

Abstract
Potassium trifluoroborates have gained significant utility as coupling partners in organic synthesis, particularly in the Suzuki-Miyaura coupling reaction. Recently, they have also been used as radical precursors under oxidative conditions to generate carbon-centered radicals. These versatile reagents have found new applications in photoredox catalysis, including radical substitution, conjugate addition reactions, and transition metal dual catalysis. In addition, this photomediated redox neutral process has enabled radical-radical coupling with persistent radicals in the absence of a metal, and this process remains to be fully explored. In this study, we report the radical-radical coupling of benzylic potassium trifluoroborate salts with isolated acyl azolium triflates, which are persistent radical precursors. The reaction is catalyzed by an organic photocatalyst and forms isolable tertiary alcohol species. These compounds can be transformed into a range of substituted ketone products by simple treatment with a mild base.
Keywords: Potassium trifluoroborate, photoredox, acyl azolium, radical-radical coupling
Graphical Abstract
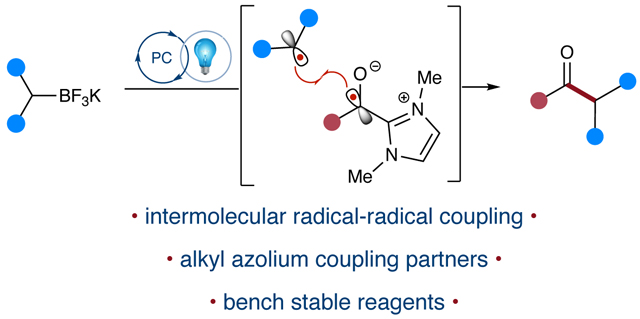
Organoboron compounds have long been recognized for providing chemists with unique strategies for carbon–carbon bond formation.(1) Recently, these methodologies have undergone significant growth, resulting in the discovery of new two-electron nucleophilic displacement, boron enolate, and transition metal coupling reactions.(2) The Suzuki-Miyaura coupling reaction has contributed significantly to the explosion of boronic acid and ester diversity, providing a rapid and efficient method for assembling diverse chemical libraries.(3) Potassium trifluoroborate salts, first reported by Chambers and coworkers in 1960, have become widely adopted bench-stable reagents.(4) Vedejs accelerated this adoption by demonstrating the facile transformation of boronic acids to trifluoroborate salts upon treatment with inexpensive KHF2.(5) More recently, the renaissance of free radical methodology development has capitalized on the unique properties of organoboron compounds, offering new directions in the field of boron chemistry.
Boronic acid derivatives are widely used as radical precursors in various chemical reactions including conjugate addition reactions, transition metal-mediated coupling reactions, and radical-radical coupling reactions (Figure 1). Baran was among the first to report the oxidation of arylboronic acids to generate aryl radicals, which then participated in Minisci reactions with electron-deficient heteroarenes (Fig. 1A).(6) Potassium trifluoroborates were reported by Fensterbank in 2010 as radical precursors, then subsequently employed by Baran and Molander to access aryl, alkyl, and alkoxy radicals.(7) Recent advances in photoredox catalysis have provided new routes to radicals, including boron-containing precursors.(8) Akita and colleagues reported the photocatalytic oxidation of a variety of borates to generate the corresponding carbon-centered radicals (Fig. 1B).(9) These organoboron-derived radicals then reacted with TEMPO and electron-deficient alkenes to form C–O and C–C bonds. Later, this work was extended to the hydroalkoxymethylation of alkenes.(10)
Figure 1.

Reaction Plan Overview: A) Boronic acid and potassium trifluoroborate as radical precursors in heteroarylation. B) Application in photoredox catalysis. C) Transition metal-photoredox dual catalysis enabled ketone synthesis. D) Radical-radical coupling of potassium triflluoroborates and isolated acyl azoliums.
