

Abstract
In breast tumors, somatic mutation frequencies in TP53 and PIK3CA vary by tumor subtype and ancestry. Emerging data suggest tumor mutation status is associated with germline variants and genetic ancestry. We aimed to identify germline variants that are associated with somatic TP53 or PIK3CA mutation status in breast tumors. A genome-wide association study was conducted in 2,850 women of European ancestry with breast cancer using TP53 and PIK3CA mutation status (positive or negative) as well as specific functional categories [e.g., TP53 gain-of-function (GOF) and loss-of-function, PIK3CA activating] as phenotypes. Germline variants showing evidence of association were selected for validation analyses and tested in multiple independent datasets. Discovery association analyses found five variants associated with TP53 mutation status with P values <1 × 10−6 and 33 variants with P values <1 × 10−5. Forty-four variants were associated with PIK3CA mutation status with P values <1 × 10−5. In validation analyses, only variants at the ESR1 locus were associated with TP53 mutation status after multiple comparisons corrections. Combined analyses in European and Malaysian populations found ESR1 locus variants rs9383938 and rs9479090 associated with the presence of TP53 mutations overall (P values 2 × 10−11 and 4.6 × 10−10, respectively). rs9383938 also showed association with TP53 GOF mutations (P value 6.1 × 10−7). rs9479090 showed suggestive evidence (P value 0.02) for association with TP53 mutation status in African ancestry populations. No other variants were significantly associated with TP53 or PIK3CA mutation status. Larger studies are needed to confirm these findings and determine if additional variants contribute to ancestry-specific differences in mutation frequency.
Significance:
Emerging data show ancestry-specific differences in TP53 and PIK3CA mutation frequency in breast tumors suggesting that germline variants may influence somatic mutational processes. This study identified variants near ESR1 associated with TP53 mutation status and identified additional loci with suggestive association which may provide biological insight into observed differences.
Introduction
TP53 and PIK3CA are among the most frequently mutated genes in breast tumors (1). The frequency of somatic mutations in these genes varies by tumor subtype as well as ancestry (1–4). Pan-cancer and breast cancer–specific studies have found that tumors arising in individuals of African ancestry (AFA), particularly West African ancestry, are more likely to have somatic TP53 mutations and less likely to have somatic PIK3CA mutations than tumors arising in individuals of European ancestry (EUR; refs. 2–9). TP53 somatic mutations are more common in triple-negative [estrogen receptor (ER) negative, progesterone receptor (PR) negative, HER2 negative] breast cancers (TNBC), while PIK3CA mutations are more common in hormone receptor (HR)-positive HER2− tumors. However, even after adjusting for breast cancer subtype, ancestral differences in TP53 and PIK3CA somatic mutation frequencies persist for some subtypes (2–4, 10). For example, one study found that 39% of HR+ HER2− tumors from individuals of AFA had TP53 alterations compared with 24% of those of EUR (P value 0.005; ref. 8). Similarly, in HR+ HER2− tumors PIK3CA somatic mutations are less frequent in individuals of AFA (26%) versus EUR (42%; P value 0.001; ref. 8). The biological mechanisms leading to the observed differences in TP53 and PIK3CA somatic mutation frequency across populations and breast tumor subtypes are not understood.
TP53 encodes transcription factor TP53 and is mutated in a high proportion of breast and other cancers, resulting in altered expression of genes important for response to cellular stress and apoptosis. Unlike many genes involved in tumorigenesis, TP53 can have either loss-of-function (LOF) mutations, which lead to total loss of the ability of the protein to transactivate, or gain-of-function (GOF) mutations, which result in TP53 binding to new promoters to activate genes not typically associated with TP53 (11, 12). TP53 is a tetramer but can also bind to related proteins TP63 and TP73 (13). Some TP53 tumor-associated mutations act in a dominant-negative manner where the mutant version of the protein interferes with the function of wildtype proteins in the tetramer. In previous studies, we found that in breast tumors with TP53 mutations, those from AFA women were less likely to have GOF mutations than those from EUR women (14). Mutations without dominant-negative activity were associated with TNBC and ER-negative (ER−) status. These data suggest that types of TP53 mutations in breast tumors differ by self-reported race and tumor subtypes which may be due to different functional consequences of these mutations within cells.
While most somatic events in tumors are likely due to exogenous or endogenous mutators, recent evidence suggests that germline variants may influence the type and burden of somatic changes. Tumor mutational burden, caused in part by somatic mutations in DNA repair genes, is a polygenic trait with an estimated 13% of the variation explained by common germline variants (15). Some tumor mutational signatures are associated with common inherited variants in genes such the apolipoprotein B mRNA editing enzyme catalytic polypeptide (APOBEC) mutation signature and variants in GNB5 (16). Pathogenic variants (PV) in high-risk cancer susceptibility genes also associate with the presence of somatic mutations and specific mutational signatures. Breast tumors arising in individuals with a germline BRCA1 PV have more frequent occurrence of somatic TP53 mutations compared with those without a BRCA1 PV (17–20). Breast and ovarian tumors arising in individuals with BRCA1 and BRCA2 PVs typically show homology directed repair deficiency signatures (20, 21).
On the basis of these studies, we hypothesized that the germline genetic background of an individual can influence specific mutational processes, tumor promotion, and/or mutations in specific cancer-related genes during tumorigenesis, any of which could lead to the observed differences in the frequency of key cancer driver mutations by ancestry (22). The goal of this study was to identify inherited common germline variants (G) that are associated with TP53 or PIK3CA somatic mutation status (M) in breast tumors using a Germline Variant by Mutation (GxM) genome-wide association study (GWAS) design to assess the influence of genetic background on mutation frequency of these genes.
Materials and Methods
Ethics Approval and Consent to Participate
This study was approved by the Ohio State University (OSU) Cancer Institutional Review Board (IRB; protocol number 2005C0082). All data and samples were from deidentified individuals who had undergone informed consent for participation in research studies.
Nigerian Study
The City of Hope (COH) IRB and the University of Chicago IRB-approved study for participants enrolled at their respective sites.
COH Latina Study
One hundred and twenty Latina patients with breast cancer seen at COH in Duarte, California were included in this study. All participants signed a written informed consent approved by the COH IRB.
Malaysian Breast Cancer Study
Malaysian Breast Cancer Study (MyBrCa) was approved by the Independent Ethics Committee, Ramsay Sime Darby Health Care (reference no: 201208.1), and the Medical Ethics Committee, University Malaya Medical Centre (reference no: 842.9).
Discovery Breast Cancer Datasets
Existing datasets of women with breast cancer from The Cancer Genome Atlas (TCGA; ref. 1), Molecular Taxonomy of Breast Cancer International Consortium (METABRIC; ref. 23), and the Welcome Trust Sanger Institute (24) were used for the discovery GxM GWAS. Each study had existing genome-level single-nucleotide variant (SNV) genotyping data, somatic mutation data for TP53 and PIK3CA, and associated clinical and tumor details such as self-reported race/ethnicity, age at diagnosis, and ER, PR and HER2 status (Supplementary Tables S1–S4).
PIK3CA and TP53 Somatic Mutation Classification
For discovery and validation analyses, PIK3CA mutation status (yes or no) was defined for the following phenotypes: any non-LOF somatic variant in PIK3CA, any activating/hotspot mutation (25), or specific activating mutations (e.g., p.E542K, p.E545K, p.H1047R/L). TP53 mutation status was classified as the presence of any somatic variant in a coding exon or splice-site (yes/no), and variants resulting in TP53 LOF or GOF as described previously (Supplementary Table S5; Supplementary Data S1; refs. 14, 26). Somatic variants that resulted in a synonymous change and were not predicted to affect splicing were not considered to be a mutation. GOF mutations displayed one or more of the following phenotypes in functional studies: interference with TP63 or TP73 activity, transactivation of genes repressed by wildtype TP53, or cooperation with oncogenes in rat or mouse embryonic fibroblasts. TP53 LOF variants were those that abolished transactivation activity and/or resulted in altered splicing, frameshift, or nonsense changes. TP53 somatic missense variants with insufficient data to functionally score as LOF or GOF were called unknown and were not included in LOF or GOF specific analyses (Supplementary Table S5). Larger copy-number loss of TP53 was not included as a mutation functional category in the analyses due to lack of annotated data for multiple datasets. Controls for each analysis were individuals with breast cancer with no somatic mutation in the gene being assessed.
