

Abstract
This study provides a detailed understanding of the preclinical pharmacokinetics and metabolism of ELP‐004, an osteoclast inhibitor in development for the treatment of bone erosion. Current treatments for arthritis, including biological disease‐modifying antirheumatic drugs, are not well‐tolerated in a substantial subset of arthritis patients and are expensive; therefore, new treatments are needed. Pharmacokinetic parameters of ELP‐004 were tested with intravenous, oral, and subcutaneous administration and found to be rapidly absorbed and distributed. We found that ELP‐004 was non‐mutagenic, did not induce chromosome aberrations, non‐cardiotoxic, and had minimal off‐target effects. Using in vitro hepatic systems, we found that ELP‐004 is primarily metabolized by CYP1A2 and CYP2B6 and predicted metabolic pathways were identified. Finally, we show that ELP‐004 inhibits osteoclast differentiation without suppressing overall T‐cell function. These preclinical data will inform future development of an oral compound as well as in vivo efficacy studies in mice.
Keywords: CYP450, metabolism, mice, osteoclast, pharmacokinetics, preclinical
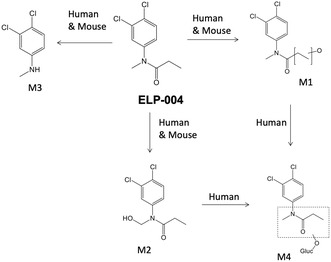
In vitro metabolic pathways of ELP‐004.
Abbreviations
- AA
peak area with automatic integration
- AUC
area under the curve
- AV
number of scans averaged
- BBB
blood–brain barrier
- bDMARDS
biological disease‐modifying antirheumatic drugs
- C 0
initial concentration
- C max
maximum concentration
- CIA
collagen‐induced arthritis
- CL
clearance
- CNS
central nervous system
- CYP
cytochrome P450
- DCA
3,4‐dichloroaniline
- DCPA
3,4‐dichloropropionanilide
- DTT
dithiothreitol
- ESI
electron spray ionization
- F
bioavailability
- HPbCD
hydroxypropyl‐beta‐cyclodextrin
- HLS9
human liver S9 fraction
- IC50
concentration that produces 50% inhibition
- ICIS
Interactive Chemical Information System peak detection and integration algorithm
- IL‐6
interleukin 6
- ITMS
ion trap mass spectrometry
- IV
intravenous
- LC
liquid chromatography
- LC–MS/MS
liquid chromatography–tandem mass spectrometry
- M
metabolite
- m/z
mass‐to‐charge ratio
- MLS9
mouse liver S9 fraction
- MS
mass spectrometry
- nH
Hill coefficients
- NL
normalization intensity level
- PAPS
3′‐phosphoadenosine‐5′‐phosphosulfate
- PK
pharmacokinetics
- PXR
pregnane X receptor
- RA
rheumatoid arthritis
- RDB
rings plus double bonds
- RT
retention time
- t max
time at maximum concentration
- TNFα
tumor necrosis factor‐alpha
- UD
unchanged drug
- UDPAG
uridine 5′‐diphosphate N‐acetyl‐glucosamine
- UDPGA
uridine 5′‐diphosphate glucuronic acid
- V ss
steady‐state volume of distribution
- V z
terminal volume of distribution
1. INTRODUCTION
Bone erosion is associated with several diseases including rheumatoid arthritis (RA), multiple myeloma, and other metastatic cancers. 1 , 2 , 3 RA is a chronic autoimmune disease associated with debilitating and painful joint destruction 3 which impacts quality of life. 4 , 5 Options to modulate disease progression include anti‐metabolites, steroids, and biologics, such as TNFα‐blocking molecules, all of which have significant efficacy tempered by debilitating side effects, particularly over the long term. 6 The biological disease‐modifying antirheumatic drugs (bDMARDs), for example, etanercept, inhibit the activity of key inflammatory cytokines, tumor necrosis factor‐alpha (TNFα), and interleukin 6 (IL‐6), 7 , 8 , 9 leading to increased infections and other symptoms indicating intolerance to bDMARDs. 7 , 10 Therefore, additional treatments without debilitating consequences are needed in the clinic to treat patients with RA.
Patients are likely to report joint pain prior to being referred to a rheumatologist. However, bone erosions develop early in the course of RA with more than 10% of patients developing erosions within 8 weeks of disease onset and up to 60% by 1 year. 11 New antirheumatic drugs that prevent bone erosion without compromising systemic immune function (e.g., no changes in cytokine production) would benefit patients if started early on.
We previously found that 3,4‐dichloropropionanilide (DCPA) functions as an Orai calcium channel antagonist, 12 inhibits mineral resorption by osteoclasts in vitro, 13 and reduces bone erosion and cartilage damage in a collagen‐induced arthritis (CIA) model. 14 However, DCPA is metabolized into two immunotoxic intermediates 15 , 16 , 17 , 18 , 19 which produce toxicities independent of its function as an Orai calcium channel antagonist. 16 Therefore, our long‐term goal was to make a derivative of DCPA with reduced toxicities and equal efficacy in reducing bone erosion in arthritis. We tested several derivatives of DCPA for their ability to inhibit the Orai channel 20 and identified ELP‐004 as a compound with decreased toxicity but similar efficacy as an Orai inhibitor at as DCPA. 21 Here we assess the pharmacological parameters of our lead compound, ELP‐004 (Figure 1A). We characterized pharmacokinetic parameters of ELP‐004 including extent of absorption (e.g., bioavailability), brain distribution, and elimination (e.g., regulation of CYP enzymes). We further assessed potential genotoxic and off‐target effects and characterized the metabolites using in vitro hepatic systems as well as determined the CYPs that likely metabolize ELP‐004. Finally, we assessed its efficacy ex vivo in primary mouse osteoclasts and T cells. These data provide the basis for moving into in vivo efficacy experiments. Future studies will focus on treatment of RA with ELP‐004 in models of established disease.
FIGURE 1.
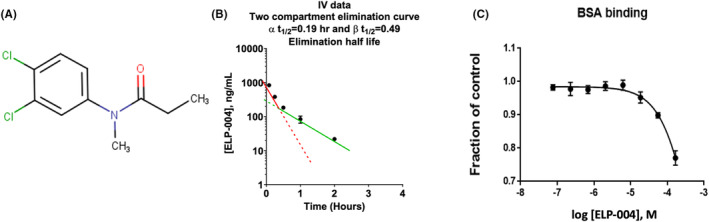
(A) Structure of ELP‐004. (B) Plasma concentration–time profiles and elimination half‐life slopes following a single i.v. dose of 2 mg/kg ELP‐004 (n = 3 per timepoint). See Table S6 for individual data values. (C) BSA binding assay.
2. MATERIALS AND METHODS
ELP‐004 was synthesized commercially (batch number ET190701‐2‐P1D; WuXi AppTec, Shanghai, China) and determined to be >99 pure by NMR (Figure S1). Buffer components were obtained from commercial vendors and were of biological grade. Organic solvents used in the analytical procedures were LC–MS/MS grade.
Animal experiments (unless otherwise noted) were conducted at WVU and approved by the WVU Institutional Animal Care and Use Committee (Protocol 1604002075). Mice were purchased from Jackson Laboratory (Bar Harbor, ME) and housed in a specific pathogen‐free barrier facility with 12 h light/dark cycles and fed normal chow.
2.1. Pharmacokinetic profile of ELP‐004 in mice
PK experiments were performed within guidelines established by the Janssen Animal Care and Use Committee and approved by the local Johnson and Johnson Ethical Committee. ELP‐004 was reconstituted in 20% hydroxypropyl‐beta‐cyclodextrin (HPbCD) and administered to female C57Bl/6 mice (3/timepoint/route of administration) in the fed state. Following i.v. (2 mg/kg), s.c. (2 mg/kg), or p.o. (10 mg/kg) administration, plasma was isolated from whole blood at 5, 15, and 30 min and at 1, 2, 4, 7, and 24 h. Plasma from p.o. was not isolated at 5 min. ELP‐004 concentration was measured by LC–MS/MS (see below). The limit of quantification was 10 ng/mL and samples below that were not used in the analyses. PK parameters were calculated using Phoenix WinNonlin (Certara, NJ, USA).
