

Abstract
Human immunodeficiency virus type 1 (HIV-1) rapidly develops resistance to lamivudine during monotherapy, typically resulting in the appearance at position 184 in reverse transcriptase (RT) of isoleucine instead of the wild-type methionine (M184I) early in therapy, which is later replaced by valine (M184V). M184V reduces viral susceptibility to drug in vitro by approximately 100-fold, but also results in a lower processivity of RT. We show that a drop in absolute viral fitness associated with the outgrowth of M184V results in a drop in viral load only in individuals with high CD4+ counts, from whom we estimate the relative fitness of M184V in the presence of drug to be approximately 10% of that of the wild type prior to therapy. The timing of emergence of the M184V mutant varies widely between infected individuals. From analysis of the frequency of M184I and M184V mutants determined at multiple time points in seven individuals during lamivudine therapy, we estimated the fitness advantage of M184V over M184I during therapy to be approximately 23% on average. We have also estimated the average ratio of the frequencies of the two mutants prior to therapy to be 0.2:1, with a range from 0.12:1 to 0.33:1. We have found that the differences between individuals in the rate of evolution of lamivudine resistance arise due to genetic drift affecting the relative frequency of M184I and M184V prior to therapy. These results show that stochastic effects can be significant in HIV evolution, even when there is large fitness difference between mutant and wild-type variants.
Resistance to the reverse transcriptase (RT) inhibitor lamivudine (3TC) involves mutations at one residue in RT, methionine (ATG) position 184 (3, 29). Typically, after about 2 weeks of 3TC monotherapy, isoleucine (ATA) appears at this position. Although this substitution confers a several-hundred-fold increase in the 50% inhibitory concentration relative to the wild type, it also dramatically reduces the replication rate of the virus by reducing the processivity of the enzyme (1). After 8 to 20 weeks, isoleucine is replaced by valine (GTG), which also confers drug resistance but has less of an impact on the processivity of RT, such that in the absence of drug, the valine mutant has a fitness intermediate to that of wild type and the isoleucine mutant in vitro.
The pattern of evolution of drug resistance to 3TC, with the initial appearance of the M184I mutant, followed by the replacement of the fitter M184V mutant, has been explained in terms of a balance between mutation and selection (26). In human immunodeficiency virus (HIV), G-to-A mutations are more common than A-to-G mutations and result in a higher production rate of M184I mutants (13, 17). Based on the classical population genetics result that the frequency of a deleterious mutation reflects a balance between mutation and selection (9), this mutational bias towards A must be large enough to overcome the higher fitness of M184V in order to result in M184I mutants being present at higher frequencies than M184V prior to therapy. This is consistent with in vitro studies which have shown the mutation rate from wild type to M184I is over four times higher than that to M184V (17), while the enzymatic efficiency of M184V (45% relative to the wild type for virion-derived RT) is less than twice that of M184I (28%) (1). The replicative advantage of M184V over M184I during therapy results in the eventual outgrowth of M184V despite its lower initial frequency. However, the timing of the appearance of the M184V mutant varies widely between individuals (26). This could be due to different levels of resistance prior to therapy or different rates of increase during therapy. Once fixed, average viral loads associated with the M184V mutant are lower than those associated with the wild type prior to therapy. However, there is considerable variation in the viral load response between individuals: some individuals show a very marked reduction, while others even show an increase in viral load.
It has been argued that the evolution of resistance is completely deterministic (5) because the number of productively infected cells within the body is very high (4). Under this assumption, between-host differences in the relative frequencies of M184V to M184I prior to therapy reflect differences in the relative fitnesses of these mutants, because the mutation rate is unlikely to vary between individuals. In contrast, it has been argued that chance effects may play an important role in generating variation in the rate of evolution of drug resistance (19, 20). These effects have been discussed by using the concept of effective population size. Although the number of infected cells may be very large, HIV may evolve as if it were a smaller population. A high variance in the number of secondary infected cells produced per infected cell could increase the importance of chance effects—an effect captured in the concept of a “variance effective population size” (6, 31). A high variance may arise due to spatial differences in the level of immune activation and spatial clustering of infected cells, such that relatively few cells have access to target cells. Additionally, selection acting in different directions on linked regions of the HIV genome can give rise to “genetic conflicts” (8), which could also lead to an apparently low effective population size and increase the amount of noise in the evolution of drug resistance.
In order to assess the relative importance of chance in the evolution of 3TC resistance, we analyzed the dynamics of M184V and M184I mutants in seven individuals on 3TC monotherapy in order to estimate the rate of outgrowth of M184V over M184I and their relative frequency prior to therapy. We have also analyzed the response of viral load to the outgrowth of 184V in order to estimate the fitness of M184V during therapy relative to that of the wild type prior to therapy. In individuals with a low CD4+ count, the decrease in viral fitness associated with 184V has no effect on the viral load.
MATERIALS AND METHODS
Study population.
