

Abstract
Biosimilars are biological drugs created from living organisms or that contain living components. They share an identical amino‐acid sequence and immunogenicity. These drugs are considered to be cost‐effective and are utilized in the treatment of cancer and other endocrine disorders. The primary aim of biosimilars is to predict biosimilarity, efficacy, and treatment costs; they are approved by the Food and Drug Administration (FDA) and have no clinical implications. They involve analytical studies to understand the similarities and dissimilarities. A biosimilar manufacturer sets up FDA‐approved reference products to evaluate biosimilarity. The contribution of next‐generation sequencing is evolving to study the organ tumor and its progression with its impactful therapeutic approach on cancer patients to showcase and target rare mutations. The study shall help to understand the future perspectives of biosimilars for use in gastro‐entero‐logic diseases, colorectal cancer, and thyroid cancer. They also help target specific organs with essential mutational categories and drug prototypes in clinical practices with blood and liquid biopsy, cell treatment, gene therapy, recombinant therapeutic proteins, and personalized medications. Biosimilar derivatives such as monoclonal antibodies like trastuzumab and rituximab are common drugs used in cancer therapy. Escherichia coli produces more than six antibodies or antibody‐derived proteins to treat cancer such as filgrastim, epoetin alfa, and so on.
Keywords: biologics, biosimilars, cancer, Escherichia coli, monoclonal antibody, next‐generation sequencing, pharmacovigilance
With crucial mutational categories, biosimilar drug prototypes, and clinical practices including blood and liquid biopsy, cell therapy, gene therapy, recombinant therapeutic proteins, and tailored pharmaceuticals, it aids in targeting certain organs. Monoclonal drugs commonly used in cancer therapy include monoclonal antibodies like trastuzumab, rituximab, infliximab, and epoetin.
Abbreviations
- cfDNA
cell‐free DNA
- CKD
chronic kidney disease
- ctDNA
circulating tumor DNA
- EGFR
epidermal growth factor receptor
- EHPPs
European Healthcare Providers and Patients
- EMA
European Medicines Agency
- EpoR
erythropoietin receptor
- EU
European Union
- FDA
Food and Drug Administration
- MS
mass spectrometry
- NGS
next‐generation sequencing
- NMA
National Medicines Agency
- PK/PD
pharmacokinetics/pharmacodynamics
- RTX
rituximab
- VEGF
vascular endothelial growth factor
- WHO
World Health Organization
1. INTRODUCTION
Biosimilars are drugs similar in structure and function to biological medicine, known as reference biologics. Biologic medicines are derived from living cells and microorganisms such as animal cells, tissues, yeast, and bacteria [1]. They are not biogeneric but are almost a direct copy of the original manufactured pharmaceutical drugs. Biosimilars are less costly for patients than the original reference biologics. They are produced differently than biologics, though, using various cell lines and purification techniques that yield various end products. Nonetheless, they play a crucial role in fostering competition in the pharmaceutical industry and granting patients access to essential medications [2].
2. GUIDELINES OF EUROPEAN MEDICINES AGENCY (EMA) AND FDA‐APPROVED BIOSIMILARS
Biosimilar drugs are effective, safe, and pure as biologics for treating many chronic diseases in patients with illnesses such as kidney failure, growth disorder, inflammatory digestive disorder, diabetes, cancers, arthritis, bowel disease, and so on [2]. Biologic medicine dates back to the 20th century and gained significance in the 21st century. The first biological medicine was approved in the 1980s, thanks to advances in biotechnology, which also produced recombinant proteins and genes to diagnose illnesses and problems in humans [3]. The Food and Drug Administration (FDA) initially approved monoclonal antibodies in 1986. Then, in 1998, the FDA authorized biomedicine for rheumatoid arthritis [4]. In 2006, omnitrope (somatropin), the first biosimilar medication, was approved by Europe [5]. In 2014, the number of biosimilar approvals in the United States and EU increased to 245 [6]. In 2021, the first biosimilar medication for ophthalmology was approved in both Europe and the United States. In 2022, the EMA began reviewing a biosimilar candidate for an orphan indication after accepting the first application for a biosimilar medication for multiple sclerosis [7]. The usage of biologics has increased significantly in several regions of the world over the past few years. In 2016, the agencies and authorized firms accepted 1357 biologics for use in human medication and therapy, of which 737 were biosimilars and the rest were designated as “bio better” [8]. In addition, 73 biological medicines were approved for use in humans from 2013 to 2016. Out of them, monoclonal antibodies gained importance; however, 23 were approved to be used in several treatments including improper bowel movement and neoplastic tumor along with the diagnostic procedures [9]. Global markets experienced tremendous expansion following the introduction of biopharmaceutical medications that had been approved. Regulators and healthcare professionals have acknowledged and expressed trust in the use of biosimilar medications. There exists a key difference between biosimilars and biological products in their originality [10]. The differences lie in originality and physical characteristics such as their composition, structure, regulations, manufacturing processes, and marketing. Biologics are more structurally complex than biosimilars due to their primary alignments between the amino acid sequence and secondary motifs that consist of complicated three‐dimensional structures. These structures can be modified by the process of glycosylation after synthesis for use in biological activities. The complexity and large size of biological characteristics pose a significant challenge to pharmaceutical companies [11]. A comparative analysis of biologics and biosimilars has been listed in Table 1.
Table 1.
| Biosimilars | Biologics |
|---|---|
| Biosimilars are complex due to their biological origin and ultimately require biotechnological processes, which require significantly higher R&D costs. | Biologics are the proteins isolated and purified from living cells whereas biosimilars are produced by imitating a biological product and therefore require many modern technologies. |
| Biosimilars offer greater safety and efficacy than reference products because the protein structure of biologics is more likely to trigger acute and chronic immune responses upon contact with disease. | Biologics are effective and work similarly to biosimilars, but their patents expire sooner. |
| Biosimilars specifically target organs and are suitable for treating conditions such as autoimmune diseases, chronic diseases, skin allergies, and gastrointestinal disorders. They have greater efficacy in treating the above diseases more efficiently than biologics or other chemically synthesized drugs. | Biological medicines also target specific organs when treating diseases, but they are not used for all diseases and are limited to a few diseases, such as inflammatory arthritis, inflammatory bowel disease, neutropenia, and so on. |
| Due to their large structure and high molecular weight, biosimilars are complex to manufacture and sensitive to changes in physical conditions. | The manufacturing process is as complex and expensive to manufacture as biosimilars. |
| Examples—Truxima (rituximab‐abbs), ogivri (trastuzumab‐dkst), zarxio (filgrastim‐sndz), and so on. | Examples—Filgrastim (therapeutic protein), adalimumab (monoclonal antibodies), vaccines such as tetanus, and so on. |
Despite available advanced techniques to characterize the structure and chemical properties of biopharmaceuticals, the inheritance complexity still remains. This makes it difficult to define these products' characteristics fully. Defining them may vary due to different processes of manufacturing. Though biologics and biosimilars have the same goal medically, to treat diseases, they differ in several different ways, starting from product development to commercial path and approval, which may affect patients' access, safety, cost, adaption in clinics, innovation, and pricing [12, 13]. The differences that separate the two from one another are their manufacturing process, types of equipment, and facilities for clinically approved drugs in terms of purity, identity, and safety. Chemically synthesized drugs can be easily analyzed during manufacturing but biosimilars are complex molecules as they contain homogenized proteins [14]. The end process of biosimilars and other drugs is to evaluate pharmacokinetics/pharmacodynamics (PK/PD) which should be FDA‐approved and possess biological characterization for taking care of the human race with safe medicines [15]. The development of biosimilar products implied pharmaceutical strategies with rules and regulations and risks in clinical applications [16]. The FDA helps to provide data and appraisals for new biosimilar drugs with effective strategies, immunogenicity assessments, and statistical considerations. Biosimilars' approval is more complicated and drastic as compared to generic drugs [17]. The EMA has demonstrated the greatest number of biosimilars with updated biologics to date over time. Through its purchasing and pricing policies, the World Health Organization (WHO) establishes regulatory guidelines for the development and approval of biologics [18].