2.2. ELP‐004 in the brain
The distribution of ELP‐004 in the brain was measured as previously described. 22 , 23 Briefly, 100 μL of ELP‐004 solution (40 mg/kg) was delivered via s.c. After 15 min or 1 h, blood was collected via cardiac puncture, and plasma was isolated (BD, 365965). Brains were homogenized with a Dounce homogenizer (A and B) using PBS pH 7.4, with 10X volume. Ethyl acetate was added for a liquid‐liquid extraction; samples were vortexed and centrifuged 14 000×g for 10 min. The ethyl acetate fraction was transferred to a new tube and dried overnight. The samples were reconstituted with neat acetonitrile and analyzed by LC–MS/MS (AB Sciex 5500). Standard matrix optimization was conducted to ensure accurate concentrations were obtained in all LC–MS/MS experiments. Brain‐corrected concentration was calculated by subtracting 2% of the plasma concentration from the measured brain concentration.
2.3. BSA binding
Bovine serum albumin (BSA) binding was measured using a fluorescent high‐throughput screening assay. 24 ELP‐004 (10 mM) was serially diluted in a 96‐well black plate to give an 8‐point data point curve. Diclofenac (10 mM) was used as a positive control, and the final concentration of BSA was 7.5 μM in a final reaction volume of 200 μL. After 1 h at room temperature, fluorescence quenching was measured at Ex 280 nm and Em 340 nm.
2.4. Off‐target pharmacology analysis
A series of 52 CEREP ultra high‐throughput assays were conducted at Janssen Pharmaceuticals. ELP‐004 was tested at 10 μM in duplicate. Compound binding was calculated as a percent inhibition of the binding of a radioactive‐labeled ligand specific for each target. Reference compound was tested concurrently with ELP‐004, and the percent inhibition was calculated. The IC50 values and Hill coefficients (nH) were determined by non‐linear regression analysis of the competition curves generated with mean replicate values using Hill equation curve fitting using software developed at CEREP (Hill software).
2.5. AMES test
AMES tests were performed according to standard protocols at IIT Research Institute (IITRI). using four Salmonella typhimurium tester strains (TA98, TA100, TA1535, and TA1537) and one Escherichia coli strain (WP2 uvrA) obtained from Molecular Toxicology Inc. (Boone, NC). Aroclor‐1254‐induced rat liver post‐mitochondrial S9 fraction (Aroclor‐S9) (Molecular Toxicology Inc.) [or PBS (pH 7.4) for plates without S9], ELP‐004 (5, 2.5, 1.0, 0.5, 0.25, 0.10, or 0.05 mg/plate) or DMSO (vehicle), and bacterial cultures were combined (n = 3 per condition) and assessed for colony growth on Vogel–Bonner Medium E (VBE) glucose‐minimal agar plates. Positive controls included 2‐aminoanthracene (2‐AA), 2‐aminofluorene (2‐AF), 4‐nitroquinoline N‐oxide (4‐NQO), acridine mutagen (ICR‐191), methyl methanesulfonate (MMS), sodium azide (NaN3), and daunomycin. The number of revertant colonies per plate were counted. Positive (or mutagenic) is defined as: (1) the mean number of revertant colonies per plate demonstrated a two‐fold increase (three‐fold increase for TA1535 and TA1537) over the mean number of revertants in the concurrent vehicle/negative control, and (2) there was a dose‐related increase over the range tested.
2.6. Structural chromosomal aberration in CHO cells
Clastogenic activity of ELP‐004 was assessed at IITRI using the structural chromosomal aberration assay in Chinese hamster ovary (CHO‐WBL; Merck Research Laboratories) cells, with and without an exogenous metabolic activation system. Cells were treated with ELP‐004 (0.008, 0.016, 0.032, 0.063, 0.13, 0.25, 0.5, 1.0, and 2.0 mg/mL), vehicle (DMSO), or positive controls [cyclophosphamide (CP) with exogenous S9 and mitomycin C (MMC) without exogenous S9] for 3 h both in the absence and presence of Aroclor‐S9. Cultures were treated with Gibco® Colcemid™ (final concentration of 0.1 μg/mL) for the final 2 h of incubation and viability was assessed using trypan blue exclusion. Cells were exposed to a hypotonic shock with 0.075 M KCl, fixed, dropped onto glass slides, air‐dried, and stained with 5% Giemsa solution.
A target of 300 cells per dose level (150 from each replicate culture, if available) was evaluated for chromosomal aberrations. At least 25 metaphases were analyzed from each culture that demonstrated >25% of cells with one or more aberrations. Percent polyploidy and endoreduplication (numerical changes) were analyzed by evaluating 100 metaphases per culture. Statistical analysis consisted of one‐way analysis of variance (ANOVA) followed, if necessary, by the post hoc Dunnett test to compare the percentage of cells with structural chromosome aberrations in test article‐treated groups in comparison to vehicle. A non‐parametric Kruskal–Wallis test was performed, with post hoc comparisons, if necessary, using Dunn's test. Statistical comparisons were performed using SigmaPlot® software (version 13.0; Systat Software Inc.; Chicago, IL). A minimum significance level of p < .05 was used.
2.7. Human ether‐a‐go‐go‐related gene (hERG) inhibition
The hERG Safety assay was performed by Evotec (Framingham, MA) using a hERG stably transfected HEK293 cell‐based assay which employs QPatch HTX System (Sophion Bioscience A/S) automated patch clamp electrophysiology measurements. The voltage protocol was run continuously during the experiment. Pre‐ and post‐compound current were measured by whole cell patch clamp. The vehicle (0.5% DMSO) was applied for 3 min followed by the test substance (ELP‐004; 0.5, 1, 5, 10, 50, or 100 μM) in triplicate. Only cells with a seal resistance greater than 100 MΩ and a pre‐compound current of at least 0.2 nA were used to evaluate hERG blockade. E‐4031, a known hERG inhibitor, was used as a positive control and DMSO is included as a negative control.
2.8. CYP reaction phenotyping of ELP‐004
CYP assays were conducted at Cyprotex (Framingham, MA) using established methodology. cDNA‐expressed human P450 enzyme preparations from baculovirus‐infected insect cells (Supersomes™, Corning, see Table S1) were preincubated with 100 mM pH 7.4 phosphate buffer, 5 mM MgCl2 and ELP‐004 (1 μM; DMSO <0.1%) and initiated with NADPH (1 mM). Control Supersomes™ (no CYPs present) were used to reveal non‐enzymatic degradation. Control compounds (Table S2) known to be metabolized specifically by each CYP isoform were included. ELP‐004 was incubated for 0, 5, 15, 30, and 45 min with each CYP isoform. An aliquot was removed from each incubation at each timepoint and mixed with an organic solvent containing an internal standard, centrifuged at 4°C to precipitate the proteins, and supernatant was transferred to a new plate for LC–MS/MS analysis (see below). A plot of ln peak area ratio (compound peak area/internal standard peak area) against time was used to determine the slope after correction for any loss in the incubations with the control Supersomes™. Intrinsic clearance (μg/min/mg microsomal protein) and t 1/2 were calculated (n = 3–5/timepoint).
2.9. Reversible inhibition of CYP enzymes by ELP‐004
ELP‐004 (0.025, 0.079, 0.25, 0.79, 2.5, 7.9, and 25 μM) was incubated with human liver microsomes (Xenotech; 1210097, mixed sex) and NADPH (1 mM) in the presence of an appropriate isoform‐specific probe substrate (Table S3) at 37°C for 10–60 min (specific to CYP tested). A selective inhibitor of each isoform was used as a positive control (Table S3). Probe substrate metabolites were analyzed by LC–MS/MS: AB Sciex API5500 MS coupled with an Agilent 1290 LC system and an Agilent 1290 Infinity HTS chilled autosampler, controlled through Analyst software (AB Sciex). Samples were analyzed using the solvent/gradient shown in Table S4. A decrease in the metabolite compared with vehicle control was used to calculate an IC50 value.
2.10. Nuclear receptor activation
Activation of Human Pregnane X Receptor (PXR) was assessed using the PXR Reporter Assay kit (Indigo Biosciences). The PXR reporter cells were dosed with increasing concentrations of ELP‐004 (0.32, 1.0, 3.2, 10, 32, 100 μM; DMSO 0.4%; n = 2), positive control (rifampicin, 0.032, 0.1, 0.32, 1.0, 3.2, and 10 μM) or vehicle control and incubated (37°C) 22–24 h. Cell viability was assessed using the Live Cell Multiplex Assay Kit (Indigo Biosciences), and luciferase activity was measured using the Luciferase Detection Reagent (PXR kit).