Ancestry and GWAS Analyses
PLINK (RRID:SCR_001757) was used to merge datasets, filter, and analyze data. Ancestry SNVs for principal component analyses (PCA) were determined using the Affymetrix annotation accomplished by subtracting the minor allele frequency (MAF) from each of four populations (Han Chinese in Beijing, Yoruba in Ibadan, Northern Europeans from Utah, Japanese in Tokyo) in the annotation in a pairwise manner and taking the top 1,000 SNVs from each comparison. This resulted in the use of 4,486 unique “ancestry” SNVs; 4,212 of those had a MAF of greater than 1%. PCA were performed on these 4,212 SNVs to identify individuals of non-EUR (e.g., those who did not cluster with the EUR group); these individuals were removed from the discovery analyses and were included in the validation studies (Supplementary Fig. S1A–S1C and S2A–S2D). There was a high concordance of ancestry assignment with self-reported race. Filtering also included removal of SNVs with MAF less than 0.01, male participants, and samples and SNVs with greater than 10% missing values. Imputed SNVs were not included. SNVs showing Hardy Weinberg equilibrium P values less than 1 × 10−50 were also removed. Association analyses were run on a final set of 2,850 females of EUR and 739,537 SNVs with PLINK using a logistic model with a covariate for study. An additive model was assumed. P values were FDR corrected and visualized using R (27). QQ Plots for each analysis were generated using R (Supplementary Fig. S3A–S3C and S4A–S4E).
Selection of Variants for Validation Analyses
Variants were prioritized for validation studies through multiple qualitative and quantitative filtering steps (Supplementary Tables S6 and S7; Fig. 1A and B). Information used to rank variants included P values <1 × 10−4, ORs, MAF greater than 10% for estimated power detection in at least one of three populations (European, African, or East Asian), allele frequency differences by ancestry, proximity to a variant identified in GWAS for breast cancer risk or other relevant phenotypes (e.g., other cancers, age at menarche, obesity), proximity to a gene showing a role in tumorigenesis, and mapping to a functionally active region (e.g., transcription start site, active chromatin markers, estimated or actual transcription factor binding site, disruption of a transcription factor binding motif, chromatin immunoprecipitation sequencing region for breast cancer cell line, characterized gene enhancer, characterized promoter region, or expression quantitative trait locus). Online resources used for in silico screening of candidate SNVs included UCSC Genome Browser (RRID:SCR_005780; ref. 28), GTEx Portal (RRID:SCR_001618; ref. 29), RegulomeDB v.2.0.3 (RRID:SCR_017905; ref. 30), HaploReg v4.0 (RRID:SCR_006796; ref. 31), and dbSNP (RRID:SCR_002338; ref. 32). In addition to variants chosen from the discovery GxM GWAS findings, additional variants were analyzed including two SNVs mapping near SETD9/MAP3K1 previously shown to be associated with PIK3CA somatic mutations in breast cancer (33), an XPC variant rs2228001 previously shown to be associated with TP53 mutation status (34), and a variant in AURKA (rs2273535) associated with somatic TP53 mutations in mouse studies (35). When a genotyping assay for a variant could not be designed for technological reasons, another variant from that locus or a variant in high linkage disequilibrium (LD; r2 > 0.8) with the original variant was included as a replacement. For validation sample sets with GWAS-level genotyping data, the original variant and the replacement variant were both included in analyses.
FIGURE 1.

Variant selection for validation studies. Flow chart for variant prioritization for validation studies is shown for TP53 mutation associations (A) and PIK3CA mutation associations (B). Variants were first filtered by P values, MAF, and location near relevant GWAS loci. Variants were then filtered by ORs, location relative to gene with role in breast cancer, TP53 or PI3K/AKT pathway and then by RegulomeDB score. Variants are only counted once but may fall within one or more categories.
Validation Genotyping
Validation genotyping for 188 SNVs of interest (95 for TP53 and 93 for PIK3CA) was completed for cohorts without existing genome-wide genotyping data including individuals from the Stefanie Spielman Breast Cancer Cohort (n = 144), OSU Total Cancer Care (TCC; n = 352) and the COH Latina Breast Cancer Study (n = 120) using a Fluidigm HD Biomark in a 96 × 96 format in the OSU Comprehensive Cancer Center (CCC) Genomics Shared Resource (GSR; Supplementary Tables S8 and S9). Each genotyping plate contained two duplicate DNA samples, three no-template controls (water), and one control DNA sample genotyped on all plates. DNAs that failed for more than 10% of SNVs from a plate were repeated and if failed again were removed from analysis. SNVs that failed for more than 10% of samples or failed to consistently form three clear genotyping groups were removed from analyses. For genetic ancestry, 96 SNVs were chosen for genotyping from existing ancestry informative marker (AIM) panels (refs. 36–38; Supplementary Tables S10–S12). Of the 96 AIM SNVs, two were removed for poor genotyping performance.
Somatic Mutational Analyses
For the validation studies, TP53 and PIK3CA mutational status from the clinical testing reports or targeted or exome sequencing of tumor DNA was available for breast cancer cases from the COH Latina Breast Cancer Study, TCGA, and a subset of the TCC cases. For cases in which mutation status was not known, tumor tissue or DNA was available from the Spielman Breast Cancer Cohort and the TCC program for mutational analysis.
Sanger Sequencing Mutational Analysis
Tumor samples lacking existing somatic mutation data (n = 126 for TP53, n = 184 for PIK3CA) were screened for somatic mutations in TP53 coding exons (exons 2–10) and PIK3CA exons 4, 9, and 20 using Sanger sequencing. Tumor DNA (10–20 ng) was PCR amplified and products were confirmed for size by gel electrophoresis (Supplementary Table S13). PCR products were Exo/SAP-IT treated and Sanger sequenced in both forward and reverse directions by the GSR. Sequence chromatograms were evaluated for mutations using DNASTAR Lasergene v.17 (RRID:SCR_000291) by two different laboratory members.
GxM Validation Analyses
Data used for validation of key findings included genotype and tumor mutational data from individuals of non-EUR from the three discovery datasets as well as samples (germline and/or tumor DNA) or existing data from 1,285 individuals of multiple ancestries from the METABRIC (n = 166), Stefanie Spielman Breast Cancer Cohort (n = 144), OSU TCC (n = 352), a Nigerian breast cancer study (n = 100), the COH Latina Breast Cancer Study (n = 120), TCGA (n = 302), and a TCGA study (“Banerji study”) of women from Mexico and Vietnam (n = 101; Supplementary Tables S2–S4 and S14–S18; refs. 3, 4, 39, 40). Genetic ancestry by PCA classified 341 women as AFA (26.5%), 572 women as EUR (44.5%), and 133 women as East Asian ancestry (EAS; 10.4%). The remainder of women (18.6%) were admixed [falling between principal component (PC) clusters], most of whom self-identified as Hispanic/Latino. Because of some missing genotypes, not every variant had data for all 1,285 individuals.
For association analyses, logistic models were employed with an additive effect for the SNV. Study and ancestry were included as covariates in the models. For the study and ancestry-specific analyses, the study analysis omitted the effect of study, and the ancestry analyses omitted the ancestry PC from the models. Because two different panels were used for ancestry determination, individuals of known ancestry (HapMap, TCGA; RRID:SCR_004563 and RRID:SCR_003193 respectively) were used as anchors for each panel. The PC1/PC2 were rotated so that the known ancestry groups overlapped and the distance from the anchor group was calculated as the PC covariables. For individuals with available genome-wide genotyping data, imputation of validation SNVs not present on the GWAS genotyping panels was performed. Imputation was carried out after removing genotypes with no calls or Y chromosome calls. Eagle (RRID:SCR_015991) was used to phase SNV, and imputation was done using Minimac3 (RRID:SCR_009292). The maximum expected error rate across imputed validation SNPs was 0.086. Formats were converted to PLINK format, and variants with greater than two alleles were removed.
Independent Validation Studies
SNVs of interest were also assessed independently in two cohorts with existing genotyping and mutation data: 859 women with breast cancer from the MyBrCa (41, 42) and 393 AFA women with TNBC from the Breast Cancer in African Americans: Understanding Somatic Mutations and Etiology (B-CAUSE) study (Supplementary Tables S19 and S20; ref. 43). Validation SNVs for the MyBrCa study were excluded if they had a MAF less than 1% in Malaysian individuals and SNVs were excluded from analyses for the MyBrCa and B-CAUSE studies if they mapped to the X chromosome as these data were unavailable. For the MyBrCa study, association tests were conducted using SNPtest adjusted to information for ancestry (four PCs), age of diagnosis, and ER status. B-CAUSE data came from women who self-identified as Black and were diagnosed with TNBC. The African-ancestry Breast Cancer Genetic (AABCG) is a large breast cancer consortium which provided genome-wide genotyping data for the B-CAUSE study. AFA was confirmed by estimating global AFA using ADMIXTURE (ref. 44; Supplementary Tables S20). As the frequency of somatic TP53 mutations in the B-CAUSE TNBC cases was high, analyses were run for TP53 GOF-associated germline variants using individuals with LOF TP53 mutations and those with no mutations as controls; conversely for TP53 LOF-associated variants, analyses were run using individuals with GOF plus those with no mutations as controls. Logistic regression was employed with a covariable for study and main effect of SNV genotype for ESR1 variants for combined analyses of Discovery/EUR validation/MyBrCa and AFR validation/B-CAUSE datasets.
