

Abstract
Barth Syndrome (BTHS) is a life-threatening genetic disorder with unknown pathogenicity caused by mutations in TAFAZZIN (TAZ) that affect remodeling of mitochondrial cardiolipin (CL). TAZ-deficiency leads to accumulation of mono-lyso-CL (MLCL), which forms a peroxidase complex with cytochrome c (cyt c) capable of oxidizing polyunsaturated fatty acids (PUFA)-containing lipids. We hypothesized that accumulation of MLCL facilitates formation of anomalous MLCL/cyt c peroxidase complexes and PUFA-phospholipids peroxidation as the primary BTHS pathogenic mechanism. Using genetic, biochemical/biophysical, redox lipidomic, and computational approaches, we reveal mechanisms of peroxidase-competent MLCL/cyt c complexation, and increased phospholipid peroxidation in different TAZ-deficient cells, animal models and in pre-transplant biopsies from BTHS patient hearts. A specific mitochondria-targeted anti-peroxidase agent inhibited MLCL/cyt c peroxidase activity, prevented phospholipid peroxidation, improved mitochondrial respiration of TAZ-deficient C2C12 myoblasts, and restored exercise endurance in the BTHS Drosophila model. Targeting MLCL/cyt c peroxidase offers therapeutic approaches to BTHS treatment.
Keywords: Barth syndrome, TAFAZZIN-deficiency, mono-lyso-cardiolipin/cytochrome c complex, anomalous peroxidase activity, phospholipid peroxidation in TAZ-deficient cells and tissues, solid-state NMR, molecular dynamics simulations, Drosophila model of Barth syndrome, restoration of exercise deficiency by triphenyl-phosphonium-imidazole-substituted oleic acid
INTRODUCTION
Barth syndrome (BTHS) is an X-linked genetic multi-system disorder caused by mutations in the TAFAZZIN (TAZ) gene which results in aberrant lipid metabolism. BTHS patients exhibit varying degrees of cardiomyopathy, neutropenia, muscle weakness, growth delay, fatigue, and exercise intolerance, thus limiting quality of life. Despite improvements in diagnosis, premature death is commonplace due to lack of effective therapies. Clinical trials for two potential BTHS drug treatments, bezafibrate and elamipretide, failed to show beneficial effects in their respective trials, with only limited improvement reported for elamipretide in a subsequent trial1, 2. These results underscore the central problem - the leading pathogenic mechanism of BTHS is unknown. TAZ mutations impair remodeling of the mitochondria-specific phospholipid, cardiolipin (CL), which is essential for the normal function of >100 mitochondrial proteins. Consequently, treating secondary effects of TAZ deficiency do not address the underlying pathophysiology. Thus, elucidation of the primary pathogenic mechanism is required to identify therapeutic targets for intervention.
CL is localized almost exclusively in the matrix-facing leaflet of the inner mitochondrial membrane (IMM), accounting for 20–25 mol% of its phospholipids3. Combinations of fatty acids in CL’s four acyl chain positions result in a myriad of distinct molecular species, with many tissues containing highly diverse CL profiles4. Surprisingly, a remarkably uniform CL profile is characteristic of some organs, particularly heart and skeletal muscles of larger vertebrates (including humans), where >80% of all CLs are represented by a single species, tetra-linoleoyl-CL (CL(L)4)5, 6. While the significance of this uniformity remains enigmatic, it is achieved via an unusual two-stage biosynthetic process: initial biosynthesis of randomly diversified CLs, followed by remodeling, yielding longer-chain polyunsaturated species with a high predominance of CL(L)47, 8.
Remodeling begins with phospholipase A (PLA)-catalyzed CL hydrolysis, producing mono-lyso-CLs (MLCL) and free fatty acids7. This is followed by re-acylation and reconstitution of the tetra-acylated form, ultimately enriching CL(L)4 in heart and skeletal muscles5. The identity of the relevant PLA is still conjectural with the notable exception of Cld1 as the confirmed CL-specific PLA in yeast9. Re-acylation is catalyzed by one of three enzymes: two of them – mono-lyso-cardiolipin acyl transferase-1 (MLCLAT-1) and lyso-cardiolipin acyltransferase-1 (ALCAT1) – utilize acyl-CoA as substrate, while the third, TAZ, utilizes phospholipids for the transacylation reaction7, 10.
While many mitochondrial deficiencies are linked to TAZ mutations, distinguishing between the primary pathogenic mechanism of BTHS and secondary consequences of CL/MLCL imbalance has proven difficult. Secondary consequences include dysregulated redox cycling leading to excessive production of reactive oxygen species (ROS) and oxidative injury11. However, antioxidant therapies have failed to yield consistently beneficial effects6. Our previous work, in model biochemical experiments, has established that CL and MLCL can form a complex with the intermembrane space (IMS) hemoprotein, cytochrome c (cyt c), and this complex acts as a peroxidase towards polyunsaturated (PUFA)-CL12, 13. Normally, low levels of MLCL and compartmentalization of CL and cyt c preclude formation of this peroxidase complex. However, TAZ deficiency causes accumulation of MLCLs in the IMS-facing leaflet of the IMM, the site of the TAZ-catalyzed reaction14, making it available to interact with IMS-localized cyt c.
We hypothesized that the formation of MLCL/cyt c complexes serves as the primary pathogenic mechanism in BTHS, realized via oxidation of PUFA-phospholipids. We further predicted that mitochondria-targeted ligands capable of suppressing the peroxidase activity of MLCL/cyt c complexes may be effective therapeutics for treating BTHS. Here, we employed genetic, biochemical, biophysical, structural, redox lipidomic, and computational modeling approaches to characterize the causative mechanisms and pathogenic role of MLCL/cyt c complexes in BTHS. We discovered that a MLCL/cyt c-mediated pro-oxidant mechanism is responsible for BTHS pathogenesis and that targeting MLCL/cyt c peroxidase offers a potential therapeutic approach to BTHS treatment.
RESULTS
High spin heme-Fe in MLCL/cyt c peroxidase complex
Binding of anionic molecules to cyt c, including anionic CL, induces rupture of the weak M80-S-Fe axial bond in the protein15. This causes a transition from the native hexa-coordinated heme-Fe to a penta-coordinated configuration16 – a requirement for cyt c peroxidase competency17. This heme-Fe is characteristic of many non-native cyt c states induced by denaturing agents18. In the common ferrous (Fe2+) and ferric (Fe3+) oxidation states, the metal ion typically adopts different configurations of the electron distributions in its d orbitals depending on the strength and number of ligand species bound. In turn, this leads to diagnostically different spectral features; for instance, in the case of the hexacoordinate “low-spin” (with 1 unpaird d electron) ferric heme and the pentacoordinate “high-spin” (with 5 unpaired d electrons) peroxidatic ferric heme. We used absorption spectroscopy to distinguish between the high spin and low spin ferric hemes in MLCL/cyt c complexes. Addition of tri-linoleoyl-MLCL (MLCL(L)3) to cyt c resulted in appearance of features typical of the high spin-state: i) a weak absorption at 610–620 nm, ii) a more-pronounced shoulder at 560 nm, and iii) a markedly increased absorbance at 495 nm (Fig. 1a). Simultaneously, we observed a decreased absorbance at 695 nm, indicating disruption of the M80-S-Fe bond (Fig. 1b). It is to be understood that these observed changes indicate incomplete reactions; full conversion of low-spin to high-spin ferric heme, for example, would be expected to lead to the complete loss of the 695 nm band rather than the observed partial reduction in intensity (Fig. 1b).
Figure 1. Electronic and structural rearrangements occurring in MLCL/cyt c peroxidase complexes.
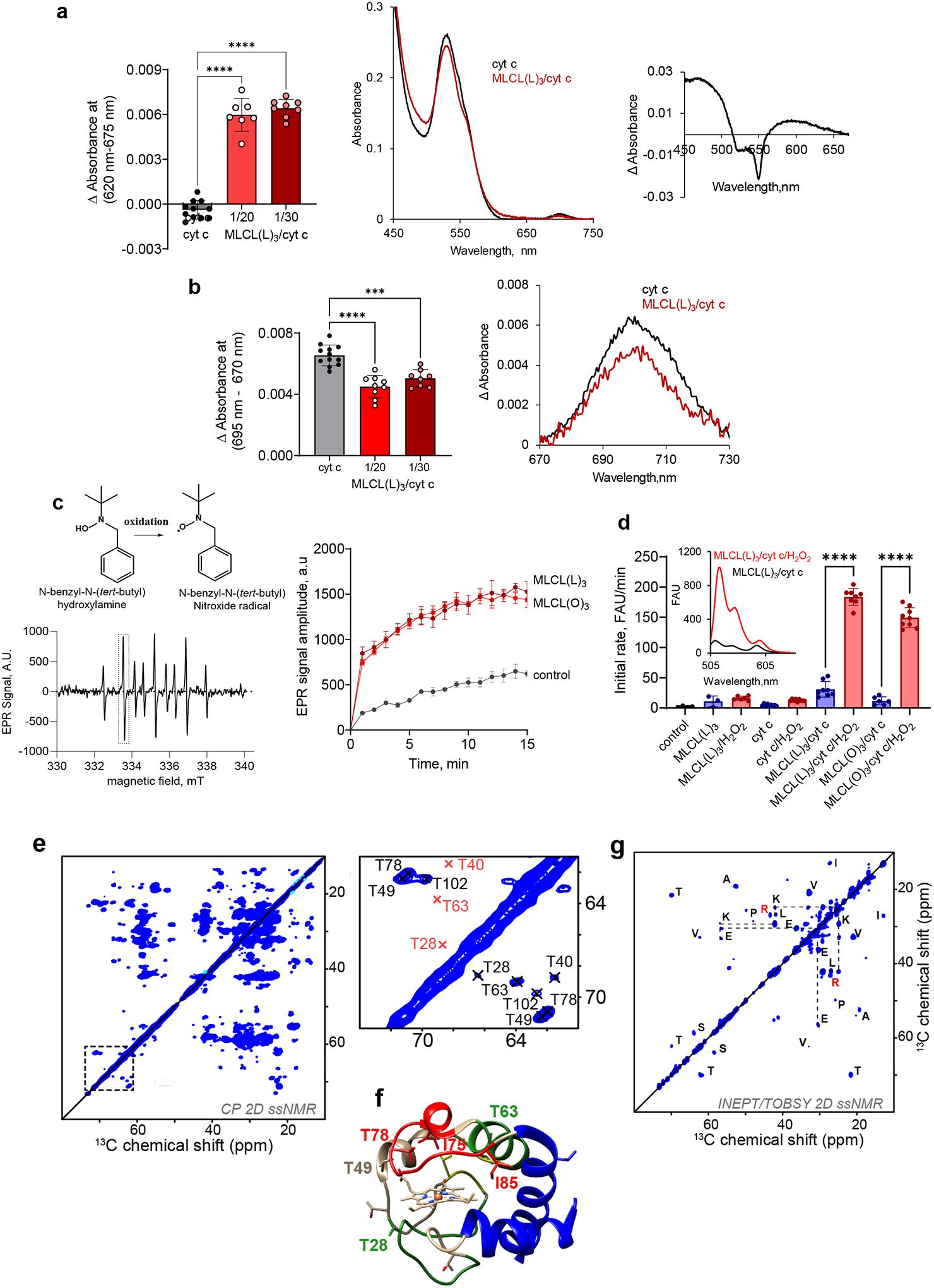
a, MLCL–dependent formation of heme-Fe high spin forms assessed by absorbance at 620nm (left panel). Data are presented as mean values ± SD, ****P<0.0001, one-way ANOVA, Tukey’s multiple comparison test. N=11 (cyt c), N=7 (MLCL(L)3/cyt c = 1/20), N=8 (MLCL(L)3/cyt c = 1/30). Each data point represents a biologically independent sample. b, Effect of MLCL on the absorption spectra of cyt c (75μM) (middle panel); the differential absorption spectrum produced by subtracting the spectrum of cyt c from the spectrum of MLCL/cyt c (right panel) MLCL induces decrease of the absorption at 695 nm indicating disruption of the M80-S-Fe coordination (left panel). Data are presented as mean values ± SD ***P=0.0001, ****P<0.0001, one-way ANOVA, Tukey’s multiple comparison test. N=12 (cyt c), N=7 (MLCL(L)3/cyt c = 1/20), N=8 (MLCL(L)3/cyt c = 1/30). Each data point represents a biologically independent sample. Absorption spectra of cyt c (75μM) with and without MLCL (right panel). c, Left panel: Typical EPR signal of N-benzyl-N-(tert-butyl)nitroxide radical generated by MLCL(O)4/cyt c complexes (lower panel). The underscored part of the spectrum was scanned repeatedly to measure the kinetics of radical formation. Structure of N-benzyl-N-(tert-butyl)nitroxide and its radical are inserted (upper panel). Rigth panel: Time courses of N-benzyl-N-(tert-butyl)nitroxide radical EPR signals generated by MLCL(L)3/cyt c, and MLCL(O)3/cyt c complexes. Control: N-benzyl-N-(tert-butyl)nitroxide radical EPR signals generated by cyt c/H2O2 in the absence of MLCL. Data are presented as mean values ± SD, N=3 (control), N=4 (MLCL(L)3), N=4 (MLCL(O)3) biologically independent samples. d, Oxidation of BODIPY-C11 by MLCL/cyt c complexes. Data are presented as mean values ± SD, ****P<0.0001, one-way ANOVA, Tukey’s multiple comparison test. N=4 (control), N=3 (MLCL(L)3), N=6 (MLCL(L)3/H2O2), N=9 (cyt c), N=9 (cyt c/H2O2), N=8 (MLCL(L)3/cyt c), N=8 (MLCL(L)3/cyt c/H2O2), N=6 (MLCL(O)3/cyt c), N=9 (MLCL(O)3/cyt c/H2O2), biologically independent experiments. Insert: Fluorescence emission spectra of BODIPY oxidized by MLCL(L)3/cyt c in the absence and presence of H2O2 (excitation = 495 nm). e, 2D CP-based ssNMR spectrum of labeled cyt c bound to DOPC:MLCL(L)3 (1:1) vesicles (left). The region marked with a dashed box show key threonine peaks, and is also shown enlarged to the right, with residue assignments indicated. f, Structure of cyt c, with color-coding showing relative stability of the native fold segments (colored red< green< blue). The heme group, threonine residues (T28, T49, T63, and T78) and Ω-loop residues I75 and I85 that line the heme binding pocket are shown as sticks. g, 2D INEPT-TOBSY ssNMR spectrum showing the assignments for observed residues with high flexibility. The ssNMR was performed at 600 MHz (1H) and 253 K (e) or 278 K (g).
Next, we used EPR to examine whether the MLCL-induced heme-Fe transition results in “reprogramming” of cyt c into a peroxidase. Incubation of either MLCL(L)3 or tri-oleoyl MLCL (MLCL(O)3) with cyt c in the presence of N-benzyl-N-(tert-butyl)hydroxylamine generated a one-electron oxidation product with the characteristic nine line electron paramagnetic resonance (EPR) spectrum of N-benzyl-N-(tert-butyl)nitroxide radical (Fig. 1c). The spectrum was interpreted by the coupling of the unpaired electron with the nitrogen nucleus and two equivalent benzylic protons; the spectroscopic parameters are a(N) = 16.76 G, a(2Hβ) = 10.62 G, (1G = 0.1 mT). Both MLCL(L)3/cyt c and MLCL(O)3/cyt c complexes generated N-benzyl-N-(tert-butyl)nitroxide radicals (Fig. 1c) with similar efficiency. We also assessed the peroxidase activity of MLCL/cyt c by monitoring the oxidation of BODIPY-C11 via fluorescence. H2O2 caused a significant increase in oxidized BODIPY-C11 fluorescence following incubation with either MLCL(O)3/cyt c or MLC(L3)/cyt c complexes. Incubation of BODIPY-C11 with H2O2 and MLCL alone had no effect on its fluorescence, suggesting that cyt c is required to confer peroxidase activity to the MLCL/cyt c complex (Fig. 1d).
Solid-state NMR (ssNMR) of cyt c binding to MLCL
Next, we probed the impact of MLCL on the molecular structure and dynamics of cyt c protein by solid-state nuclear magnetic resonance (ssNMR) spectroscopy. The ssNMR technique selectively detects the stable-isotope-labeled (15N,13C) protein in its complex with MLCL-containing lipid membranes. In the 2D ssNMR spectrum (Fig. 1e) we observed numerous off-diagonal peaks from the MLCL-bound protein. Most peak positions match those expected for the cyt c native fold, but this is only true for a subset of the peaks (Extended Data Fig. 1a)12, 19, 20. The intensities fo these peaks depend on the rigidity of the observed protein. A subset of strong peaks revealed an immobilized and folded core formed by the N- and C-terminal α-helical segments (Extended Data Fig. 1a–b). These data closely resembled our prior studies of cyt c binding to non-hydrolyzed CL, where we identified a partial unfolding of a specific part of the protein: its so-called Ω-loop D. This loop is the least stable part of the native protein, and is functionally relevant (see below). Our current data recapilate a similar result for the MLCL-cyt c complex: many expected cyt c peaks are diminished or missing, indicating a local loss of stable fold (Fig. 1 and Extended Data Fig. 1c–d). Using threonine (Thr) residues as local probes (Fig. 1e–f) we see evidence for dynamics affecting specific residues (e.g. Thr28 and Thr78) as changes in peak height and loss of cross-diagonal symmetry. Thus, different parts of the protein display different extents of motion (detected mobile residues marked red in Fig. 1f). A striking feature of MLCL-bound cyt c is that the Ω-loop becomes much more mobile. This can even be seen in special ssNMR measurements that highlight only the most flexible (i.e. not-folded) amino acids, so-called J-based ssNMR (INEPT spectroscopy; Extended Data Fig.1e)21, 22. 2D INEPT-ssNMR (Fig. 1f) shows only the signals from the labeled protein and reveals surprisingly strong backbone signals, indicating a highly flexible multiresidue segment of approximately 11 residues. This is not seen for CL-bound cyt c (Extended Data Fig. 1f). The chemical shifts and peak patterns for the detected flexible residues identify amino acids that match an extended region from the aforementioned Ω-loop D (between residues I75 and I85). As expected for flexible segments, these Ω-loop D residues were among the missing or modulated signals mentioned for the 2D spectra (Extended Data Fig. 1c–d) obtained by CP-ssNMR, where we detected the rigid or folded protein parts. We also analyzed the lipid membrane structure and dynamics, using 31P ssNMR that detects only the phosphate headgroups of the lipids (Extended Data Fig. 2). The 31P ssNMR lineshapes inform us on the phase and dynamics of the lipids. Inclusion of MLCL gave a new signal with a narrow lineshape indicative of high (headgroup) motion in a bilayer context23, which is not seen for normal CL. This bilayer mobilization effect of the lyso-lipid was independent of the effect of cyt c. In summary, ssNMR studies of the MLCL-bound protein show that cyt c retains its most stable folded core, but gains localized flexibility in a particular region (Ω-loop D), with the latter much more pronounced than the motion induced by normal CL12. Increased disorder in MLCL-containing membranes (detected by 31P NMR) may underlie these dynamic changes, while also contributing to mitochondrial membrane destabilizing and trans-bilayer CL scrambling.
Molecular dynamics simulations of cyt c binding to MLCL
To examine the interactions between cyt c and MLCL at atomic detail, we performed molecular dynamics (MD) simulations of cyt c interacting with liposomes containing DOPC, di-oleoyl-phosphatidylethanolamine (DOPE), and MLCL. MD employs physical and chemical principles to simulate biomolecular systems’ dynamics and interactions, providing detailed explanations not attained through experimentation. Among a series of initial MD trajectories, we selected ten in which cyt c diffused towards the membrane bilayer, and its positively charged residues and M80 interacted with MLCL (Extended Data Fig. 3a). Fig. 2 illustrates the summary results from these simulations. We examined cyt c-liposome interactions within two time intervals: (i) initial recognition and membrane binding (during t < 100 ns) (Movies M1, M2), which involved seven positively charged residues, K8, K13, K72, K73, and K86-K88, along with two hydrophobic residues, I81 and A83, and two polar residues, N70 and Q12 (Fig. 2a upper panel), and (ii) stabilization of interactions during the last 100 ns of simulations exhibited additional hydrophobic contacts with T78, M80, and F82 (Fig. 2a lower panel). The bar plots in the respective panels display the frequency of occurrence of different contacts (different residues along the abscissa and three types of lipid molecules color-coded as labeled) based on snapshots collected at regular (50 ps) intervals during the course of MD simulations. Notably, the initial interactions between cyt c and liposomes were primarily driven by contacts between K86-K87 and MLCL molecules (Fig. 2a upper panel), while longer time stabilization also involved various interactions with DOPE and DOPC (Fig. 2a lower panel).
Figure 2. Simulations reveal close association of M80 with MLCLs driving the opening of the heme-binding pocket.