Data were expressed as fold activation relative to vehicle control after normalizing luciferase activity against cell viability. EC50 and E max values were derived from non‐linear regression analysis of the log dose–response curves.
Activation of Aryl Hydrocarbon Receptor (AhR) and Constitutive Androstane Receptor (CAR3) were performed as described above using Indigo Biosciences Kits with methylcholanthrene (3‐MC) (0.0063, 0.02, 0.063, 0.2, 0.63, and 2 μM) or 6‐(4‐Chlorophenyl)‐imidazo[2,1‐b][1,3]thiazole‐5‐carbaldehyde O‐(3,4‐dichlorobenzyl)oxime (CITGO) (0.0079, 0.025, 0.079, 0.25, 0.79, and 2.5 μM) controls, respectively.
2.11. Liver S9 incubations and sample preparation
Experiments were conducted by Janssen Pharmaceuticals using standard protocols. Briefly, incubations were performed using either cofactor‐fortified mouse or human liver S9 (mouse liver S9: XenoTech, Male CD1, M1000 S9/Lot 0910418; mixed‐sex human liver S9: BD UltraPool, 452116, Lot 38289). For phase I metabolism, 0.1 M phosphate buffer (pH 7.4), 1 mg/mL liver S9, 1.2 mM EDTA, 6 mM MgCl2, 5 mM glutathione, 10 μM ELP‐004, and 30 μL NADPH regenerating system [25 μL of solution A (451220) and 5 μL of solution B (451200)] and 1 mM of a NAD+ (N1636, Sigma) were incubated in 0.5 mL total volume. Phase II cofactors were 6 mM uridine 5′‐diphosphoglucuronic acid triammonium salt (UDPGA, U5625, Sigma) and 1.2 mM uridine 5′‐diphospho‐N‐acetylglucosamine sodium salt (UDPAG, U4375, Sigma), 2 mM d,l‐dithiothreitol (DTT, D‐0632, Sigma), and 10 μM adenosine 3′‐phosphoadenosine 5′‐phosphosulfate lithium salt hydrate (PAPS, A1651, Sigma). Reactions were incubated at 37°C for 1 h and quenched by ultracentrifugation at 627 000×g for 15 min to pellet proteins.
LC–MS/MS analyses on the supernatant were performed using an Accela LC coupled to an LTQ XL or LTQ Orbitrap XL (ThermoScientific). See Table S5 for column and gradient details. All mass spectrometers were operated in the positive ESI mode. ELP‐004 (labeled JNJ‐64093536) was quantified using a standard curve of ELP‐004 (2–4000 ng/mL).
2.12. Osteoclast differentiation
Osteoclast precursors were isolated from the femur bone marrow of DBA/1 male mice (8–10 weeks) and differentiated as described. 21 , 25 , 26 To evaluate osteoclastogenesis, cells were replated at a density of 40 000/well in 24‐well plates (Ibidi, 82426) and stimulated with 100 ng/mL RANKL (Shenandoah Biotech, 200‐04) and 50 ng/mL mCSF (Shenandoah Biotech, 200‐08) with or without ELP‐004. TRAP staining (Sigma, 387A) was performed after 5 days according to manufacturer's instructions.
To evaluate mineral resorption, 27 cells were cultured on osteoassay mineral matrix‐coated plates (Corning, 3987) at a density of 25 000/well; resorption pit formation was imaged. 21 Briefly, media were replaced with osteoclast differentiation media plus 100 ng/mL RANKL with increasing concentrations of ELP‐004 (0–200 μM). Cells were refed/treated on day 2 (d2). On d5, osteoclast formation was observed. Osteoclasts were removed using 10% bleach; plates were rinsed with water and air‐dried.
Images of entire wells were acquired using a Lionheart imaging system (Biotek, Agilent Technologies) merging photographs of parts of the wells into single images with manufacturer's software (Gen5). Pit analysis was performed after conversion to 8‐bit greyscale format in ImageJ Software (NIH) as previously described. 28 The area resorbed was calculated using the measure feature in ImageJ.
2.13. T‐cell stimulation
Splenocytes were isolated from naïve DBA/1 mice in two independent groups separated by time, as described. 29 Splenocytes were stimulated with 5 μg/mL anti‐CD3 (clone 17A2, eBioscience, 14‐0032‐86) and 2 μg/mL anti‐CD28 (clone 37.51, eBioscience, 14‐0281‐86) ex vivo in the presence of vehicle (0.1% DMSO) or ELP‐004 (50, 100, 200, and 400 μM). Live CD3+ T cells were enumerated after 48 h by flow cytometry using counting beads and normalized to the vehicle control. Secreted cytokines were measured in the supernatant using the Meso Scale Discovery (MSD) mouse proinflammatory cytokine panel (K15048D).
2.14. Statistics
Resorption pit quantification and T cell–secreted cytokines were compared using repeated measures one‐way ANOVA with Dunnett's post hoc test for multiple comparisons using GraphPad Prism v9. Data are mean ± SD unless noted. Default statistical significance was 0.05.
3. RESULTS
3.1. Pharmacokinetics
Mice were administered the test compound ELP‐004 (Figure 1A) by i.v. (2 mg/kg), s.c. (2 mg/kg), or p.o. (10 mg/kg). Plasma concentration–time profiles and pharmacokinetic parameters are summarized in Figure 1B and Table 1, respectively. Intravenous administration at 2 mg/kg resulted in a plasma concentration at time zero (extrapolated; C 0) of 1220 ng/mL and a terminal volume of distribution (V d) of 3.65 L/kg. Elimination of the drug followed a two‐compartment model with alpha elimination t 1/2 of 0.19 h, and a beta elimination t 1/2 of 0.49 h. Following oral administration at 10 mg/kg, a C max of 211 ng/mL was reached at ~15 min (T max). An overall bioavailability (F) for oral administration was 13%. With a 2 mg/kg subcutaneous dose, the C max of 461 ng/mL was also reached at 15 min; the difference in C max concentrations can be attributed to the subcutaneous route having a bioavailability (F) of 83%. Systemic clearance and steady‐state V d (V ss) of ELP‐004 were estimated at 85.5 mL/min/kg and 2.48 L/kg, respectively. The volume of distribution is ~3.4 times higher than total body water (0.725 L/kg), indicating that the compound distributes outside the plasma.
TABLE 1.
Pharmacokinetic data of ELP‐004 administered via i.v., p.o., s.c.
| Route of administration | i.v. | p.o. | s.c. |
|---|---|---|---|
| Dose | 2 mg/kg | 10 mg/kg | 2 mg/kg |
| t 1/2 (h) | 0.493 | 1.19 | 0.445 |
| t max (h) | ‐ | 0.25 | 0.25 |
| C 0 (ng/mL) | 1220 | ‐ | ‐ |
| C max (ng/mL) | ‐ | 211 ± 42.9 | 461 ± 39.0 |
| AUClast (h*ng/mL) | 374 ± 23.2 | 227 ± 18.0 | 307 ± 31.7 |
| AUCInf (h*ng/mL) | 390 | 245 | 322 |
| AUC Extr (%) | 3.96 | 7.29 | 4.55 |
| V z (L/kg) | 3.65 | ‐ | ‐ |
| V ss (L/kg) | 2.48 | ‐ | ‐ |
| CL (mL/min/kg) | 85.5 | ‐ | ‐ |
| C 0 (μM) | 5.26 | ‐ | ‐ |
| C max (μM) | ‐ | 0.909 ± 0.185 | 1.986 ± 0.100 |
| F (%) | ‐ | 12.6 | 82.6 |
The ability of drugs to bind plasma proteins alters the free drug concentration available for target binding and potential pharmacologic effect; alternatively, increased binding can shield compounds from elimination pathways such as glomerular filtration of the kidneys and enzymatic reactions in the liver and blood as well as limit the ability of the drug to partition into tissues. 30 Albumin is the most abundant carrier protein in the blood; therefore, the ability of ELP‐004 to bind bovine serum albumin (BSA) was tested in vitro. The concentration of BSA was held at a constant concentration of 7.5 μM while the concentration of ELP‐004 was varied (75 nM–150 μM). The signal was normalized to a positive control (diclofenac) which binds strongly to albumin in circulation. BSA‐bound ELP‐004 was equal to diclofenac at concentrations ≤7.5 μM, but minimally reduced (≤25%) as compared to diclofenac at concentrations of 15–150 μM.
