

Abstract
Background
Hereditary ataxia syndromes can result in significant speech impairment, a symptom thought to be responsive to treatment. The type of speech impairment most commonly reported in hereditary ataxias is dysarthria. Dysarthria is a collective term referring to a group of movement disorders affecting the muscular control of speech. Dysarthria affects the ability of individuals to communicate and to participate in society. This in turn reduces quality of life. Given the harmful impact of speech disorder on a person's functioning, treatment of speech impairment in these conditions is important and evidence‐based interventions are needed.
Objectives
To assess the effects of interventions for speech disorder in adults and children with Friedreich ataxia and other hereditary ataxias.
Search methods
On 14 October 2013, we searched the Cochrane Neuromuscular Disease Group Specialized Register, CENTRAL, MEDLINE, EMBASE, CINAHL Plus, PsycINFO, Education Resources Information Center (ERIC), Linguistics and Language Behavior Abstracts (LLBA), Dissertation Abstracts and trials registries. We checked all references in the identified trials to identify any additional published data.
Selection criteria
We considered for inclusion randomised controlled trials (RCTs) or quasi‐RCTs that compared treatments for hereditary ataxias with no treatment, placebo or another treatment or combination of treatments, where investigators measured speech production.
Data collection and analysis
Two review authors independently selected trials for inclusion, extracted data and assessed the risk of bias of included studies using the standard methodological procedures expected by The Cochrane Collaboration. The review authors collected information on adverse effects from included studies. We did not conduct a meta‐analysis as no two studies utilised the same assessment procedures within the same treatment.
Main results
Fourteen clinical trials, involving 721 participants, met the criteria for inclusion in the review. Thirteen studies compared a pharmaceutical treatment with placebo (or a low dose of the intervention), in heterogenous groups of degenerative cerebellar ataxias. Three compounds were studied in two trials each: a levorotatory form of 5‐hydroxytryptophan (L‐5HT), idebenone and thyrotropin‐releasing hormone tartrate (TRH‐T); each of the other compounds (riluzole, varenicline, buspirone, betamethasone, coenzyme Q10 with vitamin E, α‐tocopheryl quinone and erythropoietin) were studied in one trial. The 14th trial, involving a mixed group of participants with spinocerebellar ataxia, compared the effectiveness of nonspecific physiotherapy and occupational therapy within an inpatient hospital setting to no treatment. No studies utilised traditional speech therapies. We defined the primary outcome measure in this review as the percentage change (improvement) in overall speech production immediately following completion of the intervention or later, measured by any validated speech assessment tool. None of the trials included speech as a primary outcome or examined speech using any validated speech assessment tool. Eleven studies reported speech outcomes derived from a subscale embedded within disease rating scales. The remaining three studies used alternative assessments to measure speech, including mean time to produce a standard sentence, a subjective rating of speech on a 14‐point analogue scale, patient‐reported assessment of the impact of dysarthria on activities of daily living and acoustic measures of syllable length. One study measured speech both subjectively as part of a disease rating scale and with further measures of speech timing. Three studies utilised the Short Form‐36 Health Survey (SF‐36) and one used the Child Health Questionnaire as measures of general quality of life. A further study utilised the Functional Independence Measure to assess functional health.
Five studies reported statistically significant improvement on an overall disease rating scale in which a speech subscale was included. Only three of those studies provided specific data on speech performance; all were comparisons with placebo. Improvements in overall disease severity were observed with α‐tocopheryl quinone; however, no significant changes were found on the speech subscale in a group of individuals with Friedreich ataxia. A statistically significant improvement in speech according to a speech disorders subscale was observed with betamethasone. Riluzole was found to have a statistically significant effect on speech in a group of participants with mixed hereditary, sporadic and unknown origin ataxias. No significant differences were observed between treatment and placebo in any other pharmaceutical study. A statistically significant improvement in functional independence occurred at the end of the treatment period in the rehabilitation study compared to the delayed treatment group but these effects were not present 12 to 24 weeks after treatment. Of the four studies that assessed quality of life, none found a significant effect. A variety of minor adverse events were reported for the 13 pharmaceutical therapies, including gastrointestinal side effects and nausea. Serious adverse effects were reported in two participants in one of the L‐5HT trials (participants discontinued due to gastrointestinal effects), and in four participants (three taking idebenone, one taking placebo) in the idebenone studies. Serious adverse events with idebenone were gastrointestinal side effects and, in people with a previous history of these events, chest pain and idiopathic thrombocytopenic purpura. The rehabilitation study did not report any adverse events.
We considered six studies to be at high risk of bias in some respect. We suspected inadequate blinding of participants or assessors in four studies and poor randomisation in a further two studies. There was a high risk of reporting bias in two studies and attrition bias in four studies. Only one study had a low risk of bias across all criteria. Taken together with other limitations of the studies relating to the validity of the measurement scales used, we downgraded the quality of the evidence for many of the outcomes to low or very low.
Authors' conclusions
There is insufficient and low or very low quality evidence from either RCTs or observational studies to determine the effectiveness of any treatment for speech disorder in any of the hereditary ataxia syndromes.
Keywords: Humans, Friedreich Ataxia, Friedreich Ataxia/complications, Friedreich Ataxia/therapy, Randomized Controlled Trials as Topic, Speech, Speech Disorders, Speech Disorders/drug therapy, Speech Disorders/etiology, Speech Disorders/therapy, Spinocerebellar Degenerations, Spinocerebellar Degenerations/complications, Spinocerebellar Degenerations/therapy
Plain language summary
Treatment for speech disorder in Friedreich ataxia and other hereditary ataxia syndromes (inherited disorders of movement co‐ordination)
Review question
We reviewed the evidence about the effects of treatment on speech difficulties in people with Friedreich ataxia and other hereditary ataxias.
Background
People with hereditary ataxia develop problems with co‐ordinating movement, which becomes worse over time. There are a range of other symptoms but this is the main feature of this group of diseases. Symptom onset is dependent on disease type and can begin in childhood or adulthood. Some types of hereditary ataxia appear later in life, even in middle age or older. Friedreich ataxia (FRDA) is the most common of the young onset hereditary ataxias.
Speech difficulties are a major feature of many of these disorders. People with ataxia often seek medical help because of slower speech, slurred speech or because the voice sounds harsh, or more nasal. Such difficulties can affect how well a person is able to communicate with friends, family and workmates.
Study characteristics
We searched widely for clinical trials and found 14 trials of treatments for speech problems in hereditary ataxias. The trials involved 721 participants. The duration of treatment was between two weeks and two years. Thirteen trials compared a medicine to a placebo and the 14th compared a mixed physiotherapy and occupational therapy treatment to no treatment. Ten different medicines were tested: L‐hydroxytryptophan (L‐5HT) (two studies), thyrotropin‐releasing hormone (TRH) (two studies), varenicline, riluzole, idebenone (two studies), betamethasone, coenzyme Q10 with vitamin E, buspirone, ɑ‐tocopheryl quinone and erythropoietin. We did not find any studies of traditional speech therapies. There were three ongoing trials.
Key results
When planning the review, we decided to use the percentage change in speech production after treatment as our primary measure of whether treatments were effective. None of the studies measured speech in a way that allowed us to report this. Five studies reported improvement in overall disease severity but only two studies, of riluzole in various ataxias and betamethasone in ataxia telangiectasia, demonstrated improvement of speech production. It is difficult to say whether these improvements in speech might make a meaningful difference to patients.
A variety of minor adverse events occurred with the medicines, including effects on the stomach and intestines, such as feeling sick. This kind of effect caused two people taking L‐5HT to stop treatment. Another person experienced this effect while taking idebenone. Two more people taking idebenone experienced heart or autoimmune problems; however, they each had experienced those problems earlier in their life. None of the other studies found differences in speech performance on active treatment. All trials had some problems in conduct or design that could potentially affect the findings.
Conclusions
Most of the included studies were small and looked at a mixed group of people with different forms of ataxia. The current evidence base is of low or very low quality and does not allow us to decide whether treatments for speech problems in the hereditary ataxia syndromes are effective.
The evidence is up to date to October 2013.
Summary of findings
Summary of findings for the main comparison. Hydroxytryptophan versus placebo for speech disorder resulting from hereditary ataxias.
| Hydroxytryptophan (L‐5HT) for speech disorder resulting from hereditary ataxias | ||||||
| Patient or population: people with speech disorder resulting from hereditary ataxias Settings: hospital Intervention: L‐5HT | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | L‐5HT | |||||
| Short‐term (within 1 month) percentage change (improvement) in overall speech production | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in these studies |
|
Short‐term (within 1 month) change in isolated movement, objective and subjective measures of speech production
Wessel 1995: Mean syllable duration during rapid‐syllable repetition task. Scale from: 1 to 500. Shorter durations are better. Follow‐up: 10 months Trouillas 1995: Mean time for producing a standard sentence. Shorter durations are better. Follow‐up: 6 months |
The mean short‐term (within 1 month) change in isolated movement, objective and subjective measures of speech production in the control groups was a
2 ms increase in mean syllable duration (Wessel 1995) 0.2 s increase (Trouillas 1995) |
The mean short‐term (within 1 month) change in isolated movement, objective and subjective measures of speech production in the intervention groups was
0 ms higher (CI not calculable)1 (Wessel 1995) 0.5 s lower (0.9 s lower to 0.1 lower) (Trouillas 1995) |
Not estimable | 4
(Wessel 19953,4) 19 (Trouillas 1995) |
⊕⊝⊝⊝ very low4,5,6 | Results from Trouillas 1995 and Wessel 1995 were not comparable due to difference in outcome measurement No differences were observed after treatment in either the placebo or L‐5HT conditions in either Trouillas 1995 or Wessel 1995 |
| Short‐term (within 1 month) change in quality of life scores related to communication as measured by validated communication assessments | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in Trouillas 1995 or Wessel 1995 |
| Longer‐term (minimum 1 month) change in generic quality of life scores | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in Trouillas 1995 or Wessel 1995 |
| Adverse effects (during the study) | See comment | See comment | Not estimable | 65 (2 studies) | See comment | Minor gastrointestinal side effects in 8/39 L‐5HT and 5/39 placebo participants in Wessel 1995 and 6/14 L‐5HT and 2/12 placebo participants in Trouillas 1995. Data could not be pooled because Wessel 1995 did not break down results by condition |
| Longer‐term burdens (minimum 1 month) (for example demands on caregivers, frequency of tests and restrictions on lifestyle) | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in Trouillas 1995 or Wessel 1995 |
| Economic outcomes | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in Trouillas 1995 or Wessel 1995 |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; L‐5HT: L‐hydroxytryptophan; RR: risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1CI of estimate of effect was not calculable in Wessel 1995, as no measure of variance was reported for the change. 2Cross‐over study design where the 4 participants each contributed 2 measurements (Wessel 1995). 3Only 4 of the original 19 participants with Friedreich ataxia completed the speech assessments during both arms of Wessel 1995, while only 19 of 26 participants completed Trouillas 1995. 4Missing data from Friedreich ataxia group in Wessel 1995. 5The method of allocation and blinding is not clear in Trouillas 1995. Adverse effects within the treatment arm may reduce the success of blinding of investigators or participants. 6Mean duration of a standard sentence is an insensitive measure of dysarthria (Trouillas 1995).
Summary of findings 2. Thyrotropin‐releasing hormone tartrate versus placebo for speech disorder resulting from hereditary ataxia.
| Thyrotropin‐releasing hormone tartrate (TRH‐T) for speech disorder resulting from hereditary ataxia | ||||||
| Patient or population: people with speech disorder resulting from hereditary ataxia Settings: hospital Intervention: TRH‐T | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | TRH‐T | |||||
| Short‐term (1 week) percentage change (improvement) in overall speech production | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in Filla 1988 or Sobue 1983 |
| Short‐term (1 week) change in isolated movement, objective and subjective measures of speech production Inherited Ataxias Clinical Rating Scale (IACRS) Follow‐up: mean 3.5 weeks | See comment | See comment | Not estimable | 245 (2 studies1) | ⊕⊝⊝⊝ very low2,3,4 | Raw data were not reported in either study investigating TRH‐T. Improvements in speech were observed in both treatment and placebo conditions |
| Short‐term (1 week) change in quality of life scores related to communication as measured by validated communication assessments | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in Filla 1988 or Sobue 1983 |
| Longer‐term (minimum 1 month) change in generic quality of life scores | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in Filla 1988 or Sobue 1983 |
| Adverse effects (during study) | See comment | See comment | Not estimable | 320 (2 studies) | See comment | Adverse effects included psycho‐neurologic effects (e.g. headache, dizziness and drowsiness), cardiovascular (e.g. hot feeling, flushing, palpitation and chest oppressed feeling), gastrointestinal (e.g. nausea, vomiting and abdominal pain) and other effects (e.g. urinary frequency, general malaise and sweating) in 50/101 participants on 2 mg TRH, 35/92 participants on 0.5 mg TRH and 20/97 participants on placebo (Sobue 1983) Filla 1988 reported 44 adverse effects for participants on TRH‐T, however did not report on adverse effects experienced by participants on placebo |
| Longer‐term burdens (minimum 1 month) (for example demands on caregivers, frequency of tests, restrictions on lifestyle) | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in Filla 1988 or Sobue 1983 |
| Economic outcomes | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in Filla 1988 or Sobue 1983 |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval;RR: risk ratio; TRH‐T: thyrotropin‐releasing hormone tartrate | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1Filla 1988 (n = 31) utilised a cross‐over design. 2Reported outcomes for speech do not include all participants (Sobue 1983). 3Speech was not a primary outcome measure, but a subscale on a disease severity measure (Filla 1988; Sobue 1983). 4Speech performance was evaluated via subjective clinician‐derived measures of severity (Filla 1988; Sobue 1983).
Summary of findings 3. Varenicline versus placebo for speech disorder resulting from hereditary ataxia.
| Varenicline for speech disorder resulting from hereditary ataxia | ||||||
| Patient or population: people with speech disorder resulting from hereditary ataxia Settings: hospital Intervention: varenicline | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Varenicline | |||||
| Short‐term (1 week) percentage change (improvement) in overall speech production | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in this study |
| Short‐term (1 week) change in isolated movement, objective and subjective measures of speech production SARA. Scale from: 0 to 6. Higher scores indicate more severe speech disorder. Follow‐up: mean 56 days | The mean short‐term (1 week) change in isolated movement, objective and subjective measures of speech production in the control groups was a decrease of 0.39 | The mean short‐term (1 week) change in isolated movement, objective and subjective measures of speech production in the intervention groups was a 0.06 greater decrease (no measure of variance)1 | Not calculable | 13 (1 study) | ⊕⊝⊝⊝ very low2,3,4,5 | Speech did not significantly improve in the treatment or placebo conditions |
| Short‐term (1 week) change in quality of life scores related to communication as measured by validated communication assessments | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in this study |
|
Longer‐term (minimum 1 month) change in generic quality of life scores
SF‐36
Follow‐up: mean 56 days Scale of 0 to 100. Higher score reflects less disability |
The mean longer‐term (minimum 1 month) change in generic quality of life scores in the control groups was a decrease of 1.42 | The mean longer‐term (minimum 1 month) change in generic quality of life scores in the intervention groups was 1.75 higher (no measure of variance)1 | Not calculable | 13 (1 study) | ⊕⊝⊝⊝ very low2,5 | Functional health did not significantly improve in either condition |
| Adverse effects (during study) | See comment | See comment | Not estimable | 18 (1 study) | See comment | 27 adverse effects were reported for the varenicline group and 19 adverse effects for the placebo group. 18 participants entered the treatment phase (9 in each group) |
| Longer‐term burdens (minimum 1 month) (for example, demands on caregivers, frequency of tests, restrictions on lifestyle) | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in this study |
| Economic outcomes | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in this study |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio; SARA: Scale for the Assessment and Rating of Ataxia; SF‐36: Short Form 36 Health Survey | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1CI of estimate of effect not calculable as the variance of change was not reported. 2Only 5 participants completed the placebo arm and 8 completed the varenicline arm of the study. The placebo group had higher clinical severity scores and appeared on average older compared to the varenicline group, possibly influencing their responsiveness to treatment (Filla 2012). The small sample size and lack of control for multiple comparisons may also introduce imprecision and statistical error. 3Speech was not a primary outcome measure, but a subscale on a disease severity measure. 4Speech performance was evaluated via subjective clinician‐derived measures of severity. 5Data were analysed for 9 participants in each group despite the large participant dropouts. The timing of these assessments was not stated. The second period of the experiment, which was to include a cross‐over component, was abandoned due to the high dropout rate observed in the initial period reported.
Summary of findings 4. Riluzole versus placebo for speech disorder resulting from hereditary ataxias.
| Riluzole for speech disorder resulting from hereditary ataxias | ||||||
| Patient or population: people with speech disorder resulting from hereditary ataxias Settings: hospital Intervention: riluzole | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Riluzole | |||||
| Short‐term (1 week) percentage change (improvement) in overall speech production | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in this study |
| Short‐term (within 1 month) change in isolated movement, objective and subjective measures of speech production ICARS. Scale from: 0 to 8. Higher scores indicate more severe speech disorder. Follow‐up: mean 8 weeks | The mean short‐term (within 1 month) change in isolated movement, objective and subjective measures of speech production in the control groups was 0.05 higher | The mean short‐term (within 1 month) change in isolated movement, objective and subjective measures of speech production in the intervention groups was 0.79 lower (0.43 to 1.15 lower) |
Not calculable | 38 (1 study) | ⊕⊕⊝⊝ low1,2 | Statistically significant difference between groups (P value < 0.001). Data were not divided by type of ataxia |
| Short‐term (within 1 month) change in quality of life scores related to communication as measured by validated communication assessments | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in this study |
| Longer‐term (minimum 1 month) change in generic quality of life scores | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in this study |
| Adverse effects (during study) | See comment | See comment | Not estimable | 38 (1 study) | See comment | 4 adverse events occurred: 3 in the riluzole group and 1 in the placebo group (N = 20 in each group). 2 participants in the treatment arm were found to have an increase in alanine aminotransferase 1.5 times over the normal limit |
| Longer‐term burdens (minimum 1 month) (for example, demands on caregivers, frequency of tests, restrictions on lifestyle) | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in this study |
| Economic outcomes | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in this study |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; ICARS: International Cooperative Ataxia Rating Scale; RR: risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1Speech was not the primary outcome measure, but a subscale on a disease severity rating scale.
2Speech was rated subjectively on scales of 'fluency of speech' and 'clarity of speech'.
Summary of findings 5. Idebenone versus placebo for speech disorder resulting from hereditary ataxias.
| Idebenone for speech disorder resulting from hereditary ataxias | ||||||
| Patient or population: people with speech disorder resulting from hereditary ataxias Settings: hospital Intervention: idebenone | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Idebenone | |||||
| Short‐term (1 week) percentage change (improvement) in overall speech production | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in the two studies of idebenone |
| Short‐term (within 1 month) change in isolated movement, objective and subjective measures of speech production ICARS, FARS Follow‐up: mean 6 months | See comment | See comment | Not estimable | 117 (2 studies) | very low1,2,3,4 | Speech subscales were not reported separately from overall ICARS or FARS scores in the 2 studies investigating idebenone |
| Longer‐term (minimum 1 month) change in generic quality of life scores | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in the 2 studies of idebenone |
| Adverse effects (during study) | See comment | See comment | Not estimable | 118 (2 studies) | See comment | Among the 118 participants in the 2 studies, 4 serious adverse events were reported in participants taking idebenone. Only 1, neutropenia, was reported to be related to the study drug. There were 200 non‐serious adverse events in treatment groups and 58 in the placebo group in Di Prospero 2007, while 21 participants in treatment groups and 10 placebo participants experienced adverse events in Lynch 2010. The incidence of adverse events was similar in treatment and placebo groups in both studies |
| Longer‐term burdens (minimum 1 month) (for example, demands on caregivers, frequency of tests, restrictions on lifestyle) | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in the 2 studies of idebenone |
| Economic outcomes | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in the 2 studies of idebenone |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; FARS: Friedreich Ataxia Rating Scale; ICARS: International Cooperative Ataxia Rating Scale; RR: risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1Investigators in Di Prospero 2007 implemented a second statistical analysis without non‐symptomatic and non‐ambulatory participants, with the rationale that this would remove floor and ceiling effects caused by the tests used. This group was found to experience greater improvement from the drug.
2Speech was measured as subjective impression of investigator in both Di Prospero 2007 and Lynch 2010.
3Speech was not measured directly, but as part of a larger ataxia assessment scale in both Di Prospero 2007 and Lynch 2010.
4While Di Prospero 2007 found an improvement on the ICARS, Lynch 2010 did not identify improvements due to idebenone. This was despite Lynch 2010 using similar doses of idebenone over the same timeframe as Di Prospero 2007. Lynch 2010 also excluded participants with ICARS scores lower than 10 and greater than 54 in order to identify the larger improvements within this subgroup observed in Di Prospero 2007.