Biosimilar medicines in recent years have gained great momentum as being low‐cost and effective alternatives to biologics. Different agencies have been playing significant roles in ensuring the safety, efficiency, and quality of biosimilars. These regulatory agencies have outlined guidelines to develop and improve the production of biosimilars. The FDA in its guides commits itself to upholding the standards for assessing biosimilars, encompassing analytical, clinical, immunogenicity, and pharmacological aspects, while promoting competition in the biologics market [19]. Similarly, EMA guidelines provide the requirements for comparative qualitative, preclinical, and clinical studies, as well as considering extrapolation to other indicators. On the other hand, the WHO and Health Canada contributed to the guidelines for its harmonization. WHO guidelines focus on a stepwise approach to demonstrate similarity and recommend robust postmarketing surveillance to ensure ongoing safety and efficient monitoring [20]. Health Canada aligns its guidance with international standards and underscores the importance of analytical and clinical comparability studies, emphasizing the need for robust pharmacovigilance programs to address potential risks. These global regulatory guidelines collectively aim to foster a robust and competitive biosimilar market, ensuring that patients have access to safe and effective biological medicines while maintaining the highest standards of quality and safety [21]. For biosimilars to be interchangeable, their efficacy and safety risks should not exceed the originator monoclonal antibody. They should not only demonstrate similarities to biologics but must demonstrate the same clinical result of biologics when administered to patients without healthcare provider intervention. The concept of interchangeability aims to balance between promoting competition and ensuring patient safety by establishing a higher standard for biosimilar products to be used as direct substitutes for their reference biologics. This guideline is left to the national governing authority to make a decision on it, since regulations vary from one regulatory agency to another [22]. Changing a patient's treatment from one biosimilar to another biosimilar can occur for various reasons, including cost savings, availability, or patient preference. Regulatory agencies have provided guidelines for switching from one biosimilar to another biosimilar, ensuring careful monitoring, safety, and treatment efficacy of patients. Substitution, on the other hand, involves the replacement of a prescribed reference biologic with a biosimilar by a pharmacist without the prescriber's involvement. The permissibility of substitution varies from country to country and is subject to regulations and legal frameworks. Some countries may allow or even encourage biosimilar substitution to promote cost savings, while others may require the prescriber's explicit approval. The effectiveness and safety of using biosimilars in everyday life facilitate many medical discoveries and increase positive treatment outcomes in patients which is a basic characteristic of comparing biosimilar to reference drugs. Acting toward a specific goal helps the patient build a shield against the problem. Over the years, biosimilars have had a significant impact on healthcare delivery, especially treating different types of cancer and other health‐related diseases. Biosimilar cancer care depends highly on several factors such as policies of the health system, patients' and healthcare providers' acceptance, and approval from regulatory authorities to gain prominence in the health sector. Moreover, biosimilars are not available on all biologic drugs, the specific drug and competition in the market vary with their cost savings. However, biosimilars make treatment more affordable and accessible for patients and are potentially effective at reducing the cost of cancer care at the same time generating cost‐savings for health delivery systems [23]. As more biosimilars make their way into the markets, their role in improving cancer therapies continues to grow due to their affordability and availability. They have made their way into the healthcare system with the approval of different types to treatment of different types of cancers including stomach cancer, breast cancer, and cervical cancer among other cancers. As patients experience side effects such as low blood count which increases high risk of infections of their cancer therapy, biosimilars can be a significant help for treatment [24].
Some biosimilars currently approved in the United States to treat cancer include filgrastim, and zarxio was approved in March 2015 to help fight infection in patients' bodies with low leukocyte count because patients with cancer receive transplantation and chemotherapy [25]. Bevacizumab‐awwb was approved in September 2017, as the first biosimilar to treat cancers of different body parts like colorectal, brain, cervical, and so on [26]. Trastuzumab dust was approved from 2017 to 2019, to treat breast cancers with a reference name in the market, Herceptin [27]. Pegfilgrastim‐jmdb was approved from 2018 to 2019 to fight against nonmyeloid cancer patients with a reference name of Rituxan in the market [28].
3. REGULATORY CONSIDERATIONS OF NATIONAL MEDICINES AGENCIES ON BIOSIMILARS
To provide insight into the viewpoints of regulators and the usage of biosimilars, this study looks at the guidelines and subjective assessments about biosimilar regulation supplied by the national medicines agencies (NMAs) and the EMA. The majority of the NMAs' websites did not provide any information on biosimilars nor did they provide any educational material. Of the NMAs that did provide guidance, the scope and content varied significantly. Those countries that are heavily involved in EU, or biosimilar regulatory, activities, or guidelines had more comprehensive information across the country [29]. Despite the EU's strong track record of evaluating and approving biosimilars, which has led to the availability of an abundance of biosimilar products with EU‐wide marketing authorizations, the adoption of biosimilars has been successful across healthcare systems. However, several studies have revealed a lack of knowledge and trust in biosimilars among European healthcare providers and patients (EHPPs), suggesting uncertainty and reluctance to use them. This lack of understanding and confidence in biosimilar development presents a novel paradigm of novel drugs, necessitating a deeper understanding of biological medicine and biotechnology. Access to biosimilar development information is essential for both healthcare professionals and patients. This study provides an overview of European information on biosimilar products and their use, as well as considerations on how regulatory measures can enable stakeholder confidence in the utilization of biosimilar medicines [30]. In 2019, the International Coalition of Medicines Regulatory Authorities brought together 29 medicines from other parts of the world, of which the EMA is a member.
The study shows the importance of having access to reliable and transparent information and clear instructions on how to use biosimilars to provide health professionals with a better understanding of them. Additionally, it evaluates the manner in which regulators, both at central and national levels in Europe, disseminate information and guidance regarding the use of biological products, with a particular emphasis on guidance on interchangeability [31]. Nevertheless, regulatory information serves as a foundation for subsequent comprehensive and accurate dissemination of information on biological products and their uses. The results of the study demonstrate that regulatory information on biological substances and their use, including guidance on interchangeability and related practices, vary across NMAs in terms of accessibility, safety, scope, and content [32]. As the scope of the study was limited to the investigation of regulatory guidance, no evaluation of local pricing guidance or authorities or ministries was conducted. Despite advances in cancer treatment, and the accessibility and affordability of biosimilars to treat cancer, the WHO in recent years estimated the number of increasing cancer cases by 60% [33]. In 2018, the total expenditure on all medicinal products used to treat cancer patients reached $150 billion and is projected to reach $240 billion in 2023, with an annual growth rate of 9%–12% [34]. There is a barrier created for cancer treatment due to cost, accessibility, and clinical outcomes. Drugs that are effective in the oncology circle are biological and are not accessible and affordable to patients due to their high cost which prohibits patients from accessing them. On the basis of sales data for 2019, the top‐selling cancer drugs were all biologics based on antibodies (monoclonal). The top‐selling biologics for cancer were pembroizumab; nivolumab, rituximab, bevacizumab, and trastuzumab. These drugs are expensive, which makes cancer treatment inaccessible and unaffordable for patients with various types of cancer [35]. However, with the availability of biologics as substitutes for some of the above‐mentioned reference drugs, there has been price competition among biosimilar competitors against these reference drugs, which has helped to reduce the cost of treatment. Biosimilar cost savings are estimated to be over €10 billion for the EU5 alone in the period of 2016–2022 (IQVIA Institute). It has been estimated that the cost of health care in the US could be reduced by $250 billion over the next 5 years if 11 biosimilars were to be released, including those for Avastin®, Herceptin®, Trastuzumabo®, and Rituxan®, respectively. The low cost of biosimilars could lead to a decrease in healthcare expenditure and improve access to biologic therapy for those with cancer [36]. Currently, the cost‐to‐value ratio of biosimilars to biologics is between 59.4% and 86.0%, with the latter being the most cost‐effective. The cost of using a biosimilar for 1 month is estimated to be between $322 and $7424, whereas the total cost per patient is approximately $17,578 to 38,923 [37]. This price decrease has caused an increase in prescription drug prices and has contributed to a decrease in the financial burden for patients. In the United Kingdom, the launch of the Filgrastim‐Biosimilar caused an increase of 104% in uptake [38]. In Europe, the availability of Epoetin‐Biosimilars between 2007 and 2014 resulted in a 263% increase in treatment volume and a 50% decrease in price in Romania and Bulgaria [39]. Biosimilars for certain oncology drugs have been given the green light. The EMA said that 39 major patents on certain oncology biosimilars had expired by May 2021. The EMA has approved biosimilars for filgrastim, bevacizumab, trastuzumab, pegfilgrastim, and rituximab. Since trastuzumab biosimilars first came out in Europe in 2018 [40].
4. ROLE OF BIOSIMILAR ACCEPTANCE IN THE TREATMENT OF GASTROINTESTINAL DISEASES
In the United States, there are currently six biosimilars approved for the treatment of TNF‐1, but only two of them are widely available [41]. There is some evidence to suggest that biosimilars can be used for both initial treatment initiation and as effective and potentially cost‐effective alternatives to the original biologic. A study was conducted to evaluate the use of prescribed biosimilars and treatment preferences in terms of prescribing behavior and patient attitudes toward biosimilars. The study was based on a cross‐sectional study conducted in 2015–2016 for the treatment of patients with ulcerative colitis or Crohn's disease. Twenty‐five gastroenterologists and 16 patients were recorded for the purpose of the study. Out of the sample, less than 15% of the biosimilars were biologic therapies, while more than 80% of gastroenterologists were willing to prescribe a biologic on a biosimilar. However, patients were hesitant to accept the use of biosimilars as first‐line treatment, with 79% being satisfied with the treatment and 69% satisfied with symptom control. On the other hand, 35% of the patients reported no concern when treated with a biologic or biosimilar. In addition to other data, gastroenterologists reported that biosimilars accounted for 12%–13% of biologic therapies and 4%–5% of all drugs. The majority (88%) of gastroenterologists expressed a preference for bio‐originator over biosimilars when asked about their preferences [42]. When asked why they prescribed biosimilars rather than bio‐originators, the majority (89%) and the majority (100) of gastroenterologists, respectively, expressed a desire to “experience” the new product as a medication. In addition, 44% also selected reasons such as efficacy, cost‐effectiveness, and economic stability. The results of this study suggest that there is a lack of acceptance of biosimilars and a need to educate patients on the significance of treatment and physician communications [43]. This may be a reflection of the introduction of biosimilars in the gastroenterology field, as bio‐originator‐based drugs have been available in Europe since 2008 and are estimated to have a market share of 60%–80% across five major European countries [44].