Since the V ss suggested distribution outside of the plasma, the ability of ELP‐004 to distribute to the central nervous system (CNS) was assessed. Mice were administered a high dose of 40 mg/kg ELP‐004 subcutaneously, and concentrations were assessed in the brain and plasma by LC–MS/MS after 15 min and 1 h. The ratio of brain/blood was found to be 0.70 ± 0.25 and 0.27 ± 0.09 at 15 min and 1 h, respectively (Table 2) suggesting that ELP‐004 is not preferentially concentrated in the brain.
TABLE 2.
Blood–brain barrier permeability following s.c. administration (40 mg/kg).
| Compound | Time | Plasma (μM) | Brain (corrected) (μM) | Brain: plasma |
|---|---|---|---|---|
| ELP‐004 | 15 min | 4.20 ± 2.83 | 2.44 ± 0.97 | 0.70 ± 0.25 |
| 1 h | 7.07 ± 2.92 | 2.00 ± 0.68 | 0.27 ± 0.09 |
3.2. Genotoxic evaluation
The mutagenic activity of ELP‐004 was investigated in the bacterial reverse mutation assay. Ames tests were performed as described earlier. No increase in the number of revertant colonies was observed in the five test strains at any ELP‐004 concentrations tested, in either the presence or absence of S9 mix (Table 3). No growth inhibitory effects were detected in the bacterial test strains. The clastogenic activity of ELP‐004 was evaluated in the structural chromosomal aberration assay in Chinese hamster ovary (CHO) cells, as described earlier. ELP‐004 did not increase the number of chromosomal aberrations (induce a clastogenic response) in CHO cells at any of the tested concentrations (Table 4). No clastogenic activity was seen in assays of ELP‐004 performed either with or without metabolic activation. Based on these data, we conclude that ELP‐004 is negative for clastogenic activity (induction of structural chromosome aberrations) in CHO cells.
TABLE 3.
Evaluation of the potential mutagenic activity ELP‐004 in the bacterial reverse mutation assay (AMES test).
| Revertants/plate (mean ± SD); N = 3 | |||||||
|---|---|---|---|---|---|---|---|
| Strain | |||||||
| Metabolic activation status | Material | Dose (level/plate) | TA98 | TA100 | WP2 uvr A | TA1535 | TA1537 |
| With S9 | DMSO | 0.1 mL | 32 ± 7.6 | 110 ± 5.3 | 64 ± 4.2 | 16 ± 0.6 | 13 ± 1.0 |
| ELP‐004 | 0.05 mg | 13 ± 3.1 | 89 ± 22.9 | 41 ± 9.7 | 16 ± 3.5 | 3 ± 2.6 | |
| 0.10 mg | 15 ± 2.0 | 78 ± 13.1 | 30 ± 10.6 | 18 ± 6.4 | 2 ± 1.2 | ||
| 0.25 mg | 22 ± 5.0 | 103 ± 16.4 | 33 ± 6.1 | 13 ± 2.9 | 2 ± 1.0 | ||
| 0.50 mg | 25 ± 9.5 | 79 ± 3.1 | 44 ± 5.0 | 7 ± 3.8 | 2 ± 3.2 | ||
| 1.00 mg | 10 ± 2.6 | 53 ± 4.6 | 31 ± 1.7 | 7 ± 1.2 | 3 ± 1.5 | ||
| 2.50 mg | 9 ± 2.1 | 16 ± 3.5 | 19 ± 4.7 | 12 ± 7.5 | 5 ± 2.1 | ||
| 5.00 mg | 0 ± 0.0 | 0 ± 0.0 | 0 ± 0.0 | 0 ± 0.0 | 0 ± 0.0 | ||
| Without S9 | DMSO | 0.1 mL | 27 ± 5.6 | 105 ± 17.0 | 53 ± 10.5 | 15 ± 5.5 | 16 ± 1.2 |
| ELP‐004 | 0.05 mg | 28 ± 8.1 | 86 ± 11.9 | 36 ± 7.5 | 15 ± 4.7 | 0 ± 0.0 | |
| 0.10 mg | 19 ± 6.4 | 72 ± 13.9 | 28 ± 11.2 | 15 ± 5.0 | 2 ± 2.6 | ||
| 0.25 mg | 16 ± 3.8 | 64 ± 1.0 | 39 ± 8.4 | 17 ± 3.1 | 1 ± 1.2 | ||
| 0.50 mg | 12 ± 3.5 | 59 ± 16.8 | 41 ± 8.1 | 13 ± 4.6 | 1 ± 0.6 | ||
| 1.00 mg | 5 ± 0.6 | 38 ± 20.2 | 32 ± 2.1 | 13 ± 2.5 | 1 ± 0.6 | ||
| 2.50 mg | 7 ± 3.1 | 0 ± 0.6 | 30 ± 24.3 | 1 ± 0.6 | 2 ± 1.2 | ||
| 5.00 mg | 0 ± 0.6 | 0 ± 0.0 | 0 ± 0.0 | 0 ± 0.0 | 0 ± 0.0 | ||
TABLE 4.
Evaluation of the potential clastogenic activity ELP‐004 in the structural chromosomal aberration assay in Chinese hamster ovary (CHO) cells.
| Agent | Target concentration | Replicate | Number of cells evaluated | Cells with aberrations (%) | Polyploidy (%) | Endoreduplication (%) | |
|---|---|---|---|---|---|---|---|
| One a | Multiple | ||||||
| Chromosome aberration assay without metabolic activation: summary | |||||||
| Negative control—DMSO (%) | 1 | A | 150 | 0.0 | 0.0 | 0.0 | 0.0 |
| B | 150 | 0.0 | 0.0 | 2.0 | 0.0 | ||
| A + B b | 300 | 0.0 | 0.0 | 1.0 | 0.0 | ||
| Test article—ELP‐004 (mg/mL) | 0.032 | A | 150 | 0.0 | 0.0 | 0.0 | 0.0 |
| B | 150 | 0.0 | 0.0 | 2.0 | 0.0 | ||
| A + B | 300 | 0.0 | 0.0 | 1.0 | 0.0 | ||
| 0.063 | A | 150 | 0.0 | 0.0 | 0.0 | 0.0 | |
| B | 150 | 0.0 | 0.0 | 0.0 | 0.0 | ||
| A + B | 300 | 0.0 | 0.0 | 0.0 | 0.0 | ||
| 0.13 | A | 150 | 0.0 | 0.0 | 2.0 | 0.0 | |
| B | 150 | 0.0 | 0.0 | 1.0 | 0.0 | ||
| A + B | 300 | 0.0 | 0.0 | 1.5 | 0.0 | ||
| Positive control—Mitomycin C (μg/mL) | 0.5 | A | 25 | 7.3 | 2.7 | 1.0 | 2.0 |
| B | 25 | 36.0 | 20.0 | 0.0 | 3.0 | ||
| A + B | 50 | 11.4 | 5.1 | 0.5 | 2.5 | ||
| Chromosome aberration assay with metabolic activation: summary | |||||||
| Negative control—DMSO (%) | 1 | A | 150 | 0.0 | 0.0 | 2.0 | 0.0 |
| B | 150 | 0.0 | 0.0 | 0.0 | 0.0 | ||
| A + B b | 300 | 0.0 | 0.0 | 1.0 | 0.0 | ||
| Test article—ELP‐004 (mg/mL) | 0.032 | A | 150 | 0.0 | 0.0 | 0.0 | 0.0 |
| B | 150 | 0.0 | 0.0 | 0.0 | 0.0 | ||
| A + B | 300 | 0.0 | 0.0 | 0.0 | 0.0 | ||
| 0.063 | A | 150 | 0.0 | 0.0 | 0.0 | 1.0 | |
| B | 150 | 0.0 | 0.0 | 1.0 | 0.0 | ||
| A + B | 300 | 0.0 | 0.0 | 0.5 | 0.5 | ||
| 0.13 | A | 150 | 0.0 | 0.0 | 0.0 | 2.0 | |
| B | 150 | 1.3 | 0.7 | 1.0 | 4.0 | ||
| A + B | 300 | 0.7 | 0.3 | 0.5 | 3.0 | ||
| Positive control—Mitomycin C (μg/mL) | 10 | A | 25 | 80.0 | 64.0 | 1.0 | 0.0 |
| B | 25 | 56.0 | 44.0 | 2.0 | 0.0 | ||
| A + B | 50 | 68.0 | 54.0 | 1.5 | 0.0 | ||
Parameter is analyzed by ANOVA for the test article groups versus the negative control group; No differences were observed.