Summary of findings 6. Physiotherapy and occupational therapy versus placebo for speech disorder resulting from hereditary ataxias.
| Physiotherapy and occupational therapy for speech disorder resulting from hereditary ataxias | ||||||
| Patient or population: people with speech disorder resulting from hereditary ataxias Settings: inpatient hospital rehabilitation Intervention: physiotherapy and occupational therapy | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control | Physiotherapy and occupational therapy | |||||
| Short‐term (1 week) percentage change (improvement) in overall speech production | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in this study |
| Short‐term (1 week) change in isolated movement, objective and subjective measures of speech production SARA. Scale from: 0 to 6. Higher scores indicate more severe speech disorder. Follow‐up: 4 weeks1 | The mean short‐term (1 week) change in isolated movement, objective and subjective measures of speech production in the control groups was an increase of 0.1 | The mean short‐term (1 week) change in isolated movement, objective and subjective measures of speech production in the intervention groups was 0.2 lower (0.48 lower to 0.08 higher) | 42 (1 study2) | ⊕⊝⊝⊝ very low5,6 | No statistically significant treatment response was observed | |
| Short‐term (1 week) change in quality of life scores related to communication as measured by validated communication assessments Functional Independence Measure (total). Scale from: 0 to 126. Higher scores indicate less disability Follow‐up: mean 4 weeks1 | The mean short‐term (1 week) change in quality of life scores related to communication as measured by validated communication assessments in the control groups was a decrease of 0.2 | The mean short‐term (1 week) change in quality of life scores related to communication as measured by validated communication assessments in the intervention groups was 1.4 higher (0.57 to 2.23 higher) | 42 (1 study2) | ⊕⊝⊝⊝ very low3,4 | Significant improvement in total FIM score immediately post intervention and at 4 weeks. No significant effect for treatment at 12 or 24 weeks post intervention | |
| Longer‐term (minimum 1 month) change in generic quality of life scores | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in this study |
| Adverse effects (during study) | See comment | See comment | Not estimable | 42 (1 study) | See comment | None reported |
| Longer‐term burdens (minimum 1 month) (for example, demands on caregivers, frequency of tests, restrictions on lifestyle) | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in this study |
| Economic outcomes | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in this study |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval;FIM: Functional Independence Measure; RR: risk ratio; SARA: Scale for the Assessment and Rating of Ataxia | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1Long‐term follow‐up was conducted at 12 to 24 weeks; however, all participants had completed the same treatment and were therefore not controlled after the initial 4‐week treatment or no treatment phase.
2Only the first 4 weeks of the study were randomised. Post intervention (at 12 to 24 weeks), assessments measured longer‐term treatment effects as all participants completed the same treatment. 3Ratings were based on subjective clinician‐derived measures of severity. 4The speech subscale was not reported in long‐term follow‐up. 5Speech performance was evaluated via subjective clinician‐derived measures of severity. 6Speech was not a primary outcome measure, but a subscale on a disease severity measure.
Summary of findings 7. Betamethasone versus placebo for speech disorder resulting from hereditary ataxias.
| Betamethasone (BETA) for speech disorder resulting from hereditary ataxias | ||||||
| Patient or population: people with speech disorder resulting from hereditary ataxias Settings: university Intervention: betamethasone | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | BETA | |||||
| Short‐term (1 week) percentage change (improvement) in overall speech production | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in this study |
| Short‐term (within 1 month) change in isolated movement, objective and subjective measures of speech production ICARS. Scale from: 0 to 8. Higher scores indicate more severe disorder. Follow‐up: mean 30 days | The median short‐term (within 1 month) change in isolated movement, objective and subjective measures of speech production in the control groups was a reduction of 0.5 | The median short‐term (within 1 month) change in isolated movement, objective and subjective measures of speech production in the intervention groups was a 0.1 greater reduction (0.5 lower to 2.5 lower))2 | Not estimable | 13 (1 cross‐over study) | ⊕⊕⊕⊝ moderate1 | Statistically significant difference between groups (P value = 0.02) Changes were reported as medians |
| Short‐term (within 1 month) change in quality of life scores related to communication as measured by validated communication assessments | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in this study |
| Longer‐term (minimum 1 month) change in generic quality of life scores Child Health Questionnaires Follow‐up: mean 30 days | See comment | See comment | Not calculable | 0 (0) | See comment | Data not presented. No difference reported between groups |
| Adverse effects (during study) | See comment | See comment | Not estimable | 13 (1 cross‐over study) | See comment | Mild adverse effects included asthenia (1 participant), mood swings (1 participant), moon face (8 participants), increased body weight (12 participants) |
| Longer‐term burdens (minimum 1 month) (for example, demands on caregivers, frequency of tests, restrictions on lifestyle) | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in this study |
| Economic outcomes | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in this study |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). BETA: betamethasone; CI: confidence interval;ICARS: International Cooperative Ataxia Rating Scale; RR: risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1Speech disorder was measured on a subjective, clinician‐derived severity rating scale.
2The CI was not calculable in Zannolli 2012 as the variance of change was not reported.
Summary of findings 8. High‐dose versus low‐dose coenzyme Q10 and vitamin E for speech disorder resulting from hereditary ataxia.
| High‐dose versus low‐dose coenzyme Q10 (CoQ10) and vitamin E for speech disorder resulting from hereditary ataxia | ||||||
| Patient or population: people with speech disorder resulting from hereditary ataxia Settings: hospital Intervention: CoQ10 and vitamin E (high dose versus low dose) | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| CoQ10 and vitamin E (low dose) | CoQ10 and vitamin E (high dose) | |||||
| Short‐term (1 week) percentage change (improvement) in overall speech production | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in this study |
|
Short‐term (within 1 month) change in isolated movement, objective and subjective measures of speech production
Cooper 2008 reported 3 speech measures: ICARS speech subscale. Scale from: 0 to 8. Higher scores indicate greater clinical severity Syllable repetition (number of repetitions of "pata" per 10 seconds). Higher repetitions represent lesser speech impairment Time taken (seconds) to read a standard passage. Higher scores represent greater speech impairment Follow‐up: mean 2 years |
The mean short‐term (within 1 month) change in isolated movement, objective and subjective measures of speech production in the low‐dose group was
ICARS speech subscale: a decrease of 0.05 Syllable repetition: a decrease of 0.6 Standard passage: an increase of 0.7 |
The mean short‐term (within 1 month) change in isolated movement, objective and subjective measures of speech production in the high‐ dose group was
ICARS speech subscale:
0.03 higher (0.16 lower to 0.22 higher) Syllable repetition: 0.5 higher (0.03 lower to 1.03 higher) Standard passage: 2.3 higher |
Not estimable | 43 (1 study) | ⊕⊕⊝⊝ low1,2 | No statistically significant difference was observed between groups for any speech measure |
| Short‐term (within 1 month) change in quality of life scores related to communication as measured by validated communication assessments | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in this study |
| Longer‐term (minimum 1 month) change in generic quality of life scores | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in this study |
| Adverse effects (during study) | See comment | See comment | Not estimable | 50 (1 study) | See comment | No major adverse events Minor effects included increased bowel frequency (1 participant, high‐dose group) and prolonged nausea (1 participant, low‐dose group) |
| Longer‐term burdens (minimum 1 month) (for example, demands on caregivers, frequency of tests, restrictions on lifestyle) | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in this study |
| Economic outcomes | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in this study |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; CoQ10 : coenzyme Q10; ICARS: International Cooperative Ataxia Rating Scale; RR: risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1Results are reported for both the RCT (no significant results) and for a comparison of the treated groups with a cross‐sectional data set, which was not adequately described. 2The primary outcome measure was based on a subjective measure of speech quality.
Summary of findings 9. Buspirone versus placebo for speech disorder in Friedreich ataxia and other hereditary ataxias.
| Buspirone for speech disorder in Friedreich ataxia and other hereditary ataxias | ||||||
| Patient or population: people with speech disorder in Friedreich ataxia and other hereditary ataxias Settings: hospital Intervention: buspirone | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Buspirone | |||||
| Short‐term (1 week) percentage change (improvement) in overall speech production | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in this study |
| Short‐term (within 1 month) change in isolated movement, objective and subjective measures of speech production ICARS. Follow‐up: mean 12 weeks | See comment | See comment | Not estimable | 19 (1 cross‐over study) | ⊕⊕⊝⊝ low1,2,3 | Speech subscales were not reported separately from overall ICARS scores. No difference between groups |
| Short‐term (within 1 month) change in quality of life scores related to communication as measured by validated communication assessments | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in this study |
| Longer‐term (minimum 1 month) change in generic quality of life scores | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in this study |
| Adverse effects (during study) | See comment | See comment | Not estimable | 19 (1 cross‐over study) | See comment | Minor adverse events included dizziness in 5 participants (4 buspirone, 1 placebo) and drowsiness in 4 participants (3 buspirone and 1 placebo) |
| Longer‐term burdens (minimum 1 month) (for example, demands on caregivers, frequency of tests, restrictions on lifestyle) | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in this study |
| Economic outcomes | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in this study |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; ICARS: International Cooperative Ataxia Rating Scale; RR: risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1Speech was measured on a subjective scale. 2The speech subscale of ICARS was not reported separately from the total score. 3Genetically confirmed Friedreich ataxia and SCA were analysed in same group as idiopathic ataxias. Data were not presented for genetically confirmed ataxias only.
Summary of findings 10. α‐tocopheryl quinone versus placebo for speech disorder resulting from hereditary ataxia.
| α‐tocopheryl quinone for speech disorder resulting from hereditary ataxia | ||||||
| Patient or population: people with speech disorder resulting from hereditary ataxia Settings: hospital Intervention: α‐tocopheryl quinone | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | α‐tocopheryl quinone | |||||
| Short‐term (1 week) percentage change (improvement) in overall speech production | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in Lynch 2012 |
| Short‐term (within 1 month) change in isolated movement, objective and subjective measures of speech production | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in Lynch 2012 |
| Short‐term (within 1 month) change in quality of life scores related to communication as measured by validated communication assessments | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in Lynch 2012 |
| Longer‐term (minimum 1 month) change in generic quality of life scores SF‐36 scale from: 0 to 100. Higher score indicate less disability. Follow‐up: mean 28 days | The mean longer‐term (minimum 1 month) change in generic quality of life scores in the control groups was an increase of 3.26 | The mean longer‐term (minimum 1 month) change in generic quality of life scores in the intervention groups was 3.27 lower (7.79 lower to 1.25 higher) | Not calculable | 19 (1 study) | ⊕⊕⊕⊝ moderate1 | No statistically significant difference between groups. Data presented for placebo and high‐dose (0.75 g twice daily) treatment arms only |
| Adverse effects (during study) | See comment | See comment | Not estimable | 31 (1 study) | See comment | No severe drug‐related adverse events occurred. Minor adverse events were found equally across the treatment arms |
| Longer‐term burdens (minimum 1 month) (for example, demands on caregivers, frequency of tests, restrictions on lifestyle) | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in Lynch 2012 |
| Economic outcomes | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in Lynch 2012 |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio; SF‐36: Short Form 36 Health Survey | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1The method of randomisation is not clear and did not completely match the three groups. The placebo group had higher clinical severity scores. 2Speech was measured using a subjective clinical impression.
Summary of findings 11. Erythropoietin versus placebo for speech disorder in Friedreich ataxia and other hereditary ataxias.
| Recombinant human erythropoietin (rhuEPO) for speech disorder in Friedreich ataxia and other hereditary ataxias | ||||||
| Patient or population: people with speech disorder in Friedreich ataxia and other hereditary ataxias Settings: hospital Intervention: rhuEPO | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | rhuEPO | |||||
| Short‐term (1 week) percentage change (improvement) in overall speech production | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in this study |
| Short‐term (within 1 month) change in isolated movement, objective and subjective measures of speech production SARA. Scale from: 0 to 48. Follow‐up: mean 6 months | See comment | See comment | Not calculable | 16 (1 study) | ⊕⊕⊝⊝ low1,2 | Speech subscales were not reported separately to overall SARA scores. No difference between groups |
| Short‐term (within 1 month) change in quality of life scores related to communication as measured by validated communication assessments | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in this study |
| Longer‐term (minimum 1 month) change in generic quality of life scores SF‐36. Scale from 0 to 100. Higher scores indicate less disability. Follow‐up: mean 6 months | See comment | See comment | Not calculable | 16 (1 study) | ⊕⊕⊝⊝ low3,4 | No difference between groups |
| Adverse effects (during study) | See comment | See comment | Not estimable | 16 (1 study) | See comment | No serious adverse events. 3 participants had sideropenic anaemia (2 rhuEPO group and 1 placebo group) |
| Longer‐term burdens (minimum 1 month) (for example, demands on caregivers, frequency of tests, restrictions on lifestyle) | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in this study |
| Economic outcomes | See comment | See comment | Not estimable | 0 (0) | See comment | Not an outcome in this study |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; rhuEPO: recombinant human erythropoietin; RR: risk ratio; SARA: Scale for the Assessment and Rating of Ataxia; SF‐36: Short Form 36 Health Survey | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1Speech was not measured directly but as part of an overall clinical severity scale. 2Speech was measured on a subjective, clinician‐rated, scale.
3Unclear randomisation, allocation concealment and blinding. 4Magnitude of change is not reported.
Background
Ataxias are neurological conditions in which muscle co‐ordination is impaired. Friedreich ataxia, an autosomal recessive neurodegenerative disorder, is the most common hereditary ataxia. It affects approximately 1 in 40,000 people (Delatycki 2000). Several other known autosomal dominant ataxias (for example, spinocerebellar ataxias (SCAs)) and recessive hereditary ataxias can also affect speech. In many hereditary ataxias, speech difficulties have been documented as a common outcome of disease progression (Rosen 2012), typically manifesting as dysarthria (slurred speech). In the case of Friedreich ataxia (Folker 2010) and SCA (Schalling 2007), individuals often present with a reduced rate of speech, vocal instability and imprecise consonants. Dysarthria affects the ability to communicate and participate in society, and reduces quality of life. Given the harmful impact of speech difficulties on a person's functioning, a strong body of evidence is required on which to base the treatment of speech impairment in these conditions.
Description of the condition
The major clinical features of Friedreich ataxia include progressive ataxia (100%), dysarthria (95%), scoliosis (78%), cardiomyopathy (65%), diabetes mellitus (8%) and foot deformity (74%) (Delatycki 1999). Onset generally occurs in childhood at an average age of 10 years, with the individual losing the ability to walk at an average age of 19 years. Life expectancy is markedly reduced. The many recognised SCAs vary in their clinical presentation and age of onset, and some are known to influence speech function (Schalling 2007). The prevalence of speech disorder in SCA is not yet known. A number of rare autosomal recessive hereditary ataxias also exist, where little is known about the clinical features relating to speech. Speech impairment in Friedreich ataxia and SCA varies depending on a number of factors, for example the severity of other clinical features and the stage of disease progression. At the impairment level, dysarthria arises from impaired respiratory, phonatory and articulatory subsystems underlying speech production (Duffy 2013). Perceptually, dysarthria is often characterised by a reduced vocal pitch or uncontrolled variation in pitch, a slower rate of speech, imprecise production of sounds (slurred speech) and reduced intelligibility. Deleterious consequences can also go beyond the physiological impairment level and lead to activity limitation (for example, avoiding use of the telephone) or the misperception by others that the person is cognitively impaired (Gibilisco 2013). Difficulties can be influenced by environmental factors (for example, background noise) (Hartelius 2007). Limited data exist on the speech profiles of recessive hereditary ataxias other than Friedreich ataxia.
Description of the intervention
This review focuses on the effects of treatments, including speech therapy or pharmaceutical therapies, for people with hereditary ataxias. Speech therapy may take the form of instrumental intervention, traditional drill‐based therapy techniques or a combination of both. Typically, pharmaceutical treatments are designed to alter the natural course of the disease itself. In Friedreich ataxia, for example, medications may be designed to reduce muscle tremors and spasms, treat cardiac issues or increase levels of frataxin (reduced expression of the protein frataxin is the cause of Friedreich ataxia).
How the intervention might work
The effectiveness of an intervention can best be conceptualised using the International Classification of Functioning, Disability and Health (ICFDH) (WHO 2001). At an impairment level, this means improving the capacity of people with a hereditary ataxia to communicate orally. This can be achieved in speech therapy by enhancing the production of sounds and words, by improving breath support for speech, maintaining adequate levels of intelligibility and, where possible, restoring the person's speech to pre‐morbid levels. At an activity and participation level, interventions might increase a person's ability to participate in the many social and professional activities for which effective communication skills are needed. Finally, changes can be made at an environmental level to improve communication outcomes for the person with speech difficulties (for example, by educating communication partners on effective strategies). Improvement in these three domains could enable people with hereditary ataxias to participate in society more actively and maintain personal and professional relationships.
Why it is important to do this review
Dysarthria is a primary feature of Friedreich ataxia, with estimates of prevalence ranging from 91% (Dürr 1996) to 100% (Folker 2010; Schöls 1997). A study in Friedreich ataxia by Harding 1981 showed dysarthria to be present in all participants 10 years after onset of the condition, suggesting that speech disorder is an inevitable outcome of disease progression. Speech disorder is also a key component of other hereditary ataxias including the SCAs (Schalling 2007); however, prevalence rates are not yet known. The likely presence of speech impairment in all individuals with a hereditary ataxia necessitates the development of effective and proven therapies for this aspect of these disorders.
Objectives
To assess the effects of interventions for speech disorder in adults and children with Friedreich ataxia and other hereditary ataxias.
Methods
Criteria for considering studies for this review
Types of studies
We considered all randomised controlled trials (RCTs) and quasi‐RCTs for inclusion. Quasi‐RCTs are studies in which participants are allocated to intervention groups by methods that are not truly random, such as alternate days, date of birth or case record number.
Types of participants
We considered studies with participants of any age, sex, ethnicity, stage of illness and any degree of illness severity. We included only studies in which participants had a genetically confirmed diagnosis of a hereditary ataxia, unless the studies were conducted prior to the discovery of the disease‐specific gene (i.e. Dürr 1996 for Friedreich ataxia and Orr 1993 for SCA1).
Types of interventions
Interventions in four categories of therapy to improve speech, based on intervention types described in Morgan 2008, compared to no treatment, placebo or another treatment or combination of treatments were considered for inclusion in the review. The categories of therapy were as follows.
Non‐instrumental intervention: intervention using traditional drill exercises with auditory feedback (perceptual) as the primary means of feedback. For example, exercises of the lips or tongue to increase the rate, strength, range or co‐ordination of the musculature supporting articulation; drill breathing exercises to increase respiratory/breath support for speech; and voicing drills to increase the loudness of phonation.
Instrumental approaches utilising biofeedback: interventions that use some form of instrumentation and that provide visual or other forms of biofeedback in addition to auditory feedback. For example, electropalatography; kinematics; and visual biofeedback acoustic treatment.
Pharmaceutical treatments with speech function as a primary, secondary or other outcome measure.
Any other intervention or combination of interventions.
We included interventions if they were administered for a minimum of one week and a maximum of 12 months.
Types of outcome measures
We considered both standardised and nonstandardised speech‐specific outcome measures. Outcome measures that were not speech‐specific acted as secondary assessment tools.
Primary outcomes
Our primary outcome measure was the percentage change (improvement) in overall speech production immediately following completion of the intervention or later, measured by any validated speech assessment tool).
Secondary outcomes
Secondary outcomes were the following.
Change in isolated movement, objective and subjective measures of speech production (for example, acoustic analysis of nasality; articulation; laryngeal function; respiratory function; and oral motor function), within one month post intervention.
Change in quality of life scores related to communication, measured by validated communication assessments such as the Voice Handicap Index (ordinal variables), within one month post intervention.
Generic quality of life measures (for example, Short Form‐36 Health Survey (SF‐36)), a minimum of one month post intervention.
Adverse effects (during the study).
Burdens (for example, demands on caregivers, frequency of tests and restrictions on lifestyle), a minimum of one month post intervention.
Economic outcomes (for example, cost and resource use).
Search methods for identification of studies
Electronic searches
On 14 October 2013, we searched the Cochrane Neuromuscular Disease Group Specialized Register, CENTRAL (2013, Issue 9), MEDLINE (January 1966 to September 2013), EMBASE (January 1980 to October 2013), CINAHL Plus (January 1937 to October 2013), PsycINFO (January 1806 to October 2013), Education Resources Information Center (ERIC) (January 1966 to October 2013), Linguistics and Language Behavior Abstracts (LLBA) (1973 to October 2013) and Dissertation Abstracts (1980 to October 2013). We also searched ClinicalTrials.gov (www.clinicaltrials.gov/) and the World Health Organization International Clinical Trials Registry Platform (ICTRP) (www.who.int/ictrp/en/) for ongoing trials.
The detailed search strategies are in the appendices: Neuromuscular Disease Group Specialized Register (Appendix 1), CENTRAL (Appendix 2), MEDLINE (Appendix 3), EMBASE (Appendix 4), CINAHL Plus (Appendix 5), PsycINFO (Appendix 6), ERIC Dialog (Appendix 7), ERIC ProQuest (Appendix 8), LLBA (Appendix 9), Dissertation Abstracts (Appendix 10), ClinicalTrials.gov (Appendix 11), and ICTRP (Appendix 12).
Searching other resources
We scanned conference abstracts for relevant studies. We checked all references in the identified trials to identify any additional published data.
We requested information from authors of potentially relevant trials. We requested information on unpublished data from authors of five published studies (Assadi 2007; Di Prospero 2007; Filla 1988; Lynch 2010; Mariotti 2009), but no additional data were available. We made contact with experts and information groups in the areas of linguistics and speech therapy; however, we identified no additional trials.
Data collection and analysis
Selection of studies
Two authors (AV and JF) independently screened titles and abstracts to exclude reports that were obviously irrelevant. In cases of uncertainty we evaluated the full‐text article. Two review authors (AV and JF) evaluated the full‐text article of potentially eligible studies. In the event of disagreement over inclusion of a particular paper, AV, JF and MLP reached a consensus after re‐assessing the inclusion criteria together. We selected studies without limitation as to language.
Data extraction and management
Two authors (AV and JF) performed data extraction and independently entered data onto a data extraction form. Discrepancies would have been resolved by the third author (MP) but this was not necessary. Two authors checked these data, AV entered them into Review Manager (RevMan) and JF checked the data entry.
The data extraction form included the following items.
General information: published/unpublished, title, authors, reference/source, contact address, country, language of publication, year of publication.