The original study population consisted of 20 men with asymptomatic or mildly symptomatic HIV-1 infection who were selected from a cohort of 40 men treated with different doses (0.5 to 20.0 mg/kg of body weight/day) of 3TC (Glaxo) monotherapy as part of a phase I to phase II trial (28). Nineteen of these patients had an initial decline in HIV-1 RNA load. The relative amounts of the wild type (methionine [ATG]) and resistant mutants (isoleucine [ATA] or valine [GTG]) at position 184 in RT were determined by a primer-guided nucleotide incorporation assay (14, 27).
To estimate the rate of viral decay, we analyzed those individuals who had at least two viral load measurements taken during the first week of therapy, giving a sample size of 16. In order to obtain accurate estimates of the relative fitness of the M184I and the M184V mutants, we analyzed patients for which the frequency of each resistant mutant was greater than 5% (the apparent limit of detection) (26) and the total frequency was less than 100% for at least two time points between 2 and 4 weeks of 3TC therapy. For these patients, time points where the frequency of either mutant was less than 5% or was obscured by high background levels were treated as random missing data. This reduced the sample size to seven patients. To obtain estimates of relative fitness of M184V and the wild type from the steady-state viral loads prior to and late during therapy, we analyzed 16 patients who had initial viral loads associated with <5% M184V and final viral loads associated with >95% M184V. Fourteen of these patients also had viral load data taken 1 to 2 weeks prior to therapy.
Estimation of the rate of viral decay during initial therapy.
To estimate the rate of viral decay, we fitted two linear models to the log viral load data, which allowed the initial viral load to vary by patient. The first model assumed that the rate of decay was the same in all patients, whereas the second allowed the rate of decay to vary between individuals. These two models were compared by using analysis of variance (ANOVA) and restricted maximum likelihood (REML).
Estimation of the relative fitness of M184V and M184I in vivo by using the relative rates of outgrowth.
In order to obtain estimates of the relative fitness of the M184V and M184I drug-resistant mutants in vivo during therapy from the observed frequency of resistant mutants, we employed a simple mathematical model. The relative frequency of two mutants, p1 and p2, with constant fitnesses of 1 + s1 and 1 + s2, respectively (s2 > s1, and the fitness of the wild type is 1) over time in an infinite, well-mixed population can be calculated from standard population genetics theory (equation 5.2.1 in reference 6). If time is given in generations, the relative frequency over time is given by the expression
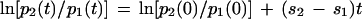 |
1 |
Hence, if ln(p2/p1) is plotted against time, the intercept gives an estimate of the relative frequency of the mutants prior to therapy and the slope gives an estimate of the relative fitness of the mutants, which can be obtained by fitting linear models. We assumed a generation time of 2.6 days (24).
We first estimated the relative fitness of M184V relative to M184I by averaging across all seven patients, allowing the initial frequency of M184V relative to M184I to vary between hosts by fitting a general linear model assuming an identity link and independent normally distributed errors. We compared this to a similar model in which both the initial frequency and the fitness of M184V were the same between all individuals, by using the generalized estimating equation approach (21). This allowed us to estimate the correlation between measurements taken for the same individual and hence also allows us to test whether the differences in the outgrowth of M184V are simply due to measurement error, which results in uncorrelated residuals. We assumed normally distributed errors, an identity link function, and a first-order autoregressive working correlation structure for the residuals. The significance of the correlation between residuals was calculated by randomizing measurements at each time point.
We also obtained estimates of the fitness of 184V, assuming that both the initial frequency, ln[p2(0)/p1(0)], and the relative fitness, s2 − s1, varied between individuals. A similar approach has been used by Havlir et al. (10) and Eastman et al. (7). Differences between hosts in the rate of increase of M184V during therapy may arise due to differences in dose or in viral genetic background. Due to the large number of extra parameters (seven patients, and so an additional six parameters) in this model, we could not statistically test the improvement in fit. Instead, we assumed that differences in the slope between individuals could be approximated by a normal distribution with a mean of 0 and a variance of ς2. This allowed us to test whether there was significant variation in the slope between individuals by only introducing one extra parameter. This was performed by fitting a linear mixed-effects model by using REML.
Estimation of the relative fitness of M184V (in the presence of drug) to wild type (in the absence of drug) by using the steady-state viral loads.
The lower viral load relative to the baseline associated with M184V suggested that the relative fitness of M184V might be estimated from the ratio of final viral load to initial viral load, which we call f. In order to test whether variation in f between patients was due to factors such as initial viral load, dose, or CD4+ count, we fitted general linear models to the data. To test for homogeneity of variance, we used restricted maximum likelihood and a power-law variance weighting function.
Statistical software.
All statistical tests were performed in the R programming language (http://www.ci.tuwien.ac.at/R/) by using the gee, boot, and lme libraries.
RESULTS
Estimates of the frequency of M184I and M184V mutants.
