

Abstract
Neurotransmitter release is triggered in microseconds by Ca2+-binding to the Synaptotagmin-1 C2 domains and by SNARE complexes that form four-helix bundles between synaptic vesicles and plasma membranes, but the coupling mechanism between Ca2+-sensing and membrane fusion is unknown. Release requires extension of SNARE helices into juxtamembrane linkers that precede transmembrane regions (linker zippering) and binding of the Synaptotagmin-1 C2B domain to SNARE complexes through a ‘primary interface’ comprising two regions (I and II). The Synaptotagmin-1 Ca2+-binding loops were believed to accelerate membrane fusion by inducing membrane curvature, perturbing lipid bilayers or helping bridge the membranes, but SNARE complex binding orients the Ca2+-binding loops away from the fusion site, hindering these putative activities. Molecular dynamics simulations now suggest that Synaptotagmin-1 C2 domains near the site of fusion hinder SNARE action, providing an explanation for this paradox and arguing against previous models of Sytnaptotagmin-1 action. NMR experiments reveal that binding of C2B domain arginines to SNARE acidic residues at region II remains after disruption of region I. These results and fluorescence resonance energy transfer assays, together with previous data, suggest that Ca2+ causes reorientation of the C2B domain on the membrane and dissociation from the SNAREs at region I but not region II. Based on these results and molecular modeling, we propose that Synaptotagmin-1 acts as a lever that pulls the SNARE complex when Ca2+ causes reorientation of the C2B domain, facilitating linker zippering and fast membrane fusion. This hypothesis is supported by the electrophysiological data described in the accompanying paper.
Keywords: synaptotagmin, SNAREs, neurotransmitter release, membrane fusion, molecular dynamics simulation, synaptic vesicle fusion
Classification: Neuroscience
Release of neurotransmitters by Ca2+-triggered synaptic vesicle exocytosis is crucial for neuronal communication. Exocytosis involves several steps, including vesicle tethering to presynaptic active zones, priming of the vesicles to a release-ready state(s) and vesicle fusion with the plasma membrane, which occurs very fast upon Ca2+ influx into a presynaptic terminal (1) [less than 60 μs in fast synapses (2)]. The basic steps that lead to exocytosis have been reconstituted with the main components of the neurotransmitter release machinery (3–6) and the functions of these components have been defined (7–9). The SNAP receptors (SNAREs) syntaxin-1, SNAP-25 and synaptobrevin form tight complexes (10) that consist of four-helix bundles (11, 12) and bring the membranes together as they assemble (zipper) from the N- to the C-terminus (13), likely inducing membrane fusion (14) by promoting lipid acyl chain encounters at the polar membrane-membrane interface (15). N-ethylmaleimide sensitive factor (NSF) and soluble NSF attachment proteins (SNAPs) disassemble SNARE complexes (10) to recycle the SNAREs (16). Munc18–1 and Munc13s organize SNARE complex formation by an NSF-SNAP-resistant mechanism (3, 17) in which Munc18–1 binds first to a closed conformation of syntaxin-1 (18, 19) and later binds to synaptobrevin, forming a template complex (20–22) for SNARE assembly while Munc13 bridges the two membranes (4, 23) and opens syntaxin-1 (24). In the resulting primed state, the SNARE complex is bound to Synaptotagmin-1 (Syt1) (25) and to complexin (26), forming a spring-loaded macromolecular assembly (27) that prevents premature fusion but is ready to trigger fast fusion when Ca2+ binds to Syt1 (28).
The cytoplasmic region of Syt1 is formed mostly by two C2 domains (C2A and C2B) that bind three and two Ca2+ ions, respectively, via loops at the tip of β-sandwich structures (29–31). While it is well established that Syt1 triggers neurotransmitter release through its C2 domains (28, 32), and that Ca2+-binding to the C2B domain is particularly crucial for release (33, 34), the underlying mechanism remains highly enigmatic. Ca2+ induces insertion of these loops into membranes (35, 36), which was proposed to facilitate membrane fusion by perturbing the bilayers (35, 37, 38) and/or inducing membrane curvature (39, 40). In addition, the Syt1 C2B domain binds to phosphatidylinositol 4,5-bisphosphate (PIP2) through a polybasic region on the side of the β-sandwich (41) and was proposed to help induce fusion by bridging the two membranes (42, 43). Many studies reported diverse interactions of Syt1 with the SNAREs that were generally enhanced by Ca2+ [reviewed in (44, 45)]. However, a fragment spanning the two Syt1 C2 domains (C2AB) binds with higher affinity to nanodiscs containing SNARE complex than to plain nanodiscs in the absence of Ca2+ while the affinity is similar in the presence of Ca2+, suggesting that Ca2+ actually induces dissociation of C2AB from membrane-anchored SNARE complexes (46, 47) or at least weakens the C2AB-SNARE interactions.
Three structures of Syt1-SNARE complexes have been described, each revealing binding through distinct surfaces of the C2B domain that are not involved in Ca2+ binding (25, 48, 49). However, strong evidence suggests that, among these three, the key functionally relevant binding mode is mediated by a so-called primary interface (25, 47, 50) formed by two regions of C2B: region I involving E295 and Y338; and region II involving R281, R398 and R399. Evidence suggested that this interface is important for vesicle priming (49, 51), which is strongly supported by the accompanying paper (52) and likely arises because Syt1 cooperates with Munc18–1 and Munc13–1 in mediating Ca2+-independent assembly of trans-SNARE complexes (17). Intriguingly, while an R398Q,R399Q mutation in region II that disrupts C2B-SNARE complex binding (47) strongly impairs neurotransmitter release (25, 53), release is also impaired by a E295A,Y338W mutation in region I (25) that enhances binding (47). Moreover, this binding mode orients the C2B Ca2+-binding loops away from the site of membrane fusion, which hinders a direct role for the loops in fusion. Note also that Ca2+-dependent binding to PIP2-containing membranes induces an approximately perpendicular orientation of the C2B domain with respect to the bilayer (54) that is incompatible with SNARE complex binding via the primary interface (47). These findings suggested that binding of Syt1 to the SNARE complex via the primary interface is important to generate the primed state but hinders membrane fusion, and Ca2+-induced dissociation of Syt1 from the SNAREs relieves the inhibition (47).
While this model explains a large amount of data and release-of-inhibition models have been popular since 30 years ago (55), there is evidence that Syt1 is not merely a SNARE inhibitor and exerts an active action to trigger release upon Ca2+ binding (7, 44). Dissociation from the SNAREs might allow the Syt1 C2 domains to reorient toward the site of fusion such that they can directly facilitate fusion through its Ca2+-binding loops (47), but this notion does not bode well for the fast speed of release and Ca2+-induced dissociation of Syt1 from membrane-anchored SNARE complexed has not been demonstrated. Moreover, it is not clear how actions of Syt1 that have been proposed to facilitate membrane fusion such as bilayer perturbation (35, 37, 38), induction of membrane curvature (39, 40) or membrane bridging (42, 43) can cooperate with the SNAREs to induce fast membrane fusion, particularly after molecular dynamics (MD) simulations revealed a natural mechanism for SNARE complexes to induce fast, microsecond-scale membrane fusion (15). In this mechanism, extension of the synaptobrevin and syntaxin-1 helices that form the four-helix bundle into the juxtamembrane (jxt) linkers that precede the transmembrane (TM) regions (referred to as jxt linker zippering) is key to catalyze encounters between the lipid acyl chains of the two bilayers that initiate fusion, but it is unclear how the Syt1 Ca2+-binding loops can facilitate these events.
To address these questions and shed light into how Syt1 triggers neurotransmitter release, we have used a combination of all-atom MD simulations with NMR and fluorescence spectroscopy assays. The simulations suggest that placing the Syt1 C2 domains near the site of fusion severely hinders the ability of SNARE complexes to bring the membranes together and induce membrane fusion, which provides an explanation for why the primary interface orients the C2B Ca2+-binding loops away from the site of fusion and argues against the notion that Syt1 facilitates membrane fusion by inducing membrane curvature, perturbing the bilayers or bridging the membranes. NMR experiments show that binding of the Syt1 C2B domain to the SNARE complex via the arginines of region II of the primary interface remains when binding through region I is disrupted. Fluorescence resonance energy transfer (FRET) show that Ca2+ does not dissociate a fragment spanning the two C2 domains of Syt1 (C2AB) from membrane-anchored SNARE complexes but induces a reorientation of the C2B domain with respect to the SNARE four-helix bundle. These results, together with previous electron paramagnetic resonance (EPR) data (54), suggest a model whereby Ca2+ binding causes dissociation of region I but not region II of the primary interface and reorientation of C2B on the membrane, which pulls the SNARE complex through ionic interactions and facilitates linker zippering to initiate fast membrane fusion. While further research will be needed to test this lever hypothesis of Syt1 action, the central notion that neurotransmitter release is triggered by Ca2+-induced re-modeling of the Syt1-SNARE primary interface is strongly supported by multiple correlations between our NMR data and electrophysiological results described in the accompanying paper (52).