5. PHARMACEUTICAL PRODUCTION OF BIOSIMILAR DRUGS
Advanced therapeutic approaches are involved in the development of biosimilars that differ from chemically driven medical drugs in terms of complex peptides, proteins, and glycoproteins targeting disabilities using entirely new ways of life‐saving medications [45]. The development is predominantly characterized to explain its variability and pharmaco‐toxicological evaluations. Preclinical developments show therapeutic equivalence and relative bioavailability including phase 1 PK and PD. If there is a biosimilar's similarity to other reference medicine demonstrations, the derivation to other demonstrations could be sustainable; however, this needs to be well justified with data from clinical, analytical, and nonclinical sources [46]. An open system of quality control checking is settled for quick and economical assessment of qualitative and quantitative analysis of biosimilar potential drugs along with originators executed with chromatography techniques, protein identification, and separation of isoforms through mass spectrometry (MS) and gel electrophoresis (Figure 1).
Figure 1.
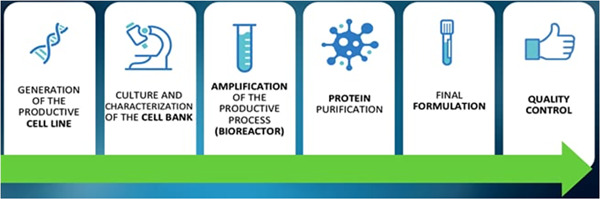
Productive process of biosimilar drugs [47]. (Creative Commons license.)
Biosimilar drug development and manufacturing is a systematic process with three main steps. The first step is to identify the attributes of the reference product, then determine the cell expansion and expression of the biological product, determine how similar the biological product is, isolate and purify the protein, and determine the formulation [48]. In the pharmaceutical industry, specific approaches are needed to make sure biosimilars are very similar to the reference products. The second step is to determine the target quality attributes, clinical effectiveness, and potency of the biosimilar drugs, and also to check how much variability the reference products have compared to each other to make sure there is a good pharmacodynamic similarity [49]. For biosimilars to be considered, the biotherapeutic must go through a strict development process and a stepwise comparability test to show that any differences have no effect on the product's clinical performance compared to the reference product. Biosimilars and generic drugs are similar as they have original brand names and provide options at a lower cost. But there are some big differences. The FDA requires biosimilars to meet strict approval standards, so patients and healthcare workers will have to rely on the biosimilar just like they would on the reference drug. Most biosimilars are made from small molecules like aspirin and are made synthetically. They are usually made in living systems like bacteria or animals using a special type of DNA technology called recombinant DNA. The basic steps for making a biological product are as follows: A gene of interest is injected into a living cell which leads to the synthesis of proteins like antibodies. Subsequently, the particular chemical is released into the culture medium as the cells continue to develop in the bioreactor. After being filtered and chromatographed, the purified product is inspected to determine its structure, functionality, and level of purity before being put into a single container for use. Generic drugs' active ingredients are usually smaller, easier to replicate, and easier to make [22]. But most biological products are made from living systems, so it is expected that there will be slight changes to the protein through the use of recombinant DNA technology as part of the manufacturing process. Basically, each batch of a reference product or biosimilar might have a mix of lots of small variations of proteins that have been isolated from the bacteria E. coli. There may be slight variations in the overall composition; however, these do not have an impact on the end product. Manufacturers of biologics provide data to demonstrate that their products exhibit similar variations as the reference products, as illustrated in Figure 2. Manufacturers must adhere to stringent safety, purity, and potency requirements. This entire process can take over a decade. However, it would be beneficial if biologic products could be made more accessible and cost‐effective to patients [51].
Figure 2.
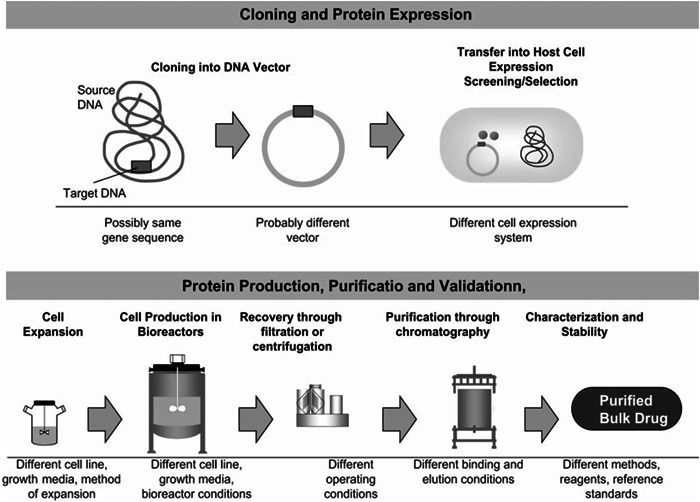
Manufacturing of recombinant protein (obtained from Escherichia coli) and extraction and purification processes in pharmaceutical industries [50], adopted with permission.
Once the original biologic is patented, it is okay for other companies to make a copy. Since the company that made the original biologic does not have to tell anyone how it is made, biosimilar makers have to start from the beginning. They have to look at the original biologic and look at published data to figure out the basic structure. Then, they have to make a three‐dimensional version of the drug with the parts that work and the parts that do not which is a complex process. Scientists have to rely on living cultures, like E. coli, to put together the drugs. Since it is made in a living body, it is impossible to know exactly how it is made. Even small differences in temperature, nutrients, and pH can make a difference in the structure, function, and safety of the drug, however, it can attach to its target just like its original biologics. The use of recombinant technology has had a big impact on biopharma production to show off therapeutic approaches and to study biological products like human growth hormones and interferons. Biosimilars are not only subset of biologic drugs and vaccines but also derivatives of monoclonal antibodies which are involved in immunotherapy, for instance, monoclonal antibodies for the treatment of cancer and neutropenia [52]. A number of drugs manufactured by E. coli are used in the treatment of cancer, human immunodeficiency virus (HIV)‐related neutropenia, leukemia, acquired immunodeficiency syndrome (AIDS)‐related Kaposi's sarcoma, and so on like filgrastim, IFN α2a, IFN α2b [53].
6. MONOCLONAL ANTIBODIES AS BIOSIMILARS
Monoclonal antibodies are employed in immunotherapy tests because of their capacity to activate the immune system against cancer. As shown in Figure 3, they can easily recognize foreign agents, such as germs, bacteria, and viruses that can cause disease, and target them for destruction. They are lab‐made proteins, specifically designed to treat cancer. For example, rituximab, in cancer cases, binds to protein CD20 on B cells (white blood cells), to target and kill cancer cells. Blinatumomab, to treat acute lymphoblastic leukemia, binds to CD19 on B cells and CD3 on T cells, helping the T cell to target and kill leukemia cells. Additional data such as in vitro and in vivo pharmacodynamic studies showed pharmacological actions that the biosimilar extrapolation efficacy is comprehensive to the reference products. Structure, functions, immunogenicity, and pharmacokinetic profiles exemplify the action of substances and receptor targets through biophysical, analytical, operational methods, and clinical studies. The factors responsible for the drug reactions include patient, disease, and immune status [55]. These antibodies can also block the molecules needed for the growth of cancer cells that interrupt the body's vulnerable system, which makes them a precious type of targeted remedy for treating cancer. For illustration, a monoclonal antibody called trastuzumab binds to a patch molecule called HER2 on the surface of cancer cells. Blocking HER2 keeps it from transferring signals, the cancer cells need to grow. Another illustration involves vascular endothelial growth factor (VEGF), which is a patch that makes blood vessels grow. A monoclonal antibody called bevacizumab blocks VEGF. Blocking can stop the growth of new blood vessels that the excrescence needs to survive. A third illustration is the monoclonal antibody pembrolizumab. Pembrolizumab attaches to motes called immune checkpoints on immune cells. Blocking vulnerable checkpoints helps the immune cells kill cancer cells. Other monoclonal antibodies treat cancer by flagging cancer cells for destruction. For illustration, when the monoclonal antibody, rituximab, attaches to a molecule called CD20 in cancer cells, it acts like a flag for vulnerable cells or immune cells [56]. The immune system sees this flag and destroys the cancer cells. They fight cancer by delivering medicines, poisons, or radioactive particles to cancer cells. For example, “Brentuximab vedotin” is a monoclonal antibody that is linked to a chemotherapy medicine. When the antibody attaches to its target on cancer cells, it delivers the chemotherapy drug which kills them [57].
Figure 3.
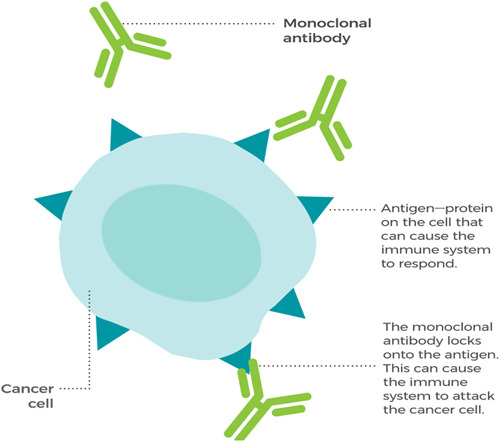
Monoclonal antibodies such as trastuzumab, infliximab, pembrolizumab, etc. help to kill cancer cells, marking the cancer cells so that the immune system can recognize the cells and destroy them [54], adopted with permission.
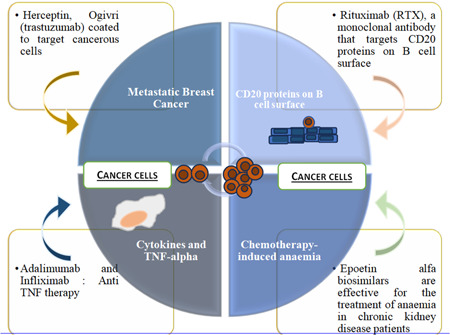
6.1. Herceptin, ogivri (trastuzumab)
Herceptin is a monoclonal antibody that can help the HER signaling that usually leads to cell growth and accumulation and can beget breast cancer. Growth in tissue, epidermal growth factors, as signaling motes, are insulin‐like growth factors and the receptors, insulin‐like growth factor receptors (HER 2). Also, the response is intermediated for cell growth, proliferation, and division [58].