Photoredox catalysis offers kinetic control over radical generation and provides a unique opportunity to create new C–C bond-forming methodologies.(11) This controlled generation of radicals also enables dual catalytic systems, as demonstrated by the work of the Sanford group and others through photoredox-transition metal dual catalysis.(12) This approach tackles a long-standing problem by eliminating the rate-limiting step of transmetalation, which hinders the ability of CSP3–CSP2/CSP3 couplings in Suzuki-Miyaura reactions.(13) The reaction pathway proceeds via oxidative addition into a variety of C–X electrophiles followed by radical capture via single electron transmetalation of the boron-derived carbon radical and reductive elimination to yield the desired C–C coupled product. An application related to this work is the nickel/photoredox dual catalytic system applied to acyl chlorides and potassium organotrifluoroborate salts to produce α-alkoxyketones and dialkyl ketones (Fig. 1C).(14)
Carbon-centered radicals are generally highly reactive and short-lived.(15) Nevertheless, conjugate acceptors and metal catalysts offer numerous opportunities for radical capture and subsequent transformations. On the other hand, direct radical-radical coupling of carbon-centered radicals presents a challenge because these organic radicals tend to undergo self-coupling and off - target degradation pathways. (16) However, under conditions where a) radicals are generated at equivalent rates, such as in photoredox-mediated reactions, and b) they exhibit variable radical lifetimes with one radical being persistent and the other transient, radical-radical coupling is favorable. This is because the persistent radical coupling partner increases in concentration over time, and subsequent generation of the transient radical leads to expedient cross-coupling. This phenomenon is known as the persistent radical effect.(17)
The recent development of persistent ketyl radicals has facilitated the development of a variety of novel radical-radical coupling methodologies for accessing tertiary alcohols and ketones.(18) Among these methods, the persistent azolium-derived ketyl radical intermediate (5b) has demonstrated remarkable utility in radical-radical coupling reactions. In 2019, Ohmiya and co-workers accessed this intermediate through the direct reduction of phthalimide-derived redox-active esters by the strongly reducing Breslow intermediate.(19) The resulting radical pair undergoes C–C bond formation via radical-radical coupling. To complement this approach, our group has developed methodologies to capitalize on the direct reduction of acyl azoliums and the corresponding persistent acyl azolium radical intermediates. Our reductive approach employed photoredox and N-heterocyclic carbene (NHC) dual catalysis to couple N-acyl imidazoles and Hantzsch esters to form ketones.(20) Shortly after, we reported the applications of isolated acyl azoliums and expanded our methods to include both stoichiometric ester azoliums and three-component radical-relay reactions, which employed potassium trifluoroborates as radical precursors in the photoredox-mediated alkoxycarbonylation of trifluoroborates and synthesis of γ-aryloxy ketones.(21) The reactivity of the azolium-derived ketyl radical has also been developed by other groups, leading to a variety of novel transformations including radical relay, 𝛼-heteroatom functionalization, and triplet ketone methodologies.(22) Herein, we utilize a stoichiometric acyl azolium to facilitate radical-radical coupling with potassium trifluoroborate-derived alkyl radicals in a photoredox-mediated process to form ketone products.
