

Abstract
Terpene cyclization, one of the most complex chemical reactions in nature, is generally catalyzed by two classes of terpene cyclases (TCs). Cytochrome P450s that act as unexpected TC-like enzymes are known but are very rare. Here, we genome-mined a cryptic bacterial terpenoid gene cluster, named ari, from the thermophilic actinomycete strain Amycolatopsis arida. By employing a heterologous production system, we isolated and characterized three highly oxidized eunicellane derived diterpenoids, aridacins A–C (1–3), that possess a 6/7/5-fused tricyclic scaffold. In vivo and in vitro experiments systematically established a non-canonical two-step biosynthetic pathway for diterpene skeleton formation. First, a class I TC (AriE) cyclizes geranylgeranyl diphosphate (GGPP) into a 6/10-fused bicyclic cis-eunicellane skeleton. Next, a cytochrome P450 (AriF) catalyzes cyclization of the eunicellane skeleton into the 6/7/5-fused tricyclic scaffold via C2-C6 bond formation. Based on the results of quantum chemical computations, hydrogen abstraction followed by electron transfer coupled to barrierless carbocation ring-closure is shown to be a viable mechanism for AriF-mediated cyclization. The biosynthetic logic of skeleton construction in the aridacins is unprecedented, expanding the catalytic capacity and diversity of P450s and setting the stage to investigate the inherent principles of carbocation generation by P450s in the biosynthesis of terpenoids.
Keywords: biosynthesis, cytochrome P450 enzymes, eunicellane diterpenoids, terpene cyclization
Entry for the Table of Contents
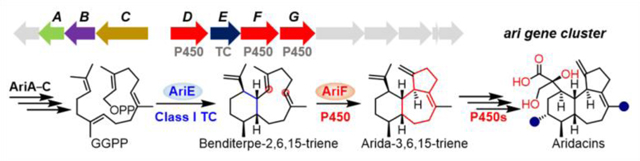
Three highly oxidized eunicellane derived 6/7/5-fused tricyclic diterpenoids, aridacins A−C, were discovered through genome mining. In vivo, in vitro and quantum chemical computations systematically established that terpene cyclase (TC) and P450 work in tandem to create the backbone of aridacins, representing an unprecedented strategy in terpene skeletal construction.
Introduction
The eunicellanes, a unique subfamily of diterpenoids that contain more than 360 examples, are mainly found in Octocorallia soft corals with only a few known members isolated from plants and bacteria.[1–8] Structurally, all eunicellanes share a 6/10-bicyclic skeleton with most representatives, particularly those from corals, exhibiting diverse oxidation patterns.[9] These structures imbue eunicellanes with promising bioactivities[1,2] with examples including the anti-inflammatory klysimplexin R,[10] the antimetastatic polyanthellin A,[11] and the potent tubulin inhibitor eleutherobin.[12] Therefore, they are fascinating targets for both chemists and biologists (Figure 1A). The biosynthetic logic of eunicellanes has remained poorly understood; the diterpene cyclases responsible for construction of the cis and trans 6/10-bicyclic skeletons were only recently disclosed from bacteria and corals.[3,4,8,13,14]
Figure 1.

(A) Representative bioactive eunicellane diterpenoids; (B) P450s serve as unexpected TCs in terpenoid biosynthesis.
Terpene cyclizations are one of the most complex chemical reactions in nature. These intriguing reactions are catalyzed by a superfamily of enzymes named terpene cyclases (TCs), which utilize nature’s library of acyclic C5n diphosphate precursors to generate complex mono- or polycyclic terpene skeletons via carbocation intermediates. TCs are grouped into two canonical classes according to their biochemical strategies to generate the initial carbocation: class I TCs generate carbocations by abstracting the diphosphate group and class II TCs protonate an alkene or epoxide of the prenyl diphosphate; the products of class II TCs often serve as substrates for class I TCs providing a two-step sequential route to structural diversity in terpenoid biosynthesis (Figure S1).[15] Lately, a number of unconventional enzymes exhibiting TC-like reactions have been unveiled, yet diverging from canonical TCs in terms of sequence and structure.[16]
Cytochrome P450 enzymes (P450s) are heme-containing monooxygenases that serve as ubiquitous tailoring enzymes that are capable of catalyzing diverse oxidation reactions including carbon-carbon bond formation in natural products biosynthesis.[17,18] P450s utilize a single-electron process to produce radical intermediates via Compound I (a high-valent oxoiron cationic radical) which readily abstracts a hydrogen from the substrate to yield reactive radical species. Typically, the radical species rapidly rebounds with the hydroxyl radical species, which is derived from molecular oxygen, to generate the hydroxylation product.[17,18] Alternatively, though rarely observed, an electron from the substrate can be transferred to the radical center resulting in a reactive carbocation, which can stimulate a series of cyclization or rearrangement reactions as seen in TC chemistry.[15,16] P450s that serve as TC-like enzymes are known but rare in the biosynthesis of terpenoids. The three known examples are (i) PenM/PntM catalyzing the final step of oxidative rearrangement via a neopentyl cation intermediate in the biosynthesis of the bacterial sesquiterpenoid pentalenolactone;[19,20] (ii) TwCYP71BE86, a plant P450, mediating a methyl shift of the abietane-type diterpene scaffold in triptonide biosynthesis;[21] and (iii) VrtK, which resembles class II TCs and initiates cyclization in the biosynthesis of the fungal meroterpenoid viridicatumtoxin (Figure 1B).[22] However, these P450s operate exclusively at the termini of their respective biosynthetic pathways; no P450s are known to be involved in the initial terpene skeleton construction prior to the tailoring steps.