Inhibition can also be attained by a monoclonal antibody, known as trastuzumab (Herceptin). The process starts with many kinds of receptors on the cell face; one is epidermal growth factor receptor (EGFR) dimer. EGFR dimer once bound to the ligand or signaling patch from the surface of the cell gets phosphorylated to the inner cytosolic point and also it activates RAS protein, RAS protein activates RAF protein, RAF activates MEK, MEK activates ERK, and eventually ERK goes inside the nexus and cause activation of specific genes [59].
The alternate growth factor receptor is the HER2/EGFR receptor; the signaling patch binds and the cytosolic point phosphorylated. Another receptor brace, both HER receptor pair (HER2/HER3), a hetero‐dimer, actuated and upon activation cytosolic point gets phosphorylated. Also, HER2/HER4 receptor dyads are also actuated and cytosolic site is phosphorylated [60].
All of the mentioned receptor dyads spark the cytosolic sphere which is also phosphorylated, activates PI3K (kinase), this kinase activates AKT and further activates dissimilar proteins that lead to the activation of transcriptional expressive genes in the nucleus that involves survival, accumulation, and cell cycle progression and that results in cellular growth, and overactivation of this path may be directed toward cell growth that can turn normal cell into tumor cell. And with growing mutations, a standard cell can become cancer cell. In abnormal HER2 breast cancer cell, plenitude of HER2 receptors present is further than the normal volume of HER2 receptors a cell can handle. And due to the amplified number of HER2 receptors, the cell signaling for growth and proliferation increases. In this case, multiple HER2 receptors send further signals causing cells to grow too rapidly. In breast cancer, Herceptin targets HER2 receptors; a drug, a monoclonal antibody prevents dimerization of HER2 receptors [61]. The binding separates from HER2 to become dimerized as shown in Figure 4; therefore, the cytosolic site phosphorylation gets averted, and it will sustain the cell from further growth. The antibody‐drug conjugate, “Trastuzumab deruxtecan,” is approved to treat cancer patients who have preliminarily entered at least two lines of treatment against HER2. A new study examines its efficacy and safety as a second‐line treatment for metastatic breast cancer. In the third trial, a multicenter, open‐label, randomized, controlled trial, 524 cases of patients were registered, metastatic disease progressed after treatment, and taxane were inked to admit either “Trastuzumab deruxtecan” or the usual second‐line treatment, “Trastuzumab emtansine” intravenously every 3 weeks. “Trastuzumab deruxtecan” showed an advantage over “Trastuzumab emtansine” with respect to that has developed after close monitoring for intestinal lung disorder and pneumonitis [62].
Figure 4.
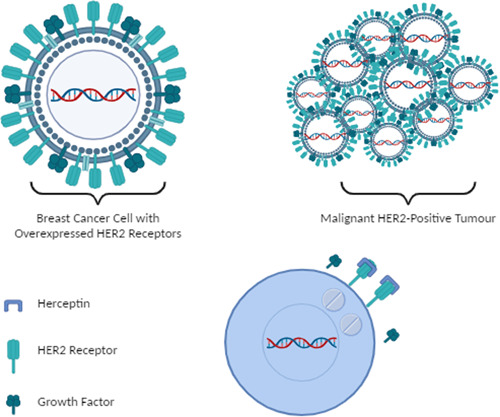
The use of trastuzumab in metastatic breast cancer as early and advanced‐stage disease which shows a picture of malignant HER2‐positive tumor coated with trastuzumab shows drug delivery response to the tumor cell. (The image is created using Biorender.com).
6.2. Rituximab (anti‐CD20 monoclonal antibody)
Rituximab (RTX) is an antibody that targets CD20 proteins on B cell surface. It is used to immunosuppress autoimmune diseases and cancers associated with B cells. It is used in conditions such as non‐Hodgkin's lymphoma, rheumatoid arthritis, and autoimmune hemolytic anemia. RTX targets CD20‐associated B cells and reduces their function [63]. There are several tests that must be done before or during treatment with RTX. These include hepatitis panel test and tuberculosis test before start of treatment. Monitoring of B cell count and quantiflobulin measures the volume of protective proteins produced by the immune system. The first dose of RTX must be administered as an infusion, either in a monitored outpatient infusion suite or in a hospital setting due to the risk of severe allergic reactions. The treatment usually consists of two infusions (2 weeks apart) and will be repeated when the disease has started to resolve or to prevent further disease progression. The duration of the infusion can range from 6 months to several years, but it will not be repeated unless a minimum of 4 months elapsed since the most recent infusion. The initial infusion typically lasts approximately 6 h, and subsequent infusions may take up to 4 h depending on the infusion reaction and the drug used [64]. During the infusion, steroids are administered, acetaminophen is administered, and an antiseptic, such as Benadryl, is administered to reduce the severity of allergic reactions (Figures 5, 6, 7).
Figure 5.
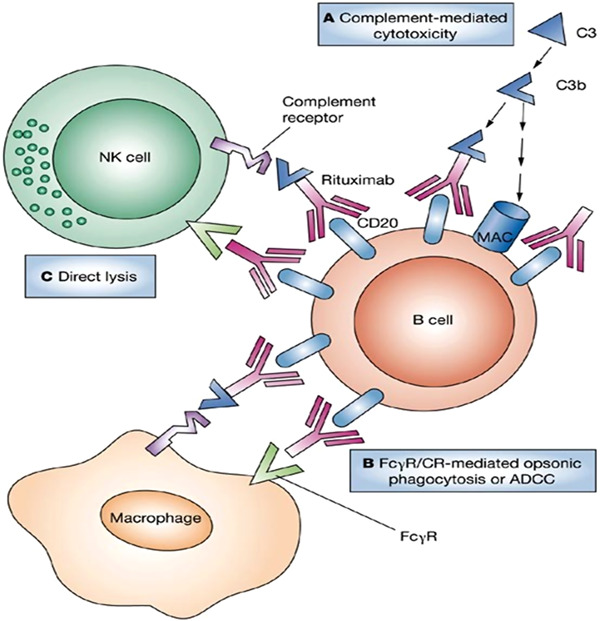
Mechanism of rituximab via binding to B cells, cause breakage; and complete depletion of B cells, rituximab diverts monocytes or macrophages from binding with tissue‐associated immune complexes [65], adopted with permission. ADCC, antibody‐dependent cellular cytotoxicity; CR, complement receptor; NK, natural killer.
Figure 6.
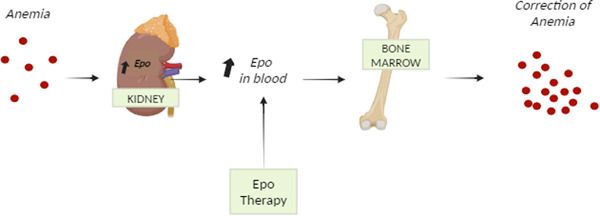
Epoetin alfa biosimilars' mechanism of action. Once bound to the erythropoietin receptor (EpoR), it promotes and proliferates red blood cells and cures anemia. (The image is created using Biorender.com).
Figure 7.
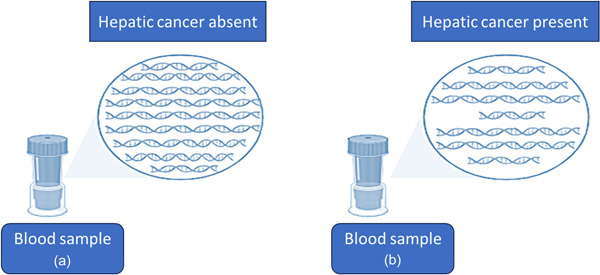
Size of DNA fragments in the blood with/without liver cancer patients. (The image is created using Biorender.com).
The immunological agent rituximab has been shown to interact with the vulnerable system, potentially raising the susceptibility of the body to illnesses such as sore throat, fevers, chickenpox, and shingles. Additionally, Rituxan has been associated with some very serious potential side effects, including a decrease in the number of thrombocytes in the blood, a condition known as platelet aggregation. The most serious of these side effects are progressive multifocal leukocytopenia, a virus‐induced brain infection that can occur in patients receiving Rituxan or those with weakened immune systems. This condition has been associated with death or severe disability, and there is no known cure, treatment, or prevention for it. Despite these potential side effects, the drug has been approved for pretreatment of patients and those with aggressive types of parasympathetic nervous system disorders and has been shown to improve response rates and survival compared to chemotherapy alone [66].
6.3. Adalimumab and infliximab (anti‐TNF therapy)
These monoclonal antibodies are designed to target cytokines and TNF‐α. TNF‐α is a cytokine that is associated with the formation of inflammations in the acute phase of reactions. It is an important cytokine that is utilized in a variety of inflammatory disorders in conditions such as inflammatory bowel disease, rheumatoid arthritis, and psoriasis. These antibodies target the cytokines, attenuate the inflammatory response, and are highly effective in the treatment of these conditions [67].
The clinical development of infliximab was initially conducted in mice, as humans have an immune response to a mouse protein. The amino acid sequences of mice were then replaced with analogous human antibody domains, resulting in the combination of the two. Common brand names for infliximab available in the market include Remicade (5 mg/kg), Remsima (6 mg/kg), Renflexis (7 mg/kg), and Avsola (8 mg/kg). TNF‐α is produced by TNF‐producing cells, and when it attaches to receptor‐presenting cells, an inflammatory response is induced. However, TNF‐α inhibitors bind to this protein and inhibit or remove its binding to its receptors, thereby interrupting the inflammation response. It is recommended that Remicade be administered as an intravenous infusion at the beginning of treatment (0–6 weeks) and that maintenance therapy should be administered at the end of treatment (8 weeks) for 2 h every 8 weeks [68].