Trial characteristics: design, duration of follow‐up, method of randomisation, allocation concealment, blinding (participants, people administering treatment and outcome assessor).
Participants: age, sex and any other recorded baseline characteristics, inclusion and exclusion criteria, total number of participants, number in each group, disease severity, withdrawals and losses to follow‐up (reasons and description).
Intervention(s) and outcome(s): placebo or control interventions included, type of speech therapy, drug dosage regimen, duration, frequency, interval, comparison intervention(s), co‐treatment(s), the number and type of adverse events, other outcomes reported in the trial.
We resolved differences in data extraction by consensus, and by referring back to the original article. Where necessary, we requested further information from the authors of the primary studies.
Assessment of risk of bias in included studies
AV and JF independently assessed all included studies for risk of bias. We graded the items according to the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011), and presented judgements for each included trial in the 'Risk of bias' summary (Figure 1). We assessed trials in the following domains: sequence generation, allocation concealment, blinding (participants and outcome assessors), incomplete outcome data (participant losses and use of intention‐to‐treat (ITT) analysis), selective outcome reporting and other sources of bias. We then made a judgement of high, low or unclear risk of bias for each domain. We would have consulted the third author in the event of disagreement or resolved disagreements by discussion, or both.
1.
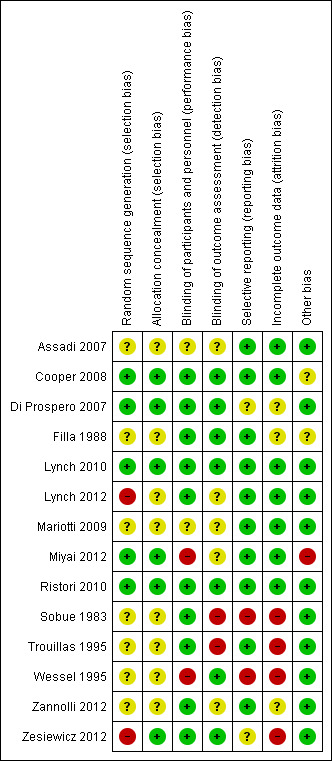
'Risk of bias 'summary: review authors' judgements about each risk of bias item for each included study.
Key: green (+) = low risk of bias; yellow (?) = unclear risk of bias; red (‐) = high risk of bias.
Measures of treatment effect
Measures of treatment effect for primary outcome measures relied on the outcome measures provided by the study authors including: improvements in isolated sound, single word, sentence or conversation level productions. We would have analysed data using the Cochrane statistical package Review Manager (RevMan) 5 (RevMan 2014), had suitable data been available.
In the protocol for this review we stated that "For dichotomous data we will derive risk ratios (RR) and 95% confidence intervals (CIs) for each outcome. For continuous variables we will calculate mean differences and 95% CIs for each outcome. We will use a fixed‐effect model to calculate pooled estimates and their 95% CIs, however, if the model yields large standard errors (i.e. the studies are not homogenous), a random‐effects model will be considered" (Vogel 2011b); however, no data were available for analysis. If studies are available in future, to enable the combination of studies measuring the same outcome using different measurement tools, we will summarise continuous data using standardised mean differences. We considered that binary outcomes were likely to be common in early reports within the field (e.g. improved outcome versus no change or worse). We planned to analyse such data by calculation of the RR with a 95% CI.
Unit of analysis issues
For cross‐over designs, the effect of conditioning represents a potential source of bias if the training period precedes no training. For this reason, if a difference in treatment effects and its standard error had been available from a cross‐over trial, we would have combined results with those of parallel‐group studies using the generic inverse variance (GIV) facility in RevMan. In the absence of these data we would have analysed only the first arm of the study.
Dealing with missing data
One review author (AV) contacted primary investigators for assistance and information in cases where data were missing within published studies.
Assessment of heterogeneity
We did not conduct a meta‐analysis as no two studies employed the same assessments for any one drug. Lynch 2010 and Di Prospero 2007 both compared the effect of idebenone or placebo in participants with Friedreich ataxia using the International Cooperative Ataxia Rating Scale (ICARS); however, data on the speech subscales of the disease rating scale were not available.
If data had been available we planned to assess consistency of results using the I2 statistic for heterogeneity (Higgins 2011). I2 is a quantity describing approximately the proportion of variation in point estimates that is due to heterogeneity of a sample rather than error in sampling of the population. For values greater than 50%, we would have examined forest plots for differences between trials which could explain the heterogeneity. We would have used a test of homogeneity to determine whether the heterogeneity was genuine. In the event of too few studies being available to make this test feasible, we would have applied a random‐effects model.
Assessment of reporting biases
There were insufficient studies to investigate publication bias and other reporting biases using funnel plots.
We had planned to evaluate funnel plot asymmetry visually and use formal tests for funnel plot asymmetry. If the plots had suggested that treatment effects may not be sampled from a symmetric distribution, as assumed by the random‐effects model, we would have performed further meta‐analyses using a fixed‐effect model.
Data synthesis
Meta‐analysis was not possible and we therefore reported the results of the trials narratively. Eight out of 11 treatments included in this review were assessed in only one study. Meta‐analyses of the studies involving the same intervention were not possible because they used different outcome measures or lacked data relating to speech outcomes.
'Summary of findings' table
We included a 'Summary of findings' table, incorporating our key primary and secondary outcome measures as follows.
Short‐term percentage change (improvement) in overall speech production.
Short‐term change in isolated movement, objective and subjective measures of speech production.
Short‐term change in quality of life scores related to communication as measured by validated communication assessments.
Longer‐term (minimum one month) change in generic quality of life scores.
Adverse effects (during study).
Economic outcomes.
The table also included information about trial characteristics (for example, design and duration of follow‐up), and participants. We assessed the quality of evidence for each outcome for each comparison using Grading of Recommendations Assessment, Development and Evaluation (GRADE) criteria: study limitations, consistency of effect, imprecision, indirectness and publication bias. We used methods and recommendations described in Section 8.5 and Chapter 12 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011), and prepared the tables using GRADEpro software (GRADEpro 2008). We included information in footnotes to justify our decisions to down‐ or up‐grade the quality of evidence.
Subgroup analysis and investigation of heterogeneity
In reference to participant characteristics, we planned to undertake subgroup analysis by modus of inheritance and causative gene or chromosomal locus, type of ataxia and the severity of dysarthria.
We would have also considered heterogeneity in reference to study design and implementation characteristics, including, but not limited to, methods of recruitment and randomisation and methods of implementing therapy. However, no such analyses were possible.
Sensitivity analysis
If studies had been suitable for meta‐analysis, we would have used sensitivity analysis to assess the robustness of the overall findings by examining the impact of study quality; for example, lack of allocation concealment or high rates of loss to follow‐up, the impact of missing data or the impact of imputations, and the rigour of eligibility criteria employed in the study. We would have also evaluated the possibility of one or more large studies dominating the results.
The methods for this systematic review were prespecified in the protocol (Vogel 2011b). We have listed deviations from protocol in Differences between protocol and review.
Results
Description of studies
See Characteristics of excluded studies and Characteristics of included studies.
Results of the search
The search conducted up until October 2013 identified 494 records and we identified a further nine records from reference lists. Table 1 reports the number of studies retrieved from each search strategy. After duplicates were removed, 425 records remained from which we retrieved 56 papers for further examination. After screening the full text of the 56 selected papers for eligibility, 14 papers were not relevant, 25 papers were excluded for methodological reasons and 14 studies met the inclusion criteria (Assadi 2007; Cooper 2008; Di Prospero 2007; Filla 1988; Lynch 2010; Lynch 2012; Mariotti 2009; Miyai 2012; Ristori 2010; Sobue 1983; Trouillas 1995; Wessel 1995; Zannolli 2012; Zesiewicz 2012). We identified three ongoing studies (EUCTR 2009‐016317‐20‐IT; EUCTR 2012‐005312‐26‐DE; Schulz 2009), which are described in Characteristics of ongoing studies. A flow diagram of the study selection process is presented in Figure 2.
2.

Study flow diagram.
Table 1
| Database | Period searched | Date searched | Number of hits |
| Cochrane Neuromuscular Disease Group Specialized Register | Up to 15 October 2013 | 15 October 2013 | 0 |
| CENTRAL | Up to 14 October 2013 | 14 October 2013 | 25 |
| MEDLINE | 1966 to October 2013 | 14 October 2013 | 96 |
| EMBASE | 1980 to October 2013 | 14 October 2013 | 75 |
| CINAHL | 1937 to October 2013 | 14 October 2013 | 35 |
| PsycINFO | 1806 to October 2013 | 14 October 2013 | 95 |
| ERIC | 1966 to October 2013 | 14 October 2013 | 31 |
| LLBA | 1973 to October 2013 | 15 October 2013 | 59 |
| Dissertation Abstracts | 1980 to October 2013 | 15 October 2013 | 62 |
| Clinical Trial Registries | Up to 15 October 2013 | 15 October 2013 | 16 |
| Additional records from reference lists of relevant studies | ‐ | ‐ | 9 |
Included studies
We included 14 studies in the qualitative analysis and these are described in the Characteristics of included studies section.
Design
Assadi 2007, Filla 1988, Wessel 1995 and Zannolli 2012 used a double‐blind, placebo‐controlled, cross‐over design. Miyai 2012 employed a single‐blinded, randomised design. Di Prospero 2007, Lynch 2010, Lynch 2012, Mariotti 2009, Ristori 2010, Sobue 1983, Trouillas 1995 and Zesiewicz 2012 were double‐blind, placebo‐controlled, randomised, parallel‐group studies. Cooper 2008 was a double‐blind, randomised trial with a low‐dose group as comparison.
Setting
Trials were carried out in Europe, Japan and the USA. Filla 1988, Mariotti 2009 and Ristori 2010 were performed in Italy, Wessel 1995 in Germany, Cooper 2008 in the UK, and Assadi 2007, Zesiewicz 2012, Lynch 2010, Lynch 2012 and Di Prospero 2007 in the USA, all in outpatient settings. Trouillas 1995 was conducted across 12 outpatient settings in France. Interventions in Miyai 2012 and Sobue 1983 were administered in an inpatient setting in Japan. Zannolli 2012 was a multicentre study that took place in six Italian universities.
Participants
There were 721 participants in the 14 included studies. Thirty‐nine participants with degenerative cerebellar ataxias, including Friedreich ataxia (19), olivopontocerebellar atrophy (7) and cerebellar atrophy (13), were recruited to Wessel 1995. We only included data on the Friedreich ataxia group in this review as the aetiology of disease for the cerebellar atrophy and olivopontocerebellar atrophy participants was unclear. Sixteen participants with Friedreich ataxia and 14 participants with various degenerative ataxias (prior to the discovery of disease‐specific genotypes) completed Filla 1988. Miyai 2012 recruited 42 participants with pure cerebellar degeneration; they included 20 people with SCA6 (genetically confirmed), six people with SCA31 and 16 participants with idiopathic cerebellar ataxia. Twenty participants with SCA3 were recruited to Zesiewicz 2012 and 13 completed the study. Ristori 2010 included 40 participants with ataxia from a range of aetiologies (eight SCA, eight Friedreich ataxia, one fragile X tremor/ataxia syndrome, 10 sporadic ataxia and 13 ataxic syndromes of unknown origin). Performance on the speech subscale was not broken down by diagnosis. Individuals with genetically confirmed Friedreich ataxia were recruited in the following studies: Cooper 2008 (50 participants), Di Prospero 2007 (48 participants), Lynch 2010 (70 participants), Lynch 2012 (31 participants) and Mariotti 2009 (16 participants). Trouillas 1995 recruited 26 participants clinically diagnosed with Friedreich ataxia (not genetically confirmed). Sobue 1983 initially recruited 290 participants with spinocerebellar degeneration (SCD) and reported on the speech outcomes of 214 participants with predominantly cerebellar forms of SCD at all time points. Zannolli 2012 recruited 13 participants with ataxia telangiectasia. Assadi 2007 involved 20 individuals with ataxia of various aetiologies, including four with Friedreich ataxia, nine with SCA, one with dentatorubral‐pallidoluysian atrophy (DRPLA) and six with idiopathic ataxia.
Interventions
Wessel 1995 administered the levorotatory form of hydroxytryptophan (L‐5HT) orally in a dose of 1000 mg/day. Each treatment phase, with L‐5HT or placebo, lasted 10 months, after which the participants crossed over to the other phase. Investigating treatment effects over 10 months in a progressive neurodegenerative disease makes delineation of treatment versus placebo effects difficult. In Trouillas 1995, L‐5HT was administered for a period of six months. Dosage was dependent upon participant weight, being 200 mg/day to 600 mg/day during the first month and 300 mg/day to 900 mg/day for the remaining five months. Filla 1988 administered a daily dose of 2 mg and 4 mg of thyrotropin‐releasing hormone (TRH) tartrate or placebo intramuscularly, each over a period of one month, in an ABCB design. In Sobue 1983, the three treatment arms received either TRH tartrate 0.5 mg, TRH tartrate 2 mg or placebo over a period of two weeks. The nonpharmaceutical trial, Miyai 2012, studied behavioural therapy with a delayed intervention (no treatment) versus an immediate intervention paradigm. The report describes the intervention as a mix of occupational therapy and physiotherapy sessions delivered every week day and for one hour on the weekend over four weeks. In Zesiewicz 2012, participants' response to varenicline (Chantrix) (four weeks for titration and four weeks at a dose of 1 mg twice daily) was compared to the response to placebo over 56 days. Ristori 2010 compared riluzole (two 50 mg tablets daily) and placebo over eight weeks. Di Prospero 2007 and Lynch 2010 compared various doses of idebenone with placebo over a period of six months. Betamethasone was compared with placebo in Zannolli 2012, a cross‐over study with two 30‐day phases. The participants were given a full dose of betamethasone (0.1 mg/kg/day) for the first and last third of the phase and a tapered dose during the middle 10 days of each phase. Cooper 2008 was a comparison of high‐ and low‐dose coenzyme Q10 (CoQ10) plus vitamin E (600 mg CoQ10 and 2100 IU vitamin E per day versus 30 mg CoQ10 and 4 IU vitamin E per day). Lynch 2012 administered both high doses (750 mg/day) and low doses (510 mg/day) of α‐tocopheryl quinone and compared each to placebo over a period of 28 days. Mariotti 2009 was a comparison of recombinant human erythropoietin (rhuEPO) versus placebo over a period of 24 weeks. Dosages of rhuEPO were 20,000 IU every three weeks for nine weeks (visits one to three), 40,000 IU every three weeks for nine weeks (visits four to six) and 40,000 IU every two weeks for six weeks (visits seven to nine). Assadi 2007 treated participants with buspirone HCl 30 mg twice daily or placebo for 12 weeks.
Outcomes
Wessel 1995 employed syllable repetition rates as an index of motor speech performance. The extended duration of each phase of the study (10 months) makes it difficult to separate the effect of the drug from the influence of disease progression. Every two months, for six months after the start of treatment, Trouillas 1995 measured speech by timing participants as they produced a standard sentence. Assadi 2007, Cooper 2008, Di Prospero 2007, Filla 1988, Lynch 2010, Lynch 2012, Mariotti 2009, Miyai 2012, Ristori 2010, Zannolli 2012 and Zesiewicz 2012 all employed subjective clinician‐derived measures of severity to evaluate speech production, which was measured at various time points between 12 weeks and two years. Speech subscales of severity scales were analysed separately from the total score in Cooper 2008, Lynch 2012, Ristori 2010 and Zannolli 2012. Cooper 2008 also assessed speech by measuring the timing of a standard passage and syllable repetition. Sobue 1983 measured speech on a 14‐point dysarthria scale as rated by the neurologist.
Lynch 2012, Mariotti 2009, Miyai 2012, Zannolli 2012 and Zesiewicz 2012 included functional health as an outcome measure.
Excluded studies
The reasons for exclusion of the 25 excluded studies are given in Characteristics of excluded studies. The predominant reason for exclusion was that the studies were not RCTs or quasi‐RCTs. We contacted the authors of five RCTs for additional information.
Risk of bias in included studies
For details of our 'Risk of bias' assessments see Figure 1.
Allocation
All studies randomly allocated participants to either treatment or placebo (or no treatment in the delayed treatment arm of Miyai 2012). The method of random selection was not clear in seven trials (Assadi 2007; Filla 1988; Mariotti 2009; Sobue 1983; Trouillas 1995; Wessel 1995; Zannolli 2012). The block randomisation and small sample size within Zesiewicz 2012 might have prevented equal distribution of age and disease severity between the trial arms. The method of randomisation was not clear in Lynch 2012 and the placebo group appears to have had a more severe mean clinical rating. As a result we considered both these trials as at high risk of bias. The method of allocation concealment was not clear in eight studies (Assadi 2007; Filla 1988; Lynch 2012; Mariotti 2009; Sobue 1983; Trouillas 1995; Wessel 1995; Zannolli 2012), but at low risk of bias in the rest of the included studies.
Blinding
We judged blinding of participants in Wessel 1995 and Miyai 2012 to be at high risk of bias. Wessel 1995 did not state whether there was blinding for the speech outcomes or whether the same individual both assessed (recorded) and analysed speech. In addition, common adverse events, such as the gastrointestinal side effects seen with L‐5HT in doses of 900 mg to 1000 mg orally per day, potentially undermine blinding. Eleven included studies assessed speech perceptually (subjectively) via clinician‐derived measures of severity. When speech assessments are conducted within standardised clinical assessment protocols, information collected as part of the clinical assessment is potentially able to influence judgements about speech function. Assessment of this nature is susceptible to assessor bias and low levels of reliability. The investigators who performed Miyai 2012 were reportedly blinded to the group allocation; however, the additional effects of inpatient stay and treatment regime were not clearly described. Data from Trouillas 1995 are at a high risk of detection bias due to the experience of adverse effects in the treatment arm. Blinding of outcome assessment may have been undermined in Sobue 1983, as it is not clear whether clinicians who assessed the safety of the treatment also rated its efficacy. The method of blinding of participants and assessors is not clear in Assadi 2007 and Mariotti 2009. The blinding of investigators is also unclear in Zannolli 2012 and Lynch 2012, although participants were adequately blinded. There is a low risk of both performance and detection bias in Cooper 2008, Di Prospero 2007, Filla 1988, Lynch 2010, Ristori 2010 and Zesiewicz 2012.
Incomplete outcome data
Four studies were at high risk of attrition bias (Sobue 1983; Trouillas 1995; Wessel 1995; Zesiewicz 2012). Wessel 1995 reported a limited data set (four out of 19 potential participants) for the only relevant subgroup (Friedreich ataxia). The reasons for these dropouts were not addressed. Only five of the 10 participants completed the placebo arm and eight of the 10 completed the varenicline arm of Zesiewicz 2012. Despite these dropouts, data from nine participants in each group were included in the outcome description. Seven out of 26 participants did not complete Trouillas 1995, two of whom withdrew due to adverse effects of the treatment. Six out of 220 participants with a predominantly cerebellar form of spinocerebellar degeneration in Sobue 1983 dropped out of the study. One participant did not complete Miyai 2012 due to death. Filla 1988 reported one dropout in the Friedreich ataxia group; we considered the risk of bias to be unclear. One participant from each of the experimental and control groups of Ristori 2010 withdrew consent prior to receiving riluzole or placebo and were therefore removed from the analysis. One participant from Di Prospero 2007 on low‐dose idebenone withdrew due to illness prior to follow‐up assessments, and we assessed the risk of bias as unclear. Two more participants were later removed as they had started rehabilitation during the trial. Results of ataxic symptom visual analogue scales were reported for only 218 participants who were classified with a diagnosis of a predominantly cerebellar form of SCD. At the conclusion of the trial, investigator‐rated speech outcomes were reported for 214 participants and participant‐rated speech outcomes were reported for 210 participants. The reasons for these reduced numbers are not addressed. Three out of 13 participants from Zannolli 2012 were excluded from the per protocol analysis; however, statistical analyses were provided for the ITT group. Of the 50 participants in Cooper 2008, four withdrew from the high‐dose group and three withdrew from the low‐dose group. One participant in the high‐dose treatment arm of Lynch 2012 discontinued treatment due to a protocol violation. One of the 20 participants in Assadi 2007 withdrew, having moved away from the treatment site. All participants completed the Lynch 2010 and Mariotti 2009 studies according to the protocol.
Selective reporting
The primary outcome related to speech (syllable duration in ms) in Wessel 1995 was reported for a small selection of the Friedreich ataxia group. Mean group scores and standard deviations were reported. It is unclear if any additional outcomes were analysed but not reported. Improvements in ataxic symptoms in Sobue 1983 were statistically tested using comparisons of the frequency of participants in each treatment arm who either improved by at least one point on the 14‐point scale or by at least two points on the 14‐point scale. These methods make interpretation of clinical significance difficult. We considered both these studies at high risk of bias from selective reporting. We considered two studies at unclear risk of bias (Di Prospero 2007; Zesiewicz 2012). The report of Zesiewicz 2012 does not state the timing of assessments. The second planned period of the experiment, which was to include a cross‐over component, was abandoned due to the high dropout rate observed in the initial period. All outcomes were reported in Di Prospero 2007; however, two statistical analyses were completed, based on the hypothesis that the second would remove floor and ceiling effects arising from the nature of the assessments used. All assessments were reported in Ristori 2010, but the study did not report on results grouped by aetiology of ataxic symptoms. All planned outcomes were reported in Assadi 2007, Cooper 2008, Filla 1988, Lynch 2010, Lynch 2012, Mariotti 2009,Miyai 2012, Ristori 2010, Trouillas 1995 and Zannolli 2012.