Herein, we first genome-mined for novel eunicellane diterpenoids by targeting the bacterial biosynthetic gene cluster ari from the thermophilic actinomycete Amycolatopis arida. We then activated this cryptic biosynthetic gene cluster (BGC) by heterologous expression in model Streptomyces hosts and isolated three highly oxidized and eunicellane derived 6/7/5-tricyclic diterpenoids, aridacins A−C (1−3). Their structures were unambiguously established by extensive spectroscopic analyses, including phenylglycine methyl ester (PGME) transformation and single crystal X-ray diffraction. Using a combination of heterologous production, in vivo inactivation, in vitro biochemical experiments, and quantum chemical calculations, we established that a P450, AriF, mediates the formation of the 6/7/5-tricyclic skeleton from the 6/10-bicyclic eunicellane substrate benditerpe-2,6,15-triene (4) via a carbocation mechanism.
Results and Discussion
Terpenoids are the largest family of natural products with over 95,000 known compounds (http://dnp.chemnetbase.com),[23] the vast majority of which have been isolated from plants and fungi; less than 1.5% are from bacteria.[24] However, the genomes of bacteria showcase extensive terpenoid biosynthetic potential that has yet to be revealed.[24] As genes from bacterial secondary metabolism tend to be clustered together, we envisioned that targeting the gene clusters with multiple P450s neighboring TCs might be a practical approach for the discovery of highly oxidized terpenoids.[25] We targeted a putative 16-kb ari BGC from a thermophilic actinomycete Amycolatopsis arida CGMCC 4.5579 (formerly known as Yuhushiella deserti, Figures 2A and S2).[26,27] The ari BGC encodes a class I TC, a UbiA family prenyltransferase, four P450s, a ferredoxin, a geranylgeranyl diphosphate (GGPP) synthase, two genes of 4-hydroxy-3-methylbut-2-enyl diphosphate reductase (IspH) and 1-deoxy-d-xylulose 5-phosphate synthase (DXS), found in the non-mevalonate pathway, and three unknown genes (Figures 2A and Table S1). The sole TC in the ari BGC (named AriE) shares 71.6% protein sequence identity with Bnd4 from the benditerpenoic acid biosynthetic pathway,[8,13] suggesting that this BGC might encode eunicellane derived diterpenoids.
Figure 2.

Biosynthesis of aridacins. (A) The ari biosynthetic gene cluster. (B) The proposed biosynthetic pathway of aridacins. Dashed arrows depict biosynthetic steps that were unable to be established by in vivo or in vitro experiments. (C) Δδ values (δ(S) – δ(R)) of the phenylglycine methyl ester (PGME) amides of 1a and 1b, and the X-ray crystal structure of 1a. (D) EIC (extracted ion chromatogram; m/z 333–349; negative mode) analysis of metabolites from engineered Streptomyces strains. S. lividans TK64 with empty pSET152 was used as a control. (E) HPLC analysis of the metabolites of ΔariD and ΔariF constructs in S. lividans. (F) HPLC analysis of the metabolites of engineered E. coli strains. DL10025 is a GGPP-overproducing E. coli strain and is used as a control. DL10026–DL10028 are DL10025 harboring ariE, ariE + ariD, and ariE + ariF, respectively.
Initial attempts to isolate ari BGC-related terpenoids from A. arida were unsuccessful, presumably due to the BGC being transcriptionally silent under laboratory culture conditions. To activate the ari BGC, we cloned and heterologously expressed it in three model Streptomyces hosts. Streptomyces lividans DL10011 (Tables S2–S4), which harbors the entire ari BGC (orf1–orf6), showed three new peaks (1–3) by HPLC-MS with m/z [M−H]– at 333, 349, and 349, respectively, when cultured in several terpenoid production media (Figures 2D and S3–S6).[28,29] PTMM medium provided higher titers of 2 and 3 while XTM medium was more conducive to produce 1.[29] Therefore, DL10011 was individually fermented on a 15-L scale in both PTMM and XTM media to isolate aridacins A–C (1–3, Figure 2B).