Data Availability
The majority of data generated or analyzed during this study are included in this published article in Supplementary Tables, in TCGA, dbGAP and/or the following data repositories as listed below. TCGA tumor mutation data and SNV genotyping data are available in dbGAP under accession numbers phs001687.v1.p1, phs000178.v11.p8, and phs002387.v1.p1. METABRIC sequencing data of tumors and SNV genotyping data are available on the European Genome-Phenome archive using accession numbers EGAD0001000164, EGAS00000000083, EGAD00010000158, EGAD00010000266, EGAS00001004518, and EGAD00001006399. The Welcome Trust Sanger Institute data are available in the European Genome-Phenome archive using accessing number EGAS00001001178 and EGAD0010000915. Sequencing data and processed genomic data from the Nigerian breast cancer cases are in dbGAP under study accession number phs001687.v1.p1. Tumor/normal whole-exome sequencing (WES) and RNA-sequencing data and accompanying phenotypic and clinical/histologic data for the COH Latina Breast Cancer Study are deposited in dbGAP (dbGaP Study Accession: phs003218; ref. 39). MyBrCa WES and shallow whole genome sequencing (sWGS) files are available on the European Genome-phenome Archive under the study accession number EGAS00001004518. Access to controlled patient data will require the approval of the MyBrCa Tumour Genomics Data Access Committee upon request to genetics@cancerresearch.my. Sequence and genotyping data for the Banerji and colleagues study (40) are available in dbGAP under accession number phs000369.v1.p1. Summary-level statistics genotyping data for the AABCG study are available at GWAS Catalog (accession number: GCST90296719, GCST90296720, GCST90296721, and GCST90296722). B-CAUSE TNBC sequencing data are in the process being deposited into dbGaP with accession number pending.
Results
To identify germline variants associated with TP53 or PIK3CA somatic mutations in tumors, we identified existing datasets with GWAS-level germline variant information, somatic mutation information for TP53 and PIK3CA, and demographic and clinical information such as age of diagnosis, tumor subtype defined by hormonal (ER and PR) status and HER2 amplification. Three datasets were identified that fit these criteria (Supplementary Tables S2–S4). After filtering for SNVs with MAF less than 1%, individuals with 10% or higher SNV genotypes missing, SNVs out of Hardy–Weinberg equilibrium (P value <1 × 10−50) and individuals of non-EUR, 2850 females of EUR with breast cancer and 739,537 SNVs were included in the discovery GWAS for variants associated with TP53 and PIK3CA mutation status.
Discovery GxM for TP53 and PIK3CA Mutation Status
Analyses for association with any TP53 mutation, GOF TP53 mutation, and LOF TP53 mutation were performed for the 2,850 women of EUR in the discovery dataset in which 30.8% of women had a TP53 somatic mutation (Table 1; Supplementary Tables S2–S5). Following analysis, no SNV met the genome-wide statistical significance threshold of a P value <5 × 10−8; four variants were identified with P values ≤1.0 × 10−6 and 34 variants had P values less than ≤1.0 × 10−5 across 22 loci (Fig. 2A–C; Supplementary Tables S21 and S22). Two variants showed P values of <1.0 × 10−5 for more than one TP53 mutation functional category: rs1561072 for any TP53 mutation and GOF TP53 mutations and rs2886631 for any TP53 mutation and LOF TP53 mutations.
TABLE 1.
TP53 and PIK3CA somatic mutation frequencies
| Study | Total N | TP53 Mutation N (%) | TP53 subtype N (%) | PIK3CA Mutation N (%) | PIK3CA subtype N (%) |
|---|---|---|---|---|---|
| Discovery | 2,850 | 879 (31%) | GOF 237 (8%) | 1,095 (38%) | Activating 858 (30%) |
| LOF 536 (19%) | p.E542K 112 (4%) | ||||
| Unknown 106 (4%) | p.E545K 193 (7%) | ||||
| p.1047R/L 387 (14%) | |||||
| Validation | 1,285 | 414 (40%) | GOF 110 (11%) | 290 (28%) | Activating 235 (23%) |
| LOF 277 (27%) | p.E542K 40 (4%) | ||||
| Unknown 27 (3%) | p.E545K 58 (6%) | ||||
| p.H1047R/L 133 (13%) | |||||
| MyBrCa | 859 | 369 (43%) | GOF 114 (13%) | 247 (29%) | Activating 217 (25%) |
| LOF 241 (28%) | p.E542K 20 (2%) | ||||
| Unknown 14 (2%) | p.E545K 55 (6%) | ||||
| p.H1047R/L 115 (13%) | |||||
| B-CAUSE | 393 | 365 (93%) | GOF 85 (22%) | 9 (2%) | Activating 4 (1%) |
| LOF 260 (66%) | p.H1047R/L 2 (0.5%) | ||||
| Unknown 20 (5%) |
Abbreviations: N, number; %, percent of total number.
FIGURE 2.
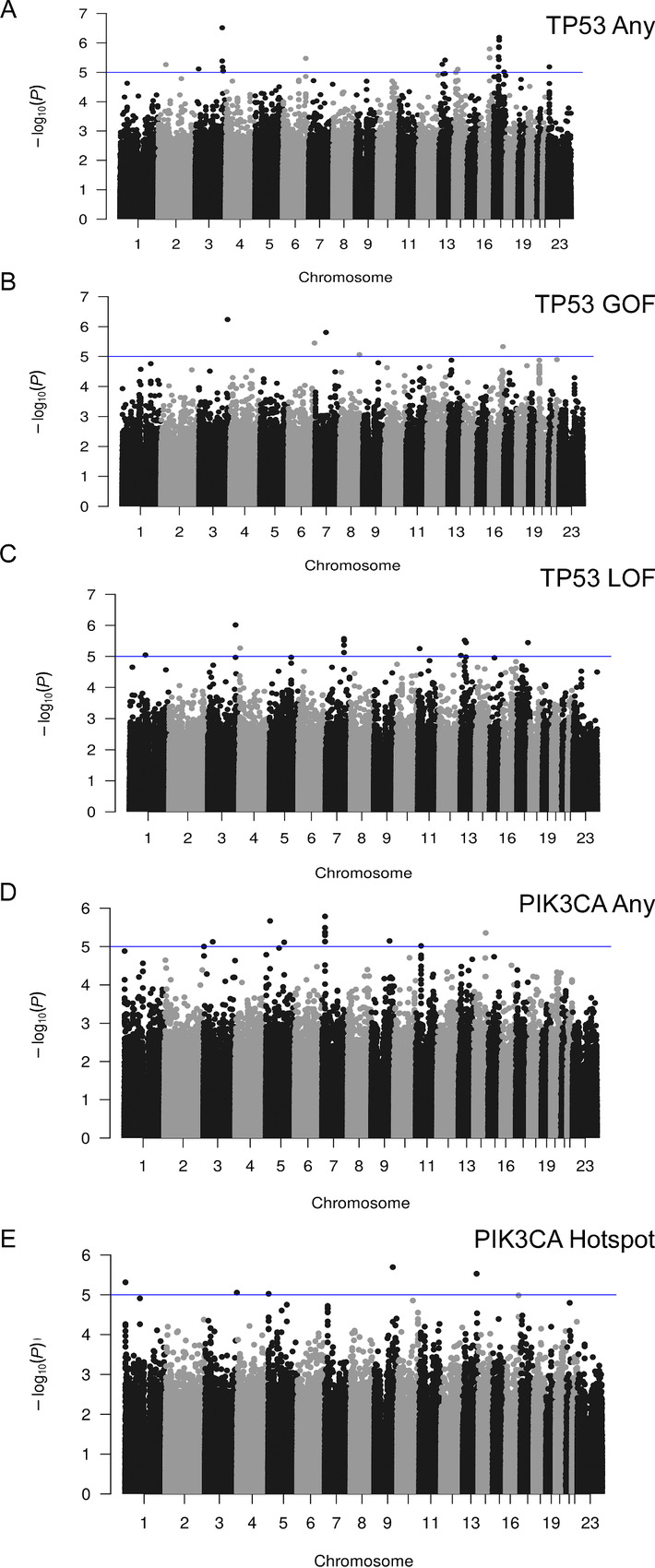
Manhattan plots for TP53 and PIK3CA discovery GxM analyses. Discovery GWAS data for 879 TP53 mutation carriers and 1,965 breast cancer cases without TP53 mutations (A), 237 cases with TP53 GOF mutations and 1,965 breast cancer cases without TP53 mutations (B), 536 cases with TP53 LOF mutations and 1,965 controls (C), 1,095 PIK3CA mutation carriers and 1,642 breast cancer cases without PIK3CA mutations (D), and 858 cases with PIK3CA activating/hotspot mutations and 1,642 breast cancer cases without PIK3CA mutations, are plotted by −log10 (P values; E). Blue lines represent P values of less than 1 × 10−5. Chromosome numbers are indicated. GXM, germline variant by mutation; GWAS, genome-wide association study; GOF, gain of function; LOF, loss of function.