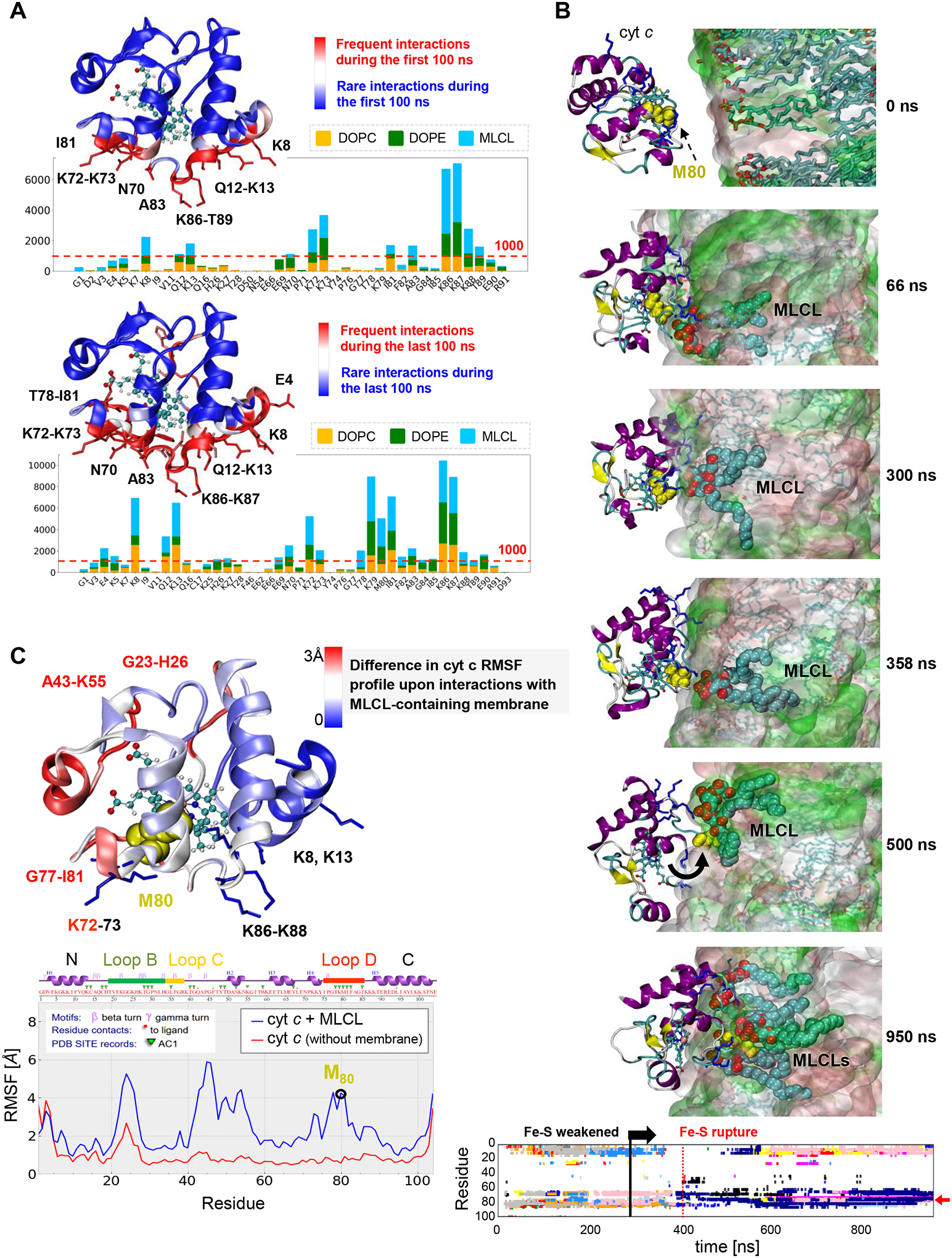
a, Distribution of contacts between cyt c residues and lipid molecules DOPE, DOPC and MLCL. Membrane-binding residues of cyt c (located within 4.5 Å from the membrane) observed during the first 100 ns (upper panel), and the last 100 ns (lower panel) of multiple 700–950 ns long MD simulations were recorded at fixed (50 ps) intervals, to quantitatively determine the most probable interactions. The bar plot shows the number of contacts made by selected cyt c residues (abscissa) with different types of lipid molecules (color-coded as labeled). Red dashed line indicates the threshold for frequent contacts; the corresponding residues are colored red in the ribbon diagram, where the heme group is also displayed in stick representation. b, Time evolution of conformational changes in the Ω-loop I75-I85 (cyan sticks/lines) containing M80 (yellow spheres) and its interactions with MCL, observed during MD simulations (0 < t < 950 ns). The top six panels depict the evolution of interactions, and the bottom plot provides a quantitative summary of the interactions between cyt c residues and multiple MLCL molecules (each shown by a different color). In the top six panels, the lipid bilayer is in surface representation with lipid molecules in sticks at t = 0 (top panel), and the MLCL is in cyan spheres (with the phosphate groups in red) in all panels except t = 0. The interaction involves various stages: (i) During the first 332 ns the Fe-S bond remains intact. It is completely ruptured at 415 ns, preceeded by small extensions at t = 332 and 368 ns, (ii) During the interval 415 < t <500 ns, M80 gains an increase in mobility, and (iii) at t ≥ 500 ns M80 is trapped by several MLCLs (bottom panel). The lower panel shows the time evolution of contacts between cyt c residues and MLCL molecules (different colors for different MLCLs). The red arrow points to interactions of M80 with multiple MLCLs (atom-atom contacts within 4.5 Å). See Movie 3 for a visualization. c, RMSF profile of cyt c residues in the last 100 ns of two sets of MD runs, one in the presence of membrane (blue curve, averaged over four runs) and the other in water (no membrane) (red curve, average over two runs). The crystal structure of cyt c is colored by the relative size of fluctuations (RMSFs in the presence of the membrane minus those in its absence, in Å). Dark red regions (G23-H26, K72, G77-I81) exhibit enhanced mobilities in the presence of a membrane upon interactions with MLCL; wherease dark blue regions (K8, K13, K86-K88) are less flexible being associated with the lipid head groups.
M80/MLCL interactions drive the opening of heme-binding pocket
Closer examination of the time evolution of cyt c – membrane interactions during MD simulations (Fig. 2b) revealed that the bond Fe-S between the catalytic iron and the S atom of M80 exhibited two types of behavior: 1) an increase in the distance between M80 and Fe upon the interaction of M80 with MLCL (the most frequent behavior); 2) a rupture of the Fe-S bond, inducing a conformational change in the Ω−loop D and insertion of M80 into the membrane to interact with multiple MLCL molecules (Movie M3, Fig. 2b bottom panel). Close association of M80 with the membrane was accompanied by partial unfolding of cyt c and exposure of the heme-binding site to the membrane (at t ≥ 500 ns). Additional simulations carried out in the absence of membrane, showed that the positions and interactions between catalytic Fe and S remained unchanged (Extended Data Fig. 3b). This underscores the role of the membrane in regulating the peroxidase activity of the complex.
The presence of MLCL-containing membrane induced robust changes in the mobility of invididual parts of cyt c (Fig. 2c). To better understand the scale of these changes, we evaluated the fluctuations of individual residues within protein structure using a measure called Root Mean Square Fluctuations (RMSF). RMSF characterizes the flexibility of individual regions within a protein’s structure in MD simulations. Assessment of the RMSF of Cα-atoms in the presence or absence of membrane revealed increased mobility in three cyt c regions primarily induced with the interactions with MLCL: (i) G23-H26 (Loop B), (ii) A43-K55, and (iii) G72-Y74 and G77-I81 (Ω−loop D). Overall, these computational results detail the MLCL-induced conversion of cyt c from the hexa- to penta-coordinated state as experimentally observed by ssNMR and electronic spectra.
MLCL/cyt c complexes peroxidize PUFA phospholipids
We next performed LC-MS measurements of phospholipid oxidation by MLCL/cyt c complexes. Oxidizable MLCL(L)3 and non-oxidizable MLCL(O)3 were incubated with unilamellar DOPC liposomes in the presence or absence of oxidizable phospholipids, including stearoyl-linoleoyl-phosphatidylethanolamine (SLPE), stearoyl-linoleoyl-phosphatidylcholine (SLPC), stearoyl-arachidonoyl-phosphatidylethanolamine (SAPE), and stearoyl-arachidonoyl-phosphatidylcholine (SAPC). MLCL(L)3/cyt c caused robust MLCL oxidation (Fig. 3a). MS/MS analysis revealed oxygenated MLCL(L)3 with 1–4 oxygens as well as oxidatively-truncated species with 9-oxo-nonanoic acid (ONA) formed via degradation of 9-hydroperoxy-(OOH)-linoleic residues (Fig. 3a, Extended Data Fig. 4a). 9-OOH-MLCL(L)3 and di-9-OOH-MLCL(L)3 were the dominant oxidation products. Thus MLCL(L)3, directly associated with cyt c, is a peroxidation substrate. When peroxidizable PUFA-phospholipids that do not bind cyt c (SLPE and SLPC) were incubated with MLCL(L)3/cyt c complexes, MLCL(L)3 oxidation was significantly suppressed (Fig. 3a). 9-OOH-MLCL(L)3 and di-9-OOH-MLCL(L)3 were not detectable in the presence of SLPE. Instead, SLPE and SLPC containing 9-OOH- and 9-hydroxy(OH)-linoleic acid as well as their oxidatively-truncated products were detected (Fig. 3b, Extended Data 4b-c). When non-oxidizable MLCL(O)3 was used instead of MLCL(L)3, PUFA-phospholipid peroxidation was enhanced (Fig. 3b). As both peroxidizable and non-peroxidizable MLCLs are present in cells, we next examined a mixture of 1:3 MLCL(L)3/MLCL(O)3 with cyt c in the presence of SAPE or SAPC. Production of OH- and OOH-products (predominantly mono-OOH- and di-OOH-derivatives (Fig. 3b)), as well as oxidatively-truncated species (5-hydroxy-8-oxo-6-octenoic acid (HOOA) formed via degradation of OOH-arachidonic acid) were observed. Thus, the peroxidase activity of MLCL/cyt c complexes can catalyze peroxidation of PUFA substrates both within the complex and in the membrane (i.e., not directly interacting with cyt c).
Figure 3. Peroxidase activity of the MLCL/cyt c complex causes phospholipid peroxidation in vitro and induces changes in the lipidome and oxy-lipidome of genetically-manipulated yeast.
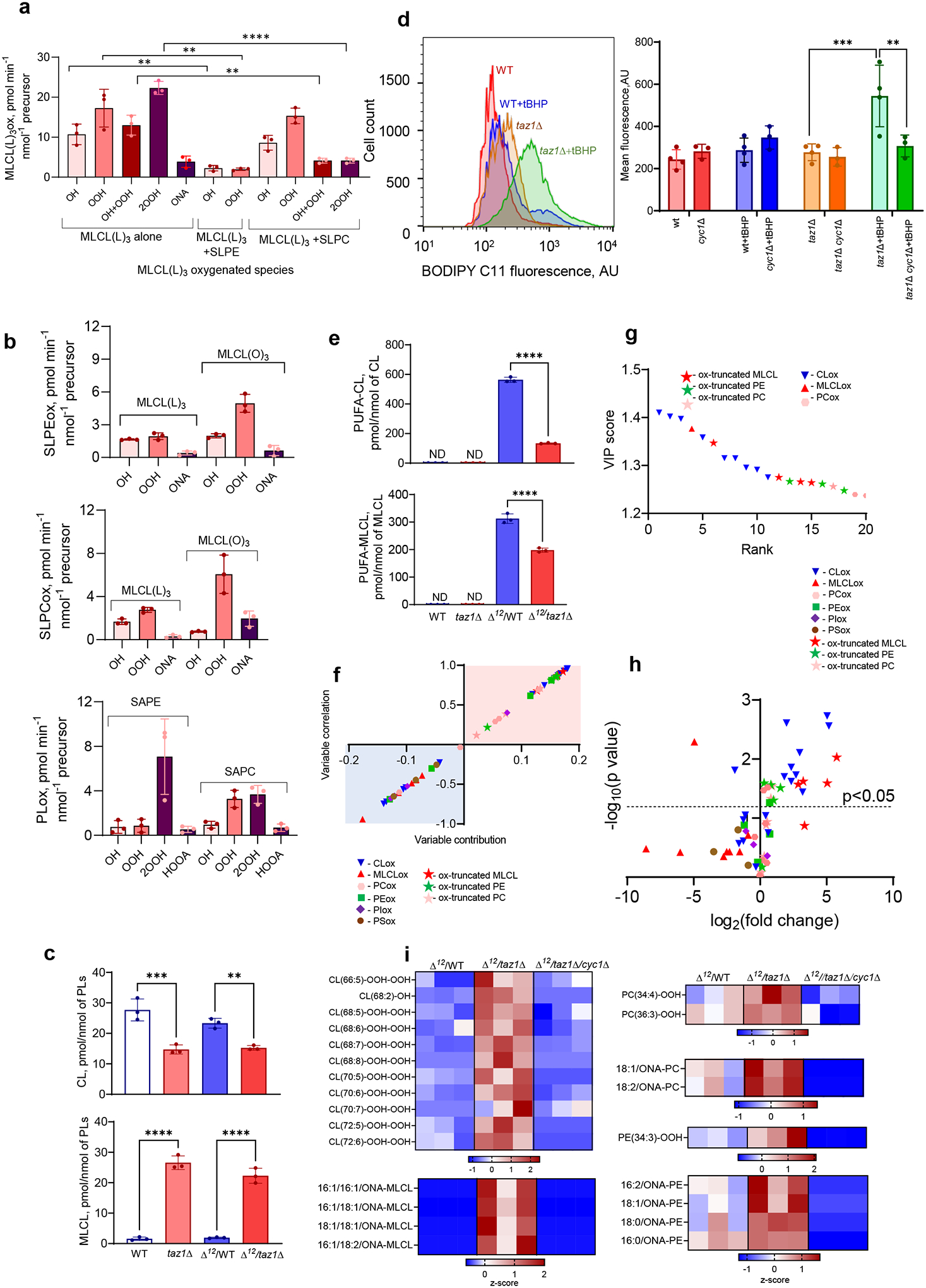
a, MLCL/cyt c complexes cause peroxidation of PUFA-phospholipids in a model system. Initial rate of formation of different MLCL(L)3 oxidation products generated by MLCL(L)3/cyt c complexes in the absence and presence of PUFA-PL (stearoyl-linoleoyl-PE, SLPE or stearoyl-linoleoyl-PC, SLPC). Data are presented as mean values ± SD. Significant differences were determined by unpaired t-test, N=3; **P=0.0054 (MLCL(L)3 alone vs MLCL(L)3+SLPE), **P=0.005 (OH MLCL(L)3 alone vs OH MLCL(L)3+SLPC), **P=0.0039 (OH/OOH MLCL(L)3 alone vs OH/OOH MLCL(L)3+SLPC), **p=0.005 (OOH MLCL(L)3 alone vs OOH MLCL(L)3+SLPC), ****p<0.0001 (2OOH MLCL(L)3 alone vs 2OOH MLCL(L)3+SLPC). Each data point represents a biologically independent experiment. b, Generation of SLPE (upper panel) and SLPC (middle panel) oxidation products by MLCL(L)3/cyt c and MLCL(O)3/cyt c complexes. Data are presented as mean values ± SD. N=3. Generation of SAPE and SAPC oxidation products by MLCL(L)3/MLCL(O)3 (1:3)/cyt c complexes (lower panel). OH (hydroxy), OH/OOH (hydroxy/hydroperoxy), OOH (hydroperoxy), 2OOH (di-hydroperoxy) species.OH (hydroxy species), OOH (hydroperoxyl species), 2OOH (di-hydroperoxy species), ONA (9-oxo-nonanoic acid) and HOOA (5-hydroxy-8-oxooct-6-enoic acid) species. N=3. Each data point represents a biologically independent experiment. c, Content of total CL (upper panel) and MLCL (lower panel) in WT, taz1Δ, Δ12/ WT, and Δ12/taz1Δ yeast cells. Data are presented as mean values ± SD. Significant differences were determined by one-way ANOVA, Tukey’s multiple comparison test. **P=0.0077, ***P=0.0004, ****P<0.0001; Each data point represents a biologically independent experiment. d, Detection of lipid peroxidation by BODIPY-C11 581/591 in yeast cells. (Gating strategy is shown on Supplementary Fig.1). Oxidation of BODIPY-C11 results in increased fluorescence emission at 510 nm, which is depicted as a rightward shift in the median green FL1 fluorescence intensity (MFI) histogram (left panel). The Y-axis corresponds to the number of cells displaying a given MFI (X-axis). Quantitative assessment of mean fluorescence of shifted peak (right panel). Data are presented as mean values ± SD. Statistical significance was analyzed using a one/two-sided Student’s t-test **p=0.0344, ***p=0.0083. N = 3–4 biologically independent samples. e, Content of PUFA-CL (upper panel) and PUFA-MLCL (lower panel) in WT, taz1Δ , Δ12/WTand Δ12/taz1Δ yeast cells. Data are presented as mean values ± SD. Significant differences were determined by one-way ANOVA, N=3; ****P<0.0001; f, S-plot of OPLS-DA analysis of Δ12/WT and Δ12- taz1Δ yeast lipidomes showing variable correlation versus variable contribution to the OPLS-DA model. g,The variable importance in projection (VIP) score plots reflect the significance of variables for the OPLS-DA models. The VIP>1 was considered to be a statistically significant difference between the two groups. Data are presented for up to 20 phospholipid molecular species. h, Volcano plot showing the changes in the levels of oxygenated phospholipid species induced by TAZ deficiency. N=3, Significant differences were determined by unpaired t-test; i, Content of oxygenated and oxidatively truncated phospholipids in Δ12/WT and Δ12/taz1Δ yeast cells. Data are presented as heat maps auto-scaled to z scores and coded blue (low values) to red (high values).
TAZ-deficient/Δ12-desaturase yeast cells peroxidize phospholipids
Next, we utilized the genetically tractable yeast model that uniquely enables manipulation of CL remodeling and PUFA-lipid composition and offers two key advantages: 1) The yeast taz1Δ mutant exhibits the biochemical manifestations of BTHS, specifically, increased MLCL and decreased CL. 2) Although yeast cells only synthesize saturated and Δ9-monounsaturated fatty acids, we generated WT and taz1Δ yeast strains capable of synthesizing PUFAs by expressing the H. brasiliensis Δ12-desaturase24. This system enabled us to determine the specific contribution of PUFA-containing phospholipids to the peroxidation mechanism.
Previously, we showed that the effects of TAZ1 deletion were CL-specific, such that levels of other phospholipids – phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidyl-glycerol (PG), phosphatidylserine (PS) – were not altered24. As expected, the level of CL was significantly decreased and MLCL content was robustly increased in taz1Δ yeast vs. WT controls (Fig. 3c, Extended Data Fig. 4d).
To test whether increased MLCL is pivotal in the generation of peroxidase activity independently of PUFA-PLs, we first employed BODIPY-C11 as an alternative peroxidase substrate and assessed its tert-butyl hydroperoxide (tBHP)-induced oxidation in taz1Δ vs. WT yeast cells. With tBHP as a source of oxidizing equivalents, the peroxidase activity was significantly higher in taz1Δ cells, as demonstrated by increased content of oxidized BODIPY-C11 (Fig. 3d). This directly demonstrated that peroxidase activity in taz1Δ cells resulted from the mitochondrial accumulation of MLCL in spite of the absence of PUFA-MLCL and other peroxidizable PUFA-PLs. To directly genetically test the importance of cyt c in the formation of the peroxidase complex with MLCL, we monitored oxidation of BODIPY-C11 in TAZ-deficient yeast lacking functional cyt c. Exposure of taz1Δ cells lacking cyt c (cyc1Δ) to tBHP resulted in a significant reduction in BODIPY-C11 oxidation vs. samples containing functional cyt c (Fig. 3d).
Next, we examined differences in the oxy-lipidome of WT and taz1Δ yeast cells expressing Δ12-desaturase (Δ12/WT, Δ12/taz1Δ) and hence capable of biosynthesizing PUFA-PLs. Peroxidizable PUFA-phospholipids containing C16:2 and C18:2 fatty acids, including CL and MLCL, were present only in Δ12-desaturase-expressing cells (Fig. 3e, Extended Data Fig. 4e–f). Biologically relevant changes were determined using orthogonal projection of latent structures-discriminant analysis (OPLS-DA) (Extended Data Fig. 4g). We found that oxygenated CL and oxidatively-truncated PL species, MLCL, PE, and PC, were significantly elevated in Δ12/taz1Δ cells and had a high variable importance in projection (VIP) score (Fig. 3f–j). The oxy-lipidome of Δ12-desaturase-expressing cells was represented by 60 oxygenated phospholipid species, including 16 oxidatively-truncated species (Extended Data Fig. 3h). Of these, 22 species were increased in Δ12/taz1Δ cells relative to Δ12/WT cells (Fig. 3h). Oxidized CL was predominant and mainly contained four oxygens, while levels of two OOH-PC and one OOH-PE species were also elevated in taz1Δ (Fig. 3i). Additionally, the levels of four truncated-MLCL, four truncated-PE, and two truncated-PC species were significantly elevated (Fig. 3i). All oxidatively-truncated phospholipids contained ONA, a product of 9-OOH-linoleic acid degradation. Peroxidation of PS – lacking from mitochondria25 – was not altered in Δ12/taz1Δ cells. Cyt c-deficientΔ12/taz1Δ cells neither oxidized PUFA-phospholipids nor generated oxidatively truncated phospholipids. This indicates that cyt c/MLCL peroxidase activity is responsible for the accumulation of oxygenated phospholipid species in TAZ-deficient cells (Fig. 3i). Thus, elevated MLCL resulting from TAZ deficiency drives increased mitochondrial phospholipid peroxidation.
PUFA-phospholipids peroxidation in TAZ-deficiency
The Δ12/taz1Δ yeast model produces only two types of oxidizable PUFA-phospholipids (containing either C16:2 or C18:2), simplifying the identification of peroxidation products. Because redox lipidomics of TAZ-deficient mammalian tissues with their diverse PUFA-phospholipid profile has not been performed before, we examined individual peroxidation products in major phospholipid classes in several types of cells and tissues from mouse and human samples. Mouse samples included myoblasts from WT and TAZ-KO C2C12 cells26, as well as hearts from WT and TAZ KD mice27. Human samples included lymphoblast lines derived from three affected BTHS individuals and three demographically-matched healthy controls, along with heart tissue samples from healthy controls, patients with non-BTHS-associated heart failure (NBHF), and patient with BTHS-associated heart failure (Extended Data Fig. 5a). In all mouse and human samples, TAZ deficiency was associated with decreased CL and elevated MLCL (Fig. 4a). In human heart samples, BTHS tissue exhibited the characteristic decrease in CL and increase in MLCL compared to both control and NBHF samples, which did not differ in their CL profile (Fig. 4a). Thus, decreased CL and increased MLCL are the direct biochemical outcomes of TAZ-deficiency.
Figure 4. TAZ deficiency induces changes in the phospholipidome of mouse and human cells and tissues.