7. CHEMOTHERAPY‐INDUCED ANEMIA AND EPOETIN FOR CANCER
Epoetin alfa biosimilars are made by preclinical characterization and pharmacodynamic studies and are effective for the treatment of anemia with proper pharmacosurveillance and monitoring. The severity of anemia in chronic kidney disease patients is high and twice as ubiquitous in health‐related quality of life disability. The characterizations of biosimilars with a series of constructive analyses are administered to ensure the differences and similarities with the reference medicine followed by pharmaco‐toxico‐logical assessments [69]. Clinical trials complementing immunocytochemistry and nonclinical findings with cell studies for biosimilar epoetins were adequately similar to the reference medicine. Here and now, the ongoing studies have provided substitutional options for pharmacodynamic developments with biologically made drugs. Following the severity of renal anemia, clinicians use recombinant human erythropoietin restricted to hemodialysis patients. The erythropoietin efficiency includes qualitative and/or quantitative analysis in which differences can be detected between the two in comparative animal studies. Thermo Scientific DionexUltiMATE 3000 provides solutions and excellent chromatographic performance to examine recombinant human erythropoietin [70]. Erythropoietin (Epo) and its receptor (EpoR) interaction are hypercritical to promote and proliferate the cell survival and differentiation in red blood cell production. To observe the activity of cytokines and reagents on the human EpoR gene, erythroleukemia cell line is used to understand the biosimilar action on erythroid progenitors [71].
8. ONCOLOGY PRACTICE OF BIOSIMILAR DRUGS AND BIOMARKER DISCOVERY
Biosimilar variants are reassigned to new treatment areas. The two biosimilar drugs, filgrastim and epoetin, with cancer‐fighting abilities could expand accessibility to improve patient(s) health. The only concerned factor for oncologists switching from original biological products to biosimilars is the loss of efficacy and adverse events while clinical alteration in the biological drugs that can induce immunogenicity. Despite many studies, biosimilars have proven safe and effective not only for the treatment of cancer but also for other diseases such as Crohn's disease, colitis, and so on [72].
One significant point of using biosimilar drugs is its cost advantage over reference products. The combinations of immunotherapies with pembrolizumab and obinutuzumab show supportive care in prophylaxis patterns. Filgrastim is the first biosimilar agent for prophylaxis of chemotherapy‐induced (febrile) neutropenia; obinutuzumab to treat follicular lymphoma; and pembrolizumab for lung cancer [73]. Therapeutic oncology biosimilars have demonstrated random trials in patients with comparable tumor response and efficacy results to its reference drugs. Trastuzumab biosimilar is the second cancer treatment biosimilar for breast cancer or metastatic gastric adenocarcinoma for 24 weeks. The biosimilar response has been approved after the third trial [74].
A preliminary study of high‐throughput computational analyses and a comprehension of blood test with new “omics” study called fragmentomics have paved the way to detect cancer in the liver. It looks at the DNA fragments. To detect a large number of DNA fragments, a machine‐learning approach, developed by a group of scientists to observe the early cancer growth in the body and fragmentation patterns of prenatal cell‐free DNA (cfDNA) using supervised machine‐learning to distinguish cancer subtypes, named after the evaluation of DNA fragments, that is, DELFI [75]. It discovers the link to predict tissue‐ and tumor‐specific gene expression for multicancer screening. cfDNA molecules are a promising approach to evaluate tumors on the allelic status of mutations or single‐nucleotide polymorphisms for genetic aberrations via deep whole‐genome sequencing. In vivo tumor modes, xenograft models, and quantitative polymerase chain reaction optimized to improve the efficiency of largely set DNA fragments, with the influence of next‐generation sequencing (NGS) technology, to indicate higher cancer‐derived fragmentation, circulating tumor DNA (ctDNA). cfDNA screening monitors for mutations in AKT and PIK3CA genes and tumor cells to prevent resistance for the mild trial as a biomarker [76].
The liquid biopsy test for early detection methods combined fragmentomics with mutations and epigenetic changes for large sets of clinical studies to detect the cancers; lung cancer, colon cancer, breast cancer, and so on and early measuring the consequences of abnormal mitosis via multicancer screening [77]. cfDNA analysis shows the clinical applications to detect the location of tumor or metastatic lesions using single‐nucleotide substitution mutations [78].
cfDNA analysis is a fast‐growing approach for early diagnosis and can be accessible in peripheral blood with tumor‐specific variations in the germline DNA and acts as a biomarker. The applications of cfDNA analysis, as shown in Figure 8, insights into molecular evolution, detecting cancer recurrence, and mutational changes in cervical cancer patients, and screen diseases before clinical onset [79].
Figure 8.
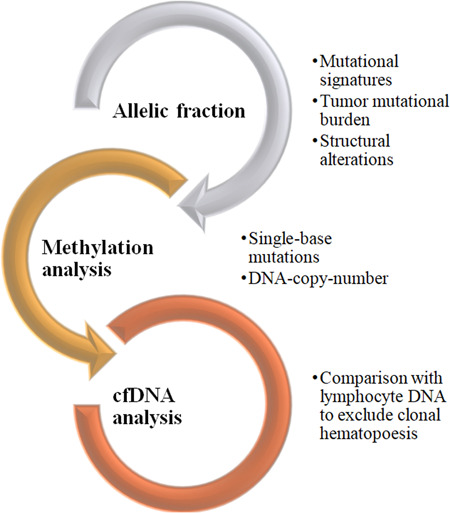
Current methods for cfDNA analysis to detect metastatic lesions using mutational signature analysis, chromosomal translocations, and tumor heterogeneity evaluation using allelic fraction calculation, tumor mutational burden count, and methylation pattern analysis to evaluate alterations due to clonal hematopoiesis. cfDNA, cell‐free DNA. (Created using MS Word smart art.)
9. INFLUENCE OF NGS TO DIAGNOSE CANCER
The progression of tumor in cancer patients can be analyzed with large‐scale sequencing and high‐throughput computational analyses with the multiple sets of omics studies brought oncology to a new model from tumor‐site classification to cancer‐molecular classification with the expansion of transcriptomics, proteomics, and genomics. It involves isolation of nucleic acids, fragmenting DNA/RNA, library preparation and sequencing, amplification, and data annotation [80]. The process involves high‐throughput sequencing for multiple genes and detecting rare variations, NGS makes the patient classification for genetic variations possible and easier to find mutations [81]. They are designed to improve personalized medicine and treatment of autoimmune disorders and cancer, which depends on the patient's unique genetics, with sequencing of whole genome and whole exome to guide therapy. There are different parameters for in vitro diagnostic category, including alignment and read quality to monitor assay quality and types of sources and competency of DNA (ctDNA or formalin‐fixed paraffin‐embedded and tumor cell content) that need to be standardized or sequenced for clinical utility [82]. NGS can be used for tumor multigene indication in ovarian cancers to determine somatic mutations, determining tumor mutational burden in cervical cancer, thyroid cancer, vulva cancer, and salivary cancer [83] (Figure 9).
Figure 9.
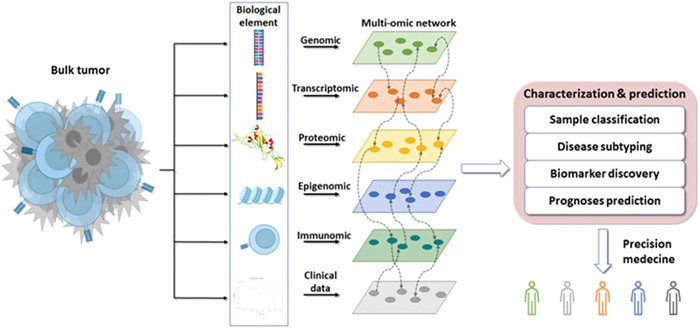
The analysis of bulk tumor with multiomic networks of next‐generation sequencing such as genomic, transcriptomics, proteomics, epigenomics, immunomics, and clinical data to molecularly characterize the subtypes of cancer and treatment brochure to promote precision medicine [84]. (Creative Commons license.)
They expand their popularity in oncology with the detection of cancer cells and interaction between the tumor and host immune system. These interactions defined the cancer progression, molecular mechanism of cancer, and biomarker‐derived studies for tumor immune microenvironment. Trastuzumab, sunitinib (metastatic renal carcinoma), paclitaxel (lung cancer) targeting immune, and inflammatory signaling checkpoints in multiple specialized microenvironments. In the context of cancer research, multiomics data helps to predict the disease subtypes and classification to disease progression [85]. The phenotypic characteristics are being used in multivariable models to predict outcomes toward precision medicine. Precision medicine will enhance treatment probability to 10 times with multiomics profiling analyses [86]. The battle and treatment against cancer have been an ongoing process for decades; with scientists and researchers striving continually to develop innovative therapies to combat this disease. In recent years, one significant advancement has been the emergence of biosimilars as a viable option for treating cancer cells. Biosimilars ushered in a new era in cancer treatment, offering cost‐effective, accessible, and affordable alternatives to reference biologics while maintaining comparable efficacy and safety profiles.