In all patients studied, the percentage of resistant mutants increased over the duration of therapy, with a shift from M184I to M184V (Fig. 1). However, there was considerable variation among the seven individuals studied in terms of the rate of evolution of resistance. For example, after 2 weeks of therapy, in patient 22, the frequencies of M184I and M184V were 70 and 8%, respectively, while in patient 17, the frequencies were 31% for M184I and 66% for M184V. At the initiation of therapy, six out of the seven patients had undetectable levels (0%) of both mutants. In patient 17, before therapy, M184I was at a frequency of 3% and M184V was at a frequency of 1%, although these low estimates are liable to considerable measurement error.
FIG. 1.

Estimates of the percentage of methionine (M [white]), isoleucine (I [gray]), and valine (V [black]) at position 184 of RT after approximately 2, 3, and 4 weeks of 3TC monotherapy.
Estimating the rate of viral decay.
In order to determine whether there were differences in drug efficacy between individuals, we estimated the rate of decay of plasma virus during the first week of therapy by fitting a single slope to the log viral load data for all individuals as suggested by previous studies. This model was a very good fit to the data (ANOVA, 85.8% of variation explained) and gave a decay rate of −0.31 (0.03 standard error [SE]), which equates to a half-life of 2.2 days, similar to the rates of decay seen in nevirapine and ritonavir monotherapy (12, 24, 30). Similar estimates of decay rate were obtained by using restricted maximum likelihood, allowing for random variation in initial log viral load.
A model which allowed for variation in the viral decay rate between individuals did not give a significantly better fit (accumulated ANOVA, F15,10 = 1.02, P = 0.5; REML, deviance [1 df] = 0.15, P = 0.7). We concluded that there was no difference between individuals in the effect of drug on the replication rate of wild-type virus.
Estimating fitness of resistant virus.
It was not possible directly to estimate the relative fitness of M184V to the wild type with the small number of data points available for each patient. However, we could estimate the relative fitness of the two resistant strains of virus to each other from their relative proportions over time, because the rate of replacement of M184I by M184V was slow enough to be estimated with samples taken a week apart. When the log of the ratio of frequency of M184V relative to M184I, ln(p2/p1), was plotted against time, we observed substantial differences in frequency between patients at any given time point, although the rate of increase in 184V was relatively consistent between individuals (Fig. 2). From this, we inferred that the starting frequencies of M184V relative to M184I may differ among patients, but the fitness of M184V was relatively constant between individuals. A linear model with uniform slope but allowing for different intercepts was therefore fitted to the data from all seven patients. The estimated mean fitness difference was 22.57% (3.66% SE), and the estimates of the ratio of initial frequencies ranged from 0.12:1 to 0.33:1 (mean, 0.20:1; coefficient of variation [CV], 0.43). This model fitted the data well, accounting for 77.9% of the observed variation.
FIG. 2.

Plot of the log ratio of the frequency of the valine to the isoleucine mutants, ln(M184V/M184I), over time in generations, assuming a generation time of 2.6 days for each patient.
To test whether it was necessary to allow the initial frequency of M184V relative to M184I to vary between hosts, we also obtained parameter estimates assuming that both fitness and prior frequency of M184V were the same between hosts. This model gave a slightly lower estimate of the fitness advantage of M184V (14.99%, 5.73% SE), and a higher average prior frequency (0.37:1), but it gave a much poorer fit to the data, accounting for only 28% of the observed variation, significantly less than the model which allowed initial frequency to vary between hosts (accumulated ANOVA, P = 0.011).
The comparison of these models is hampered by the small sample size (seven individuals) and measurement error. If the apparent differences in the initial frequency arose due to high measurement error, then the deviations from the average increase should not be correlated over time; however, if there were real underlying differences in the initial frequency, we would expect to see a significant positive correlation (i.e., patients that have higher-than-average frequencies of M184V early in therapy also have higher-than-average frequencies of M184V late into therapy. We found a strong positive correlation between measurements taken a week apart (r = 0.696). To test the significance of the observed correlation, we estimated the correlation coefficient for 1,000 data sets where measurements were randomized within time points. The observed correlation was significantly higher than those calculated from the randomized data set (mean = −0.19, P = 0.001). This argues strongly that the deviations in initial frequency are not due to measurement error.
Estimates of fitness and initial frequency treating patients separately.
We also fitted the linear model to the data from each patient separately. Estimates of relative fitness and initial frequency obtained in this way varied widely between patients (Table 1). Estimates of fitness ranged from 7.2% (patient 15) to 34.5% (patient 22), with an average across individuals of 23.3%. The relative frequency prior to therapy again varied widely between individuals, from 0.042:1 in patient 22 to 0.85:1 in patient 17. Although this model accounted for a large amount of the variation (96.9%), this was not a significantly better fit than assuming no variation in fitness (accumulated ANOVA, P = 0.103). A similar analysis using restricted maximum likelihood, but where variation in the fitness of M184V was assumed to be distributed normally between individuals, gave a P value close to significance (P = 0.06).
TABLE 1.