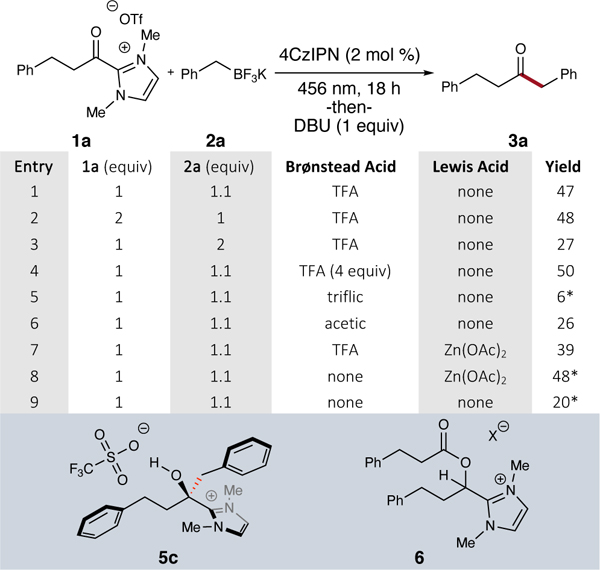
Our initial investigations into this coupling utilized hydrocinnamyl acyl azolium (1a) and benzyl potassium trifluoroborate (2a) to afford the benzylated ketone product (3a, Table 1). We initially investigated the reaction with a (1:1.1,1a:2a) stoichiometry to enable monitoring reaction by disappearance of (1a) via LC-MS analysis. We observed that the borates were undetectable or underwent degradation under this analysis. The reaction was monitored via LC-MS for the appearance of tetrahedral intermediate (5c), which was later confirmed by single crystal X-ray crystallography. This intermediate undergoes rapid transformation to the ketone product upon treatment with DBU. The selection of 4CzIPN as photocatalyst and MeCN as reaction solvents were informed by our previous studies (Entry 9). Monitoring the reaction by LC-MS revealed a byproduct (6) whose structure was assigned as the transesterification/secondary alcohol based on MS data and NMR spectroscopic characterization of a related compound (6, Figure 4). Given that acyl azoliums are competent acylating agents in the presence of alcohols, this was not surprising, but clearly shutting this pathway down was deemed critical. We attempted to attenuate the nucleophilicity of the key ketyl radical oxyanion with Brønsted and Lewis acid additives. Increases in yield were observed with trifluoroacetic acid (TFA) and Zn(OAc)2; however, the addition of Zn(OAc)2 caused the formation of an undesirable gel over the course of the reaction. We proceeded with optimization using TFA (Entry 1) to maintain reaction homogeneity, isolating 47% of ketone 3a using these conditions. To optimize further, we considered that the process may be impacted by the acidity or concentration of the acid additive. TFA outperformed triflic and acetic acids (Entry 5 and Entry 6), while 4 equivalents of TFA (Entry 4) provided a comparable yield at 50%. Furthermore, modulation of the equivalency was explored: 2 equivalents of 1a (Entry 2) provided similar yields to our initial stoichiometry, yet 2 equivalents of 2a (Entry 3) proved deleterious to the yield. The reaction was determined to proceed sufficiently well in a slight excess of borate in the presence of stoichiometric Brønsted acid additive and 4CzIPN photocatalyst at 0.2 M in MeCN. With optimized conditions in hand, we next sought to explore the scope of trifluoroborate and azolium coupling partners.(23)
Table 1.
Optimization of reaction conditions. 1 equiv of Brønsted and Lewis acid additives employed unless noted otherwise. Reactions performed at 0.2 M. *1H NMR yield of product vs 1,3,5-trimethoxybenzene in CDCl3.