Aridacin A (1) had the molecular formula C20H30O4 as deduced by the (–)-HRESIMS ion at m/z 333.2063 (calcd 333.2071, [M – H]–), indicating six degrees of unsaturation (Figure S7). The 1H NMR (Figure S8 and Table S5) exhibited characteristic signals for two olefinic protons at δH (4.90 and 4.81) and two methyl groups at δH (1.61 and 0.90). Its 13C NMR and DEPT spectra showed 20 carbon resonances including two methyls, eight methylenes, five methines, and five quaternary carbons (Figures S9 and S10). The planar structure of 1, a 6/7/5-fused tricyclic diene, was elucidated by the spin systems in the 1H-1H COSY spectrum of H2-4/H2-5 and H2-8/H2-9/H1-10/H1-11/H2-12/H2-13/H1-14/H1-1/H1-2, and the key HMBC correlations of H-1 with C-15, H2-17 with C-15 and C-16, and H2-20 with C-2, C-3, and C-4 (Figures S11–S13). The key ROESY correlations of H-17a/H-1, H-17a/H-10, along with H-2/H-11 suggested that H-10 and H-11 were β- and α-orientated, respectively (Figure S14). Analyses of the NMR data of 2 and 3 concluded they were analogs of 1 (Figures S15–S28 and Tables S6 and S7). In comparison with 1, aridacins B (2) and C (3) have additional hydroxyl groups at C-19 and C-12, respectively (Figure 2B). To determine the stereochemical configuration of C-15 of the glyceric acid moiety in 1, two derivatives, 1a and 1b, were synthesized using (R)- and (S)-PGME (Figures S29–S35 and Table S8);[30] the stereogenic center at C-15 was determined to be S configured based on the Δδ-values (δ(S) – δ(R)) shown in Figure 2C. The absolute configuration of 1a was further determined by X-ray crystal structure (CCDC 2216506, Figure 2C).[31]
Aridacins A–C (1–3) are bona fide bacterial diterpenoids possessing a 6/7/5-fused tricyclic scaffold, despite the scaffold being previously seen in TC studies (Figure S36). Odyverdiene B was produced by heterologously expressing nd90_0354 from Streptomyces sp. ND90 in a Streptomyces host;[32,33] isocatenula-2,14-diene was produced by incubating GGPP with diterpene cyclase CaCS from Catenulispora acidiphila.[34] The highly oxidized glyceric acid moiety, which may originate from a six-electron oxidation of the C-17 methyl group and sequential epoxidation and hydrolysis of the C-15 alkene, is also uncommon in terpenoids.[35] Aridacins A–C were screened for cytotoxicity in five cancer cells and antibacterial activity in a panel of Gram-positive and Gram-negative bacteria. However, 1–3 showed no significant biological activities (Tables S9 and S10).
To probe the biosynthetic pathway of 1–3, we first identified the boundary of ari BGC (Figures S37 and S38). We heterologously expressed ariA–G in S. lividans TK64 to yield S. lividans DL10016, which, based on HPLC-MS analysis, produced equal amounts of 1–3 with that of S. lividans DL10011 (Figure 2D, panels iii and iv). This result supported that (i) ariA–G can be assigned putative roles in the biosynthesis of aridacins; (ii) the UbiA-like gene (orf1), of which several members of this family are known as TCs, is not involved in cyclization or any other step, and (iii) three P450s genes, ariD, ariF and ariG, are sufficient to decorate the diterpene skeleton. When ariA–C were removed (i.e., ariD–G), the engineered S. lividans DL10017 strain produced significantly less (<10%) of 2–3 than DL10011, suggestive of their important roles in precursor flux but not essential roles in the biosynthesis of 1–3 (Figure 2D, panel v). To further determine the function of AriD–G, we constructed a series of heterologous expression strains, each lacking one of the ariA–G genes in S. lividans DL10016 (ΔariD, ΔariE, ΔariF, and ΔariG; Figures 2D, panels vi–ix, and S39). In the ariE knockout strain (S. lividans DL10019), 1–3 was expectedly abolished. The ΔariG mutant (S. lividans DL10021) only accumulated 1 and 2, indicating AriG is responsible for installing the C-12 hydroxyl group of 3. Both ΔariD (S. lividans DL10018) and ΔariF mutants (S. lividans DL10020) completely abolished the production of 1–3, but intriguingly produced the highly hydrophobic products 4 and 5, respectively (Figures 2E and S40). Due to low titers of 4 and 5 from these heterologous hosts, however, we were unable to accumulate enough material for structural elucidation.