Estimates of the relative fitness and frequency of M184V relative to M184I on a patient-by-patient basis
| Patient identification no. | Relative fitness advantage of M184V over M184I (s2 − s1) | Initial M184V/M184I ratio |
|---|---|---|
| 11 | 0.276 | 0.079 |
| 15 | 0.072 | 0.681 |
| 17 | 0.082 | 0.846 |
| 21 | 0.302 | 0.197 |
| 22 | 0.345 | 0.042 |
| 26 | 0.345 | 0.045 |
| 34 | 0.208 | 0.350 |
The evidence of variation in fitness from the maximum-likelihood analysis of the full model, where both prior frequency and relative fitness were assumed to vary between individuals, was equivocal. Hence, we also analyzed the variation in the estimates of prior frequency and fitness for the full model to determine whether they were biologically reasonable. Surprisingly, a highly significant negative correlation was found between the estimates of initial frequency and fitness (Spearman's rank correlation = −0.93; exact two-sided test, P = 0.007). Under the assumption that differences in the initial frequency and rates of increase would be due to differences in viral genetic background, this makes little sense, because it suggests that the rarer (i.e., less fit) a mutant is prior to therapy, the fitter it is during therapy. This is unlikely for two reasons. First, this hypothesis implies significant genetic variation with respect to 3TC resistance in HIV-1 RT among different infected individuals, although no site with a large effect other than 184 has consistently emerged during monotherapy. Second, given that the M184V mutant is fitter than M184I even in the absence of drug (1), it is extremely unlikely that the greater the fitness difference between M184V and M184I, the rarer M184V is prior to therapy. In contrast, such a negative correlation could easily arise as a statistical artifact, because estimates of the slope and intercept of a straight line are strongly negatively correlated. We conclude that there are large differences in the ratio of initial frequencies, but the evidence that these are related to fitness is weak.
A simple stochastic model of the relative frequency of M184V and M184I prior to therapy.
One explanation for the variation in prior frequency is that the frequencies of M184V and M184I fluctuate due to random genetic drift. To explore this possibility, we developed a simple stochastic model which describes the relative frequency of M184V to M184I prior to therapy under different assumptions of the variance effective population size, Ne (6, 31). Although at steady state, each productively infected cell gives rise, on average, to one daughter productively infected cell, there is likely to be a very high variance in the number of such daughter infected cells. Many mechanisms can contribute to this, including poor mixing of infected cells within the body and variation in the levels of immune stimulation, such that some cells will have contact with more uninfected target cells than others. The higher the variance, the more important genetic drift will be.
We assume that, for each patient, Ne is constant and the mutation rates are μ1 and μ2 per replication cycle for the production of M184I and M184V mutants, respectively, from the wild type, with negligible mutation between them or back to the wild type. We also assume the M184I and M184V mutants carry a cost of resistance pretherapy of s1pre and s2pre, respectively. From the theory of univariate birth-death processes (16), it follows that the distribution of the number of cells infected with a single resistant mutant in a group of individuals follows a negative binomial distribution with mean μNe/spre, variance μNe/spre2, and index μNe/(1 −spre). Hence the relative frequency of M184V to M184I prior to therapy in an individual is the ratio of two random negative binomial variables. The mean, E[p2/p1], and variance, Var[p2/p1], of the ratio of M184V to M184I in a group of individuals can be approximated by the delta method (18) to give
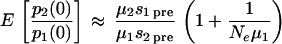 |
2 |
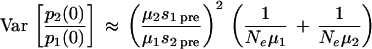 |
3 |
The CV, CV[p2(0)/p1(0)], is approximately
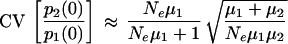 |
4 |
Under this model, significant levels of variation prior to therapy could occur solely by genetic drift. From equation 4, it can be seen that the CV is independent of the cost of resistance, only depending on the relative mutation rates from wild-type virus and the effective population size. For reasonable estimates of the relative mutation rates (fourfold) (17) and costs of resistance in the absence of therapy (based on the relative enzymatic efficiency of RT) (1), significant variation in the relative frequencies of M184V to M184I can be obtained for an effective population size as high as 106 (Fig. 3). Recent estimates of Ne for HIV have been substantially lower than this (19, 23), which would magnify the effect.
FIG. 3.

Approximate values for the mean and the CV of the frequency of M184V relative to M184I prior to therapy, p2(0)/p1(0), for effective population sizes between 105 and 108. The parameter values assumed were μ1 = 6 × 10−5, μ2 = 1.5 × 10−5, s1 = 0.45, and s2 = 0.28. Similar results were obtained with a wide range of parameter values.
Our approach assumes that the rate of increase in M184V over the period of study is deterministic, which is a reasonable approximation given a fitness advantage for M184V over M184I of around 20%, because chance effects have little impact on mutations with such a large fitness advantage. There are two ways in which the rise of M184V during the period of study could deviate from a simple logistic increase. First, the variance effective population size might be very low. In order for this to be an important effect, the product of effective population size and the selective advantage of M184V, Ne(s2 − s1), must be small (<1). Even assuming a smaller fitness advantage of 10%, Ne would have to be extremely small (∼10) to result in significant deviations from a deterministic rise. Secondly, the fitness of an M184V mutant may be affected by selection on genetic variation linked to position 184, resulting in an apparently stochastic increase in frequency rather than a smooth logistic increase in M184V. For this to be an important effect, the probability that a mutant with a selective advantage of more than 20% over M184I (i.e., s2 − s1) emerges over the 2-week study period would have to be unrealistically high to generate such variation between hosts.