Results
Syt1 C2 domains close to the fusion site hinder SNARE action
Since all-atom MD simulations have provided a powerful tool to visualize the neurotransmitter release machinery bridging two membranes (27, 56) and understand how the SNAREs mediate membrane fusion (15), we carried out several all-atom MD simulations to investigate the merits of various models that have been proposed for how Syt1 accelerates membrane fusion. Similar to our previous studies (15, 27), the systems used for all simulations contained four trans-SNARE complexes bridging a vesicle and a flat bilayer with lipid compositions that resemble those of synaptic vesicles and synaptic plasma membranes, respectively (57, 58), with or without Syt1 C2AB molecules (Table S1). Complexin was not included in these simulations for simplicity, as fast neurotransmitter release is impaired but not abolished in the absence of complexins (59) and we wanted to focus on how the functions of Syt1 and the SNAREs are coupled.
In a first simulation, we used a system of four trans-SNARE complexes that were zippered to distinct extents at the C-terminus, and included four C2AB molecules bound to five Ca2+ ions each and with the Ca2+-binding loops pointing toward the flat bilayer without contacting it (Fig. S1A) to investigate whether the Syt1 C2 domains spontaneously insert into the bilayers, perturb or bridge the bilayers, and perhaps even initiate membrane fusion (referred to as cac2absc simulation). The SNARE complexes were in the same configurations and positions as those of a simulation of the primed state described previously [prsg simulation in (27)], with different extents of assembly. During the 668 ns of this simulation, the two membranes were brought into contact (Fig. 1A), but there was no substantial progress in zippering of the SNARE complexes and the C2 domains of the distinct C2AB molecules exhibited different orientations with respect to each other, to the SNARE complexes and to the membranes (Fig. S1B,C). One of the C2B domains adopted an approximately perpendicular orientation with respect to the flat bilayer, inserting the Ca2+-binding loops into this bilayer and binding to the vesicle through the opposite end of the domain that contains R398 and R399 (Fig. 2A), as predicted in membrane-bridging models of Syt1 function (42, 43). However, the Ca2+-binding loops inserted only to a small extent into the acyl region of the bilayer, and there was no overt perturbation of the bilayer or induction of curvature (Fig. 2A). The other C2 domains adopted more slanted or even parallel orientations with respect to the flat bilayer (Fig. S1B,C). One of the C2B domains had the Ca2+-binding loops oriented toward the center of the membrane-membrane interface and was located next to the C-terminus of a SNARE complex that was almost fully assembled from the beginning of the simulation (Fig. S1D), which could facilitate cooperation of the SNAREs and the C2B domain in inducing membrane fusion. We note however that this configuration was already present early in the simulation (at 180 ns), but there was no initiation of fusion during the rest of the simulation.
Figure 1.

The Syt1 C2 domains hinder SNARE action when located near the site of fusion. (A-D) Diagrams showing thin slices of frames taken at 668 ns of the cac2absc simulation (A), 510 ns of the fusiong simulation (B), 570 ns of the sytfusion2g simulation (C) and 450 ns of the sytfusion3 simulation (D). Lipids are shown as stick models with nitrogen atoms in dark blue, oxygens in red, phosphorus in orange and carbon atoms in yellow (vesicle) or light blue (flat bilayer). Phosphorous atoms of phospholipids and the oxygen atoms of cholesterol molecules are shown as spheres to illustrate the approximate locations of lipid head groups. Proteins are represented by ribbon diagrams, with SNARE complexes in salmon color, Syt1 C2A domain in slate blue and Syt1 C2B domain in green. Ca2+ ions are shown as yellow spheres. Note that, because the slices shown are thin, only portions of some of the proteins, if any, are seen in the slices. The same color-coding was used in all the figures except when noted otherwise.
Figure 2.

Syt1 C2 domain Ca2+-binding loops do not insert deeply into lipid bilayers and cause limited bilayer perturbation. The diagrams show examples of C2 domains with their Ca2+-binding loops interacting with the flat bilayer from frames taken at 668 ns of the cac2absc simulation (A), 510 ns of the fusiong simulation (B) and 450 ns of the sytfusion3 simulation (C). Lipids are shown as stick models. Ca2+ ions are shown as yellow spheres. SNARE complexes are represented by ribbon diagrams and Syt1 C2 domains by ribbon diagrams and stick models with nitrogen atoms in dark blue, oxygen in red, sulfur in yellow orange and carbon colored in slate blue (C2A) and green (C2B). Other color coding is as in Fig. 1. The hydrophobic residues at the tips of the Ca2+-binding loops that insert into the flat bilayer are shown as spheres and labeled. R398 and R399 at the opposite end of the C2B domain are labeled in (A, C) to illustrate how the C2B domain can bridge two membranes as predicted (42).
We reasoned that the probability of observing membrane fusion in this simulation might have been hindered because three SNARE complexes were partially assembled. Thus, we generated a similar system but with the four SNARE four-helix bundles almost fully assembled and four C2AB molecules with the C2B domain Ca2+-binding loops oriented toward the center of the membrane-membrane interface (fusiong simulation) (Fig. S2A). We performed an MD simulation of this system for 510 ns, but we again did not observe initiation of fusion (Fig. 1B and S2B).
To examine the possibility that the C2AB molecules might actually hinder SNARE action in the fusiong simulation, we performed a parallel 510 ns simulation with an identical system that lacked the four C2AB molecules (nosytfusion simulation) (Fig. S3A). There was again no fusion in this simulation (Fig. S3B), but the two membranes came into contact more quickly than in the presence of C2AB molecules (Fig. S2C and S3C). It is also noteworthy that at the end of the nosytfusion simulation the contact between the two bilayers (Fig. S3D) was considerably more extensive than at the end of the fusiong simulation (Fig. 1B). Such extended interfaces were observed in a previous simulation of a similar system (27) and in cryo-electron microscopy (cryo-EM) images of SNARE-mediated liposome fusion reactions (60). Hence, our results suggest that the C2AB molecules of the fusiong simulation hindered the action of the SNAREs in bringing the membranes together and the formation of extended interfaces.
We also explored the notion that the Ca2+-binding loops of both Syt1 C2 domains might play a direct role in membrane fusion if the C2 domains are located between the two membranes with the Ca2+-binding loops oriented toward the center of the membrane-membrane interface such that one of the loops can bind to the vesicle and the other to the flat bilayer. In this configuration, Ca2+-binding might favor movement of lipids toward the Ca2+-binding sites to destabilize the bilayers and initiate fusion. To test this idea, we performed a 570 ns MD simulation of a system analogous to the nosytfusion system but including two C2AB molecules between the membranes (sytfusion2g simulation) (Fig. S4A). We did not observe the postulated lipid movements during the simulation and it became clear that the C2AB molecules hindered the action of the SNARE complexes in bringing the membranes together (Fig. 1C and S4B).
In these simulations, the SNARE complexes may have been too far from the center of the bilayer-bilayer interface to effectively induce membrane fusion. Placing them closer to the center might facilitate fusion and would prevent the C2 domains from coming close to the site of fusion because of steric hindrance. However, the C2 domains could be located further from the center, where they could bridge the two membranes as predicted in some models of Syt1 function (42, 43). In such positions, the C2 domains might act as wedges that prevent the membranes from coming closer while the SNARE complexes pull the membranes together in the center, resulting in a torque that could help to bend the membranes to initiate fusion [as proposed previously for Munc18–1 function (61)]. To test this model, we used a system with four trans-SNARE complexes closer to the center that we generated previously [fusion2g in (15)], increased the separation between the flat membrane and the vesicle by 1.6 nm to make room for the bridging C2 domains, used a restrained simulation to move the syntaxin-1 TM regions to their positions in the translated flat bilayer, and included four C2AB molecules in positions in which the two C2 domains were poised to bridge the two membranes (Fig. S5A). We ran a 450 ns MD simulation (sytfusion3) and observed that the two membranes came closer to each other but were not brought into contact because of steric hindrance caused by the bridging C2 domains, and the SNARE complexes were unable to pull the bilayers together in the center (Fig. 1D and S5B).
The results of all these simulations need to be interpreted with caution because of the limited time of the simulations and hence do not rule out the various models tested. However, the overall results do suggest that C2 domains near the site of fusion hinder the action of SNARE complexes in bringing the membranes together. Importantly, throughout these simulations we observed that the hydrophobic residues at the tips of the C2 domain Ca2+-binding loops often contacted the hydrophobic acyl region of the flat bilayer but did not insert deeply (e.g. Fig. 2A–C), consistent with results from previous MD simulations [e.g. (56)]. Such insertion causes local perturbations but not major alterations of the bilayer structure, and does not induce membrane curvature. Fluorescent probes attached to cysteines in these positions are expected to insert more deeply into the acyl region because they are larger, which has led to the assumption than that the Ca2+-binding loops penetrate into the membrane [reviewed in (44, 61)] more than they actually do. Note also that electron microscopy experiments supporting the notion that Syt1 induces membrane curvature used negative stain, which strongly perturbs membranes, and were performed with artificially high protein-to-lipid ratios (1:40) (39, 40) such that proteins should cover most of the lipid surface. All these observations suggest that popular models postulating that Syt1 facilitates membrane fusion by bridging the membranes, perturbing the bilayers or inducing membrane curvature are likely incorrect. Thus, while it seemed paradoxical that binding of the Syt1 C2B domain to the SNARE complex via the primary interface orients its Ca2+-binding loops away from the fusion site (25), it now appears that this feature makes a lot of sense because it keeps Syt1 away from the fusion site where it would hinder the actions of the SNAREs that induce membrane fusion.