Reported value is a sum for the column entitled “Number of Cells Evaluated.” For all other columns, an average value is presented.
3.3. Off‐target toxicities
Identification of off‐target interactions can help predict potential side effects and safety issues of compounds under development. ELP‐004 was screened at 10 μM for its potential to interfere with the binding of native ligands as described earlier. Inhibition greater than 50% represent significant effect of the test compound, whereas an inhibition lower than 10% is not considered significant. Nine targets had inhibition greater than 10% after ELP‐004 treatment, with only one target (dopamine transporter) having greater than 50% (Table 5). Most receptors did not demonstrate inhibition over the assay background (Table S7).
TABLE 5.
Off‐target pharmacological profiling of ELP‐004 [10 μM] with >10% inhibition.
| Radioligand assays | Concentration (M) | % of inhibition | Reference compound |
|---|---|---|---|
| A1 (h) (antagonist) | 1.0E‐05 | 21.9 | DPCPX |
| CCK1 (CCKA) (h) (agonist) | 1.0E‐05 | 10.3 | CCK‐8s |
| CCR1 (h) (agonist) | 1.0E‐05 | 16.2 | MIP‐1α |
| MT1 (ML1A) (h) (agonist radioligand) | 1.0E‐05 | 25.8 | Melatonin |
| Kappa (KOP) (agonist) | 1.0E‐05 | 15.2 | U 50488 |
| 5‐HT1A (h) (agonist) | 1.0E‐05 | 20.7 | 8‐OH‐DPAT |
| 5‐HT2B (h) (agonist) | 1.0E‐05 | 23 | (±)DOI |
| 5‐HT3 (h) (antagonist) | 1.0E‐05 | 12.4 | MDL 72222 |
| Dopamine transporter (h) (antagonist) | 1.0E‐05 | 82.2 | BTCP |
Note: Data are presented as mean values; n = 2. The results are expressed as percent inhibition of control‐specific binding.
Abbreviation: h, human.
To screen for potential cardiotoxicity, ELP‐004 [0.5–100 μM] was evaluated for its ability to inhibit hERG (human Ether‐a‐go‐go‐related gene) channel activity using whole cell patch clamp. ELP‐004 is not a potent inhibitor hERG activity, as indicated by its low pIC50 of <4 and inability to inhibit the channel by greater than 30% at all tested concentrations (Table 6).
TABLE 6.
hERG channel inhibition of ELP‐004 [0.5–100 μM].
| hERG channel inhibition | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| % Mean inhibition | IC50 determination | |||||||||
| Test compound: ELP‐004 | ||||||||||
| Compound | 0.5 μM | 1 μM | 5 μM | 10 μM | 50 μM | 100 μM | IC50 (μM) | pIC50 | pIC50 SE | N |
| 2.11 | 1.44 | 2.03 | 3 | 13.96 | 27.87 | >100 | <4 | ‐ | 4 | |
| Negative control: Vehicle (0.5% DMSO) | ||||||||||
| Compound | 1st addition | 2nd addition | 3rd addition | 4th addition | 5th addition | 6th addition | IC50 (μM) | pIC50 | pIC50 SE | N |
| 3.8 | 4.97 | 5.39 | 3.95 | 3.37 | 4.03 | ‐ | ‐ | ‐ | 3 | |
| Positive control: E‐4031 | ||||||||||
| Compound | 0.001 μM | 0.003 μM | 0.011 μM | 0.033 μM | 0.1 μM | 0.3 μM | IC50 (μM) | pIC50 | pIC50 SE | N |
| 0.79 | 5.36 | 26.5 | 52.71 | 80.06 | 91.55 | 0.03 | 7.53 | 0.03 | 5 | |
3.4. P450 phenotyping and inhibition
The stability of ELP‐004 was tested in the presence of recombinant enzymes CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4. ELP‐004 (1 μM) was incubated with recombinant enzymes and NADPH. The amount of ELP‐004 remaining at each timepoint was analyzed by LC–MS/MS. Incubations in control recombinant enzymes were also performed to assess non‐enzymatic mediated stability. The intrinsic clearance of ELP‐004 was 2.85, 0.815, 0.219, 0.279, and 0.193 μg/min/mg microsomal protein in the presence of CYP1A2, CYP2B6, CYP2C8, CYP2C19, and CYP3A4 recombinant enzymes, respectively (Table 7). ELP‐004 was not metabolized by CYP2C9 and CYP2D6 recombinant enzymes.
TABLE 7.
Cytochrome P450 phenotyping and inhibition by ELP‐004.
| Isoform | CYP1A2 | CYP2B6 | CYP2C8 | CYP2C9 | CYP2C19 | CYP2D6 | CYP3A4 |
|---|---|---|---|---|---|---|---|
| ELP‐004 t 1/2 (min) | 9.73 | 8.51 | 127 | >180 | 99.2 | >180 | 143 |
| Recombinant CYP CLINT (μL/min/mg) | 2.85 | 0.815 | 0.219 | <0.154 | 0.279 | <0.154 | 0.193 |
| Inhibition IC50 (μM) | >25 | >25 | >25 | >25 | >25 | >25 | >25 a |
Abbreviation: CLINT, intrinsic clearance.
Midazolam and testosterone.
Next, the ability of ELP‐004 to reversibly inhibit these CYP isoforms was assessed in human liver microsomes. A decrease in the formation of the metabolite compared to vehicle control is used to calculate an IC50 value. Metabolite peak area ratio response as a percentage of vehicle control is plotted against the test compound concentration. ELP‐004 did not inhibit any CYP over the concentration range assessed (0.02–25 μM), and therefore no IC50 values were calculated (Table 7).
3.5. Nuclear receptor activation
Nuclear receptors involved in the induction of drug‐metabolizing enzymes include PXR, AhR, and CAR which are known to regulate CYP3A4, CYP1A2, and CYP2B6, respectively. Induction was assessed by incubating reporter cells with increasing concentrations of ELP‐004 (0.32–100 μM) or a corresponding standard and measuring luminescent signal. Fold activation for each concentration is shown in Table 8. PXR expression increased 2.28‐fold at 100 μM ELP‐004; whereas AhR expression increased 4.94‐fold at 10 μM ELP‐004 and 26‐fold at 100 μM ELP‐004. Human PXR and AhR were both induced with EC50 of 38 μM. CAR3 expression was not altered at any dose tested.
TABLE 8.
Nuclear receptor activation.
| Fold activation | PXR | AhR | CAR3 |
|---|---|---|---|
| 100 μM ELP‐004 | 2.28 | 26.00 | 1.00 |
| 32 μM ELP‐004 | 1.60 | 11.18 | 0.06 |
| 10 μM ELP‐004 | 0.86 | 4.94 | 0.69 |
| 3.2 μM ELP‐004 | 1.18 | 1.86 | 0.66 |
| 1.0 μM ELP‐004 | 0.48 | 1.41 | 0.82 |
| 0.32 μM ELP‐004 | 1.21 | 1.09 | 0.48 |
| EC50 (μM) | 38.06 | 38.07 | ‐ |
3.6. Metabolite identification
To evaluate the metabolism of ELP‐004 in vitro, ELP‐004 was incubated with boiled (denatured) or cofactor‐supplemented human or mouse liver S9 fractions. Cofactors included NADPH, NADH, UDPGA, DTT, and PAPS to ensure Phases 1 and 2 metabolism. Figure 2A shows the MS traces for human (left) and mouse (right) liver S9 fractions, respectively. In boiled and unboiled samples, the unchanged drug (UD) had a retention time of 26 min. By comparing the amount of UD from liver S9 samples to the boiled liver S9 samples, the turnover of ELP‐004 was found to be 24.2% humans and 14.3% in mice (Figure S2).
FIGURE 2.