Our simple stochastic model, together with consideration of the size of the fitness difference in the 3TC-resistant mutants, suggested that estimates of fitness and frequency based on our hypothesis that there is a combination of genetic drift prior to therapy, with an approximately deterministic increase during therapy, are biologically plausible. We conclude that this model is likely to be a good fit to the increase of M184V relative to M184I over the period of study. However, these mutants may be subject to genetic drift during the early stages of therapy. Because the rarer mutant (M184V) is more subject to genetic drift, it will take longer to enter a phase of deterministic increase, and, hence, we are likely to underestimate the relative frequency of M184V prior to therapy, even if the absolute fitness of virus remains constant during therapy.
Estimation of the relative fitness of M184V compared to that of the wild type.
Although we could not estimate the relative fitness of resistant virus compared to that of the wild type by using the rate of viral outgrowth during early therapy, the lower viral load associated with M184V at the equilibrium reached during drug therapy suggested that its fitness relative to that of the wild type might be estimated from the difference in equilibrium viral load between baseline and late therapy. We used the ratio of final viral load to baseline viral load, which we called f, as a measure of the change in viral load associated with the outgrowth of M184V. Data on the viral load in the period prior to therapy allow us to determine whether variation in viral load response is different from “background” variation.
On average, the viral load associated with M184V was 64.3% that at baseline, although there was significant variation between individuals, with some individuals showing a marked decrease and others even showing an increase (range of f = 7.3 to 191%). We analyzed this variation by fitting a linear model. Neither dose nor log initial viral load accounted for significant variation in the decrease in viral load (dose: F1,14 = 0.18, P = 0.68; viral load, F1,14 = 0.02, P = 0.90, respectively). However, we found a significant negative correlation between the decrease in viral load and CD4+ count, with CD4+ count explaining 28.1% of the variation (F1,14 = 6.85, P = 0.02). When the regression was plotted, it appeared that variation in the reduction in viral load was higher for lower CD4+ counts (Fig. 4), which was statistically significant (REML, power law weighting function, exponent = 1.2; P = 0.04).
FIG. 4.

Change in viral load (VL) associated with the outgrowth of M184V for 16 individuals, together with the best fit obtained from maximum likelihood.
To investigate the interaction between the reduction in viral load and CD4+ count, we split individuals into two groups: those that had CD4+ counts of <200 (n = 8) and those that had CD4+ counts of 200 or more (n = 8). Seven individuals in each group also had viral load measurements taken 1 or 2 weeks prior to the initiation of therapy. This allowed us to compare the variation in the reduction in viral load seen during therapy with that prior to therapy (Fig. 5).
FIG. 5.

Changes in the viral load (VL) before therapy (obtained by dividing initial viral load by viral load obtained 1 to 2 weeks prior to therapy) compared to the changes in viral load during therapy (obtained by dividing viral load 12 weeks into therapy by initial viral load) for individuals with low (<200) and high (≥200) CD4+ counts.
For individuals with a low CD4+ count, the drop in viral fitness due to the outgrowth of M184V appeared to have no effect on the viral load, with no significant differences in the mean (two-tailed paired t test, P = 0.47). Three out of the seven patients showed a decrease in viral load during therapy, but five showed a decrease in viral load prior to therapy as well, which was not significantly different from an equal chance of a rise or a fall of viral load (exact B test, two sided, P = 0.42). In contrast, for individuals with a high CD4+ count, the ratio of final to initial viral load was significantly less than 1, and it was significantly less than the change in viral load seen prior to therapy in the absence of changes in viral fitness (two-sided approximate t test, unequal variances, P = 0.036). All eight individuals with a high CD4+ count showed a decrease in viral load during therapy, compared to four out of eight with a low CD4+ count (exact B test, two sided, P = 0.027).
Based on the reduction in viral load in individuals with a CD4+ count of 200 or more, the relative fitness of M184V (during therapy) compared to that of the wild type (in the absence of therapy) was approximately 30%. This is consistent with the difference in the enzymatic efficiency of RT as assayed by using virion-derived RT (45%) (1, 17).
The fitness of M184V relative to that of the wild type may be much lower than suggested by simply taking the average viral load reduction for individuals with a CD4+ count greater than 200. Figure 4 shows that the variance of the viral load reduction decreases as CD4+ count increases. This is a consequence of the reduction in viral load swamping the error in measuring individual viral loads as the reduction in viral load increases. In the two individuals with the highest CD4+ count, the viral load is reduced to 10% of that prior to therapy, implying that the relative fitness of M184V is approximately 10% of that of the wild type. Because target cell compensation may still occur at high CD4+ cell counts, this estimate of relative fitness may be an overestimate and is much lower than expected based on in vitro assays of enzyme reactivity.