Ca2+ does not cause dissociation of Syt1 C2AB from membrane-anchored SNARE complex
The observation that Syt1 C2AB binds to nanodiscs containing SNARE complex more tightly to SNARE-free nanodiscs in the absence of Ca2+ whereas the affinity is similar in the presence of Ca2+ suggested that Ca2+ induces dissociation of C2AB from membrane-anchored SNARE complex (47), but this notion was not tested directly. To examine whether Ca2+ indeed induces such dissociation, we used an approach involving labeling of SNARE complex with bimane on residue 59 of SNAP-25 and replacement of T285 of Syt1 with a tryptophan. This design led to tryptophan-induced quenching of the bimane fluorescence upon binding of Syt1 C2B domain to liposome-anchored SC-59-bimane in the absence of Ca2+ due to the predicted proximity of the tryptophan to the bimane in the primary interface (62). Correspondingly, we observed strong quenching of the fluorescence of nanodisc-anchored SC-59-bimane upon addition of C2AB-T285W in the absence of Ca2+ (Fig. 3). Importantly, the quenching remained upon addition of Ca2+ (Fig. 3), showing that Ca2+ does not induce dissociation of Syt1 C2AB from membrane-anchored SNARE complex under the conditions of these experiments.
Figure 3.
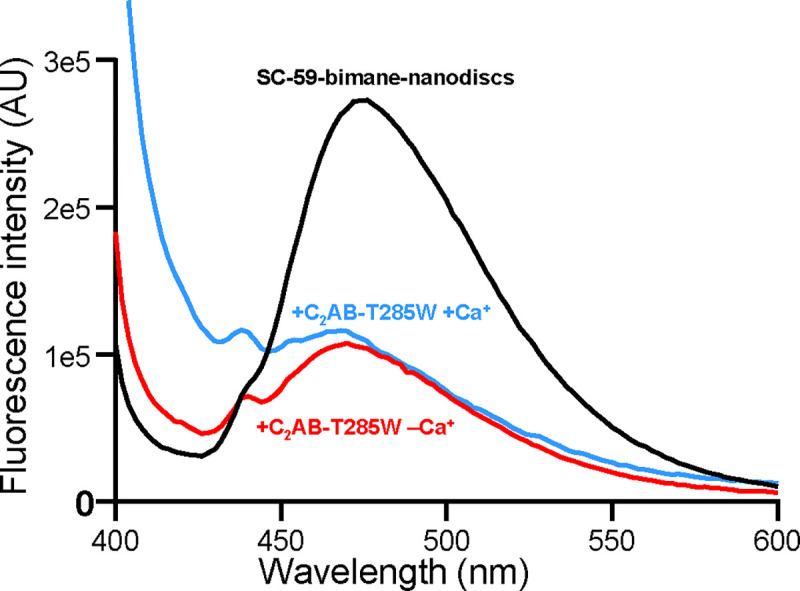
Ca2+ does not induce dissociation of Syt1 C2AB from membrane-anchored SNARE complex. The graph shows fluorescence spectra of nanodisc-anchored SC-59-bimane alone (black curve), or in the presence of C2AB-T285W and Mg-ATP plus 1 mM EGTA (red curve) or 1 mM Ca2+ (blue curve). Mg-ATP was included in the samples to hinder non-specific C2AB-SNARE complex interactions (47).
Region II of the Syt1-SNARE complex remains bound upon disruption of region I
In parallel with our MD simulations and bimane fluorescence experiments, we studied the effects of mutations in the primary interface on Syt1 C2B domain-SNARE complex binding in solution using NMR spectroscopy to probe the energy landscape of the primary interface and to correlate binding through this interface with Syt1 function in the electrophysiological data (52). For this purpose, we acquired transverse relaxation optimized spectroscopy (TROSY)-enhanced 1H-15N heteronuclear single quantum coherence (HSQC) spectra of wild type (WT) and mutant versions of 2H,15N-labeled C2B domain specifically 13CH3-labeled at the Ile δ1 and Met methyl groups (here referred to as 15N-C2B for simplicity), and analyzed the cross-peak perturbations caused by titration with SNARE complex four-helix bundle bound to a complexin-1 fragment that spans residues 26–83 and prevents aggregation of C2B-SNARE complexes (referred to as CpxSC). Previous studies using this methodology showed that, in solution: i) the WT C2B domain binds to CpxSC via both the primary interface and the polybasic region that interacts with lipids when the SNARE complex is membrane-anchored; ii) an R322E/K325E mutation in the polybasic region (REKE) abolishes CpxSC binding through this region; iii) an R398Q/R399Q mutation in region II of the primary interface (RQRQ) abolishes binding through this interface; iv) an R322E/K325E/R398Q/R399Q mutation abolishes all binding; and v) an E295A/Y338W mutation in region I enhances the affinity of binding through the primary interface (47).
Superpositions of full 1H-15N TROSY-HSQC spectra of WT and mutant 15N-C2B titrated with CpxSC are shown in Fig. S6–S14. The positions of residues corresponding to diagnostic cross-peaks that reflect binding to region I or II of the primary interface are illustrated in red on the ribbon diagram of Fig. S15A, which displays the side chains of the C2B domain that were mutated in stick models. R281 is one of the three arginines in region II and was mutated to compare its contributions to C2B-CpxSC binding with those of R398 and R399; A402 is located between regions I and II; and E295 and Y338 are the key residues of C2B that form region I. These four residues were replaced individually in mutations that suppress the dominant negative lethality caused by overexpression of Syt1 with disrupted C2B domain Ca2+ binding sites in Drosophila (50). In initial experiments we analyzed the effects of a double E295K/Y338A mutation in the background of WT C2B domain and observed that the V283 and K288 cross-peaks still shifted upon titration with CpxSC, showing that binding to region II still persisted (Fig. S7, S15B). Since Y338 was replaced by aspartate in the mutant that suppressed lethality in the Drosophila screen, and an A402T mutation also suppressed lethality (50), we titrated a double Y338D/A204T 15N-C2B domain mutant but still observed binding to region II (Fig. S8, S15B). In these experiments it was difficult to assess the effects on binding to region I because of the limited cross-peak shifts for residues of this region observed for WT C2B (Fig. S6, S15B). The limited nature of these changes arises because of interference by binding modes involving the polybasic region and, consequently, full binding to the primary interface is better observed for the REKE mutant, which exhibits more overt shifts for many cross-peaks, including some from residues in region I (47) such as those of K297 and L299 (Fig. 4, S9; compare with WT C2B in Fig. S6, S15B). Hence, additional mutations in the primary interface were performed in the background of the REKE mutant.
Figure 4.

The Syt1 C2B domain still binds to the SNARE complex through region II of the primary interface when binding through region I is abolished. The diagrams show expansions of 1H-15N TROSY HSQC spectra of 15N-C2B domain mutants (as indicated above) acquired in isolation (black contours) or increasing concentrations of CpxSC (rainbow colours). The residues corresponding to the cross-peaks shown in the expansions and the regions where they are located are indicated on the left. The following concentrations of 15N-C2B mutant and CpxSC (μM/μM) were used for the different mutants (from black to purple): REKE 32/0, 30/10, 28/19, 26/28, 24/36, 20/51, 17/64, 12/85; REKE-Y338W 32/0, 30/10, 28/19, 26/28, 24/36, 20/53, 17/67, 12/88; REKE-E295A 32/0, 30/10, 28/19, 26/28, 24/36, 20/53, 17/65, 12/88; REKE-A402T 32/0, 30/10, 28/19, 26/28, 24/36, 20/53, 17/67, 12/88; REKE-Y338D 32/0, 30/10, 28/19, 26/28, 24/36, 20/53, 17/68, 12/88. For the REKE-R281A mutant the concentrations were 32/0 (black), 26/28 (yellow) and 13/98 (purple). The intensities of cross-peaks decreased as CpxSC was added because 15N-C2B was diluted and binding to CpxSC causes cross-peak broadening. Contour levels were adjusted to compensate for these decreased intensities, but after doing comparable adjustments for the spectra of the REKE-E295A mutant the cross-peaks in the middle of the titration are still very weak because of chemical exchange broadening.