Other potential sources of bias
We judged other potential sources of bias (such as bias related to the study design, analysis used or some other problem) as unclear for Filla 1988. We assessed Miyai 2012 as at high risk of bias in this category as the participants were treated in an inpatient setting within a hospital and may have been exposed to additional therapeutic care during their stay. Any clinical discrepancies between comparison groups may influence group responsiveness to treatment. A failure to control for multiple comparisons can also introduce error into statistical outcomes. Cooper 2008 reported the results of both a randomised trial and a comparison of a cross‐sectional data set of untreated patients. The clinical details of this cross‐sectional data set were not described and we considered the risk of other bias to be unclear in this study. We identified no additional sources of bias in the 11 other trials (Assadi 2007; Di Prospero 2007; Lynch 2010; Lynch 2012; Mariotti 2009; Ristori 2010; Sobue 1983; Trouillas 1995; Wessel 1995; Zannolli 2012; Zesiewicz 2012).
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7; Table 8; Table 9; Table 10; Table 11
Hydroxytryptophan (L‐5HT) versus placebo
Two studies compared L‐5HT to placebo in participants with degenerative cerebellar disorders (Trouillas 1995; Wessel 1995). See Characteristics of included studies and Table 1.
Primary outcome: Percentage change (improvement) in overall speech production immediately following completion of the intervention or later, measured by any validated speech assessment tool
Not reported.
Secondary outcomes
Change in isolated movement, objective and subjective measures of speech production within one month post intervention
Wessel 1995 measured repetition rates for syllables as an index of motor speech performance. The participants were asked to produce chains of consonant‐vowel syllables at maximum speed. The three syllables /pa/, /ta/ and /ka/, involving movements of three different articulators, were used. Each task was performed twice in the course of a more comprehensive articulation protocol, resulting in a total of six repetition tasks. Mean syllable durations were calculated from smoothed sound pressure level contours of the speech wave. The lowest value that was obtained throughout the six tasks, that is, each participant's optimum performance, was used as the dependent variable. Syllable length was not significantly different between the L‐5HT and placebo conditions, with a 2 ms (standard deviation range of 38 ms to 73 ms) increase in syllable duration in both groups. This analysis was based on only four of 19 participants with Friedreich ataxia.
Trouillas 1995 examined the rate of speech production among 19 participants by measuring the time required to produce a standard sentence. The mean time for the treatment arm reduced by 0.3 s and the mean time for the control arm increased by 0.2 s. This difference was not statistically significant. There was no meaningful difference between groups.
Meta‐analysis of the two studies was not possible due to the heterogeneity of outcome measures. Results are provided in parallel in Table 1. Confidence intervals (CIs) were not calculable in Wessel 1995, as the report did not provide a measure of variance of the change following treatment.
Change in quality of life scores related to communication within one month post intervention
Not reported.
Generic quality of life measures a minimum of one month post intervention
Not reported.
Adverse effects
Wessel 1995 (19 participants) reported minor gastrointestinal adverse effects in eight participants on L‐5HT. Five participants who received the placebo treatment complained of minor gastrointestinal side effects. A risk ratio (RR) was not calculable as data were provided for all participants and not broken down by type of disorder.
Trouillas 1995 (19 participants) reported minor gastrointestinal side effects in six participants on L‐5HT and two participants from the control group. Two of those participants on L‐5HT left the study as a result of the adverse effects.
Burdens
Not reported.
Economic outcomes
Not reported.
Thyrotropin‐releasing hormone tartrate (TRH‐T) versus placebo
Two studies compared TRH‐T versus placebo in participants with degenerative cerebellar disorders (Filla 1988; Sobue 1983). See Characteristics of included studies and Table 2.
Primary outcome: Percentage change (improvement) in overall speech production immediately following completion of the intervention or later, measured by any validated speech assessment tool
Not reported.
Secondary outcomes
Change in isolated movement, objective and subjective measures of speech production within one month post intervention
Filla 1988 employed a subjective rating (on a scale of zero to four) of speech impairment based on clinical impression during administration of the Inherited Ataxias Clinical Rating Scale (IACRS). We requested data on speech performance from the study authors, but data were not available. The report did not describe specific data on speech changes; however, repeated measures ANOVA (n = 30, F = 4.69, df = 4,116, P value < 0.001) showed significant changes between speech ratings before and after treatment with TRH‐T. The Neuman‐Keuls method was used to identify sample means that were statistically significantly different between groups and time points. Significant differences were observed on both 2 mg of TRH (at two months P value < 0.05) and 4 mg of TRH (at four months P value < 0.01) as well as the placebo condition at four months (P value < 0.05). Raw or summative data on speech were not provided. No longer‐term outcomes were described beyond the washout period of the compound.
Sobue 1983 reported results of investigator‐rated speech assessment and participant‐rated impact of dysarthria on activities of daily living (ADL) for a subset of participants (n = 214) with a predominantly cerebellar form of SCD. Statistical tests of the symptoms of SCD were based on the numbers of participants in each treatment arm who recorded improvements either greater than one point or greater than two points on a 14‐point scale. Data from the investigator‐rated scales showed statistically significantly differences across placebo, 0.5 mg of TRH‐T and 2 mg of TRH‐T after one and two weeks of treatment respectively. Statistically significant differences were not observed on either measure one week after treatment concluded. Participant ratings of the impact of dysarthria on ADL were not statistically significantly different on any dose or placebo at any stage of the trial.
The two studies investigating TRH‐T provided neither raw data nor measures of variance, therefore CI could not be calculated.
Change in quality of life scores related to communication within one month post intervention
Not reported.
Generic quality of life measures a minimum of one month post intervention
Not reported.
Adverse effects
Filla 1988 (30 participants) reported a combination of adverse effects on the TRH‐T treatment: nausea (18), vomiting (three), hot flushes (eight), sweating (three), headaches (four), dizziness (four), palpitations (one) and urgent micturition (three). The number of adverse effects was not reported for participants taking the placebo. All adverse effects occurred immediately after administration of the drug and lasted a few minutes. A risk ratio (RR) was not calculable as studies provided data on the number of adverse effects, not the number of participants experiencing adverse effects.
Sobue 1983 (256 participants) reported 50 adverse effects among 101 participants on 2 mg TRH‐T, including 14 psycho‐neurologic effects (e.g. headache, dizziness, drowsiness), 14 cardiovascular effects (e.g. hot feeling, flushing, palpitation, chest oppressed feeling), 28 gastrointestinal effects (e.g. nausea, vomiting, abdominal pain) and 20 other effects (e.g. urinary frequency, general malaise, sweating). Of the 92 participants on 0.5 mg TRH‐T, 35 experienced adverse effects (four psycho‐neurologic, 12 cardiovascular, 29 gastrointestinal, 13 other) and 20/97 participants on placebo experienced adverse effects (eight psycho‐neurologic, nine cardiovascular, two gastrointestinal, six other). Two participants experienced severe adverse effects (not described) but were able to continue the treatment.
Burdens
Not reported.
Economic outcomes
Not reported.
Varenicline versus placebo
One study compared varenicline versus placebo in participants with spinocerebellar ataxia (SCA) type 3 (Zesiewicz 2012). See Characteristics of included studies and Table 3.
Primary outcome: Percentage change (improvement) in overall speech production immediately following completion of the intervention or later, measured by any validated speech assessment tool
Not reported.
Secondary outcomes
Change in isolated movement, objective and subjective measures of speech production within one month post intervention
Subjective rating of speech impairment based on clinical impression formed during administration of the Scale for the Rating and Assessment of Ataxia (SARA). The SARA contains a subscale focusing on speech function (a scale of zero to six where the lower value indicates an improvement). Speech did not statistically significantly improve (n = 13, P value = 0.11) in either condition on the speech subscale of the SARA. The control group demonstrated a mean decrease of 0.39 on the speech subscale of the SARA and the intervention group had a mean decrease of 0.45. CIs could not be calculated as variance in change was not reported.
Change in quality of life scores related to communication within one month post intervention
Not reported.
Generic quality of life measures a minimum of one month post intervention
Functional health and wellbeing was assessed using the SF‐36. No statistically significant improvement in functional health occurred in either condition, with a 0.33 mean increase in the varenicline condition and a 1.42 mean decrease in the placebo condition (n = 13).
Adverse effects
The treatment and placebo arms contained nine participants each. In the varenicline arm, six participants reported nausea, one vomiting, one constipation, one disturbed sleep, one fatigue, two vivid dreams, one irritability, one auditory hallucinations, one spasticity, one increased stiffness in lower extremities, one increased dizziness, one increased tremor, two tingling in legs, one increased imbalance, one dizziness, one shaky legs, one leg cramps, one shivering, one blotchy feet and one cold feet. Two participants receiving varenicline decreased their dosage during the trial because of adverse events. In total there were 27 reported adverse effects for the varenicline group and 19 reported adverse effects in the placebo group, including one serious case of urosepsis. Four participants in the placebo condition discontinued the study: one for urosepsis, one for muscle pain and two for noncompliance. One participant in the varenicline group discontinued the study because of auditory hallucinations, which were later attributed to a sleep disorder. A RR was not calculable as the study provided data on the number of adverse effects, but not the number of participants experiencing adverse effects in total.
Burdens
Not reported.
Economic outcomes
Not reported.
Riluzole versus placebo
One study compared riluzole versus placebo in a group of people with mixed hereditary ataxias (Ristori 2010). See Characteristics of included studies and Table 4.
Primary outcome: Percentage change (improvement) in overall speech production immediately following completion of the intervention or later, measured by any validated speech assessment tool
Not reported.
Secondary outcomes
Change in isolated movement, objective and subjective measures of speech production within one month post intervention
The subjective rating of speech impairment was based on a clinical impression formed during administration of the dysarthria component of the International Cooperative Ataxia Rating Scale (ICARS) (scale zero to eight): speech statistically significantly improved in the riluzole condition (n = 38). The mean change in the ICARS dysarthria subscale was ‐0.74 (standard deviation (SD) 0.81) versus 0.05 (SD 0.40) in the placebo group (P value < 0.001). Data were provided for the participants as a whole and not by disease type.
We requested additional data on speech performance from the study authors, but data were not available.
Change in quality of life scores related to communication within one month post intervention
Not reported.
Generic quality of life measures a minimum of one month post intervention
Not reported.
Adverse effects
In the riluzole arm (20 participants), two participants were found to have an increase in alanine aminotransferase, a measure of liver health (1.5 times above normal), and one participant presented with transient vertigo. In total there were three reported adverse effects for the riluzole group and one reported adverse effect (transient vertigo) in the placebo group (20 participants). A RR was not calculable as only one adverse event was observed in the placebo group.
Burdens
Not reported.
Economic outcomes
Not reported.
Idebenone versus placebo
Two studies compared idebenone versus placebo in participants with Friedreich ataxia (Di Prospero 2007; Lynch 2010). Di Prospero 2007 compared low, intermediate and high doses of idebenone with placebo and Lynch 2010 compared low and high doses of idebenone with placebo. See Characteristics of included studies and Table 5.
Primary outcome: Percentage change (improvement) in overall speech production immediately following completion of the intervention or later, measured by any validated speech assessment tool
Not reported.
Secondary outcomes
Change in isolated movement, objective and subjective measures of speech production within one month post intervention
Di Prospero 2007 measured speech subjectively on a subscale of the ICARS. A Jonckheere trend test on all participants who completed the study (n = 47) revealed a statistically significant improvement in overall ICARS scores, with the authors reporting that eye movement and speech subsections were responsible for the majority of observed variance. A subgroup of participants not including non‐symptomatic and non‐ambulatory participants also revealed a statistically significant difference in overall ICARS scores on both Jonckheere and ANCOVA statistical tests. The estimated difference in the change in ICARS score for the subset of participants was 1.99 (95% CI ‐3.57 to 7.54) for the low‐dose group (11 participants), 6.24 (95% CI 1.60 to 10.89) for the intermediate‐dose group (13 participants) and 7.76 (95% CI 2.96 to 12.56) for the high‐dose group (12 participants). Speech subscales were not reported separately from overall ICARS scores.
By contrast, Lynch 2010 found no statistically significant differences between idebenone and placebo groups in scores on the ICARS or Friedreich Ataxia Rating Scale (FARS) (n = 70). Improvements (reductions) in mean ICARS scores for each group were ‐2.8 (standard error of the mean (SEM) 1.07) for placebo (n = 24), ‐4.3 (SEM 1.22) for the 450/900 mg dose (n = 22) and ‐2.8 (SEM 1.43) for the 1350/2250 mg dose (n = 24).
We requested data on speech performance from the respective study authors, but data were not available. We were therefore unable to report measures of change.
Change in quality of life scores related to communication within one month post intervention
Not reported.
Generic quality of life measures a minimum of one month post intervention
Not reported.
Adverse effects
There were 200 adverse effects reported in the treatment groups and 58 in the placebo group of Di Prospero 2007 (48 participants). Two adverse events were serious enough to result in hospitalisation. One serious adverse event involved chest pain and occurred in the placebo group. The second event involved nausea, vomiting and dehydration, which occurred in the low‐dose idebenone group three weeks after the completion of the trial and was judged to be unrelated to the study medication. Only one adverse event was thought to be related to the study drug. This was a case of neutropenia in a male participant in the high‐dose idebenone group. The neutropenia occurred after six months of treatment and resolved within a week of discontinuation of idebenone. Statistical analysis revealed no statistically significant difference in the incidence of adverse events between the four groups.
Lynch 2010 (70 participants) reported two serious adverse events. One participant reported chest pain unrelated to cardiac involvement and another experienced idiopathic thrombocytopenic purpura; however, both participants had a prior history of the respective condition. Both incidents spontaneously resolved while the participants were still taking study medication and the events were considered unrelated to treatment. Less serious adverse effects occurred at a comparable rate in idebenone and placebo groups; except for gastrointestinal tract irritations which were more frequent in the high‐dose idebenone group, though this difference was not statistically significant. Adverse events affected 14 participants in the high‐dose treatment group, seven participants in the low‐dose group and 10 participants in the placebo group.
Burdens
Not reported.
Economic outcomes
Not reported.
Physiotherapy and occupational therapy versus no treatment
One study compared physiotherapy and occupational therapy versus no (delayed) treatment in participants with SCA6 and SCA31 (and idiopathic SCA) (Miyai 2012). See Characteristics of included studies and Table 6.
Primary outcome: Percentage change (improvement) in overall speech production immediately following completion of the intervention or later, measured by any validated speech assessment tool
Not reported.
Secondary outcomes
Change in isolated movement, objective and subjective measures of speech production within one month post intervention
Miyai 2012 assessed speech using a subjective rating based on clinical impression during administration of the SARA. The SARA contains a subscale focusing on speech function (the subscale ranges from zero to six, where the lower value indicates an improvement). A mean decrease of 0.1 (standard error (SE) 0.1) on the speech subscale was observed in the intervention group and a mean increase of 0.1 (SE 0.1) was observed in the control group after treatment (four weeks); neither change was statistically significant; the between‐group mean difference (MD) was ‐0.20 (95% CI ‐0.48 to 0.08). The analysis was based on the results of all 42 participants who were recruited.
Change in quality of life scores related to communication within one month post intervention
Not reported.
Generic quality of life measures a minimum of one month post intervention
Miyai 2012 assessed functional independence using the Functional Independence Measure (FIM). Significant differences were observed between baseline, post intervention and four‐week post intervention scores on the total FIM and FIM motor scores, with a mean 1.2 (SE 0.3) increase in the treatment condition and a mean 0.2 (SE 0.3) decrease in the no treatment condition on the total FIM at four weeks (MD 1.40, 95% CI 0.57 to 2.23) and a mean 1.1 (SE 0.3) increase with treatment and a mean 0.1 (SE 0.3) decrease with placebo on the motor FIM at four weeks (MD 1.20, 95% CI 0.37 to 2.03).
Adverse effects
None reported. One participant died at week 17 (after the randomised period of the study) from cerebral haemorrhage. This event was not reported as a consequence of the intervention.
Burdens
Not reported.
Economic outcomes
Not reported.
Betamethasone (BETA) versus placebo
One study compared BETA (betamethasone disodium phosphate) with placebo in participants with ataxia telangiectasia, in a randomised, double‐blind, placebo‐controlled, cross‐over trial (Zannolli 2012). See Characteristics of included studies and Table 7.
Primary outcome: Percentage change (improvement) in overall speech production immediately following completion of the intervention or later, measured by any validated speech assessment tool
Not reported.
Secondary outcomes
Change in isolated movement, objective and subjective measures of speech production within one month post intervention
Zannolli 2012 measured speech using a subscale of the ICARS and compared BETA and placebo treatments with baseline. The median reduction in severity on this subscale was one point greater with BETA than the median reduction for placebo, within the intention‐to‐treat (ITT) population (MD ‐1, 95% CI ‐2.5 to ‐0.5, n = 13).
Change in quality of life scores related to communication within one month post intervention
Not reported.
Generic quality of life measures a minimum of one month post intervention
No significant difference was identified using quality of life measures (Child Health Questionnaire) (n = 10). The magnitude of change was not reported.
Adverse effects
One participant in Zannolli 2012 (total of 13 enrolled participants) experienced asthenia during drug tapering, which did not require medical intervention. One further participant experienced mild mood swings and depressed attitude. Moon face was present in eight participants on BETA. Small increases in body weight occurred in 12 participants on BETA and four participants on placebo.
Burdens
Not reported.
Economic outcomes
Not reported.
Coenzyme Q10 (CoQ10) and vitamin E in high dose and low dose
Cooper 2008 compared high and low doses of CoQ10 and vitamin E in participants with genetically confirmed Friedreich ataxia, in a double‐blind, randomised trial. See Characteristics of included studies and Table 8.
Primary outcome: Percentage change (improvement) in overall speech production immediately following completion of the intervention or later, measured by any validated speech assessment tool
Not reported.
Secondary outcomes
Change in isolated movement, objective and subjective measures of speech production, measured within one month post intervention
Cooper 2008 measured ICARS scores of 43 participants in the high‐dose (n = 22) and low‐dose (n = 21) groups and found no difference in change between the two. The ICARS speech subscale demonstrated a 0.05 decrease in the low‐dose group and a 0.02 decrease in the high‐dose group. Syllable repetitions decreased by 0.6 in the low‐dose group and 0.1 in the high‐dose group. Time taken (s) to read a standard passage increased in the low‐dose group by 0.7 and by 3.0 s in the high‐dose group. The rate of change of the ICARS speech subscale was reported but not significantly different between groups. Post hoc analysis compared the two groups on high and low doses with cross‐sectional data from 77 untreated people with Friedreich ataxia. Participants were divided into four groups based on the size of GAA1 repeat length. Post hoc analysis found that 10 participants in the low‐dose group (n = 21) and 11 participants in the high‐dose group (n = 22) had changes in ICARS scores over two years, which were below the 95% CIs of the cross‐sectional data. Sixteen of these participants had an absolute improvement in ICARS score. Speech was also measured by the time taken to read a standard passage and the repetitions of the syllables /pa/ and /ta/. None of these measures signified a statistically significant improvement for low‐dose versus high‐dose groups.
Change in quality of life scores related to communication within one month post intervention
Not reported.
Generic quality of life measures a minimum of one month post intervention
Not reported.
Adverse effects
No serious adverse events occurred among the 50 participants who started the trial. Minor adverse events included one participant with increased bowel frequency (high‐dose group) and another participant with prolonged nausea (low‐dose group).
Burdens
Not reported.
Economic outcomes
Not reported.
Buspirone versus placebo
Assadi 2007 compared buspirone with placebo in participants with a variety of hereditary and idiopathic SCAs in a randomised, double‐blind, placebo‐controlled trial. See Characteristics of included studies and Table 9.
Primary outcome: Percentage change (improvement) in overall speech production immediately following completion of the intervention or later, measured by any validated speech assessment tool
Not reported.
Secondary outcomes
Change in isolated movement, objective and subjective measures of speech production, within one month post intervention
Buspirone had no effect on total ICARS scores (n = 19). The speech subscale was not reported separately as an outcome. We requested data on speech performance from the study authors, but data were not available.
Change in quality of life scores related to communication within one month post intervention
Not reported.
Generic quality of life measures a minimum of one month post intervention
Not reported.
Adverse effects
Minor adverse events included dizziness (one participant on placebo, four on buspirone) and drowsiness (one participant on placebo, three on treatment). No serious adverse events occurred.
Burdens
Not reported.
Economic outcomes
Not reported.
α‐tocopheryl quinone versus placebo
Lynch 2012 compared two doses of the antioxidant α‐tocopheryl quinone with placebo, in 31 participants with Friedreich ataxia. See Characteristics of included studies and Table 10.
Primary outcome: Percentage change (improvement) in overall speech production immediately following completion of the intervention or later, measured by any validated speech assessment tool
Not reported.
Secondary outcomes
Change in isolated movement, objective and subjective measures of speech production, measured within one month post intervention
Speech was measured using a subscale of the FARS within the bulbar section of the exam. Data on speech outcomes within the FARS were requested from the authors but not available.
Change in quality of life scores related to communication within one month post intervention
Not reported.
Generic quality of life measures a minimum of one month post intervention
The SF‐36 (scored out of 100, higher is better) demonstrated an increase in self reported quality of life for the placebo group (change of +3.26 (SD ± 5.0)) on day 28 compared to baseline, a decrease for the low‐dose group (mean change of ‐4.77 (SD ± 5.7) versus baseline) and a marginal decrease for the high‐dose group (change of ‐0.01 (SD ± 5.31) versus baseline). These results were not statistically significant.
Adverse effects
No severe drug‐related adverse events occurred. Minor adverse events occurred equally across the treatment arms.
Burdens
Not reported.
Economic outcomes
Not reported.
Erythropoietin (rhuEPO) versus placebo
Mariotti 2009 compared rhuEPO and placebo in a group of 16 individuals with genetically confirmed Friedreich ataxia, in a randomised, placebo‐controlled, double‐blind, dose‐response pilot trial. See Characteristics of included studies and Table 11.