10. FUTURE PERSPECTIVES
The complexity of biological products makes biosimilars more complex than chemical drugs. Manufacturers and prescribers must be cognizant of the need for post‐market vigilance and its efficacy rate in analytical surveys conducted on patients. Additionally, various studies, such as pharmacokinetics/pharmacodynamics, clinical efficacy, biophysical studies, and safety findings, must be conducted. It is now widely accepted that the practices must be significantly altered to facilitate the availability of biosimilars. Of the 33 biosimilars currently in the market, 17 are used in cancer treatments, three are used in autoimmune conditions, and one is used in diabetes. Animal testing is an old practice for new drugs to ensure that they are not toxic to humans. The main way biological drugs work is by attaching to receptors. Usually, animal testing protocols call for a high dose to get a toxic response. In the near future, the demonstration of biosimilars will no longer necessitate animal testing or clinical efficacy testing; however, the clinical pharmacological testing will gradually decrease as the regulatory agencies become more confident in the safety and effectiveness of the biosimilars. The most widely used products are erythropoietin (erythromycin), infliximab (rituximab), and so on. However, the FDA has begun to regulate products that are treated as drugs, such as insulin and other hormones as biologics, at the beginning of 2020. Because biosimilars are immunogenic, there have been several safety concerns raised about them. This is the case when an individual's immune system malfunctions or they respond to foreign proteins. This often occurs with a single injection may result in the patient needing to discontinue therapy altogether or in the medication's efficacy being diminished. Antibodies against biosimilars can be produced in two major methods, the most prevalent of which appears to be through the agglomeration of proteins and contaminants. Determining if there is a parallel between the reference biologic and the biosimilar is challenging due to the larger molecular sizes and complexity of the biosimilars. While capillary electroluminescence, peptide mapping, and other methods have been launched recently, high‐performance liquid chromatographies, MSs, and nuclear magnetic resonances remain the primary methods of analysis. Though it is unclear if they can identify all structural variations or whether they have an impact on clinical efficacy and safety, all of these developments have enhanced our understanding of molecules. It is challenging to compare data between various labs since the assessment of receptor binding and cell responses is not standardized. Since both biosimilars and targeted treatment have benefits and drawbacks of their own, it is impossible to compare the efficacy of the two. Drugs used in targeted treatment selectively target and act on the chemicals and proteins that fuel the development of cancer cells; this is a deliberate action. However, biosimilars are used to treat a variety of illnesses and have the special ability to occasionally provide tailored therapy when necessary, which also lessens side effects. They are able to duplicate medicines that are specifically designed to impede the development and spread of cancer cells. Bevacizumab is one such biosimilar that limits tumor development in ovarian, lung, and colorectal malignancies by inhibiting angiogenesis. These biosimilars support the sustainability of healthcare systems in addition to improving access to medicines that can save lives.
11. CONCLUSION
The development of biosimilars stands from the high cost of biological drugs and the need for patients to gain access to life‐saving drugs that would be a remedy to their illnesses. Changes in the expression systems used to make a drug can affect its biological properties, clinical performance, and toxicity. It is important to note that the identical gene product encoded by the same amino acid sequence could be extracted from animal tissue or used through recombinant DNA techniques. Many analysis tests have been performed on biosimilars to prove their confirmation of the reference biologics to avoid clinical indifference to their effectiveness, safety, purity, or how they work when administered to patients. As clinical data accumulates, confidence in biosimilars will increase, which may lead to broader acceptance and increased use in cancer treatment. Ongoing research aims to optimize biosimilar development processes, improve product quality, and discover new biological targets, thereby enriching the therapeutic landscape. The challenge lies in the distinctions between generic products and biosimilars in clinical practice. The specific drug administered to the patient must be clearly identified and reconfirmed. On the other hand, with the advent of next‐generation sequencing, which is being utilized to treat a wide range of cancer types, thus broadening treatment options and increasing patient access to innovative therapies. The future of biosimilars in cancer treatment holds considerable promise in expanding indications as more biosimilars receive regulatory approval, and their utilization is likely to expand to cover a broader spectrum of cancer types and therapeutic indications.
AUTHOR CONTRIBUTIONS
Shilpa Malakar: Conceptualization (equal); investigation (equal); methodology (equal); validation (equal); writing—original draft (equal); Emmanuel Nuah Gontor: Conceptualization (equal); investigation (equal); methodology (equal); writing—original draft (equal). Moses Y. Dugbaye: Data curation (equal); validation (equal); writing—original draft (equal). Kamal Shah: Data curation (equal); formal analysis (equal); methodology (equal); supervision (equal); writing—review and editing (equal). Sakshi Sinha: Methodology (equal); project administration (equal); supervision (equal); writing–original draft (equal); writing—review and editing (equal). Priya Sutaoney: Conceptualization (equal); formal analysis (equal); investigation (equal); resources (equal); supervision (equal); validation (equal); writing—original draft (equal); writing—review and editing (equal). Nagendra Singh Chauhan: Data curation (equal); investigation (equal); supervision (equal); visualization (equal); writing—review and editing (equal).
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
ETHICS STATEMENT
Not applicable.
INFORMED CONSENT
Not applicable.
ACKNOWLEDGMENTS
An extension of gratitude is due to the management of Kalinga University. The authors are grateful to the chairman Dr. Rajeev Kumar, chancellor Dr. Sandeep Arora; director general Dr. Byju John, and registrar Dr. Sandeep Gandhi for their valuable help and support.
Malakar S, Gontor EN, Dugbaye MY, Shah K, Sinha S, Sutaoney P, et al. Cancer treatment with biosimilar drugs: a review. Cancer Innov. 2024;3:e115. 10.1002/cai2.115
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no data sets were generated or analyzed during the current study.
REFERENCES
- 1. Valderrama‐Rincon JD, Fisher AC, Merritt JH, Fan YY, Reading CA, Chhiba K, et al. An engineered eukaryotic protein glycosylation pathway in Escherichia coli. Nat Chem Biol. 2012;8(5):434–436. 10.1038/nchembio.921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alfonso‐Cristancho R, Armstrong N, Arjunji R, Riemsma R, Worthy G, Ganguly R, et al. Comparative effectiveness of biologics for the management of rheumatoid arthritis: systematic review and network meta‐analysis. Clin Rheumatol. 2017;36(1):25–34. 10.1007/s10067-016-3435-2 [DOI] [PubMed] [Google Scholar]
- 3. Tiwari G, Tiwari R, Bannerjee S, Bhati L, Pandey S, Pandey P, et al. Drug delivery systems: an updated review. Int J Pharm Investig. 2012;2(1):2–11. 10.4103/2230-973X.96920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liu JKH. The history of monoclonal antibody development—progress, remaining challenges and future innovations. Ann Med Surg. 2014;3(4):113–116. 10.1016/j.amsu.2014.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. De Keyser F. Choice of biologic therapy for patients with rheumatoid arthritis: the infection perspective. Curr Rheumatol Rev. 2011;7(1):77–87. 10.2174/157339711794474620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vandenplas Y, Simoens S, Van Wilder P, Vulto AG, Huys I. Informing patients about biosimilar medicines: the role of European Patient Associations. Pharmaceuticals. 2021;14(2):117. 10.3390/ph14020117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Walsh G, Walsh E. Biopharmaceutical benchmarks 2022. Nat Biotechnol. 2022;40(12):1722–1760. 10.1038/s41587-022-01582-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schiestl M, Zabransky M, Sörgel F. Ten years of biosimilars in Europe: development and evolution of the regulatory pathways. Drug Des Devel Ther. 2017;11:1509–1515. 10.2147/DDDT.S130318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gherghescu I, Delgado‐Charro MB. The biosimilar landscape: an overview of regulatory approvals by the EMA and FDA. Pharmaceutics. 2020;13(1):48. 10.3390/pharmaceutics13010048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Beck A, Reichert JM. Approval of the first biosimilar antibodies in Europe: a major landmark for the biopharmaceutical industry. MAbs. 2013;5(5):621–623. 10.4161/mabs.25864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dowlat HA. The opportunities and challenges of biosimilar orphans. Exp Opin Orphan Drugs. 2016;4(6):563–566. 10.1517/21678707.2016.1171142 [DOI] [Google Scholar]