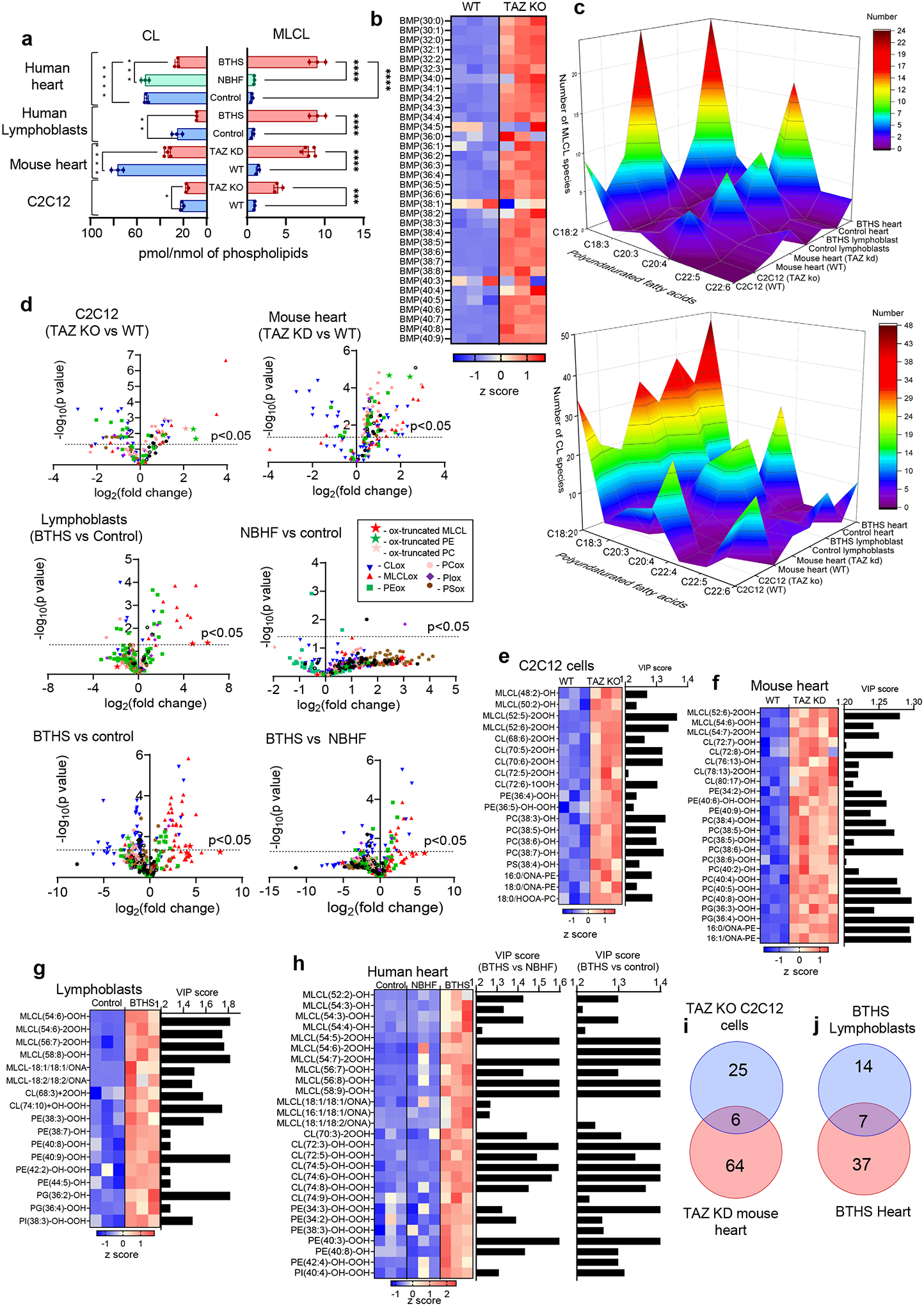
a, Content of MLCL and CL in C2C12 cells, mouse heart, human lymphoblasts, and human heart samples. 2-tailed t-test, For MLCL: ***p=0.0002, ****p<0.0001, one-way ANOVA, Tukey’s multiple comparison test. For CL: *p=0.0189 (unpaired 2 tailed-t test), **p=0.0009, ***p=0.0002, ****p<0.0001, one-way ANOVA, Tukey’s multiple comparison test. Data are presented as mean values ± SD. N=3 biologicaly independent cells, N=3 and N=5 biologicaly indedendent control and TAZ ko mice, respectively, N=3 biologicaly independent human lymphoblasts and N=3 biologicaly independent human heart biopsy samples. b, Content of bis-monoacylglycerophosphate (BMP) in WT and TAZ-deficient cells. Data are presented as heat maps auto-scaled to z scores and coded blue (low values) to red (high values). c, Number of PUFA-MLCL (upper panel) and PUFA-CL (lower panel) species in C2C12 cells, mouse heart,human lymphoblasts, and human heart. d, Volcano plots showing significant changes in the levels of oxygenated phospholipid species induced by either TAZ deficiency in cells and mouse heart or TAZ mutation in human lymphoblasts and heart samples from BTHS patients. N=3–5; Significant differences were determined by unpaired two-tailed t-test. Content of oxygenated phospholipids in WT and TAZ-deficient cells (e), heart tissue obtained from WT and TAZ-KD mice (f), control and BTHS lymphoblasts (g) and control, NBHF, and BTHS heart samples (h). Data are presented as heat maps auto-scaled to z scores and coded blue (low values) to red (high values) (left panels), and bar graphs of the variable importance in projection (VIP) score plots that reflect the significance of variables for the OPLS-DA models (right panels). i, The two-way Venn diagram congregated oxygenated molecular species of phospholipids detected in TAZ-KO C2C12 myoblasts in vitro and heart of TAZ-KD mice in vivo. Six species (MLCL(52:6)-2OOH, PE(36:4)-OOH, PC(38:5)-OH, PE-16:0/ONA, PC(38:6)-OH, PC(40:7)-OH) common to TAZ deficiency were identified. The value in each area indicates the number of oxygenated molecular phospholipid species with significantly higher levels in TAZ groups vs WT by unpaired two-tailed t-test. j, The two-way Venn diagram congregated oxygenated molecular species of phospholipids detected in BTHS lymphoblasts and BTHS hearts. Seven MLCL species common to BTHS (MLCL(54:6)-2OOH; MLCL(54:7)-2OOH; MLCL(56:7)-OOH; MLCL(56:8)-OOH; MLCL(54:5)-2OOH; MLCL(58:9)-OOH; MLCL(18:1/18:1/ONA) were identified. The value in each area indicates the number of oxygenated molecular phospholipid species with significantly higher levels in BTHS groups vs respective control by unpaired twotailed t-test.
We next examined levels of bis(monoacylglycero)-phosphates (BMPs), which are excellent biomarkers of autophagy activation as they appear during the fusion of late-endosomes with lysosomes28, in TAZ-KO vs WT C2C12 cells. We observed accumulation of essentially all BMP molecular species in two major clusters, non-oxidizable (stearic (C18:0), oleic (C18:1)) and highly oxidizable (C20:4) species (Fig. 4b). These data demonstrate a strong activation of autophageal machinery in TAZ-KO vs WT C2C12 cells29.
A striking increase in the molecular diversification of MLCL and CL was observed in all TAZ-deficient samples, whereby the most characteristic feature was the emergence of polyunsaturated species, especially those containing arachidonoyl-residues (C20:4), which are excellent peroxidation substrates (Fig. 4c, Extended Data Fig. 5b–e, Supplementary Tables 1–2). Specifically, TAZ-KO myoblasts exhibited a reduction of C18:2-species and the appearance of MLCL and CL species containing C20:4 (Fig. 4c, Extended Data Fig. 5b, Supplementary Tables 1–2). Hearts from TAZ KD mice also had increased MLCL and CL species containing mostly C20:4, along with linolenic (C18:3) and C18:2 acids (Fig. 4c, Extended Data Fig. 5c, Supplementary Tables 1–2). BTHS lymphoblasts contained a higher number of MLCL and CL species predominantly containing C20:4 and C18:3 (Fig. 4b, Extended Data Fig. 5d, Supplementary Tables 1–2). Similarly, CL and MLCL species in BTHS hearts were more diverse compared to control and NBHF hearts, including enrichment with C20:4 and C18:3 (Fig. 4c, Extended Data Fig. 5e, Supplementary Tables 1–2). An increase in MLCL species with docosapentaenoic (C22:5) and docosahexaenoic (C22:6) acids was observed in hearts from TAZ KD mice, BTHS lymphoblasts, and BTHS heart samples (Fig. 4c). Collectively, TAZ deficiency resulted in increased levels of readily-oxidizable PUFA-containing CL and MLCL species.
We reasoned that MLCL/cyt c peroxidase activity and increased peroxidizable lipid substrates in TAZ-deficient cells should lead to the accumulation of peroxidized PUFA phospholipids (Fig. 4d, Extended Data Fig. 5f). Comparison of the oxi-lipidomes of WT and TAZ-KO mouse myoblasts and WT and TAZ KD mouse heart samples using OPLS-DA (Extended Data Fig. 5g) showed that the levels of 32 species were significantly elevated in TAZ-KO cells compared to WT, and more than half of these were also upregulated in TAZ KD hearts (Fig. 4d). Human samples showed a similar pattern of oxidized lipid enrichment (Extended Data Fig. 5g). Specifically, 25 oxygenated species were upregulated in BTHS lymphoblasts, and 43 and 23 species were upregulated in BTHS hearts compared to control or NBHF tissue, respectively (Fig. 4c). Control and NBHF tissue samples did not differ significantly with respect to oxidized lipids.
In TAZ-deficient mouse myoblasts and heart tissues (Fig. 4e–f) as well as in BTHS lymphoblasts and heart biopsy samples (Fig. 4g–h), oxygenated phospholipids were mainly represented by MLCL, CL, PE, and PC. Among these, oxygenated species of MLCL, CL, and PE differed the most between BTHS and controls, resulting in a high VIP score (>1.2) (Fig. 4e–h). Oxygenated MLCL and CL originated predominantly from C20:4-containing species. No significant differences in oxygenated PS were detected in any of the TAZ-deficient samples. Because PS is localized primarily outside of mitochondria, this suggests that BTHS-associated lipid oxidation takes place mainly in mitochondria. The predominance of peroxidized MLCL suggests that peroxidation occurs primarily within MLCL/cyt c complexes, while the presence of other oxidized phospholipids indicates that peroxidation also occurs outside of the complex. While the quantitative assessment of non-oxidized major phospholipids, PC and PE, revealed small changes in their content, the variety of individual molecular species and their distribution were cell and tissue dependent (Extended Data Fig. 6).
Next, we identified overlapping lipid species that were enriched across sample types using a two-way Venn diagram. Analysis of oxygenated lipids from mouse myoblasts and hearts revealed six shared species of oxygenated lipids elevated in both TAZ-deficient models relative to controls, including MLCL(52:6)-2OOH, PE(36:4)-OOH, PC(38:5)-OH, PE-16:0/ONA, PC(38:6)-OH, PC(40:7)-OH (Fig. 4i). A similar analysis of overlapping species in human samples revealed seven oxygenated species common to both BTHS lymphoblasts and BTHS heart tissue: MLCL(54:6)-2OOH; MLCL(54:7)-2OOH; MLCL(56:7)-OOH; MLCL(56:8)-OOH; MLCL(54:5)-2OOH; MLCL(58:9)-OOH; MLCL(18:1/18:1/ONA) (Fig. 4j). No other oxygenated phospholipids were common to BTHS lymphoblasts and BTHS heart tissue. Thus, MLCL oxidation – likely occurring within MLCL/cyt c complexes – is a hallmark of the BTHS lipidome and may serve as a BTHS biomarker.
IOA suppresses MLCL/cyt c peroxidase activity
If MLCL/cyt c peroxidase activity is the primary driver of BTHS pathogenesis, its inhibitors should prevent the injurious effects of TAZ deficiency. We previously established that a mitochondria-targeted Fe-ligand, imidazole oleic acid (IOA), can reconstitute the hexa-coordinated low-spin Fe in the complex and inhibit its peroxidase activity30. Therefore, we tested whether IOA can suppress the MLCL-driven conversion of cyt c into a peroxidase.
IOA prevents MLCL/cyt c phospholipid peroxidation
We first asked whether IOA could convert the MLCL/cyt c complex to some non-peroxidase state in biochemical models. IOA attenuated the MLCL(L)3-induced changes in absorbance at 620 nm by 35% (Fig. 5a), consistent with interaction of the IOA imidazole moiety with the heme-Fe, and decreased the absorbance at 695 nm, suggesting that liganding of heme-Fe by imidazole occurred rather than the native Fe-M80 interaction (Fig. 5b). We demonstrated that effective inhibition of CL/cyt c peroxidase activity requires targeted delivery of IOA into mitochondria by conjugating it with triphenyl-phosphonium (TPP)30. TPP-IOA acted similarly to IOA alone, indicating that TPP had a negligible effect on the interaction of IOA with both MLCL(O)3/cyt c and MLCL(L)3/cyt c complexes (Extended Data Fig. 7a–b). Consequently, oxidation of N-benzyl-N-(tert-butyl)hydroxylamine by MLCL(O)3/cyt c peroxidase activity was also strongly suppressed by IOA (Fig. 5c). Furthermore, IOA and TPP-IOA effectively inhibited the MLCL(L)3/cyt c-induced accumulation of phospholipid oxidation products in a concentration-dependent manner, including HOO-MLCL(L)3 and HOO-PE as well as their oxidatively-truncated species (Fig. 5d, Extended Data Fig. 7c).
Figure 5. Effect of TPP-IOA on the structure of MLCL/cyt c peroxidase complexes, lipid oxidation.
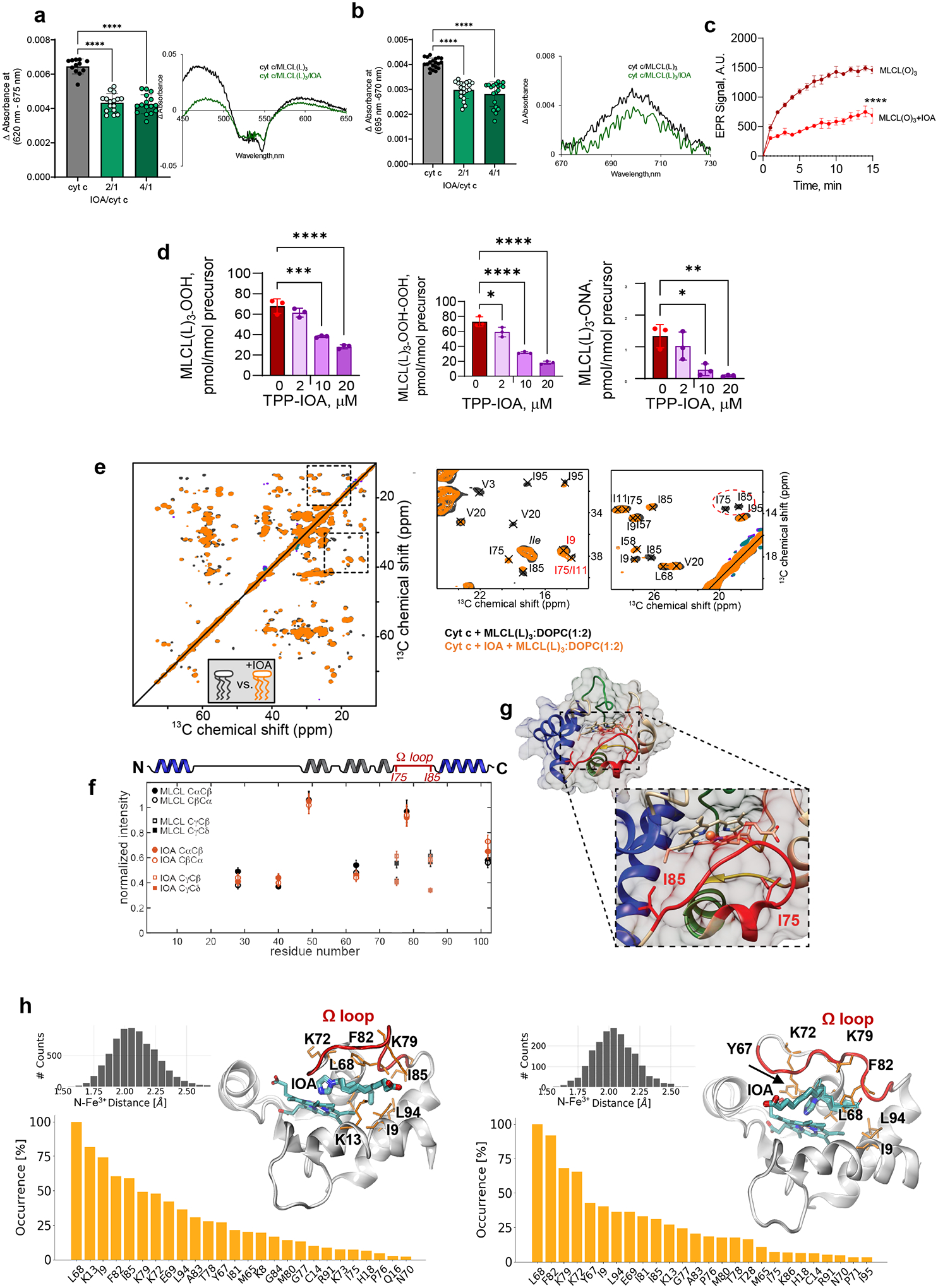
Effect of IOA on MLCL-dependent formation of heme-Fe high-spin forms (a) and the absorbance at 695 nm b. Insert: representative absorption spectra of MLCL/cyt c with or without IOA. Data are presented as mean values ± SD. N=11 (cyt c), N=7 (IOA/cyt c = 2/1), N=8 (IOA/cyt c = 4/1). Each data point represents a biologically independent sample. ****P<0.0001. One-way ANOVA, Tukey’s multiple comparison test. c, Time-courses of N-benzyl-N-(tert-butyl)nitroxide radical EPR signal formed by MLCL(O)3/cyt c in the absence (dark red) and presence (light red) of IOA (10μM). Data are presented as mean values ± SD. N=3. One-way ANOVA, Tukey’s multiple comparison test. ****p<0.001. Each data point represents a biologically independent sample. d, TPP-IOA inhibits accumulation of hydroproxy-MLCL species - MLCL(L)3-OOH (left panel), dihidroperoxy MLCL species MLCL(L)3-OOH (middle panel) and oxidatively truncated MLCL(L)3-ONA species (right panel) formed in the MLCL(L)3/cyt c driven reaction. N=3. Data are presented as mean values ± SD. One-way ANOVA, Tukey’s multiple comparison test. For MLCL(L)3-OOH ***p=0.001,****p<0.0001. For MLCL(L)3-OOH-OOH *p=0.032, ****p<0.0001. For MLCL(L)3-ONA *p=0.0109, **p=0.0042. Each data point represents a biologically independent sample. e, 2D CP-ssNMR spectrum (left) and zoomed regions (right) for labeled cyt c bound to DOPC:MLCL (2:1) vesicles in absence (black) and presence (orange) of 4x excess IOA. Dashed oval (right-most panel) marks peaks for Ile75/Ile85 that are missing in the IOA sample. f, Normalized peak intensity in the absence (black) and presence (orange) of IOA, for Thr Cα/β peaks and Cβ/γ/δ of Ile75 and Ile85. Differences indicate changes in local molecular motion. g, Image of the unbound fold of cyt c, with inset showing Ile75 and Ile85 (in sticks) and the Ω-loop (red) inbetween. h, Two different binding poses of IOA, bound to cyt c, obtained by docking simulations followed by MD runs: type 1 (7 runs, left panel) and type 2 (3 runs, right panel). Type 2 occasionally exhibited a flip to type 1 (the thin sticks showing the alternative conformation). The residues that were distinguished by their high contact frequencies (normalized with respect to the top-ranking residue) are displayed in the bar plots, in decreasing frequency of contacts. The distance between the N atom of the imidazole and Fe3+ ion is shown in the black histograms (inset: the last 100 ns of each set of simulations).
ssNMR reveals IOA localization at heme cavity
In the presence of IOA, 2D ssNMR revealed a specific set of residue-specific spectral changes in MLCL-bound cyt c (Fig. 5e; orange) compared to control (black). Most notable were changes in peak position and intensity for residues I9, I75, and I85 (dashed ovals). Changes in peak position indicate changes in local structure and/or dynamics, whereas the intensity changes stem from a change in local motion. Elsewhere in the protein, the (Thr) peak intensities (Fig. 5f, Extended Data Fig. 7d) revealed minimal changes, meaning that the IOA did not modulate overall protein motion and showing the high reproducibility in the measurements. In contrast, the clear IOA-induced reduction in the intensities of the I75 and I85 peaks indicates increased local motion near those amino acids located in the Ω-loop D. Thus, we were able to see by NMR the localized impact of IOA on hydrophobic residues in the protein’s Ω-loop D (I75-I85), near the heme cavity (shown in red in Fig. 5g).
MD simulations of MLCL/cyt c complexes plus IOA
We further analyzed the conformational dynamics of the cyt c-IOA complex (Fig. 5h). The initial configuration of the system was obtained by molecular docking simulations of IOA onto an open conformation of cyt c (Fig. 2b, 950 ns). Docking simulations usually identify the binding poses of small molecules onto target proteins provided that those sites are solvent-accessible, but those binding events involving the insertion of the small molecule into relatively buried (cryptic) pockets require prior opening of the target protein pocket to expose the binding site. In this case, the disruption of the Fe-S bond was required for the insertion of IOA into the close proximity of the catalytic Fe; hence we used the partially relaxed/unfolded conformation of cyt c obtained at the end of the 950 ns simulations (see Fig. 2b). This permitted us to target the vicinity of the Fe-S bond, and visualize the mechanism of binding of IOA to cyt c. Similar to a previous study30, we observed that L68, K13, and Ω-loop D had the most significant effect on the coordination of IOA by cyt c residues. However, current simulations indicated a shorter distance of 2.1 ± 0.2 Å between the imidazole N and the catalytic Fe in two alternative bound conformations of IOA (types 1 and 2 binding, with the negatively charged end of IOA facing opposite directions; see the left and right panels in Figure 5h, inset). This distance is even shorter than that of the Fe-S bond (~2.5 ± 0.1 Å). This indicates that IOA interacts with cyt c by utilizing the imidazole group as the sixth ligand to the heme-Fe, and that the conformational flexibility near the Ω-loop D and the overall relaxation of cyt c achieved in MD simulations is critical to expose additional crucial residues for IOA coordination, such as I9, K72, K79, and Y67 (type 1).
Mitochondria-targeted TPP-IOA rescues fatigue in TAZ-deficient flies
The pivotal test of our hypothesis was to determine if suppression of MLCL/cyt c peroxidase activity ameliorates the pathological outcome of BTHS. Therefore, we examined the potential of TPP-IOA to rescue motor dysfunction in the Drosophila model of BTHS. Previous studies reported that Drosophila TAZ mutants exhibit motor weakness31, and this powerful functional model has been utilized previously to test therapeutifc modalities for BTHS32. TAZ889 mutant flies showed reduced endurance relative to W1118 control flies (Fig. 6a). Detailed oxy-lipidome comparison between WT and mutant flies indicated that impaired endurance was associated with a decrease in CL and increase in MLCL (Fig. 6b, Extended Data Fig. 8a–b). TAZ deficiency resulted in a greater diversity of oxidizable PUFA-CL/MLCL species. Specifically, an increased number of CL and MLCL species containing C18:2 and C18:3 fatty acids were detected in TAZ-deficient flies (Fig. 6c, Extended Data Fig. 8c–d). Furthermore, mutant flies showed an accumulation of lipid peroxidation products, including oxygenated MLCL, PE, PC (Fig. 6d), and oxidatively truncated MLCL and PE species (Fig. 6e). Importantly, TPP-IOA had a strong positive effect on the endurance of flies. TAZ889 flies fed TPP-IOA for either 10 or 17 days showed markedly enhanced endurance relative to vehicle-fed controls (Fig. 6f, upper and lower panel, respectively). TPP-IOA did not affect the endurance of control flies (Extended Data Fig. 8e). TPP-IOA also restored levels of both CL and MLCL in mutant flies (Fig. 6g) and reduced levels of lipid peroxidation products (Fig. 6h). In contrast, TPP-IOA administration had no effect on CL and MLCL content in control flies (Extended Data Fig. 8f). No toxic effects of TPP-IOA were observed in experiments with Drosophila flies, and we noted no apparent loss of endurance in WT flies that received TPP-IOA, suggesting that oxidative phosphorylation was not inhibited by our treatment regimen.