|
A variety of substituted benzyl potassium trifluoroborates were employed with three different acyl azoliums to access hydrocinnamyl (3a-f),(24) cyclohexyl (3g-3j),(25) and benzoyl products (3k-3p, Scheme 1).(26) The structure of product 3c was confirmed by single crystal X-ray diffraction (see SI 34). These studies indicated that alkyl- and aryl-substituted benzylic trifluoroborates (3b-3d, 3h, 3l-3n) performed comparably to the standard unsubstituted products (3a, 3g, 3k). However, the electron-rich p-methoxy substituted salts performed poorly in the reaction (3e, 3i, 3o) with the dimerized p-methoxy benzyl product observed by GC-MS indicating a possible electronic mismatch of the azolium and p-methoxy benzyl radicals.(27) The π-withdrawing p-CO2Me-substituted trifluoroborate provided the ketone products in reduced yields (3f, 3j, 3p), and significant azolium starting material remained after 24 hours, suggesting the absence of redox reactivity, and was attributed to a reduced rate of oxidation as the inductive effect of the electron deficient arene increases the oxidation potential. Fortunately, transitioning to the secondary potassium trifluoroborates provided improved yields of the products (3q-3s) in 55%, 53% and 66% yields, respectively. Furthermore, ester-bearing 2º borates offering a handle for further diversification provided moderate yields of 43%, 52%, and 62% for (3t, 3u, 3v). Finally, naphthylic potassium trifluoroborates were utilized to explore the effects of the resonance-stabilized radical in the synthesis of (3w, 3x) and resulted in a notable reduction in yield in comparison to (3a) and (3m). To broaden the scope of the reaction, the non-stabilized cyclohexyl, cyclopentyl, and tert-butyl aliphatic potassium trifluoroborates were subjected to the standard reaction conditions. However, the reactions failed to yield the desired intermediates. These examples in addition to the poor yielding p-methoxy and p-CO2ME substrates demonstrate the challenges in the selection of coupling partners for radical-radical coupling which are a less explored class of reactions in comparison to radical addition reactions with proposed trends in radical reactivity.(28)
Scheme 1.

Scope of potassium trifluoroborate salts. Reaction conditions: azolium (1 equiv, 0.5 mmol), potassium trifluoroborate (1.1 equiv), TFA (1 equiv), 4CzIPN (2 mol %), MeCN (0.2 M), irradiated w/456 nm LEDs for 24 -or- 48 h, then DBU (1 equiv).
Following the investigation of the potassium trifluoroborate substrates, we sought to explore a variety of different acyl azolium coupling partners (Scheme 2). Initially, a variety of arene substitution patterns were employed in the reaction with alkyl (4a), electron-poor (4b-4e), and electron-rich (4f, 4g) substituents well tolerated in the reaction. It is notable that the para-methoxy substituted azolium (4g) performed well in the reaction, achieving 73% yield, albeit requiring 48 h to reach completion. The extended π-system of the naphthalene-substituted azolium (4h) resulted in a reduced yield in comparison to (3k). The alkyl substituents of the 2º aliphatic azolium were expanded to include cyclopentyl (4i) and phenpropyl (4j) which performed in appreciable yields. Finally, we employed FDA-approved drug gemfibrozil to synthesize the 3º acyl azolium (1n) that was coupled with the 2º potassium trifluoroborate (2h) to furnish the ester-bearing analog (4m).
Scheme 2.

Scope of acyl azolium triflate salts. Reaction conditions: azolium (1 equiv, 0.5 mmol), potassium trifluoroborate (1.1 equiv), TFA (1 equiv), 4CzIPN (2 mol %), MeCN (0.2 M), irradiated w/456 nm LEDs for 24 -or- 48 h, then DBU (1 equiv).
The proposed mechanism of this transformation proceeds through a reductive quenching process where the photocatalyst 4CzIPN is excited by blue light to 4CzIPN* (Figure 2). The activated photocatalyst subsequently oxidizes the benzyl potassium trifluoroborate (2a) and liberates the transient benzyl radical (5a). The resulting photocatalyst radical anion, 4CzIPN•–, performs a single electron reduction of acyl azolium (2b) to access the persistent azolium radical (5b). This reduction may occur by single electron transfer (SET) followed by fast protonation from TFA, or by a potential proton coupled electron transfer (PCET) process to yield (5b) and TFA–. It is difficult to fully determine whether SET and protonation or PCET is the dominant pathway, as both processes may be operative.(29) The two resulting radical partners then undergo radical-radical coupling to form (5c), a tertiary alcohol intermediate that was isolated and confirmed by single crystal X-ray crystallography.(21b) This stable intermediate is then transformed to the desired ketone product by treatment with DBU, liberating the neutral azolium (5d) and the desired ketone product (3a). Importantly, the experiment was run in the absence of a photocatalyst and no intermediate formation and subsequent product was observed. Addition of Brønsted acid leads to a divergence in reaction profile, primarily the complete disappearance of (6), a major byproduct (based on HRMS analysis, see SI 31 for details).(30) The formation of the byproduct is consistent with a reduction to the ketyl radical anion followed by transesterification and a hydrogen atom abstraction or an electron transfer/proton transfer event. A radical trapping experiment was also performed with 3 equivalents of TEMPO, eliminating the formation of the intermediate (5d) and corresponding product (3a). Additionally, trapping of (5e) the benzyl adduct was observed by high-resolution mass spectrometry.