We next cloned ariE into our GGPP-overproduction E. coli system[36,37] to yield E. coli DL10026, and produced 20 mg of 4. 1H and 13C NMR analyses confirmed its structure as the cis-eunicellane benditerpe-2,6,15-triene (Figures 2F and S41–S43).[8] We also cloned and heterologously produced ariE in E. coli (Figure S44). AriE, incubated with GGPP and Mg2+, resulted in the production of 4 as well as the known 14-membered (R)-(–)-cembrene A (6, Figures 3B and S45–S48).[38,39] Compound 4 was easily transformed into a new cis, trans-6/6/6-fused tricyclic product gersemiene C (7) under mild acidic conditions (Figures 3A and S49–S56, and Table S11);[40] a similar transformation was seen for the trans-eunicellane albireticulene resulting in two trans, trans-6/6/6 isomers.[14] This non-enzymatic cyclization occurs via protonation at C-6 of 4 followed by 2,7-annulation and deprotonation at C-20 to access 7 (Figure S49). Taken together, AriE merely produces the 6/10-fused bicyclic eunicellane diterpene skeleton but not the 6/7/5-fused tricyclic scaffold seen in 1–3.
Figure 3.

Biochemical characterization of TC AriE and P450 AriF. (A) The comprehensive functions of AriE and AriF. The almond area represents the native pathway of ari BGC, while the blanched light grey area represents a shunt pathway. (B) HPLC profiles of the in vitro reactions of AriE with GGPP. (C) HPLC profiles of the in vitro reactions of AriF with benditerpe-2,6,15-triene (4) and the redox partner CamA/CamB. (D) EIC (m/z 301 and 317; negative mode) analysis of metabolites from S.lividans DL10011. S.lividans TK64 with empty pSET152 was used as a control. (E) EIC (m/z 301 and 317; negative mode) analysis of S.albus DL10029 (harboring ariF) that was supplemented with 4. (F) EIC (m/z 301 and 317; negative mode) analysis of the in vitro reaction of AriF with 8 and the redox partner FdR/Fdx.
Given the accumulation of 4 in the ΔariF mutant, we proposed that the P450 AriF catalyzes the third ring formation immediately after terpene cyclization. To validate this hypothesis, we cloned a codon-optimized version of ariF into E. coli DL10026, the producer of 4, with the redox partner RhFRed to yield E. coli DL10028. Besides 4, DL10028 produced a new compound with an identical retention time with that of 5 seen in the ΔariF mutant (Figure 2F). A large-scale (30-L) fermentation was performed to collect 3 mg of 5. The GC-MS of 5 showed a molecular ion peak at m/z 270.17 (Figure S57), corresponding to a diterpene hydrocarbon of molecular formula C20H30 with six degrees of unsaturation. Its 1D and 2D NMR spectra suggested that 5 was an analogue of aridacin A (1) with an isopropenyl group in place of the glyceric acid side chain in 1 (Figures S58–S63 and Table S12). This structural difference was supported by 1H NMR data (δH 4.84 and 4.81, CH216; 1.65, CH317) and 13C NMR data (δC 148.6, C15; 111.6, CH216; and 22.6, CH317). Thus, 5 was assigned as a 6/7/5-fused tricyclic diterpene and named arida-3,6,15-triene.
To obtain direct evidence that AriF catalyzes the unusual cyclization of 4 into 5, we sought to confirm enzymatic activity via in vitro experiments. Unfortunately, due to unsuccessful expression of either wild-type or codon-optimized ariF in E. coli, we produced AriF in S. albus J1074 (Figure S64). AriF was assayed in the presence of 4, NADPH, and potential redox partners (CamA/CamB, RhfRed, or FdR/Fdx).[41] AriF produced a single enzymatic product 5 when paired with either CamA/CamB or RhfRed, with much higher production of 5 observed with CamA/CamB (Figures 3C and S65). Overall, these data conclusively support that the nascent 6/7/5-fused tricyclic skeleton of 1–3 is created by a step-wise combination of a class I TC (AriE) and P450 (AriF), an unprecedented event in the biosynthesis of core terpene skeletons.
In a recent study, Hu et al. demonstrated heterologous expression of ariD–G genes in S. albus J1074M, leading to the production of a 6/10-fused eunicellane diterpenoid (9, Figure 3A), albeit without the detection of aridacins (1–3).[42] To elucidate the biosynthetic control of 6/7/5-tricyclic versus 6/10-bicyclic diterpenoids by the ari BGC, we reanalyzed the metabolites from our engineered strains. Based on HPLC-MS analysis, besides the aridacins, which were major products, S. lividans DL10011 also produced trace amounts of 8–11 (Figures 3D, S66, and S67), whereas S. albus DL10015 only produced 9 and 11 (Figure S68). Both ΔariE and ΔariF mutants completely abolished the production of 1–3 and 8–11. Significantly, the ΔariD mutant yielded traces of 8–11 (Figures S37, S38, S66, and S67). Then, S. lividans DL10011 was fermented on a 41-L scale to accumulate 8−11. The 1D and 2D NMR data supported that the structure of 8 is highly similar to that of 9, except for the substitution of the hydroxymethyl group at C-19 in 9 with a methyl group in 8 (Figures S69–S75 and Table S13). Compared to 8, 10 features an additional ethylene oxide at C-15–C-16 instead of a terminal double bond (Figures S76–S82 and Table S14); 11 possesses a hydroxymethyl group at C-17 (Figures S83–S89 and Table S15).