- 12. Watts MJ, Addison I, Long SG, Hartley S, Warrington S, Boyce M, et al. Crossover study of the haematological effects and pharmacokinetics of glycosylated and non‐glycosylated G‐CSF in healthy volunteers. Br J Haematol. 1997;98(2):474–479. 10.1046/j.1365-2141.1997.2393053.x [DOI] [PubMed] [Google Scholar]
- 13. McKenna RM, Oberg KE. Antibodies to Interferon‐α in treated cancer patients: incidence and significance. J Interferon Cytokine Res. 1997;17(3):141–143. 10.1089/jir.1997.17.141 [DOI] [PubMed] [Google Scholar]
- 14. Öberg K, Alm G. The incidence and clinical significance of antibodies to interferon‐a in patients with solid tumors. Biotherapy. 1997;10(1):1–5. 10.1007/BF02678211 [DOI] [PubMed] [Google Scholar]
- 15. Maschio G. Keynote lecture: erythropoietin and systemic hypertension. Nephrol Dial Transplant. 1995;10(Suppl 2):74–79. 10.1093/ndt/10.supp2.74 [DOI] [PubMed] [Google Scholar]
- 16. DiMasi JA, Grabowski HG, Hansen RW. Innovation in the pharmaceutical industry: new estimates of R&D costs. J Health Econ. 2016;47:20–33. 10.1016/j.jhealeco.2016.01.012 [DOI] [PubMed] [Google Scholar]
- 17. Koutnik‐Fotopoulos E. FDA commissioner outlines new plan to increase biosimilars by balancing innovation and competition. Am Health Drug Benefits. 2018;11(8):427–428. [PMC free article] [PubMed] [Google Scholar]
- 18. Agostini C, Canonica GW, Maggi E. European medicines agency guideline for biological medicinal products: a further step for a safe use of biosimilars. Clin Mol Allergy. 2015;13(1):3. 10.1186/s12948-015-0010-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kurki P, Kang HN, Ekman N, Knezevic I, Weise M, Wolff‐Holz E. Regulatory evaluation of biosimilars: refinement of principles based on the scientific evidence and clinical experience. BioDrugs. 2022;36(3):359–371. 10.1007/s40259-022-00533-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ventola CL. Biosimilars: part 1: proposed regulatory criteria for FDA approval. P&T. 2013;38(5):270–287. [PMC free article] [PubMed] [Google Scholar]
- 21. Bui LA, Hurst S, Finch GL, Ingram B, Jacobs IA, Kirchhoff CF, et al. Key considerations in the preclinical development of biosimilars. Drug Discov Today. 2015;20(Suppl 1):3–15. 10.1016/j.drudis.2015.03.011 [DOI] [PubMed] [Google Scholar]
- 22. Vulto AG, Jaquez OA. The process defines the product: what really matters in biosimilar design and production? Rheumatology. 2017;56(56):iv14–iv29. 10.1093/rheumatology/kex278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Funaki A, Hirata I, Matsui H, Isezaki T, Funakoshi R. Factors affecting patients' acceptance of switching to biosimilars are disease‐dependent: a cross‐sectional study. Biol Pharm Bull. 2023;46(1):128–132. 10.1248/bpb.b22-00429 [DOI] [PubMed] [Google Scholar]
- 24. Gasteiger C, Lobo M, Dalbeth N, Petrie KJ. Patients' beliefs and behaviours are associated with perceptions of safety and concerns in a hypothetical biosimilar switch. Rheumatol Int. 2021;41(1):163–171. 10.1007/s00296-020-04576-7 [DOI] [PubMed] [Google Scholar]
- 25. Raedler LA. Zarxio (Filgrastim‐sndz): first biosimilar approved in the United States. Am Health Drug Benefits. 2016;9(Spec Feature):150–154. [PMC free article] [PubMed] [Google Scholar]
- 26. Kazazi‐Hyseni F, Beijnen JH, Schellens JHM. Bevacizumab. Oncologist. 2010;15(8):819–825. 10.1634/theoncologist.2009-0317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Triantafyllidi E, Triantafillidis JK. Systematic review on the use of biosimilars of trastuzumab in HER2+ breast cancer. Biomedicines. 2022;10(8):2045. 10.3390/biomedicines10082045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Burris HA, Belani CP, Kaufman PA, Gordon AN, Schwartzberg LS, Paroly WS, et al. Pegfilgrastim on the same day versus next day of chemotherapy in patients with breast cancer, non‐small‐cell lung cancer, ovarian cancer, and non‐Hodgkin's lymphoma: results of four multicenter, double‐blind, randomized phase II studies. J Oncol Pract. 2010;6(3):133–140. 10.1200/JOP.091094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kos IA, Azevedo VF, Neto DE, Kowalski SC. The biosimilars journey: current status and ongoing challenges. Drugs Context. 2018;7:212543. 10.7573/dic.212543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Barbier L, Mbuaki A, Simoens S, Declerck P, Vulto AG, Huys I. Regulatory information and guidance on biosimilars and their use across Europe: a call for strengthened one voice messaging. Front Med. 2022;9:820755. 10.3389/fmed.2022.820755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. O'Callaghan J, Barry SP, Bermingham M, Morris JM, Griffin BT. Regulation of biosimilar medicines and current perspectives on interchangeability and policy. Eur J Clin Pharmacol. 2019;75(1):1–11. 10.1007/s00228-018-2542 [DOI] [PubMed] [Google Scholar]
- 32. Daller J. Biosimilars: a consideration of the regulations in the United States and European Union. Regul Toxicol Pharmacol. 2016;76:199–208. 10.1016/j.yrtph.2015.12.013 [DOI] [PubMed] [Google Scholar]
- 33. Guymer RH, Campbell TG. Age‐related macular degeneration. Lancet. 2023;401(10386):1459–1472. 10.1016/S0140-6736(22)02609-5 [DOI] [PubMed] [Google Scholar]
- 34. de Oliveira Avellar W, Ferreira ÉA, Vieira ACRA, de Melo AC, Aran V. Clinical cancer research in South America and potential health economic impacts. Healthcare. 2023;11(12):1753. 10.3390/healthcare11121753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Papautsky EL, Carlson M, Johnson SM, Montague H, Attai DJ, Lustberg MB. Characterizing experiences of non‐medical switching to trastuzumab biosimilars using data from internet‐based surveys with US‐based oncologists and breast cancer patients. Breast Cancer Res Treat. 2022;194(1):25–33. 10.1007/s10549-022-06615-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tabernero J, Vyas M, Giuliani R, Arnold D, Cardoso F, Casali PG, et al. Biosimilars: a position paper of the European Society for Medical Oncology, with particular reference to oncology prescribers. ESMO Open. 2016;1(6):e000142. 10.1136/esmoopen-2016-000142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bhardwaj K, Bangarurajan K, Naved T, Rajput S. Perspective, perceptions, and promulgation of biosimilars: a questionnaire‐based study to assess and understand the current challenges of biosimilars to the potential and intended users. J Pharm Bioallied Sci. 2020;12(2):124–130. 10.4103/jpbs.JPBS_11_20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tinsley SM, Grande C, Olson K, Plato L, Jacobs I. Potential of biosimilars to increase access to biologics: considerations for advanced practice providers in oncology. J Adv Pract Oncol. 2018;9(7):699–716. 10.6004/JADPRO.2018.9.7.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Maksabedian Hernandez EJ, Graf M, Portelli A, Shafrin J. Estimating the impact of biosimilar entry on prices and expenditures in rheumatoid arthritis: a case study of targeted immune modulators. J Med Econ. 2022;25(1):1118–1126. 10.1080/13696998.2022.2113252 [DOI] [PubMed] [Google Scholar]
- 40. Peeters M, Planchard D, Pegram M, Gonçalves J, Bocquet F, Jang H. Biosimilars in an era of rising oncology treatment options. Future Oncol. 2021;17(29):3881–3892. 10.2217/fon-2021-0546 [DOI] [PubMed] [Google Scholar]
- 41. Dey M, Zhao SS, Moots RJ. Anti‐TNF biosimilars in rheumatology: the end of an era? Expert Opin Biol Ther. 2021;21(1):29–36. 10.1080/14712598.2020.1802421 [DOI] [PubMed] [Google Scholar]
- 42. Rudrapatna VA, Velayos F. Biosimilars for the treatment of inflammatory bowel disease. Pract Gastroenterol. 2019;43(4):84–91. [PMC free article] [PubMed] [Google Scholar]
- 43. Meyer A, Rudant J, Drouin J, Weill A, Carbonnel F, Coste J. Effectiveness and safety of reference infliximab and biosimilar in crohn disease: a French equivalence study. Ann Intern Med. 2019;170(2):99–107. 10.7326/M18-1512 [DOI] [PubMed] [Google Scholar]
- 44. Danese S, Gomollon F; Governing Board and Operational Board of ECCO . ECCO position statement: the use of biosimilar medicines in the treatment of inflammatory bowel disease (IBD). J Crohn's Colitis. 2013;7(7):586–589. 10.1016/j.crohns.2013.03.011 [DOI] [PubMed] [Google Scholar]
- 45. McCamish M, Woollett G. The state of the art in the development of biosimilars. Clin Pharm Ther. 2012;91(3):405–417. 10.1038/clpt.2011.343 [DOI] [PubMed] [Google Scholar]
- 46. Ishii‐Watabe A, Kuwabara T. Biosimilarity assessment of biosimilar therapeutic monoclonal antibodies. Drug Metab Pharmacokinet. 2019;34(1):64–70. 10.1016/j.dmpk.2018.11.004 [DOI] [PubMed] [Google Scholar]
- 47. Debiasi M, Pimentel FF, Pereira PJS, Barrios CH. Biosimilars in Brazil: the beginning of an era of broader access. J Cancer Ther. 2017;8:814–826. 10.4236/jct.2017.89071 [DOI] [Google Scholar]