Following association analyses for PIK3CA mutation status for the 2,850 women in the discovery set, 38% of whom had a PIK3CA somatic mutation, no SNV met genome-wide significance of P value of <5 × 10−8 (Fig. 2D; Table 1). Forty-four SNVs were associated with one or more PIK3CA mutation functional category with P value <1 × 10−5 (Fig. 2E; Supplementary Table S23 and S24). Of these, rs2026801 showed evidence of association (P value <1 × 10−5) for any PIK3CA mutation and activating PIK3CA mutations, and rs1712829 showed evidence of association with both p.H1047R and any PIK3CA mutation.
Selection of Variants for Validation Studies
Using in silico filtering approaches, all variants with P values < 1 × 10−4 for any somatic mutation functional category were evaluated for potential inclusion in validation studies. Variants were prioritized for further evaluation by allele frequency in one or more ancestral group (MAF > 10%), potential function using in silico prediction models, location near a known GWAS hit for breast cancer or related phenotype (e.g., age of menarche, obesity), location near a gene involved in tumor development, or known relationship to TP53 or PI3K pathways (Fig. 1A and B; Supplementary Tables S6 and S17). Of these, 188 variants from TP53 (n = 95) and PIK3CA (n = 93) GxM analyses were chosen for validation studies and successfully genotyped in multi-ancestral populations (Supplementary Table S25). For individuals with GWAS-level genotyping data, 119 variants for TP53 and 106 variants for PIK3CA were tested (Supplementary Tables S26–S31).
Mutation Status and Ancestry in Validation Populations
In the validation datasets, ancestry classifications by PCA yielded 340 AFA individuals, 602 EUR individuals and 134 EAS individuals. The remainder of study individuals (n = 209) were considered admixed and not assigned to a specific group; these included individuals of Hispanic/Latino background who demonstrated a high degree of admixture. In the validation datasets, 40% had a TP53 somatic mutation, and 28% had a PIK3CA somatic mutation in their breast tumor (Table 1). The MyBrCa study included 859 women from Malaysia with breast cancer, of whom 43% carried a somatic TP53 mutation (43%) and 29% had a somatic PIK3CA mutation (Table 1). Of the 393 women of AFA with TNBC in the B-CAUSE study, 93% had a TP53 somatic mutation and only 2.3% had any PIK3CA somatic mutations (Table 1).
Association of Variants at the ESR1 Locus and TP53 Mutation Status
Association analyses of validation SNVs were performed separately by ancestry and study. After multiple comparison corrections, variants at the ESR1 locus were the only ones showing statistically significant evidence of association with TP53 mutations in at least one validation dataset. In the MyBrCa study, ESR1 variant rs9383938 showed association with having a TP53 mutation (OR = 1.81; P value 9.8 × 10−8) and TP53 GOF mutation status (P value 8.4 × 10−6; Table 2; Supplementary Tables S27 and S32). Another ESR1 locus variant, rs9479090, was also associated with TP53 mutations in this population (P value 2.8 × 10−7). Combined analyses of the discovery, EUR validation and MyBrCa studies completed for three variants at the ESR1 locus, rs9397436, rs9383938, and rs9479090, all showed evidence for association with having one or more TP53 mutation functional categories after multiple comparisons corrections (Table 2). AFA-specific analyses for these variants showed a trend for association with rs9479090 and having any TP53 mutation (OR = 1.33, P value 0.02; Table 3). None of these variants showed evidence of association in admixed individuals mapping between the European and Asian PCA clusters, most of whom self-identified as Hispanic. Of note, the TP53-associated alleles showed lower allele frequency in the EUR and Hispanic populations.
TABLE 2.
ESR1 locus variants and TP53 mutation associations
| SNV Ref Allele | rs9397436 T | rs9383938 G | rs9479090 A | |||
|---|---|---|---|---|---|---|
| TP53 Mutation | Any | LOF | GOF | Any | GOF | Any |
| Discovery Cases/Controls | 853/1,797 | 516/1,797 | 236/1,797 | 1,073/2,242 | 292/2,242 | 1,106/2,303 |
| Discovery OR | 1.53 | 1.48 | 1.79 | 1.46 | 1.71 | 1.44 |
| Discovery P | 1.39E-05 | 6.9E-04 | 1.1E-04 | 6.8E-05 | 3.5E-05 | 5.4E-05 |
| Discovery MAF | 8.4% | 10.0% | 11.0% | |||
| EUR Valid Cases/Controls | 217/356 | 132/356 | 67/356 | 130/227 | 34/227 | 215/357 |
| EUR Valid OR | 1.14 | 1.04 | 1.17 | 1.04 | 0.96 | 1.06 |
| EUR Valid P | 0.55 | 0.89 | 0.66 | 0.87 | 0.92 | 0.8 |
| EUR MAF | 9.4% | 9.4% | 10.7% | |||
| MyBrCa Cases/Controls | 369/490 | 241/490 | 114/490 | 369/490 | 114/490 | 369/490 |
| MyBrCa OR | 1.43 | 1.35 | 1.51 | 1.81 | 2.07 | 1.76 |
| MyBrCa P | 0.001 | 0.02 | 0.01 | 9.8E-08* | 8.4E-06* | 2.8E-07* |
| MyBrCa MAF | 37.3% | 37.7% | 38.1% | |||
| Combined Cases/Controls | 1,439/2,643 | 889/2,643 | 417/2,643 | 1,572/2,959 | 440/2,959 | 1,690/3,150 |
| Combined OR (95% CI) | 1.35 (1.17–1.54) | 1.24 (1.06–1.46) | 1.53 (1.25–1.88) | 1.47 (1.31–1.66) | 1.59 (1.32–1.90) | 1.42 (1.27–1.59) |
| Combined P | 1.8E-05a | 0.007 | 3.5E-05a | 2.0E-10a | 6.07E-07a | 4.6E-10a |
Abbreviations: 95% CI, 95% confidence interval; EUR Valid, European ancestry Validation Study; GOF, gain of function; LOF, loss of function; MAF, minor allele frequency; OR, odds ratio; P, P values; Ref allele, reference allele.
a Significant after multiple comparisons corrections.
TABLE 3.
ESR1 variant associations in combined AFA datasets
| SNV Ref Allele | rs9397436 T | rs9383938 G | rs9479090 A | |||
|---|---|---|---|---|---|---|
| TP53 Mutation | Any | GOF | LOF | Any | GOF | Any |
| Cases/Control | 427/333 | 85/333 | 296/333 | 247/146 | 49/146 | 401/331 |
| AFA MAF | 31% | 15% | 27% | |||
| OR (95% CI) | 0.97 (0.69–1.41) | 0.59 (0.26–1.17) | 1.12 (0.75–1.65) | 1.45 (0.94–2.26) | 1.42 (0.72–2.75) | 1.33 (1.05–1.68) |
| P | 0.91 | 0.16 | 0.58 | 0.097 | 0.3 | 0.02 |
Abbreviations: 95% CI, 95% confidence interval; AFA MAF, minor allele frequency in combined African Ancestry datasets; OR, odds ratio, P, P-value; Ref allele; reference allele.
Association of Other Loci with TP53 and PIK3CA Mutations
After correcting for multiple comparisons, no other variants were significantly associated with any TP53 mutation functional category in any of the validation datasets (Supplementary Tables S26–S28, S32, and S33). Variants showing a nonsignificant trend for association in more than one dataset included rs10931697 for TP53 GOF in the EUR validation, AFR validation, and MyBrCa studies (P values 0.008, 0.02, and 0.09, respectively), and rs6709393 (P values 0.003 and 0.16) in the MyBrCa and B-CAUSE studies. No SNVs were significantly associated with any PIK3CA mutation type in the validation datasets, MyBrCA study or B-CAUSE study (Supplementary Tables S29–S31, S34, and S35).
Discussion
To our knowledge, this is the first genome-wide breast cancer–specific study to identify germline variants that are associated with TP53 or PIK3CA somatic mutation status. As different types of mutations may have differential effects on cancer-related phenotypes, we also tested for association of specific subcategories of TP53 (any, LOF, GOF) and PIK3CA (any, activating, specific site) mutations with common SNVs. Five variants from the discovery analyses of women of EUR showed suggestive evidence (P value <1 × 10−6) for association with TP53 mutation status. Analyses of candidate variants in a Malaysian study, MyBrCa, and combined analyses confirmed that variants at the ESR1 locus were associated with multiple TP53 mutation classifications and remained significant after corrections of multiple comparisons.
