

Summary
The causality between circulating proteins and thyroid cancer (TC) remains unclear. We employed five large-scale circulating proteomic genome-wide association studies (GWASs) with up to 100,000 participants and a TC meta-GWAS (nCase = 3,418, nControl = 292,703) to conduct proteome-wide Mendelian randomization (MR) and Bayesian colocalization analysis. Protein and gene expressions were validated in thyroid tissue. Through MR analysis, we identified 26 circulating proteins with a putative causal relationship with TCs, among which NANS protein passed multiple corrections (PBH = 3.28e-5, 0.05/1,525). These proteins were involved in amino acids and organic acid synthesis pathways. Colocalization analysis further identified six proteins associated with TCs (VCAM1, LGMN, NPTX1, PLEKHA7, TNFAIP3, and BMP1). Tissue validation confirmed BMP1, LGMN, and PLEKHA7’s differential expression between normal and TC tissues. We found limited evidence for linking circulating proteins and the risk of TCs. Our study highlighted the contribution of proteins, particularly those involved in amino acid metabolism, to TCs.
Subject areas: computational bioinformatics, cancer
Graphical abstract
Highlights
-
•
We assessed the causal relationship between circulating proteins and TC
-
•
Proteome-wide MR identified 26 proteins and colocalized with 6 shared genetic variants
-
•
The expression of proteins was verified in both the public database and our cohort
-
•
Proteins are associated to amino acids, carboxylic acids, and organic acid synthesis
Computational bioinformatics; Cancer
Introduction
Thyroid Cancer (TC), recognized as one of the most prevalent endocrine malignancies,1 significantly impacts the endocrine system and physiological functions and markedly diminishes patients’ quality of life. TC can be divided into several types: papillary TC (PTC), follicular TC (FTC), medullary TC (MTC), poorly differentiated TC (PDTC), and anaplastic TC (ATC).2 PTC and FTC are collectively known as differentiated TC (DTC), which has a high incidence, accounting for almost 90% of all TC pathological types.3 Over recent decades, the global incidence of TC has escalated, positioning it as a critical public health issue.4,5 Comprehensive genome-wide association studies (GWASs) have identified over 30 single-nucleotide polymorphisms (SNPs) associated with increased risk for TC, predominantly in gene regions related to FOXE1,6 PCNXL2,7 and the TERT-CLPTM1L.8 Despite these advances in previous studies in elucidating the causes of TC, the underlying pathogenesis of TC has remained largely elusive. The interplay between genetic predispositions and environmental risk factors in the pathogenesis of TC is still largely unexplored. Therefore, deeper understanding of the biological pathways involved in TC is crucial for discovering potential therapeutic interventions.
Proteomics, employing various sample types such as serum, plasma, and tissue, has become an integral part of TC research. This surge is largely attributed to the continual advancements in mass spectrometry and other state-of-the-art technologies.9 These developments play a pivotal role in identifying protein markers, elucidating pathogenesis, and predicting risks associated with TC.10,11,12 In observational studies, previous research has identified certain proteins linked to TC susceptibility, such as LCAT, GPX3, and leukotriene B4.10,13 However, current studies have not definitively determined whether these proteins act as intermediaries between risk genes and TC susceptibility or are merely reflections of changes induced by TC. And traditional observational epidemiological association studies are prone to reverse causation and bias due to confounding variables.
Mendelian randomization (MR), a genetic epidemiological method, overcomes the limitations of traditional observational studies by using genetic instrumental variables to assess the presumed causal relationship between exposure and outcome. In MR analysis, genetic risk alleles and non-risk alleles are hypothesized to be randomly assigned at conception, mirroring the random assignment of participants to treatment and control groups in randomized clinical trials. In two-sample MR studies, proteomic quantitative trait loci (pQTLs) and GWASs of TC are utilized independently to evaluate the putative causal relationship between proteins and TC. Advanced genetic analytical techniques like Bayesian colocalization methods and transcriptome-wide association studies could identify shared causal genetic loci between complex traits or diseases and explore the associations between protein-coding genes and TC.
Therefore, we aimed to explore the putative causal relationship between circulating proteins and TC, through integrating data from five large-scale proteomic studies across multiple ancestries with a meta-analyzed TC cohort for genetic analysis. Initially, we assessed the correlations and variabilities in the proteomes across different protein cohorts. Secondly, we performed a comprehensive proteome-wide MR analysis to examine the putative causal relationship and identified shared causal variant loci between proteins and TC. Thirdly, the relationships between the genes encoding these proteins and thyroid tumorigenesis were investigated. Finally, the protein and gene expression data from our cohort and public databases were used to conduct external validation, aiming to elucidate the actual biological mechanisms of the special proteins in TC pathogenesis.
Results
Multi-ancestry pQTL comparability validation
All analyzed data were based on the summarized information listed in Table 1. Among the five different proteomic cohorts, a total of 315 pQTLs were consistently detected across each cohort (Figure S1). The results of the protein correlation analysis between different cohorts (Figure S2) showed Pearson correlations ranging from 0.37 to 0.69 (p range: 3.57e-58 to 6.27e-11). The Atherosclerosis Risk in Communities (ARIC) cohort showed higher correlations compared to other cohorts, showing a correlation of 0.61 with the deCODE cohort (p = 1.12e-165) and 0.69 with the INTERVAL cohort (p = 2.98e-81). Consistent correlations were also observed between proteins detected in plasma and serum (Pearson: 0.37–0.57, p range: 1.39e-43 to 9.94e-21). In terms of protein variability across different cohorts (Figure S3), only the deCODE and UK Biobank Pharmaceutical Proteomics Project (UKB-PPP) cohorts were relatively balanced (deCODE: 54.8% vs. UKB-PPP: 45.2%). This result confirms the comparability of pQTL data across different populations.
Table 1.