Primary outcome: Percentage change (improvement) in overall speech production immediately following completion of the intervention or later, measured by any validated speech assessment tool
Not reported.
Secondary outcomes
Change in isolated movement, objective and subjective measures of speech production within one month post intervention
Speech was measured as part of the SARA. The speech subscale scores were not reported separately. No difference was found on the SARA between rhuEPO and placebo groups (P value = 0.60, n = 16). The CI could not be calculated for Mariotti 2009, as no measure of the variance of change was reported. We requested data on speech performance from the study authors, but none were available.
Change in quality of life scores related to communication within one month post intervention
Not reported.
Generic quality of life measures a minimum of one month post intervention
Quality of life was measured with the SF‐36. No difference was observed between the rhuEPO and placebo groups (n = 16, P value = 0.18).
Adverse effects
No serious adverse events occurred. Three female participants demonstrated sideropenic anaemia (two in the rhuEPO group and one in the placebo group). All participants underwent iron therapy.
Burdens
Not reported.
Economic outcomes
Not reported.
Discussion
There are currently no studies describing an effective and clinically significant treatment for speech disorder (dysarthria) in any hereditary ataxia syndrome. Fourteen studies met the inclusion criteria for the current review (Assadi 2007; Cooper 2008; Di Prospero 2007; Filla 1988; Lynch 2010; Lynch 2012; Mariotti 2009; Miyai 2012; Ristori 2010; Sobue 1983; Trouillas 1995; Wessel 1995; Zannolli 2012; Zesiewicz 2012). There were no studies employing behavioural or instrumental interventions designed specifically to improve speech. One double‐blind, placebo‐controlled, randomised study of the drug riluzole demonstrated slight improvements in overall dysarthria in a cohort of 40 individuals with cerebellar ataxia of mixed aetiologies (Ristori 2010). The investigators utilised a subjective assessment of speech to measure change and included sporadic ataxia and ataxia of unknown origin alongside hereditary ataxias. A second study, Zannolli 2012, demonstrated slight improvements in dysarthria rating with betamethasone (BETA), in a cohort of 13 individuals with ataxia telangiectasia using a subjective measure of speech. Di Prospero 2007 identified improvements in International Cooperative Ataxia Rating Scale (ICARS) score in a study of 48 people with Friedreich ataxia taking idebenone; however, improvements were small and a larger idebenone study involving 70 people with Friedreich ataxia by Lynch 2010 did not find any improvement, despite mirroring the dose and length of the Di Prospero 2007 study. Sobue 1983 identified slight, but not clinically meaningful, improvements on a subjective measure of speech performance in a thyrotropin‐releasing hormone (TRH) tartrate trial of 290 participants with spinocerebellar degeneration. Conflicting findings were reported by Filla 1988, who did not find a significant effect with the same drug in a study of 16 Friedreich ataxia and spinocerebellar ataxia (SCA) participants. Assadi 2007, Cooper 2008, Mariotti 2009, Trouillas 1995, Wessel 1995 and Zesiewicz 2012 did not find statistically significant differences between intervention and placebo in each of their pharmaceutical trials. Miyai 2012 employed a combination of physiotherapy and occupational therapy within an inpatient setting. Short‐term improvements in 'functional independence' were documented in the intervention group (compared to the no intervention group) but treatment effects were not present 12 weeks post intervention.
Assadi 2007, Wessel 1995, Zannolli 2012 and Filla 1988 utilised a double‐blind, cross‐over, placebo‐controlled methodology. Di Prospero 2007, Lynch 2010, Lynch 2012, Mariotti 2009, Ristori 2010, Sobue 1983, Trouillas 1995 and Zesiewicz 2012 were double‐blind but not cross‐over studies and Miyai 2012 was single‐blinded. Cooper 2008 utilised a double‐blind randomised design with a low‐dose group as placebo. The trials aimed to determine whether treatment with pharmacotherapy (L‐5HT in Wessel 1995 and Trouillas 1995; TRH‐T in Filla 1988 and Sobue 1983; varenicline in Zesiewicz 2012; riluzole in Ristori 2010; idebenone in Di Prospero 2007 and Lynch 2010; betamethasone in Zannolli 2012; CoQ10 with vitamin E in Cooper 2008; buspirone in Assadi 2007; a‐tocopheryl quinone in Lynch 2012; and rhuEPO in Mariotti 2009) or physiotherapy (Miyai 2012) could improve the conditions of people with degenerative cerebellar ataxia. Only three of these studies employed an objective motor speech outcome measure (repetition rates for syllables or timing of standard passages) (Cooper 2008; Trouillas 1995; Wessel 1995). The remaining studies evaluated speech performance via subjective clinician‐derived measures of severity as a component of an ataxia disease rating scales. Subjective speech assessments lack adequate levels of intra‐ and inter‐rater reliability and are generally not psychometrically appropriate for monitoring change (Vogel 2010; Vogel 2011; Vogel 2014).
Summary of main results
Given the paucity of empirical data on treatment for speech disorder in Friedreich ataxia and other hereditary ataxia syndromes, it was not possible to determine whether any treatment is clearly effective or if one type of treatment is more effective than another for improving speech production. No behavioural or instrumental treatment studies specifically targeting speech were found within the search results.
Overall completeness and applicability of evidence
There is no evidence of an effective treatment for speech disorder in any hereditary ataxia syndrome. The majority of included trials employed subjective assessments of speech production and no studies formally considered overall intelligibility or alternative metrics of performance (for example, standardised assessment tools). To date, the available evidence for treatment of speech disorder in hereditary ataxia is incomplete and not applicable in a clinical setting.
Quality of the evidence
The very high attrition rate within the Friedreich ataxia subgroup in Wessel 1995 limits the interpretability of these data. The study was also at high risk of bias due to the selective reporting of participant performance on a limited outcome regime. Sobue 1983 is at a high risk of attrition bias as speech outcomes for several participants were not reported, with no explanation. Trouillas 1995, Wessel 1995 and Zesiewicz 2012 are also at risk of attrition bias. Only three studies, Cooper 2008, Trouillas 1995 and Wessel 1995, employed an objective motor speech outcome measure. The use of a subjective speech assessment conducted by potentially poorly blinded examiners limits the accuracy of data in Assadi 2007, Lynch 2012, Mariotti 2009, Miyai 2012 and Zannolli 2012. Speech outcomes within several studies were further confounded as researchers used subjective measures of speech, which were not reported separately to the total clinical severity rating (Assadi 2007; Di Prospero 2007; Filla 1988; Lynch 2010; Mariotti 2009; Miyai 2012; Zesiewicz 2012). Perceptual (subjective) evaluation of speech is restricted by a number of psychometric limitations including poor intra‐ or inter‐rater reliability, floor and ceiling effects, and the use of discrete rather than continuous variables (Vogel 2010; Vogel 2011; Vogel 2014). This means that the quality of the current evidence base for treatment of speech disorder in hereditary ataxia is no better than low.
Potential biases in the review process
This review only utilised published data available through searches of electronic databases. We contacted the authors of five methodologically appropriate randomised controlled trials (RCTs) (Di Prospero 2007; Filla 1988; Lynch 2010; Lynch 2012; Ristori 2010), with the aim of acquiring unpublished speech data; however, no further data were available. This limited the information available to us, as additional data might have provided new insights into the outcomes of the treatments. The Filla 1988 trial authors provided a translation from the original Italian into English.
Agreements and disagreements with other studies or reviews
One other systematic review has been published on treatments for Friedreich ataxia; however, it did not deal with speech production but rather overall clinical functioning. Kearney 2012 evaluated the evidence for antioxidants and other pharmaceutical treatments for Friedreich ataxia and concluded that no RCT using idebenone or any other pharmacological treatment has shown significant benefit for the treatment of neurological symptoms associated with Friedreich ataxia.
Authors' conclusions
Implications for practice.
The absence of treatment trials, let alone randomised controlled trials (RCTs), of effective therapies that improve speech production in hereditary ataxia demonstrates the critical and urgent need for more research in this area. There is at present no high quality evidence on which to base practice.
Implications for research.
This review highlights the paucity of evidence for treatments of speech disorder in hereditary ataxias. The reasons for the clear lack of evidence are unknown; however, several factors may be responsible:
Speech deficits in neurodegenerative diseases are often considered secondary to other more life‐threatening comorbidities (for example, cardiomyopathy) or adjunct signs affecting mobility, and do not therefore receive priority within patient care plans.
Speech clinicians often employ non‐standardised treatment regimes tailored to individual needs in response to areas of deficit, making wider comparisons difficult.
The three studies that included an objective measure of speech production (Cooper 2008; Trouillas 1995; Wessel 1995), did not include broader and potentially more meaningful assessments looking at intelligibility or overall speech production. The remaining 11 studies employed subjective clinician‐derived measures of severity, which typically lack reliability and sensitivity to change (Vogel 2011). In order for accurate decisions to be made about the effectiveness of any treatment, assessment models need to be tailored to monitoring change, rather than identifying impairment. As stated, the speech assessment tools used in 11 of the 14 included studies used measures that were designed and well‐suited for classification or for identification of impairment. However, evidence from the clinical trials literature in related cognitive domains has shown that a different practical, methodological and statistical framework needs to be adopted in the assessment of behaviour to guide decisions about change (Collie 2003). Similar evidence in the speech literature has demonstrated the need for stable, reliable and sensitive assessment protocols in trials where speech is changing as a function of disease progression in Friedreich ataxia (Rosen 2012), Huntington's disease (Vogel 2012), treatment for depression (Mundt 2012), or induced neurophysiological change resulting from sustained wakefulness (Vogel 2010b). Briefly, assessments examining change need to be sensitive to impairment, while simultaneously remaining stable in the absence of true change. Tasks should be designed to limit the impact of practice and familiarity by remaining brief, easy to complete and suitably motivating. They should preferably have alternate forms, and be partnered with appropriate statistical models. Similarly, assessments that fail to satisfy assumptions of normality or do not utilise continuous variables may produce data with floor or ceiling effects, again limiting their sensitivity to change (Vogel 2014). In light of these considerations, the use of subjective evaluation tools in the included studies may mean that the true impact of the tested therapies is unknown.
Another methodological difficulty that can arise in clinical populations relates to the willingness of participants to be randomly allocated into either a treatment group or non‐intervention control group.
Mobility is often a significant barrier to participation. Individuals who rely on caregivers for transport may have limited capacity to participate in a treatment trial, which may be run intensively over consecutive weeks (for example, Lee Silverman Voice Treatment (Ramig 2001)).
The rare nature of many hereditary ataxias, combined with the varied diagnostic types and severity of the disease, makes it difficult to recruit large numbers of sufficiently similar participants to take part in trials.
For these barriers to be overcome, large, potentially multinational, multicentre randomised clinical trials need to be established to determine the effectiveness of specific treatment options.
Acknowledgements
We wish to thank the members of the Cochrane Neuromuscular Disease Review Group for their assistance and acknowledge the patients of the Friedreich Ataxia Clinic, Monash Medical Centre, Melbourne, Australia.
The editorial base of the Neuromuscular Disease Group receives support from the MRC Centre for Neuromuscular Diseases.
Appendices
Appendix 1. NMD Register (CRS) search strategy
#1 friedreich or freidreich or "spinocerebellar ataxia*" or "cerebellar ataxia*" or "spinocerebellar degeneration*" or "degenerative cerebellar" [REFERENCE] [STANDARD] #2 "dentatorubral‐pallidoluysian atrophy" or "myoclonic epilepsies" or "episodic ataxia*" [REFERENCE] [STANDARD] #3 ataxia* and Charlevoix‐Saguenay [REFERENCE] [STANDARD] #4 arsacs or Marinesco‐Sjogren [REFERENCE] [STANDARD] #5 MeSH DESCRIPTOR Spinocerebellar Degenerations Explode All [REFERENCE] [STANDARD] #6 Ataxia* and "oculomotor apraxia" [REFERENCE] [STANDARD] #7 Ataxia* and "vitamin E deficiency" [REFERENCE] [STANDARD] #8 "Ataxia telangiectasia" or Joubert NEAR syndrome* [REFERENCE] [STANDARD] #9 Joubert NEAR syndrome* [REFERENCE] [STANDARD] #10 ataxia* NEAR (hereditary or genetic*) [REFERENCE] [STANDARD] #11 ataxia* NEAR autosomal [REFERENCE] [STANDARD] #12 #1 or #2 or #3 or #4 or #5 or #6 or #7 or #8 or #9 or #10 or #11 [REFERENCE] [STANDARD] #13 speech or voice or vocal or dysarth* or dysphon* or anarth* or dyspros* [REFERENCE] [STANDARD] #14 "activities of daily living" or "quality of life" [REFERENCE] [STANDARD] #15 fars or sara or icars [REFERENCE] [STANDARD] #16 #13 or #14 or #15 [REFERENCE] [STANDARD] #17 #12 and #16 [REFERENCE] [STANDARD]
Appendix 2. CENTRAL search strategy
#1 " friedreich ataxia" or "freidreich ataxia" or "dentatorubral pallidoluysian atrophy" or arsacs or "Spinocerebellar Degeneration*" or "Marinesco Sjogren" #2 "cerebellar ataxia" or "cerebellar degeneration" or "spinocerebellar ataxia*" or "episodic ataxia*" or "Ataxia Telangiectasia" #3 Myoclonic near Epileps* #4 ataxia* and Charlevoix near Saguenay #5 Ataxia* and oculomotor near apraxia #6 Ataxia* and "vitamin E" near deficiency #7 Joubert near syndrome #8 ataxia* near (hereditary or genetic*) #9 #1 or #2 or #3 or #4 or #5 or #6 or #7 or #8 #10 #1 or #2 or #3 or #4 or #5 or #6 or #7 or #8 #11 speech or articulat* or voice or vocal or communicat* or dysarth* or dysphon* or anarth* or dyspros* #12 FARS or SARA or ICARS or "activities of daily living" or "quality of life" #13 #11 or #12 #14 #10 and #13
Appendix 3. MEDLINE (OvidSP) search strategy
Database: Ovid MEDLINE(R) <1946 to October Week 1 2013> Search strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 randomized controlled trial.pt. (387734) 2 controlled clinical trial.pt. (89736) 3 randomized.ab. (285393) 4 placebo.ab. (156181) 5 drug therapy.fs. (1760424) 6 randomly.ab. (198338) 7 trial.ab. (300539) 8 groups.ab. (1270218) 9 or/1‐8 (3284645) 10 exp animals/ not humans.sh. (4050082) 11 9 not 10 (2796869) 12 (Friedreich$ ataxia or Freidreich$ ataxia).tw. or Friedreich Ataxia/ (2698) 13 spinocerebellar ataxia$.mp. or exp cerebellar Ataxias/ (9673) 14 (cerebellar ataxia$1 or spinocerebellar degeneration$1 or degenerative cerebellar).tw. (4270) 15 dentatorubral‐pallidoluysian atrophy.mp. or Myoclonic Epilepsies, Progressive/ (495) 16 episodic ataxia$.mp. (390) 17 (ataxia$ and Charlevoix‐Saguenay).mp. (99) 18 arsacs.mp. (70) 19 exp Spinocerebellar Degenerations/ or Marinesco‐Sjogren.mp. (6420) 20 (Ataxia$ and oculomotor apraxia).mp. (168) 21 (Ataxia$ and vitamin E deficiency).mp. (207) 22 Ataxia Telangiectasia/ or Ataxia‐telangiectasia.mp. (7467) 23 (Joubert adj5 syndrome$).mp. (476) 24 (ataxia$ adj5 (hereditary or genetic$)).mp. (1635) 25 (ataxia$ adj5 autosomal).mp. (1267) 26 or/12‐25 (20850) 27 (speech or articulat$ or voice or vocal or communicat$).mp. (371122) 28 Speech/ (17897) 29 Voice Disorders/ or Voice/ (10517) 30 Vocal Cord Paralysis/ or Vocal Cords/ (11395) 31 (dysarth$ or dysphon$ or anarth$ or dyspros$).mp. (8780) 32 Dysarthria/ (1850) 33 Dysphonia/ (702) 34 Speech Disorders/ (9857) 35 exp "rehabilitation of speech and language disorders"/ (8688) 36 "Activities of Daily Living"/ (50732) 37 "Quality of Life"/ (119526) 38 (fars or sara or icars).tw. (1218) 39 or/27‐38 (531229) 40 11 and 26 and 39 (181) 41 remove duplicates from 40 (167)
Appendix 4. EMBASE (OvidSP) search strategy
Database: EMBASE <1980 to 2013 Week 41> Search strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 crossover‐procedure.sh. (38658) 2 double‐blind procedure.sh. (118100) 3 single‐blind procedure.sh. (18340) 4 randomized controlled trial.sh. (357811) 5 (random$ or crossover$ or cross over$ or placebo$ or (doubl$ adj blind$) or allocat$).tw,ot. (1006930) 6 trial.ti. (153871) 7 or/1‐6 (1144144) 8 exp animal/ or exp invertebrate/ or animal.hw. or non human/ or nonhuman/ (20103289) 9 human/ or human cell/ or human tissue/ or normal human/ (14948140) 10 8 not 9 (5187786) 11 7 not 10 (1004080) 12 limit 11 to embase (768408) 13 (Friedreich$ ataxia or freidereich$ ataxia).mp. or Friedreich Ataxia/ (3533) 14 cerebellar ataxia/ or cerebellar ataxia.tw. (6821) 15 (spinocerebellar ataxia$ or spinocerebellar degeneration$).mp. or Spinocerebellar degeneration/ (5278) 16 degenerative cerebellar.tw. (82) 17 dentatorubral‐pallidoluysian atrophy.mp. or myoclonus epilepsy/ (4909) 18 episodic ataxia$.mp. (507) 19 (ataxia$ and Charlevoix‐Saguenay).mp. (137) 20 arsacs.mp. (95) 21 Marinesco‐Sjogren.mp. (199) 22 (Ataxia$ and oculomotor apraxia).mp. (254) 23 (Ataxia$ and vitamin E deficiency).mp. (225) 24 Ataxia Telangiectasia/ or Ataxia‐telangiectasia.mp. (6540) 25 (Joubert adj5 syndrome$).mp. (804) 26 (ataxia$ adj5 (hereditary or genetic$)).mp. (5470) 27 (ataxia$ adj5 autosomal).mp. (2013) 28 or/13‐25 (26177) 29 speech/ (29407) 30 dysphonia/ (5750) 31 voice/ (12713) 32 vocal cord/ (9089) 33 vocal cord paralysis/ (5764) 34 dysarthria/ (8738) 35 speech disorder/ (18605) 36 exp speech rehabilitation/ (12766) 37 (speech or articulat$ or voice or vocal or communicat$ or dysarth$ or dysphon$ or anarth$ or dyspros$).mp. (536778) 38 daily life activity/ (54451) 39 activities of daily living.tw. (18670) 40 (sara or fars or icars).tw. (1696) 41 or/29‐40 (594658) 42 12 and 28 and 41 (65) 43 remove duplicates from 42 (65)
Appendix 5. CINAHL Plus (EBSCOhost) search strategy
14 October 2013, 7:55:00 AM S32 S31 Limiters ‐ Exclude MEDLINE 9 S31 S18 AND S27 AND S3065 S30 S28 OR S29233,568 S29 FARS or SARA or ICARS or activities of daily living or quality of life93,620 S28 (speech or articulat* or voice or vocal or communicat* or dysarth* or dysphon* or anarth* or dyspros*)144,347 S27 S19 or S20 or S21 or S22 or S23 or S24 or S25 or S261,180 S26 cerebellar ataxia* or spinocerebellar degeneration* or degenerative cerebellar326 S25 ataxia* N5 genetic*149 S24 ataxia* N5 hereditary29 S23 (Ataxia* and oculomotor apraxia) or (Ataxia* and vitamin E deficiency) or Ataxia telangiectasia or (ataxia* N5 autosomal)197 S22 episodic ataxia* or (ataxia* and Charlevoix‐Saguenay) or arsacs or Marinesco‐Sjogren or (Joubert N5 syndrome*)142 S21 Friedreich* ataxia or Freidreich* ataxia or spinocerebellar ataxia* or dentatorubral pallidoluysian atrophy or (Myoclon* Epilep*)623 S20 (MH "Epilepsy, Juvenile Myoclonic")12 S19 (MH "Ataxia Telangiectasia") OR (MH "Spinocerebellar Degenerations+")295 S18 S1 or S2 or S3 or S4 or S5 or S6 or S7 or S8 or S9 or S10 or S11 or S12 or S13 or S14 or S15 or S16 or S17632,216 S17 ABAB design*81 S16 TI random* or AB random*127,116 S15 ( TI (cross?over or placebo* or control* or factorial or sham? or dummy) ) or ( AB (cross?over or placebo* or control* or factorial or sham? or dummy) )258,734 S14 ( TI (clin* or intervention* or compar* or experiment* or preventive or therapeutic) or AB (clin* or intervention* or compar* or experiment* or preventive or therapeutic) ) and ( TI (trial*) or AB (trial*) )88,979 S13 ( TI (meta?analys* or systematic review*) ) or ( AB (meta?analys* or systematic review*) )28,367 S12 ( TI (single* or doubl* or tripl* or trebl*) or AB (single* or doubl* or tripl* or trebl*) ) and ( TI (blind* or mask*) or AB (blind* or mask*) )20,304 S11 PT ("clinical trial" or "systematic review")112,699 S10 (MH "Factorial Design")881 S9 (MH "Concurrent Prospective Studies") or (MH "Prospective Studies")216,259 S8 (MH "Meta Analysis")17,466 S7 (MH "Solomon Four‐Group Design") or (MH "Static Group Comparison")32 S6 (MH "Quasi‐Experimental Studies")6,241 S5 (MH "Placebos")8,399 S4 (MH "Double‐Blind Studies") or (MH "Triple‐Blind Studies")27,689 S3 (MH "Clinical Trials+")166,844 S2 (MH "Crossover Design")10,966 S1 (MH "Random Assignment") or (MH "Random Sample") or (MH "Simple Random Sample") or (MH "Stratified Random Sample") or (MH "Systematic Random Sample")62,816
Appendix 6. PsycInfo (OvidSP) search strategy
Database: PsycINFO <1806 to October Week 2 2013> Search strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 ataxia/ (1908) 2 (friedreich$ ataxia or freidreich$ ataxia).mp. (263) 3 spinocerebellar ataxia.mp. (487) 4 Spinocerebellar degeneration.mp. (55) 5 (cerebellar ataxia or degenerative cerebellar).mp. (691) 6 myoclonus/ (616) 7 epilepsy/ (16675) 8 6 and 7 (234) 9 myoclon$ epilepsy.mp. (501) 10 dentatorubral‐pallidoluysian atrophy.mp. (33) 11 episodic ataxia$.mp. (91) 12 (ataxia$ and Charlevoix‐Saguenay).mp. (26) 13 arsacs.mp. (20) 14 Marinesco‐Sjogren.mp. (10) 15 (Ataxia$ and oculomotor apraxia).mp. (47) 16 (Ataxia$ and vitamin E deficiency).mp. (15) 17 Ataxia telangiectasia.mp. (92) 18 (Joubert adj5 syndrome$).mp. (63) 19 (ataxia$ adj5 (hereditary or genetic$)).mp. (292) 20 (ataxia$ adj5 autosomal).mp. (210) 21 or/1‐5,8‐20 (3015) 22 exp Oral Communication/ (26849) 23 exp vocalization/ (13458) 24 exp speech disorders/ (8996) 25 vocal cords/ (221) 26 (speech or articulat$ or voice or vocal or communicat$ or dysarth$ or dysphon$ or anarth$ or dyspros$).mp. (281719) 27 activities of daily living.mp. (9814) 28 (syllable$ or pronounc$).mp. (25558) 29 (sara or fars or icars or ataxia rating scale).mp. (1369) 30 or/22‐29 (321645) 31 21 and 30 (502) 32 (random$ or rct or cct or factorial$ or crossover$ or cross over$ or cross‐over$ or placebo$ or (doubl$ adj blind$) or (singl$ adj blind$) or assign$ or allocat$ or volunteer$).tw. (234648) 33 31 and 32 (24)
Appendix 7. ERIC Dialog/Datastar search strategy
1 FRIEDREICH ADJ ATAXIA OR FRDA OR SPINOCEREBELLAR ADJ ATAXIA$
2 dentatorubral ADJ pallidoluysian ADJ atrophy
3 myclon$ ADJ epileps$
4 episodic ADJ ataxia$ OR arsacs OR Spinocerebellar ADJ Degeneration$ OR Marinesco ADJ Sjogren
5 Ataxia$ AND oculomotor ADJ apraxia
6 Ataxia$ AND vitamin ADJ E ADJ deficiency
7 Ataxia ADJ Telangiectasia
8 Joubert WITH syndrome$
9 ataxia$ WITH (hereditary OR genetic$ OR autosomal)
10 ataxia$ WITH Charlevoix ADJ Saguenay
11 1 OR 2 OR 3 OR 4 OR 5 OR 6 OR 7 OR 8 OR 9 OR 10
12 speech OR articulat$ OR voice OR vocal OR communicat$ OR dysarth$ OR dysphon$ OR anarth$ OR dyspros$
13 11 AND 12
Appendix 8. ERIC (ProQuest) search strategy
(Friedreich ADJ ataxia OR freidreich ADJ ataxia OR FRDA OR spinocerebellar ADJ ataxia) OR (dentatorubral ADJ pallidoluysian ADJ atrophy OR myclon$ ADJ epileps$) OR (episodic ADJ ataxia$ OR arsacs OR Spinocerebellar ADJ Degeneration$ OR Marinesco ADJ Sjogren OR Ataxia$ AND oculomotor ADJ apraxia) OR (Ataxia$ AND vitamin ADJ E ADJ deficiency OR Ataxia ADJ Telangiectasia) OR (Joubert WITH syndrome$ OR ataxia$ WITH (hereditary OR genetic$ OR autosomal)) OR (ataxia$ WITH Charlevoix ADJ Saguenay OR cerebellar ataxia OR degenerative cerebellar) AND (speech OR articulat$ OR voice OR vocal OR communicat$ OR dysarth$ OR dysphon$ OR anarth$ OR dyspros$ OR FARS OR SARA OR ICARS OR activities of daily living OR quality of life)
Appendix 9. Linguistics & Language Behavior Abstracts
all(speech) AND all((ataxia OR friedreich)) OR all(spinocerebellar)
Appendix 10. Dissertation Abstracts
ataxia AND ab((dysarthria OR speech))
Appendix 11. ClinicalTrials.gov
ataxia AND (dysarthria OR speech)
Appendix 12. International Clinical Trials Registry Platform
ataxia AND (dysarthria OR speech)
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Assadi 2007.
