Csr global regulatory system. Environ Microbiol 15:313–324. doi: 10.1111/j.1462-2920.2012.02794.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Leistra AN, Gelderman G, Sowa SW, Moon-Walker A, Salis HM, Contreras LM. 2018. A canonical biophysical model of the CsrA global regulator suggests flexible regulator-target interactions. Sci Rep 8:9892. doi: 10.1038/s41598-018-27474-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dubey AK, Baker CS, Romeo T, Babitzke P. 2005. RNA sequence and secondary structure participate in high-affinity CsrA–RNA interaction. RNA 11:1579–1587. doi: 10.1261/rna.2990205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang X, Dubey AK, Suzuki K, Baker CS, Babitzke P, Romeo T. 2005. CsrA post-transcriptionally represses pgaABCD, responsible for synthesis of a biofilm polysaccharide adhesin of Escherichia coli. Mol Microbiol 56:1648–1663. doi: 10.1111/j.1365-2958.2005.04648.x [DOI] [PubMed] [Google Scholar]
- 12. Park H, Yakhnin H, Connolly M, Romeo T, Babitzke P. 2015. CsrA participates in a PNPase autoregulatory mechanism by selectively repressing translation of pnp transcripts that have been previously processed by RNase III and PNPase. J Bacteriol 197:3751–3759. doi: 10.1128/JB.00721-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Renda A, Poly S, Lai Y-J, Pannuri A, Yakhnin H, Potts AH, Bevilacqua PC, Romeo T, Babitzke P. 2020. CsrA-mediated translational activation of ymdA expression in Escherichia coli. mBio 11:e00849-20. doi: 10.1128/mBio.00849-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Baker CS, Morozov I, Suzuki K, Romeo T, Babitzke P. 2002. CsrA regulates glycogen biosynthesis by preventing translation of glgC in Escherichia coli. Mol Microbiol 44:1599–1610. doi: 10.1046/j.1365-2958.2002.02982.x [DOI] [PubMed] [Google Scholar]
- 15. Dubey AK, Baker CS, Suzuki K, Jones AD, Pandit P, Romeo T, Babitzke P. 2003. CsrA regulates translation of the Escherichia coli carbon starvation gene, cstA, by blocking ribosome access to the cstA transcript. J Bacteriol 185:4450–4460. doi: 10.1128/JB.185.15.4450-4460.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Figueroa-Bossi N, Schwartz A, Guillemardet B, D’Heygère F, Bossi L, Boudvillain M. 2014. RNA remodeling by bacterial global regulator CsrA promotes Rho-dependent transcription termination. Genes Dev 28:1239–1251. doi: 10.1101/gad.240192.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Aoyama JJ, Raina M, Zhong A, Storz G. 2022. Dual-function Spot 42 RNA encodes a 15-amino acid protein that regulates the CRP transcription factor. Proc Natl Acad Sci U S A 119:e2119866119. doi: 10.1073/pnas.2119866119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Patterson-Fortin LM, Vakulskas CA, Yakhnin H, Babitzke P, Romeo T. 2013. Dual posttranscriptional regulation via a cofactor-responsive mRNA leader. J Mol Biol 425:3662–3677. doi: 10.1016/j.jmb.2012.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yakhnin H, Yakhnin AV, Baker CS, Sineva E, Berezin I, Romeo T, Babitzke P. 2011. Complex regulation of the global regulatory gene csrA: CsrA-mediated translational repression, transcription from five promoters by Eσ70 and EσS, and indirect transcriptional activation by CsrA. Mol Microbiol 81:689–704. doi: 10.1111/j.1365-2958.2011.07723.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Babitzke P, Romeo T. 2007. CsrB sRNA family: sequestration of RNA-binding regulatory proteins. Curr Opin Microbiol 10:156–163. doi: 10.1016/j.mib.2007.03.007 [DOI] [PubMed] [Google Scholar]