Figure 6. . TAZ-deficiency induces changes in Drosophila endurance and the Drosophila lipidome.
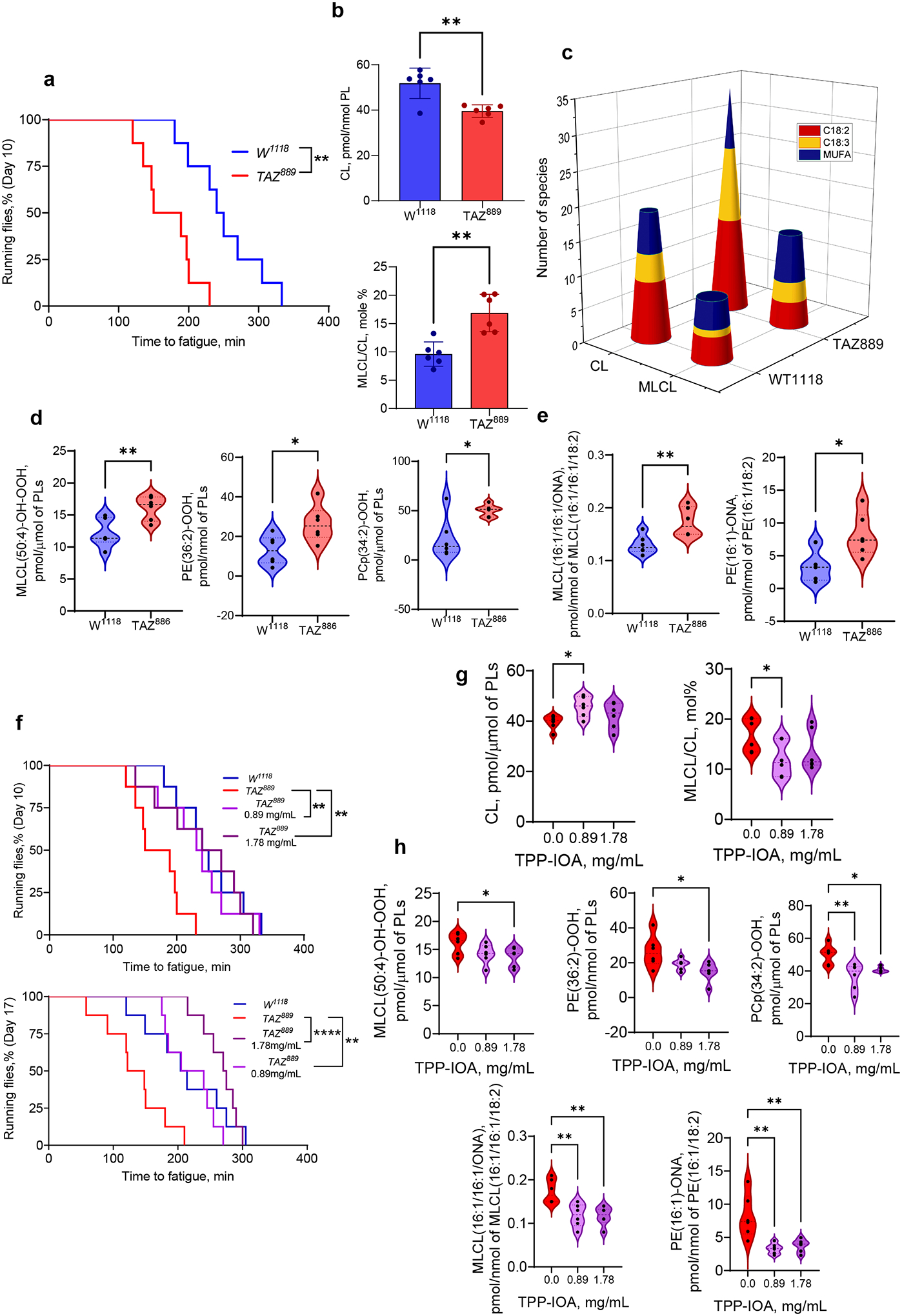
a, TAZ889 mutants have reduced endurance relative to control w1118 flies. Time to fatigue was quantified as the time when less than 20% of flies in a vial were still running. Each vial of 20 flies was treated statistically as a single independent replicate (N = 8 biological replicates for each group). Significance was determined by log-rank analysis. **p=0.0027. b, Content of CL (upper panel) and MLCL (lower panel) in control w1118 and TAZ889-deficient flies. Data are presented as mean values ± SD. CL, *P=0.0020; MLCL/CL-OOH,*P=0.0022, unaired two-tailed t-test. c, Quantitative characterization of CL and MLCL molecular species containing poorly oxidizable monounsaturated (MUFA) fatty acids and readily oxidizable polyunsaturated fatty acids (PUFA), C18:2 and C18:3 in control w1118 and TAZ889-deficient flies. TAZ deficiency results in accumulation of oxygenated (d) and oxidatively truncated phospholipids (e). Data are presented as mean values ± SD. MLCL(50:4)-OH-OOH, **P=0.0069; PE(36:2)-OOH, **P=0.0190; PCp(34:2)-OOH, *P=0.0112; MLCL(16:1/16:1/ONA), **P=0.0087; PE(16:1)-ONA, *P=0.0215, unpaired t-test. f, TPP-IOA improves the endurance of TAZ889 mutants. Each vial of 20 flies was treated statistically as a single independent replicate (N = 8 biological replicates for each group). Significance was determined by log-rank analysis. Age day 10: p-value for 0.89mg/ml = 0.0097, P-value for 1.78 mg/ml = 0.0257. Age day 17: p-value for 0.89 mg/ml = 0.0032, p-value for 1.78 mg/ml = <.0001. In graph, , **p<0.01,****p<0.0001 g, Content of CL (left panel) and MLCL (right panel) in TAZ889-deficient flies after feeding TPP-IOA. Data are presented as mean values ± SD. CL, *P=0.0121; MLCL, *P=0.0313, unpaired two-tailed t-test; h, TPP-IOA protects lipids against oxidation induced by TAZ-deficiency. Data are presented as mean values ± SD. One-way ANOVA, Tukey’s multiple comparison test. MLCL(50:4)-OH-OOH, *P=0.0458; PE(36:2)-OOH,*P=0.0213; PCp(34:2)-OOH, *P=0.0339, **P=0.0032; MLCL(16:1/16:1/ONA), **P=0.0049 vs TPP-IOA 0.89, **P=0.0039 vs TPP-IOA 1.78; PE(16:1)-ONA,**P=0.0022 vs TPP-IOA 0.89, **P=0.0046 vs TPP-IOA 1.78. Lipidomic analysis was performed using 6 vials (N=20 fly torsos per vial). For all violin plots presented, individual points including maximal and minimal are shown as black circles. Dashed black line indicates median and doted lines indicate quartiles.
IOA improves respiratory parameters in TAZ KO cells
In agreement with our hypothesis, we found that TAZ-deficiency was associated with impairment of several mitochondrial respiratory parameters, which were positively affected by the treatment of TAZ-KO C2C12 cells with the peroxidase inhibitor, IOA (Fig. 7a–e). Notably, increases in these parameters were also detected in WT C2C12 cells (Fig. 7). No toxicity was observed after treatment of C2C12 cells with IOA in sub-micromolar concentrations (employed as a complex with fatty acid free BSA) over the course of three days. To further characterize functional impairments of mitochondria in TAZ-deficient C2C12 cells, we performed measurements of the mitochondrial membrane potential of WT and TAZ-KO cells using fluorescence of tetramethylrhodamine methyl ester (TMRM), a cell-permeant dye that accumulates in mitochondria in proportion to their membrane potential33. Using flow-cytometry, we found a significant decrease in the membrane potential of TAZ-KO C2C12 cells, detected as a shift in the number of cells with lowered mean fluorescence intensity compared to WT cells (Fig. 7f). Treatment with IOA decreased this shift in both WT and TAZ-KO cells, reflecting an increase in the number of cells with a higher membrane potential (Fig. 7f–g).
Figure 7. Bioenergetic characteristics, mitochondrial membrane potential of WT and TAZ-KO cells and the effect of treatment with MLCL/cyt c peroxidase complex inhibitor IOA.
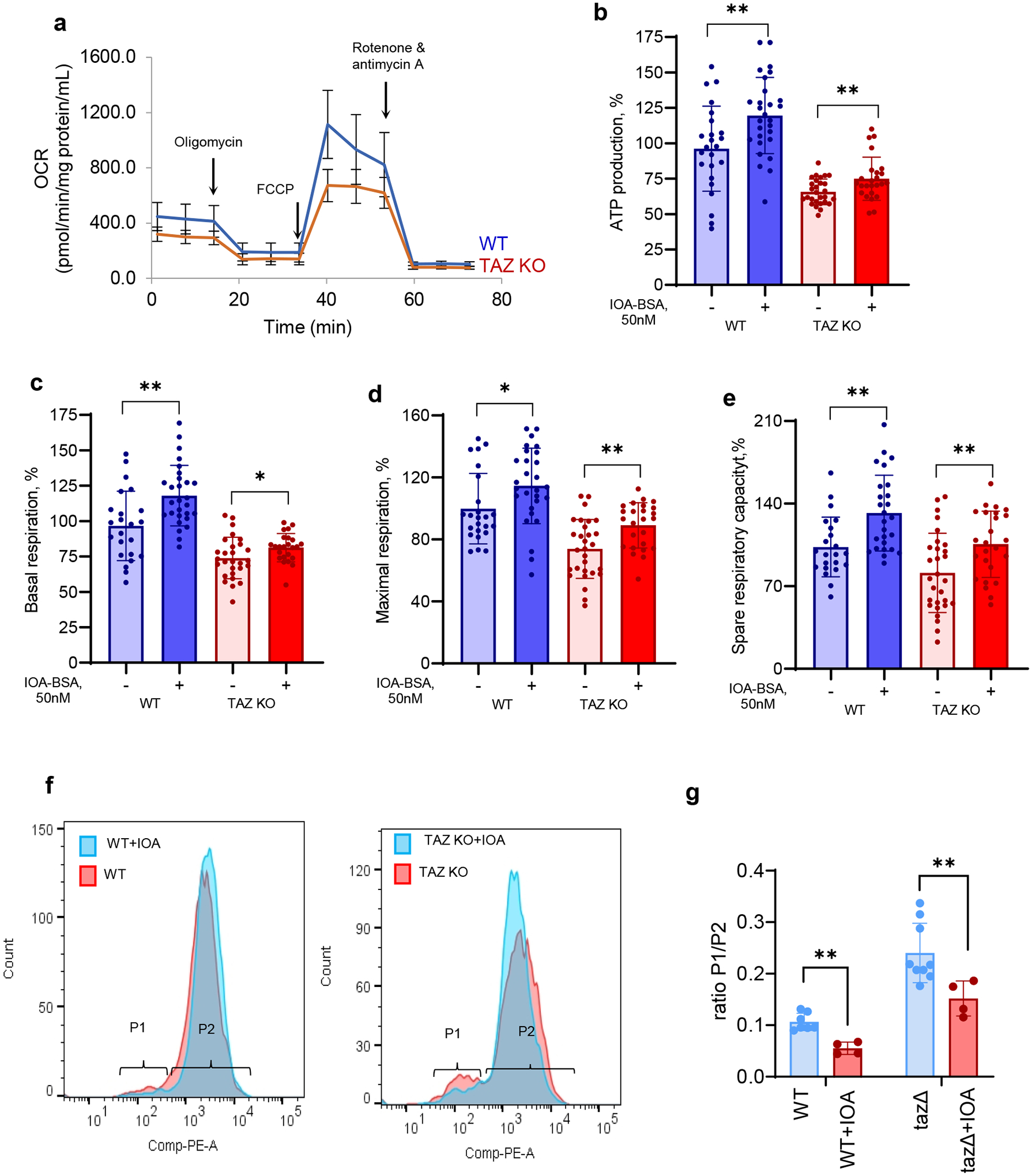
a, Oxygen consumption rates (OCR) assessed by Seahorse XFe96 Extracellular Flux Analyzer in wild type and TAZ KO C2C12 myoblasts. b, ATP-linked respiration (ATP production), represented by a decrease in OCR following inhibition of ATP synthase by oligomycin, was lower in TAZ-KO vs WT cells (p<0.0001). IOA increased ATP production in WT and TAZ KO cells vs respective untreated cells. c, Compared to WT, TAZ-KO cells had significantly lower basal respiration (p=0.0201) measured before the addition of the ATP synthase inhibitor, oligomycin. IOA increased basal respiration in WT and TAZ KO cells vs respective untreated cells. d, Maximal respiration measured after the addition of the uncoupler carbonyl cyanide-p-trifluoromethoxyphenyl-hydrazone (FCCP), was decreased in TAZ KO cells vs WT (p<0.0001). IOA increased maximal respiration in WT and TAZ KO cells vs respective untreated cells. e, Spare respiratory capacity calculated as the difference between maximal respiration and ATP-linked respiration was diminished in TAZ KO vs WT cells (p=0.0055). IOA improved spare respiratory capacity in WT and TAZ KO cells vs their untreated counterparts. Data are presented as mean values ± SD. *P< 0.05, **P<0.01, 2-tailed unpaired t-test, between untreated and treated cells from the same group (WT and TAZ-KO); two-way ANOVA followed by Šídák’s multiple comparisons test for comparisons between WT and TAZ-KO untreated cells (adjusted P values); a, N = at least 12 biological replicates (microplate wells seeded with 5,000 cells) from one experiment, 12 OCR readings over time for each well, 3 reading repeats at each point; N = at least 24 biological independent samples (microplate wells seeded each with 5,000 cells) from 2 independent experiments, 12 OCR readings over time for each well, 3 reading repeats at each point, each data point is the average of 3 X 3 = 9 readings, OCR values normalized by the protein content of each well independently and then expresses as a percentage of the mean of at least 25 WT wells with no treatment. f, Effect of IOA (1 μM) on mitochondrial membrane potential in WT and TAZ K/O C2C12 cells assessed by TMRM. g, Quantification was performed using percentage of cells in populations P1 and P2. Data are presented as mean values ± SD. Each data point represents a biologically independent sample. Statistical significance was analyzed using unpaire two-tailed t-test **p=0.0018 (WT vs WT+IOA), **p=0.028 (tazΔ vs tazΔ+IOA).
DISCUSSION
Many mitochondrial impairments have been implicated in the overall pathogenic mechanism of BTHS. However, these studies failed to identify a direct (i.e., primary) mechanistic link between them and the root cause of BTHS – deficient tafazzin transacylase activity – and instead only invoke disrupted mitochondrial structure/function as the precipitating cause. The main barrier to the development of effective therapies has been the gap in understanding the primary pathogenic mechanism underlying the disorder. Consequently, current therapies only reduce symptoms without curing the disease. The loss of CL and accumulation of MLCL - the major pathognomonic metabolic abnormality in BTHS - leading to ROS production and oxidative stress has been associated with the disease34. However, the mechanism linking these conditions to the pathology has not been identified and genotype/phenotype correlations have not been demonstrated. A recent elamipretide clinical trial1 showed no significant beneficial effect of the drug following the randomized, double-blind, placebo-controlled Part 1, although a limited beneficial effect was reported following the Part 2 open-label extension, in which all participants received the drug with no placebo control group35.
Our work discovered that the massive enrichment of MLCL makes it available to form complexes with the IMS hemoprotein, cyt c, thus converting the latter into an anomalous peroxidase capable of catalyzing peroxidation of mitochondrial PUFA-phospholipids. We identified the mechanisms that initiate MLCL/cyt c complex peroxidase competency. Normally, cyt c maintains the hexagonal organization of its heme-Fe, which does not support peroxidase activity, but is optimized to act as a shuttle of electrons between mitochondrial complexes III and IV36. Disordering or denaturing of the protein (e.g., by nitration, phosphorylation, and sulfoxidation of specific amino acids, high temperature, acids, bases, and anionic lipids) results in the transition of the protein into a penta-coordinate high-spin form whereby the heme-Fe is capable of interactions with H2O2 and organic (including lipid) hydroperoxides37–42. This is accompanied by a gain of peroxidase function43, 44. Interestingly, negatively charged hydroperoxy-phospholipids, particularly CL and MLCL, are effective in causing these structural re-arrangements of cyt c17, 45. Furthermore, hydroperoxy-CL can also be utilized as a source of oxidizing equivalents, feeding the peroxidase reaction of the cyt c/CL complexes46. As a result, peroxidized CL (and MLCL) formed by the peroxidase activity of the complex may effectively propagate the peroxidation reaction without any additional supplementation with H2O2.
We demonstrated that MLCL-induced changes in cyt c dynamics are accompanied by rupture of the Fe-S-M80 bond. This promotes the transition of the heme-Fe to a penta-coordinated, high spin configuration that permits access of H2O2 and other hydroperoxides (including hydroperoxy-lipids) to the heme catalytic site47, 48. ssNMR results and computational MD simulations support the dynamic structural impact of MLCL binding, most notably mobilization of the M80-containing Ω−loop of cyt c. Formation of MLCL/cyt c complexes with anomalous peroxidase activity leads to toxic phospholipid peroxidation compatible with two specific features of TAZ deficiency established by redox lipidomics: i) molecular diversification of MLCL and CL with appearance of readily oxidizable PUFA species, and ii) accumulation of peroxidation products, including primary hydroperoxy-derivatives and secondary oxidatively-truncated degradation products. Formation and accumulation of MLCL and CL peroxidation products in which the hydrophobic acyl chains contain polar oxygen functionalities disrupt normal hydrophobic interactions within the membrane. Furthermore, oxidatively-truncated CL and MLCL species covalently modify mitochondrial proteins causing their “lipoxidation”49, 50.
Not only does MLCL accumulate in TAZ-deficient cells, but its localization also differs from that of CL in WT cells, making it more readily accessible to form peroxidase complexes with cyt c. Given the location of TAZ on the outer leaflet of the IMM, it is assumed that CL must be flipped from the inner leaflet of the IMM (where it is synthesized) to the outer leaflet to undergo remodeling by TAZ51, 52. The absence of functional TAZ may preclude further translocation of its substrate, MLCL, away from the site where it is typically processed. This suggests that in TAZ-deficient cells, MLCL may accumulate in the outer leaflet of the IMM, immediately adjacent to the location of cyt c in the IMS. In contrast, CL is thought to be enriched in the inner leaflet of the IMM, facing away from cyt c53. Moreover, prior work has shown that MLCL is not as potent as CL in binding to mitochondrial membrane protein complexes54, 55. One implication of such a reduced binding affinity is that MLCL would be more freely available in the IMM (unlike CL that is mostly sequestered within mitochondrial protein complexes). The increased amount of unbound MLCL contributes to destabilization of the IMM, facilitating enhanced lipid scrambling due to increased dynamic disorder as detected also by our 31P ssNMR measurements.
Our study shows that one of the primary substrates of MLCL/cyt c peroxidase complexes is CL, yielding peroxidized CL molecules (CLox). CLox species have been identified as apoptosis cell death signals17 and production of CLox has been shown to disrupt multiple cellular pathways/ functions. For example, CL in the IMM binds and stabilizes respiratory chain complexes, but when CL becomes oxidized, this interaction is diminished, resulting in a concomitant reduction in respiratory chain function56. Similarly, increased levels of CLox have been shown to disrupt other fundamental mitochondrial parameters that have been implicated in BTHS pathology, including regulation of cyt c-mediated apoptosis, mitochondrial membrane potential and structure57–59.
TAZ-deficiency associated peroxidation (catalyzed by the MLCL/cyt c peroxidase) is not limited to CL and MLCL but also affects other mitochondrial PUFA-phospholipids, including PE, PC, PI and PG. Comparison of the oxidized PUFA-phospholipids profiles in BTHS vs. control cells showed that: a) in human lymphoblasts, which contain a limited number of mitochondria, only 8 out of 17 oxygenated phospholipid species were represented by oxidized MLCL and CL species; b) in human heart where myocytes are enriched with mitochondria, 20 out of 27 oxygenated species were represented by oxidized MLCL and CL species. Notably, in a model system, while oxygenated species of PUFA-PE and PUFA-PC were formed in PUFA-MLCL/cyt c catalyzed reaction, oxidative metabolites of PUFA-MLCL were predominant. When PUFA-MLCL(L)3 was substituted with non-oxidizable MLCL(O)3, the oxidation of PUFA-PE and PUFA-PC was significantly increased. Thus, MLCL/cyt c complexes formed in BTHS mitochondria can oxidize not only MLCL and CL but also other membrane PUFA-phospholipids. Oxygenated phospholipids and their downstream degradation products modulate a host of cellular functions/pathways implicated in BTHS pathology.
Mitochondria damaged by oxidatively-modified phospholipids are usually eliminated by mitophagy in which externalization of CL to the mitochondrial surface play a prominent role60. The pool of “free” CL, markedly depleted by TAZ-deficiency, may be insufficient for the fulfillment of this essential signaling function, resulting in the observed defective mitophagy61, 62 and documented by massive enlargement of lysosomes and accumulation of autophagic vacuoles wrapped by mitochondria in hearts of TAZ deficient mice62. In line with this, our data revealed elevated levels of endolysosomal phospholipid biomarkers, BMP29 in TAZ-deficient C2C12 myoblasts. Of note, enhanced biosynthesis of BMP was observed in lymphoblasts of BTHS patients63. Failure to eliminate damaged mitochondria in an attempt to rescue TAZ-deficient cells may prompt regulated death pathways, particularly apoptosis, as has been described in TAZ deficient mice64. This may be further enhanced by high levels of MLCL known to interact with pro-apoptotic tBID65.
Tissue specificity for the proposed peroxidation mechanism likely results from two factors: 1) tissue-based differences in mitochondrial abundance and expression of tafazzin66, and 2) the relative diversity/abundance of PUFA-phospholipids in various tissues. The highest levels of tafazzin expression are normally seen in cardiac/skeletal tissue, bone marrow, spleen, and brain66, which can translate into more MLCL species in these tissues as a result of TAZ deficiency. The MLCL:CL ratio is both significantly increased and diversified in a tissue-specific manner in BTHS patient cells relative to control cells (0.0–0.2 in control cells vs. 0.4–100 in BTHS patient cells)54, 67. Therefore, accumulation of peroxidation products, would also differ among tissue types and thus cause tissue-selective responses to the MLCL/cyt c activity seen in BTHS. This indicates that generation of MLCL/cyt c complexes can lead to tissue-specific responses in BTHS.