Figure 2.

Proposed mechanism for the 4CzIPN-dependent radical-radical coupling of benzyl potassium trifluoroborate salts and aliphatic acyl azoliums. Proposed structure of ester byproduct observed by LC-MS when reaction run in the absence of TFA. TEMPO trapping adduct 5f observed by HRMS, TEMPO (3 equiv) added to standard reaction conditions.
In summary, this work describes the use of potassium trifluoroborate salts as radical precursors for use in radical-radical coupling with persistent azolium-derived ketyl radicals. This reaction utilizes a pair of readily available and bench-stable salts derived from organohalides and esters, an easily-accessed organophotocatalyst, and visible light. By taking advantage of the persistent radical character of azolium intermediates in ketone synthesis, we have employed a new coupling partner class with well-established potassium trifluoroborates. This catalytic method provides a metal-free route to a variety of substituted ketone products, including drug derivatives. Overall, the development of this new transformation of potassium trifluoroborate salts broadens their utility in organic chemistry and further substantiates the early fundamental studies by the pioneers of boron chemistry.
Supplementary Material
Acknowledgment
The authors thank Northwestern University for support of this work. We also thank Charlotte Stern (NU) for X-ray crystallographic analysis.
Funding Information
We gratefully acknowledge support from the National Institute of General Medical Sciences (NIH) for support of this work (R35 GM136440). D.Y. and E.J.F. thank Northwestern for Undergraduate Research Grants. D.Y. thanks the Chemistry of Life Processes Institute at Northwestern for support in the form of the Lambert Fellowship.
Footnotes
Supporting Information
See link
Conflict of Interest
The authors declare no conflict of interest.
References and Notes
- (1) (a).Matteson DS; Mah RW H. J. Am. Chem. Soc 1963, 85,2599–2603; [Google Scholar]; (b) Evans DA; Vogel E; Nelson JV J. Am. Chem. Soc 1979, 101, 6120–6123; [Google Scholar]; (c) Miyaura N; Yanagi T; Suzuki A. Synth. Commun 1981, 11, 513–519. [Google Scholar]
- (2) (a).Cowden CJ; Paterson I, Asymmetric Aldol Reactions Using Boron Enolates. In Organic Reactions, ed.; 2004; pp 1–200; [Google Scholar]; (b) Doucet H. Eur. J. Org. Chem 2008, 2008, 2013–2030; [Google Scholar]; (c) Sandford C; Aggarwal VK Chem. Commun 2017, 53, 5481–5494; [DOI] [PubMed] [Google Scholar]; (d) Hall DG Chem. Soc. Rev 2019, 48, 3475–3496; [DOI] [PubMed] [Google Scholar]; (e) Abiko A, The Chemistry of Boron Enolate: Synthesis and Reactivity. In PATAI’S Chemistry of Functional Groups, ed.; 2020; pp 1–75. [Google Scholar]
- (3) (a).Miyaura N; Suzuki A. Chem. Rev 1995, 95, 2457–2483; [Google Scholar]; (b) Dombrowski AW; Aguirre AL; Shrestha A; Sarris KA; Wang YJ Org. Chem 2022, 87, 1880–1897. [DOI] [PubMed] [Google Scholar]
- (4) (a).Chambers RD; Clark HC; Willis CJ J. Am. Chem. Soc 1960, 82, 5298–5301; [Google Scholar]; (b) Molander GA; Sandrock DL Curr. Opin. Drug Discov. Dev 2009, 12, 811–23; [PMC free article] [PubMed] [Google Scholar]; (c) Lennox AJJ; Lloyd-Jones G [Google Scholar]; C. Chem. Soc. Rev 2014, 43, 412–443. [DOI] [PubMed] [Google Scholar]