| Methods | Randomised, double‐blind, placebo‐controlled, cross‐over trial | |
| Participants | 20 individuals with SCA or Friedreich ataxia were recruited and 19 completed the protocol. Of these, 14 were genetically confirmed (4 Friedreich ataxia, 1 SCA1, 4 SCA2, 2 SCA3, 1 SCA6, 1 SCA17, 1 dentatorubral‐pallidoluysian atrophy (DRPLA)) and 6 were idiopathic. Participants had clinically symptomatic ataxia and either cerebellar or brainstem atrophy on imaging studies or genetic confirmation of a hereditary SCA | |
| Interventions | Participants received either buspirone hydrochloride 30 mg twice daily or placebo for 12 weeks. A titration period was implemented in the 1st 2 weeks of each arm (10 mg buspirone twice daily in week 1; 20 mg buspirone twice daily in week 2). A 4‐week washout period followed the 1st treatment phase, after which participants were crossed into the alternative treatment arm | |
| Outcomes | ICARS, which includes an 8‐point speech subscale, was used to evaluate clinical features at baseline and at the end of each treatment phase (12 weeks' duration). The speech subscale was not reported separately to the total ICARS score | |
| Notes | Several participants (6/20) were diagnosed with an idiopathic type of cerebellar degeneration (without a confirmed genetic diagnosis) Mr and Mrs Dennis Culnan provided the funding for the study. The provider of buspirone and placebo is not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation is not clear |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment is not clear |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Blinding of participants and personnel is not clear |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Blinding of investigators at assessment is not clear |
| Selective reporting (reporting bias) | Low risk | All planned outcomes are reported |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 1 participant withdrew from the protocol after moving away from the treatment site |
| Other bias | Low risk | None identified |
Cooper 2008.
| Methods | Double‐blind randomised trial with high‐dose and low‐dose groups | |
| Participants | 50 participants with a diagnosis of Friedreich ataxia (genetically confirmed) | |
| Interventions | Participants were split into high‐dose and low‐dose groups. The high‐dose group received 2100 IU/day vitamin E and 600 mg/day coenzyme Q10 (CoQ10) (participants under 18 years of age received 30 IU/kg/day vitamin E and 9 mg/kg/day CoQ10). The low‐dose group received 30 mg CoQ10 and placebo tablets containing 4 IU/day of vitamin E, which were indistinguishable from active vitamin E tablets. Participants received treatment over a 2‐year study period | |
| Outcomes | Primary outcome measure: ICARS (including an 8‐point speech subscale) Further outcome measures included ICARS subscales (including speech), speech tests (passage test, PaTa test), limb co‐ordination, heart function and an ADL questionnaire. Testing occurred at baseline and every 6 months for 2 years |
|
| Notes | Pharma Nord, Morpeth, UK provided vitamin E and CoQ10. The study was supported by grants from Ataxia UK, the Wellcome Trust and the Medical Research Council | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A statistician external to the study randomised participants to give 2 groups of 24 and 26 participants matched for age, GAA1 repeat size, cardiac hypertrophy and clinical severity (ICARS) |
| Allocation concealment (selection bias) | Low risk | The researchers were not involved in the allocation sequence, which was devised by an external statistician |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The treatment was independently dispensed by the hospital pharmacy |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Assessors were blind to the treatment received by participants |
| Selective reporting (reporting bias) | Low risk | All outcomes reported either in the article or in supplementary online tables |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 22/26 participants completed the high‐dose treatment 21/24 participants completed the low‐dose treatment |
| Other bias | Unclear risk | Results of the randomised controlled (low‐dose group) segment of the study were not significant. Post hoc analysis was completed using a cross‐sectional data set of untreated participants, which is not adequately described |
Di Prospero 2007.
| Methods | Randomised, double‐blind, placebo‐controlled study with 3 treatment groups and 1 control group | |
| Participants | 48 participants with Friedreich ataxia (genetically confirmed) aged 9 to 17 years of age and weighing 30 kg to 80 kg 47 participants (24 male, 23 female) were assessed post treatment with a mean age of 13.4 years (SD 2.4). A subgroup excluding non‐symptomatic and non‐ambulatory participants involved 33 participants (16 male, 17 female) with a mean age of 12.8 years (SD 2.3) Participants were not exposed to idebenone, coenzyme Q10 or other dietary supplements for a period of 1 month prior to enrolment |
|
| Interventions | Participants took either a low dose, intermediate dose or high dose of idebenone over a period of 6 months. Doses were stratified according to weight (≤ 45 kg or > 45 kg): low dose (180 mg or 360 mg), intermediate dose (450 mg or 900 mg) and high dose (1350 mg or 2250 mg). The total daily dose was divided and taken 3 times each day | |
| Outcomes | Participants were scored on ICARS, which contains a subjective dysarthria rating scale and FARS, which includes a rating of speech within a neurological examination subscore. An ADL survey developed alongside the FARS was also completed. Scores were obtained at baseline and after 6 months' idebenone or placebo, by a single rater | |
| Notes | Idebenone and placebo were provided by Santhera Pharmaceuticals (Liestal, Switzerland). The study was supported by intramural NIH research funds | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random allocation, stratified by body weight (to maintain dose range) and the shorter GAA repeat length (to control for disease progression) |
| Allocation concealment (selection bias) | Low risk | The list of randomised numbers and corresponding treatment numbers was computer‐generated by a third party (Hesperion Ltd, Allschwil, Switzerland). Participants and investigators were blind to the allocated study treatment. The allocations were maintained by the 3rd party and only made available when the trial was complete |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants were blinded to their treatment allocation. The manufacturer provided idebenone or placebo in prepackaged kits marked with treatment numbers |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Investigators were blinded to the treatment groups |
| Selective reporting (reporting bias) | Unclear risk | All outcomes were reported. The second statistical analysis, which was conducted to remove potential ceiling and floor effects, involved seemingly arbitrary cut‐off points (including baseline scores between 10 and 54 on a 100‐point scale). The process effectively removed non‐ambulatory participants and 1 participant with no symptoms |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | One participant from the low‐dose idebenone group did not complete the follow‐up assessment due to "intercurrent illness" |
| Other bias | Low risk | None identified |
Filla 1988.
| Methods | Double‐blind, placebo‐controlled, cross‐over study using an ABCB design | |
| Participants | 17 participants with Friedreich ataxia (not genetically confirmed) (8 female, 9 male, mean age 23.4 years (± 8.1 years)) and 14 participants with other SCAs (6 female, 8 male, mean age 47.1 years (± 13.4 years)) including: 10 with autosomal dominant cerebellar ataxia, 2 with idiopathic late onset cerebellar ataxia (not genetically confirmed), 1 with autosomal recessive late onset cerebellar ataxia (no formal diagnosis) and 1 with early onset cerebellar ataxia with retained tendon reflexes (not genetically confirmed) | |
| Interventions | Thyrotropin‐releasing hormone tartrate (TRH‐T) or placebo were administered intramuscularly. There were 2 sequences (groups of participants) within the study: sequence I began the study in the placebo arm, sequence II in the active treatment arm (TRH‐T). 9 participants with Friedreich ataxia and 6 participants with SCA underwent sequence I (1st month with placebo). 7 participants with Friedreich ataxia and 9 participants with SCA underwent sequence II (1st month with TRH 2 mg). Each treatment was administered for 1 month in a double cross‐over design. Participants were allocated randomly to either sequence. The daily dose of TRH‐T was 2 mg for the 1st month and 4 mg for the 2nd active treatment phase | |
| Outcomes | Neurological and clinical function were evaluated using the Inherited Ataxias Clinical Rating Scale (IACRS) which contains a speech component. The study neurologist, blinded to the treatment condition, evaluated speech subjectively using a categorical scale where 0 reflected 'normal' function and 4 was considered 'not understandable' | |
| Notes | 6 participants remained on their pre‐existing pharmaceutical treatment regime throughout the experiment. Diagnosis was not genetically confirmed for the cohort as the genes for the respective disorders had not yet been identified (Campuzano 1996; Orr 1993) Cyanamid Italia, Italy provided TRH‐T and matching placebo |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly allocated to TRH‐T or placebo. Given the relatively even distribution of participants with Friedreich ataxia and SCA in the 2 groups, the random nature of the selection is unclear |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants blinded to treatment condition |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Assessor blinded to treatment condition. The same experimenter conducted the assessments over the course of the trial |
| Selective reporting (reporting bias) | Low risk | All planned outcomes reported |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 1 participant in the Friedreich ataxia group dropped out |
| Other bias | Unclear risk | Dose strength was not randomised (2 mg in the 1st treatment phase and 4 mg in the 2nd) |
Lynch 2010.
| Methods | Randomised, double‐blind, placebo‐controlled trial with 3 treatment arms: low‐dose idebenone, high‐dose idebenone and placebo | |
| Participants | 70 ambulatory children (33 male, 37 female) aged 8 to 18 years (mean age 13.7 years, SD 2.8) with Friedreich ataxia (with confirmed GAA expansion mutations). Children with an ICARS scores less than 10 or greater than 54 were excluded | |
| Interventions | 3 treatment arms received either idebenone or placebo for 24 weeks. Group A received a low dose of either 450 mg/day (body weight ≤ 45 kg) or 900 mg/day (body weight > 45 kg). Group B received a high dose of either 1350 mg/day (body weight ≤ 45 kg) or 2250 mg/day (body weight > 45 kg). Group C received placebo. Daily dosages were divided into 3 tablets per day | |
| Outcomes | Outcomes were measured at baseline, 12 weeks and 24 weeks after the beginning of treatment. Participants were scored on the ICARS, which contains a speech subscale, and the FARS, which includes a subjective rating of speech within a neurological examination subscore | |
| Notes | Santhera Pharmaceuticals (Liestal, Switzerland) provided idebenone and funding | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Allocation to treatment arms was computer‐generated |
| Allocation concealment (selection bias) | Low risk | A third party (Fischer Services, Allschwil, Switzerland) conducted random allocation to treatment arms |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants were blinded to their allocation and received prepackaged kits of either placebo or idebenone marked with their treatment number |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Treatment assignments were maintained by the third party and not revealed to investigators until the trial was completed |
| Selective reporting (reporting bias) | Low risk | All planned outcomes were reported |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants who were allocated to study arms completed all assessments |
| Other bias | Low risk | None identified |
Lynch 2012.
| Methods | Double‐blind, placebo‐controlled, randomised trial involving 3 treatment arms (placebo, low‐dose α‐tocopheryl quinone (510 mg/day) or high‐dose α‐tocopheryl quinone (EPI‐A0001) (750 mg/day)) | |
| Participants | 31 participants with genetically confirmed Friedreich ataxia aged between 18 and 60 years. 10 participants were randomised into the placebo arm, 11 into the low‐dose α‐tocopheryl quinone arm and 10 into the high‐dose α‐tocopheryl quinone arm | |
| Interventions | Participants received capsules containing either 250 mg or 170 mg of α‐tocopheryl quinone in olive oil 3 times daily with meals. The placebo group received identical capsules containing 0.01% beta‐carotene in olive oil. Treatment was provided for 28 days | |
| Outcomes | Outcomes were measured at baseline, at 14 days of treatment and at 28 days of treatment. The primary study outcome was a measure of diabetic tendency. Secondary outcome measures included the FARS and SF‐36 | |
| Notes | The Friedreich Ataxia Research Alliance and Penwest Pharmaceutical provided funding Authors Lynch and Willi received grant support from Penwest Pharmaceutical to undertake the study. Authors Hawi and Sciascia were employed by Penwest during the study. Authors Miller and Shrader held stock in Edison Pharmaceuticals (owner of EPI‐A0001). Author Miller was an employee and received 100% of his compensation from Edison Pharmaceuticals. Author Shrader was a compensated consultant for Edison Pharmaceuticals. Author Rioux had been previously employed by Edison Pharmaceuticals. Provider of the drug for this study is not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | The method of randomisation is not clear and did not completely match the 3 groups. The placebo group had higher clinical severity scores |
| Allocation concealment (selection bias) | Unclear risk | The method of allocation concealment is not clear |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants were blinded to the treatment by the use of identical capsules for α‐tocopheryl quinone and placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It is not clear how examiners were blinded to the treatments |
| Selective reporting (reporting bias) | Low risk | All planned outcomes were reported |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | One participant from the high‐dose α‐tocopheryl quinone arm discontinued treatment due to a major protocol violation |
| Other bias | Low risk | None identified |
Mariotti 2009.
| Methods | Randomised, placebo‐controlled, double‐blind, dose‐response pilot trial | |
| Participants | 16 participants (9 female, 7 male) with genetically confirmed Friedreich ataxia, aged 18 to 40 years. 11 received recombinant human erythropoietin (rhuEPO) and 5 received placebo | |
| Interventions | Participants received either rhuEPO or placebo over a period of 24 weeks. Dosage was 20,000 IU every 3 weeks for 9 weeks (visits 1 to 3), 40,000 IU every 3 weeks for 9 weeks (visits 4 to 6) and 40,000 IU every 2 weeks for 6 weeks (visits 7 to 9) | |
| Outcomes | Primary outcome measures were of safety and tolerability of rhuEPO and efficacy of rhuEPO in increasing frataxin in peripheral lymphocytes. Secondary outcome measures included the SARA and SF‐36 | |
| Notes | The study was funded by the Italian Agency for Pharmaceutics (Agenzia Italiana del Farmaco; AIFA grant FARM6H95MJ; to F.T.) EPREX, Janssen‐Cilag, Cologno Monzese, Milan, Italy provided RhuEPO | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to rhuEPO or placebo at a ratio of 2:1. The method of randomisation is unclear given the similarity of the genetic and clinical characteristics of the participants in each group |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment is not clear |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Participants reportedly blinded to treatment conditions, however the method used is not described |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Assessor reportedly blinded to treatment conditions, however the method used is not described |
| Selective reporting (reporting bias) | Low risk | All planned outcomes reported |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All enrolled participants completed the protocol |
| Other bias | Low risk | None identified |
Miyai 2012.
| Methods | Single‐blinded randomised design with delayed intervention arm versus immediate intervention arm | |
| Participants | 20 participants with SCA type 6 (genetically confirmed), 6 participants with SCA31 (genetically confirmed) and 16 participants with ICA presenting with pure cerebellar ataxia (negative DNA result for known SCAs) Immediate treatment group: 8 SCA6, 2 SCA31 and 11 ICA participants; 8 female, 13 male; mean age 63.5 years (standard error (SE) 2.4 years), mean disease duration 9.3 years (SE 1.3 years) Delayed treatment group: 12 SCA6, 4 SCA31 and 5 ICA, 12 female, 9 male, mean age 61.5 years (SE 2.3 years), mean disease duration 10.3 years (SE 1.4 years) |
|
| Interventions | Inpatient hospital setting over the course of 4 weeks. Immediate treatment group: 2 hours of physical therapy (focusing on posture and gait) and occupational therapy (focusing on ADL) on weekdays and 1 hour on weekends. Delayed treatment group (control): the same intervention after a 4‐week delay |
|
| Outcomes | Primary outcome measures were derived from changes in baseline end point scores (at completion of intervention, and 4, 12 and 24 weeks post intervention) of the SARA. The SARA contains a subscale focusing on speech function. The study protocol also included a standardised scale of functional independence, the Functional Independence Measure (FIM). Only data from the short‐term component of the trial (up to 4 weeks) was randomised, all subsequent time points were part of the follow‐up observational study and not included in this review | |
| Notes | Description of the interventions and their relationship with the services provided in an inpatient hospital setting were not clearly described. A large proportion of participants (16/42) were diagnosed with an idiopathic type of cerebellar degeneration (without a confirmed genetic diagnosis) The study author(s) declared no potential conflicts of interest. Grants from various governmental agencies declared as sources of support |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomly allocated to immediate or delayed entry groups. We believe this process was adhered to given the uneven (and potentially confounding) distribution of disease types in the 2 groups |
| Allocation concealment (selection bias) | Low risk | Sealed envelopes with allocation information on groups (immediate or delayed entry control group) were randomly selected by a person who was not involved in this study |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No sham or alternative treatment was included. Participants would have known they were being treated; however, they may have been blinded to the type of treatment |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It is difficult to determine whether those assessing the study participants were in contact with the treating clinicians. Assessments were conducted by physicians or therapists familiar with the measures. They were reportedly blinded to the group allocation but the additional effect of inpatient stay, in conjunction to the treatment regime, was not clearly described |
| Selective reporting (reporting bias) | Low risk | All planned outcomes reported |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All randomised participants completed the trial. 1 participant died during the follow‐up observational component of the study as a result of cerebral haemorrhage at 17 weeks (outside the randomisation period) |
| Other bias | High risk | Participants were treated in an inpatient setting within a hospital and may have been exposed to additional therapeutic care during their stay |
Ristori 2010.