Given the very high level of phospholpid polyunsaturation in the brain, one may suggest that TAZ deficiency should have neurological consequences. Indeed, robust accumulation of CL/MLCL metabolites along with significant memory deficiency have been observed in the brain of Taz-KD mice68. Moreover, several studies have demonstrated major neuropsychological and neurophysiological phenotypes in BTHS patients, including difficulties in sensory perception, fatigue, and cognitive and psychological challenges69, 70. BTHS patients exhibit specific cognitive phenotypes such as lower visual spatial skills and impaired mathematical ability. Moreover, mathematical weakness seemed to worsen with age71, 72.
There are several models of BTHS. Although each model has unique characteristics, all models show mitochondrial dysfunction. Homozygous flies lacking full length tafazzin transcripts exhibit mitochondrial cristae abnormalities and a nearly 80% reduction in CL levels, similar to that observed in BTHS patients31. As observed in BTHS cells, CL and MLCL in the Drosophila BTHS model are represented by oxidizable PUFA-phospholipids with two and three double bonds. In addition, the tafazzin-deficient Drosophila model is characterized by pronounced motor weakness and reduced endurance – two of the most impactful clinical phenotypes reported by BTHS patients73, 74. Using this model, we demonstrated that TPP-IOA was effective in suppressing PUFA-phospholipid peroxidation and preventing fatique.
The nature and specific role of the CL re-acylation machinery is not fully understood. While tafazzin is acyl-CoA-independent acyltransferase75, acyl-CoA-driven exchange mechanisms are also utilized for CL remodeling, including MLCLAT-1 localized to mitochondria76, ALCAT-1 localized to ER and mitochondria77, and mitochondrial trifunctional protein hydratase subunit A78. Expression of ALCAT-1 is associated with oxidative stress and implicated in the mitochondrial etiology of diseases related to mitochondria dysfunction79. It has also been suggested that α/β hydrolase domain 2 plays a role in the synthesis, turnover, or remodeling of phospholipids, including CL80. Therefore, one or more acyl-CoA-dependent acylation mechanisms may be involved in restoration of CL/MLCL content in TPP-IOA treated tafazzin-deficient flies.
Our work established that enhanced lipid peroxidation, initiated by MLCL/cyt c complexation, is characteristic of all models of TAZ-deficiency employed in the study and engages all major classes of mitochondrial membrane phospholipids. Notably, lipid peroxidation was determined to be a phenotypic characteristic of the BTHS fly model. The fact that IOA has a protective effect on endurance in TAZ-deficient flies strongly suggests that inhibiting lipid peroxidation leads to an increased capacity of skeletal/cardiac muscle activity and decreased fatigue. This is further supported by our demonstration that physiologically relevant concentrations of IOA’s preserve bioenergetic functions of mitochondria in TAZ-deficient C2C12 myoblasts. Notably, high supra-physiological TPP-IOA concentrations (>5 μM), can perturb oxidative phosphorylation and reduce phosphorylation coupling efficiency in vitro as has been noted in our work and found by other workers in the field81–85.
In conclusion, the robust effects of IOA and its mitochondria-targeted derivative TPP-IOA, including mitigation of phospholipid peroxidation and mitochondrial respiration in both in vitro and in vivo models, improved endurance in the in vivo fly model along with mechanistic data showing the striking effectiveness of these compounds in suppressing MLCL/cyt c complex peroxidase activity, strongly support our hypothesis that formation of MLCL/cyt c complexes acts as the primary pathogenic mechanism of BTHS and highlights these molecules as a prototype for new therapies.
METHODS
Saccharomyces cerevisiae strains
The S. cerevisiae WT, taz1Δ, and cyc1Δ yeast strains used in this study were from the BY4741 genetic background (MATa) obtained from the Invitrogen yeast deletion collection (cat. #: 95401.H2). All mutant strains were authenticated by PCR prior to performing the experiments. taz1Δcyc1Δ mutant strains were generated through genetic crossing between taz1Δ and cyc1Δ yeast cells. The resulting taz1Δcyc1Δ mutants were further confirmed by PCR. Yeast expression plasmids pYES2-EV (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) (empty vector) and pYES2-desa (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) (containing the H. brasiliensis Δ12-desaturase gene) were kind gifts from Dr. Gasparovic (Rudjer Boskovic Institute, Zagreb, Croatia).
Mouse C2C12 myoblasts
The TAZ-KO cell line was generated by CRISPR-mediated inactivation of TAZ in a commercially available mouse C2C12 myoblast cell line (ATCC, cat. #: CRL-1772). Disruption of TAZ exon 1 was validated by PCR and genomic sequencing, and loss of TAZ protein expression was validated by immunoblotting prior to performing experiments. Wild-type C2C12 cells (from which the TAZ-KO mutant was derived) represent an isogenic control for the TAZ-KO cell line. Generation and validation of the TAZ-KO C2C12 cell line have been previously described19, 24.
Drosophila melanogaster strains
TAZ889 flies were generated using CRISPR/Cas-9 mediated genomic editing by homology-dependent repair resulting in deletion of nucleotides 8–896 of the TAFAZZIN gene. In place of the deletion, a red fluorescent protein marker was knocked in to track the presence of the mutation during cross schemes. The CRISPR plasmid containing the deletion was injected into a w1118 background and that progenitor line was assessed next to TAZ889 as a genetic background control. The line was validated by PCR and genomic sequencing and BLAST results confirmed the deletion and RFP insertion. Generation and validation of the Drosophila TAZ889 strain have been previously described74.
Fly Maintenance: Flies were kept on a standard 10% yeast and 10% sugar diet and fed either drug or vehicle food. All experimental flies were collected within a 72-hour period and within 24 h of enclosing (N=160 per experimental group). Males were used exclusively for these experiments because the automated exercise machine is optimized for males When flies were 4 days old they were switched to drug food (1.78 mg/mL or 0.89mg/mL) or vehicle food. Drug food was created by dissolving 8.9 mg of TTP-IOA in 1 mL of water with 25% ethanol. Dilutions were made from the stock (1:5 and a 1:10) to achieve 1.78 mg/mL and a 0.89mg/mL solution. 50μl of either concentration was dropped onto the surface of the food. Flies were fed drug or vehicle food for a full five days and on age Day 10 their endurance was tested.
Endurance: The Power Tower is an automated machine that stimulates the flies to run upwards in their vial. Endurance was measured by allowing flies to run, using an automated machine that stimulates them to run upwards in their vial until exhaustion. A vial was considered fatigued when less than 20% of the flies were still running upwards in the vial. Sample sizes of 8 vials for each cohort were not chosen by power analysis but are consistent with numbers from previous experiments73, 74. Pair-wise log-rank tests were performed to determined the significance.
Data distribution was assumed to be normal but was not formally tested. Vials were scored for fatigue by experimenters who were blinded to the treatment and genotype group.
BTHS lymphoblast cell lines
Three BTHS lymphoblast cell lines (GM22164, GM22150, and GM22129) derived from clinically affected patients and characterized by loss-of-function mutations in TAZ, along with three lymphoblast lines derived from apparently healthy demographic-matched control individuals (AG14840, AG14798, AG14725) were obtained from the Coriell Institute for Medical Research (Camden, NJ). These cell lines were validated prior to submission to the Coriell Institute.The TAFAZZIN mutation associated with each BTHS patient cell line is as follows: GM22164: Gln233Ter; GM22150: Trp79Ter; and GM22129: IVS1+5G>A.
Mouse model of BTHS and heart tissue
The mouse experiments were performed with approval of the University of Manitoba Animal Policy and Welfare Committee which adheres to the principles for biomedical research involving animals developed by the Canadian Council on Animal Care and the Council for International Organizations of Medical Sciences and was reported in accordance with ARRIVE guidelines. The animal strain used was C57BL/6J mice, the animal protocol number from the University of Manitoba is B2019–030/1 (AC11468), 4 month old animals were used for all studies and randomized and pooled from separate litters usually 10 per group (WT and TazKD), sex was considered in the design of the experiments as only male mice with one X chromosome develop Taz deficiency. Mice were maintained in an environmentally controlled facility (22°C, 37% humidity, 12 h light/dark cycle) with access to food (diet: TD.01306 from Harlan, Indianapolis, Indiana, USA) and water ad libitum. TAZ deficient animals were generated by mating male transgenic mice (strain B6.Cg-Gt(ROSA)26Sortm1(H1/tetO-RNAi:Taz,CAG-tetR)Bsf/ZkhuJ, Jackson Laboratory, Bar Harbour, ME) containing a doxycycline (DOX) inducible TAZ-specific short-hair-pin RNA (shRNA) with female C57BL/6J mice (Jackson Laboratory, Bar Harbour, ME). Knockdown of TAZ was induced in utero by administration of DOX (625 mg of DOX/kg of chow) as part of the standard low-fat 6% (w/w) DOX-containing rodent chow (Envigo Teklad, Rodent diet cat# TD.01306) to female C57BL/6 mice both before mating and during pregnancy. TAZ-deficiency is initiated in utero to maximize the duration of TAZ knockdown27. Dams with litters continued to receive the DOX diet to maintain TAZ knockdown in the offspring for the entire suckling period. Male offspring were then weaned at 3 weeks of age onto the DOX-containing diet. In addition, some dams and corresponding male offspring were fed a low-fat (6%, w/w) standard rodent chow not treated with DOX and served as standard wild-type animals. Male mice positive for the TAZ shRNA transgene were identified by PCR using primers (5’-CCATGGAATTCGAACGCTGACGTC-3’ forward, 5’-TATGGGCTATGAACTAATGACCC-3’ reverse). At 4 months of age hearts from wild-type and TAZ knockdown mice were harvested and frozen at −80°C prior to analysis. No animals and data points were excluded from the analysis.
Human heart tissue samples
Ascertainment of human myocardial samples was approved by the University of Pittsburgh Institutional Review Board (IRB) protocol STUDY19070300. Patients and patient caregivers provided informed written consent. Discarded human myocardial samples were collected from patients undergoing heart surgery or heart transplantation. In brief, samples were resected from the explantated diseased heart after heart transplantation. Samples from donor hearts that ended up not being used for transplantation were collected without recording of any patient identifiers. In these cases, clinical information was retrieved through an honest broker (approval through IRB-P00000126). Available clinical and demographic characteristics of the non-failing controls and patients are presented in Extended Data Fig 5a.
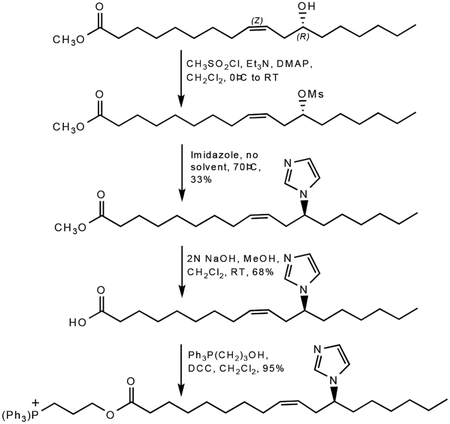
Synthesis of imidazole-substituted fatty acid TPP-IOA
The synthesis of TPP-IOA relied on the ready availability of the naturally occurring fatty acid (9Z,12R)-(+)-ricinoleic acid. The 12-OH group offered a site for heterocycle substitution on a long chain fatty acid without having to prepare new starting materials. Briefly, methyl rincoleic acid was mesylated on the 12-hydroxy group (CH3SO2Cl, Et3N, 4-dimethylaminopyridine (DMAP); CH2Cl2, 0°C to RT). The crude mesylates were used without purification as they tended to decompose when chromatographed on silica. Substitution of the mesylate by imidazole could be affected in tetrahydrofuran (THF) solution using potassium tert-butoxide and 18-crown-6, but almost identical yields could be obtained by simply heating the mesylates with 2–3 equivalents of imidazole at ~ 80–90°C in the absence of solvent. Hydrolysis of the methyl esters with NaOH in a mixture of MeOH/CH2Cl2 afforded the free carboxylic acids that could then be esterified with 3-hydroxypropyltriphenylphosphonium bromide (DCC, CH2Cl2). Overall yields were about 20% from the starting fatty acid methyl esters. The synthetic scheme for TPP-IOA is shown below.
9-(Z)-(3-(12-imidazol-1-yl)octadeca-9-enoyloxy)propyl)triphenylphosphonium bromide
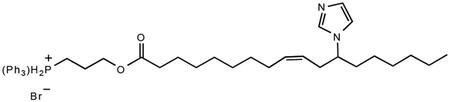
3-Hydroxypropyltriphenylphosphine (876.5 mg, 2.46 mmol), N,Ń-dicyclohexylcarbodiimide (485.6 mg, 2.35 mmol) and 12-imidazol-1-yl-(Z)-9-octadecaenoate (816.7 mg, 2.34 mmol) were dissolved in 50 mL dichloromethane. The mixture was allowed to stir at room temperature overnight. The mixture was filtered using Celite to remove precipitated urea and evaporated to provide a crude product that was purified via column chromatography (dichloromethane : methanol 10:1) to yield 1.54 g (2.23 mmol, 95.3%) of a light yellow, thick oil. Rf = 0.58 (dichloromethane : methanol 10:1). 1H NMR (300 MHz, CDCl3) δ 7.90–7.65 (m, 15H), 7.54 (s, 1H), 7.03 (s, 1H), 6.93 (s, 1H), 5.37 (m, 1H), 5.14 (m, 1H), 4.34 (t, 2H), 3.98 (m, 3H), 2.43 (m, 2H), 2.22 (t, 2H), 1.98 (m, 2H), 1.85–1.72 (m, 4H), 1.51 (m, 2H), 1.25–1.15 (m, 18H), 0.81 (t, 3H); 13C NMR (CDCl3) δ 172.9, 134.82, 134.78, 133.2, 133.1, 133.0, 130.2, 130.0, 128.1, 123.4, 117.9, 116.8, 116.4, 62.7, 62.5, 58.4, 49.4, 34.9, 33.7, 33.6, 31.1, 28.8, 28.6, 28.5, 28.3, 26.7, 25.5, 24.3, 22.0, 21.8, 19.6, 18.9, 13.5 ; MS (FAB) m/z 651 (M+, 100%), 375 (11%), 319 (15%), 303 (12%), 289 (11%).
Preparation of liposomes
To study the peroxidase activity of cyt c/MLCL complexes, we utilized model systems with liposomes of different phospholipid composition. The three major goals of these experiments were to establish whether: i) oxidizable PUFA MLCL in the complex with cyt c are preferred substrates of peroxidation; ii) oxidizable PUFA phospholipids that do not readily form complexes with MLCL can be peroxidized by cyt c complexes with non-oxidizable MLCL species, and iii) PUFA phospholipids can be co-oxidized by peroxidase activity of cyt c complexes with PUFA-MLCL. Experimentally, these goals were addressed using three types of liposomes: liposomes of group I contained: DOPC/MLCL(L3) (2:1 ratio), liposomes of group II contained: DOPC/MLCL(O)3/SLPC (2:1:1 ratio), DOPC/MLCL(O)3/SLPE, DOPC/MLCL(O)3/SAPC (2:1:1 ratio), DOPC/MLCL(O)3/SAPE, (2:1:1 ratio), liposomes of group III contained: DOPC/MLCL(L3)/SLPC (2:1:1 ratio), DOPC/MLCL(L)3/SLPE, DOPC/MLCL(L)3/SAPC (2:1:1 ratio), DOPC/MLCL(L)3/SAPE, DOPC/MLCL(L)3//MLCL(O)3/SAPC (2:1:1:1 ratio), DOPC/MLCL(L)3//MLCL(O)3/SAPE (2:1:1:1 ratio). All liposomes were prepared by extrusion through a membrane with 100 nm pores in 25 mM HEPES buffer (pH 7.4) to a concentration of 100 μM in DTPA using a mini-extruder (Avanti Polar Lipids). Liposomes (100 μM) were incubated in the presence of cyt c (10 μM) and H2O2 (25 μM was added every 10 min) at 37°C. At the end of incubation, lipids were extracted and analyzed by LC/MS.
Absorption spectroscopy
Optical spectra were recorded using a UV160U spectrophotometer (Shimadzu, Japan). Samples in 25mM HEPES buffer (pH 7.4) 100 μM in DTPA were contained in a 50 μl cuvette, 0.3 mm pathlength. The final concentration of cyt c in the experiments was 75 μM. Incubations were performed at room temperature. Imidazole oleic acid (IOA) and TPP-imidazole oleic acid (TPP-IOA) dissolved in DMSO was added in ratios IOA/cyt c of 2 to 1 and 4 to 1. Cyt c was pre-incubated with IOA for 10 min before addition of liposomes. The baseline was subtracted from each individual spectrum before obtaining the differential spectra to minimize the interference of light scattering. For assessment of the formation of high-spin iron we calculated the absorbane at 620 nm by subtraction of the absorbance reading at 675 nm from the absorbance reading at 620 nm.
Oxidation of BODIPY C11 by cyt c/MLCL.
Cyt c (2 μM) was mixed with DOPC/MLCL liposomes containing bovine heart MLCL consist of 90% of MLCL(L)3 or MLCL(O)3 (MLCL/cyt c ratio=20:1) and incubated with BODIPY-C11 (1 μM) and H2O2 (25 μM) in 25 mM HEPES buffer (pH 7.4) containing 100 μM DTPA. Imaging reader Cytation 5 and UV160U spectrophotometer (Shimadzu, Japan) were used to mesure fluorescence of BODIPY-C11 and record its spectra at emission wavelength 495 nm after excitation at 515 nm, respectively.
WT, taz1Δ, cyc1Δ and taz1Δcyc1Δ (N = 4) yeast cells were grown overnight to the mid log phase. Cells (OD 1.0) were washed with PBS twice, stained with 2 μM BODIPY C11(Thermo Fisher Scientific) in PBS, treated with 50 μM tert-Butyl hydroperoxide (tBHP, Millipore-Sigma) and incubated at 30 °C for 30 minutes. Fluorescence intensity was measured using flow cytometry (Cytek Northern Lights) at the Microscopy, Imaging and Cytometry Resources Core, Wayne State University, School of Medicine. Median fluorescence intensity of single cells was analyzed by FlowJo software v10. No statistical methods were used to pre-determine sample sizes, but a sample size of three to four biological replicates has been deemed sufficient for statistical analysis of most flow cytometery data86. The gaiting strategy is presented on Supplementary Fig.1. Data distribution was assumed to be normal but this was not formally tested. Data collection and analysis were not performed blind to the conditions of the experiments.
Mitochondrial bioenergetics
WT and TAZ-KO C2C12 cellswere seeded in 6-well plates (150,000 cells/well) and treated with IOA (50 nM) in complete DMEM for 2 days. To prevent possible toxicity of IOA, it was delivered to cells as a complex with fatty-acid-free albumin (IOA:BSA, 3:1) – which was used in all incubations and assessments of bioenergetic parameters. Cells were then harvested and plated in a 96-well Seahorse assay plate (5,000 cells/well) and drug treatment was continued for an additional 3 days. On the day of the assay, medium was removed, and cells were treated with fresh 1 μM IOA solution in complete DMEM for 4 h. Controls were subjected to the same procedures with drug-free medium. For the Seahorse assay on day 5, complete DMEM was removed, cells (experimental and controls) were washed with XF DMEM assay medium without drug and incubated for 1 h in a non-CO2 incubator. Oxygen consumption rate (OCR) was measured in accordance with the XF Cell Mito Stress Test Kit protocol in a Seahorse XFe96 Extracellular Flux Analyzer. All media were devoid of pyruvate and contained glucose and L-glutamine as energy sources. Complete DMEM also contained 10% fetal bovine serum and antibiotics (penicillin + streptomycin).
Mitochondrial membrane potential
WT and TAZ- KO C2C12 cells were seeded in 24-well plates and grown in medium with or without IOA (1 μM) in the presence of fatty acid free bovine serum albumin (IOA:BSA, 3:1) for 3 days. On the day of the experiment, cells were detached by trypsinization, resuspended in DMEM without fetal bovine serum, and incubated in a CO2 incubator with 100 nM TMRM for 30 min. Cells were then washed one time with DMEM and resuspended in 500 μL of DMEM without fetal bovine serum. Cells were analyzed using a BD FACSCanto flow cytometer with excitation at 488 nm and an emission filter at 585 ± 42 nm (PE equivalent bandpass filter). Two independent experiments were performed with at least 24 biological replicates (microplate wells seeded each with 5,000 cells). For each well, there were 12 oxygen consumption rate (OCR) readings over time, with 3 reading repeats at each of these 12 points. OCR values were normalized by the protein content of each well independently and then expressed as a percentage of the mean of at least 25 WT wells with no treatment.
Redox phospholipidomics
Lipids were extracted using the Folch procedure, and phosphorus was determined by a micro‐method. Phospholipids were analyzed by LC/MS using a Dionex Ultimate 3000 HPLC system coupled on‐line to a Orbitrap Fusion Lumos mass spectrometer (ThermoFisher) using a normal phase column (Luna 3 μm Silica (2) 100 Å, 150 × 2.0 mm, (Phenomenex)). The analysis was performed using gradient solvents (A and B) containing 10 mM ammonium formate at a flow rate of 0.2 ml/min. Solvent A contained isopropanol/hexane/water (285:215:5, v/v/v), and solvent B contained isopropanol/hexane/water (285:215:40, v/v/v). The column was eluted for 0‐23 min with a linear gradient from 10% to 32% B; 23‐32 min with a linear gradient of 32%–65% B; 32‐35 min with a linear gradient of 65%–100% B; 35‐62 min held at 100% B; 62‐64 min with a linear gradient from 100% to 10% B; followed by an equilibration from 64‐80 min at 10% B. Analysis was performed in negative ion mode at a resolution of 140,000 for the full MS scan in a data‐dependent mode. Analysis of raw LC/MS data was performed using software package Compound Discoverer™ 2.1 (ThermoFisher) with an in‐house generated analysis workflow and oxidized PL database. Briefly, peaks with S/N ratio of more than 3 were identified and searched against oxidized PL database. Lipids were further filtered by retention time and confirmed by a fragmentation mass spectrum. Deuterated phospholipids (Avanti Polar Lipids) were used as internal standards. Values for m/z were matched within 5 ppm to identify the lipid species. All samples were analysed randomly.
EPR measurements of peroxidase activity
EPR spectra of N-benzyl-N-(tert-butyl)nitroxide were recorded at room temperature in gas-permeable Teflon tubing (inner diameter, 0.8 mm; thickness, 0.013; Alpha Wire Corp., Elizabeth, NJ) on a JES-FA 100 ESR spectrometer (JEOL, Kyoto, Japan) at X-band (9.4 GHz). The tubing (length, 20 cm) was filled with 90 μL of sample, double-folded, and placed in an open 3.0 mm (internal diameter) EPR quartz tube. The spectra were recorded under the following EPR conditions: center field, 335.08 mT; sweep width, 5 mT; microwave power, 10 mW; width modulation, 0.06 mT; time constant, 0.03 s; scan time, 30 sec. The magnitude of the EPR signal of N-benzyl-N-(tert-butyl)nitroxide was measured 1 min after the addition of 20μM H2O2 to the 5 μM cyt c, 100μM phospholipid and 100 μM N-benzyl-N-(tert-butyl)hydroxylamine in buffer solution (25 mM HEPES buffer, pH 7.4). Concentration of (Z)-12-(1H-imidazol-1-yl)octadec-9-enoic acid (IOFA) was 10 μM (phospholipid/IOFA/cyt c (20:2:1)).