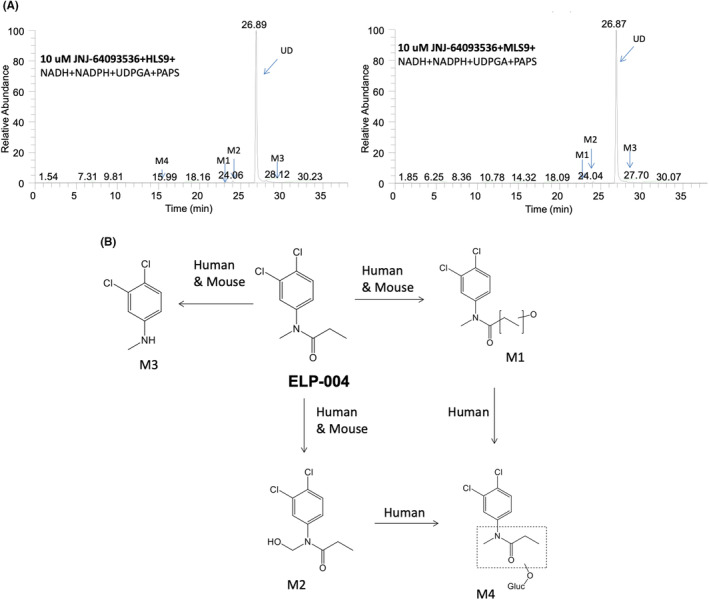
(A) Reconstructed ion chromatograms displaying the unchanged drug and its metabolites from LC–MS/MS analysis following 1‐h incubation of 10 μM of ELP‐004 in NADPH, NADH, UDPGA, DTT, PAPS fortified liver S9 from human (HL, left), and mouse (ML, right). (B) In vitro metabolic pathways of ELP‐004 in human and mouse.
Incubation with liver S9 fractions led to the production of metabolites of ELP‐004 (Table 9). The UD incubated in cofactor‐fortified human liver S9 as full spectrum is shown in Figure S3, with the m/z of 232.02. This spectrum corresponded with the ELP‐004 standard is shown in Figure S4. The products of metabolism observed with incubation of ELP‐004 and murine/human liver S9 were identified from fragments seen in the product ion mass spectra (Figures S5–S8). Four metabolites (M1, M2, M3, and M4) were identified in human liver S9, while only three major metabolites (M1, M2, and M3) were observed in mouse liver S9. Figure 2B shows the predicted in vitro metabolic pathways for ELP‐004 in both mouse and human liver S9 fractions.
TABLE 9.
Molecular ions and characteristic fragment ions of ELP‐004 and metabolites.
| ID | Retention time (min) | [M + H]+ m/z | Key fragment ions m/z |
|---|---|---|---|
| ELP‐004 | 26.89 | 232.02901 | |
| M1 | 22.90 | 248.02406 | 174, 202 |
| M2 | 24.06 | 248.02407 | 180, 195 |
| M3 | 28.12 | 176.00282 | 161 |
| M4 | 15.99 | 424.05616 |
3.7. Effects of ELP‐004 on osteoclast differentiation/function and T‐cell cytokine production
Finally, the ability of ELP‐004 to inhibit osteoclast differentiation and function ex vivo was evaluated. Bone marrow cells were harvested from male DBA/1 mice (n = 3) and grown in mCSF‐ and RANKL‐supplemented media to induce osteoclast differentiation with or without ELP‐004. Multinucleated TRAP+ cells are the prominent in the vehicle‐treated cultures at d5, but multinucleation was inhibited by ELP‐004 ~50% at 50 μM and was totally blocked at 100 μM (Figure 3A, top). Cells were then cultured and differentiated on mineralized matrix to assess the ability of ELP‐004 to inhibit osteoclast function. Lighter regions in the well are indicative of matrix resorption pits (Figure 3A, bottom). ELP‐004 inhibited the degradation of matrix at concentrations equal to its effects on multinucleation (Figure 3A, right; p < .05).
FIGURE 3.
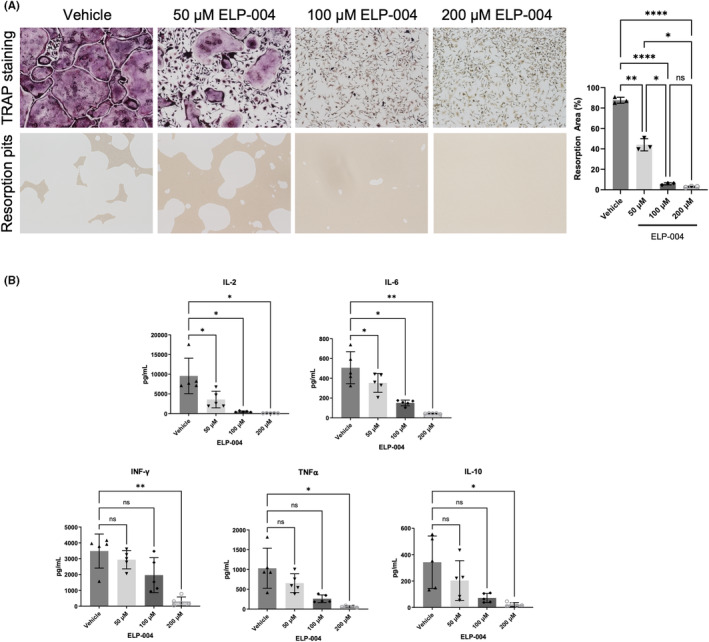
(A) Monocytes were isolated from the bone marrow of male DBA/1 mice (n = 3). Cells were stimulated with RANKL and mCSF and treated with escalating doses of ELP‐004 ex vivo. Cells were stained for the TRAP expression (top) or plated on matrix‐coated plates to assess bone resorption activity (bottom). Area of resorption pits was quantified (right). Groups were analyzed using repeated measures ANOVA with the Geisser–Greenhouse correction; multiple comparisons were performed using Tukey's post hoc test. (B) Single cell suspensions of splenocytes were prepared from male DBA/1 mice (n = 5). T cells were stimulated ex vivo with anti‐CD3 and anti‐CD28 and treated with increasing doses of ELP‐004. Secreted cytokines were assessed using a multi‐analyte ELISA (MSD plate). Cytokine concentrations were using compared using repeated measures one‐way ANOVA with a Dunnett post hoc test for multiple comparisons. Data are presented as mean ± SD. *p < .05; **p < .01; ****p < .0001.
The effects of ELP‐004 treatment on primary T‐cell number and function were also assessed ex vivo. Splenocytes were isolated from DBA/1 mice (n = 5), and the T cells were stimulated in culture with anti‐CD3/CD28 in presence or absence of ELP‐004 (0–400 μM). After 48 h of treatment and stimulation, live CD3+ T cells in the 400 μM ELP‐004 treatment were reduced to 63% of vehicle (p < .05); live cells were unaffected by other concentrations of ELP‐004 (Figure S9A). Secreted cytokines were measured and compared with the basal expression in their respective vehicle control for each mouse. IL‐2 and IL‐6 were decreased at all concentrations of ELP‐004 (50, 100, and 200 μM) while IFN‐γ, TNFα, and IL‐10 showed a dose‐dependent inhibition with significant inhibition observed at the highest dose (Figure 3B). All other cytokines measured (IL‐12p70, IL‐1β, CXCL1 [KC/GRO], IL‐5, and IL‐4) were not consistently affected by ELP‐004 (Figure S9B). Together, these data suggest that ELP‐004 reduces osteoclast activity without altering overall T‐cell cytokine production.
4. DISCUSSION
Our goal is to develop a safe and effective small molecule to prevent the destruction of bone. Our lead compound, ELP‐004, shows promising effects on osteoclasts inhibition in vitro without inducing genotoxic or extensive off‐target toxicities. The core structure of ELP‐004 is an aniline, a class of molecules with a high risk of forming reactive metabolites. However, ELP‐004 is predicted to preclude the formation of reactive metabolites by methylation of the amide nitrogen. The t 1/2 and t max of both p.o. and s.c. administration indicate that ELP‐004 is both rapidly absorbed and removed in vivo, suggesting that further manipulation (e.g., time‐release encapsulation) may be necessary to improve overall compound stability and efficacy.
Potential off‐target inhibitions were analyzed and only nine possible off‐target inhibitions were identified (Table 3). Only the dopamine transporter (DAT) is of major concern. DATs are well‐known for their involvement in brain function as transmitter imbalances cause psychiatric and neurodegenerative diseases 31 ; however, they are also thought to play an important role in gastrointestinal disorders. 32 We found that ELP‐004 does not preferentially accumulate in the brain. We have previously demonstrated that DAT inhibitors 33 , 34 , 35 result in locomotor hyperactivity and increased levels of motor stereotypes as expected; however, we did not observe these symptoms in mice treated acutely with high doses of ELP‐004 (Figure S10). Furthermore, the half‐life of ELP‐004 is approximately 30 min, indicating the drug is eliminated within a few hours. Given the short half‐life and the time to C max, the dopaminergic side effects would likely occur within 1–2 h of administration. Other medications known to bind DAT, for example, bupropion, have an 8 to 12‐h timeframe in the body. Together, these data suggest that the in vitro DAT inhibition by ELP‐004 may not be of large concern in vivo, but this will be assessed in future studies.