Subsequently, we fed 4 into S. albus DL10029 (harboring ariF), which resulted in the complete conversion of 4 to 5 and 8–11 (Figures 3E and S90). However, the previous in vitro experiment of AriF with 4 showed that only 5 was observed. The challenge in converting 4 to 8, or in achieving further oxidation of products 9–11 in an in vitro assay, might be attributed to the limited activity of AriF with unsuitable redox partners. Nonetheless, upon incubation of AriF with 8, NADPH, and redox partners CamA/CamB, RhfRed, or Fdx/FdR, the presence of 9–11 was observed across all three redox partner systems (Figures 3F and S91). Despite their relatively small amounts, these compounds could be discerned via EIC. Hence, these data suggested that 8–11 emerge as shunt products within the ari BGC, with their formation orchestrated by the P450 AriF. It remains to be seen which diterpenoids the ari BGC produces in its native host, A. arida.
We initially proposed three possible pathways for the formation of arida-3,6,15-triene (5) mediated by AriF (Figure 4). In pathway (i), AriF abstracts a hydrogen from CH3-20 of 4 via Compound I to yield radical intermediate a, which undergoes electron transfer rather than oxygen rebound to form allylic carbocation b. A subsequent 2,6-ring closure yields the tricyclic skeleton with deprotonation at C-6 providing 5. In pathway (ii), hydrogen abstraction and cation formation occur at C-4 followed by a 1,3-hydride shift to form intermediate b; cyclization and deprotonation then follow as in pathway (i). In pathway (iii), a 2,6-ring closure of a generates radical intermediate f, which can be oxidized and deprotonated to form 5. In addition, a radical rebound mechanism employed by AriF could generate the shunt product 8 (pathway (iv)).
Figure 4:

Possible catalytic mechanisms for P450 AriF. Three plausible pathways (i, red), (ii, green) and (iii, blue) for the formation of the 6/7/5-fused tricyclic scaffold from 4. Pathway (iv, black) represents the formation of 8. SET = single electron transfer, HAT = hydrogen atom transfer.
Quantum chemical computations were carried out to examine the three possible cyclization mechanisms. Geometry optimizations and energies were obtained using the mPW1PW91/6–31+G(d,p) density functional theory method,[43–46] which has been used extensively for modeling terpene-forming carbocation reactions.[47–49] Structures for radicals a and d were readily located. While a structure for carbocation c was readily located, attempts to locate b and e with the relative stereochemical configuration corresponding to 5 led directly to structures with 6/7/5-fused tricyclic frameworks, i.e., c was formed when attempting to locate b and its analog when attempting to locate e. These results suggest that the cations formed from radicals a and d have their π-bonds and cation centers so close in space that there is no barrier for them to combine. In addition, since product 5 is observed, rather than a product of direct cyclization of carbocation e (i.e., 6/7/5 tricycle with an alkene between C-3 and C-4), it seems plausible that initial hydrogen abstraction occurs at C-20, i.e., pathway (ii) is unlikely.
Reactions in the presence of the [Fe]=O cofactor were then examined (Figure 5). For these calculations, the B3LYP-D3/6–31+G(d,p)-LANL2DZ(Fe) method was used.[43,50–52] At this level of theory, the [Fe]=O species has doublet and quartet states that are very close in energy — the quartet is predicted to be only ~0.2 kcal/mol higher. The free energy barrier for H-transfer to form a from 4 in the doublet state is predicted to be 23 kcal/mol and that reaction is predicted to be exergonic by approximately 5 kcal/mol. Radical a can then undergo multiple reactions (Figure 4). Ring closure to form f is predicted to have a barrier from doublet complex a of approximately 15 kcal/mol and be exergonic by approximately 10 kcal/mol, making pathway (iii) toward 5 energetically viable.
Figure 5.

Computed energetics for the formation of intermediate a and its reactions. Geometries of key transition structures are shown (distances in Å).
Radical rebound of a (pathway (iv)) was also examined. This process was predicted to have a barrier of approximately 4 kcal/mol and be quite exergonic in the quartet state (Figure 5). Rebound is expected to be barrierless in the doublet state from productive [Fe]-OH conformations,[53] and we were unable to find a transition structure for rebound of doublet a, in line with this expectation. Both of these observations are consistent with the observation of hydroxylated products.