Data sources for studied phenotypes
| Study | Phenotype | Cases | Controls | Seq-platform | Adjustment |
|---|---|---|---|---|---|
| deCODE14 | Plasma proteins | 35,559 | – | Illumina | age, gender, and sample age |
| UKB-PPP15 | Plasma proteins | 54,219 | – | Illumina | age, gender, batch, UKB center, UKB genetic array, time between blood sampling and measurement, and the first 20 genetic principal components |
| ARIC16 | Plasma proteins | 7,213 | – | Affymetrix | age, gender, 10 genetic principal components (PCs), and study sites. |
| AGES17 | Serum proteins | 5,457 | – | Illumina | age and gender |
| INTERVAL18 | Plasma proteins | 3,622 | – | Affymetrix | age, gender, duration between blood draw and processing (binary, ≤1 day/>1day), and the first three principal components of ancestry from multi-dimensional scaling |
| UKBB19 | Thyroid cancer | 986 | 5,135 | Illumina | age, gender, race, assessment center, and year of enrollment |
| FinnGen R9 | Thyroid cancer | 1783 | 287137 | Illumina and Affymetrix | age, gender, 10 PCs, genotyping batch. |
| Aleksandra et al., GWAS20 | Thyroid cancer | 649 | 431 | Illumina | age, gender, and genotyping batch |
| TCGA21 | Tissue transcriptions | 502 | 58 | Illumina | – |
| GTEx22 | Tissue transcriptions | 279 | – | Illumina | – |
| GEO23,24 | Tissue transcriptions | 105 | 73 | Affymetrix | – |
| Our multi-omic cohort | Tissue proteins and Transcriptions | 50 | 50 | Illumina | – |
ARIC, the Atherosclerosis Risk in Communities study; NA, not available; UKB-PPP, The UK Biobank Pharma Proteomics Project; TCGA, The Cancer Genome Atlas Program; GTEx, Genotype-Tissue Expression Project; GEO, Gene Expression Omnibus.
Putative causal associations between proteins and TC
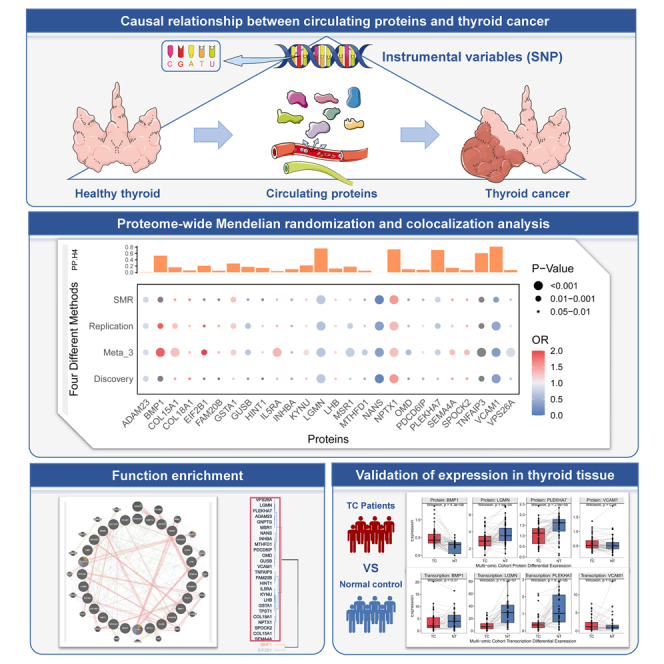
In total, 1,525 proteins were found to be replicated across the discovery and replication cohorts (Figure 1A), and detailed results of F, R2, and MR analysis for all cis-pQTLs are provided in Data S1 and S2. Based on the Bonferroni significance threshold (p < 3.28e-5), MR analysis (Figures 1B and 1C) revealed that only the N-acetylneuraminate synthase (NANS) protein was associated with a reduced risk of developing TC. In the discovery cohort, for a 1-standard deviation increase in NANS expression, the odds ratio (OR) for TC is 0.33 (95% confidence interval [CI] = 0.20–0.52, p = 2.92e-6). And in the replication cohort, the OR is 0.11 (95% CI = 0.04–0.28, p = 2.94e-6). The Steiger test and reverse MR analysis did not reveal any opposite causal relationship.
Figure 1.

Mendelian randomization and colocalization analysis results
(A) Ven diagram depicting proteins in discovery only, replication only, or in both.
(B) Proteome-wide MR volcano map results in the discovery cohort.
(C) Proteome-wide MR volcano map results in the replication cohort.
(D) Proteome-wide MR analyzed by four different methods and colocalization analysis (PP.H4) results.
To identify as many potentially positive proteins as possible, we adopted a suggestive p threshold of 0.05 for further screening. Additionally, we integrated the analysis methods of Summary-data MR(SMR) and random effects model meta-analysis across three independent cohort MR results. Proteins that were significant in all four methods with consistent beta directions were selected for subsequent analysis.
We identified 26 proteins potentially related to the etiology of TC (Figure 1D). The pooled results showed that higher levels of 14 proteins are associated with an increased risk of TC occurrence, including BMP1, COL15A1, COL18A1, EIF2B1, FAM20B, GSTA1, HINT1, IL5RA, KYNU, LHB, NPTX1, SEMA4A, SPOCK2, and TNFAIP3. The three proteins that most significantly increased the risk were BMP1 (ORReplication = 1.86, 95% CI = 1.26–2.75, p = 1.88e-3), NPTX1 (ORReplication = 1.35, 95% CI = 1.16–1.58, p = 1.49e-4), and TNFAIP3 (ORReplication = 3.45, 95% CI = 1.58–7.53, p = 1.92e-3). Conversely, higher levels of 12 proteins are associated with decreased risk of TC occurrence, such as NANS, GUSB, VCAM1, OMD, INHBA, MTHFD1, PDCD6IP, LGMN, PLEKHA7, VPS26A, MSR1, and ADAM23. The three proteins that most significantly decreased the TC risk were NANS (ORReplication = 0.33, 95% CI = 0.20–0.52, p = 2.92e-6), VCAM1 (ORReplication = 0.41, 95% CI = 0.24–0.68, p = 6.72e-4), and LGMN (ORReplication = 0.62, 95% CI = 0.47–0.82, p = 8.29e-4). All passed the HEIDI test (p > 0.05) (Data S3). The results of weighted mode, weighted median, MR Egger, and inverse-variance weighted method are shown in Data S4.
Colocalization analysis
The results of colocalization analysis (Table S1) for the 26 circulating proteins in relation to TC indicated that only one protein (Figure 1D), VCAM1 (coloc.analysis using Bayes factors posterior probability H4 (abf-PP.H4) = 0.82), showed strong evidence of colocalization with TC. Additionally, moderate evidence of colocalization was observed for five proteins: LGMN, NPTX1, PLEKHA7, TNFAIP3, and BMP1. The remaining proteins, which showed limited colocalization evidence, were identified as low-intensity colocalization targets.
Function and network prediction of TC proteins
The identified 26 proteins associated with TC formed a co-regulatory network, characterized by common expression and physical interactions (Figure 2). In the Gene Ontology (GO) enrichment analysis, these proteins were involved in several biological processes (BPs), including the biosynthesis of dicarboxylic acids, proteoglycan metabolism, and chondroitin sulfate metabolism. As for cellular components (CCs), they are associated with collagen trimers, the Golgi lumen, and the lysosomal lumen. In terms of molecular functions (MFs), these proteins relate to the extracellular matrix’s structural constituent conferring tensile strength, cysteine-type peptidase activity, and receptor ligand activity (Figure S4A). Analysis via the Kyoto Encyclopedia of Genes and Genomes (KEGG) showed that these genes are mainly involved in the biosynthesis of cofactors and the regulation of nuclear factor κB (NF-κB) and tumor necrosis factor (TNF) signaling pathways (Figure S4B). The PPI network revealed interactions among these proteins (Figure S5).