Species‐specific differences were observed in the turnover of ELP‐004 (24.2% in humans and 14.3% in mice after 1 h) as well as the number of metabolites produced (four in humans and three in mice). In vitro phenotyping revealed that ELP‐004 was primarily metabolized by human CYP1A2 and CYP2B6. The CYP enzyme family is the most important enzyme system catalyzing the Phase 1 metabolism of pharmaceuticals and other xenobiotics. P450 induction and inhibition are essential to predicting possible drug interactions. Here, we measured nuclear receptor upregulation as a surrogate of CYP induction. Human nuclear receptors involved in the induction of drug‐metabolizing enzymes include PXR, AhR, and CAR, which are known to regulate CYP3A4, CYP1A2, and CYP2B6, respectively. While CYP3A4 was not found to be a major metabolizer of ELP‐004, it appears to be induced in response to high doses (100 μM) as indicated by PXR upregulation. This dose is greater needed to inhibit osteoclast function; therefore, it is unlikely that CYP3A4 will be induced at doses used in vivo. As CYP3A4 is involved in the metabolism of approximately 50% of all marketed medications, 36 the lack of induction by ELP‐004 at clinically relevant concentrations is important. CYP1A2 both metabolizes ELP‐004 and the upregulation of AhR suggests that it is induced at concentrations >10 μM. We do not anticipate upregulation of CYP1A2 at ELP‐004 concentrations used in vivo; however, this will be monitored in future studies. CYP2B6, the other major player in ELP‐004 metabolism, is not induced at any ELP‐004 concentration tested. Furthermore, no P450s were found to be inhibited at any of the concentrations tested in our studies.
Ex vivo treatment of primary monocytes with ELP‐004 significantly reduced osteoclast differentiation and function beginning at 50 μM with almost complete inhibition at 100 and 200 μM. At these same concentrations, ex vivo IL‐2 and IL‐6 secretion was inhibited in stimulated T cells. These data are consistent with our previous study demonstrating that Orai inhibition by DCPA (the parent compound) decreased Ca2+ influx resulting in decreased NFAT‐dependent IL‐2 secretion in Jurkat cells. 15 At high doses of ELP‐004 (200 μM), we also observed decreased concentrations of secreted proinflammatory (Th1) cytokines, IFN‐γ and TNFα, as well as the anti‐inflammatory (Th2) cytokine IL‐10 suggesting that overall T‐cell function is suppressed at high concentrations. The reduction of IL‐6 secretion by T cells stimulated ex vivo was of interest due to its known roles in autoimmune diseases (reviewed in [37]). Elevated serum and tissue IL‐6 levels are associated with RA 38 and drugs targeting the IL‐6 signaling are approved or are in clinical trials for the treatment of RA. 39 , 40 , 41
In summary, these data provide an important and comprehensive characterization of ELP‐004 pharmacokinetics, genotoxic and off‐target effects, and metabolism. We further demonstrate that ELP‐004 is effective in reducing osteoclast formation and function in vitro. The overall goal is to formulate this compound for oral administration which can be used to reduce disease‐induced bone erosions. Therefore, imminent studies will focus on improving the bioavailability and prolonging the half‐life of ELP‐004 in vivo. The data from this study will also be used to inform future efficacy studies with ELP‐004 in mouse models of established arthritis as well as other disease models that are associated with bone erosions.
AUTHOR CONTRIBUTIONS
John B. Barnett, Harry C. Blair, Jonathan Soboloff, Jamie L. McCall, Werner J. Geldenhuys, Björn C. G. Söderberg, and Lisa J. Robinson participated in research design. Jamie L. McCall, Werner J. Geldenhuys, Lisa J. Robinson, and Michelle R. Witt conducted the experiments. Peter M. Gannett and Björn C. G. Söderberg contributed to new reagents. Werner J. Geldenhuys, Jamie L. McCall, Michelle R. Witt, Lisa J. Robinson, and Harry C. Blair performed the data analysis. Werner J. Geldenhuys and Jamie L. McCall wrote or contributed to the writing of the manuscript.
CONFLICT OF INTEREST STATEMENT
JBB is the President and Chief Science Officer of ExesaLibero Pharma, Inc. JBB, WJG, BCGS, HCB, and JS serve on the Scientific Advisory Board of ExesaLibero Pharma, Inc. Drs. JBB, JS, and HCB have a patent [US 10,682,320 B2] on methods for inhibiting osteoclast development which includes the use of ELP‐004. JLM is an employee of ExesaLibero Pharma, Inc. LJR, PMG, and MRW declare no competing interests.
ETHICS STATEMENT
Procedures on animals carried out at WVU were approved by the WVU Institutional Animal Care and Use Committee (Protocol 1604002075). All WVU IACUC policies, guidance, and procedures are in full compliance with all federal regulations and consistent with the Guide for the Care and Use of Laboratory Animals. PK experiments were performed within guidelines established by the Janssen Animal Care and Use Committee and approved by the local Johnson and Johnson Ethical Committee.
Supporting information
Data S1.
ACKNOWLEDGMENTS
This work was funded in part by the National Institutes of Health (NIH) STTR [Grant R42AR074812] to WJG (WVU) and JBB (ExesaLibero Pharma, Inc). Cytokine data were obtained at the WVU Flow Cytometry & Single Cell Core Facility (RRID:SCR_017738) which is supported by the Institutional Development Awards (IDeA) from the National Institute of General Medical Sciences of the NIH under grant numbers P30GM121322 (TME CoBRE) and P20GM103434 (INBRE). LC/MS/MS analyses performed at WVU Metabolomics & Small Molecule Analysis Facility was supported by the WVU Cancer Institute, the WVU HSC Office of Research & Graduate Education, and NIH grant P20GM103434. The authors would like to thank Thomas Henson (West Virginia University) and Irina L. Tourkova (University of Pittsburgh) for their assistance in performing tasks associated with the data collection as well as Dr. Edward Lawson and Fran Xu at Janssen Research & Development, LLC and Terrence McManus at WVU for their assistance with the PK and metabolite data. Finally, we would like to thank Dr. Paul Lockman at WVU for his careful review of the manuscript.
McCall JL, Geldenhuys WJ, Robinson LJ, et al. Preclinical evaluation of ELP‐004 in mice. Pharmacol Res Perspect. 2024;12:e1230. doi: 10.1002/prp2.1230
DATA AVAILABILITY STATEMENT
The authors declare that all the data supporting the findings of this study are available within the paper and its Supplemental Data.