Replacement of R281 with alanine (REKE-R281A mutant) strongly impaired overall binding of the C2B domain to CpxSC, although some binding remained at high CpxSC concentrations (Fig. 4, S10). These effects are comparable to those caused by individual R398Q or R399Q mutations but milder than those caused by the double R398Q/R399Q mutation (47). These results suggest that each arginine has comparable contributions to SNARE binding, consistent with the similar phenotypes caused by point mutations in the arginines (52). We also analyzed REKE-Y338W and REKE-E295A 15N-C2B mutants to dissect the contributions of each residue substitution to the enhancement in CpxSc binding affinity caused by the E295A/Y338W mutation in region I of the C2B domain (47). We found that the cross-peaks shifts observed for REKE-Y338W were similar to those observed for REKE (Fig. 4, S11), consistent with the lack of a phenotype caused by this mutation (52). In contrast, the E295A mutant exhibited more extensive shifts throughout the 1H-15N TROSY-HSQC spectrum than the REKE mutant Fig. S9, S12), indicating a more intimate interaction, and the shifts in diagnostic cross-peaks were larger earlier in the titration than observed for REKE (Fig. 4). We even observed clear chemical exchange broadening for some cross-peaks of the E295A mutant (e.g. the R398 cross-peak, Fig. 4) and broadening beyond detection of the W404 side chain cross-peak (Fig. S12). These observations show that the E295A mutation enhances the affinity of the C2B domain for Cpx-SC whereas the Y338W mutation has little effect. Indeed, the KDs between C2B and CpxSC estimated by fitting the changes in the 1H chemical shift of V283 and in the 15N chemical shift of K288 to a single binding site model (Fig. S16) were 34.4 ± 9.3 and 33.2 ± 4.6 μM for the REKE mutant, 30.4 ± 4.4 and 33.0 ± 8.5 μM for REKE-Y338W, and 7.3 ± 2.0 and 4.6 ± 0.4 μM for REKE-E295A. The about 5 to 6-fold enhancement in the affinity of REKE-E295A derived from these results correlates exquisitely well with the 4-fold decrease in spontaneous release caused by the E295A mutation (52), strongly supporting the notion that Syt1-SNARE binding through the primary interface inhibits spontaneous release. Moreover, the E295A and E295/Y338W mutations cause similar phenotypes, including enhanced vesicle priming and severe impairments in Ca2+-triggered release (52), showing that the phenotypes of the double mutant observed earlier (25) are caused by the E295A substitution.
No changes were caused by CpxSc on the K297 and L299 cross-peaks of 15N REKE-C2B bearing either A402T or Y338D mutations (REKE-A402T and REKE-Y338D) (Fig. 4, S13–S14), showing that the A402T and Y338D mutations practically abrogate binding to region I. Intriguingly, the changes in the V283 and K288 cross-peaks of both mutants were comparable to those observed for REKE-C2B, showing that robust binding to region II remained for these mutants, albeit the binding mode was likely altered given the different direction of the movements of the R398 cross-peak for REKE-A402T and REKE-Y338D compared to REKE (Fig. 4). Hence, binding through region II remains even in the absence of binding through region I.
Analysis of interactions between C2B domain arginines and SNARE acidic residues at region II by MD simulations
The model postulating that Ca2+-induces dissociation of Syt1 from the SNARE complex to trigger neurotransmitter release arose in part because of the observation that the E295A/Y338W mutation in region I of the primary interface impairs release (25) but enhances C2B domain-SNARE complex binding (47). Our NMR results showing that disruption of region I does not lead to dissociation of region II, together with the fluorescence data showing that Ca2+ does not dissociate C2AB from liposome-anchored SNARE complex (Fig. 3), bring the possibility that neurotransmitter release is triggered by a Ca2+-induced re-arrangement of the primary interface in which region I dissociates but region II does not. To develop a hypothesis of how such re-arrangement could trigger release, it is important to have a clear picture of the configuration of the C2B domain with respect to the SNARE complex and the membrane. Such a model could be derived from the multiple crystal structures of Syt1-SNARE complexes that have been described (25, 49), which revealed similar configurations of the primary interface. There was some variability in the interactions involving the three C2B arginines of region II (R281, R398, R399) in the various structures, but the arginines were generally close to multiple acidic residues of both SNAP-25 and syntaxin-1 (Table S2) (Fig. S17A,B). However, MD simulations of primed Syt1 C2AB-SNARE-complexin-1 complexes bridging a vesicle and a flat bilayer suggested that additional configurations exist in which the three arginines interact primarily with acidic residues of SNAP-25, perhaps favored by simultaneous interactions of the C2B domain with the flat bilayer (27). A concern about these simulations is their limited length and the fact that the SNARE four-helix bundles were in distinct stages of assembly, which could affect the interactions with Syt1.
To further examine the consistency of these results, we set up a system with four almost fully assembled SNAREs complexes bridging a vesicle and a flat bilayer, and bound to Syt1 C2AB through the primary interface as in the crystal structure corresponding to PDB code 5KJ7 (Fig. S18A). Complexin-1 was omitted to examine whether the previously observed Syt1-SNARE binding modes (27) are altered in its absence. We ran a 596 ns simulation (referred to as s1action2), which led to similar configurations of the C2B domain with respect to the SNARE complex and the flat bilayer (Fig. S18B–E) that resembled those observed in the previous simulations (27). This conclusion was supported by analysis of the distances between the guanidine carbon (CZ) of R281, R398 and R399 of the four C2AB molecules and the carboxyl carbon of the nearby acidic residues (CG for aspartates, CD for glutamates) of syntaxin-1 or SNAP-25 along the trajectories (Fig. S19). These plots and the trajectory-averaged distances calculated for each C2AB-SNARE complex (Table S3) again showed a predominance of configurations in which the three arginines interact primarily with the SNAP-25 acidic residues, as illustrated by a representative pose shown in Fig. S17C,D), although there were also frequent interactions with syntaxin-1 acidic residues. The similarity of the distances averaged over the four C2AB-SNARE complexes in the s1action2 simulation and those calculated for the prsg simulation of ref. (27) (Table S3) further shows the consistency of these results and suggest that the Syt1-SNARE binding modes are not substantially affected by complexin-1.
A lever hypothesis for Syt1 action
How can Ca2+ binding to the C2B domain play such a critical role in triggering fast neurotransmitter release (33, 34) if its Ca2+-binding loops are pointing away from the site of fusion in the primed state? The hypothesis that we propose to answer this question emerged from the realization that zippering of the jxt linkers separating the SNARE motifs and TM regions of synaptobrevin and syntaxin-1 is critical for release (63, 64) and liposome fusion (65), which was reinforced by our recent MD simulations (15). The simulations showed that, because the SNARE four-helix bundle is oriented parallel to the bilayers and the TM regions are approximately perpendicular, the jxt linkers must form kinked helices (e.g. Fig. 5A) or become partially unstructured (e.g. Fig. S18B–E) to accommodate the changes in direction. Linker zippering led to fast (microsecond scale) fusion in the simulations (15) and is in principle energetically favorable (66). However, linker zippering is expected to be hindered by substantial energy barriers because the zippering pulls the hydrophobic TM regions into the polar bilayer interface (67), and the abundant basic and aromatic residues of the jxt linkers have high propensities to interact with the lipids (15, 27). Our hypothesis predicts that Ca2+ binding to the Syt1 C2B domain helps to overcome these energy barriers because it induces reorientation of the domain with respect to the flat membrane, from the parallel orientation existing in the primed state (modeled in Fig. 5A) to an approximately perpendicular orientation that allows insertion of both of its Ca2+ binding loops into the membrane as observed by EPR (54) (modeled in Fig. 5B). During this reorientation, the C2B domain is predicted to act as a lever, as region I of the primary interface dissociates but the three arginines of region II remain electrostatically bound to the SNARE acidic residues and pull the SNARE four-helix bundle away from the fusion site, facilitating linker zippering (Fig. 5B) and fast membrane fusion. This model explains how Syt1 can act remotely from the site of fusion without inserting its Ca2+-binding loops near this site, where Syt1 would hinder the action of the SNAREs that trigger membrane fusion. The model also allows rationalization of the various phenotypes caused by mutations in the primary interface, most notably the findings that neurotransmitter release is impaired by mutations that impair or enhance binding through this interface and is differentially affected by mutations in the SNARE acidic residues (25, 52, 53, 68) (see discussion).
Figure 5.

Lever hypothesis of Syt1 function. (A) Model of the primed state before Ca2+ influx. Lipids are shown as stick models with the same color coding as in Fig. 1. The SNARE complex is represented by a ribbon diagram in salmon color except for the jxt linkers, which are colored in dark gray. The Syt1 C2B domain is represented by a green ribbon diagram with key residues at the primary interface shown as spheres: E295 and Y338 (region I) are in magenta; R281, R398 and R399 (region II) are in blue. The SNARE complex, flat bilayer and vesicle configurations were extracted from one of the frames of the fusion2g simulation described in (15) to illustrate a potential configuration of the primed state. The C2B domain was placed at a location analogous to that in one of the primed complexes at the end of the s1action2 simulation. The Syt1 C2A domain and complexin are also part of the primed state but are not shown for simplicity. (B) Model of a potential Ca2+-activated state built manually in Pymol using the configuration of panel (A) as starting point, rotating the C2B domain to an approximately perpendicular orientation with respect to the flat bilayer that mimics the plasma membrane, as observed by EPR (54), and moving the SNARE four-helix bundle to keep ionic interactions of the SNARE acidic residues with the arginines of region II of the C2B domain. The synaptobrevin and syntaxin-1 jxt linkers were modeled as helices that extend from the four-helix bundle based on the crystal structure of the SNARE complex (77), and the TM regions were pulled toward the polar membrane-membrane interface to maintain the continuity of the polypeptide chains. Ca2+ ions are shown as yellow spheres. (C, D) Close-up views of the primary interface in panel (A) and (B), respectively, showing the C2B arginines and SNARE acidic residues as stick models to show the interactions between these residues in the proposed initial configuration and in one of many potential configurations of the Ca2+-activated state.