DISCUSSION
It is widely recognized that the high mutation rate of HIV will generate a background of resistance-associated mutations within all patients. These mutants clearly are less fit than the wild type in the absence of antiretroviral drugs, and the expected frequency reached by any individual mutation can be approximated by the balance between the rate of mutation generating them and the intensity of selection removing them from the population (9). Coffin (5) applied this deterministic relationship explicitly to the evolution of drug resistance in HIV, assuming that the population size of HIV within a patient was effectively infinite, such that there was no variation between individuals due to genetic drift around the frequency at the mutation-selection balance.
The preexisting frequency of resistance-associated mutations is of immediate significance for the efficacy of antiretroviral therapy, and two previous studies have estimated the frequency of nevirapine and ritonavir resistance mutations, respectively, prior to therapy (7, 10). These studies suggested that both the frequency of resistance prior to therapy and the rate of increase during therapy varied dramatically between individuals, but the number of patients studied was too few to establish the levels of variability between patients. We have used frequency data of two 3TC-resistant mutations, M184I and M184V, from seven patients on 3TC monotherapy, to describe the between-host variation in these parameters in more detail. The simple genetic mechanism of 3TC resistance makes it unlikely that the rate of outgrowth of M184V will be confounded by potential interactions between the primary resistance mutation and variation at other sites.
Resistance to 3TC is conferred by two amino acid substitutions at a single site, which each have a substantial fitness advantage over the wild type in the presence of drug, leading to a rapid replacement of the wild type in patients on monotherapy. Our results confirm that the frequency of M184V relative to M184I increases approximately deterministically during therapy and that the fitness advantage is approximately 20%. However, prior to initiation of therapy, the ratio of the initial frequencies ranged widely from 0.12:1 to 0.33:1 among the seven patients. Our model accounted for 78% of the variation in the data. A simpler model which did not allow the frequency of M184V relative to M184I to vary between individuals gave a much poorer fit to the data. A more complex model which allowed the fitness of M184V to also vary between individuals gave a biologically unrealistic negative correlation between prior frequency and fitness. Analysis of an explicit mathematical model indicated that genetic drift is likely to be important before therapy when both mutations are rare, and it may also play a part during the early stages of therapy: because the M184V mutant is rarer than M184I, it will be more subject to sampling effects.
On average, the steady-state viral load when M184V is fixed in the population is lower than the pretreatment viral load, and this has been hypothesized to stem from the lower fitness of M184V relative to the wild type (in the absence of drug). However, the reduction in viral load is highly variable between individuals. We have shown that a reduction in viral load is not seen in individuals with a low CD4+ count. One possible explanation for this observation is suggested by a theoretical study by Bonhoeffer et al. (2). They showed that under simple models of virus dynamics, changes in viral fitness may be compensated for by changes in target cell availability. Individuals with high CD4+ counts do show a dramatic drop in viral load: more robust immune responses in these individuals could potentially weaken the negative feedback between viral fitness and target cell availability. The remaining variation in the reduction in viral load can be explained simply by the apparently stochastic variation in the viral load over time. Based upon individuals with a high CD4+ count, where the viral burden is most likely to reflect viral fitness, we estimate that the fitness of M184V in the presence of drug is approximately 10% of that of wild-type virus in the absence of drug.
The role of chance in HIV evolution is currently a topic of much debate, which has major implications for the evolution of drug resistance. Given the large number (107 to 108) (4) of infected cells in the body, it has been proposed that chance effects are not important (5). However, it has been proposed from analysis of variation in env sequences that the effective population size (Ne) may be substantially lower than this (19). This could arise if the variance in the number of infected cells produced per infected cells is high (which can arise due to poor mixing in the body) or from conflicting selective pressures (8). The dramatic between-host variations observed in the timing and pattern of HIV drug resistance mutations during therapy are also consistent with a role for stochastic evolution (20, 23). Recently, Rouzine and Coffin (25) have argued that the effective population size within the host must be greater than 105, based on a study of variation in HIV-1 protease, and concluded that stochastic effects will be unimportant for the evolution of single mutations. In contrast, we have shown that even at effective population sizes as high as 106, genetic drift is sufficient to generate variation in the frequency of resistant mutants prior to therapy, which arises due to genetic drift having a larger effect on the relative numbers of rare mutants than on each mutant individually. Even small differences between individuals in the relative frequency of rare mutants can result in dramatic differences in the timing of the outgrowth of the fittest resistant mutant. We conclude that stochastic effects are important for single point mutants when these mutants are competing with each other.