Protein production, liposome preparation and solid-state NMR measurements
Human cyt c was produced in E. coli BL21 (DE3) cells using the pBTR-1 plasmid (AddGene Plasmid #22468), which comprises the CYCS and CYC3 genes, coding for human cyt c and yeast cyt c heme lyase, the latter needed for the correct heme integration in c-type cytochromes87. Uniformly 13C, 15N enriched cyt c was produced according to previously described protocols87. Cells were grown in minimal media containing 0.1% (w/v) 15NH4Cl and 0.4% (w/v) 13C d-glucose, 50 μM FeCl3 and 100 μM unlabeled δ-aminolevulinic acid. Briefly, protein purification was done using an HiTrap SP HP ion exchange chromatography column equilibrated with 20 mM sodium borate pH 8.5. Protein elution was performed with 1M NaCl. HPLC-ESI-MS analysis was used to confirm the intact molecular mass of the obtained protein. Oxidized cyt c was obtained by addition of a five-fold molar excess of K3Fe(CN)6 in 20 mM HEPES (pH 7.4) at room temperature for 20 min, followed by buffer exchange into 20 mM HEPES (pH 7.4) using Amicon ultracentrifugation filters with a 3 kDa cutoff. Protein concentration and completion of oxidation was determined by UV-Vis spectrophotometry analysis based on molar extinction coefficients Dioleoyl phosphatidylcholine (1,2-dioleoyl-sn-glycero-3-phosphocholine, DOPC; C18:1) and both bovine heart monolysocardiolipin (MLCL) and bovine heart cardiolipin (CL) were purchased from Avanti Polar Lipids (Birmingham, AL, USA).
Mixtures of MLCL/DOPC or CL/DOPC phospholipids were used to prepare unilamellar liposomes by extrusion,88. Stocks of phospholipids in chloroform were mixed in a 1:1 (or 1:2) molar ratio, dried under N2 gas and placed under vacuum overnight. The dried lipid films were resuspended in 20 mM HEPES buffer (pH 7.4), vortexed and exposed to 5 freeze-thaw cycles at −78°C (liquid-nitrogen bath) and 56°C (water bath). Finally, the lipid solutions were extruded using an Avanti mini-extruder through 200 nm polycarbonate membrane 21 times, to obtain large unilamellar vesicles (LUVs). LUVs were slowly added into cyt c stock solution to a final 1:25 P:L (protein-to-lipid) molar ratio. The resulting suspension was incubated and vortexed for ~15 min at room temperature. The protein-lipid complexes were transferred into 3.2 mm regular-wall MAS ssNMR rotors using a home-built packing tool89 via an ultracentrifugation step at 175,000 g, for 3 h at 4 °C (Optima L-100 XP ultracentrifuge with a SW-32 Ti rotor, Beckman Coulter). The same procedure was followed to prepare Cyt c/MLCL-containing liposomes complexes (1:40 P:L molar ratio) with and without the IOA inhibitor. IOA was added to the protein in a four-fold molar excess prior to its binding to the lipids, and MLCL/DOPC phospholipids were mixed in a 1:2 molar ratio.
The ssNMR measurements were performed on a AVANCE NEO 600 MHz (14.1 T) spectrometer from Bruker Biospin (Billerica, MA, USA). The 2D 13C-13C cross polarization (CP) dipolar-assisted rotational resonance (DARR) solid-state NMR spectra were acquired using a 3.2-mm Bruker EFree HCN MAS probe, at 10 kHz MAS and 253 K temperature. A 70%−100% ramped 1H–13C CP step, 25-ms DARR 13C–13C mixing, and 5 μs 90°carbon pulse and 2.5 μs 90° proton pulse were used. Two-pulse phase-modulated (TPPM) proton decoupling during acquisition was around 83 kHz, recycle delay set to 2.7 s and number of scans 64 per datapoint. 2D 15N-13C NCA spectra were acquired at 10 kHz MAS and 253 K temperature, using 100 kHz TPPM proton decoupling, 3 s recycle delay and 256 scans per datapoint. Here, the first step was a CP-MAS transfer from 1H to 15N with a 70–100% ramp on 1H, using rf fields of approx. 75 (1H) and 50 (15N) kHz, and optimized contact time of 1 ms. The magnetization was transferred from 15N to 13C in a second CP step using a 90%–100% ramp on 13C, and selectivity was achieved by setting the carrier frequency to be on-resonance with Cα and with an optimized contact time of 5 ms. A two-dimensional grid search was used to optimize the power level of the 13C and 15N channels. 2D 13C–13C spectra to detect the highly mobile carbons were obtained by combining refocused INEPT 1H–13C transfers with the P913 total through bond correlation spectroscopy (TOBSY) pulse sequence at 10 kHz MAS and 278 K22. 1H decoupling around 70 kHz was applied during evolution and acquisition, 2.5 s recycle delay and 64 scans per datapoint were used. The 1D 13C direct excitation, INEPT, and CP spectra were measured using 50 kHz 13C nutation frequency, 2.5 sec repetition delay, 512 scans, 0.015 sec acquisition time, 10 kHz spinning frequency and temperature set to 278K. During the acquisition time, proton TPPM decoupling was set at 83 kHz. 13C-1H 70–100 ramped MAS CP experiment was done using 1 ms contact time. The ssNMR spectra were processed and peaks were integrated with the NMRPipe software package90. Chemical shifts were referenced to 4,4-dimethyl-4-silapentane-1 sulfonic acid and liquid ammonia using adamantane 13C chemical shifts as an external reference88. NMR spectral analysis and peak assignment were performed in the CcpNmr software suite version 2.420 and NMRPipe software tools88. The molecular structure of cyt c was visualized using the UCSF Chimera program91.
Computational Methods
Molecular Dynamics Simulations and molecular docking
Full-atomic molecular dynamics (MD) simulations of horse heart cytochrome c structure (PDB code: 1hrc92) were performed in the presence of membrane, containing 50% 1,2-dioleoy-sn-glycero-3-phosphocholine (DOPC), 30% 1,2-dioleoy-sn-glycero-3-phosphoethanolamine (DOPE) and 20% monolysocardiolipin (MLC(L)3, MLCL) molecules. The initial system (Movie 1, initial frame) and inputs were prepared using CHARMM-GUI server93. The structure and parameters of MLCL structure were prepared based on the CL TLCL2 parameters for lipids defined in the CHARMM force field and heme group parameters were obtained from Autenrieth et al.94. MD simulations of cyt c were performed using the NAMD95 software. Simulation setup consisted of cyt c placed at a distance of 15 Å from the membrane and solvated with TIP3P water molecules. At the initial stage, we performed twenty independent MD runs for MLCL (100 ns each). Each independent MD run was first energy-minimized using steepest descent algorithm, followed by a short equilibration where the backbone atoms of the protein were restrained and the remaining atoms were allowed to relax for 2.5 nanoseconds (ns). This was followed by a production run of 100 ns each at constant temperature of 303 K and a pressure coupling of 1 barMD systems at 100 ns were sorted based on the binding interface with the membrane. Ten cyt c conformations in which the molecule diffused towards the bilayer and its positively charged amino acids and M80 interacted with MLCLs (Extended Data Fig. 2a, labeled in red) were further selected to be extended to 300 ns. In the simulations in which we observed strong interactions between M80 and MLCL, and given that the bond between M80 and heme-Fe is disturbed upon membrane binding46, we drastically weakened the Fe-S bond between M80 and the catalytic iron and extended simulations up to 950 ns (Extended Data Fig. 3c) which led to a complete rupture of the Fe-S bond.
Apart from the simulations in explicit membrane, we carried out four independent MD runs of cyt c in explicit water, including two in which the Fe-S bond was removed (Extended Dat Fig, 3b). Prior to productive runs which were 100 ns long, the following protocol was adopted: 0.2 ns of water equilibration, 10,000 steps of minimization, 0.35 ns of heating from 0 to 300 K, and 0.15 ns equilibration of the whole system. Simulations were performed with a cutoff of 12 Å for non-bonded interactions and Langevin piston algorithm to maintain the temperature at 300K and pressure at 1 atm.
Docking simulations of IOA inhibitor to cyt c were performed using SMINA96. Five runs were performed and the one with the lowest binding energy (highest affinity) was selected for further investigation. Among the conformations, we selected the one with the shortest distance between Fe3+ and N atom of imidazole (~3.5 Å). We further performed 200 ns MD simulations (2 independent runs) for cyt c-IOA complex on which we applied temporary restraints between Fe3+ and N atom of IOA imidazole so the IOA could optimize its interaction with the protein structure. After 200 ns, the restraints were removed and we performed ten additional 300 ns runs for the complex where we could observe stable interactions between the IOA imidazole and the heme group. Non-standard parameters were set based on the default CHARMM ff lipid paramaters (for MLCL), and Swiss-param97 with quantum chemistry calculations. We used and our in-house developed scripts implemented in the ProDy98 API for trajectory analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
Comparisons of means were performed using unpaired two-tailed Student’s test and one-way ANOVA with Tukey post hoc test. Dara are reported as mean ± S.D. unless otherwise stated.
Statistical analyses were performed using Graphpad Prism. SIMCA 16.1 software (Sartorius) was used to perform orthogonal projection of latent structures - discriminant analysis (OPLS-DA) of lipidomes data. The data were presented as a OPLS-DA score plot, S-plot derived from OPLS-DA that is indicated important variables and a variable importance in projection (VIP) score that is a measure of importance of variable in the OPLS-DA model. A variables with a VIP score close to or greater than 1.2 were considered important in given model.
Extended Data
Extended Data Fig. 1. SSNMR analysis of structural and dynamic rearrangements in the cyt c peroxidase complex with MLCL.
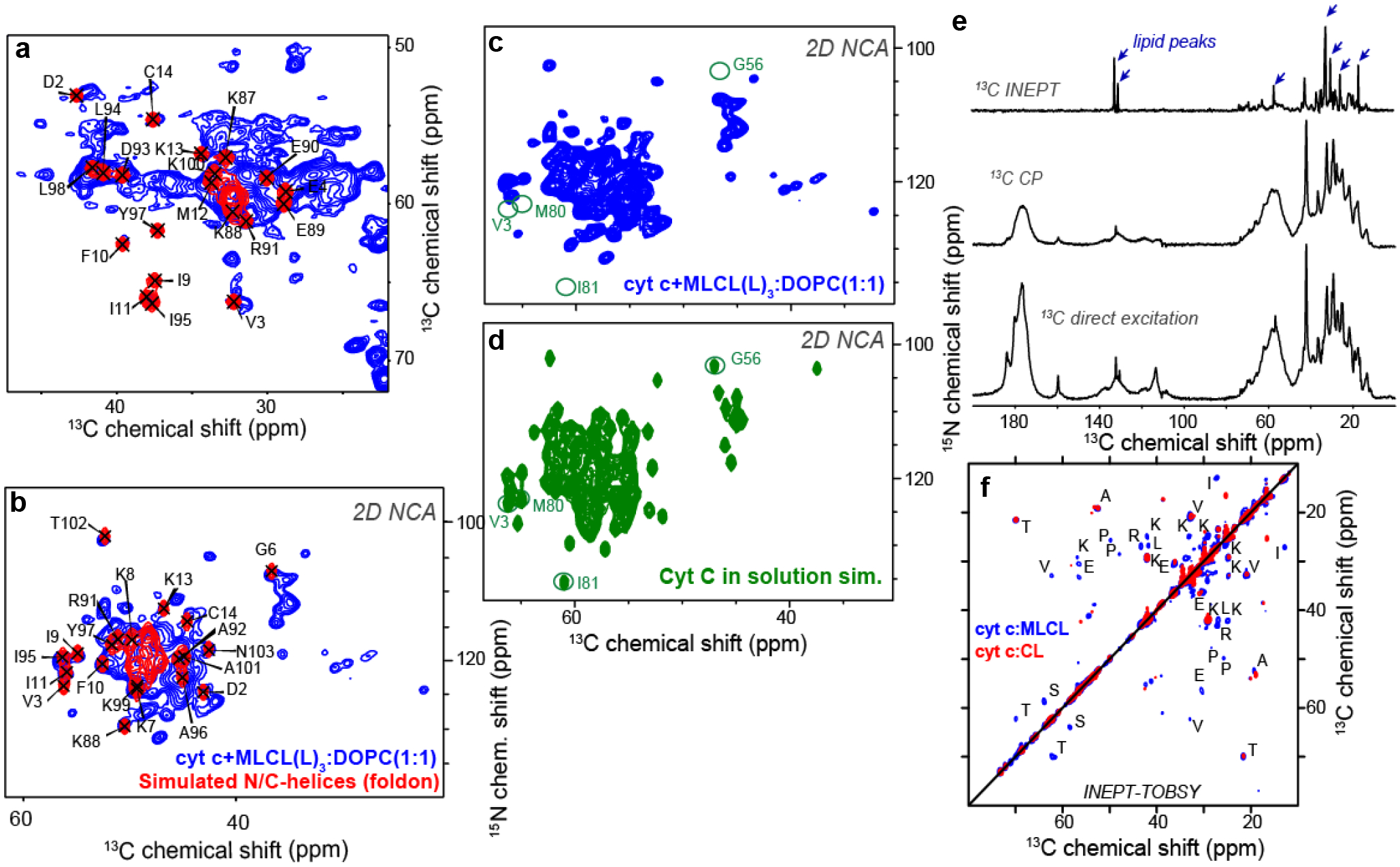
a, Region from the 2D CP-DARR spectrum of 15N,13C labeled cyt c bound to DOPC:MLCL(L)3 (1:1; blue). Overlaid red contours show simulated peak patterns constructed from the solution NMR shifts of the N- and C-terminal a-helices (BMRB ID 25908). b, 2D 15N-13C NCA ssNMR spectrum of the same sample, along with a corresponding simulated spectrum for the N- and C-terminal a-helices. The simulated peaks from the blue foldon coincide with strong peaks in the experimental ssNMR spectra. c, 2D (CP-based) 15N-13C NCA ssNMR spectrum of membrane-bound cyt c, alongside the analogous simulated spectrum (d) for cyt c in solution (BMRB ID 25908). Key resonances missing in the experimental spectrum (c) are indicated with green circles; these mostly belong to the Ω-loop D (e.g. Met80 and Ile81). These 2D NCA ssNMR spectra were acquired at 253 K and 10 kHz MAS. e, 1D 13C INEPT, CP, and direct excitation spectra of 15N,13C labeled cyt c bound to DOPC:MLCL(L)3 (1:1), measured at 278K. These DYSE ssNMR spectra detect flexible (INEPT), rigid (CP), or both (direct excitation) sample components. The liquid crystalline lipids contribute many peaks to the INEPT spectrum (marked with blue arrows), while the labeled protein dominates the other two spectra. f, Overlay of 2D INEPT-TOBSY spectra for 15N,13C-labeled cyt c bound to MLCL/DOPC (blue) and CL/DOPC (red) vesicles. Cross-peaks stem from the labeled protein and must reflect flexible residues. Many more (and stronger) peaks are seen for MLCL-bound cyt c.
Extended Data Fig. 2. 31P ssNMR analysis of MLCL-induced structural and dynamic changes in the lipid membrane.
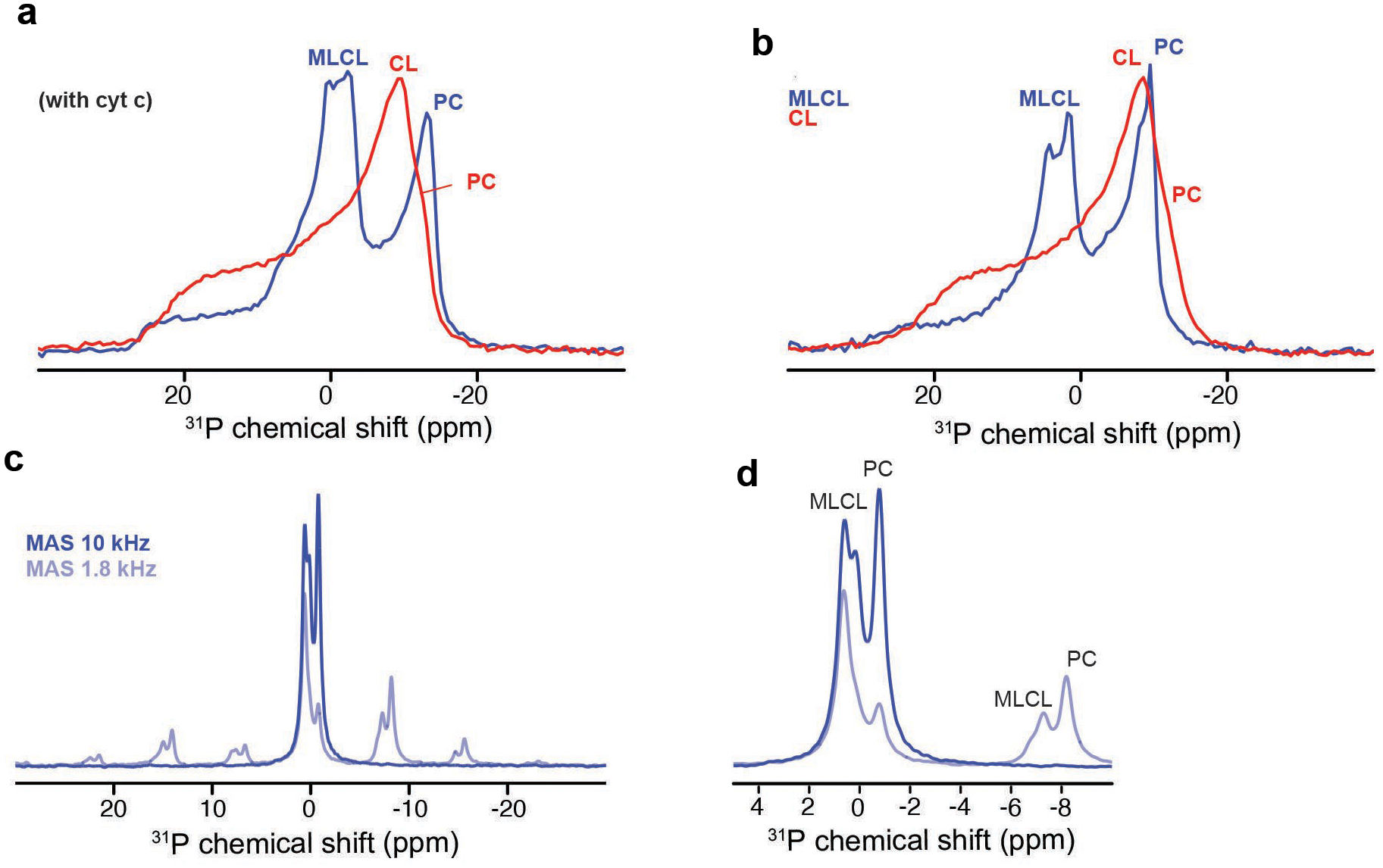
a, 1D 31P ssNMR static spectra of DOPC:MLCL (1:1) (blue) and DOPC:CL (1:1) (red) vesicles with with bound cyt c. b, Analogous 31P NMR spectra in absence of cyt c. Panels (a,b) show that the presence of MLCL results in a narrower extra component, that is neither isotropic nor reflects a typical non-bilayer phase. As previously discussed, (Powell and Marsh, 1985), these signals reflect MLCL in the liquid crystalline bilayer, undergoing distinct, increased dynamics (of the phosphate groups) compared to normal CL. c, 1D 31P ssNMR MAS spectra of DOPC:MLCL (1:1) at 10 kHz (blue curve) and 1.8 kHz (light blue curve) MAS rates. d, Zoomed-in region from (C) showing the isotropic peak (left) and first sideband (at 1.8 kHz MAS). Peak assignments are indicated, based on prior publications13,21 (Powell and Marsh, 1985; Li et al., 2019). Note that in the side bands, the PC signals are strong, one MLCL signal is partly retained and one MLCL signal is attenuated or missing. This is consistent with the shown assignments and cited literature.
Extended Data Fig. 3. Final shapshots from simulations of cyt c – membrane interactions and superposition of the conformations of cyt c reached after 100 ns runs.
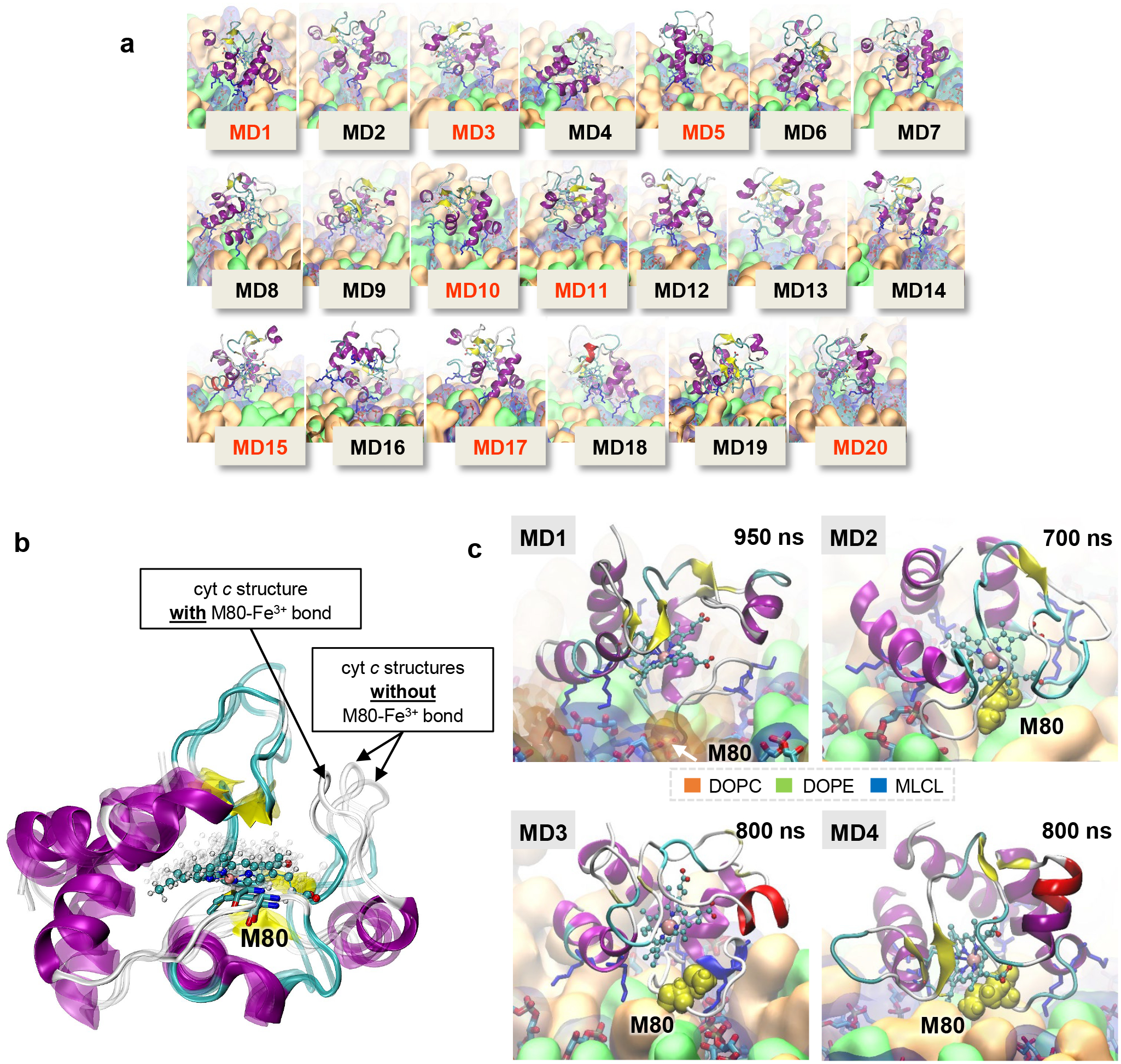
a, Final shapshots from simulations of cyt c – membrane interactions, and residues making frequent contacts with MLCL. Final conformations of cyt c reached after 100 ns in twenty independent MD trajectories. Red labels denote the systems which were further extended to 300 ns. The colors of the components of the membrane are following: DOPC – in orange, DOPE – in green, MLCL – in transparent blue. b, Superposition of the conformations of cyt c reached after 100 ns runs, in a system without membrane which contained Fe-S bond (sharp structure) and two independent runs where the bond was drastically weakened (transparent structures). All final conformations of heme group are displayed as grey shadow balls-sticks. c, Final conformations of cyt c after association with the membrane, observed in four runs MD1-MD4 (labeled). The white arrow in the upper left panel shows the residue M80 which inserted deep into the membrane, being trapped by several MLCL molecules, also shown in Fig. 2b.