REFERENCES
- 1. Hameed A, Brady JJ, Dowling P, Clynes M, O'Gorman P. Bone disease in multiple myeloma: pathophysiology and management. Cancer Growth Metastasis. 2014;7:33‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang M, Xia F, Wei Y, Wei X. Molecular mechanisms and clinical management of cancer bone metastasis. Bone Res. 2020;8(1):30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Choy E. Understanding the dynamics: pathways involved in the pathogenesis of rheumatoid arthritis. Rheumatology (Oxford). 2012;51(Suppl 5):v3‐v11. [DOI] [PubMed] [Google Scholar]
- 4. Druce KL, Cordingley L, Short V, et al. Quality of life, sleep and rheumatoid arthritis (QUASAR): a protocol for a prospective UK mHealth study to investigate the relationship between sleep and quality of life in adults with rheumatoid arthritis. BMJ Open. 2018;8(1):e018752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Matcham F, Scott IC, Rayner L, et al. The impact of rheumatoid arthritis on quality‐of‐life assessed using the SF‐36: a systematic review and meta‐analysis. Semin Arthritis Rheum. 2014;44(2):123‐130. [DOI] [PubMed] [Google Scholar]
- 6. Costello R, David T, Jani M. Impact of adverse events associated with medications in the treatment and prevention of rheumatoid arthritis. Clin Ther. 2019;41(7):1376‐1396. [DOI] [PubMed] [Google Scholar]
- 7. Bongartz T, Sutton AJ, Sweeting MJ, Buchan I, Matteson EL, Montori V. Anti‐TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies: systematic review and meta‐analysis of rare harmful effects in randomized controlled trials. JAMA. 2006;295(19):2275‐2285. [DOI] [PubMed] [Google Scholar]
- 8. Umeda M, Koga T, Ichinose K, et al. Efficacy of infliximab as a switched biologic in rheumatoid arthritis patients in daily clinical practice. Immunol Med. 2018;41(4):181‐186. [DOI] [PubMed] [Google Scholar]
- 9. Yip RML, Yim CW. Role of interleukin 6 inhibitors in the management of rheumatoid arthritis. J Clin Rheumatol. 2021;27(8):e516‐e524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sartori NS, Picon P, Papke A, Neyeloff JL, da Silva Chakr RM. A population‐based study of tuberculosis incidence among rheumatic disease patients under anti‐TNF treatment. PLoS One. 2019;14(12):e0224963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Machold KP, Stamm TA, Nell VPK, et al. Very recent onset rheumatoid arthritis: clinical and serological patient characteristics associated with radiographic progression over the first years of disease. Rheumatology (Oxford). 2007;46(2):342‐349. [DOI] [PubMed] [Google Scholar]
- 12. Zhou Y, Lewis TL, Robinson LJ, et al. The role of calcium release activated calcium channels in osteoclast differentiation. J Cell Physiol. 2011;226(4):1082‐1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Robinson LJ, Mancarella S, Songsawad D, et al. Gene disruption of the calcium channel Orai1 results in inhibition of osteoclast and osteoblast differentiation and impairs skeletal development. Lab Investig. 2012;92(7):1071‐1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Blair HC, Soboloff J, Robinson LJ, et al. Suppression of arthritis‐induced bone erosion by a CRAC channel antagonist. RMD Open. 2016;2(1):e000093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lewis TL, Brundage KM, Brundage RA, Barnett JB. 3,4‐Dichloropropionanilide (DCPA) inhibits T‐cell activation by altering the intracellular calcium concentration following store depletion. Toxicol Sci. 2008;103(1):97‐107. [DOI] [PubMed] [Google Scholar]
- 16. Lewis TL, Holaskova I, Barnett JB. The toxicity of the N‐hydroxy and 6‐hydroxy metabolites of 3,4‐dichloropropionanilide does not depend on calcium release‐activated calcium channel inhibition. Toxicol Sci. 2013;131(2):395‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Barnett JB, Gandy J, Wilbourn D, Theus SA. Comparison of the immunotoxicity of propanil and its metabolite, 3,4‐dichloroaniline, in C57Bl/6 mice. Fundam Appl Toxicol. 1992;18(4):628‐631. [DOI] [PubMed] [Google Scholar]
- 18. Brundage KM, Barnett JB, Mahaney JE. The amide class herbicide 3,4‐dichloropropionanilide (DCPA) alters the mobility of hydrocarbon chains in T‐lymphocyte but not macrophage membranes. J Toxicol Environ Health A. 2003;66(23):2253‐2265. [DOI] [PubMed] [Google Scholar]
- 19. McMillan DC, Leakey JEA, Arlotto MP, McMillan JEM, Hinson JA. Metabolism of the arylamide herbicide propanil. II. Effects of propanil and its derivatives on hepatic microsomal drug‐metabolizing enzymes in the rat. Toxicol Appl Pharmacol. 1990;103(1):102‐112. [DOI] [PubMed] [Google Scholar]
- 20. Barnett JB, Blair HC, Soboloff J. Method for inhibiting osteoclast development. US 11,654,123 B2; 2023.
- 21. Robinson LJ, Soboloff J, Tourkova IL, et al. The function of the calcium channel Orai1 in osteoclast development. FASEB J. 2021;35(6):e21653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Saralkar P, Mdzinarishvili A, Arsiwala TA, et al. The mitochondrial mitoNEET ligand NL‐1 is protective in a murine model of transient cerebral ischemic stroke. Pharm Res. 2021;38(5):803‐817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vijikumar A, Saralkar P, Saylor SD, Sullivan PG, Huber JD, Geldenhuys WJ. Novel mitoNEET ligand NL‐1 improves therapeutic outcomes in an aged rat model of cerebral ischemia/reperfusion injury. Exp Neurol. 2022;355:114128. [DOI] [PubMed] [Google Scholar]
- 24. Sułkowska A. Interaction of drugs with bovine and human serum albumin. J Mol Struct. 2002;614(1):227‐232. [Google Scholar]
- 25. Swamydas M, Lionakis MS. Isolation, purification and labeling of mouse bone marrow neutrophils for functional studies and adoptive transfer experiments. J Vis Exp. 2013;77:e50586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tourkova IL, Dobrowolski SF, Secunda C, et al. The high‐density lipoprotein receptor Scarb1 is required for normal bone differentiation in vivo and in vitro. Lab Investig. 2019;99(12):1850‐1860. [DOI] [PubMed] [Google Scholar]
- 27. Boraschi‐Diaz I, Komarova SV. The protocol for the isolation and cryopreservation of osteoclast precursors from mouse bone marrow and spleen. Cytotechnology. 2016;68(1):105‐114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gritsaenko T, Pierrefite‐Carle V, Creff G, Vidaud C, Carle G, Santucci‐Darmanin S. Methods for analyzing the impacts of natural uranium on in vitro osteoclastogenesis. J Vis Exp. 2018;131:e56499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McCall JL, Blair HC, Blethen KE, Hall C, Elliott M, Barnett JB. Prenatal cadmium exposure does not induce greater incidence or earlier onset of autoimmunity in the offspring. PLoS One. 2021;16(9):e0249442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wanat K. Biological barriers, and the influence of protein binding on the passage of drugs across them. Mol Biol Rep. 2020;47(4):3221‐3231. [DOI] [PubMed] [Google Scholar]
- 31. Salatino‐Oliveira A, Rohde LA, Hutz MH. The dopamine transporter role in psychiatric phenotypes. Am J Med Genet B Neuropsychiatr Genet. 2018;177(2):211‐231. [DOI] [PubMed] [Google Scholar]
- 32. Cerantola S, Caputi V, Contarini G, et al. Dopamine transporter genetic reduction induces Morpho‐functional changes in the enteric nervous system. Biomedicine. 2021;9(5):465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Buletko AB, Dluzen DE, McDermott JL, Darvesh AS, Geldenhuys WJ. Markers associated with testosterone enhancement of methamphetamine‐induced striatal dopaminergic neurotoxicity. Neurotoxicol Teratol. 2012;34(3):338‐343. [DOI] [PubMed] [Google Scholar]
- 34. Geldenhuys WJ, Bezuidenhout LM, Dluzen DE. Effects of a novel dopamine uptake inhibitor upon extracellular dopamine from superfused murine striatal tissue. Eur J Pharmacol. 2009;619(1–3):38‐43. [DOI] [PubMed] [Google Scholar]
- 35. Geldenhuys WJ, Darvesh AS, Dluzen DE. Dimebon attenuates methamphetamine, but not MPTP, striatal dopamine depletion. Neurochem Int. 2012;60(8):806‐808. [DOI] [PubMed] [Google Scholar]
- 36. Zhou S‐F. Drugs behave as substrates, inhibitors and inducers of human cytochrome P450 3A4. Curr Drug Metab. 2008;9(4):310‐322. [DOI] [PubMed] [Google Scholar]
- 37. Jones BE, Maerz MD, Buckner JH. IL‐6: a cytokine at the crossroads of autoimmunity. Curr Opin Immunol. 2018;55:9‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hirano T, Matsuda T, Turner M, et al. Excessive production of interleukin 6/B cell stimulatory factor‐2 in rheumatoid arthritis. Eur J Immunol. 1988;18(11):1797‐1801. [DOI] [PubMed] [Google Scholar]
- 39. Genovese MC, Fleischmann R, Kivitz AJ, et al. Sarilumab plus methotrexate in patients with active rheumatoid arthritis and inadequate response to methotrexate: results of a phase III study. Arthritis Rheumatol. 2015;67(6):1424‐1437. [DOI] [PubMed] [Google Scholar]
- 40. Kivitz AJ, Gutierrez‐Ureña SR, Poiley J, et al. Peficitinib, a JAK inhibitor, in the treatment of moderate‐to‐severe rheumatoid arthritis in patients with an inadequate response to methotrexate. Arthritis Rheumatol. 2017;69(4):709‐719. [DOI] [PubMed] [Google Scholar]
- 41. Nishimoto N, Yoshizaki K, Miyasaka N, et al. Treatment of rheumatoid arthritis with humanized anti‐interleukin‐6 receptor antibody: a multicenter, double‐blind, placebo‐controlled trial. Arthritis Rheum. 2004;50(6):1761‐1769. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1.
Data Availability Statement
The authors declare that all the data supporting the findings of this study are available within the paper and its Supplemental Data.