Figure 2.

GeneMANIA predictions of functions and networks for cis-pQTL related to TC
Thyroid tissue transcription-wide association
We conducted thyroid tissue-specific SMR analysis for genes encoding the identified 26 proteins, focusing on the 13 genes with complete expression quantitative trait loci (eQTL) data from thyroid tissues in the Genotype-Tissue Expression Project(GTEx) v8 database (Data S5). In thyroid tissues, genes predicted to have higher transcription levels, such as FAM20B, HINT1, KYNU, and SEMA4A, are associated with increased risk of TC. Conversely, lower levels of the proteins GUSB, VCAM1, PDCD6IP, and ADAM23 were related to higher risk of TC. All eight genes passed the HEIDI test (p > 0.05) and exhibited directions of effect consistent with proteins.
Tissue-specific proteins and gene expression validation
We performed expression validation for proteins with moderate to strong colocalization in our multi-omics TC cohort (Data S6). NPTX1 and TNFAIP3 proteins were not detectable in over 70% of our cohort, with expression levels at zero. Therefore, validation was conducted only for BMP1, LGMN, PLEKHA7, and VCAM1 (Figure 3A). Compared to normal thyroid tissue, TC tissue showed a significant upregulation of BMP1 protein expression in TC patients (Log2 fold change [log2FC] = 0.23; p = 4.3e-8; Figure 3B), and downregulation was observed for LGMN (log2FC = −0.37; p = 6.0e-4) and PLEKHA7 (log2FC = -0.37; p = 2.9e-5) proteins. No significant difference was observed in VCAM1 protein expression between the two groups (p = 0.54). In terms of gene expression, LGMN (p = 6.7e-7) and PLEKHA7 (p = 9.7e-5) showed downregulation in TC tissues.
Figure 3.

Visual validation of protein and transcriptional expression boxplots
(A) Putative causal proteins of MR and colocalization analysis (PP.H4>0.5) results of TC.
(B) Box map results of differential expression of proteins and transcriptomes between TC patients (red) and normal tissues (blue) in our multi-omics thyroid tumor-specific cohort (a) protein expression (b) gene expression.
(C) Differential expression of thyroid cancer transcriptomes in public database between TC patients (orange) and healthy controls (wathet) (a) TCGA (b) TCGA and GTEx (c) GEO:GSE33630 (d) GEO:GSE60542. NS, no significance, ∗: 0.01≤p<0.05, ∗∗:0.001≤p<0.01, ∗∗∗:p<0.001
Further external validation in public databases (Figure 3C), including The Cancer Genome Atlas (TCGA), GTEx, and Gene Expression Omnibus(GEO) thyroid cohorts, revealed similar findings. BMP1 gene expression was significantly upregulated in the TCGA and GEO cohorts, while LGMN and PLEKHA7 showed consistent upregulation across all four public cohorts (Data S7).
Discussion
Our findings indicate moderate level of consistency in pQTLs across diverse ancestries and proteomic platforms. We employed proteome-wide MR approach to assess the putative causal relationships between proteins and TC. After rigorous multiple testing, we identified 26 proteins that potentially exert causal influence on TC. Further, using Bayesian colocalization analysis, we pinpointed six shared causal variant loci with moderate to strong evidence of association with TC, including BMP1, VCAM, LGMN, and PLEKHA7. We substantiated the reliability of these identified proteins and their encoding genes by validating their dual expression in both TC and normal tissues.
Despite previous reports of associations between TC risk SNPs and circulating proteins, no study has yet employed MR approach. In this study, we confirmed associations between several proteins previously identified and thyroid tumors, such as BMP1,25 COL18A1,26 and GUSB.27 However, apart from VCAM1, LGMN, NPTX1, PLEKHA7, TNFAIP3, and BMP1, we observed weaker colocalization support for associations with TC, suggesting these links might be influenced by linkage disequilibrium (LD). The lack of strong colocalization support could be attributed to insufficient statistical power. Despite our colocalization analysis being based on large-scale TC meta-GWAS, further research is required to validate our findings.
The strong colocalization of VCAM128 and moderate evidence for LGMN29 and PLEKHA730 suggest their significant roles in TC pathogenesis. VCAM1 is a cell adhesion molecule that helps regulate inflammation-associated vascular adhesion and the transendothelial migration of leukocytes.31 LGMN has been demonstrated to be overexpressed not just in breast, prostate, and liver tumor cells but also in the macrophages that compose the tumor microenvironment.29,32 LGMN may promote the progression of TC through the miR-495/autophagy axis,33 and VCAM1 can promote the migration and invasion of TC cells.34 PLEKHA7 contributes to the trafficking and retention of transmembrane proteins, including nectins, Tspan33, and the copper pump ATP7A, at cell-cell junctions and lateral membranes.35 These proteins’ involvement in endothelial function could relate to angiogenesis in TC, a key factor in tumor growth and metastasis.