ESR1 Locus Variants, Breast Cancer Risk, and Association with TP53 Mutation Status
We found evidence that multiple ESR1 locus variants were associated with TP53 mutation status. In our discovery study, ten variants at this locus showed a trend toward association (P value <1 × 10−4) for one or more of the three functional categories of TP53 mutations. From breast cancer GWAS, multiple variants near ESR1 have been associated with breast cancer of all subtypes as well as ER− tumors (43, 45–48). Some variants at the ESR1 locus have been reported to exhibit ancestry-specific association with breast cancer risk (48–50). For example, ESR1 variant rs140068132 which is thought to have originated in Indigenous Americans, is protective for breast cancer risk (50). In gnomAD, the MAF of ESR1 variants showing association with having a TP53 mutation in our study are lowest in individuals of European, Latin American, and South Asian ancestry and are higher in individuals of African and EAS which may explain in part the higher proportion of breast tumors in these populations with TP53 mutations in these populations.
Variants at the ESR1 locus were among the first to be associated with breast cancer risk in GWAS (48, 51) and are associated with breast cancer in multiple populations including Chinese, Indian, Nigerian, African American, Malaysian, Latina/Hispanic. European, and Korean (47, 49, 50, 52). These include variants rs9397436 and rs9383938 which were associated with having a tumor with a TP53 mutation our study (53, 54). Some variants show ancestry-specific differences ORs. Rs2046210, which was originally discovered to be associated with breast cancer in Asian populations, showed a per-allele OR of 1.36 in EAS but ORs close to 1 in EUR and AFA populations (55–57). ESR1 variants are also associated with specific breast tumor subtypes in GWAS. In EUR-based studies, rs2747652 was associated with HER2-positive/nonluminal breast cancer (58) and rs2757318, rs2046210, and rs9383938 were associated with ER− breast cancer (53, 59). Interestingly, association of rs2046210 with ER− tumors appears to be more pronounced in EUR than EAS (55). Functional mapping of variants across the ESR1 locus found that multiple variants, including those found in our study, overlap with enhancer regions or show association with ESR1 expression (45).
In ER− breast tumors, TP53 and ESR1 mutations tend to be mutually exclusive (60). This may be due in part to the regulatory relationship between TP53 and ESR1. Mutant TP53 is correlated with lower ESR1 gene expression which is thought to be due in part to TP53 binding to the ESR1 promoter to activate expression (61). Mutant TP53 tumors have lower estrogen response signatures compared with TP53 wildtype tumors which may be caused by both decreased transcriptional activation of ESR1 by mutant TP53 and increased levels of ESR1-targeting miRNAs (60). These studies suggest the possibility that mutation of TP53 may be an early event that promotes lineage toward ER− breast tumors; it is possible that variants at the ESR1 locus may enhance or reverse this association. Further functional studies are warranted to understand the connection between ESR1 variants, TP53 mutational status, and breast cancer subtypes.
Variants Associated with TP53 Mutation Status
Other variants in our study showing suggestive evidence of association with TP53 mutation status included rs17103093 which was associated with any TP53 mutation phenotype (discovery OR 1.54, P value 3.3 × 10−5 and combined validation analysis OR 1.4, P value 0.03). Rs17103093 maps to an intron of TACC2. This variant did not show evidence of association with TP53 mutations in the MyBrCa study. TACC2 encodes one of three homologous coiled-coiled proteins; it shows increased expression in higher grade breast tumors and is associated with local recurrence and reduced survival (62, 63). Variants at the TACC2 locus are associated with risk of low-grade breast cancer, overall breast cancer, and epithelial ovarian cancer (48, 64, 65). Two variants at other loci, rs6703393 and rs6890674, showed consistent direction of association for TP53 GOF mutations in the discovery analyses (OR 0.79, P value 7.5 × 10−5) and the MyBrCa study (OR 0.28, P value 0.003) but had no evidence of association in the combined validation analyses (P values 0.99 and 0.83, respectively). rs6709393 maps near the RAB17 gene which encodes for a small GTPase associated with invasion (66). rs6890674 is located in the 3′ untranslated region of CD180, an orphan Toll-like receptor that is expressed on B cells and is involved in inflammatory and autoimmune diseases (67). Additional studies are needed to determine if these represent real associations.
Ancestral Differences in TP53 and PIK3CA Mutation Frequencies Across Cancer Types
Associations with genetic ancestry and specific somatic driver mutations have been observed in other cancer types (23, 68). Genetic ancestry is associated with specific somatic driver mutations in EGFR, KRAS, and STK11 in lung cancer in individuals of Indigenous American ancestry relative to those of EUR or EAS ancestry (69, 70). TP53 mutations are found at a higher frequency in individuals of AFA relative to individuals of EUR tumors in multiple tumor types (lung, colon, gastric, human papilloma virus–negative head and neck), suggesting that genetic background and/or differences in exposures/socio-determinants of health may influence selection of TP53 somatic mutations (71–74). PIK3CA somatic mutations also show differences by ancestry in different tumor types. For example, PIK3CA mutations have been observed at lower frequencies in bladder tumors arising in EAS individuals and in head and neck squamous cell carcinomas from AFA individuals (74, 75). Conversely, PIK3CA mutations are more often observed in colorectal tumors from AFA individuals (76). Variants identified in this study may have utility in explaining TP53 and PIK3CA somatic mutation frequencies arising in different tissues that differ by genetic ancestry. We did not observe any significant AFA-specific associations at the ESR1 locus after corrections for multiple comparisons, but rs9479090 showed suggestive evidence (P value < 0.05).
Study Limitations
There are limitations to this study. Our discovery analyses were performed in non-Hispanic individuals of EUR, which means that variants enriched in or specific to non-European populations may not have been identified. We were underpowered to determine whether our GxM findings were responsible for the observed differences in breast cancer TP53 and PIK3CA mutation frequency for individuals of non-European populations and for variants associated with specific PIK3CA mutations (e.g., p.E542K, p.E545K, and p.H1047R/L). In our validation study, we did not genotype all variants/loci with P values of less than 1 × 10−4 observed in our discovery set, some of which were not included because of low MAF in one or more populations. As such, we may have missed key variants/loci associated with TP53 or PIK3CA mutation status. The source of somatic mutation information varied widely with some information coming from clinical reports, some from whole genome/WES of tumors, some from targeted sequencing studies, and some from in-house Sanger sequencing studies. Next-generation sequencing is more sensitive than Sanger sequencing for somatic mutations that are present in fewer than 20% of cells or for tumors with a high degree of immune or stromal infiltrate. Our study was based on the premise that TP53 and PIK3CA mutations would be early driver events in tumor development, and mutations in these genes should be present in a high proportion of tumor cells. In a previous study, in which we evaluated types of TP53 mutation by self-reported race and ethnicity, we found no differences in TP53 mutation frequency across studies by modality of somatic variation detection suggesting that Sanger sequencing is reasonable for mutation detection of early driver events present in a large proportion of cells (14). Copy number information was not available for a large proportion of tumors; thus, TP53 mutations due to larger deletions (e.g., chromosome 17p loss) were not included. We expect that a subset of tumors defined as not having a mutation in TP53 may have had large copy number losses at that locus resulting in the missing of individuals with LOF mutations due to larger deletions.
Across populations, somatic mutations in TP53 are more common in TNBC and HER2+ tumors; conversely, somatic mutations in PIK3CA are much more frequent in ER-positive (ER+) tumors and luminal breast cancers (2, 4). Even with adjustment based on tumor subtype, it is difficult to sort out the association of the SNV with somatic mutation versus association of the SNV with tumor subtype. Previous studies stratifying by ER− and ER+ tumor status have found ancestry differences in mutation frequency for these genes, but this was not the case for all studies stratifying by tumor subtype (4, 8). Future mechanistic studies are needed to determine whether germline variants help drive tumor subtypes that are characterized by certain gene mutations and/or whether germline variants impact a cellular context in which a particular mutation is more likely to be selected and the mutation is important for determining tumor subtype.
Conclusions
This study provides evidence that ESR1 germline variants may shape somatic mutation processes or mutation selection of TP53 in breast tumors. In the future, polygenic risk scores could identify individuals who are at increased risk of mutations in specific genes should they develop breast cancer which may ultimately inform prevention strategies, such as potential vaccination-based prevention for high-risk individuals more likely to carry a specific somatic mutation. Larger multi-ancestry studies are warranted to confirm the study findings and determine whether germline variants explain some of the differences in TP53 and PIK3CA breast cancer mutation frequencies by genetic ancestry. Functional and mechanistic studies are needed to understand the target genes and pathways for variants associated with these mutations in breast tumors.