Extended Data Fig. 4. Peroxidase activity of cyt c/MLCL complex causes phospholipid peroxidation in vitro and induces changes in lipidome and oxy-lipidome of genetically manipulated yeast.
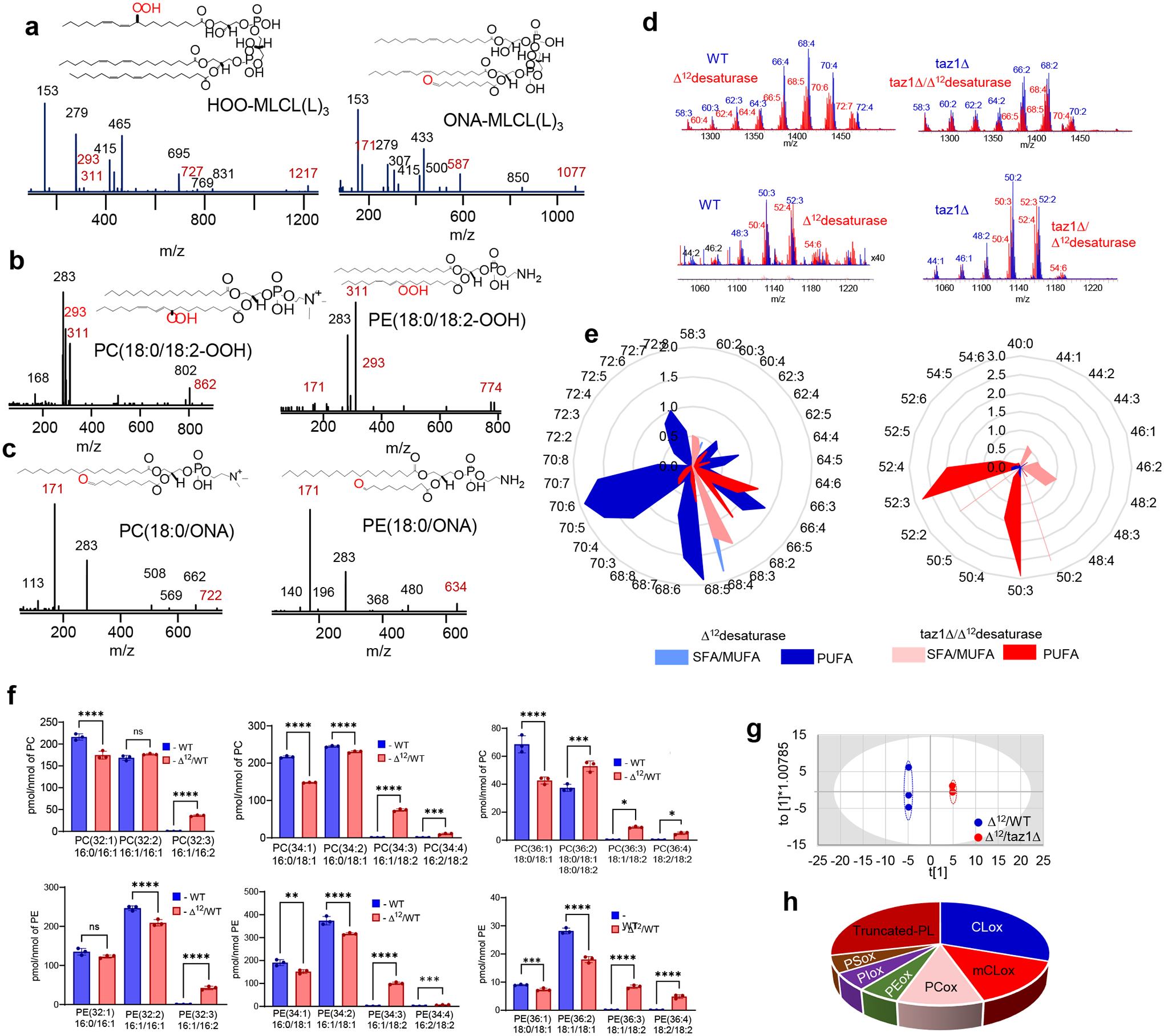
MS/MS spectra of (a) HOO-MLCL(L)3 (left panel) and oxidatively truncated ONA-MLCL(L)3 (right panel), (b) PC(18:0/18:2-OOH) (left panel) and PE(18:0/18:2-OOH) (right panel), (c) PC(18:0/ONA) (left panel) and PE(18:0/ONA) (right panel). Possible structures of oxidation products are inserted. ONA- 9-oxo nonanoic acid. d, Typical MS spectra of CL (upper panels) and MLCL (lower panels) obtained from yeast cells. e, Effect of D12desaturase on composition of CL (left panel) and MCL (right panel) in WT and taz1D yeasts. SFA – saturated fatty acids, MUFA – monounsaturated fatty acids, PUFA – polyunsaturated fatty acids. f, Content of PC (upper panels) and PE (lower panels) molecular species in WT and D12/taz1D cells. Data are presented as mean values ± SD. Each data point represents a biologically independent sample. Upper panels: left ****P<0.0001, middle ****P<0.0001, ***P=0.0004, right *P=0.0202, ***P=0.0001. ****P<0.0001. One-way ANOVA, Tukey’s multiple comparison test. Lower panels; left ****P<0.0001, middle **P=0.0115. ***P=0.0006, unpaired two-tailed t-test, ***P<0.0001, ****P<0.0001, One-way ANOVA, Tukey’s multiple comparison test. right ***P=0.0056, unpaired two-tailed t-test, ****P<0.0001, One-way ANOVA, Tukey’s multiple comparison test. g, OPLS-DA analysis showing the differences in oxy-lipidomes of D12 and D12/taz1D yeast cells; h, Pie plots showing the number of oxidatively modified phospholipid species. In total, 60 oxygenated phospholipid species were detected in yeast cells.
Extended Data Fig. 5. TAZ deficiency induces the changes in phospholipidome of mouse myoblasts and human lymphoblasts in vitro and mouse and human heart in vivo.
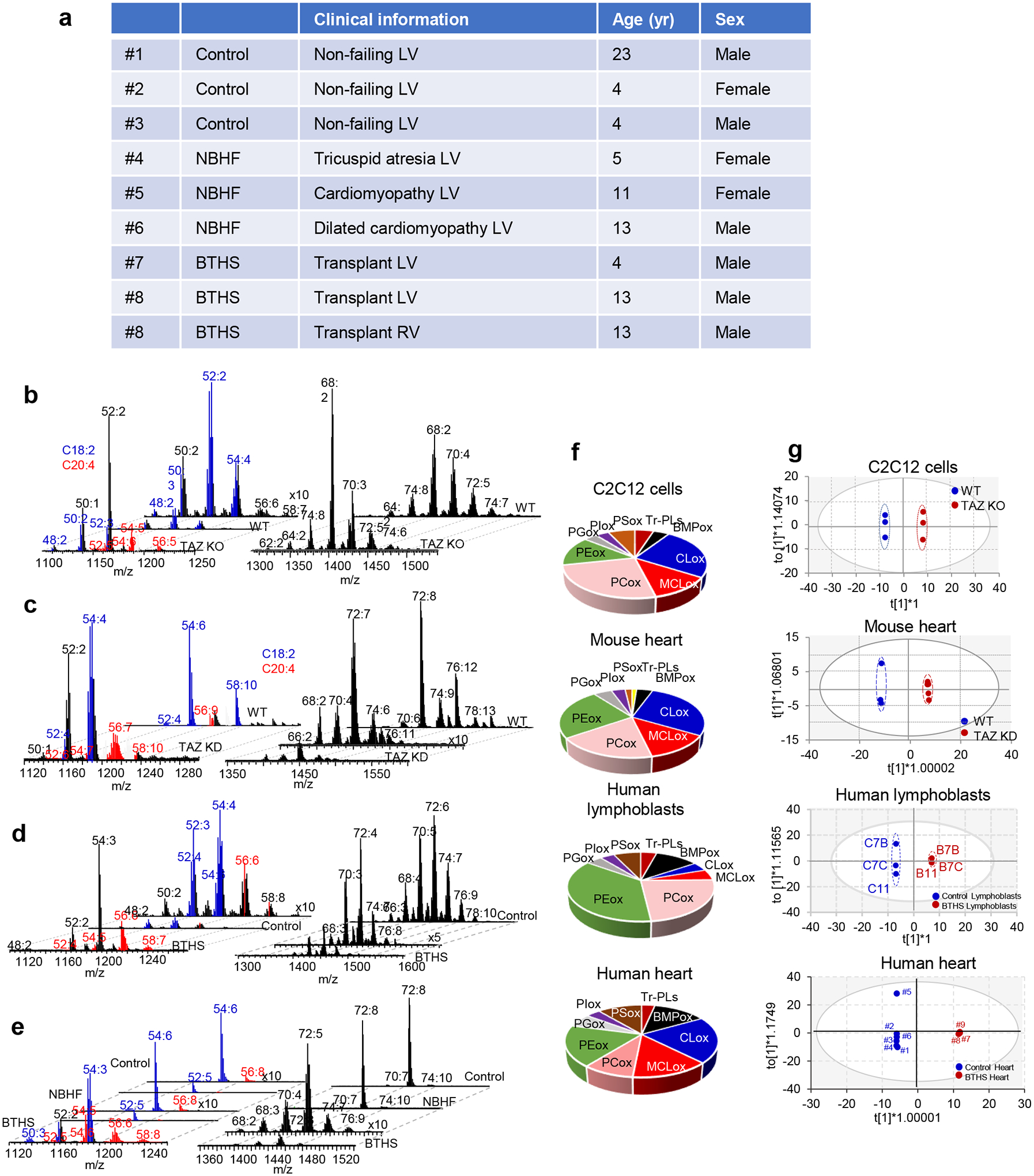
a, Table: Human heart biopsy sample description. Typical spectra of MLCL (left panels) and CL (right panels) obtained from WT and TAZ KO C2C12 cells (b), WT and TAZ KD mice (c), human lymphoblasts (d) and heart biopsy samples from control, non-BTHS-associated heart failure (NBHF) and BTHS-associated heart failure (BTHS) patients (e). TAZ deficiency in cells and mice and TAZ mutation in human results in a decrease of MLCL molecular species with C18:2 and appearance of MLCL molecular species containing C20:4 (shown in red); (f) Pie plots showing the total number of MLCL species in C2C12 cells, mouse heart, human lymphoblasts and heart samples from BTHD patients. (g) The score plots of OPLS-DA analysis show the differences in oxy-lipidomes in WT and TAZ ko cells, WT and TAZ KS mice, human control and BTHS lymphoblasts and control heart samples and BTHS heart samples.
Extended Data Fig. 6. Content of PE and PC species in cells, mouse heart, human lymphoblasts and human heart biopsy samples.
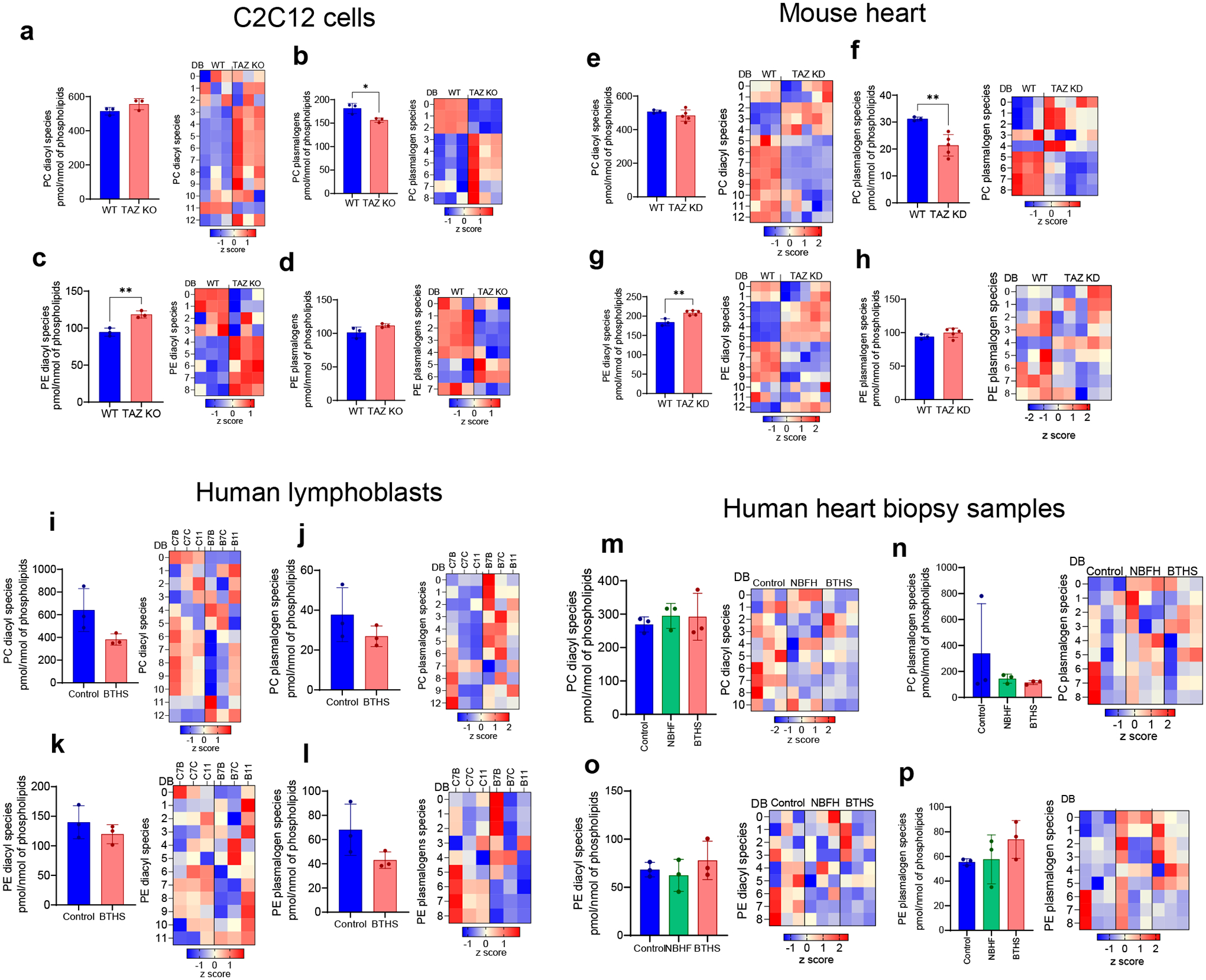
Extended Data Fig. 6. C2C12 cells a-d: a, Content of total diacyl-PC (left panel) and its molecular species (right panel); b, Content of total plasmalogen-PC (left panel) and its molecular species (right panel); c, Content of total diacyl-PE (left panel) and its molecular species (right panel); d, Content of total plasmalogen-PE (left panel) and its molecular species (right panel). Mouse heart (E-H): e, Content of total diacyl-PC (left panel) and its molecular species (right panel); f, Content of total plasmalogen-PC (left panel) and its molecular species (right panel); g, Content of total diacyl-PE (left panel) and its molecular species (right panel); h, Content of total plasmalogen-PE (left panel) and its molecular species (right panel). Human lymphocytes (i-l): i, Content of total diacyl-PC (left panel) and its molecular species (right panel); j, Content of total plasmalogen-PC (left panel) and its molecular species (right panel); k, Content of total diacyl-PE (left panel) and its molecular species (right panel); l, Content of total plasmalogen-PE (left panel) and its molecular species (right panel). Human heart biopsy samples (m-p): m, Content of total diacyl-PC (left panel) and its molecular species (right panel); n, Content of total plasmalogen-PC (left panel) and its molecular species (right panel); o, Content of total diacyl-PE (left panel) and its molecular species (right panel); p, Content of total plasmalogen-PE (left panel) and its molecular species (right panel).*p<0.05, **p<0.01. DB - double bond number.
Extended Data Fig. 7. Effect of TPP-IOA on structure of cyt c/MLCL peroxidase complex, with MLCL, lipid oxidation and the endurance of Drosophila melanogaster.
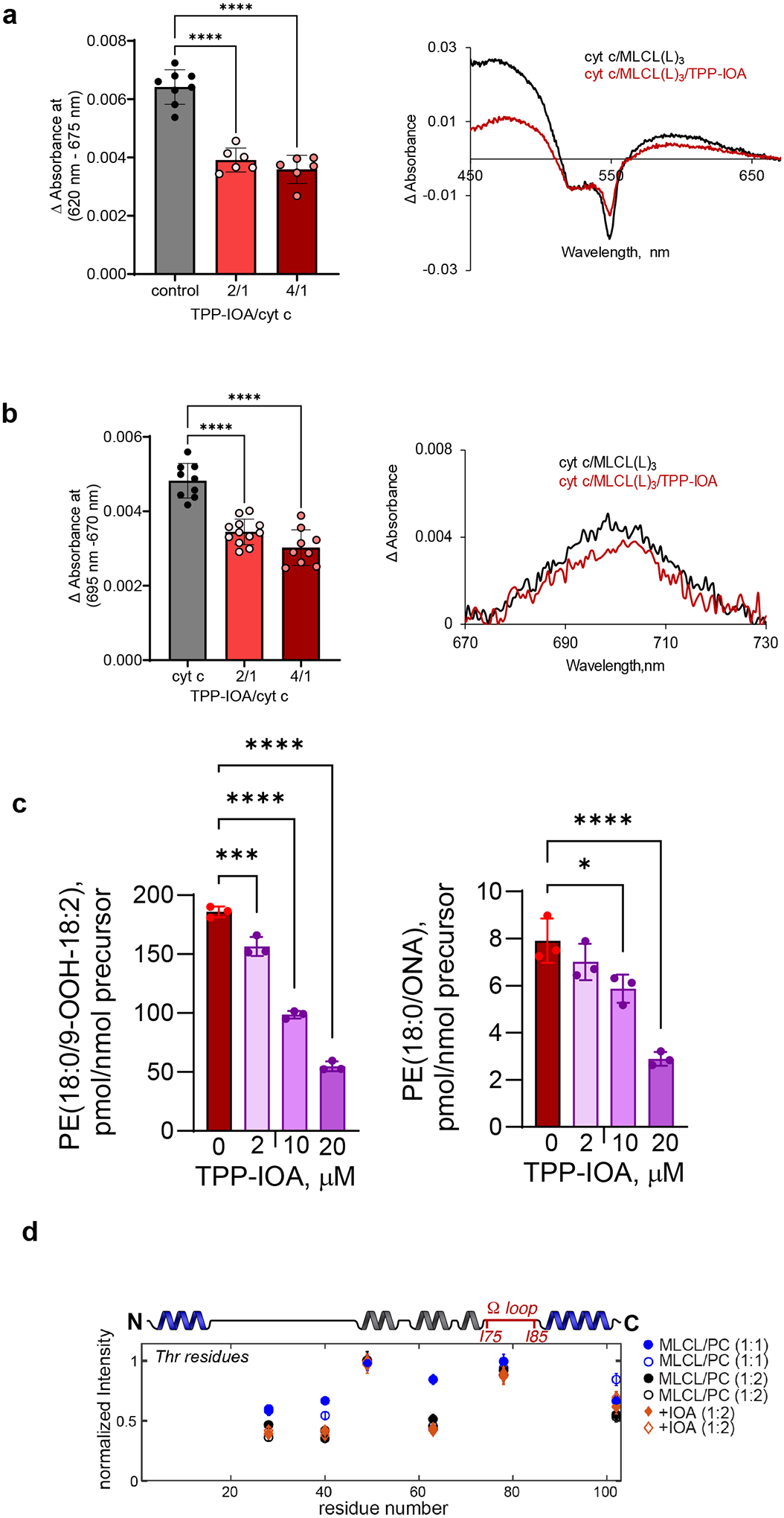
Effect of TPP-imidazole-oleic acid (TPP-IOA) on MLCL(L)3-dependent formation of heme iron high-spin forms assessed by absorbance at 620 nm (a) and absorbace at 695 nm (b). Inserts: the differential absorption spectra (a) and representative absorption spectra (b) of MLCL/cyt c with or without TPP-IOA. Data are presented as mean values ± SD. Each data point represents a biologically independent sample. ****p<0.0001, One-way ANOVA, Tukey’s multiple comparison test. (a) N=8 (control), N=6 (TPP-IOA/cyt c = 2/1), N=6 (TPP-IOA/cyt c = 4/1). (b) N=8 (control), N=10 (TPP-IOA/cyt c = 2/1), N=9 (TPP-IOA/cyt c = 4/1).(c) TPP-IOA inhibits accumulation of HOO-PE (left panel) and oxidatively truncated PE (right panel) species formed in MLCL(L)3/cyt c driven reaction. Data are presented as mean values ± SD. N=3. Each data point represents a biologically independent sample. Right panel: *p=0.0287, ****p<0.0001, Left panel: ***p=0.0007, ****p<0.0001. One-way ANOVA, Tukey’s multiple comparison test. (d) Normalized peak intensity values from 13C-13C ssNMR spectra of cyt c bound to DOPC:MLCL(L)3 (1:1; blue), or DOPC:MLCL(L)3 (2:1) LUVs in the absence (black) and presence (orange) of a four-fold excess of IOA with respect to protein (1:40 P/L ratio). Shown are the Ca/Cb (open) and Cb/Ca (closed) peaks for Thr residues. Each residue shows two datapoints: one for either side of the diagonal: see Fig. 1f). Increasing the ratio of MLCL (over PC) caused a net decrease in peak intensities, signifying increased protein motion. Addition of the IOA inhibitor had a much more modest effect of the Thr peak volumes.