The evolution of resistance to 3TC monotherapy, where amino acid substitutions at a single site confer 100-fold reductions in susceptibility to the drug, is at first sight a classical example of deterministic evolution, and yet we have shown that even in this case, the ratio of the frequency of two rare mutants, such as the M184V and M184I mutants considered here, varies significantly between patients as well as within a patient over time (results not shown), with direct consequences for the time taken for maximum resistance to be seen. We have also shown that random fluctuations in measurements of viral load can give the false impression that viral fitness varies between individuals and that the impact of a given viral fitness may differ between individuals. In the current clinical context, 3TC is used as a component of multidrug regimens, which usually include zidovudine. Resistance to both drugs involves multiple substitutions at additional sites, which include site 210 in addition to changes seen under single-drug therapy at 184 and 215 (15, 22): although not well understood, the fitness differences between the different combinations are almost certainly smaller than the fitness differences observed here between M184V and M184I. In addition, multiple mutants will also occur at a lower frequency than single mutants. The relevance of stochastic variation over time and between patients in the evolution of resistance to combination therapy is therefore likely to be substantially greater than in the simple system described here.
ACKNOWLEDGMENTS
This work was supported by a Medical Research Fellowship (G81/298) to S.D.W.F. and by an unrestricted grant from Roche Molecular Systems, Alameda, Calif.
REFERENCES
- 1.Back N K T, Nijhuis M, Keulen W, Boucher C A B, Essink B B O, van Kuilenburg A B P, van Gennip A H, Berkhout B. Reduced replication of 3TC-resistant HIV-1 variants in primary cells due to a processivity defect of the reverse transcriptase enzyme. EMBO J. 1996;15:4040–4049. [PMC free article] [PubMed] [Google Scholar]
- 2.Bonhoeffer S, Coffin J M, Nowak M A. Human immunodeficiency virus drug therapy and virus load. J Virol. 1997;71:3275–3278. doi: 10.1128/jvi.71.4.3275-3278.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boucher C A B, Cammack N, Schipper P, Schuurman R, Rouse P, Wainberg M A, Cameron J M. High-level resistance to (−) enantiomeric 2′-deoxy-3′-thiacytidine in vitro is due to one amino acid substitution in the catalytic site of human immunodeficiency virus type 1 reverse transcriptase. Antimicrob Agents Chemother. 1993;37:2231–2234. doi: 10.1128/aac.37.10.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chun T W, Carruth L, Finzi D, Shen X F, Di Giuseppe J A, Taylor H, Hermankova M, Chadwick K, Margolick J, Quinn T C, Kuo Y H, Brookmeyer R, Zeiger M A, Barditch Crovo P, Siliciano R F. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature. 1997;387:183–188. doi: 10.1038/387183a0. [DOI] [PubMed] [Google Scholar]
- 5.Coffin J M. HIV population dynamics in vivo—implications for genetic variation, pathogenesis, and therapy. Science. 1995;267:483–489. doi: 10.1126/science.7824947. [DOI] [PubMed] [Google Scholar]
- 6.Crow J F, Kimura M. An introduction to population genetics theory. New York, N.Y: Harper & Row; 1970. [Google Scholar]
- 7.Eastman P S, Mittler J, Kelso R, Gee C, Boyer E, Kolberg J, Urdea M, Leonard J M, Norbeck D W, Mo H, Markowitz M. Genotypic changes in human immunodeficiency virus type 1 associated with loss of suppression of plasma viral RNA levels in subjects treated with ritonavir (norvir) monotherapy. J Virol. 1998;72:5154–5164. doi: 10.1128/jvi.72.6.5154-5164.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gerrish P J, Lenski R E. The fate of competing beneficial mutations in an asexual population. Genetica. 1998;103:127–144. [PubMed] [Google Scholar]
- 9.Haldane J B S. A mathematical theory of natural and artificial selection. Proc Camb Phil Soc. 1927;28:838–844. [Google Scholar]
- 10.Havlir D V, Eastman S, Gamst A, Richman D D. Nevirapine-resistant human immunodeficiency virus: kinetics of replication and estimated prevalence in untreated patients. J Virol. 1996;70:7894–7899. doi: 10.1128/jvi.70.11.7894-7899.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hill W G, Robertson A. The effect of linkage on limits to artificial selection. Genet Res. 1966;8:269–294. [PubMed] [Google Scholar]
- 12.Ho D D, Neumann A U, Perelson A S, Chen W, Leonard J M, Markowitz M. Rapid turnover of plasma virions and CD4+ lymphocytes in HIV-1 cells. Nature. 1995;373:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- 13.Ji J, Loeb L A. Fidelity of HIV-1 reverse transcriptase copying a hypervariable region of the HIV-1 env gene. Virology. 1994;199:323–330. doi: 10.1006/viro.1994.1130. [DOI] [PubMed] [Google Scholar]