Ca2+ -induces reorientation of the Syt1 C2B domain bound to membrane-anchored SNARE complex
Testing our hypothesis is hindered by several factors. First, the proposed Ca2+-induced reorientation of the C2B domain may occur transiently to trigger release in microseconds and hence may be difficult to observe with experiments performed under equilibrium conditions. Second, the reorientation may be difficult to monitor because it may be much less pronounced than depicted in Fig. 5 (see discussion). Third, FRET studies showed that Syt1 C2AB interacts with nanodisc-anchored SNARE complex in part through the primary interface, but the binding is weak and there are also non-specific binding modes that muddle the interpretation of the data (47). Buffers with physiological ionic strength and including ATP hinder such unwanted interactions but also decrease the overall affinity of Ca2+-free C2AB for nanodisc-anchored SNARE complex. Ca2+ enhances the overall affinity, but the enhancement arises because Ca2+ strongly increases the affinity of C2AB for the lipids. Interactions between C2AB and SNARE complex anchored on nanodiscs are actually weakened by Ca2+, as shown by the fact that C2AB binds with similar affinity to nanodiscs containing or lacking SNARE complex (47). Fourth, use of bimane fluorescence quenching by a nearby tryptophan (Fig. 3) to monitor the postulated Ca2+-induced reorientation is hindered by the need to place the tryptophan near the bimane group, which might alter the native binding mode and potentially enhance affinity due to tryoptophan-bimane interactions.
Taking these concerns into account, we studied whether Ca2+ induces reorientation of the C2B domain bound to liposome-anchored SNARE complex using FRET experiments with Syt1 C2AB covalently linked through a 37-residue sequence to the C-terminal SNARE motif of SNAP-25, an approach that was used to stabilize Syt1-SNARE complex interactions in the X-ray studies that revealed the primary interface (25). The covalent link facilitates quantitative binding of C2AB to the SNARE complex through the primary interface in the absence of Ca2+ and thus prevents FRET changes that could arise from a Ca2+-induced increase of the overall affinity of C2AB for the SNARE complex-containing liposomes rather than from C2B reorientation. We also include complexin-1 in these experiments because it hinders non-specific interactions between Syt1 and the SNARE complex (47). A photostable Cy5-Trolox acceptor probe was attached to residue 412 of C2AB in the fusion protein, which was used to form liposome-anchored SNARE complexes that were also labeled with a Cy3-Trolox donor probe on residue 27 or 34 of the SNAP-25 N-terminal SNARE motif. These positions were chosen because the distances between donor and acceptor probes are predicted to change substantially based on the model of Fig. 5 (see also Fig. 6A). To have reference spectra without FRET, we acquired fluorescence spectra with analogous samples in the presence of detergent to disrupt the liposomes and of NSF plus αSNAP to disassemble the SNARE complex.
Figure 6.

Ca2+ induces reorientation of the Syt1 C2B domain with respect to membrane-anchored SNARE complex. (A) Models of the Syt1 C2B domain-SNARE complex in the primed state before Ca2+ influx and in a potential Ca2+-activated state, analogous to those of Fig. 5A,B and with the same color code. Residues of C2B and SNAP-25 that were mutated to cysteines to attach fluorescent probes are shown as spheres and labeled with the residue number. In these models, the following changes are predicted for the distances between the approximate points of attachment of the fluorescent probes in the absence or presence of Ca2+: SNAP-25 Q34 CD carbon - Syt1 E412 CD carbon from 34 Å (−Ca2+) to 48 Å (+Ca2+); SNAP-25 E27 CD carbon - Syt1 E412 CD carbon from 41 Å (−Ca2+) to 52 Å (+Ca2+); SNAP-25 Q34 CD carbon - Syt1 E391 CB carbon 31 Å (−Ca2+) to 28 Å (+Ca2+). Note that there is a large uncertainty in the distance changes because the configuration of the Ca2+-activated state is unknown and the 27–412 and 34–412 distances depend strongly on the angle of rotation (Supplementary Discussion). (B-D) Fluorescence spectra of liposome-anchored C2AB-SNARE-complexin-1 complex with stabilized Cy3 and Cy5 probes attached to the following residues of SNAP-25 and the C2B domain, respectively: 27–412 (B), 34–412 (C) and 34–391 (D). The spectra were obtained in the presence of Mg-ATP plus 1 mM EGTA (red curves), 1 mM Ca2+ (blue curves) or 1% β-OG plus NSF and αSNAP as a control to disrupt the Syt1-SNARE complex (black curves). In the experiments of (B-D), Mg-ATP was included in the samples to hinder non-specific C2AB-SNARE complex interactions (47). (E) Quantification of the FRET difference between the spectra obtained in 1 mM Ca2+ and 1 mM EGTA normalized with the FRET observed in the presence of 1 mM EGTA. Bars represent mean values and error bars represent standard deviations from experiments performed at least in triplicate. Statistical significance and p values were determined by one-way analysis of variance (ANOVA) with the Holm-Sidak test (*** p < 0.001).
Importantly, comparison of the spectra obtained in EGTA with the reference spectra revealed robust FRET for the samples of liposome-anchored C2AB-SNARE-complexin-1 complex with the 27–412 and 34–412 FRET pairs and Ca2+ induced reproducible decreases in FRET (Fig. 6B,C), which was verified with experiments performed on different days with different samples. As a negative control, we also performed FRET experiments with Cy5-Trolox attached to residue 391 of C2B in the fusion protein and Cy3-Trolox attached to residue 34 of SNAP-25, since the distance between the two probes was predicted to change little upon Ca2+ binding (Fig. 6A). Indeed, we did not observe appreciable changes in FRET for the 34–391 pair (Fig. 6D), supporting the prediction and confirming that Ca2+ does not induce dissociation of Syt1 from the liposome-anchored SNARE-complexin-1 complex under these conditions. Quantification of the Ca2+-induced changes in donor fluorescence for repeat experiments showed the statistical significance of these results (Fig. 6E). We note that the changes in distances between the fluorescent probes predicted by the model are approximate given the considerable size of the probes and the uncertainty in the configuration of the Ca2+-bound state (Supplementary Discussion). Regardless of this uncertainty, these FRET data clearly support the notion that Ca2+ induces reorientation of the C2B domain bound to membrane-anchored SNARE complex, which is at the heart of the model of Syt1 action that we propose.
Discussion
The function of Syt1 as the Ca2+ sensor that triggers neurotransmitter release is well established, but its mechanism of action has remained enigmatic despite intense research for over three decades. Models postulating that Syt1 cooperates with the SNAREs in triggering membrane fusion by perturbing bilayers, inducing membrane curvature or bridging the two membranes were attractive, but appeared to be incongruous with the finding that Syt1 binds to the SNARE complex through the primary interface (25), which orients the C2B domain Ca2+-binding loops away from the fusion site. The proposal that Ca2+ induces dissociation of Syt1 from the SNARE complex, which would allow the C2 domains to reorient toward the site of fusion, provided a potential solution to this paradox (47) but did not bode well for the fast speed of release. Moreover, it was unclear how these models of Syt1 function can be reconciled with our MD simulation revealing microsecond scale fusion induced by SNARE complexes upon jxt linker zippering (15). The MD simulations presented here now cast further doubt on these models and suggest that Syt1 does not act directly at the site of fusion. Our NMR titrations, together with previous EPR data (54), lead naturally to the hypothesis that Ca2+ binding induces reorientation of the C2B domain on the membrane and partial rather than full dissociation from the SNAREs such that ionic interactions involving the C2B arginine cluster pull the SNARE complex and facilitate jxt linker zippering to induce fast membrane fusion. Although further research will be required to test this lever hypothesis and the underlying details, the notion that Ca2+-induced remodeling of Syt1-SNARE interactions at the primary interface is crucial for neurotransmitter release is supported by our FRET assays and by the correlation of our MD simulation and NMR results with the electrophysiological data described in the accompanying paper (52).
Need to revise current models of Syt1 action.
MD simulations need to be interpreted with caution because force fields are not perfect and because of the limited simulation lengths that can be achieved, particularly for multimillion atom systems such as those studied here. However, all-atom MD simulations have provided a powerful tool to visualize the neurotransmitter release machinery (27, 56) and to elucidate how the SNAREs triggers fast membrane fusion, showing how linker zippering pulls the hydrophobic TM region to the polar membrane-membrane interface where they catalyze lipid acyl chain encounters at the interface, initiating bilayer merger (15). This mechanism makes a lot of sense from a physicochemical perspective and explains a large amount of experimental data (15) but does not involve a central role for membrane curvature in initiating fusion, in contrast to assumptions made from theoretical calculations (69). Hence, there is no need for neurotransmitter release to depend on induction of membrane curvature by Syt1, as proposed by some models (39, 40). Other models postulated that insertion of the Syt1 C2 domains perturbs bilayers and causes lipid disorder (35, 37, 38), which can facilitate fusion, but bilayer perturbation is efficiently caused by the events that ensue after SNARE jxt linker zippering without the need for Syt1 (15). In fact, our MD simulations strongly suggest that, when placed near the site of fusion, the Syt1 C2 domains hinder SNARE action [Fig. 1, S1–S5; see also (27)]. Note also that the experiments supporting these models of Syt1 function used much larger protein-to-lipid ratios than the Syt-to-lipid ratios present in synaptic vesicles (57), and our MD simulations indicate that a few Syt1 molecules induce rather limited perturbation of lipid bilayers (e.g. Fig. 2).