What of the carbocation cyclization pathway (pathway (i))? Estimating the energetics for electron transfer is difficult at best. Shaik et al. have argued against the feasibility of electron transfers in P450s to form allylic cations from allylic radicals.[53] Along these lines, using separate [Fe]–OH and terpenyl species (i.e., not complexes [and mPW1PW91 energies for the cations here]), we predict that electron-transfer from a to form b is endothermic by ~60 kcal/mol (note that this estimate uses the triplet for the neutral [Fe]–OH, which is predicted to be ~11 kcal/mol lower in energy than the singlet, and c, because b is not a minimum). But these energetics neglect the benefits of ion-pairing; as also noted by Shaik et al., the electron affinity of [Fe]–OH can differ by approximately 40 kcal/mol between the gas phase and a “polarizing medium” with a modest dielectric constant. Immersing our structures in a chloroform continuum (using SMD),[54] which is sometimes used as a very crude model of the polarizable environment of an enzyme active site, leads to the prediction that electron-transfer from a to c is exothermic by approximately 9 kcal/mol. In these calculations, however, the structures were not re-optimized. Ion pairing was still neglected here and was estimated by Shaik et al. be worth approximately 14 kcal/mol,[53] which would lead to a prediction that the electron transfer is more than 20 kcal/mol downhill. Why would electron transfer be so favorable here if Shaik et al. argue against electron transfer for analogous cases with analogous polarizable continuum environments? It seems the reason is that electron-transfer and cyclization (formation of a new C–C δ-bond) are coupled in this case.
In summary, electron transfer from radical a to form a carbocation (pathway (i)) appears to be possible, as it is driven by being coupled to ring-closure. Direct ring-closure from radical a (pathway (iii)) has a barrier that is feasible but likely quite a bit higher. Altogether, pathway (i) is appears to be the most likely mechanistic route for AriF cyclization of the cis-eunicellane skeleton of 4 into arida-3,6,15-triene (5).
Conclusion
Most of the skeletal diversity of terpenes in nature are generated by TCs, enzymes that generate structurally and stereochemically complex polycyclic hydrocarbons from acyclic precursors. However, the enzymatic repertoire capable of performing terpene cyclization is far more diverse than a single enzyme family.[16] P450s that serve as TC-like enzymes are known but rare in the biosynthesis of terpenoids. In this study, we used genome mining and discovered a new class of highly oxidized eunicellane derived 6/7/5-fused tricyclic diterpenoids aridacins A−C (1–3) and the shunt benditerpenoic acid-like eunicellane diterpenoids (8–11) from the thermophilic actinomycete A. arida. Heterologous production, in vivo inactivation, and in vitro enzyme characterization systematically established that a TC and P450, AriE and AriF, respectively, successively generate the 6/10-fused bicyclic cis-eunicellane skeleton and the 6/7/5-fused tricyclic scaffold. Furthermore, AriF was also demonstrated to mediate hydroxylation and epoxidation in shunt products (8–11) formation, suggesting that its potential utility as a catalytic tool in the production of structurally diverse terpenoids. Quantum chemical computations are consistent with the proposal that AriF promotes generation of a carbocation via an electron transfer coupled to a barrierless cyclization event – to our knowledge, an unprecedented mechanistic scheme in terpene biosynthesis.[20–22,55] Although three P450s have been previously reported to mediate terpene rearrangements or cyclization in (mero)terpenoid biosynthesis, they all function toward the end of their biosynthetic pathways. In the biosynthesis of the aridacins, a TC and P450 work in tandem to initially create the backbone of the aridacins, an unprecedented strategy in terpene skeletal construction. Our discovery expands the catalytic capacity and diversity of P450s and sets the stage for future efforts to investigate the inherent principles of carbocation generation by P450s in the biosynthesis of terpenoids.
Supplementary Material
Acknowledgements
We wish to thank Prof. Ning-Hua Tan from China Pharmaceutical University for generous access to the GC-MS equipment. We sincerely thank Prof. Hui Ming Ge from Nanjing University for providing compound 9 as a standard and access to the HRESIMS equipment. We are grateful to Profs. Shengying Li from Shandong University and Hans Renata from Rice University for kind providing plasmids pET28a(+)-Fdx and pET28a(+)-FdR, and pET28a(+)-RhfRed and pETduet-1-Opt13, respectively. D.J.T. gratefully acknowledges the NSF XSEDE program. This work is supported in part by the National Natural Science Foundation of China Grant 82073746 (L.-B.D.), the National Institutes of Health Grants R00 GM124461 (J.D.R.) and R35 GM142574 (J.D.R.), the Thousand Youth Talents Program of China (L.-B.D.), the Jiangsu Specially Appointed Professor Program (L.-B.D.), the Jiangsu Funding Program for Excellent Postdoctoral Talent Program (Q.Y.), and the Postgraduate Research & Practice Innovation Program of Jiangsu Province (Z.W.).