- (5) (a).Vedejs E; Chapman RW; Fields SC; Lin S; Schrimpf MR J. Org. Chem 1995, 60, 3020–3027; [Google Scholar]; (b) Clay JM; Vedejs EJ Am. Chem. Soc 2005, 127, 5766–5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Seiple IB; Su S; Rodriguez RA; Gianatassio R; Fujiwara Y; Sobel AL; Baran PS J. Am. Chem. Soc 2010, 132, 13194–13196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7) (a).Sorin G; Martinez Mallorquin R; Contie Y; Baralle A; Malacria M; Goddard J-P; Fensterbank L. Angew. Chem. Int. Ed 2010, 49, 8721–8723; [DOI] [PubMed] [Google Scholar]; (b) Fujiwara Y; Domingo V; Seiple IB; Gianatassio R; Del Bel M; Baran PS J. Am. Chem. Soc 2011, 133, 3292–3295; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Molander GA; Colombel V; Braz VA Org. Lett 2011, 13, 1852–1855; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Lockner JW; Dixon DD; Risgaard R; Baran PS Org. Lett 2011, 13, 5628–5631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Matsui JK; Lang SB; Heitz DR; Molander GA ACS Catal. 2017, 7, 2563–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Yasu Y; Koike T; Akita M. Adv. Synth. Catal 2012, 354, 3414–3420. [Google Scholar]
- (10).Miyazawa K; Yasu Y; Koike T; Akita M. Chem. Commun 2013, 49, 7249–7251. [DOI] [PubMed] [Google Scholar]
- (11).Xie J; Jin H; Hashmi ASK Chem. Soc. Rev 2017, 46, 5193–5203. [DOI] [PubMed] [Google Scholar]
- (12) (a).Kalyani D; McMurtrey KB; Neufeldt SR; Sanford MS J. Am. Chem. Soc 2011, 133, 18566–18569; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ye Y; Sanford MS J. Am. Chem. Soc 2012, 134, 9034–9037; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Tellis JC; Primer DN; Molander GA Science 2014, 345, 433–436; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zuo Z; Ahneman DT; Chu L; Terrett JA; Doyle AG; MacMillan DWC Science 2014, 345, 437–440; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Tellis JC; Kelly CB; Primer DN; Jouffroy M; Patel NR; Molander GA Acc. Chem. Res 2016, 49, 1429–1439; [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Skubi KL; Blum TR; Yoon TP Chem. Rev 2016, 116, 10035–10074; [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Chan AY; Perry IB; Bissonnette NB; Buksh BF; Edwards GA; Frye LI; Garry OL; Lavagnino MN; Li BX; Liang Y; Mao E; Millet A; Oakley JV; Reed NL; Sakai HA; Seath CP; MacMillan DWC Chem. Rev 2022, 122, 1485–1542. [DOI] [PubMed] [Google Scholar]
- (13).Hartwig JF; Collman JP, Organotransition metal chemistry: from bonding to catalysis. ed.; Springer: 2010; Vol. 1. [Google Scholar]
- (14) (a).Amani J; Sodagar E; Molander GA Org. Lett 2016, 18, 732–735; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Amani J; Molander GA J. Org. Chem 2017, 82, 1856–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Studer A; Curran DP Angew. Chem. Int. Ed 2016, 55, 58–102. [DOI] [PubMed] [Google Scholar]
- (16).Fischer H. Chem. Rev 2001, 101, 3581–3610. [DOI] [PubMed] [Google Scholar]
- (17) (a).Daikh BE; Finke RG J. Am. Chem. Soc 1992, 114, 2938–2943; [Google Scholar]; (b) Leifert D; Studer A. Angew. Chem. Int. Ed 2020, 59, 74–108. [DOI] [PubMed] [Google Scholar]
- (18) (a).Zhang L; Chu Y; Ma P; Zhao S; Li Q; Chen B; Hong X; Sun J. Org. Biomol. Chem 2020, 18, 1073–1077; [DOI] [PubMed] [Google Scholar]; (b) Ota K; Nagao K; Ohmiya H. Org. Lett 2021, 23, 4420–4425; [DOI] [PubMed] [Google Scholar]; (c) Jiang H-L; Yang Y-H; He Y-H; Guan Z. Org. Lett 2022, 24, 4258–4263; [DOI] [PubMed] [Google Scholar]; (d) Bay AV; Scheidt KA Trends Chem. 2022, 4, 277–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ishii T; Kakeno Y; Nagao K; Ohmiya HJ Am. Chem. Soc 2019, 141, 3854–3858. [DOI] [PubMed] [Google Scholar]