Supplementary Material
Supplemental Information: References for Supplemental Table S5
Supplemental Tables S1-S35
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Acknowledgments
This work was supported in part by NCI R01 CA215151-01 (A.E. Toland). J. Ramroop was supported by a Pelotonia Postdoctoral Fellowship, N.P. Tjader was supported by a Pelotonia Graduate Research Fellowship, and T. Gandhi was supported by a Pelotonia Undergraduate Research Fellowship. M. Paredes was supported by an OSU CCC CREATES Program Fellowship. The OSU CCC GSR and TCC were funded in part by NCI grant P30 CA016058. The Spielman Breast Bank was funded in part by the Stefanie Spielman Fund for Breast Cancer Research. For the COH Latina Breast Cancer study, the work was funded by the NCI (R01CA184585, K24CA169004), the National Institute on Minority Health and Health Disparities (NIMHD) Division of Intramural Research, and the California Initiative to Advance Precision Medicine (OPR18111). Research reported in this publication included work performed in the COH Integrative Genomics Core and the Pathology Core supported by the NCI of the NIH under grant number P30CA033572. S.L. Neuhausen and this research were partially funded by the Morris and Horowitz Families Professorship. Sample collection and data were collected under support from NIH R01CA184585 (S.L. Neuhausen and E. Ziv). The Nigerian Breast Cancer study was funded in part by NCI grant R01CA228198 and NIMHD grant R01 MD013452 (D. Huo). MyBrCa was funded by the Newton-Ungku Omar Fund (grant no: MR/P012930/1), Wellcome Trust (grant no: v203477/Z/16/Z), Scientex Foundation, Estée Lauder Companies, Yayasan PETRONAS, and Yayasan Sime Darby. The B-CAUSE study was supported in part by NCI R01 CA228156 (Yao, Palmer, Zheng, Carpten), NCI R01 CA164974 (Palmer), NCI R01 CA255242 (Zheng), NCI U24 CA232979, and NCI U24 CA274159 (Liu). The genome-wide genotype was supported in part by NIH grant R01 CA202981 (Zheng). Sample preparation and genotyping assays at Vanderbilt University Medical Center were conducted at the Survey and Biospecimen Shared Resources and Vanderbilt Technologies for Advanced Genomics, which are supported in part by the Vanderbilt-Ingram Cancer Center NCI grant (P30CA068485). The authors would like to acknowledge contributions from central cancer registries supported through the Centers for Disease Control and Prevention's National Program of Cancer Registries (NPCR) and/or the NCI's Surveillance, Epidemiology, and End Results (SEER) Program. Central registries may also be supported by state agencies, universities, and cancer centers. Participating central cancer registries include the following: AL, AR, AZ, CA, CO, CT, DE, DC, FL, GA, HI, IA, IL, IN, KY, LA, MD, MA, MI, MO, MS, NE, NJ, NM, NY, NC, OH, OK, OR, PA, SC, TN, TX, VA, WA, WI. The content of this study is solely the responsibility of the authors and does not necessarily represent the official views of the U.S. Department of Health and Human Services, the NIH, the NCI, or the state cancer registries.
The Stefanie Spielman breast bank and TCC from the OSU CCC Biospecimen Services Shared Resource provided deidentified samples and data for the multi-ancestry validation studies. The OSU GSR performed the targeted genotyping studies and Sanger sequencing of tumor samples. TCGA Data: The results published here are in whole or part based upon data generated by TCGA managed by the NCI and NHGRI. Information about TCGA can be found at http://cancergenome.nih.gov. METABRIC: This study makes use of data generated by the Molecular Taxonomy of Breast Cancer International Consortium. Funding for the project was provided by Cancer Research UK and the British Columbia Cancer Agency Branch. TCGA Banerji Study: This work was a collaboration of the Broad Institute in Cambridge, MA and the National Institute of Genomic Medicine (INMEGEN) in Mexico City, Mexico. The work was conducted as part of the Slim Initiative for Genomic Medicine, a project funded by the Carlos Slim Health Institute in Mexico. Nigerian Breast Cancer Data: We are greatly indebted to all the patients who agreed to participate in this study and graciously donated their biological materials. COH Latina Breast Cancer Study: We want to thank the New York Genome Center for the quality of the sequencing and analytic services provided. We also want to thank QIAGEN for their generous donation of PAXgene Tissue Containers and DNA Extraction Kits for this study. This work is dedicated to the memory of the late Mrs. Anne Olorunde (project officer from LASUTH) and Dr. Olayiwola Oluwasola. MyBrCa: The Malaysian Breast Cancer Genetic study thanks all the study participants and all research staff at Cancer Research Malaysia, University Malaya, and Subang Jaya Medical Centre who assisted in recruitment, interviews, and samples/data processing. MyBrCa also thanks the Breast Cancer Association Consortium (BCAC), University of Cambridge and Genome Quebec for their genotyping related work and Caldas Lab and the Core Genomics Facility at the CRUK Cambridge Institute for their sequencing work. AABCG: Data analyses for the AABCG were conducted using the Advanced Computing Center for Research and Education (ACCRE) at Vanderbilt University.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Communications Online (https://aacrjournals.org/cancerrescommun/).
Authors’ Disclosures
N.P. Tjader reports grants from Pelotonia during the conduct of the study. S.L. Neuhausen reports grants from NCI during the conduct of the study. H. Hampel reports personal fees from Natera and Carelon; other from LS Cancer Diag OY; personal fees and other from GI OnDemand and Genome Medical outside the submitted work. J.R. Palmer reports grants from NIH during the conduct of the study. J.D. Carpten reports other from American Association of Cancer Research outside the submitted work. J.P. McElroy reports grants from NIH during the conduct of the study. A.E. Toland reports grants from NIH during the conduct of the study. No disclosures were reported by the other authors.
Authors’ Contributions
N.P. Tjader: Investigation, methodology, writing-review and editing. A.J. Beer: Investigation, writing-review and editing. J. Ramroop: Investigation, methodology, writing-review and editing. M.-C. Tai: Resources, formal analysis, investigation, writing-review and editing. J. Ping: Resources, formal analysis, investigation, writing-review and editing. T. Gandhi: Investigation, writing-review and editing. C. Dauch: Investigation, writing-review and editing. S.L. Neuhausen: Resources, investigation, writing-review and editing. E. Ziv: Resources, writing-review and editing. N. Sotelo: Investigation, writing-review and editing. S. Ghanekar: Investigation, writing-review and editing. O. Meadows: Investigation, writing-review and editing. M. Paredes: Investigation, writing-review and editing. J.L. Gillespie: Formal analysis, investigation, writing-review and editing. A.M. Aeilts: Resources, writing-review and editing. H. Hampel: Resources, writing-review and editing. W. Zheng: Resources, supervision, writing-review and editing. G. Jia: Formal analysis, investigation, writing-review and editing. Q. Hu: Formal analysis, investigation, writing-review and editing. L. Wei: Formal analysis, investigation, writing-review and editing. S. Liu: Formal analysis, investigation, writing-review and editing. C.B. Ambrosone: Resources, writing-review and editing. J.R. Palmer: Resources, writing-review and editing. J.D. Carpten: Resources, writing-review and editing. S. Yao: Resources, supervision, methodology, writing-review and editing. P. Stevens: Formal analysis, writing-review and editing. W.-K. Ho: Formal analysis, writing-review and editing. J.W. Pan: Investigation, writing-review and editing. P. Fadda: Investigation, writing-review and editing. D. Huo: Resources, writing-review and editing. S.-H. Teo: Conceptualization, supervision, writing-review and editing. J.P. McElroy: Conceptualization, data curation, formal analysis, writing-review and editing. A.E. Toland: Conceptualization, supervision, funding acquisition, investigation, visualization, methodology, writing-original draft, project administration, writing-review and editing.