- 21. Liu MY, Gui G, Wei B, Preston JF, Oakford L, Yüksel U, Giedroc DP, Romeo T. 1997. The RNA molecule CsrB binds to the global regulatory protein CsrA and antagonizes its activity in Escherichia coli. J Biol Chem 272:17502–17510. doi: 10.1074/jbc.272.28.17502 [DOI] [PubMed] [Google Scholar]
- 22. Alvarez AF, Rodríguez C, González-Chávez R, Georgellis D. 2021. The E. coli two-component signal sensor BarA binds protonated acetate via a conserved hydrophobic binding pocket. J Biol Chem 297:101383. doi: 10.1016/j.jbc.2021.101383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Suzuki K, Wang X, Weilbacher T, Pernestig A-K, Melefors O, Georgellis D, Babitzke P, Romeo T. 2002. Regulatory circuitry of the CsrA/CsrB and BarA/UvrY systems of Escherichia coli. J Bacteriol 184:5130–5140. doi: 10.1128/JB.184.18.5130-5140.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Potts AH, Leng Y, Babitzke P, Romeo T. 2018. Examination of Csr regulatory circuitry using epistasis analysis with RNA-seq (Epi-seq) confirms that CsrD affects gene expression via CsrA, CsrB and CsrC. Sci Rep 8:5373. doi: 10.1038/s41598-018-23713-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Suzuki K, Babitzke P, Kushner SR, Romeo T. 2006. Identification of a novel regulatory protein (CsrD) that targets the global regulatory RNAs CsrB and CsrC for degradation by RNase E. Genes Dev 20:2605–2617. doi: 10.1101/gad.1461606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vakulskas CA, Leng Y, Abe H, Amaki T, Okayama A, Babitzke P, Suzuki K, Romeo T. 2016. Antagonistic control of the turnover pathway for the global regulatory sRNA CsrB by the CsrA and CsrD proteins. Nucleic Acids Res 44:7896–7910. doi: 10.1093/nar/gkw484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kharadi RR, Sundin GW. 2022. CsrD regulates amylovoran biosynthesis and virulence in Erwinia amylovora in a novel cyclic-di-GMP dependent manner. Mol Plant Pathol 23:1154–1169. doi: 10.1111/mpp.13217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sowa SW, Gelderman G, Leistra AN, Buvanendiran A, Lipp S, Pitaktong A, Vakulskas CA, Romeo T, Baldea M, Contreras LM. 2017. Integrative FourD omics approach profiles the target network of the carbon storage regulatory system. Nucleic Acids Res 45:1673–1686. doi: 10.1093/nar/gkx048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Edwards AN, Patterson-Fortin LM, Vakulskas CA, Mercante JW, Potrykus K, Vinella D, Camacho MI, Fields JA, Thompson SA, Georgellis D, Cashel M, Babitzke P, Romeo T. 2011. Circuitry linking the Csr and stringent response global regulatory systems. Mol Microbiol 80:1561–1580. doi: 10.1111/j.1365-2958.2011.07663.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kulkarni PR, Jia T, Kuehne SA, Kerkering TM, Morris ER, Searle MS, Heeb S, Rao J, Kulkarni RV. 2014. A sequence-based approach for prediction of CsrA/RsmA targets in bacteria with experimental validation in Pseudomonas aeruginosa. Nucleic Acids Res 42:6811–6825. doi: 10.1093/nar/gku309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Foster JW. 2004. Escherichia coli acid resistance: tales of an amateur acidophile. Nat Rev Microbiol 2:898–907. doi: 10.1038/nrmicro1021 [DOI] [PubMed] [Google Scholar]
- 32. Lund P, Tramonti A, De Biase D. 2014. Coping with low pH: molecular strategies in neutralophilic bacteria. FEMS Microbiol Rev 38:1091–1125. doi: 10.1111/1574-6976.12076 [DOI] [PubMed] [Google Scholar]
- 33. De Biase D, Lund PA. 2015. The Escherichia coli acid stress response and its significance for pathogenesis, p 49–88. In Sariaslani S, Gadd GM (ed), Advances in applied Microbiology. Academic Press. [DOI] [PubMed] [Google Scholar]
- 34. Teunis P, Takumi K, Shinagawa K. 2004. Dose response for infection by Escherichia coli O157:H7 from outbreak data. Risk Anal 24:401–407. doi: 10.1111/j.0272-4332.2004.00441.x [DOI] [PubMed] [Google Scholar]