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Supporting information for this article is given via a link at the end of the document.
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.
References
- [1].Li G, Dickschat JS, Guo Y-W, Nat. Prod. Rep 2020, 37, 1367–1383. [DOI] [PubMed] [Google Scholar]
- [2].Welford AJ, Collins I, J. Nat. Prod 2011, 74, 2318–2328. [DOI] [PubMed] [Google Scholar]
- [3].Burkhardt I, de Rond T, Chen PY-T, Moore BS, Nat. Chem. Biol 2022, 18, 664–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Scesa PD, Lin Z, Schmidt EW, Nat. Chem. Biol 2022, 18, 659–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Pinto AC, Pizzolatti MG, Epifanio RDA, Frankmölle W, Fenical W, Tetrahedron 1997, 53, 2005–2012. [Google Scholar]
- [6].Zhang B-Y, Wang H, Luo X-D, Du Z-Z, Shen J-W, Wu H-F, Zhang X-F, Helv. Chim. Acta 2012, 95, 1672–1679. [Google Scholar]
- [7].Ma L-F, Chen M-J, Liang D-E, Shi L-M, Ying Y-M, Shan W-G, Li G-Q, Zhan Z-J, J. Nat. Prod 2020, 83, 1641–1645. [DOI] [PubMed] [Google Scholar]
- [8].Zhu C, Xu B, Adpressa DA, Rudolf JD, Loesgen S, Angew. Chem. Int. Ed 2021, 60, 14163–14170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Scesa PD, Schmidt EW, Angew. Chem. Int. Ed 2023, e202311406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chen B-W, Chao C-H, Su J-H, Tsai C-W, Wang W-H, Wen Z-H, Huang C-Y, Sung P-J, Wu Y-C, Sheu J-H, Org. Biomol. Chem 2011, 9, 834–844. [DOI] [PubMed] [Google Scholar]
- [11].Hassan HM, Khanfar MA, Elnagar AY, Mohammed R, Shaala LA, Youssef DTA, Hifnawy MS, El Sayed KA, J. Nat. Prod 2010, 73, 848–853. [DOI] [PubMed] [Google Scholar]
- [12].Chen X-T, Bhattacharya SK, Zhou B, Gutteridge CE, Pettus TRR, Danishefsky SJ, J. Am. Chem. Soc 1999, 121, 6563–6579. [Google Scholar]
- [13].Xu B, Tantillo DJ, Rudolf JD, Angew. Chem. Int. Ed 2021, 60, 23159–23163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Li Z, Xu B, Kojasoy V, Ortega T, Adpressa DA, Ning W, Wei X, Liu J, Tantillo DJ, Loesgen S, Rudolf JD, Chem. 2023, 3, 698–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Christianson DW, Chem. Rev 2017, 117, 11570–11648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Rudolf JD, Chang C-Y, Nat. Prod. Rep 2020, 37, 425–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Tang M-C, Zou Y, Watanabe K, Walsh CT, Tang Y, Chem. Rev 2017, 117, 5226–5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Rudolf JD, Chang C-Y, Ma M, Shen B, Nat. Prod. Rep 2017, 34, 1141–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhu D, Seo M-J, Ikeda H, Cane DE, J. Am. Chem. Soc 2011, 133, 2128–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Duan L, Jogl G, Cane DE, J. Am. Chem. Soc 2016, 138, 12678–12689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hansen NL, Kjaerulff L, Heck QK, Forman V, Staerk D, Møller BL, Andersen-Ranberg J, Nat. Commun 2022, 13, 5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chooi YH, Hong YJ, Cacho RA, Tantillo DJ, Tang Y, J. Am. Chem. Soc 2013, 135, 16805–16808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Faylo JL, Ronnebaum TA, Christianson DW, Acc. Chem. Res 2021, 54, 3780–3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Rudolf JD, Alsup TA, Xu B, Li Z, Nat. Prod. Rep 2021, 38, 905–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wilson ZE, Brimble MA, Nat. Prod. Rep 2021, 38, 24–82. [DOI] [PubMed] [Google Scholar]
- [26].Mao J, Wang J, Dai HQ, Zhang ZD, Tang QY, Ren B, Yang N, Goodfellow M, Zhang LX, Liu ZH, Int. J. Syst. Evol. Microbiol 2011, 61, 621–630. [DOI] [PubMed] [Google Scholar]