References
- 1. The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012;490:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Keenan T, Moy B, Mroz EA, Ross K, Niemierko A, Rocco JW, et al. Comparison of the genomic landscape between primary breast cancer in African American versus white women and the association of racial differences with tumor recurrence. J Clin Oncol 2015;33:3621–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pitt JJ, Riester M, Zheng Y, Yoshimatsu TF, Sanni A, Oluwasola O, et al. Characterization of Nigerian breast cancer reveals prevalent homologous recombination deficiency and aggressive molecular features. Nat Commun 2018;9:4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Huo D, Hu H, Rhie SK, Gamazon ER, Cherniack AD, Liu J, et al. Comparison of breast cancer molecular features and survival by African and European ancestry in the Cancer Genome Atlas. JAMA Oncol 2017;3:1654–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ademuyiwa FO, Tao Y, Luo J, Weilbaecher K, Ma CX. Differences in the mutational landscape of triple-negative breast cancer in African Americans and Caucasians. Breast Cancer Res Treat 2017;161:491–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yuan J, Hu Z, Mahal BA, Zhao SD, Kensler KH, Pi J, et al. Integrated analysis of genetic ancestry and genomic alterations across cancers. Cancer Cell 2018;34:549–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kamran SC, Xie J, Cheung ATM, Mavura MY, Song H, Palapattu EL, et al. Tumor mutations across racial groups in a real-world data registry. JCO Precis Oncol 2021;5:1654–8. [DOI] [PubMed] [Google Scholar]
- 8. Arora K, Tran TN, Kemel Y, Mehine M, Liu YL, Nandakumar S, et al. Genetic ancestry correlates with somatic differences in a real-world clinical cancer sequencing cohort. Cancer Discov 2022;12:2552–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Miyashita M, Bell JSK, Wenric S, Karaesmen E, Rhead B, Kase M, et al. Molecular profiling of a real-world breast cancer cohort with genetically inferred ancestries reveals actionable tumor biology differences between European ancestry and African ancestry patient populations. Breast Cancer Res 2023;25:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Omilian AR, Wei L, Hong CC, Bandera EV, Liu S, Khoury T, et al. Somatic mutations of triple negative breast cancer: a comparison of Black and White women. Breast Cancer Res Treat 2020;182:503–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yamamoto S, Iwakuma T. Regulators of oncogenic mutant TP53 gain of function. Cancers 2018;11:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Alexandrova EM, Mirza SA, Xu S, Schulz-Heddergott R, Marchenko ND, Moll UM. p53 loss-of-heterozygosity is a necessary prerequisite for mutant p53 stabilization and gain-of-function in vivo. Cell Death Dis 2017;8:e2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Billant O, Léon A, Le Guellec S, Friocourt G, Blondel M, Voisset C. The dominant-negative interplay between p53, p63 and p73: a family affair. Oncotarget 2016;7:69549–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pollock NC, Ramroop JR, Hampel H, Troester MA, Conway K, Hu JH, et al. Differences in somatic TP53 mutation type in breast tumors by race and receptor status. Breast Cancer Res Treat 2022;192:639–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sun X, Xue A, Qi T, Chen D, Shi D, Wu Y, et al. Tumor mutational burden is polygenic and genetically associated with complex traits and diseases. Cancer Res 2021;81:1230–9. [DOI] [PubMed] [Google Scholar]
- 16. Wang S, Pitt JJ, Zheng Y, Yoshimatsu TF, Gao G, Sanni A, et al. Germline variants and somatic mutation signature of breast cancer across populations of African and European ancestry in the US and Nigeria. Int J Cancer 2019;145:3321–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Greenblatt MS, Chappuis PO, Bond JP, Hamel N, Foulkes WD. TP53 mutations in breast cancer associated with BRCA1 or BRCA2 germ-line mutations: distinctive spectrum and structural distribution. Cancer Res 2001;61:4092–7. [PubMed] [Google Scholar]
- 18. Manié E, Vincent-Salomon A, Lehmann-Che J, Pierron G, Turpin E, Warcoin M, et al. High frequency of TP53 mutation in BRCA1 and sporadic basal-like carcinomas but not in BRCA1 luminal breast tumors. Cancer Res 2009;69:663–71. [DOI] [PubMed] [Google Scholar]
- 19. Holstege H, Joosse SA, van Oostrom CT, Nederlof PM, de Vries A, Jonkers J. High incidence of protein-truncating TP53 mutations in BRCA1-related breast cancer. Cancer Res 2009;69:3625–33. [DOI] [PubMed] [Google Scholar]
- 20. Natrajan R, Mackay A, Lambros MB, Weigelt B, Wilkerson PM, Manie E, et al. A whole-genome massively parallel sequencing analysis of BRCA1 mutant oestrogen receptor-negative and -positive breast cancers. J Pathol 2012;227:29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen CC, Feng W, Lim PX, Kass EM, Jasin M. Homology-directed repair and the role of BRCA1, BRCA2 and related proteins in genome integrity and cancer. Annu Rev Cancer Biol 2018;2:313–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ramroop JR, Gerber MM, Toland AE. Germline variants impact somatic events during tumorigenesis. Trends Genet 2019;35:515–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA, et al. The somatic mutation profiles of 2,433 breast cancer refines their genomic and transcriptomic landscapes. Nat Commun 2016;7:11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016;534:47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tharin Z, Richard C, Derangère V, Ilie A, Arnould L, Ghiringhelli F, et al. PIK3CA and PIK3R1 tumor mutational landscape in a pan-cancer patient cohort and its association with pathway activation and treatment efficacy. Sci Rep 2023;13:4467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. The TP53 database. Available from: tp53.isb-cgc.org.
- 27. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B 1995;57:289–300. [Google Scholar]
- 28. UCSC Genome Browser. Available from: https://genome.ucsc.edu/.
- 29. GTEx Portal. Available from: https://gtexportal.org/home/.
- 30. RegulomeDB v.2.0.3. Available from: https://www.regulomedb.org/regulome-search/.
- 31. HaploReg v4.0. Available from: https://pubs.broadinstitute.org/mammals/haploreg/haploreg.php.
- 32. NCBI dbSNP. Available from: https://www.ncbi.nlm.nih.gov/snp.
- 33. Puzone R, Pfeffer U. SNP variants at the MAPK1/SETD9 locus 5q11.2 associate with somatic PIK3CA variants in breast cancers. Eur J Hum Genet 2017;25:384–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Smith TR, Liu-Mares W, Van Emburgh BO, Levine EA, Allen GO, Hill JW, et al. Genetic polymorphisms of multiple DNA repair pathways impact age of diagnosis and TP53 mutations in breast cancer. Carcinogenesis 2011;32:1354–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mao JH, Wu D, Perez-Losada J, Jian T, Li Q, Neve RM, et al. Crosstalk between Aurora-A and p53: frequent deletion or downregulation of Aurora-A in tumors from p53 null mice. Cancer Cell 2007;11:161–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kosoy R, Nassir R, Tian CWhite PA, Butler LM, Silva G, et al. Ancestry informative marker sets for determining continental origin and admixture proportions in common populations in America. Hum Mutat 2009;30:69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nassir R, Kosoy R, Tian C, White PA, Butler LM, Silva G, et al. An ancestry informative marker set for determining continental origin: validation and extension using human genome diversity panels. BMC Genet 2009;10:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kidd JR, Friedlaender FR, Speed WC, Pakstis AJ, De LVega FM, et al. Analyses of a set of 128 ancestry informative single-nucleotide polymorphisms in a global set of 119 population samples. Investig Genet 2011;2:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ding YC, Song H, Adamson AW, Schmolze D, Hu D, Huntsman S, et al. Profiling the somatic mutational landscape of breast tumors from Hispanic/Latina women reveals conserved and unique characteristics. Cancer Res 2023;83:2600–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM, et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature 2012;486:405–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pan JW, Zabidi MMA, Ng PS, Meng MY, Hasan SN, Sandey B, et al. The molecular landscape of Asian breast cancers reveals clinically relevant population-specific differences. Nat Commun 2020;11:6433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ragu ME, Lim JMC, Ng PS, Yip CH, Rajadurai P, Teo SH, et al. TP53 somatic mutations in Asian breast cancer are associated with subtype-specific effects. Breast Cancer Res 2023;25:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jia G, Ping J, Guo X, Yang Y, Tao R, Li B, et al. Genome-wide association analyses of breast cancer in women of African ancestry identify new susceptibility loci and improve risk prediction Nat Genet 2024;56:819–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res 2009;19:1655–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dunning AM, Michailidou K, Kuchenbaecker KB, Thompson D, French JD, Beesley J, et al. Breast cancer risk variants at 6q25 display different phenotype associations and regulate ESR1, RMND1, and CCDC170. Nat Genet 2016;48:374–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Michailidou K, Lindström S, Dennis J, Beesley J, Hui S, Kar S, et al. Association analysis identifies 65 new breast cancer risk loci. Nature 2017;551:92–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Milne