- 35. Yu X-C, Yang C, Ding J, Niu X, Hu Y, Jin C. 2017. Characterizations of the interactions between Escherichia coli periplasmic chaperone HdeA and its native substrates during acid stress. Biochemistry 56:5748–5757. doi: 10.1021/acs.biochem.7b00724 [DOI] [PubMed] [Google Scholar]
- 36. Bianco CM, Fröhlich KS, Vanderpool CK. 2019. Bacterial cyclopropane fatty acid synthase mRNA is targeted by activating and repressing small RNAs. J Bacteriol 201:e00461-19. doi: 10.1128/JB.00461-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Diez-Gonzalez F, Karaibrahimoglu Y. 2004. Comparison of the glutamate-, arginine- and lysine-dependent acid resistance systems in Escherichia coli O157:H7. J Appl Microbiol 96:1237–1244. doi: 10.1111/j.1365-2672.2004.02251.x [DOI] [PubMed] [Google Scholar]
- 38. Castanie-Cornet M-P, Penfound TA, Smith D, Elliott JF, Foster JW. 1999. Control of acid resistance in Escherichia coli. J Bacteriol 181:3525–3535. doi: 10.1128/JB.181.11.3525-3535.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ma Z, Masuda N, Foster JW. 2004. Characterization of EvgAS-YdeO-GadE branched regulatory circuit governing glutamate-dependent acid resistance in Escherichia coli. J Bacteriol 186:7378–7389. doi: 10.1128/JB.186.21.7378-7389.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Capitani G, De Biase D, Aurizi C, Gut H, Bossa F, Grütter MG. 2003. Crystal structure and functional analysis of Escherichia coli glutamate decarboxylase. EMBO J 22:4027–4037. doi: 10.1093/emboj/cdg403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ma D, Lu P, Shi Y. 2013. Substrate selectivity of the acid-activated glutamate/γ-aminobutyric acid (GABA) antiporter GadC from Escherichia coli. J Biol Chem 288:15148–15153. doi: 10.1074/jbc.M113.474502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ma D, Lu P, Yan C, Fan C, Yin P, Wang J, Shi Y. 2012. Structure and mechanism of a glutamate–GABA antiporter. Nature 483:632–636. doi: 10.1038/nature10917 [DOI] [PubMed] [Google Scholar]
- 43. Gut H, Pennacchietti E, John RA, Bossa F, Capitani G, De Biase D, Grütter MG. 2006. Escherichia coli acid resistance: pH-sensing, activation by chloride and autoinhibition in GadB. EMBO J 25:2643–2651. doi: 10.1038/sj.emboj.7601107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Strandwitz P, Kim KH, Terekhova D, Liu JK, Sharma A, Levering J, McDonald D, Dietrich D, Ramadhar TR, Lekbua A, Mroue N, Liston C, Stewart EJ, Dubin MJ, Zengler K, Knight R, Gilbert JA, Clardy J, Lewis K. 2019. GABA modulating bacteria of the human gut microbiota. Nat Microbiol 4:396–403. doi: 10.1038/s41564-018-0307-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ren W, Yin J, Xiao H, Chen S, Liu G, Tan B, Li N, Peng Y, Li T, Zeng B, Li W, Wei H, Yin Z, Wu G, Hardwidge PR, Yin Y. 2016. Intestinal microbiota-derived GABA mediates interleukin-17 expression during enterotoxigenic Escherichia coli infection. Front Immunol 7:685. doi: 10.3389/fimmu.2016.00685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Quillin SJ, Tran P, Prindle A. 2021. Potential roles for gamma-aminobutyric acid signaling in bacterial communities. Bioelectricity 3:120–125. doi: 10.1089/bioe.2021.0012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Johnson MD, Bell J, Clarke K, Chandler R, Pathak P, Xia Y, Marshall RL, Weinstock GM, Loman NJ, Winn PJ, Lund PA. 2014. Characterization of mutations in the PAS domain of the EvgS sensor kinase selected by laboratory evolution for acid resistance in Escherichia coli. Mol Microbiol 93:911–927. doi: 10.1111/mmi.12704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Masuda N, Church GM. 2003. Regulatory network of acid resistance genes in Escherichia coli. Mol Microbiol 48:699–712. doi: 10.1046/j.1365-2958.2003.03477.x [DOI] [PubMed] [Google Scholar]