An additional model predicted that the Syt1 C2 domains bridge the two membranes, helping the SNAREs to bring the membranes together (42, 43). This notion was inspired in part by the observation that Syt1 C2AB can indeed bridge two membranes and in part by the overall idea that substantial energy is required to bring two membranes together (42, 70). However, the extended membrane-membrane interfaces induced by SNARE complexes, even when full zippering is prevented (60), and our previous MD simulations (15, 27), indicate that bringing membranes into contact is easier than previously thought. Moreover, the Syt1 C2 domains clearly hindered the ability of the SNAREs to bring the membranes together in our sytfusion3 simulation (Fig. 1D, S5). In summary, previous models of Syt1 function were attractive but are not supported by the new perspective on membrane fusion emerging from the MD simulations. Moreover, the fact that binding of the Syt1 C2B domain to the SNARE complex through the primary interface places its Ca2+-binding loops away from the site of fusion argues strongly against these models.
Multiple roles of the primary interface.
Previous studies (25, 50, 53), together with the correlations between our binding data (47) (Fig. 4, S6–S16) and electrophysiological results (52), provide overwhelming evidence that the primary interface plays central roles in vesicle priming, clamping of spontaneous release and Ca2+ triggering of release. The strong disruption of vesicle priming and evoked release caused by the R398Q/R399Q mutation (25, 52, 53) that abolishes binding through region II of the primary interface (47) shows the critical functional importance of this region. The Y338D and A402T mutations that abolish C2B-SNARE binding through region I (Fig. 4, S13, S14) also impair priming and evoked release (52), but the E295A and E295A/Y338W mutations in region I that enhance overall binding (47) (Fig. 4, S12) strongly impair evoked release while enhancing priming (25, 52). These findings show that interactions involving region I mediate priming but need to be dissociated for Ca2+ triggering of release. The primary interface is also important to inhibit spontaneous release, as mutations in this interface that impair binding generally lead to enhanced spontaneous release, and the E295A mutation causes a 5–6-fold enhancement in binding (Fig. 4, S16) and a 4-fold decrease in spontaneous release (52). The finding that the E295A substitution mediates the enhanced binding and the phenotypes caused by the double E295A/Y338W mutation (52) (Fig. S4, S11, S12) might seem surprising because E295 forms a salt bridge in crystal structures of Syt1-SNARE complexes (25, 49), but can be attributed to the increase in the positive electrostatic potential of C2B caused by the mutation, as overall electrostatics play a key role in binding of the highly basic C2B domain to the highly acidic SNARE complex (71).
The lever hypothesis.
The proposal that Ca2+ induces dissociation of Syt1 from the SNARE complex arose in part because the perpendicular orientation of the C2B domain with respect to the membrane induced by Ca2+ (54) is incompatible with binding of C2B to the SNARE complex via the full primary interface, and in part to explain the impairment of release caused by the E295A/Y338W mutation despite enhancing binding (47). The observation that robust C2B-SNARE complex binding through region II of the primary interface remains even upon disruption of region I with the Y338D mutation (Fig. 4, S14) provided a key clue suggesting that Ca2+ induces partial rather than full dissociation of the primary interface. Thus, reorientation of C2B with respect to the membrane can occur while interactions mediated by region I are released but those involving region II remain. In support of this notion, our bimane fluorescence quenching results (Fig. 3) and the FRET data obtained with the 34–391 pair (Fig. 6D) clearly show that Ca2+ does not dissociate the C2AB fragment from membrane-anchored SNARE complex, and the FRET data obtained with the 27–412 and 34–412 pairs (Fig. 6B,C) show that Ca2+ indeed alters the orientation of the C2B domain with respect to the SNARE complex. The exact nature and extent of the reorientation is still unclear (Supplementary discussion) but, since only one of the two Ca2+-binding loops is inserted into the membrane in the absence of Ca2+ (Fig. 5A), the reorientation is most likely driven by insertion of the other Ca2+-binding loop into the membrane (Fig. 5B) together with Ca2+-phospholipid interactions that complete the coordination spheres of the Ca2+ ions (36).
Ca2+-induced reorientation of the C2B domain is expected to pull the SNARE complex away from the fusion site through electrostatic interactions of the three C2B arginines of region II with acidic residues of the SNARE complex. The importance of these interactions for Ca2+ triggering of release is supported by the fact that individual R281A, R398Q and R399Q mutations in Syt1 and D51N and E55Q mutations in SNAP-25 severely impair evoked release without affecting vesicle priming, thus lowering the vesicle release probability (52). Intriguingly, mutations in E52 of SNAP-25 or acidic residues of syntaxin-1 (E228, D231, E234 and E238) increase the release probability (52, 72). These distinct phenotypes can be rationalized by the observation that region II of the primary interface has two faces. D51 and E55 are located along one side of the SNAP-25 N-terminal SNARE motif helix, whereas SNAP-25 E52 and the syntaxin-1 acidic residues are located on the other side of the helix (Fig. S17A,C). In the configurations most populated during our MD simulations of the primed state, the C2B arginines are generally closer to the former face, and R281 interacts with D51 and E55 while R398 interacts with E55, D58 and E62 (Fig. S17C,D, S19, Table S3). The observed phenotypes indicate that this is the active face involved in the pulling action of Syt1 that triggers evoked release. Configurations with C2B arginines closer to the syntaxin-1 acidic residues on the other face of region II also occur in our simulations (Fig. S19) and are observed in crystal structures (Fig. S17A,B, Table S2), but manual inspection suggests that the syntaxin-1 helix needs to move away from the C2B domain arginines as C2B reorients and pulls the SNARE complex (Fig. 5D). Hence, this face of region II may hinder activation of evoked release by Syt1, which would explain the increased release probabilities caused by mutations in SNAP-25 E52 or acidic residues of syntaxin-1. We note that R399 is oriented toward this inhibitory face (Fig. 17A,C) and yet is important for evoked release (52). Since interactions of the arginines must be remodeled during reorientation of the C2B domain, it is plausible that remodeling leads to new interactions of R399 that are key for the pulling action that triggers release. For instance, modeling suggests that R399 may interact with D166 and E170 in the active state (Fig. 5D), which might explain the strong impairments in neurotransmitter release caused by mutations in these residues (68).
Pulling the SNARE complex upon reorientation of C2B should increase the angle between the long axis of the four-helix bundle and the plasma membrane, consistent with data showing that the angle between the SNARE complex and a support lipid bilayer is increased by binding to Ca2+-saturated Syt1 C2AB (73). Such tilting may enable formation of a continuous helix in syntaxin-1 (Fig. 5B), which might facilitate fusion. However, it is most likely that the key consequence of the C2B domain reorientation is that pulling the SNARE complex away from the site of fusion facilitates extension of the syntaxin-1 and synaptobrevin SNARE motif helices into the jxt linkers, which is critical for neurotransmitter release (63, 64) and liposome fusion (65). This notion emerged in part from MD simulations showing that jxt linker zippering can mediate fast, microsecond scale membrane fusion, but zippering is hindered by the natural geometry of the system, which dictates that the directions of the polypeptide chains turn the corner at the jxt linkers (15) (Fig. 5A). It is unclear whether the jxt linkers form kinked helices in the primed state, as in Fig. 5A, or adopt more disordered structures before Ca2+ influx, but there is little doubt that the abundant basic and aromatic residues in the linkers interact with the lipids in the primed state (27) and that such interactions need to be rearranged for the linkers to zipper. The central aspect of our model is that the pulling force of the Syt1 C2B domain on the SNARE complex lowers the energy barrier for such rearrangement, facilitating linker zippering. This model naturally explains why the C2B domain Ca2+ binding loops are pointing away from the site of fusion when Syt1 binds to the SNARE complex through the primary interface, helping to trigger fast fusion remotely instead of being close to the fusion site where the C2 domains would hinder SNARE action (Fig. 1, S1, S2, S4, S5). We note that Ca2+ binding was proposed to induce a movement ‘en bloc’ of the Syt1-SNARE complex that deforms the membranes (25). While this model is inconsistent with the effects of mutations in region I of the primary interface on C2B-SNARE binding (Fig. 4) and neurotransmitter release (25, 52), the seminal crystal structure described in this study was crucial to develop the model proposed here.