RL, Kuchenbaecker KB, Michailidou K, Beesley J, Kar S, Lindström S, et al. Identification of ten variants associated with risk of estrogen-receptor-negative breast cancer. Nat Genet 2017;49:1767–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zheng W, Long J, Gao YT, Li C, Zheng Y, Xiang YB, et al. Genome-wide association study identified a new breast cancer susceptibility locus at 6q25.1. Nat Genet 2009;41:324–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hoffman J, Fejerman L, Hu D, Huntsman S, Li M, John EM, et al. Identification of novel common breast cancer risk variants at the 6q25 locus among Latinas. Breast Cancer Res 2019;21:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fejerman L, Ahmadiyeh N, Hu D, Huntsman S, Beckman KB, Caswell JL, et al. Genome-wide association study of breast cancer in Latinas identified novel protective variants on 6q25. Nat Commun 2014;5:5260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Stacey SN, Sulem P, Zanon C, Gudjonsson SA, Thorleifsson G, Helgason A, et al. Ancestry-shift refinement mapping of the C6orf97-ESR1 breast cancer susceptibility locus. PLoS Genet 2010;6:e1001029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fejerman L, Chen GK, Eng C, Huntsman S, Hu D, Williams A, et al. Admixture mapping identifies a locus on 6q25 associated with breast cancer risk in US Latinas. Hum Mol Genet 2012;21:1907–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Siddiq A, Couch FJ, Chen GK, Lindstrom S, Eccles D, Millikan RC, et al. A meta-analysis of genome-wide association studies of breast cancer identifies two novel susceptibility loci at 6q14 and 20q11. Hum Mol Genet 2012;21:5373–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rashkin SR, Graff RE, Kachuri L, Thai KK, Alexeeff SE, Blatchins MA, et al. Pan-cancer study detects genetic risk variants and shared genetic basis in two large cohorts. Nat Commun 2020;11:4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hein R, Maranian M, Hopper JL, Kapuscinski MK, Southey MC, Park DJ, et al. Comparison of 6q25 breast cancer hits from Asian and European genome wide association studies in the breast cancer association consortium (BCAC). PLoS One 2012;7:e42380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ruiz-Narváez EA, Rosenberg L, Yao S, Rotimi CN, Cupples AL, Bandera EV, et al. Fine-mapping of the 6q25 locus identifies a novel SNP associated with breast cancer risk in African-American women. Carcinogenesis 2013;34:287–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cai Q, Wen W, Qu S, Li G, Egan KM, Chen K, et al. Replication and functional genomic analyses of the breast cancer susceptibility locus at 6q25.1 generalize its importance in women of Chinese, Japanese and European ancestry. Cancer Res 2011;71:1344–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ahearn TU, Zhang H, Michailidou K, Milne RL, Bolla MK, Dennis J, et al. Common variants in breast cancer risk loci predispose to distinct tumor subtypes. Breast Cancer Res 2022;24:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Garcia-Closas M, Couch FJ, Lindstrom S, Michailidou K, Schmidt MK, Brook MN, et al. Genome-wide association studies identify four ER negative-specific breast cancer risk loci. Nat Genet 2013;45:392–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Li Z, Spoelstra NS, Sikora MJ, Sams SB, Elias A, Richer JK, et al. Mutual exclusivity of ESR1 and TP53 mutations in endocrine resistant metastatic breast cancer. NPJ Breast Cancer 2022;8:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rasti M, Arabsolghar R, Khatooni Z, Mostafavi-Pour Z. P53 binds to estrogen receptor 1 promoter in human breast cancer cells. Pathol Oncol Res 2012;18:169–75. [DOI] [PubMed] [Google Scholar]
- 62. Cheng S, Douglas-Jones A, Yang X, Mansel RE, Jiang WG. Transforming acidic coiled-coil-containing protein 2 (TACC2) in human breast cancer, expression pattern and clinical/prognostic relevance. Cancer Genomics Proteomics 2010;7:67–73. [PubMed] [Google Scholar]
- 63. Onodera Y, Takagi K, Miki Y, Takayama K, Shibahara Y, Watanabe M, et al. TACC2 (transforming acidic coiled-coil protein 2) in breast carcinoma as a potent prognostic predictor associated with cell proliferation. Cancer Med 2016;5:1973–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Purrington KS, Slettedahl S, Bolla MK, Michailidou K, Czene K, Nevanlinna H, et al. Genetic variation in mitotic regulatory pathway genes is associated with breast tumor grade. Hum Mol Genet 2014;23:6034–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lawrenson K, Song F, Hazelett DJ, Kar SP, Tyrer J, Phelan CM, et al. Genome-wide association studies identify susceptibility loci for epithelial ovarian cancer in east Asian women. Gynecol Oncol 2019;153:343–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. von Thun A, Birtwistle M, Kalna G, Grindlay J, Strachan D, Kolch W, et al. ERK2 drives tumour cell migration in three-dimensional microenvironments by suppressing expression of Rab17 and liprin-B2. J Cell Sci 2012;125:1465–77. [DOI] [PubMed] [Google Scholar]
- 67. Edwards K, Lydyard PM, Kulikova N, Tsertsvadze T, Volpi EV, Chiorazzi N, et al. The role of CD180 in hematological malignancies and inflammatory disorders. Mol Med 2023;29:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Henderson BE, Lee NH, Seewaldt V, Shen H. The influence of race and ethnicity on the biology of cancer. Nat Rev Cancer 2012;12:648–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Gomez F, Griffith M, Griffith OL. Genetic ancestry correlations with driver mutations suggest complex interactions between somatic and germline variation in cancer. Cancer Discov 2021;11:534–6. [DOI] [PubMed] [Google Scholar]
- 70. Carrot-Zhang J, Soca-Chafre G, Patterson N, Thorner AR, Nag A, Watson J, et al. Genetic ancestry contributes to somatic mutations in lung cancer from admixed Latin American populations. Cancer Discov 2021;11:591–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kytola V, Topaloglu U, Miller LD, Bitting RL, Goodman MM, Agostino RBD Jr, et al. Mutational landscapes of smoking-related cancers in Caucasians and African Americans: precision oncology perspectives at Wake Forest Baptist Comprehensive Cancer Center. Theranostics 2017;7:2914–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. van Beek EJAH, Hernandez JM, Goldman DA, Davis JL, McLaughlin K, Ripley RT, et al. Rates of TP53 mutation are significantly elevated in African American Patients with gastric cancer. Ann Surg Oncol 2018;25:2027–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Cavagna RO, Pinto IA, de Paula FE, Berardinelli GN, Sant'Anna D, Santana I, et al. Disruptive and truncating TP53 mutations are associated with African-ancestry and worse prognosis in Brazilian patients with lung adenocarcinoma. Pathobiology 2023;90:344–55. [DOI] [PubMed] [Google Scholar]
- 74. Mezghani N, Yao A, Vasilyeva D, Kaplan N, Shackelford A, Yoon A, et al. Molecular subtypes of head and neck cancer in patients of African ancestry. Clin Cancer Res 2023;29:910–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Peak T, Spiess PE, Li R, Grivas P, Necchi A, Pavlick D, et al. Comparative genomic landscape of urothelial carcinoma of the bladder among patients of East and South Asian genomic ancestry. Oncologist 2023;28:e910–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Myer PA, Lee JK, Madison RW, Pradhan K, Newberg JY, Isasi CR, et al. The genomics of colorectal cancer in populations with African and European ancestry. Cancer Discov 2022;12:1282–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Information: References for Supplemental Table S5
Supplemental Tables S1-S35
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Data Availability Statement
The majority of data generated or analyzed during this study are included in this published article in Supplementary Tables, in TCGA, dbGAP and/or the following data repositories as listed below. TCGA tumor mutation data and SNV genotyping data are available in dbGAP under accession numbers phs001687.v1.p1, phs000178.v11.p8, and phs002387.v1.p1. METABRIC sequencing data of tumors and SNV genotyping data are available on the European Genome-Phenome archive using accession numbers EGAD0001000164, EGAS00000000083, EGAD00010000158, EGAD00010000266, EGAS00001004518, and EGAD00001006399. The Welcome Trust Sanger Institute data are available in the European Genome-Phenome archive using accessing number EGAS00001001178 and EGAD0010000915. Sequencing data and processed genomic data from the Nigerian breast cancer cases are in dbGAP under study accession number phs001687.v1.p1. Tumor/normal whole-exome sequencing (WES) and RNA-sequencing data and accompanying phenotypic and clinical/histologic data for the COH Latina Breast Cancer Study are deposited in dbGAP (dbGaP Study Accession: phs003218; ref. 39). MyBrCa WES and shallow whole genome sequencing (sWGS) files are available on the European Genome-phenome Archive under the study accession number EGAS00001004518. Access to controlled patient data will require the approval of the MyBrCa Tumour Genomics Data Access Committee upon request to genetics@cancerresearch.my. Sequence and genotyping data for the Banerji and colleagues study (40) are available in dbGAP under accession number phs000369.v1.p1. Summary-level statistics genotyping data for the AABCG study are available at GWAS Catalog (accession number: GCST90296719, GCST90296720, GCST90296721, and GCST90296722). B-CAUSE TNBC sequencing data are in the process being deposited into dbGaP with accession number pending.