- 49. Masuda N, Church GM. 2002. Escherichia coli gene expression responsive to levels of the response regulator EvgA. J Bacteriol 184:6225–6234. doi: 10.1128/JB.184.22.6225-6234.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Burton NA, Johnson MD, Antczak P, Robinson A, Lund PA. 2010. Novel aspects of the acid response network of E. coli K-12 are revealed by a study of transcriptional dynamics. J Mol Biol 401:726–742. doi: 10.1016/j.jmb.2010.06.054 [DOI] [PubMed] [Google Scholar]
- 51. Eguchi Y, Utsumi R. 2014. Alkali metals in addition to acidic pH activate the EvgS histidine kinase sensor in Escherichia coli. J Bacteriol 196:3140–3149. doi: 10.1128/JB.01742-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Utsumi R, Kawamoto K, Yamazaki K, Taniguchi M, Yoshioka S, Tanabe H. 1996. Characterization of the signal transduction via EvgS And EvgA in Escherichia coli. J Gen Appl Microbiol 42:155–162. doi: 10.2323/jgam.42.155 [DOI] [Google Scholar]
- 53. Nishino K, Inazumi Y, Yamaguchi A. 2003. Global analysis of genes regulated by EvgA of the two-component regulatory system in Escherichia coli. J Bacteriol 185:2667–2672. doi: 10.1128/JB.185.8.2667-2672.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yamanaka Y, Oshima T, Ishihama A, Yamamoto K. 2014. Characterization of the YdeO regulon in Escherichia coli. PLOS ONE 9:e111962. doi: 10.1371/journal.pone.0111962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sayed AK, Odom C, Foster JW. 2007. The Escherichia coli AraC-family regulators GadX and GadW activate gadE, the central activator of glutamate-dependent acid resistance. Microbiology (Reading) 153:2584–2592. doi: 10.1099/mic.0.2007/007005-0 [DOI] [PubMed] [Google Scholar]
- 56. Heuveling J, Possling A, Hengge R. 2008. A role for Lon protease in the control of the acid resistance genes of Escherichia coli. Mol Microbiol 69:534–547. doi: 10.1111/j.1365-2958.2008.06306.x [DOI] [PubMed] [Google Scholar]
- 57. Aiso T, Kamiya S, Yonezawa H, Gamou S. 2014. Overexpression of an antisense RNA, ArrS, increases the acid resistance of Escherichia coli. Microbiology (Reading) 160:954–961. doi: 10.1099/mic.0.075994-0 [DOI] [PubMed] [Google Scholar]
- 58. Kinoshita-Kikuta E, Kinoshita E, Eguchi Y, Yanagihara S, Edahiro K, Inoue Y, Taniguchi M, Yoshida M, Yamamoto K, Takahashi H, Sawasaki T, Utsumi R, Koike T. 2015. Functional characterization of the receiver domain for phosphorelay control in hybrid sensor kinases. PLoS ONE 10:e0132598. doi: 10.1371/journal.pone.0132598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kinoshita-Kikuta E, Kinoshita E, Eguchi Y, Koike T. 2016. Validation of cis and trans modes in multistep phosphotransfer signaling of bacterial tripartite sensor kinases by using phos-tag SDS-PAGE. PLoS ONE 11:e0148294. doi: 10.1371/journal.pone.0148294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhu Y, Qin L, Yoshida T, Inouye M. 2000. Phosphatase activity of histidine kinase EnvZ without kinase catalytic domain. Proc Natl Acad Sci U S A 97:7808–7813. doi: 10.1073/pnas.97.14.7808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Seo SW, Kim D, O’Brien EJ, Szubin R, Palsson BO. 2015. Decoding genome-wide GadEWX-transcriptional regulatory networks reveals multifaceted cellular responses to acid stress in Escherichia coli. Nat Commun 6:7970. doi: 10.1038/ncomms8970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Pannuri A, Yakhnin H, Vakulskas CA, Edwards AN, Babitzke P, Romeo T. 