- [27].Nouioui I, Carro L, García-López M, Meier-Kolthoff JP, Woyke T, Kyrpides NC, Pukall R, Klenk H-P, Goodfellow M, Göker M, Front. Microbiol 2018, 9, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Dong L-B, Zhang X, Rudolf JD, Deng M-R, Kalkreuter E, Cepeda AJ, Renata H, Shen B, J. Am. Chem. Soc 2019, 141, 4043–4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Dong L-B, Rudolf JD, Deng M-R, Yan X, Shen B, ChemBioChem 2018, 19, 1727–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yabuuchi T, Kusumi T, J. Org. Chem 2000, 65, 397–404. [DOI] [PubMed] [Google Scholar]
- [31].Deposition Number 2216506 (for 1a) contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- [32].Yamada Y, Arima S, Nagamitsu T, Johmoto K, Uekusa H, Eguchi T, Shin-ya K, Cane DE, Ikeda H, J. Antibiot 2015, 68, 385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Yamada Y, Kuzuyama T, Komatsu M, Shin-Ya K, Omura S, Cane DE, Ikeda H, Proc. Natl. Acad. Sci. U.S.A 2015, 112, 857–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Li G, Guo YW, Dickschat JS, Angew. Chem. Int. Ed 2021, 60, 1488–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Vyry Wouatsa NA, Misra LN, Venkatesh Kumar R, Darokar MP, Tchoumbougnang F, Nat. Prod. Res 2013, 27, 1994–1998. [DOI] [PubMed] [Google Scholar]
- [36].Li F-R, Lin X, Yang Q, Tan N-H, Dong L-B, Beilstein J. Org. Chem 2022, 18, 881–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Pan X, Du W, Zhang X, Lin X, Li F-R, Yang Q, Wang H, Rudolf JD, Zhang B, Dong L-B, J. Am. Chem. Soc 2022. 144, 22067–22074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Schwabe R, Farkas I, Pfander H, Helv. Chim. Acta 1988, 71, 292–297. [Google Scholar]
- [39].Rinkel J, Lauterbach L, Rabe P, Dickschat JS, Angew. Chem. Int. Ed 2018, 57, 3238–3241. [DOI] [PubMed] [Google Scholar]
- [40].Angulo-Preckler C, Genta-Jouve G, Mahajan N, de la Cruz M, de Pedro N, Reyes F, Iken K, Avila C, Thomas OP, J. Nat. Prod 2016, 79, 1132–1136. [DOI] [PubMed] [Google Scholar]
- [41].Li S, Du L, Bernhardt R, Trends Microbiol. 2020, 28, 445–454. [DOI] [PubMed] [Google Scholar]
- [42].Hu YL, Zhang Q, Liu SH, Sun JL, Yin FZ, Wang ZR, Shi J, Jiao RH, Ge HM, Chem. Sci 2023,14, 3661–3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lee C, Yang W, Parr RG, Phys. Rev. B 1988, 37, 785–789. [DOI] [PubMed] [Google Scholar]
- [44].Adamo C, Barone V, J. Chem. Phys 1998, 108, 664–675. [Google Scholar]
- [45].Matsuda SP, Wilson WK, Xiong Q, Org. Biomol. Chem 2006, 4, 530–543. [DOI] [PubMed] [Google Scholar]
- [46].Gaussian 16, Revision C.01, Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich AV, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams-Young D, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery JA Jr., Peralta JE, Ogliaro F, Bearpark MJ, Heyd JJ, Brothers EN, Kudin KN, Staroverov VN, Keith TA, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB, and Fox DJ, Gaussian, Inc., Wallingford CT, 2016. [Google Scholar]
- [47].Tantillo DJ, Nat. Prod. Rep 2011, 28, 1035–1053. [DOI] [PubMed] [Google Scholar]
- [48].Tantillo DJ in Comprehensive Natural Products III: Chemistry and Biology (Eds.: Liu H-W, Begley TP), Elsevier, Amsterdam, 2020, pp. 644–653. [Google Scholar]
- [49].A data set collection of computational results is available in the IoChem-BD repository and can be accessed via 10.19061/iochem-bd-6-224. [DOI] [Google Scholar]
- [50].Becke AD, Phys. Rev. A 1988, 38, 3098–3100. [DOI] [PubMed] [Google Scholar]
- [51].Becke AD, J. Chem. Phys 1993, 98, 1372–1377. [Google Scholar]
- [52].Grimme S, WIREs Comput. Mol. Sci 2011, 1, 211–228. [Google Scholar]
- [53].Shaik S, Cohen S, de Visser SP, Sharma PK, Kumar D, Kozuch S, Ogliaro F, Danovich D, Eur. J. Inorg. Chem 2004, 2004, 207–226. [Google Scholar]
- [54].Marenich AV, Cramer CJ, Truhlar DG, J. Phys. Chem. B 2009, 113, 6378–6396. [DOI] [PubMed] [Google Scholar]
- [55].Soler J, Gergel S, Klaus C, Hammer SC, Garcia-Borràs M, J. Am. Chem. Soc 2022, 144, 15954–15968. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.