BMP1 association with increased risk of TC offers insights into the role of bone morphogenetic proteins in cancer.36 BMP1, multifunctional cytokines and members of the transforming growth factor β (TGF-β) family,37 are involved in extracellular matrix organization, which might influence tumor microenvironment and malignant phenotypes in TC.38
Collagens like COL15A1 and COL18A1 are integral to tissue structure. Their upregulation in TC may reflect altered extracellular matrix remodeling, a key factor in cancer invasion39 Specifically, COL15A1, non-fibrillar collagen associated with basement membranes, has been found to be involved in smooth muscle cell proliferation and atherosclerosis.40 Similarly, COL18A1, another non-fibrillar collagen, has been associated with various BPs, including angiogenesis and tumorigenesis.41 EIF2B1 in protein synthesis suggests its involvement in the abnormal proliferation characteristic of TC. Its increased levels in TC might be linked to heightened translational activity in cancer cells.42
GSTA1 and HINT1 are involved in detoxification and stress responses.43 GSTA1 is part of the glutathione S-transferase (GST) family, which are key phase II detoxification enzymes.44 These enzymes were involved in the metabolism of endogenous reactive compounds as well as xenobiotics, including toxicants and drugs.45 GSTs also exhibit significant ligand-binding properties, and several GST isoenzymes have been shown to interact with stress kinases during regulation of cell signaling pathways responsible for stress response, cell proliferation, and apoptosis.46 Their elevated levels in TC could be a response to oxidative stress in the tumor microenvironment.47
IL5RA and KYNU are intriguing due to their roles in immune response and tryptophan metabolism,48 respectively. IL5RA has been shown to be involved in immunogenic cell death, a form of cell death that stimulates an immune response against dead cell antigens.49 It has been associated with various diseases, including asthma and multiple myeloma, and its expression has been found to be upregulated in these conditions.49,50 KYNU is an enzyme involved in the kynurenine pathway.51,52 Dysregulation of this pathway has been associated with various diseases, including cancer and neurodegenerative disorders.51 Their association with TC could imply an altered immune response and amino acid metabolism in TC, as seen in colorectal cancer.53
NPTX1, SEMA4A, SPOCK2, and TNFAIP3 are linked to neural development and inflammation.54,55,56,57,58,59 NPTX1 may play a key role in synaptic loss, neurite damage, and neuronal apoptosis, mechanisms evoked by Aβ1.57 Their roles in TC might reflect the involvement of inflammatory pathways and nerve interactions in TC progression. SEMA4A, a class IV transmembrane semaphorin, plays crucial roles in dendritic cells, macrophages, and T cells.60 Moreover, emerging literature describes the role of immunoregulatory semaphorins and their receptors, plexins and neuropilins, as modulators of innate immunity and diseases defined by acute injury to the kidneys, abdomen, heart, and lungs.55 TNFAIP3 plays a crucial role in controlling the NF-κB signaling pathway, which promotes cell activation and can lead to autoinflammatory and autoimmune diseases.61
The lower levels of NANS, GUSB, INHBA, MTHFD1, PDCD6IP, and ADAM23 associated with higher risk of TC raise questions. NANS is involved in the biosynthesis of sialic acids, which are important for brain development and synaptic plasticity. However, its specific role in TC is not well studied and requires further research. GUSB is an enzyme that degrades glycosaminoglycans. It has been found to be overexpressed in some cancers. A study found that increased GUSB inhibits the expression of PD-L1, leading to primary resistance to anti-PD1 therapy in hepatocellular carcinoma.62 INHBA is a member of the TGF-β superfamily and has been shown to be differentially expressed in various cancer types, including cervical cancer.63 MTHFD1 are substrates for methionine, thymidylate, and de novo purine syntheses.64 One study suggested that the functional alteration caused by the 1958G>A polymorphism in MTHFD1 may influence DNA synthesis reactions and cell development, eventually affecting carcinogenesis.65 A study found that the Long non-coding RNA (lncRNA), ARHGEF26-AS1, acts as a miR-372-3p sponge that regulates the neuropeptide LGI1 receptor PDCD6IP and expression, thereby inhibiting the proliferation and migration of cancer cells.66 In summary, these proteins play significant roles in various cellular processes and their dysregulation could potentially contribute to the pathogenesis of TC. However, the exact mechanisms and implications of these proteins’ roles in TC require further research. Understanding these processes could provide valuable insights into the development and progression of TC and potentially guide the development of new therapeutic strategies.
The genes corresponding to these proteins have different biological functions, but 14 GO entries including “extracellular exosome” (GO:0070062) are associated with 14 of them, and 13 GO entries including “protein binding’ (GO:0005515) are associated with 13 of them. Both of these GO entries are associated with extracellular localization, molecular interactions, and signal transduction. Extracellular vesicles, as small vesicles in the extracellular space, contain various biomolecules, and the protein-binding activity within them plays a crucial role in intercellular interactions and signal transduction. These observations suggest that these 26 proteins may have certain associations or regulatory roles in the occurrence and development of TC.
In conclusion, the study conducted an extensive integrative analysis of multi-omics data concerning the risk of TC. It identified proteins with a putative causal impact on TC and the shared causal genetic variants between these proteins and TC, offering insights into the pathogenesis of TC.
Limitations of the study
Firstly, in the MR analysis, one concern is the pleiotropic effects of the instrumental variables, where genetic variations might influence TC independently of the protein exposure. To address this, we demonstrated that different MR sensitivity analysis provided consistent estimations and directions. Secondly, as the protein measurements in this study originate from diverse populations, biological samples, and measurement platforms, the findings may not be universally applicable to all circulating proteins across populations. Therefore, we repeated the validation in different cohorts and tissues, providing evidence for the ubiquity of TC-related proteins in different ancestors. Thirdly, this study only focused on cis-pQTL and lacked the results of trans-pQTL, which limited the interpretation of the results. Moreover, in reverse MR Analysis, most of the SNPs of TC were located in the trans-pQTL range. Therefore, we also supplemented Steiger’s test for direction verification, which assumes that instrumental variables influence exposure more than outcomes, helping to avoid reverse causality bias. Finally, the majority of study participants were of non-African descent. To gain more comprehensive understanding of the impact of genetic variations on metabolite measurements and TC risk, additional studies including individuals of Asian and African ancestry are necessary.
STAR★Methods
Key resources table
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Prof. Xiaobo Yang (yangx@gxmu.edu.cn).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
This paper analyzes existing, publicly available data. These access URLs for the datasets are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
This study adheres to the reporting guidelines of the Strengthening the Reporting of Observational Studies in Epidemiology using MR (STROBE-MR).74
Design
Our study's overall design is illustrated in the figure below, related to Table 1. Briefly, we utilized pQTL data from five large-scale European cohorts to explore causal relationship with TC. Bayesian colocalization analysis was employed to enhance the robustness of our findings. Subsequently, targeted gene enrichment analysis of protein-coding genes was conducted in thyroid tissues. Tissue-specific validation through proteomic and transcriptomic analysis was performed based on our multi-omic cohort and public databases, aiming to identify potential targets for TC treatment.

Overall study flowchart
Exposures
We selected SNPs associated with circulating cis-pQTL levels as instrumental variables from five major proteomic cohorts: deCODE14 health study, UK Biobank's Pharmaceutical Proteomics Project (UKB-PPP),15 ARIC,16 AGES,17 and INTERVAL.75 The deCODE and UKB-PPP cohorts acted as discovery sets for this study, with ARIC, AGES, and INTERVAL serving as replication sets. Complete summary data were available for four of the studies.14,15,16,75 Three of these studies14,15,16 employed the Illumina genotyping platform, while the other two16,75 used the Affymetrix genotyping platform. The SOMAscan platform was widely used for proteomic detection in the studies,14,16,17,75 except for the UKB-PPP cohort,15 which utilized the Olink platform. Detailed GWAS information is available in the original publications.