- (20) (a).Bay AV; Fitzpatrick KP; Betori RC; Scheidt KA Angew. Chem. Int. Ed 2020, 59, 9143–9148; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bay AV; Fitzpatrick KP; González-Montiel GA; Farah AO; Cheong PH-Y; Scheidt KA Angew. Chem. Int. Ed 2021, 60, 17925–17931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21) (a).Bayly AA; McDonald BR; Mrksich M; Scheidt KA Proc. Natl. Acad. Sci. U.S.A 2020, 117, 13261–13266; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rourke MW, C.; Schull C; Scheidt K,. ChemRxiv 2022; [Google Scholar]; (c) Zhu JL; Schull CR; Tam AT; Rentería-Gómez Á; Gogoi AR; Gutierrez O; Scheidt KA J. Am. Chem. Soc 2023, 145, 1535–1541; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zhu JL; Scheidt KA Tetrahedron 2021, 92; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Wang P; Fitzpatrick KP; Scheidt KA Adv. Synth. Catal 2022, 364, 518–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22) (a).Mavroskoufis A; Rajes K; Golz P; Agrawal A; Ruß V; Götze JP; Hopkinson MN Angew. Chem. Int. Ed 2020, 59, 3190–3194; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Meng Q-Y; Döben N; Studer A. Angew. Chem. Int. Ed 2020, 59, 19956–19960; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Liu K; Studer AJ Am. Chem. Soc 2021, 143, 4903–4909; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Meng Q-Y; Lezius L; Studer A. Nat. Commun 2021, 12, 1–8; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Sato Y; Goto Y; Nakamura K; Miyamoto Y; Sumida Y; Ohmiya H. ACS Catal. 2021, 11, 12886–12892; [Google Scholar]; (f) Ren S-C; Lv W-X; Yang X; Yan J-L; Xu J; Wang F-X; Hao L; Chai H; Jin Z; Chi YR ACS Catal. 2021, 11, 2925–2934; [Google Scholar]; (g) Mavroskoufis A; Rieck A; Hopkinson MN Tetrahedron 2021, 100, 132497; [Google Scholar]; (h) Wang X; Zhu B; Liu Y; Wang Q. ACS Catal. 2022, 12, 2522–2531. [Google Scholar]
- (23).General procedure for the coupling of potassium trifluoroborates and acyl azolium triflates: In an N2 inert atmosphere glovebox, 2,4,5,6-Tetrakis(9H-carbazol-9-yl), isophthalonitrile (4CzIPN, 2 mol %), the corresponding acyl azolium (1 equiv., 0.5 mmol), and R-BF3K salt (1.1 equiv), were sequentially added to a flame dried 2-dram vial fitted with a magnetic stir bar and suspended in anhydrous freeze-pump-thaw degassed acetonitrile (MeCN, 2.5 mL, 0.2 M). Then, 2,2,2-trifluoroacetic acid (TFA, 1.0 equiv) was added to the reaction mixture. The vial was sealed and irradiated with stirring using four Kessil PhotoReaction PR 160L 456 nm LEDS at 100 % intensity radially arranged from the vial. After 18–24 hours of irradiation, the vial was removed from the light source, and the reaction was monitored by LC-MS for the disappearance of the acyl azolium and formation of the subsequent tetrahedral intermediate analogous to 5c. Then, 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU, 1.0 equiv) was added, accompanied by a darkening of the reaction mixture under stirring. The disappearance of the tetrahedral intermediate was monitored by LC-MS (typically 10 minutes). The reaction was diluted with ethyl acetate (12.5 mL), washed once with an equivalent volume of saturated aqueous ammonium chloride solution, and the organic layer dried via brine wash. Aqueous layers were back extracted with ethyl acetate, and the combined organic layers were dried over sodium sulfate, concentrated in vacuo, and purified by flash column chromatography (20:1 Hexanes: EtOAc) to afford the desired ketone product.