2012. Translational repression of NhaR, a novel pathway for multi-tier regulation of biofilm circuitry by CsrA. J Bacteriol 194:79–89. doi: 10.1128/JB.06209-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Weaver J, Mohammad F, Buskirk AR, Storz G, Vogel J. 2019. Identifying small proteins by ribosome profiling with stalled initiation complexes. mBio 10:e02819-18. doi: 10.1128/mBio.02819-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Park H, McGibbon LC, Potts AH, Yakhnin H, Romeo T, Babitzke P. 2017. Translational repression of the RpoS antiadapter IraD by CsrA is mediated via translational coupling to a short upstream open reading frame. mBio 8:e01355-17. doi: 10.1128/mBio.01355-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yakhnin H, Babitzke P. 2022. Toeprint assays for detecting RNA structure and protein-RNA interactions. Methods Mol Biol 2516:305–316. doi: 10.1007/978-1-0716-2413-5_16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tanabe H, Yamasaki K, Katoh A, Yoshioka S, Utsumi R. 1998. Identification of the promoter region and the transcriptional regulatory sequence of the evgAS operon of Escherichia coli. Biosci Biotechnol Biochem 62:286–290. doi: 10.1271/bbb.62.286 [DOI] [PubMed] [Google Scholar]
- 67. Sayed AK, Foster JW. 2009. A 750 bp sensory integration region directs global control of the Escherichia coli GadE acid resistance regulator. Mol Microbiol 71:1435–1450. doi: 10.1111/j.1365-2958.2009.06614.x [DOI] [PubMed] [Google Scholar]
- 68. Wong SQ, Jones A, Dodd S, Grimes D, Barclay JW, Marson AG, Cunliffe VT, Burgoyne RD, Sills GJ, Morgan A. 2018. A Caenorhabditis elegans assay of seizure-like activity optimised for identifying antiepileptic drugs and their mechanisms of action. J Neurosci Methods 309:132–142. doi: 10.1016/j.jneumeth.2018.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Löscher W. 2011. Critical review of current animal models of seizures and epilepsy used in the discovery and development of new antiepileptic drugs. Seizure 20:359–368. doi: 10.1016/j.seizure.2011.01.003 [DOI] [PubMed] [Google Scholar]
- 70. Dhir A. 2012. Pentylenetetrazol (PTZ) kindling model of epilepsy. Curr Protoc Neurosci Chapter 9:Unit9.37. doi: 10.1002/0471142301.ns0937s58 [DOI] [PubMed] [Google Scholar]
- 71. Thapliyal S, Babu K. 2018. Pentylenetetrazole (PTZ)-induced convulsion assay to determine GABAergic defects in Caenorhabditis elegans. Bio Protoc 8:e2989. doi: 10.21769/BioProtoc.2989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Jin Y, Jorgensen E, Hartwieg E, Horvitz HR. 1999. The Caenorhabditis elegans gene unc-25 encodes glutamic acid decarboxylase and is required for synaptic transmission but not synaptic development. J Neurosci 19:539–548. doi: 10.1523/JNEUROSCI.19-02-00539.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Schuske K, Beg AA, Jorgensen EM. 2004. The GABA nervous system in C. elegans. Trends Neurosci 27:407–414. doi: 10.1016/j.tins.2004.05.005 [DOI] [PubMed] [Google Scholar]
- 74. Tramonti A, Santis F, Pennacchietti E, De Biase D. 2017. The yhiM gene codes for an inner membrane protein involved in GABA export in Escherichia coli. AIMS Microbiol 3:71–87. doi: 10.3934/microbiol.2017.1.71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Du B, Olson CA, Sastry AV, Fang X, Phaneuf PV, Chen K, Wu M, Szubin R, Xu S, Gao Y, Hefner Y, Feist AM, Palsson BO. 2020. Adaptive laboratory evolution of Escherichia coli under acid stress. Microbiology (Reading) 166:141–148. doi: 10.1099/mic.0.000867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Harden MM, He A, Creamer K, Clark MW, Hamdallah I, Martinez KA, Kresslein RL, Bush SP, Slonczewski JL. 2015. Acid-adapted strains of Escherichia coli K-12 obtained by experimental evolution. Appl Environ Microbiol 81:1932–1941. doi: 10.1128/AEM.03494-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Tramonti A, De Canio M, De Biase D. 2008. GadX/GadW-dependent regulation of the Escherichia coli acid fitness island: transcriptional control at the gadY–gadW divergent promoters and identification of four novel 42 bp GadX/GadW-specific binding sites. Mol Microbiol 70:965–982. doi: 10.1111/j.1365-2958.2008.06458.x [DOI] [PubMed] [Google Scholar]