Outcome
The Meta GWAS of TC in European populations was extracted from one of our previously published articles,19 with the population details, genotyping, and imputation specifics described in another paper. The study encompassed three cohorts (Aleksandra et al., UKBB, and FinnGen), totaling 296,121 individuals (nCase=3,418; nControl=292,703). Notably, the study incorporated the latest FinnGen R9 release (nCase=1,783; nControl=287,137) building upon Feng et al.19 The GWAS meta-analysis was conducted using the Metal software73 as depicted in Figure S6. Additionally, the genomic inflation factor for each individual study and the LD score regression was calculated using the “LDlink” package, detailed in Table S2.
Method details
Mendelian randomization
We conducted Proteome-wide MR analysis between circulating proteins and TC using the “twosampleMR” package. When only single independent cis-pQTL locus was present, Wald ratio method was employed to assess changes in the log2FC odds ratio of TC risk per standard deviation (SD) increase in circulating protein levels represented by the instrumental variable. For proteins with multiple independent cis-pQTL loci, analysis was carried out using the independent lead SNP (p-minimum in LD-clumping windows of 1 Mb, using a European population-based LD reference panel in the 1000Genomes Project.76 Details of the cis-pQTLs for each cohort are provided in Data S8, S9, S10, S11, and S12. Sensitivity analysis was performed utilizing inverse-variance weighted, simple mode, weighted mode, and weighted median to verify the stability of the results. Heterogeneity was tested using Q values. Recognizing that a single meta-analysis result may not fully reflect the specificity of each independent study, to capture association signals that may be diluted or missed in the meta-analysis due to inter-study heterogeneity, we conducted second round of MR analysis on the pre-meta statistical data from each independent TC GWAS. The results of each independent MR analysis were combined using random-effects model. Heterogeneity was tested using the I2 statistic.
Summary-data MR(SMR)72 (https://yanglab.westlake.edu.cn/software/smr) has been developed to estimate pleiotropic associations between genetically determined traits and complex traits. It distinguishes shared genetic variation from associations driven by LD through the aggregation of multiple SNPs within range.77 Heterogeneity was tested using the HEIDI test, where a P > 0.05 indicates that the association between the protein and TC is not driven by LD.
Multiple testing correction was conducted using Bonferroni adjustment, setting the threshold for significance at P < 3.28e-5 (0.05/1525), with suggestive P range between 3.28e-5 and 0.05. Proteins within this range may have causal relationship with TC. Replication MR analysis based on pQTL data from the replication cohorts were further conducted for the identified proteins, where a P < 0.05 was considered indicative of replication significance.
We selected instrumental variables from the GWAS summary data for TC, adhering to the same pQTL selection criteria, to conduct reverse MR analysis. Four different MR methods were employed to estimate the effects, and we also carried out Steiger test to ensure the directionality of the association between the proteins and TCs. Results with a P < 0.05 were considered statistically significant.
Colocalization analysis
To assess whether proteins and TC risk involve shared causal variant loci, we conducted Bayesian colocalization analysis using “coloc” package (https://chr1swallace.github.io/coloc/index.html). The analysis encompassed five hypotheses: 1) no association with either trait; 2) association with only trait 1; 3) association with only trait 2; 4) associations with both traits but involving different causal variants; 5) associations with both traits involving the same causal variant. Posterior probabilities for each hypothesis test (H0, H1, H2, H3, and H4) were provided.78 A posterior probability (PP.H4) greater than 0.8 indicates strong support for colocalization, while a value between 0.5 and 0.8 suggests moderate evidence of colocalization. Colocalization analysis was performed within a 250kb window around the lead SNP of the cis-pQTL.79 Visualization of the colocalized regions was conducted using the “LocusCompareR” package.80
Function annotation
We utilized GeneMANIA (http://www.genemania.org) to predict the functions and networks of cis-genes associated with proteins relevant to TC. GO (Gene Ontology) and KEGG (Kyoto Encyclopedia of Genes and Genomes) enrichment analysis were employed to explore the functions and pathways of genes encoding these proteins. Additionally, potential protein-protein interaction (PPI) networks related to TC risk were investigated using the STRING database.
Tissue protein and gene expression validation
To further validate the specific expression of these proteins in thyroid tissues and their potential functional significance, we utilized thyroid tissue eQTLs from the GTEx (v8) database to perform SMR analysis. Subsequently, from the Thyroid Tumor Precision Prevention and Control research cohort, which has included 1000 pairs of papillary thyroid carcinoma (PTC), nodular goiter (NG), and normal controls as of December 2023, we randomly selected 100 healthy control matched 1:1 with PTC patients for age (±5 years) and gender to conduct protein and gene expression assays, with the following inclusion and exclusion criteria: (1) PTC patients were diagnosed by an experienced pathologist after postoperative pathological diagnosis; (2) healthy controls were those who had no thyroid nodules detected by neck ultrasonography, self-reported no history of thyroid disease, and had normal thyroid function; (3) not taking antithyroid or thyroid hormone drugs; (4) no history of other malignant tumors; and (5) complete epidemiological investigation data as well as blood samples were available. Additionally, RNA-seq data for normal thyroid and TC tissues were extracted from the TCGA-THCA21 and GTEX22 datasets available on the Xena platform (https://xena.ucsc.edu/), as well as from the GEO database (GSE60542; GSE33630). Differential analysis was conducted using the “limma” package,81 with false discovery rate (FDR) adjustments.
Quantification and statistical analysis
Instrumental variable selection
Prior to analysis, we harmonized SNP positioning to the Human Genome Build 37 (GRCH37) using the “MungeSumstats” package.82 Annotations were performed utilizing the GRCH37 release from the GENCODE database (https://www.gencodegenes.org/). Cis-QTLs were defined as SNPs within a 1MB range of the gene encoding the protein,83 whereas those outside this region were classified as trans-QTLs. We used beta and se value of cis-pQTL as instrumental variables to carry out the correlation and variation degree among different cohorts. Inclusion criteria for cis-pQTLs in the analysis were as follows: (1) genome-wide significant association threshold (P < 5e-8); (2) loci within the major histocompatibility complex (MHC) region on chromosome 6 (26-34MB) were excluded due to their complex LD patterns; (3) only independent loci were considered (1000kb; r2<0.0001); and (4) R2 and the F-statistic (R2=2×EAF×(1-EAF)×betaˆ2; F=R2×(N− 2)/(1−R2))84 were used to estimate the strength of the genetic instruments, where R2 represents the proportion of protein level variation explained by each genetic tool. For repeated proteins within the study, the protein with the maximum R2 was selected.85 Missing information for QTLs, such as allele frequencies, was imputed using reference panel constructed from the human 1000 Genomes Project.