- (24).Analytical data for 1,4-diphenylbutan-2-one (3a). 1H NMR (500 MHz, CDCl3) δ 7.35 – 7.30 (m, 2H), 7.29 – 7.23 (m, 3H), 7.23 – 7.16 (m, 3H), 7.13 (d, J = 6.8 Hz, 2H), 3.67 (s, 2H), 2.88 (t, J = 7.2 Hz, 2H), 2.77 (d, J = 7.2 Hz, 2H).13C NMR (126 MHz, CDCl3) δ 207.6, 141.1, 134.2, 129.5, 128.9, 128.6, 128.5, 127.2, 126.2, 77.4, 77.2, 76.9, 50.5, 43.6, 29.9.
- (25).Analytical data for 1-cyclohexyl-2-phenylethan-1-one (3g). 1H NMR (500 MHz, CDCl3) δ 7.34 – 7.29 (m, 2H), 7.28 – 7.23 (m, 1H), 7.20 – 7.14 (m, 2H), 3.73 (s, 2H), 2.46 (tt, J = 11.5, 3.5 Hz, 1H), 1.86 – 1.72 (m, 4H), 1.69 – 1.62 (m, 1H), 1.41 – 1.31 (m, 2H), 1.31 – 1.13 (m, 3H).13C NMR (126 MHz, CDCl3) δ 211.4, 134.6, 129.6, 128.7, 127.0, 50.3, 48.0, 28.7, 26.0, 25.8.
- (26).1,2-diphenylethan-1-one (3k). 1H NMR (500 MHz, CDCl3) δ 8.00 – 7.95 (m, 2H), 7.55 – 7.48 (m, 1H), 7.45 – 7.38 (m, 2H), 7.32 – 7.26 (m, 2H), 7.24 – 7.18 (m, 3H), 4.25 (s, 1H).13C NMR (126 MHz, CDCl3) δ 197.7, 136.7, 134.7, 133.3, 129.6, 128.8, 128.8, 128.7, 127.0, 45.6.
- (27).De Vleeschouwer F; Van Speybroeck V; Waroquier M; Geerlings P; De Proft F. Org. Lett 2007, 9, 2721–2724. [DOI] [PubMed] [Google Scholar]
- (28).Parsaee F; Senarathna MC; Kannangara PB; Alexander SN; Arche PDE; Welin ER Nature Reviews Chemistry 2021, 5, 486–499. [DOI] [PubMed] [Google Scholar]
- (29).Tarantino KT; Liu P; Knowles RR J. Am. Chem. Soc 2013, 135, 10022–10025. [DOI] [PubMed] [Google Scholar]
- (30).Du J; Espelt LR; Guzei IA; Yoon TP Chem. Sci 2011, 2, 2115–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.