- 48. Kirchhoff CF, Wang XM, Conlon HD, Anderson S, Ryan AM, Bose A. Biosimilars: key regulatory considerations and similarity assessment tools. Biotechnol Bioeng. 2017;114(12):2696–2705. 10.1002/bit.26438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Al‐Sabbagh A, Olech E, McClellan JE, Kirchhoff CF. Development of biosimilars. Semin Arthritis Rheum. 2016;45(5 Suppl):S11–S18. 10.1016/j.semarthrit.2016.01.002 [DOI] [PubMed] [Google Scholar]
- 50. Mellstedt H, Niederwieser D, Ludwig H. The challenge of biosimilars. Ann Oncol. 2008;19(3):411–419. 10.1093/annonc/mdm345 [DOI] [PubMed] [Google Scholar]
- 51. Ferrer‐Miralles N, Domingo‐Espín J, Corchero JL, Vázquez E, Villaverde A. Microbial factories for recombinant pharmaceuticals. Microb Cell Fact. 2009;8:17. 10.1186/1475-2859-8-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lucio S. The complexities of biosimilars and the regulatory approval process. Am J Manag Care. 2018;24(11 Suppl):S231–S236. 10.1093/annonc/mdm345 [DOI] [PubMed] [Google Scholar]
- 53. Baeshen MN, Al‐Hejin AM, Bora RS, Ahmed MM, Ramadan HA, Saini KS, et al. Production of biopharmaceuticals in E. coli: current scenario and future perspectives. J Microbiol Biotechnol. 2015;25(7):953–962. 10.4014/jmb.1412.12079 [DOI] [PubMed] [Google Scholar]
- 54.Monoclonal Antibodies. LiverTox: Clinical and Research Information on Drug‐Induced Liver Injury [Internet]. National Institute of Diabetes and Digestive and Kidney Diseases; 2023. https://www.ncbi.nlm.nih.gov/books/NBK548844/ [PubMed]
- 55. Weise M, Kurki P, Wolff‐Holz E, Bielsky MC, Schneider CK. Biosimilars: the science of extrapolation. Blood. 2014;124(22):3191–3196. 10.1182/blood-2014-06-583617 [DOI] [PubMed] [Google Scholar]
- 56. Lu RM, Hwang YC, Liu IJ, Lee CC, Tsai HZ, Li HJ, et al. Development of therapeutic antibodies for the treatment of diseases. J Biomed Sci. 2020;27(1):1. 10.1186/s12929-019-0592-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bradley AM, Devine M, DeRemer D. Brentuximab vedotin: an anti‐CD30 antibody‐drug conjugate. Am J Health Syst Pharm. 2013;70(7):589–597. 10.2146/ajhp110608 [DOI] [PubMed] [Google Scholar]
- 58. Jones KL, Buzdar AU. Evolving novel anti‐HER2 strategies. Lancet Oncol. 2009;10(12):1179–1187. 10.1016/S1470-2045(09)70315-8 [DOI] [PubMed] [Google Scholar]
- 59. Nielsen DL, Andersson M, Kamby C. HER2‐targeted therapy in breast cancer. Monoclonal antibodies and tyrosine kinase inhibitors. Cancer Treat Rev. 2009;35(2):121–136. 10.1016/j.ctrv.2008.09.003 [DOI] [PubMed] [Google Scholar]
- 60. Watanabe S, Yonesaka K, Tanizaki J, Nonagase Y, Takegawa N, Haratani K, et al. Targeting of the HER2/HER3 signaling axis overcomes ligand‐mediated resistance to trastuzumab in HER2‐positive breast cancer. Cancer Med. 2019;8(3):1258–1268. 10.1002/cam4.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Valabrega G, Montemurro F, Aglietta M. Trastuzumab: mechanism of action, resistance and future perspectives in HER2‐overexpressing breast cancer. Ann Oncol. 2007;18(6):977–984. 10.1093/annonc/mdl475 [DOI] [PubMed] [Google Scholar]
- 62. Cortés J, Kim SB, Chung WP, Im SA, Park YH, Hegg R, et al. Trastuzumab deruxtecan versus trastuzumab emtansine for breast cancer. N Engl J Med. 2022;386(12):1143–1154. 10.1056/NEJMoa2115022 [DOI] [PubMed] [Google Scholar]
- 63. Mohammed R, Milne A, Kayani K, Ojha U. How the discovery of rituximab impacted the treatment of B‐cell non‐Hodgkin's lymphomas. J Blood Med. 2019;10:71–84. 10.2147/JBM.S190784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Randall KL. Rituximab in autoimmune diseases. Aust Prescr. 2016;39(4):131–134. 10.18773/austprescr.2016.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Taylor RP, Lindorfer MA. Drug insight: the mechanism of action of rituximab in autoimmune disease—the immune complex decoy hypothesis. Nat Clin Pract Rheumatol. 2007;3(2):86–95. 10.1038/ncprheum0424 [DOI] [PubMed] [Google Scholar]
- 66. Hanif N, Anwer F. Rituximab. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK564374/
- 67. Reinisch W, Gecse K, Halfvarson J, Irving PM, Jahnsen J, Peyrin‐Biroulet L, et al. Clinical practice of adalimumab and infliximab biosimilar treatment in adult patients with Crohn's disease. Inflamm Bowel Dis. 2021;27(1):106–122. 10.1093/ibd/izaa078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Carmona L. Terapias anti‐TNF y neoplasias. Reumatol Clín. 2010;6(2):102–105. 10.1016/j.reuma.2009.01.013 [DOI] [PubMed] [Google Scholar]
- 69. Goldsmith D, Dellanna F, Schiestl M, Krendyukov A, Combe C. Epoetin biosimilars in the treatment of renal anemia: what have we learned from a decade of European experience? Clin Drug Invest. 2018;38(6):481–490. 10.1007/s40261-018-0637-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gianoncelli A, Bonini SA, Bertuzzi M, Guarienti M, Vezzoli S, Kumar R, et al. An integrated approach for a structural and functional evaluation of biosimilars: implications for erythropoietin. BioDrugs. 2015;29(4):285–300. 10.1007/s40259-015-0136-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Winter SS, Howard T, Ware RE. Regulation of expression of the human erythropoietin receptor gene. Blood Cells Mol Dis. 1996;22(3):214–224. 10.1006/bcmd.1996.0102. [DOI] [PubMed] [Google Scholar]
- 72. Joshi D, Khursheed R, Gupta S, Wadhwa D, Singh TG, Sharma S, et al. Biosimilars in oncology: latest trends and regulatory status. Pharmaceutics. 2022;14(12):2721. 10.3390/pharmaceutics14122721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. McBride A, Balu S, Campbell K, Bikkina M, MacDonald K, Abraham I. Expanded access to cancer treatments from conversion to neutropenia prophylaxis with biosimilar filgrastim‐sndz. Future Oncol. 2017;13(25):2285–2295. 10.2217/fon-2017-0374 [DOI] [PubMed] [Google Scholar]
- 74. Nahleh Z, Lyman GH, Schilsky RL, Peterson DE, Tagawa ST, Chavez‐MacGregor M, et al. Use of biosimilar medications in oncology. JCO Oncol Pract. 2022;18(3):177–186. 10.1200/OP.21.00771 [DOI] [PubMed] [Google Scholar]
- 75. Lo YMD, Han DSC, Jiang P, Chiu RWK. Epigenetics, fragmentomics, and topology of cell‐free DNA in liquid biopsies. Science. 2021;372(6538):eaaw3616. 10.1126/science.aaw3616 [DOI] [PubMed] [Google Scholar]
- 76. Liu Y. At the dawn: cell‐free DNA fragmentomics and gene regulation. Br J Cancer. 2022;126(3):379–390. 10.1038/s41416-021-01635-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Gao Q, Zeng Q, Wang Z, Li C, Xu Y, Cui P, et al. Circulating cell‐free DNA for cancer early detection. Innovation (Camb). 2022;3(4):100259. 10.1016/j.xinn.2022.100259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Szilágyi M, Pös O, Márton É, Buglyó G, Soltész B, Keserű J, et al. Circulating cell‐free nucleic acids: main characteristics and clinical application. Int J Mol Sci. 2020;21(18):6827. 10.3390/ijms21186827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Cisneros‐Villanueva M, Hidalgo‐Pérez L, Rios‐Romero M, Cedro‐Tanda A, Ruiz‐Villavicencio CA, Page K, et al. Cell‐free DNA analysis in current cancer clinical trials: a review. Br J Cancer. 2022;126(3):391–400. 10.1038/s41416-021-01696-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Idris SF, Ahmad SS, Scott MA, Vassiliou GS, Hadfield J. The role of high‐throughput technologies in clinical cancer genomics. Expert Rev Mol Diagn. 2014;13(2):167–181. 10.1586/erm.13.1 [DOI] [PubMed] [Google Scholar]
- 81. Guan YF, Li GR, Wang RJ, Yi YT, Yang L, Jiang D, et al. Application of next‐generation sequencing in clinical oncology to advance personalized treatment of cancer. Chin J Cancer. 2012;31(10):463–470. 10.5732/cjc.012.10216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kusama H, Shimoda M, Miyake T, Tanei T, Kagara N, Naoi Y, et al. Prognostic value of tumor cell DNA content determined by flow cytometry using formalin‐fixed paraffin‐embedded breast cancer tissues. Breast Cancer Res Treat. 2019;176(1):75–85. 10.1007/s10549-019-05222-y [DOI] [PubMed] [Google Scholar]
- 83. Mosele F, Remon J, Mateo J, Westphalen CB, Barlesi F, Lolkema MP, et al. Recommendations for the use of next‐generation sequencing (NGS) for patients with metastatic cancers: a report from the ESMO Precision Medicine Working Group. Ann Oncol. 2020;31(11):1491–1505. 10.1016/j.annonc.2020.07.014 [DOI] [PubMed] [Google Scholar]
- 84. Raufaste‐Cazavieille V, Santiago R, Droit A. Multi‐omics analysis: paving the path toward achieving precision medicine in cancer treatment and immuno‐oncology. Front Mol Biosci. 2022;9:962743. 10.3389/fmolb.2022.962743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Tsimberidou AM, Fountzilas E, Nikanjam M, Kurzrock R. Review of precision cancer medicine: evolution of the treatment paradigm. Cancer Treat Rev. 2020;86:102019. 10.1016/j.ctrv.2020.102019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Tebani A, Afonso C, Marret S, Bekri S. Omics‐based strategies in precision medicine: toward a paradigm shift in inborn errors of metabolism investigations. Int J Mol Sci. 2016;17(9):1555. 10.3390/ijms17091555 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no data sets were generated or analyzed during the current study.