Acknowledgments
All articles used in this study are publicly available summary-level data, with the relevant studies cited. Please contact our corresponding author for consent to our individual data. All study procedures were approved by the Medical Ethics Committee of the Second Affiliated Hospital of Guangxi University of Science and Technology (Liuzhou, China); all participants signed informed consent (approval no. 2022-1). This study was supported by the National Key R&D Program of China (grant no. 2023YFC3606300 and 2020YFE0201600), the National Natural Science Foundation of China (grant no. U22A20404), the Guangxi Key Research and Development Program (grant no. 2021AB07015), Research Basic Ability Improvement Project of Young and Middle-aged Teachers in Guangxi Universities (grant no. 2023KY0093) and Specific Research Project of Guangxi for Research Bases and Talents (grant no. AD23026163).
Author contributions
Conceptualization and formal analysis, Q.F. and S.W.; funding acquisition, F.W. and X.Y.; methodology, Y.Z. and X.F.; software and visualization, W.Z., X.L., and Y.L.; writing – original draft, S.Z., K.X., H.J., and H.T.; writing – review and editing, X.Z., Y.G., F.W., and X.Y.; critical revision and final approval, all authors.
Declaration of interests
The authors declare no competing interests.
Published: May 20, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.109961.
Contributor Information
Fei Wang, Email: wangfei@gxmu.edu.cn.
Xiaobo Yang, Email: yangx@gxmu.edu.cn.
Supplemental information
References
- 1.Chen D.W., Lang B.H.H., McLeod D.S.A., Newbold K., Haymart M.R. Thyroid cancer. Lancet. 2023;401:1531–1544. doi: 10.1016/s0140-6736(23)00020-x. [DOI] [PubMed] [Google Scholar]
- 2.Chmielik E., Rusinek D., Oczko-Wojciechowska M., Jarzab M., Krajewska J., Czarniecka A., Jarzab B. Heterogeneity of Thyroid Cancer. Pathobiology. 2018;85:117–129. doi: 10.1159/000486422. [DOI] [PubMed] [Google Scholar]
- 3.Seib C.D., Sosa J.A. Evolving Understanding of the Epidemiology of Thyroid Cancer. Endocrinol. Metab. Clin. North Am. 2019;48:23–35. doi: 10.1016/j.ecl.2018.10.002. [DOI] [PubMed] [Google Scholar]
- 4.Sung H., Ferlay J., Siegel R.L., Laversanne M., Soerjomataram I., Jemal A., Bray F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 5.Pizzato M., Li M., Vignat J., Laversanne M., Singh D., La Vecchia C., Vaccarella S. The epidemiological landscape of thyroid cancer worldwide: GLOBOCAN estimates for incidence and mortality rates in 2020. Lancet Diabetes Endocrinol. 2022;10:264–272. doi: 10.1016/s2213-8587(22)00035-3. [DOI] [PubMed] [Google Scholar]
- 6.Jendrzejewski J.P., Sworczak K., Comiskey D.F., de la Chapelle A. Clinical implications of GWAS variants associated with differentiated thyroid cancer. Endokrynol. Pol. 2019;70:423–429. doi: 10.5603/EP.a2019.0027. [DOI] [PubMed] [Google Scholar]
- 7.Gudmundsson J., Thorleifsson G., Sigurdsson J.K., Stefansdottir L., Jonasson J.G., Gudjonsson S.A., Gudbjartsson D.F., Masson G., Johannsdottir H., Halldorsson G.H., et al. A genome-wide association study yields five novel thyroid cancer risk loci. Nat. Commun. 2017;8 doi: 10.1038/ncomms14517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ge M., Shi M., An C., Yang W., Nie X., Zhang J., Lv Z., Li J., Zhou L., Du Z., Yang M. Functional evaluation of TERT-CLPTM1L genetic variants associated with susceptibility of papillary thyroid carcinoma. Sci. Rep. 2016;6 doi: 10.1038/srep26037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aebersold R., Mann M. Mass-spectrometric exploration of proteome structure and function. Nature. 2016;537:347–355. doi: 10.1038/nature19949. [DOI] [PubMed] [Google Scholar]
- 10.Navas-Carrillo D., Rodriguez J.M., Montoro-García S., Orenes-Piñero E. High-resolution proteomics and metabolomics in thyroid cancer: Deciphering novel biomarkers. Crit. Rev. Clin. Lab Sci. 2017;54:446–457. doi: 10.1080/10408363.2017.1394266. [DOI] [PubMed] [Google Scholar]
- 11.Carpi A., Mechanick J.I., Saussez S., Nicolini A. Thyroid tumor marker genomics and proteomics: diagnostic and clinical implications. J. Cell. Physiol. 2010;224:612–619. doi: 10.1002/jcp.22187. [DOI] [PubMed] [Google Scholar]
- 12.Gawin M., Wojakowska A., Pietrowska M., Marczak Ł., Chekan M., Jelonek K., Lange D., Jaksik R., Gruca A., Widłak P. Proteome profiles of different types of thyroid cancers. Mol. Cell. Endocrinol. 2018;472:68–79. doi: 10.1016/j.mce.2017.11.020. [DOI] [PubMed] [Google Scholar]
- 13.Sun Z., Feng D., Jiang L., Tian J., Wang J., Zhu W. Integrated proteomic and metabolomic analysis of plasma reveals regulatory pathways and key elements in thyroid cancer. Mol. Omics. 2023;19:800–809. doi: 10.1039/d3mo00142c. [DOI] [PubMed] [Google Scholar]
- 14.Ferkingstad E., Sulem P., Atlason B.A., Sveinbjornsson G., Magnusson M.I., Styrmisdottir E.L., Gunnarsdottir K., Helgason A., Oddsson A., Halldorsson B.V., et al. Large-scale integration of the plasma proteome with genetics and disease. Nat. Genet. 2021;53:1712–1721. doi: 10.1038/s41588-021-00978-w. [DOI] [PubMed] [Google Scholar]