Clearly, multiple aspects of our model are speculative and further research will be required to examine these aspects in detail and test the overall lever hypothesis of Syt1 function. For instance, the nature of the C2B domain reorientation is unclear and it might be transient or much less pronounced than predicted in Fig. 5, as small structural changes may be sufficient to overcome the energy barrier that hinders Ca2+-evoked release (Supplementary Discussion). An additional question is what is the role of the Syt1 C2A domain, as Ca2+ binding to this domain contributes to trigger neurotransmitter release even if it is not as critical as Ca2+ binding to the C2B domain (28, 32, 34). In three of the complexes of our MD simulations of the primed state, the C2A domain Ca2+ binding loops are next to those of the C2B domain, poised to bind concomitantly to the membrane upon Ca2+ binding (Fig. S18C–E), and in two of the complexes there are ionic interactions between acidic residues of SNAP-25 and a polybasic region of the C2A domain (K189-K192) (e.g. Fig. S20). Hence, the C2A domain could cooperate with the C2B domain in pulling the SNARE complex upon Ca2+-dependent membrane binding, and could provide a second anchor point on the membrane to exert force without sliding on the membrane surface, but these ideas need to be tested. Note also that other interactions of Syt1 have described, such as binding of the C2B domain to the SNARE complex through a so-called tripartite interface (49) or Ca2+-independent oligomerization (74). Although we and others have been unable to observe the tripartite interface in solution (47, 75) or the oligomerization by cryo-EM (7, 23, 76), the relevance of these interactions and their compatibility with our model need further investigation. The lever hypothesis and the underlying ideas presented here provide a framework to address these questions and elucidate how Syt1 triggers neurotransmitter release.
Methods
Molecular dynamics simulations were performed using the same methodology employed in our previous simulations (15, 27). Methods used for site directed mutagenesis, expression and purification of SNARE proteins and Syt1 fragments, NMR spectroscopy, protein labeling with fluorescent probes and fluorescence spectroscopy were basically as described (47, 75). Specific details for all these methods are described in the SI Appendix.
Supplementary Material
Significance statement.
Neurotransmitter release requires SNARE complexes that fuse synaptic vesicles with the plasma membrane and the Ca2+-sensor synaptotagmin-1, which was thought to facilitate membrane fusion directly through its Ca2+-binding loops. However, binding of Synaptotagmin-1 to SNARE complexes orients these loops away from the fusion site. Using molecular dynamics simulations, we show that placing Synaptotagmin-1 at the fusion site hinders the action of SNARE complexes. Spectroscopic studies show that Ca2+ binding to Synaptotagmin-1 can change its interactions with SNARE complexes and, together with molecular modeling, suggest that Synaptotagmin-1 acts as a lever, pulling SNARE complexes and thus facilitating their action on the membranes to induce fusion. Functional studies described in the accompanying paper support this hypothesis.
Acknowledgements
We thank Thomas Südhof amd Axel Brunger for extensive discussions. Most of the molecular dynamics simulations presented in this paper were performed through Pathways and Leadership Computing Resource allocations on Frontera at the Texas Advanced Computing Center of The University of Texas at Austin (URL: http://www.tacc.utexas.edu) (projects MCB20033 and IBN23002). This research also used computational resources provided by the BioHPC supercomputing facility located in the Lyda Hill Department of Bioinformatics, UT Southwestern Medical Center, TX (URL: https://portal.biohpc.swmed.edu). This work was supported by grant I-1304 from the Welch Foundation (to JR), by NIH Research Project Award R35 NS097333 (to JR), and by project 278001972-TRR186 from the Deutsche Forschungsgemeinschaft (DFG; German Research Foundation) (to CR).
Footnotes
Declaration of interests
The authors declare no competing interests.
Data availability
All files corresponding to the experimental data presented in this paper and most files corresponding to the molecular dynamics simulations are being deposited in the dryad database. Because of the very large size of trajectory files, it was not practical to deposit them in this database, but these files are available from the corresponding author upon request.
References
- 1.Sudhof T. C., Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron 80, 675–690 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sabatini B. L., Regehr W. G., Timing of neurotransmission at fast synapses in the mammalian brain. Nature 384, 170–172 (1996). [DOI] [PubMed] [Google Scholar]
- 3.Ma C., Su L., Seven A. B., Xu Y., Rizo J., Reconstitution of the vital functions of Munc18 and Munc13 in neurotransmitter release. Science 339, 421–425 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu X. et al. , Functional synergy between the Munc13 C-terminal C1 and C2 domains. elife 5, e13696 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lai Y. et al. , Molecular Mechanisms of Synaptic Vesicle Priming by Munc13 and Munc18. Neuron 95, 591–607 e510 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stepien K. P., Rizo J., Synaptotagmin-1-, Munc18–1-, and Munc13–1-dependent liposome fusion with a few neuronal SNAREs. Proc Natl Acad Sci U S A 118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rizo J., Molecular Mechanisms Underlying Neurotransmitter Release. Annu Rev Biophys 51, 377–408 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brunger A. T., Leitz J., The Core Complex of the Ca(2+)-Triggered Presynaptic Fusion Machinery. J Mol Biol 435, 167853 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jahn R., Cafiso D. C., Tamm L. K., Mechanisms of SNARE proteins in membrane fusion. Nat Rev Mol Cell Biol 25, 101–118 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sollner T., Bennett M. K., Whiteheart S. W., Scheller R. H., Rothman J. E., A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell 75, 409–418 (1993). [DOI] [PubMed] [Google Scholar]
- 11.Poirier M. A. et al. , The synaptic SNARE complex is a parallel four-stranded helical bundle. Nat. Struct. Biol 5, 765–769 (1998). [DOI] [PubMed] [Google Scholar]
- 12.Sutton R. B., Fasshauer D., Jahn R., Brunger A. T., Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature 395, 347–353 (1998). [DOI] [PubMed] [Google Scholar]
- 13.Hanson P. I., Roth R., Morisaki H., Jahn R., Heuser J. E., Structure and conformational changes in NSF and its membrane receptor complexes visualized by quick-freeze/deep-etch electron microscopy. Cell 90, 523–535 (1997). [DOI] [PubMed] [Google Scholar]
- 14.Weber T. et al. , SNAREpins: minimal machinery for membrane fusion. Cell 92, 759–772 (1998). [DOI] [PubMed] [Google Scholar]
- 15.Rizo J., Sari L., Jaczynska K., Rosenmund C., Lin M. M., Molecular mechanism underlying SNARE-mediated membrane fusion enlightened by all-atom molecular dynamics simulations. Proc Natl Acad Sci U S A 121, e2321447121 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mayer A., Wickner W., Haas A., Sec18p (NSF)-driven release of Sec17p (alpha-SNAP) can precede docking and fusion of yeast vacuoles. Cell 85, 83–94 (1996). [DOI] [PubMed] [Google Scholar]
- 17.Prinslow E. A., Stepien K. P., Pan Y. Z., Xu J., Rizo J., Multiple factors maintain assembled trans-SNARE complexes in the presence of NSF and alphaSNAP. Elife 8, e38880 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dulubova I. et al. , A conformational switch in syntaxin during exocytosis: role of munc18. EMBO J 18, 4372–4382 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Misura K. M., Scheller R. H., Weis W. I., Three-dimensional structure of the neuronal-Sec1-syntaxin 1a complex. Nature 404, 355–362 (2000). [DOI] [PubMed] [Google Scholar]
- 20.Parisotto D. et al. , An extended helical conformation in domain 3a of Munc18–1 provides a template for SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) complex assembly. J. Biol. Chem 289, 9639–9650 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baker R. W. et al. , A direct role for the Sec1/Munc18-family protein Vps33 as a template for SNARE assembly. Science 349, 1111–1114 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stepien K. P., Xu J., Zhang X., Bai X. C., Rizo J., SNARE assembly enlightened by cryo-EM structures of a synaptobrevin-Munc18–1-syntaxin-1 complex. Sci Adv 8, eabo5272 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Quade B. et al. , Membrane bridging by Munc13–1 is crucial for neurotransmitter release. Elife 8, e42806 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma C., Li W., Xu Y., Rizo J., Munc13 mediates the transition from the closed syntaxin-Munc18 complex to the SNARE complex. Nat. Struct. Mol. Biol 18, 542–549 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou Q. et al. , Architecture of the synaptotagmin-SNARE machinery for neuronal exocytosis. Nature 525, 62–67 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen X. et al. , Three-dimensional structure of the complexin/SNARE complex. Neuron 33, 397–409 (2002). [DOI] [PubMed] [Google Scholar]
- 27.Rizo J., Sari L., Qi Y., Im W., Lin M. M., All-atom molecular dynamics simulations of Synaptotagmin-SNARE-complexin complexes bridging a vesicle and a flat lipid bilayer. Elife 11, e76356 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fernandez-Chacon R. et al. , Synaptotagmin I functions as a calcium regulator of release probability. Nature 410, 41–49 (2001). [DOI] [PubMed] [Google Scholar]
- 29.Sutton R. B., Davletov B. A., Berghuis A. M., Sudhof T. C., Sprang S. R., Structure of the first C2 domain of synaptotagmin I: a novel Ca2+/phospholipid-binding fold. Cell 80, 929–938 (1995). [DOI] [PubMed] [Google Scholar]