- 78. Opdyke JA, Kang J-G, Storz G. 2004. GadY, a small-RNA regulator of acid response genes in Escherichia coli. J Bacteriol 186:6698–6705. doi: 10.1128/JB.186.20.6698-6705.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Fröhlich KS, Papenfort K, Fekete A, Vogel J. 2013. A small RNA activates CFA synthase by isoform-specific mRNA stabilization. EMBO J 32:2963–2979. doi: 10.1038/emboj.2013.222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Mercante J, Edwards AN, Dubey AK, Babitzke P, Romeo T. 2009. Molecular geometry of CsrA (RsmA) binding to RNA and its implications for regulated expression. J Mol Biol 392:511–528. doi: 10.1016/j.jmb.2009.07.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. De Biase D, Pennacchietti E. 2012. Glutamate decarboxylase-dependent acid resistance in orally acquired bacteria: function, distribution and biomedical implications of the gadBC operon. Mol Microbiol 86:770–786. doi: 10.1111/mmi.12020 [DOI] [PubMed] [Google Scholar]
- 82. De Biase D, Tramonti A, John RA, Bossa F. 1996. Isolation, overexpression, and biochemical characterization of the two isoforms of glutamic acid decarboxylase from Escherichia coli. Protein Expr Purif 8:430–438. doi: 10.1006/prep.1996.0121 [DOI] [PubMed] [Google Scholar]
- 83. Dunne KA, Chaudhuri RR, Rossiter AE, Beriotto I, Browning DF, Squire D, Cunningham AF, Cole JA, Loman N, Henderson IR. 2017. Sequencing a piece of history: complete genome sequence of the original Escherichia coli strain. Microb Genom 3:mgen000106. doi: 10.1099/mgen.0.000106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Aquino P, Honda B, Jaini S, Lyubetskaya A, Hosur K, Chiu JG, Ekladious I, Hu D, Jin L, Sayeg MK, et al. 2017. Coordinated regulation of acid resistance in Escherichia coli. BMC Syst Biol 11:1. doi: 10.1186/s12918-016-0376-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Jakob V, Zoller BGE, Rinkes J, Wu Y, Kiefer AF, Hust M, Polten S, White AM, Harvey PJ, Durek T, Craik DJ, Siebert A, Kazmaier U, Empting M. 2022. Phage display-based discovery of cyclic peptides against the broad spectrum bacterial anti-virulence target CsrA. Eur J Med Chem 231:114148. doi: 10.1016/j.ejmech.2022.114148 [DOI] [PubMed] [Google Scholar]
- 86. Maurer CK, Fruth M, Empting M, Avrutina O, Hoßmann J, Nadmid S, Gorges J, Herrmann J, Kazmaier U, Dersch P, Müller R, Hartmann RW. 2016. Discovery of the first small-molecule CsrA–RNA interaction inhibitors using biophysical screening technologies. Future Med Chem 8:931–947. doi: 10.4155/fmc-2016-0033 [DOI] [PubMed] [Google Scholar]
- 87. Ricci V, Attah V, Overton T, Grainger DC, Piddock LJV. 2017. CsrA maximizes expression of the AcrAB multidrug resistance transporter. Nucleic Acids Res 45:12798–12807. doi: 10.1093/nar/gkx929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Barnard FM, Loughlin MF, Fainberg HP, Messenger MP, Ussery DW, Williams P, Jenks PJ. 2004. Global regulation of virulence and the stress response by CsrA in the highly adapted human gastric pathogen Helicobacter pylori. Mol Microbiol 51:15–32. doi: 10.1046/j.1365-2958.2003.03788.x [DOI] [PubMed] [Google Scholar]