- 15.Sun B.B., Chiou J., Traylor M., Benner C., Hsu Y.H., Richardson T.G., Surendran P., Mahajan A., Robins C., Vasquez-Grinnell S.G., et al. Plasma proteomic associations with genetics and health in the UK Biobank. Nature. 2023;622:329–338. doi: 10.1038/s41586-023-06592-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang J., Dutta D., Köttgen A., Tin A., Schlosser P., Grams M.E., Harvey B., CKDGen Consortium. Yu B., Boerwinkle E., et al. Plasma proteome analyses in individuals of European and African ancestry identify cis-pQTLs and models for proteome-wide association studies. Nat. Genet. 2022;54:593–602. doi: 10.1038/s41588-022-01051-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Emilsson V., Ilkov M., Lamb J.R., Finkel N., Gudmundsson E.F., Pitts R., Hoover H., Gudmundsdottir V., Horman S.R., Aspelund T., et al. Co-regulatory networks of human serum proteins link genetics to disease. Science. 2018;361:769–773. doi: 10.1126/science.aaq1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun B.B., Maranville J.C., Peters J.E., Stacey D., Staley J.R., Blackshaw J., Burgess S., Jiang T., Paige E., Surendran P., et al. Genomic atlas of the human plasma proteome. Nature. 2018;558:73–79. doi: 10.1038/s41586-018-0175-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feng X., Wang F., Yang W., Zheng Y., Liu C., Huang L., Li L., Cheng H., Cai H., Li X., et al. Association Between Genetic Risk, Adherence to Healthy Lifestyle Behavior, and Thyroid Cancer Risk. JAMA Netw. Open. 2022;5 doi: 10.1001/jamanetworkopen.2022.46311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Köhler A., Chen B., Gemignani F., Elisei R., Romei C., Figlioli G., Cipollini M., Cristaudo A., Bambi F., Hoffmann P., et al. Genome-wide association study on differentiated thyroid cancer. J. Clin. Endocrinol. Metab. 2013;98:E1674–E1681. doi: 10.1210/jc.2013-1941. [DOI] [PubMed] [Google Scholar]
- 21.Sanchez-Vega F., Mina M., Armenia J., Chatila W.K., Luna A., La K.C., Dimitriadoy S., Liu D.L., Kantheti H.S., Saghafinia S., et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell. 2018;173:321–337.e10. doi: 10.1016/j.cell.2018.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li X., Kim Y., Tsang E.K., Davis J.R., Damani F.N., Chiang C., Hess G.T., Zappala Z., Strober B.J., Scott A.J., et al. The impact of rare variation on gene expression across tissues. Nature. 2017;550:239–243. doi: 10.1038/nature24267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tomás G., Tarabichi M., Gacquer D., Hébrant A., Dom G., Dumont J.E., Keutgen X., Fahey T.J., 3rd, Maenhaut C., Detours V. A general method to derive robust organ-specific gene expression-based differentiation indices: application to thyroid cancer diagnostic. Oncogene. 2012;31:4490–4498. doi: 10.1038/onc.2011.626. [DOI] [PubMed] [Google Scholar]
- 24.Tarabichi M., Saiselet M., Trésallet C., Hoang C., Larsimont D., Andry G., Maenhaut C., Detours V. Revisiting the transcriptional analysis of primary tumours and associated nodal metastases with enhanced biological and statistical controls: application to thyroid cancer. Br. J. Cancer. 2015;112:1665–1674. doi: 10.1038/bjc.2014.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bai Y., Zhou G., Nakamura M., Ozaki T., Mori I., Taniguchi E., Miyauchi A., Ito Y., Kakudo K. Survival impact of psammoma body, stromal calcification, and bone formation in papillary thyroid carcinoma. Mod. Pathol. 2009;22:887–894. doi: 10.1038/modpathol.2009.38. [DOI] [PubMed] [Google Scholar]
- 26.Niu J., Guo W., Chen Y.Z., Jiang N. Identification of the collagen family as prognostic biomarkers in papillary thyroid carcinoma. Endocrine. 2022;78:491–506. doi: 10.1007/s12020-022-03175-9. [DOI] [PubMed] [Google Scholar]
- 27.Razavi S.A., Afsharpad M., Modarressi M.H., Zarkesh M., Yaghmaei P., Nasiri S., Tavangar S.M., Gholami H., Daneshafrooz A., Hedayati M. Validation of Reference Genes for Normalization of Relative qRT-PCR Studies in Papillary Thyroid Carcinoma. Sci. Rep. 2019;9 doi: 10.1038/s41598-019-49247-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maneshi P., Mason J., Dongre M., Öhlund D. Targeting Tumor-Stromal Interactions in Pancreatic Cancer: Impact of Collagens and Mechanical Traits. Front. Cell Dev. Biol. 2021;9 doi: 10.3389/fcell.2021.787485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reddy B.D., Beeraka N.M., Chitturi C.M.K., Madhunapantula S.V. An Overview of Targeting Legumain for Inhibiting Cancers. Curr. Pharm. Des. 2021;27:3337–3348. doi: 10.2174/1381612826666201125111625. [DOI] [PubMed] [Google Scholar]
- 30.Shah J., Guerrera D., Vasileva E., Sluysmans S., Bertels E., Citi S. PLEKHA7: Cytoskeletal adaptor protein at center stage in junctional organization and signaling. Int. J. Biochem. Cell Biol. 2016;75:112–116. doi: 10.1016/j.biocel.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 31.Kong D.H., Kim Y.K., Kim M.R., Jang J.H., Lee S. Emerging Roles of Vascular Cell Adhesion Molecule-1 (VCAM-1) in Immunological Disorders and Cancer. Int. J. Mol. Sci. 2018;19 doi: 10.3390/ijms19041057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khan S.U., Khan I.M., Khan M.U., Ud Din M.A., Khan M.Z., Khan N.M., Liu Y. Role of LGMN in tumor development and its progression and connection with the tumor microenvironment. Front. Mol. Biosci. 2023;10 doi: 10.3389/fmolb.2023.1121964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun J., Peng Y., Liu J., Zhou H., Sun L., He Q., Yu E. Pseudogene legumain promotes thyroid carcinoma progression via the microRNA-495/autophagy pathway. Oncol. Lett. 2021;22:616. doi: 10.3892/ol.2021.12877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen S., Su X., Jiang X., Zhang T., Min I., Ding Y., Wang X., Mao Z., Cao J., Teng X., et al. VCAM-1 Upregulation Contributes to Insensitivity of Vemurafenib in BRAF-Mutant Thyroid Cancer. Transl. Oncol. 2020;13:441–451. doi: 10.1016/j.tranon.2019.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sluysmans S., Méan I., Jond L., Citi S. WW, PH and C-Terminal Domains Cooperate to Direct the Subcellular Localizations of PLEKHA5, PLEKHA6 and PLEKHA7. Front. Cell Dev. Biol. 2021;9 doi: 10.3389/fcell.2021.729444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou Y., Xu S., Sun Q., Dong Y. An Analysis of BMP1 Associated with m6A Modification and Immune Infiltration in Pancancer. Dis. Markers. 2022;2022 doi: 10.1155/2022/7899961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ehata S., Miyazono K. Bone Morphogenetic Protein Signaling in Cancer; Some Topics in the Recent 10 Years. Front. Cell Dev. Biol. 2022;10 doi: 10.3389/fcell.2022.883523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rafi J.H., Jafar T., Pathan M.T., Reza R., Islam S., Sourna I.J., Alam R., Samad A., Ahammad F. High expression of bone morphogenetic protein 1 (BMP1) is associated with a poor survival rate in human gastric cancer, a dataset approaches. Genomics. 2021;113:1141–1154. doi: 10.1016/j.ygeno.2020.11.012. [DOI] [PubMed] [Google Scholar]