Extended Data Fig. 8. TAZ-deficiency induces changes in Drosophila melanogaster lipidome.
Typical mass spectra of MLCL (a) and CL (b) obtained from control (W1118) and (TAZ889) mutant flies. Content MLCL containing C18:2 (c) and C18:3 (d) in control w1118 and TAZ889 deficient flies. Lipidomics analysis was performed using 6 vials (n=20 fly torsos per vial). (c) *P=0.0401, **P<0.0015, ***P=0.0005 unpaired two-tailed t-test. (d) *P=0.0378, **P<0.0019, ****p<0.0001 unpaired two-tailed t-test. (e) TTP-IOA did not significantly affect the endurance of control flies. Endurance was measure using 8 vials (N=20 flies) and significance was determined by log-rank analysis., ns=not significant. (f) Content of CL (left panel) and MLCL (right panel) in control W1118 flies after feeding with TPP-IOA. Lipidomics analysis was performed using 6 vials (n=20 fly torsos per vial). For all violin plots presented, individual points including maximal and minimal are shown as black circles. Dashed black line indicates median and doted lines indicate quartiles.
Supplementary Material
Acknowledgments:
Supported by the NIH GM134715 (V.E.K. and M.L.G), HL117880 (M.L.G), NS076511 (V.E.K. and H.B.), AG059683 (R.W.), the Polish National Science Centre (2019/35/D/ST4/02203) (K.M-R), and the Natural Sciences and Engineering Research Council of Canada (RGPIN/5368-2019) (G.M.H). G.M.H. is the Canada Research Chair in Molecular Cardiolipin Metabolism.
Footnotes
Credit author contributions:
Conceptualization, V.E.K., H.B,. M.L.G.;
Methodology, V.E.K., Y.Y.T., V.A.T., A.A.K., A.A.A., J.P., J.J., R.W., M.L.G., P.C.W., A.L., G.M.H., E.V.N.;
Software, Y.Y.T., K.M-R., P.C.W.;
Validation, V.E.K., M.L.G.;
Formal Analysis, Y.Y.T., V.A.T., A.A.K., A.A.A., S.N.S., M.A.A., G.K.V., E-K.B., D.D., J.J., A.R.; Computational Modeling, K.M-R., I.B.;
Investigation, Y.Y.T., V.A.T., A.A.K., M.A.A., S.N.S., H.H.D., A.B.S., A.K., Z.L., D.D., J.J., A.R., P.L., Z.L., A.L., P.R., L.K.C., E.V.N., K.K.;
Resources, K.M.R., M.W.S., B.K., G.M.H., A.N., H.B., J.A., J.V.;
Data Curation, Y.Y.T., K.M-R., P.C.W.;
Writing, Original Draft, V.E.K., H.B., M.L.G.;
Writing, Review & Editing, Y.Y.T., K.M-R., P.C.W., M.W.S. I.B., G.M.H., E.V.N.;
Visualization, Y.Y.T., K.M-R., A.R., A.L.;
Supervision, V.E.K., R.W., M.L.G., P.C.W., J.V.;
Project Administration, V.E.K., M.L.G.;
Funding Acquisition, V.E.K., M.L.G.
Declaration of interests: “The authors declare no competing interests.”
Code Availability Statement: No custom code was generated for this study
The Data Availability Statement:
Source data for Figures 1–7 and Extendent Data Figures 4–8 are provided with the manuscript. Raw flow cytometry data can be accessed from the FlowRepository database (Repository ID: FR-FCM-Z6XG) (http://flowrepository.org/id/FR-FCM-Z6XG). Mass spectrometry data are available upon request.
References:
- 1.Reid Thompson W et al. A phase 2/3 randomized clinical trial followed by an open-label extension to evaluate the effectiveness of elamipretide in Barth syndrome, a genetic disorder of mitochondrial cardiolipin metabolism. Genet Med 23, 471–478 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dabner L et al. Treatment of Barth Syndrome by Cardiolipin Manipulation (CARDIOMAN) With Bezafibrate: Protocol for a Randomized Placebo-Controlled Pilot Trial Conducted in the Nationally Commissioned Barth Syndrome Service. JMIR Res Protoc 10, e22533 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kagan VE, Chu CT, Tyurina YY, Cheikhi A & Bayir H Cardiolipin asymmetry, oxidation and signaling. Chemistry and Physics of Lipids 179, 64–69 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oemer G et al. Molecular structural diversity of mitochondrial cardiolipins. Proc Natl Acad Sci U S A 115, 4158–4163 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maguire JJ et al. Known unknowns of cardiolipin signaling: The best is yet to come. Biochim Biophys Acta Mol Cell Biol Lipids 1862, 8–24 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ren M, Miller PC, Schlame M & Phoon CKL A critical appraisal of the tafazzin knockdown mouse model of Barth syndrome: what have we learned about pathogenesis and potential treatments? Am J Physiol Heart Circ Physiol 317, H1183–H1193 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schlame M & Greenberg ML Biosynthesis, remodeling and turnover of mitochondrial cardiolipin. Biochim Biophys Acta Mol Cell Biol Lipids 1862, 3–7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Claypool SM & Koehler CM The complexity of cardiolipin in health and disease. Trends Biochem Sci 37, 32–41 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beranek A et al. Identification of a cardiolipin-specific phospholipase encoded by the gene CLD1 (YGR110W) in yeast. J Biol Chem 284, 11572–11578 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sparagna GC & Lesnefsky EJ Cardiolipin remodeling in the heart. J Cardiovasc Pharmacol 53, 290–301 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Ji J & Greenberg ML Cardiolipin function in the yeast S. cerevisiae and the lessons learned for Barth syndrome. J Inherit Metab Dis 45, 60–71 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li M et al. Activation of Cytochrome C Peroxidase Function Through Coordinated Foldon Loop Dynamics upon Interaction with Anionic Lipids. J Mol Biol 433, 167057 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li M et al. Surface-Binding to Cardiolipin Nanodomains Triggers Cytochrome c Pro-apoptotic Peroxidase Activity via Localized Dynamics. Structure 27, 806–815 e804 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baile MG, Whited K & Claypool SM Deacylation on the matrix side of the mitochondrial inner membrane regulates cardiolipin remodeling. Molecular Biology of the Cell 24, 2008–2020 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tuominen EK, Wallace CJ & Kinnunen PK Phospholipid-cytochrome c interaction: evidence for the extended lipid anchorage. J Biol Chem 277, 8822–8826 (2002). [DOI] [PubMed] [Google Scholar]
- 16.Nantes IL, Zucchi MR, Nascimento OR & Faljoni-Alario A Effect of heme iron valence state on the conformation of cytochrome c and its association with membrane interfaces. A CD and EPR investigation. J Biol Chem 276, 153–158 (2001). [DOI] [PubMed] [Google Scholar]
- 17.Kagan VE et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol 1, 223–232 (2005). [DOI] [PubMed] [Google Scholar]
- 18.Battistuzzi G et al. Role of Met80 and Tyr67 in the low-pH conformational equilibria of cytochrome c. Biochemistry 51, 5967–5978 (2012). [DOI] [PubMed] [Google Scholar]
- 19.Li Y et al. Cardiolipin-induced activation of pyruvate dehydrogenase links mitochondrial lipid biosynthesis to TCA cycle function. J Biol Chem 294, 11568–11578 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Imai M et al. Investigation of the redox-dependent modulation of structure and dynamics in human cytochrome c. Biochem Biophys Res Commun 469, 978–984 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Matlahov I & van der Wel PCA Hidden motions and motion-induced invisibility: Dynamics-based spectral editing in solid-state NMR. Methods 148, 123–135 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andronesi OC et al. Determination of membrane protein structure and dynamics by magic-angle-spinning solid-state NMR spectroscopy. J Am Chem Soc 127, 12965–12974 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Powell GL & Marsh D Polymorphic phase behavior of cardiolipin derivatives studied by 31P NMR and X-ray diffraction. Biochemistry 24, 2902–2908 (1985). [DOI] [PubMed] [Google Scholar]
- 24.Lou W et al. Genetic re-engineering of polyunsaturated phospholipid profile of Saccharomyces cerevisiae identifies a novel role for Cld1 in mitigating the effects of cardiolipin peroxidation. Biochim Biophys Acta Mol Cell Biol Lipids 1863, 1354–1368 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Horvath SE & Daum G Lipids of mitochondria. Prog Lipid Res 52, 590–614 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Lou W et al. Loss of tafazzin results in decreased myoblast differentiation in C2C12 cells: A myoblast model of Barth syndrome and cardiolipin deficiency. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids 1863, 857–865 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Acehan D et al. Cardiac and skeletal muscle defects in a mouse model of human Barth syndrome. J Biol Chem 286, 899–908 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kobayashi T et al. A lipid associated with the antiphospholipid syndrome regulates endosome structure and function. Nature 392, 193–197 (1998). [DOI] [PubMed] [Google Scholar]
- 29.Showalter MR et al. The Emerging and Diverse Roles of Bis(monoacylglycero) Phosphate Lipids in Cellular Physiology and Disease. Int J Mol Sci 21 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Atkinson J et al. A mitochondria-targeted inhibitor of cytochrome c peroxidase mitigates radiation-induced death. Nat Commun 2, 497 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu Y et al. A Drosophila model of Barth syndrome. Proc Natl Acad Sci U S A 103, 11584–11588 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Malhotra A et al. Role of calcium-independent phospholipase A2 in the pathogenesis of Barth syndrome. Proceedings of the National Academy of Sciences 106, 2337–2341 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Creed S & McKenzie M Measurement of Mitochondrial Membrane Potential with the Fluorescent Dye Tetramethylrhodamine Methyl Ester (TMRM). Methods Mol Biol 1928, 69–76 (2019). [DOI] [PubMed] [Google Scholar]
- 34.Pang J, Bao Y, Mitchell-Silbaugh K, Veevers J & Fang X Barth Syndrome Cardiomyopathy: An Update. Genes (Basel) 13 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hornby B et al. Natural history comparison study to assess the efficacy of elamipretide in patients with Barth syndrome. Orphanet J Rare Dis 17, 336 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scott RA, Mauk AG Cytochrome c: A Multidisciplinary Approach. (University Science Books, Sausalito, CA; 1996). [Google Scholar]
- 37.Alvarez-Paggi D et al. Multifunctional Cytochrome c: Learning New Tricks from an Old Dog. Chem Rev 117, 13382–13460 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Kagan VE et al. Cytochrome c/cardiolipin relations in mitochondria: a kiss of death. Free Radic Biol Med 46, 1439–1453 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pinheiro TJ & Watts A Lipid specificity in the interaction of cytochrome c with anionic phospholipid bilayers revealed by solid-state 31P NMR. Biochemistry 33, 2451–2458 (1994). [DOI] [PubMed] [Google Scholar]
- 40.Cassina AM et al. Cytochrome c nitration by peroxynitrite. J Biol Chem 275, 21409–21415 (2000). [DOI] [PubMed] [Google Scholar]
- 41.Hannibal L et al. Alternative Conformations of Cytochrome c: Structure, Function, and Detection. Biochemistry 55, 407–428 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Schweitzer-Stenner R Relating the multi-functionality of cytochrome c to membrane binding and structural conversion. Biophys Rev 10, 1151–1185 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Droghetti E, Oellerich S, Hildebrandt P & Smulevich G Heme coordination states of unfolded ferrous cytochrome C. Biophys J 91, 3022–3031 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dunford HB, Stillman JS On the function and mechanism of action of peroxidases. Coord Chem Reviews 19, 187–251 (1976). [Google Scholar]
- 45.Basova LV et al. Cardiolipin switch in mitochondria: shutting off the reduction of cytochrome c and turning on the peroxidase activity. Biochemistry 46, 3423–3434 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Belikova NA et al. Peroxidase activity and structural transitions of cytochrome c bound to cardiolipin-containing membranes. Biochemistry 45, 4998–5009 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barr DP & Mason RP Mechanism of radical production from the reaction of cytochrome c with organic hydroperoxides. An ESR spin trapping investigation. J Biol Chem 270, 12709–12716 (1995). [DOI] [PubMed] [Google Scholar]
- 48.Diederix RE et al. Kinetic stability of the peroxidase activity of unfolded cytochrome c: heme degradation and catalyst inactivation by hydrogen peroxide. Inorg Chem 42, 7249–7257 (2003). [DOI] [PubMed] [Google Scholar]
- 49.Chicco AJ & Sparagna GC Role of cardiolipin alterations in mitochondrial dysfunction and disease. Am J Physiol Cell Physiol 292, C33–44 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Amoscato AA et al. Formation of protein adducts with Hydroperoxy-PE electrophilic cleavage products during ferroptosis. Redox Biol 63, 102758 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Claypool SM, McCaffery JM & Koehler CM Mitochondrial mislocalization and altered assembly of a cluster of Barth syndrome mutant tafazzins. J Cell Biol 174, 379–390 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schlame M Cardiolipin synthesis for the assembly of bacterial and mitochondrial membranes. J Lipid Res 49, 1607–1620 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ge Y, Boopathy S, Nguyen TH, Lugo CM & Chao LH Absence of Cardiolipin From the Outer Leaflet of a Mitochondrial Inner Membrane Mimic Restricts Opa1-Mediated Fusion. Front Mol Biosci 8, 769135 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Duncan AL Monolysocardiolipin (MLCL) interactions with mitochondrial membrane proteins. Biochem Soc Trans 48, 993–1004 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu Y et al. Loss of protein association causes cardiolipin degradation in Barth syndrome. Nat Chem Biol 12, 641–647 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Musatov A Contribution of peroxidized cardiolipin to inactivation of bovine heart cytochrome c oxidase. Free Radic Biol Med 41, 238–246 (2006). [DOI] [PubMed] [Google Scholar]
- 57.Petrosillo G, Casanova G, Matera M, Ruggiero FM & Paradies G Interaction of peroxidized cardiolipin with rat-heart mitochondrial membranes: induction of permeability transition and cytochrome c release. FEBS Lett 580, 6311–6316 (2006). [DOI] [PubMed] [Google Scholar]
- 58.Clarke SL et al. Barth syndrome. Orphanet Journal of Rare Diseases 8, 23 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gonzalvez F et al. Barth syndrome: cellular compensation of mitochondrial dysfunction and apoptosis inhibition due to changes in cardiolipin remodeling linked to tafazzin (TAZ) gene mutation. Biochim Biophys Acta 1832, 1194–1206 (2013). [DOI] [PubMed] [Google Scholar]
- 60.Chu CT et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol 15, 1197–1205 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang J, Liu X, Nie J & Shi Y Restoration of mitophagy ameliorates cardiomyopathy in Barth syndrome. Autophagy 18, 2134–2149 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hsu P et al. Cardiolipin remodeling by TAZ/tafazzin is selectively required for the initiation of mitophagy. Autophagy 11, 643–652 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hullin-Matsuda F et al. De novo biosynthesis of the late endosome lipid, bis(monoacylglycero)phosphate. J Lipid Res 48, 1997–2008 (2007). [DOI] [PubMed] [Google Scholar]
- 64.Sohn J et al. A new murine model of Barth syndrome neutropenia links TAFAZZIN deficiency to increased ER stress-induced apoptosis. Blood Adv 6, 2557–2577 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Esposti MD, Cristea IM, Gaskell SJ, Nakao Y & Dive C Proapoptotic Bid binds to monolysocardiolipin, a new molecular connection between mitochondrial membranes and cell death. Cell Death Differ 10, 1300–1309 (2003). [DOI] [PubMed] [Google Scholar]
- 66.Human Protein Atlas. (https://www.proteinatlas.org/ENSG00000102125-TAZ/tissue).
- 67.Houtkooper RH et al. Cardiolipin and monolysocardiolipin analysis in fibroblasts, lymphocytes, and tissues using high-performance liquid chromatography-mass spectrometry as a diagnostic test for Barth syndrome. Anal Biochem 387, 230–237 (2009). [DOI] [PubMed] [Google Scholar]
- 68.Cole LK et al. Aberrant cardiolipin metabolism is associated with cognitive deficiency and hippocampal alteration in tafazzin knockdown mice. Biochim Biophys Acta Mol Basis Dis 1864, 3353–3367 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Olivar-Villanueva M, Ren M & Phoon CKL Neurological & psychological aspects of Barth syndrome: Clinical manifestations and potential pathogenic mechanisms. Mitochondrion 61, 188–195 (2021). [DOI] [PubMed] [Google Scholar]
- 70.Reynolds S Successful management of Barth syndrome: a systematic review highlighting the importance of a flexible and multidisciplinary approach. J Multidiscip Healthc 8, 345–358 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Raches D & Mazzocco MM Emergence and nature of mathematical difficulties in young children with Barth syndrome. J Dev Behav Pediatr 33, 328–335 (2012). [DOI] [PubMed] [Google Scholar]
- 72.Mazzocco MM, Henry AE & Kelly RI Barth syndrome is associated with a cognitive phenotype. J Dev Behav Pediatr 28, 22–30 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Damschroder D, Reynolds C & Wessells R Drosophila tafazzin mutants have impaired exercise capacity. Physiol Rep 6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Damschroder D et al. Stimulating the sir2-spargel axis rescues exercise capacity and mitochondrial respiration in a Drosophila model of Barth syndrome. Dis Model Mech 15 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schlame M, Xu Y & Ren M The Basis for Acyl Specificity in the Tafazzin Reaction. J Biol Chem 292, 5499–5506 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Taylor WA & Hatch GM Identification of the human mitochondrial linoleoyl-coenzyme A monolysocardiolipin acyltransferase (MLCL AT-1). J Biol Chem 284, 30360–30371 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rieger B, Krajcova A, Duwe P & Busch KB ALCAT1 Overexpression Affects Supercomplex Formation and Increases ROS in Respiring Mitochondria. Oxid Med Cell Longev 2019, 9186469 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Miklas JW et al. TFPa/HADHA is required for fatty acid beta-oxidation and cardiolipin re-modeling in human cardiomyocytes. Nat Commun 10, 4671 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li J et al. Cardiolipin remodeling by ALCAT1 links oxidative stress and mitochondrial dysfunction to obesity. Cell Metab 12, 154–165 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Price TR et al. Lipidomic QTL in Diversity Outbred mice identifies a novel function for alpha/beta hydrolase domain 2 ( Abhd2 ) as an enzyme that metabolizes phosphatidylcholine and cardiolipin. bioRxiv (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Maddalena LA, Ghelfi M, Atkinson J & Stuart JA The mitochondria-targeted imidazole substituted oleic acid ‘TPP-IOA’ affects mitochondrial bioenergetics and its protective efficacy in cells is influenced by cellular dependence on aerobic metabolism. Biochim Biophys Acta Bioenerg 1858, 73–85 (2017). [DOI] [PubMed] [Google Scholar]
- 82.Reily C et al. Mitochondrially targeted compounds and their impact on cellular bioenergetics. Redox Biol 1, 86–93 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Trnka J, Elkalaf M & Andel M Lipophilic triphenylphosphonium cations inhibit mitochondrial electron transport chain and induce mitochondrial proton leak. PLoS One 10, e0121837 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Di Paola M & Lorusso M Interaction of free fatty acids with mitochondria: coupling, uncoupling and permeability transition. Biochim Biophys Acta 1757, 1330–1337 (2006). [DOI] [PubMed] [Google Scholar]
- 85.Severin FF et al. Penetrating cation/fatty acid anion pair as a mitochondria-targeted protonophore. Proc Natl Acad Sci U S A 107, 663–668 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Davis BH et al. Determination of optimal replicate number for validation of imprecision using fluorescence cell-based assays: proposed practical method. Cytometry B Clin Cytom 84, 329–337 (2013). [DOI] [PubMed] [Google Scholar]
- 87.Olteanu A et al. Stability and apoptotic activity of recombinant human cytochrome c. Biochem Biophys Res Commun 312, 733–740 (2003). [DOI] [PubMed] [Google Scholar]
- 88.Mandal A et al. Structural Changes and Proapoptotic Peroxidase Activity of Cardiolipin-Bound Mitochondrial Cytochrome c. Biophys J 109, 1873–1884 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mandal A, Boatz JC, Wheeler TB & van der Wel PCA On the use of ultracentrifugal devices for routine sample preparation in biomolecular magic-angle-spinning NMR. J Biomol Nmr 67, 165–178 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Delaglio F et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol Nmr 6, 277–293 (1995). [DOI] [PubMed] [Google Scholar]
- 91.Pettersen EF et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 92.Bushnell GW, Louie GV & Brayer GD High-resolution three-dimensional structure of horse heart cytochrome c. J Mol Biol 214, 585–595 (1990). [DOI] [PubMed] [Google Scholar]
- 93.Jo S, Kim T, Iyer VG & Im W CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comp. Chem 29, 1859–1865 (2008). [DOI] [PubMed] [Google Scholar]
- 94.Autenrieth F, Tajkhorshid E, Baudry J & Luthey‐Schulten Z Classical force field parameters for the heme prosthetic group of cytochrome c. Journal of computational chemistry 25, 1613–1622 (2004). [DOI] [PubMed] [Google Scholar]
- 95.Phillips J et al. Scalable molecular dynamics with NAMD. J. Comput. Chem 26, 1781–1802 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Koes DR, Baumgartner MP & Camacho CJ Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J. Chem. Inf. Mod 53, 1893–1904 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zoete V, Cuendet MA, Grosdidier A & Michielin O SwissParam: a fast force field generation tool for small organic molecules. J. Comp. Chem 32, 2359–2368 (2011). [DOI] [PubMed] [Google Scholar]
- 98.Bakan A, Meireles LM & Bahar I ProDy: protein dynamics inferred from theory and experiments. Bioinformatics 27, 1575–1577 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source data for Figures 1–7 and Extendent Data Figures 4–8 are provided with the manuscript. Raw flow cytometry data can be accessed from the FlowRepository database (Repository ID: FR-FCM-Z6XG) (http://flowrepository.org/id/FR-FCM-Z6XG). Mass spectrometry data are available upon request.