- 14.Kaye S, Loveday C, Tedder R S. A microtitre format point mutation assay—application to the detection of drug resistance in human immunodeficiency virus type 1 infected patients treated with zidovudine. J Med Virol. 1992;37:241–246. doi: 10.1002/jmv.1890370402. [DOI] [PubMed] [Google Scholar]
- 15.Kemp S D, Shi C, Bloor S, Harrigan P R, Mellors J W, Larder B A. A novel polymorphism at codon 333 of human immunodeficiency virus type 1 reverse transcriptase can facilitate dual resistance to zidovudine and l-2′,3′-dideoxy-3′-thiacytidine. J Virol. 1998;72:5093–5098. doi: 10.1128/jvi.72.6.5093-5098.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kendall D G. On some modes of population growth leading to R. A. Fisher's logarithmic series distribution. Biometrika. 1948;35:6–15. [PubMed] [Google Scholar]
- 17.Keulen W, Back N K T, van Wijk A, Boucher C A B, Berkhout B. Initial appearance of the 184Ile variant in lamivudine-treated patients is caused by the mutational bias of human immunodeficiency virus type 1 reverse transcriptase. J Virol. 1997;71:3346–3350. doi: 10.1128/jvi.71.4.3346-3350.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kotz S, Johnson N L, editors. Encyclopedia of statistical science. New York, N.Y: Wiley; 1989. [Google Scholar]
- 19.Leigh Brown A J. Analysis of HIV-1 env gene sequences reveals evidence for a low effective number in the viral population. Proc Natl Acad Sci USA. 1997;94:1862–1865. doi: 10.1073/pnas.94.5.1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leigh Brown A J, Richman D D. HIV-1: gambling on the evolution of drug resistance? Nat Med. 1997;3:268–271. doi: 10.1038/nm0397-268. [DOI] [PubMed] [Google Scholar]
- 21.Liang K Y, Zeger S L. Longitudinal data analysis using generalized linear models. Biometrika. 1986;73:13–22. [Google Scholar]
- 22.Nijhuis M, Schuurman R, de Jong D, van Leeuwen R, Lange J, Danner S, Keulen W, de Groot T, Boucher C A B. Lamivudine-resistant human immunodeficiency virus type 1 variants (184V) require multiple amino acid changes to become co-resistant to zidovudine in vivo. J Infect Dis. 1997;176:398–405. doi: 10.1086/514056. [DOI] [PubMed] [Google Scholar]
- 23.Nijhuis M, Boucher C A B, Schipper P, Leitner T, Schuurman R, Albert J. Stochastic processes strongly influence HIV-1 evolution during suboptimal protease inhibitor therapy. Proc Natl Acad Sci USA. 1998;95:14441–14446. doi: 10.1073/pnas.95.24.14441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perelson A S, Neumann A U, Markowitz M, Leonard J M, Ho D D. HIV-1 dynamics in vivo: virion clearance rate, infected cell lifespan, and viral generation time. Science. 1996;271:1582–1586. doi: 10.1126/science.271.5255.1582. [DOI] [PubMed] [Google Scholar]
- 25.Rouzine I M, Coffin J M. Linkage disequilibrium test implies a large effective population number for HIV in vivo. Proc Natl Acad Sci USA. 1999;96:10758–10763. doi: 10.1073/pnas.96.19.10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schuurman R, Nijhuis M, van Leeuwen R, Schipper P, de Jong D, Collis P, Danner S A, Mulder J, Loveday C, Christopherson C, Kwok S, Sninsky J, Boucher C A B. Rapid changes in human immunodeficiency virus type 1 RNA load and appearance of drug-resistant populations in persons treated with lamivudine (3TC) J Infect Dis. 1995;171:1411–1419. doi: 10.1093/infdis/171.6.1411. [DOI] [PubMed] [Google Scholar]
- 27.Syvanen A C, Aalto-Setala K, Harju L, Kontula K, Soderlund H. A primer-guided nucleotide incorporation assay in the genotyping of apoliprotein-E. Genomics. 1990;8:684–692. doi: 10.1016/0888-7543(90)90255-s. [DOI] [PubMed] [Google Scholar]
- 28.Van Leeuwen R, Katlama C, Kitchen V, Boucher C A B, Tubiana R, McBride M, Ingrand D, Weber J, Hill A, McDade H, Danner S A. Evaluation of safety and efficacy of 3TC (lamivudine) in patients with asymptomatic or mildly symptomatic human immunodeficiency virus infection—a phase I/II study. J Infect Dis. 1995;171:1166–1171. doi: 10.1093/infdis/171.5.1166. [DOI] [PubMed] [Google Scholar]
- 29.Wainberg M A, Hsu M, Gu Z X, Borkow G, Parniak M A. Effectiveness of 3TC in HIV clinical trials may be due in part to the M184V substitution in 3TC resistant HIV-1 reverse transcriptase. AIDS. 1996;10(Suppl. 5):S3–S10. doi: 10.1097/00002030-199612005-00002. [DOI] [PubMed] [Google Scholar]
- 30.Wei X, Ghosh S K, Taylor M E, Johnson V A, Emini E A, Deutsch P, Lifson J D, Bonhoeffer S, Nowak M A, Hahn B H, Saag M S, Shaw G M. Viral dynamics in human immunodeficiency type 1 infection. Nature. 1995;373:117–122. doi: 10.1038/373117a0. [DOI] [PubMed] [Google Scholar]
- 31.Wright S. Evolution and the genetics of populations: a treatise. 2. The theory of gene frequencies. Chicago, Ill: University of Chicago Press; 1969. [Google Scholar]