- 89. Lan Y-J, Tan S-I, Cheng S-Y, Ting W-W, Xue C, Lin T-H, Cai M-Z, Chen P-T, Ng I-S. 2021. Development of Escherichia coli Nissle 1917 derivative by CRISPR/Cas9 and application for gamma-aminobutyric acid (GABA) production in antibiotic-free system. Biochem Eng J 168:107952. doi: 10.1016/j.bej.2021.107952 [DOI] [Google Scholar]
- 90. Fan L-Q, Li M-W, Qiu Y-J, Chen Q-M, Jiang S-J, Shang Y-J, Zhao L-M. 2018. Increasing thermal stability of glutamate decarboxylase from Escherichia coli by site-directed saturation mutagenesis and its application in GABA production. J Biotechnol 278:1–9. doi: 10.1016/j.jbiotec.2018.04.009 [DOI] [PubMed] [Google Scholar]
- 91. Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006. doi: 10.1038/msb4100050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Cherepanov PP, Wackernagel W. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9–14. doi: 10.1016/0378-1119(95)00193-a [DOI] [PubMed] [Google Scholar]
- 93. Kitagawa M, Ara T, Arifuzzaman M, Ioka-Nakamichi T, Inamoto E, Toyonaga H, Mori H. 2005. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res 12:291–299. doi: 10.1093/dnares/dsi012 [DOI] [PubMed] [Google Scholar]
- 94. Timmermans J, Van Melderen L. 2009. Conditional essentiality of the csrA gene in Escherichia coli. J Bacteriol 191:1722–1724. doi: 10.1128/JB.01573-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Yakhnin AV, Baker CS, Vakulskas CA, Yakhnin H, Berezin I, Romeo T, Babitzke P. 2013. CsrA activates flhDC expression by protecting flhDC mRNA from RNase E-mediated cleavage. Mol Microbiol 87:851–866. doi: 10.1111/mmi.12136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Haldimann A, Wanner BL. 2001. Conditional-replication, integration, excision, and retrieval plasmid-host systems for gene structure-function studies of bacteria. J Bacteriol 183:6384–6393. doi: 10.1128/JB.183.21.6384-6393.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Pannuri A, Vakulskas CA, Zere T, McGibbon LC, Edwards AN, Georgellis D, Babitzke P, Romeo T. 2016. Circuitry linking the catabolite repression and Csr global regulatory systems of Escherichia coli. J Bacteriol 198:3000–3015. doi: 10.1128/JB.00454-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Bolivar F, Rodriguez RL, Greene PJ, Betlach MC, Heyneker HL, Boyer HW, Crosa JH, Falkow S. 1977. Construction and characterization of new cloning vehicles. II. a multipurpose cloning system. Gene 2:95–113. doi: 10.1016/0378-1119(77)90000-2 [DOI] [PubMed] [Google Scholar]
- 99. Lai Y-J, Yakhnin H, Pannuri A, Pourciau C, Babitzke P, Romeo T. 2022. CsrA regulation via binding to the base-pairing small RNA Spot 42. Mol Microbiol 117:32–53. doi: 10.1111/mmi.14769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Walker AC, Bhargava R, Vaziriyan-Sani AS, Pourciau C, Donahue ET, Dove AS, Gebhardt MJ, Ellward GL, Romeo T, Czyż DM. 2021. Colonization of the Caenorhabditis elegans gut with human enteric bacterial pathogens leads to proteostasis disruption that is rescued by butyrate. PLoS Pathog 17:e1009510. doi: 10.1371/journal.ppat.1009510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Brenner S. 1974. The genetics of Caenorhabditis elegans. Genetics 77:71–94. doi: 10.1093/genetics/77.1.71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. McIntire SL, Jorgensen E, Horvitz HR. 1993. Genes required for GABA function in Caenorhabditis elegans. Nature 364:334–337. doi: 10.1038/364334a0 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1 to S4 and Figures S1 to S6.
Data Availability Statement
The authors confirm all supporting data, code, and protocols have been provided within the article or through supplementary data files.