| Methods | Double‐blind, placebo‐controlled, randomised study | |
| Participants | 40 participants (between 18 and 80 years) with chronic cerebellar ataxia (bilateral involvement of static and kinetic functions, as well as dysarthria and oculomotor dysfunction), irrespective of aetiology Riluzole group: 20 participants (8 male, 12 female), mean age 48.9 (SD 16.8) Control group: 20 participants (7 male, 13 female), mean age 44.1 (SD 13.1) The riluzole group included 10 participants with hereditary ataxia (6 SCA, 3 Friedreich ataxia, 1 fragile X tremor/ataxia syndrome), 5 with sporadic ataxia (3 with probable multiple system atrophy type C (MSA‐C), 1 with anti‐GAD antibodies, 1 with anti‐Yo antibodies) and 5 with ataxic syndromes of unknown origin The placebo group included 7 participants with hereditary ataxia (2 SCA, 5 Friedreich ataxia), 5 with sporadic ataxia (3 with probable MSA‐C, 2 with multiple sclerosis) and 8 with ataxic syndromes of unknown origin |
|
| Interventions | Participants received riluzole (50 mg tablets, twice daily) or placebo for a period of 8 weeks. Participants suspended any pharmacological or physical therapy for ataxia for 2 weeks prior to enrolment | |
| Outcomes | Participants were assessed at baseline, 4 weeks and 8 weeks after the beginning of treatment, for symptoms, physical and neurological signs, ICARS score, electrocardiogram and complete standard laboratory safety tests. Treatment efficacy was measured by differences between riluzole and placebo groups in:
The ICARS assessment contains a dysarthria subscale which was only reported separately for the mean change of ICARS scores from baseline to 8 weeks after beginning of treatment |
|
| Notes | Only 17 out of 40 participants presented with symptoms caused by hereditary ataxia. Outcomes of the dysarthria subscale only were not reported for either individual participants; or groups according to aetiology or diagnosis Sanofi‐Aventis (Milan, Italy) provided riluzole and matching placebo |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomly assigned in a 1:1 ratio to riluzole or placebo groups. Given the relatively uneven distribution of participants with different diagnoses in the 2 groups, the random nature of the allocation appears clear |
| Allocation concealment (selection bias) | Low risk | A list of randomisation numbers and corresponding treatment numbers was computer‐generated before the start of the study. This procedure was centrally performed by personnel not involved in the study measurements |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Both examiner and participant were blinded to treatment |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | A treating investigator assessed the safety of riluzole and took all the medical decisions on the basis of the clinical and laboratory findings. A second examining investigator had access to the ICARS score but was unaware of the treatment groups until all the data had been collected and analysed (data were first entered into a paper case report form, then into electronic databases for analysis). This prevented the blinding procedure from being broken as a result of observed efficacy, adverse events or changes in laboratory tests |
| Selective reporting (reporting bias) | Low risk | All planned outcomes reported |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 2 participants (1 in the riluzole and the other in the placebo arm) withdrew their consent before receiving riluzole or placebo and were excluded from the final analysis |
| Other bias | Low risk | None identified |
Sobue 1983.
| Methods | Randomised, double‐blind, placebo‐controlled study | |
| Participants | 290 participants with SCD. Participants were all hospital inpatients with an age range of 15 to 79 years. Of the 290 people originally recruited, 256 met criteria for evaluation of efficacy. Of these, 220 were diagnosed with a predominantly cerebellar form of SCD (i.e. parenchymatous cerebellar degeneration, including 80 participants with late onset cerebellar cortical atrophy (LCCA) and 140 participants with olivopontocerebellar atrophy (OPCA)), 19 were diagnosed with cerebellospinal form of SCD and 17 had a diagnosis which was unclassified. Of these with a predominantly cerebellar form of SCD, 214 completed all speech assessments | |
| Interventions | Participants received intramuscular thyrotropin‐releasing hormone tartrate (TRH‐T) or placebo. Participants were randomised into 1 of 3 arms: 2 mg of TRH‐T, 0.5 mg of TRH‐T or placebo (5% sorbitol solution). Each arm received these doses once a day for 2 weeks. Participants were not permitted to continue with concomitant medications which may have affected ataxic symptoms, however 'routine rehabilitation' was allowed to continue | |
| Outcomes | Participants were assessed 4 times: at baseline, 1 week after commencing treatment, at the end of treatment (2 weeks) and 1 week after completing the treatment. Participants underwent a neurological examination and subjective rating of ataxic symptoms, with each symptom rated on a 14‐point VAS. At the end of the trial, the investigating physician made a 'global improvement rating' and an 'ataxia improvement rating', each of which involved a 7‐point scale from 'markedly improved' to 'markedly aggravated'. Speech was rated on the 'speed precision and rhythm of tongue‐twisters' on a 14‐point VAS. Participants completed a self rating of the impact of ataxic symptoms on ADL, including a subscale for speech | |
| Notes | This work was supported by a grant for the new drug development of the Ministry of Health and Welfare of Japan. Takeda Chemical Industries, Ltd., Osaka, Japan provided TRH‐T | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The method of randomisation was not described. Distribution of demographics and diagnostic characteristics (age, sex, duration of illness, hereditary factors) were reportedly similar in the 3 treatment arms |
| Allocation concealment (selection bias) | Unclear risk | The method of allocation is not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants were blinded to treatment allocation |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Investigators were reportedly blinded to the treatment provided; however, it is not clear if clinicians rating the efficacy were also assessing safety and therefore aware of adverse effects of treatment, which would have undermined blinding |
| Selective reporting (reporting bias) | High risk | Results of the 'global improvement rating' were reported for the 3 groups stratified by diagnosis (predominantly cerebellar SCD, cerebellospinal SCD or unclassified); however, the 'ataxia improvement rating', 14‐point VAS for each ataxic symptom and participant‐rated ADL were only reported for the predominantly cerebellar SCD group Mean scores of the 14‐point scales were not reported. Instead an improvement ratio demonstrated the rate of responses which had improved by either 1 point on the 14‐point scale or 2 points on the 14‐point scale. This creates difficulty in interpreting the overall clinical significance of any statistically significant results |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Only 254 participants satisfied criteria for inclusion in the efficacy trial. 2 more participants were excluded from the trial as they had begun rehabilitation at the same time as the trial. VAS results of specific symptoms (e.g. dysarthria) were only reported for 218 participants with a predominantly cerebellar form of SCD, as this was the only diagnostic group to demonstrate statistically significant differences on the 'global improvement rating'. The results of the investigator‐rated dysarthria scale were reported for 216 participants at the 1‐week time point, 214 participants at 2 weeks and 214 participants at 3 weeks. Participant‐rated dysarthria was reported for 212 participants at 1 week, 210 participants at 2 weeks and 210 participants at 3 weeks. No explanation is provided by the authors for these latter reductions in participant numbers |
| Other bias | Low risk | None identified |
Trouillas 1995.
| Methods | Randomised, double‐blind, placebo‐controlled trial | |
| Participants | 26 participants with Friedreich ataxia (diagnosed by clinical symptoms) were recruited from 12 centres in France. Only 19 participants (mean age 25.9 years, SD 8.1) from 8 centres completed the study. 11 participants with a mean age of 28.5 years (SD 9.4) received the levorotatory form of 5‐hydroxytryptophan (L‐5HT) and 8 participants with a mean age of 22.3 years (SD 4.1) received placebo | |
| Interventions | Participants received L‐5HT or placebo for 6 months. Dose was progressively increased in the treatment arm based on participant weight as follows: weight < 50 kg, 200 mg/day for the 1st month, 600 mg/day for the remaining 5 months; weight > 50 kg, 300 mg/day for the 1st month, then 900 mg/day for the remaining 5 months Participants in the control arm received gelules of the same size and number as their counterparts in the treatment arm |
|
| Outcomes | Post treatment assessments versus baseline were made every 2 months for 6 months Clinical symptoms were evaluated with a modified ataxia rating scale involving kinetic and static tasks, which did not include speech tasks. Quantitative values were obtained including: mean time for writing a standard sentence, mean time for pronouncing a standard sentence. 3 tests measured the time of standing in a natural position |
|
| Notes | Dose was not consistent for all participants randomised in the treatment arm The provider of L‐5HT is not stated. Panmedica Laboratories, Carros, France funded the study |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The method of random allocation is not described |
| Allocation concealment (selection bias) | Unclear risk | The method of allocation concealment is not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants in the control arm received gelules of same size and number as the treatment group |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | The method of blinding of investigators is not outlined. Experience of adverse effects in the treatment arm may have weakened the blinding process |
| Selective reporting (reporting bias) | Low risk | All planned outcomes reported |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Only 19/26 participants completed the study. 4 did not complete from the placebo group (due to "secondary refusal of consent", "no respect of regimen", "cardiac flutter" and "general discomfort") and 3 from the treatment group (2 due to "vomiting and gastralgia" and 1 to "no respect of regimen") |
| Other bias | Low risk | None identified |
Wessel 1995.
| Methods | Double‐blind, placebo‐controlled, cross‐over study | |
| Participants | 19 participants with Friedreich ataxia, 13 with cerebellar atrophy and 7 with olivopontocerebellar atrophy. Age and sex of participants are not provided. Mean age of onset of symptoms for the Freidreich ataxia participants was 20 years. Mean age of onset of the other conditions is not stated | |
| Interventions | Oral L‐5HT 1000 mg/day or placebo. "Each treatment phase, with L‐5HT or placebo, lasted 10 months, after which the treatment of participants was crossed over to the other phase." | |
| Outcomes | "Ataxia was documented and quantified by using a clinical score, posturography, measurement of grip force and the rapid‐syllable repetition rate." | |
| Notes | Only 4 of the original 19 participants with Friedreich ataxia completed the speech assessments during both arms of the experiment Provider of L‐5HT is not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly allocated to either placebo or L‐5HT. The distribution of participants within groups is not stated |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | There is a possibility of unblinding due to adverse events. L‐5HT in daily oral doses of 900 mg to 1000 mg commonly has gastrointestinal adverse effects |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Multisite study utilising clinician‐derived measures of severity which rely on clinician judgement. The assessor was blinded to the treatment condition |
| Selective reporting (reporting bias) | High risk | The primary outcome related to speech was only reported for a small group (4 out of 19) of participants with Friedreich ataxia. It is unclear if other outcomes were reported |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Data for Friedreich ataxia participants were reported separately and showed that only a small group of randomised participants completed the study |
| Other bias | Low risk | None identified |
Zannolli 2012.
| Methods | Double‐blind, randomised, placebo‐controlled, cross‐over study | |
| Participants | 13 participants (6 female, 7 male) with either a molecular diagnosis of ataxia telangiectasia based on ATM gene mutations or an alpha‐fetoprotein level more than twice the normal upper limit and an ATM protein deficiency. All participants were ambulatory | |
| Interventions | Participants were provided with an oral course of BETA (betamethasone disodium phosphate drops (1 drop, 0.0125 mg)) or placebo in a 2‐phase cross‐over design. Each treatment phase lasted 30 days. During the 1st 10 and last 10 days, participants received the full dose of 0.1 mg/kg every 24 hours. During the 10 days in the middle of the treatment phase, participants received a tapered dose of three‐quarters the daily dose for 4 days, half the daily dose for 4 days and a quarter the daily dose for 2 days. Each phase was followed by a washout period of 30 days | |
| Outcomes | Primary outcome measure: the total ICARS score. Secondary outcomes included ICARS subscale scores, a quality of life measure (Child Health Questionnaire), vital signs and biochemistry. Results are provided for both ITT and per protocol participants. Results of the ITT population are used for the purpose of this review ICARS outcomes were measured by calculating the difference between the changes in ICARS scores between BETA and placebo treatments |
|
| Notes | The study was funded by the nonprofit organisation Fondazione Monte Paschi Siena (FMPS). The provider of BETA is not stated | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The trial was centrally randomised in block sizes of 4. Group allocation is not provided alongside participants' details |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | BETA and placebo were issued to participants in identical packaging |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Unclear how the examiners were blinded to the treatment |
| Selective reporting (reporting bias) | Low risk | All planned outcomes reported |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 1 participant voluntarily discontinued treatment at the end of the 1st treatment phase, and a further 2 participants were excluded from the per protocol analysis because their plasma levels of BETA were below the lower limit of quantitation at each assessment. Results were provided for both ITT and per protocol populations |
| Other bias | Low risk | None identified |
Zesiewicz 2012.
| Methods | Double‐blind, placebo‐controlled, randomised study | |
| Participants | 20 participants with a genetically confirmed diagnosis of SCA3 were recruited and 18 entered the first phase. The mean age of participants in the varenicline group (5 female, 4 male (1 additional participant omitted from analyses)) was 47.44 years (± 10.83 years). The mean disease duration was 14 years (± 9.82 years). The mean age of the placebo group (3 female, 7 male randomised) was 53.8 years (± 11.2 years) | |
| Interventions | Participants received oral varenicline (1 mg twice daily) or placebo. The treatment phase lasted 56 days with a subsequent washout phase of 57 to 83 days for both placebo and varenicline. Participants were allowed to continue taking all concomitant medications for the duration of the study | |
| Outcomes | Primary outcome measures were derived from changes in baseline to end point scores of the SARA. The SARA contains a subscale focusing on speech function. The study protocol also included a standardised scale of functional health and wellbeing, the SF‐36 | |
| Notes | No power calculations were made to determine the required number of participants to accurately determine drug efficacy. Only 5 participants completed the placebo arm and 8 completed the varenicline arm of the study. There was no correction for multiple comparisons due to small sample size. Clinical presentation at baseline assessment was statistically significantly worse for the placebo group on the 'sitting' subscale in SARA Varenicline and matching placebo were provided by Pfizer Inc., USA. The National Ataxia Foundation, USA, and the Bobby Allison Ataxia Research Center (BAARC), USA provided funding |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | A 1:1 randomisation schedule was generated with Statistical Analysis System (SAS) 9.2 using a block size of 10. Participants were randomly allocated to either placebo or varenicline; however, the small sample size and blocked randomisation may have prevented equal distribution between groups. The placebo group had higher clinical severity scores and appeared on average older than the varenicline group, possibly influencing their responsiveness to treatment (Filla 2012) |
| Allocation concealment (selection bias) | Low risk | Sealed envelopes containing the treatment assignment for each subject were held by a member of the study team who was not involved in study assessments. All envelopes remained sealed at the end of the study. Study personnel involved in participant assessments (investigators and co‐ordinators), participants and caregivers were blinded to the treatment assignment |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants were blinded to treatment condition |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | All individuals involved in the assessments, including the investigators and participants, were blinded to the treatment assignment |
| Selective reporting (reporting bias) | Unclear risk | The 2nd period of the experiment, which was to include a cross‐over component, was abandoned due to the high dropout rate observed in the initial period reported |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Only 5 participants completed the placebo arm and 8 completed the varenicline arm of the study. Data were analysed for 9 participants in each group despite the large number of participant dropouts. The time points at which the participants withdrew from the study were not stated, neither was the time at which the final (end point) analyses were performed |
| Other bias | Low risk | None identified |
ADL: activities of daily living
FARS: Friedreich Ataxia Rating Scale
ICA: idiopathic cerebellar ataxia
ICARS: International Cooperative Ataxia Rating Scale
ITT: intention‐to‐treat
L‐5HT: L‐hydroxytryptophan
rhuEPO: recombinant human erythropoietin
SARA: Scale for Assessment and Rating of Ataxia
SCA: spinocerebellar ataxia
SCD: spinocerebellar degeneration
SD: standard deviation
TRH‐T: thyrotropin‐releasing hormone tartrate
VAS: visual analogue scale
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Arnold 2006 | No control group or randomisation of participants |
| Artuch 2002 | No control group or randomisation of participants |
| Artuch 2006 | Single case study |
| Boesch 2007 | No control group or randomisation of participants |
| Boesch 2008 | No control group or randomisation of participants. Open‐label clinical pilot study |
| Bonnan 2008 | No control group or randomisation |
| Botez 1996 | Speech not included as an outcome measure |
| Botez 1997 | Single case study |
| Broccoletti 2008 | No control group or randomisation of participants |
| Ershova 2007 | No control group |
| Heo 2008 | No control group |
| Ilg 2012 | No randomisation or placebo |
| Lagedrost 2011 | 6‐month extension of Lynch 2010, focused on cardiac outcomes |
| Meier 2012 | No control group or randomisation of participants in the open label extension of Lynch 2010 and Lagedrost 2011 (the subsequent 12 months) |
| Melancon 1982 | No randomisation |
| Nakamura 2009 | No control group or randomisation of participants |
| Ogawa 2003 | No randomisation. Single‐blinded only. Heterogenous group of spinocerebellar degeneration |
| Pineda 2008 | No randomisation. Open‐label design |
| Shimizu 1999 | No control group or placebo |
| Sobue 1980 | No control group or placebo |
| Strupp 2011 | No relevant outcome measures included |
| Trouillas 1984 | No randomisation |
| Trouillas 1997 | Unclear clinical diagnosis. All participants presented with a sporadic form of pure cerebellar cortical atrophy. Speech not reported as an outcome measure |
| Velasco‐Sanchez 2011 | No control group or randomisation of participants |
| Yabe 1999 | No control group or randomisation of participants |
Characteristics of ongoing studies [ordered by study ID]
EUCTR 2009‐016317‐20‐IT.
| Trial name or title | Pilot study to assess safety and tolerability of lithium on spinocerebellar ataxia of type 2 ‐ lithium in SCA2 |
| Methods | Double‐blind, placebo‐controlled, parallel‐group, randomised pilot clinical trial |
| Participants | Inclusion criteria: molecular diagnosis of SCA2, age > 18 years |
| Interventions | Hard capsule oral lithium carbonate 300 mg or placebo |
| Outcomes | Main objective: safety and tolerability of lithium in SCA2 Primary end point(s):
Secondary objective: effect of lithium on clinical measures |
| Starting date | 26 October 2009 |
| Contact information | Not provided |
| Notes | — |
EUCTR 2012‐005312‐26‐DE.
| Trial name or title | Sustained release 4‐aminopyridine (Fampyra®) in cerebellar gait disorder |
| Methods | Randomised, double‐blind, placebo‐controlled, cross‐over trial |
| Participants | Men or women aged 18 to 80 with a clinically evaluated diagnosis of cerebellar ataxia with at least 2 points on the SARA |
| Interventions | Sustained released 4‐aminopyridine (Fampyra) or placebo |
| Outcomes | Primary outcomes:
Secondary outcomes:
|
| Starting date | 18 March 2013 |
| Contact information | IFB LMU, Marchioninistr. 15, 81377, Munich, Germany. FACEG.studie@med.uni‐muenchen.de Hospital of the University of Munich |
| Notes | — |
Schulz 2009.
| Trial name or title | 12‐month European phase III clinical study of SNT‐MC17/idebenone in the treatment of Friedreich's ataxia: baseline neurology data and interim safety results |
| Methods | Double‐blind, placebo‐controlled, multicentre study |
| Participants | 232 people with Friedreich ataxia were randomised. Participants were aged 8 years and older. Mean age was 30 years |
| Interventions | Participants randomly assigned to placebo or to 1 of 3 body weight‐adjusted doses of idebenone (i.e. 180, 450 or 1350 mg/day for participants weighing ≤ 45 kg and 360, 900 or 2250 mg/day for participants weighing > 45 kg) |
| Outcomes | Efficacy end points include absolute change in ICARS score from baseline to week 52 (primary end point), as well as changes in the FARS, left ventricular mass index, and other measures of cardiac and neurological function |
| Starting date | Not reported |
| Contact information | Not reported |
| Notes | This study was funded by Santhera Pharmaceuticals. Published abstract available |
ECG: electrocardiogram
NYHA: New York Heart Association
SCA: spinocerebellar ataxia
Differences between protocol and review
The original protocol required that all participants have a genetically confirmed diagnosis to be included in this review. However, this excluded all studies conducted prior to the discovery of relevant disease‐specific genes. Therefore, we have included studies that were conducted prior to gene discovery techniques (for example, before 1996 for Friedreich ataxia). We included interventions if they were administered for a minimum of one week and a maximum of 12 months, instead of six months. We altered the timing of outcome measures to include assessments conducted immediately post intervention. For short‐term outcome measures, this change meant that the review included assessments conducted immediately post intervention to one month post intervention.
Professor Bruce Murdoch retired from authorship and Matthew Poole replaced him.
Contributions of authors
APV developed the original protocol, reviewed the abstracts and full‐length articles and wrote the first draft of the review. JF also reviewed the abstracts. JF and MLP evaluated the full‐length versions of the relevant articles and edited later versions of the review. The authors developed the search strategy in concert with the Trials Search Co‐ordinator of the Cochrane Neuromuscular Disease Review Group, Angela Gunn, who ran the searches.
Sources of support
Internal sources
None, Other.
External sources
-
National Health and Medical Research Council, Australia.
Adam Vogel was supported by an Early Career Research Fellowship
Declarations of interest
AV: no known conflicts of interest to declare. JF: no known conflicts of interest to declare. MLP: no known conflicts of interest to declare.