- 30.Ubach J., Zhang X., Shao X., Sudhof T. C., Rizo J., Ca2+ binding to synaptotagmin: how many Ca2+ ions bind to the tip of a C2-domain? EMBO J 17, 3921–3930 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fernandez I. et al. , Three-dimensional structure of the synaptotagmin 1 c(2)b-domain. Synaptotagmin 1 as a phospholipid binding machine. Neuron 32, 1057–1069 (2001). [DOI] [PubMed] [Google Scholar]
- 32.Rhee J. S. et al. , Augmenting neurotransmitter release by enhancing the apparent Ca2+ affinity of synaptotagmin 1. Proc. Natl. Acad. Sci. U. S. A 102, 18664–18669 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mackler J. M., Drummond J. A., Loewen C. A., Robinson I. M., Reist N. E., The C(2)B Ca(2+)-binding motif of synaptotagmin is required for synaptic transmission in vivo. Nature 418, 340–344 (2002). [DOI] [PubMed] [Google Scholar]
- 34.Shin O. H., Xu J., Rizo J., Sudhof T. C., Differential but convergent functions of Ca2+ binding to synaptotagmin-1 C2 domains mediate neurotransmitter release. Proc. Natl. Acad. Sci. U. S. A 106, 16469–16474 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chapman E. R., Davis A. F., Direct interaction of a Ca2+-binding loop of synaptotagmin with lipid bilayers. J. Biol. Chem 273, 13995–14001 (1998). [DOI] [PubMed] [Google Scholar]
- 36.Zhang X., Rizo J., Sudhof T. C., Mechanism of phospholipid binding by the C2A-domain of synaptotagmin I. Biochemistry 37, 12395–12403 (1998). [DOI] [PubMed] [Google Scholar]
- 37.Lai A. L., Tamm L. K., Ellena J. F., Cafiso D. S., Synaptotagmin 1 modulates lipid acyl chain order in lipid bilayers by demixing phosphatidylserine. J. Biol. Chem 286, 25291–25300 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wickner W., Rizo J., A cascade of multiple proteins and lipids catalyzes membrane fusion. Mol Biol Cell 28, 707–711 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martens S., Kozlov M. M., McMahon H. T., How synaptotagmin promotes membrane fusion. Science 316, 1205–1208 (2007). [DOI] [PubMed] [Google Scholar]
- 40.Hui E., Johnson C. P., Yao J., Dunning F. M., Chapman E. R., Synaptotagmin-mediated bending of the target membrane is a critical step in Ca(2+)-regulated fusion. Cell 138, 709–721 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bai J., Tucker W. C., Chapman E. R., PIP2 increases the speed of response of synaptotagmin and steers its membrane-penetration activity toward the plasma membrane. Nat. Struct. Mol. Biol 11, 36–44 (2004). [DOI] [PubMed] [Google Scholar]
- 42.Arac D. et al. , Close membrane-membrane proximity induced by Ca(2+)-dependent multivalent binding of synaptotagmin-1 to phospholipids. Nat. Struct. Mol. Biol 13, 209–217 (2006). [DOI] [PubMed] [Google Scholar]
- 43.van den B. G. et al. , Synaptotagmin-1 may be a distance regulator acting upstream of SNARE nucleation. Nat. Struct. Mol. Biol 18, 805–812 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chapman E. R., How Does Synaptotagmin Trigger Neurotransmitter Release? Annu. Rev. Biochem (2008). [DOI] [PubMed] [Google Scholar]
- 45.Rizo J., Xu J., The Synaptic Vesicle Release Machinery. Annu. Rev. Biophys 44, 339–367 (2015). [DOI] [PubMed] [Google Scholar]
- 46.Park Y. et al. , Synaptotagmin-1 binds to PIP(2)-containing membrane but not to SNAREs at physiological ionic strength. Nat Struct Mol Biol 22, 815–823 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Voleti R., Jaczynska K., Rizo J., Ca(2+)-dependent release of Synaptotagmin-1 from the SNARE complex on phosphatidylinositol 4,5-bisphosphate-containing membranes. Elife 9, e57154 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brewer K. D. et al. , Dynamic binding mode of a Synaptotagmin-1-SNARE complex in solution. Nat. Struct. Mol. Biol 22, 555–564 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou Q. et al. , The primed SNARE-complexin-synaptotagmin complex for neuronal exocytosis. Nature 548, 420–425 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guan Z. et al. , A synaptotagmin suppressor screen indicates SNARE binding controls the timing and Ca(2+) cooperativity of vesicle fusion. Elife 6, e28409 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chang S., Trimbuch T., Rosenmund C., Synaptotagmin-1 drives synchronous Ca(2+)-triggered fusion by C2B-domain-mediated synaptic-vesicle-membrane attachment. Nat Neurosci 21, 33–40 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Toulme E., Salazar Lazaro A., Trimbuch T., Rizo J., Rosenmund C., Neurotransmitter release is triggered by a calcium-induced rearrangement in the Synaptotagmin-1/SNARE complex primary interface. Accompanying manuscript (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xue M., Ma C., Craig T. K., Rosenmund C., Rizo J., The Janus-faced nature of the C(2)B domain is fundamental for synaptotagmin-1 function. Nat. Struct. Mol. Biol 15, 1160–1168 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuo W., Herrick D. Z., Cafiso D. S., Phosphatidylinositol 4,5-bisphosphate alters synaptotagmin 1 membrane docking and drives opposing bilayers closer together. Biochemistry 50, 2633–2641 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bommert K. et al. , Inhibition of neurotransmitter release by C2-domain peptides implicates synaptotagmin in exocytosis. Nature 363, 163–165 (1993). [DOI] [PubMed] [Google Scholar]
- 56.Bykhovskaia M., SNARE complex alters the interactions of the Ca(2+) sensor synaptotagmin 1 with lipid bilayers. Biophys J 120, 642–661 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Takamori S. et al. , Molecular anatomy of a trafficking organelle. Cell 127, 831–846 (2006). [DOI] [PubMed] [Google Scholar]
- 58.Chan R. B. et al. , Comparative lipidomic analysis of mouse and human brain with Alzheimer disease. J Biol Chem 287, 2678–2688 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reim K. et al. , Complexins regulate a late step in Ca2+-dependent neurotransmitter release. Cell 104, 71–81 (2001). [DOI] [PubMed] [Google Scholar]
- 60.Hernandez J. M. et al. , Membrane fusion intermediates via directional and full assembly of the SNARE complex. Science 336, 1581–1584 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rizo J., Chen X., Arac D., Unraveling the mechanisms of synaptotagmin and SNARE function in neurotransmitter release. Trends Cell Biol 16, 339–350 (2006). [DOI] [PubMed] [Google Scholar]
- 62.Wang S., Li Y., Ma C., Synaptotagmin-1 C2B domain interacts simultaneously with SNAREs and membranes to promote membrane fusion. Elife 5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vardar G., Salazar-Lazaro A., Zobel S., Trimbuch T., Rosenmund C., Syntaxin-1A modulates vesicle fusion in mammalian neurons via juxtamembrane domain dependent palmitoylation of its transmembrane domain. Elife 11, e78182 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhou P., Bacaj T., Yang X., Pang Z. P., Sudhof T. C., Lipid-anchored SNAREs lacking transmembrane regions fully support membrane fusion during neurotransmitter release. Neuron 80, 470–483 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hu Y., Zhu L., Ma C., Structural Roles for the Juxtamembrane Linker Region and Transmembrane Region of Synaptobrevin 2 in Membrane Fusion. Front Cell Dev Biol 8, 609708 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gao Y. et al. , Single reconstituted neuronal SNARE complexes zipper in three distinct stages. Science 337, 1340–1343 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ngatchou A. N. et al. , Role of the synaptobrevin C terminus in fusion pore formation. Proc Natl Acad Sci U S A 107, 18463–18468 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schupp M. et al. , Interactions Between SNAP-25 and Synaptotagmin-1 Are Involved in Vesicle Priming, Clamping Spontaneous and Stimulating Evoked Neurotransmission. J Neurosci 36, 11865–11880 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chernomordik L. V., Kozlov M. M., Mechanics of membrane fusion. Nat. Struct. Mol. Biol 15, 675–683 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zimmerberg J., Chernomordik L. V., Membrane fusion. Adv. Drug Deliv. Rev 38, 197–205 (1999). [DOI] [PubMed] [Google Scholar]
- 71.Zhou A., Brewer K. D., Rizo J., Analysis of SNARE complex/synaptotagmin-1 interactions by one-dimensional NMR spectroscopy. Biochemistry 52, 3446–3456 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Salazar Lazaro A., Trimbuch T., Vardar G., Rosenmund C., The stability of the primed pool of synaptic vesicles and the clamping of spontaneous neurotransmitter release rely on the integrity of the C-terminal half of the SNARE domain of syntaxin-1 A. Elife 12 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kiessling V. et al. , A molecular mechanism for calcium-mediated synaptotagmin-triggered exocytosis. Nat Struct Mol Biol 25, 911–917 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang J. et al. , Calcium sensitive ring-like oligomers formed by synaptotagmin. Proc Natl Acad Sci U S A 111, 13966–13971 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jaczynska K. et al. , Analysis of tripartite Synaptotagmin-1-SNARE-complexin-1 complexes in solution. FEBS Open Bio 13, 26–50 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gipson P. et al. , Morphologies of synaptic protein membrane fusion interfaces. Proc Natl Acad Sci U S A 114, 9110–9115 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stein A., Weber G., Wahl M. C., Jahn R., Helical extension of the neuronal SNARE complex into the membrane. Nature 460, 525–528 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All files corresponding to the experimental data presented in this paper and most files corresponding to the molecular dynamics simulations are being deposited in the dryad database. Because of the very large size of trajectory files, it was not practical to deposit them in this database, but these files are available from the corresponding author upon request.