New
References
References to studies included in this review
Assadi 2007 {published data only}
- Assadi M, Campellone JV, Janson CG, Veloski JJ, Schwartzman RJ, Leone P. Treatment of spinocerebellar ataxia with buspirone. Journal of the Neurological Sciences 2007;260(1‐2):143‐6. [PUBMED: 17512011] [DOI] [PubMed] [Google Scholar]
Cooper 2008 {published data only}
- Cooper JM, Korlipara LVP, Hart PE, Bradley JL, Schapira AHV. Coenzyme Q10 and vitamin E deficiency in Friedreich's ataxia: predictor of efficacy of vitamin E and coenzyme Q10 therapy. European Journal of Neurology 2008;15(12):1371‐9. [PUBMED: 19049556] [DOI] [PubMed] [Google Scholar]
Di Prospero 2007 {published data only (unpublished sought but not used)}
- Prospero NA, Baker A, Jeffries N, Fischbeck KH. Neurological effects of high‐dose idebenone in patients with Friedreich's ataxia: a randomised, placebo‐controlled trial. Lancet Neurology 2007;6(10):878‐86. [PUBMED: 17826341] [DOI] [PubMed] [Google Scholar]
Filla 1988 {published data only (unpublished sought but not used)}
- Filla A, Michele G, Martino L, Mengano A, Iorio L, Maggio MA, et al. Chronic experimentation with TRH administered intramuscularly in spinocerebellar degeneration. Double‐blind cross‐over study in 30 subjects. In: Agnoli A, Delwaide PJ, Prange AJ, Scapagnini U editor(s). Protirelin Tartrate (TRH‐T): Pharmacological and Clinical Studies: Recent Advances and Perspectives. London: John Libbey, 1988:199‐209. [Google Scholar]
- Filla A, Michele G, Martino L, Mengano A, Iorio L, Maggio MA, et al. Chronic experimentation with TRH administered intramuscularly in spinocerebellar degeneration. Double‐blind cross‐over study in 30 subjects. Rivista di Neurologia 1989;59(2):83‐8. [PUBMED: 2505370] [PubMed] [Google Scholar]
Lynch 2010 {published data only (unpublished sought but not used)}
- Lynch DR, Perlman SL, Meier T. A phase 3, double‐blind, placebo‐controlled trial of idebenone in Friedreich ataxia. Archives of Neurology 2010;67(8):941‐7. [PUBMED: 20697044] [DOI] [PubMed] [Google Scholar]
Lynch 2012 {published data only}
- Lynch DR, Willi SM, Wilson RB, Cotticelli MG, Brigatti KW, Deutsch EC, et al. A0001 in Friedreich ataxia: biochemical characterization and effects in a clinical trial. Movement Disorders 2012;27(8):1026‐33. [PUBMED: 22744651] [DOI] [PubMed] [Google Scholar]
Mariotti 2009 {published data only}
- Mariotti C, Fancellu R, Caldarazzo S, Nanetti L, Bella D, Plumari M, et al. Erythropoietin in Friedreich ataxia: no effect on frataxin in a randomized controlled trial. Movement Disorders 2012;27(3):446‐9. [DOI] [PubMed] [Google Scholar]
Miyai 2012 {published data only}
- Miyai I, Ito M, Hattori N, Mihara M, Hatakenaka M, Yagura H, et al. Cerebellar ataxia rehabilitation trial in degenerative cerebellar diseases. Neurorehabilitation and Neural Repair 2012;26(5):515‐22. [PUBMED: 22140200] [DOI] [PubMed] [Google Scholar]
Ristori 2010 {published data only}
- Ristori G, Romano S, Visconti A, Cannoni S, Spadaro M, Frontali M, et al. Riluzole in cerebellar ataxia: a randomized, double‐blind, placebo‐controlled pilot trial. Neurology 2010;74(10):839‐45. [PUBMED: 20211908] [DOI] [PubMed] [Google Scholar]
Sobue 1983 {published data only (unpublished sought but not used)}
- Sobue I, Takayanagi T, Nakanishi T, Tsubaki T, Uono M, Kinoshita M, et al. Controlled trial of thyrotropin releasing hormone tartrate in ataxia of spinocerebellar degenerations. Journal of the Neurological Sciences 1983;61(2):235‐48. [PUBMED: 6417282] [DOI] [PubMed] [Google Scholar]
Trouillas 1995 {published data only (unpublished sought but not used)}
- Trouillas P, Serratrice G, Laplane D, Rascol A, Augustin P, Barroche G, et al. Levorotatory form of 5‐hydroxytryptophan in Friedreich's ataxia. Archives of Neurology 1995;52(5):456‐60. [PUBMED: 7733839] [DOI] [PubMed] [Google Scholar]
Wessel 1995 {published data only}
- Wessel K, Hermsdörfer J, Deger K, Herzog T, Huss GP, Kömpf D, et al. Double‐blind crossover study with levorotatory form of hydroxytryptophan in patients with degenerative cerebellar diseases. Archives of Neurology 1995;52(5):451‐5. [PUBMED: 7733838] [DOI] [PubMed] [Google Scholar]
Zannolli 2012 {published data only}
- Zannolli R, Buoni S, Betti G, Salvucci S, Plebani A, Soresina A, et al. A randomized trial of oral betamethasone to reduce ataxia symptoms in ataxia telangiectasia. Movement Disorders 2012;27(10):1312‐6. [PUBMED: 22927201] [DOI] [PubMed] [Google Scholar]
Zesiewicz 2012 {published data only}
- Zesiewicz TA, Greenstein PE, Sullivan KL, Wecker L, Miller A, Jahan I, et al. A randomized trial of varenicline (Chantix) for the treatment of spinocerebellar ataxia type 3. Neurology 2012;78(8):545‐50. [PUBMED: 22323747] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Arnold 2006 {published data only}
- Arnold P, Boulat O, Malre R, Kuntzer T. Expanding view of phenotype and oxidative stress in Friedreich's ataxia patients with and without idebenone. Schweizer Archiv für Neurologie und Psychiatrie 2006;157(4):169‐76. [Google Scholar]
Artuch 2002 {published data only}
- Artuch R, Aracil A, Mas A, Colomé C, Rissech M, Monrós E, et al. Friedreich's ataxia: idebenone treatment in early stage patients. Neuropediatrics 2002;33(4):190‐3. [DOI] [PubMed] [Google Scholar]
Artuch 2006 {published data only}
- Artuch R, Brea‐Calvo G, Briones P, Aracil A, Galván M, Espinós C, et al. Cerebellar ataxia with coenzyme Q10 deficiency: diagnosis and follow‐up after coenzyme Q10 supplementation. Journal of the Neurological Sciences 2006;246(1‐2):153‐8. [DOI] [PubMed] [Google Scholar]
Boesch 2007 {published data only}
- Boesch S, Sturm B, Hering S, Goldenberg H, Poewe W, Scheiber‐Mojdehkar B. Friedreich's ataxia: clinical pilot trial with recombinant human erythropoietin. Annals of Neurology 2007;62(5):521‐4. [DOI] [PubMed] [Google Scholar]
Boesch 2008 {published data only}
- Boesch S, Sturm B, Hering S, Scheiber‐Mojdehkar B, Steinkellner H, Goldenberg H, et al. Neurological effects of recombinant human erythropoietin in Friedreich's ataxia: a clinical pilot trial. Movement Disorders 2008;23(13):1940‐4. [DOI] [PubMed] [Google Scholar]
Bonnan 2008 {published data only}
- Bonnan M, Cabre P, Olindo S, Signate A, Saint‐Vil M, Smadja D. Steroid treatment in four cases of anti‐GAD cerebellar ataxia [Traitement des ataxies cérébelleuses à anticorps anti‐GAD par cures séquentielles de corticoïdes]. Revue Neurologique 2008;164(5):427‐33. [DOI] [PubMed] [Google Scholar]
Botez 1996 {published data only}
- Botez M, Botez‐Marquard T, Elie R, Pedraza O‐L, Goyette K, Lalonde R. Amantadine hydrochloride treatment in heredodegenerative ataxias: a double blind study. Journal of Neurology, Neurosurgery & Psychiatry 1996;61(3):259‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
Botez 1997 {published data only}
- Botez MI, Mayer P, Bellemare F, Couture J. Can we treat respiratory failure in Friedreich ataxia?. Archives of Neurology 1997;54(8):1030‐3. [DOI] [PubMed] [Google Scholar]
Broccoletti 2008 {published data only}
- Broccoletti T, Giudice E, Amorosi S, Russo I, Bonito M, Imperati F, et al. Steroid‐induced improvement of neurological signs in ataxia‐telangiectasia patients. European Journal of Neurology 2008;15(3):223‐8. [DOI] [PubMed] [Google Scholar]
Ershova 2007 {published data only}
- Ershova MV, Illarioshkin SN, Sukhorukov VS. The use of noben for correction of mitochondrial disorders in Friedrich's disease. Zhurnal Nevrologii i Psikhiatrii Imeni S.S. Korsakova 2007;107(9):32‐7. [PubMed] [Google Scholar]
Heo 2008 {published data only}
- Heo JH, Lee ST, Chu K, Kim M. The efficacy of combined estrogen and buspirone treatment in olivopontocerebellar atrophy. Journal of the Neurological Sciences 2008;271(1‐2):87‐90. [DOI] [PubMed] [Google Scholar]
Ilg 2012 {published data only}
- Ilg W, Schatton C, Schicks J, Giese MA, Schöls L, Synofzik M. Video game–based coordinative training improves ataxia in children with degenerative ataxia. Neurology 2012;79(20):2056‐60. [DOI] [PubMed] [Google Scholar]
Lagedrost 2011 {published data only}
- Lagedrost SJ, Sutton MS, Cohen MS, Satou GM, Kaufman BD, Perlman SL, et al. Idebenone in Friedreich ataxia cardiomyopathy—results from a 6‐month phase III study (IONIA). American Heart Journal 2011;161(3):639‐45. [10.1016/j.ahj.2010.10.038] [DOI] [PubMed] [Google Scholar]
Meier 2012 {published data only}
- Meier T, Perlman SL, Rummey C, Coppard NJ, Lynch DR. Assessment of neurological efficacy of idebenone in pediatric patients with Friedreich's ataxia: data from a 6‐month controlled study followed by a 12‐month open‐label extension study. Journal of Neurology 2012;259(2):284‐91. [10.1007/s00415‐011‐6174‐y] [DOI] [PubMed] [Google Scholar]
Melancon 1982 {published data only}
- Melancon SB, Vanasse M, Geoffroy G, Barabe L, Proulx A, Fontaine G, et al. Oral lecithin and linoleic acid in Friedreich's ataxia: II. Clinical results. Canadian Journal of Neurological Sciences 1982;9(2):155‐64. [DOI] [PubMed] [Google Scholar]
Nakamura 2009 {published data only}
- Nakamura K, Yoshida K, Miyazaki D, Morita H, Ikeda S. Spinocerebellar ataxia type 6 (SCA6): clinical pilot trial with gabapentin. Journal of the Neurological Sciences 2009;278(1‐2):107‐11. [DOI: 10.1016/j.jns.2008.12.017] [DOI] [PubMed] [Google Scholar]
Ogawa 2003 {published data only}
- Ogawa M, Shigeto H, Yamamoto T, Oya Y, Wada K, Nishikawa T, et al. D‐cycloserine for the treatment of ataxia in spinocerebellar degeneration. Journal of the Neurological Sciences 2003;210(1‐2):53‐6. [DOI] [PubMed] [Google Scholar]
Pineda 2008 {published data only}
- Pineda M, Arpa J, Montero R, Aracil A, Domínguez F, Galván M, et al. Idebenone treatment in paediatric and adult patients with Friedreich ataxia: long‐term follow‐up. European Journal of Paediatric Neurology 2008;12(6):470‐5. [DOI: 10.1016/j.ejpn.2007.11.006] [DOI] [PubMed] [Google Scholar]
Shimizu 1999 {published data only}
- Shimizu H, Tsuda T, Shiga Y, Miyazawa K, Onodera Y, Matsuzaki M, et al. Therapeutic efficacy of transcranial magnetic stimulation for hereditary spinocerebellar degeneration. Tohoku Journal of Experimental Medicine 1999;189(3):203‐11. [DOI] [PubMed] [Google Scholar]
Sobue 1980 {published data only}
- Sobue I, Yamamoto H, Konagaya M, Iida M, Takayanagi T. Effect of thyrotropin‐releasing hormone on ataxia of spinocerebellar degeneration. Lancet 1980;315(8165):418‐9. [DOI] [PubMed] [Google Scholar]
Strupp 2011 {published data only}
- Strupp M, Kalla R, Claassen J, Adrion C, Mansmann U, Klopstock T, et al. A randomized trial of 4‐aminopyridine in EA2 and related familial episodic ataxias. Neurology 2011;77(3):269‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Trouillas 1984 {published data only}
- Trouillas P. Regression of cerebellar syndrome with long‐term administration of 5‐HTP or the combination 5‐HTP‐benserazide. Italian Journal of Neurological Sciences 1984;5(3):253‐66. [DOI] [PubMed] [Google Scholar]
Trouillas 1997 {published data only}
- Trouillas P, Xie J, Adeleine P, Michel D, Vighetto A, Honnorat J, et al. Buspirone, a 5‐hydroxytryptamine1A agonist, is active in cerebellar ataxia. Results of a double‐blind drug placebo study in patients with cerebellar cortical atrophy. Archives of Neurology 1997;54(6):749‐52. [DOI] [PubMed] [Google Scholar]
Velasco‐Sanchez 2011 {published data only}
- Velasco‐Sánchez D, Aracil A, Montero R, Mas A, Jiménez L, O'Callaghan M, et al. Combined therapy with idebenone and deferiprone in patients with Friedreich's ataxia. Cerebellum 2011;10(1):1‐8. [DOI: 10.1007/s12311-010-0212-7] [DOI] [PubMed] [Google Scholar]
Yabe 1999 {published data only (unpublished sought but not used)}
- Yabe I, Sasaki H, Yamashita I, Takei A, Fukazawa T, Hamada T, et al. A clinical trial of acetazolamide for SCA6. Rinsho Shinkeigaku (Clinical Neurology) 1999;39(8):793‐9. [PubMed] [Google Scholar]
References to ongoing studies
EUCTR 2009‐016317‐20‐IT {published data only (unpublished sought but not used)}
- EUCTR 2009‐016317‐20‐IT. Pilot study to assess safety and tolerability of lithium on spinocerebellar ataxia of type 2 ‐ lithium in SCA2. http://apps.who.int/trialsearch/Trial2.aspx?TrialID=EUCTR2009‐016317‐20‐IT (accessed 21 March 2014).
EUCTR 2012‐005312‐26‐DE {unpublished data only}
- EUCTR 2012‐005312‐26‐DE. Sustained release 4‐aminopyridine (Fampyra®) in cerebellar gait disorder. http://apps.who.int/trialsearch/Trial2.aspx?TrialID=EUCTR2012‐005312‐26‐DE (accessed 21 March 2014).
Schulz 2009 {published data only}
- Schulz J, Holder G, Meier T, on behalf of the MICONOS Study Investigators. 12‐month European phase III clinical study of SNT‐MC17/idebenone in the treatment of Friedreich’s ataxia: baseline neurology data and interim safety results. Journal of Neurology 2009;256 Suppl 2:S153. [Google Scholar]
Additional references
Campuzano 1996
- Campuzano V, Montermini L, Moltò MD, Pianese L, Cossée M, Cavalcanti F, et al. Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996;271(5254):1423‐7. [DOI] [PubMed] [Google Scholar]
Collie 2003
- Collie A, Maruff P, Darby DG, McStephen M. The effects of practice on the cognitive test performance of neurologically normal individuals assessed at brief test‐retest intervals. Journal of the International Neuropsychological Society 2003;9(3):419‐28. [DOI] [PubMed] [Google Scholar]
Delatycki 1999
- Delatycki MB, Paris DB, Gardner RJ, Nicholson GA, Nassif N, Storey E, et al. Clinical and genetic study of Friedreich ataxia in an Australian population. American Journal of Medical Genetics 1999;87(2):168‐74. [DOI] [PubMed] [Google Scholar]
Delatycki 2000
- Delatycki MB, Williamson R, Forrest SM. Friedreich ataxia: an overview. Journal of Medical Genetics 2000;37(1):1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Duffy 2013
- Duffy JR. Motor Speech Disorders: Substrates, Differential Diagnosis, and Management. 3rd Edition. St Louis, MO: Elsevier Mosby, 2013. [Google Scholar]
Dürr 1996
- Dürr A, Cossee M, Agid Y, Campuzano V, Mignard C, Penet C, et al. Clinical and genetic abnormalities in patients with Friedreich's ataxia. New England Journal of Medicine 1996;335(16):1169‐75. [DOI] [PubMed] [Google Scholar]
Filla 2012
- Filla A, Sacca F, Michele G. A randomized trial of varenicline (Chantix) for the treatment of spinocerebellar ataxia type 3. Neurology 2012;78(19):1538. [DOI] [PubMed] [Google Scholar]
Folker 2010
- Folker JE, Murdoch BE, Cahill LM, Delatycki MB, Corben LA, Vogel AP. Dysarthria in Friedreich’s ataxia: a perceptual analysis. Folia Phoniatrica et Logopaedia 2010;62(3):97‐103. [DOI] [PubMed] [Google Scholar]
Gibilisco 2013
- Gibilisco P, Vogel AP. Friedreich ataxia. BMJ 2013;347:f7062. [DOI: 10.1136/bmj.f7062] [DOI] [PubMed] [Google Scholar]
GRADEpro 2008 [Computer program]
- Jan Brozek, Andrew Oxman, Holger Schünemann. GRADEpro. Version 3.2 for Windows. Jan Brozek, Andrew Oxman, Holger Schünemann, 2008.
Harding 1981
- Harding AE. Friedreich's ataxia: a clinical and genetic study of 90 families with an analysis of early diagnostic criteria and intrafamilial clustering of clinical features. Brain 1981;104(3):589‐620. [DOI] [PubMed] [Google Scholar]
Hartelius 2007
- Hartelius L, Elmberg M, Holm R, Lövberg AS, Nikolaidis S. Living with dysarthria: evaluation of a self‐report questionnaire. Folia Phoniatrica et Logopaedica 2007;60(1):11‐9. [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Kearney 2012
- Kearney M, Orrell RW, Fahey M, Pandolfo M. Antioxidants and other pharmacological treatments for Friedreich ataxia. Cochrane Database of Systematic Reviews 2012, Issue 4. [DOI: 10.1002/14651858.CD007791.pub2] [DOI] [PubMed] [Google Scholar]
Morgan 2008
- Morgan A, Vogel A. Intervention for dysarthria associated with acquired brain injury in children and adolescents. Cochrane Database of Systematic Reviews 2008, Issue 3. [DOI: 10.1002/14651858.CD006279.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Mundt 2012
- Mundt JC, Vogel AP, Feltner DE, Lenderking WR. Vocal acoustic biomarkers of depression severity and treatment response. Biological Psychiatry 2012;72(7):580‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Orr 1993
- Orr HT, Chung MY, Banfi S, Kwiatkowski TJ Jr, Servadio A, Beaudet AL. Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type‐1. Nature Genetics 1993;4(3):221‐6. [DOI] [PubMed] [Google Scholar]
Ramig 2001
- Ramig LO, Sapir S, Countryman S, Pawlas AA, O'Brien C, Hoehn M, et al. Intensive voice treatment (LSVT) for patients with Parkinson's disease: a 2 year follow up. Journal of Neurology, Neurosurgery & Psychiatry 2001;71(4):493‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
RevMan 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Rosen 2012
- Rosen KM, Folker JE, Vogel AP, Corben LA, Murdoch BE, Delatycki MB. Longitudinal change in dysarthria associated with Friedreich ataxia: a potential clinical endpoint. Journal of Neurology 2012;259(11):2471‐7. [DOI] [PubMed] [Google Scholar]
Schalling 2007
- Schalling E, Hammarberg B, Hartelius L. Perceptual and acoustic analysis of speech in individuals with spinocerebellar ataxia (SCA). Logopedics, Phoniatrics, Vocology 2007;32(1):31‐46. [DOI] [PubMed] [Google Scholar]
Schöls 1997
- Schöls L, Amoiridis G, Przuntek H, Frank G, Epplen JT, Epplen C. Friedreich's ataxia. Revision of the phenotype according to molecular genetics. Brain 1997;120(Pt 12):2131‐40. [DOI] [PubMed] [Google Scholar]
Vogel 2010
- Vogel AP, Morgan AT. Assessment of impairment or monitoring change in Friedreich Ataxia. Movement Disorders 2010;25(11):1753‐4. [DOI: 10.1002/mds.23103] [DOI] [PubMed] [Google Scholar]
Vogel 2010b
- Vogel AP, Fletcher J, Maruff P. Acoustic analysis of the effect of sustained wakefulness on speech. Journal of the Acoustical Society of America 2010;128(6):3747‐56. [DOI] [PubMed] [Google Scholar]
Vogel 2011
- Vogel AP, Fletcher J, Snyder PJ, Fredrickson A, Maruff P. Reliability, stability, and sensitivity to change and impairment in acoustic measures of timing and frequency. Journal of Voice 2011;25(2):137‐49. [DOI] [PubMed] [Google Scholar]
Vogel 2011b
- Vogel AP, Folker J, Murdoch B. Treatment for speech disorder in Friedreich ataxia and other hereditary ataxia syndromes. Cochrane Database of Systematic Reviews 2011, Issue 1. [DOI: 10.1002/14651858.CD008953] [DOI] [PMC free article] [PubMed] [Google Scholar]
Vogel 2012
- Vogel AP, Shirbin C, Churchyard AJ, Stout JC. Speech acoustic markers of early stage and prodromal Huntington's disease: a marker of disease onset?. Neuropsychologia 2012;50(14):3273‐8. [DOI] [PubMed] [Google Scholar]
Vogel 2014
- Vogel AP, Maruff P. Monitoring change requires a re‐think of assessment practices in voice and speech. Logopedics, Phoniatrics, Vocology 2014;39(2):56‐61. [DOI: ] [DOI] [PubMed] [Google Scholar]
WHO 2001
- World Health Organization. International Classification of Functioning, Disability and Health (ICF). Geneva: World Health Organization 2001.