- 39.Martínez-Nieto G., Heljasvaara R., Heikkinen A., Kaski H.K., Devarajan R., Rinne O., Henriksson C., Thomson E., von Hertzen C., Miinalainen I., et al. Deletion of Col15a1 Modulates the Tumour Extracellular Matrix and Leads to Increased Tumour Growth in the MMTV-PyMT Mouse Mammary Carcinoma Model. Int. J. Mol. Sci. 2021;22:9978. doi: 10.3390/ijms22189978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Connelly J.J., Cherepanova O.A., Doss J.F., Karaoli T., Lillard T.S., Markunas C.A., Nelson S., Wang T., Ellis P.D., Langford C.F., et al. Epigenetic regulation of COL15A1 in smooth muscle cell replicative aging and atherosclerosis. Hum. Mol. Genet. 2013;22:5107–5120. doi: 10.1093/hmg/ddt365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bretaud S., Guillon E., Karppinen S.M., Pihlajaniemi T., Ruggiero F. Collagen XV, a multifaceted multiplexin present across tissues and species. Matrix Biol. 2020;6–7 doi: 10.1016/j.mbplus.2020.100023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adomavicius T., Guaita M., Zhou Y., Jennings M.D., Latif Z., Roseman A.M., Pavitt G.D. The structural basis of translational control by eIF2 phosphorylation. Nat. Commun. 2019;10:2136. doi: 10.1038/s41467-019-10167-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li S., Tan H.Y., Wang N., Zhang Z.J., Lao L., Wong C.W., Feng Y. The Role of Oxidative Stress and Antioxidants in Liver Diseases. Int. J. Mol. Sci. 2015;16:26087–26124. doi: 10.3390/ijms161125942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Blondis T.A., Accardo P.J., Snow J.H. Measures of attention deficit. Part II: Clinical perspectives and test interpretation. Clin. Pediatr. 1989;28:268–276. doi: 10.1177/000992288902800607. [DOI] [PubMed] [Google Scholar]
- 45.Sánchez-Gómez F.J., Díez-Dacal B., García-Martín E., Agúndez J.A.G., Pajares M.A., Pérez-Sala D. Detoxifying Enzymes at the Cross-Roads of Inflammation, Oxidative Stress, and Drug Hypersensitivity: Role of Glutathione Transferase P1-1 and Aldose Reductase. Front. Pharmacol. 2016;7:237. doi: 10.3389/fphar.2016.00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mazari A.M.A., Zhang L., Ye Z.W., Zhang J., Tew K.D., Townsend D.M. The Multifaceted Role of Glutathione S-Transferases in Health and Disease. Biomolecules. 2023;13 doi: 10.3390/biom13040688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baghban R., Roshangar L., Jahanban-Esfahlan R., Seidi K., Ebrahimi-Kalan A., Jaymand M., Kolahian S., Javaheri T., Zare P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020;18:59. doi: 10.1186/s12964-020-0530-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abd El-Fattah E.E. IDO/kynurenine pathway in cancer: possible therapeutic approaches. J. Transl. Med. 2022;20:347. doi: 10.1186/s12967-022-03554-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu C., Gao M., Zhang J., Fu Y. IL5RA as an immunogenic cell death-related predictor in progression and therapeutic response of multiple myeloma. Sci. Rep. 2023;13:8528. doi: 10.1038/s41598-023-35378-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Elena-Pérez S., Heredero-Jung D.H., García-Sánchez A., Estravís M., Martin M.J., Ramos-González J., Triviño J.C., Isidoro-García M., Sanz C., Dávila I. Molecular Analysis of IL-5 Receptor Subunit Alpha as a Possible Pharmacogenetic Biomarker in Asthma. Front. Med. 2020;7 doi: 10.3389/fmed.2020.624576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen L.M., Bao C.H., Wu Y., Liang S.H., Wang D., Wu L.Y., Huang Y., Liu H.R., Wu H.G. Tryptophan-kynurenine metabolism: a link between the gut and brain for depression in inflammatory bowel disease. J. Neuroinflammation. 2021;18:135. doi: 10.1186/s12974-021-02175-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hyland N.P., Cavanaugh C.R., Hornby P.J. Emerging effects of tryptophan pathway metabolites and intestinal microbiota on metabolism and intestinal function. Amino Acids. 2022;54:57–70. doi: 10.1007/s00726-022-03123-x. [DOI] [PubMed] [Google Scholar]
- 53.Peng X., Zheng T., Guo Y., Zhu Y. Amino acid metabolism genes associated with immunotherapy responses and clinical prognosis of colorectal cancer. Front. Mol. Biosci. 2022;9 doi: 10.3389/fmolb.2022.955705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhao Y., Yu Y., Zhao W., You S., Feng M., Xie C., Chi X., Zhang Y., Wang X. As a downstream target of the AKT pathway, NPTX1 inhibits proliferation and promotes apoptosis in hepatocellular carcinoma. Biosci. Rep. 2019;39 doi: 10.1042/bsr20181662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kanth S.M., Gairhe S., Torabi-Parizi P. The Role of Semaphorins and Their Receptors in Innate Immune Responses and Clinical Diseases of Acute Inflammation. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.672441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jiao M., Sun W., Li L., Li C., Zhou J., Li Q., Duan L. Clinical significance of SPOCK2 expression signature for high-grade serous ovarian cancer patients. Front. Genet. 2022;13 doi: 10.3389/fgene.2022.878123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gómez de San José N., Massa F., Halbgebauer S., Oeckl P., Steinacker P., Otto M. Neuronal pentraxins as biomarkers of synaptic activity: from physiological functions to pathological changes in neurodegeneration. J. Neural. Transm. 2022;129:207–230. doi: 10.1007/s00702-021-02411-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gao M., Li X., Yang M., Feng W., Lin Y., He T. TNFAIP3 mediates FGFR1 activation-induced breast cancer angiogenesis by promoting VEGFA expression and secretion. Clin. Transl. Oncol. 2022;24:2453–2465. doi: 10.1007/s12094-022-02918-4. [DOI] [PubMed] [Google Scholar]
- 59.Rossi M.N., Federici S., Uva A., Passarelli C., Celani C., Caiello I., Matteo V., Petrocchi S., Mortari E.P., De Benedetti F., et al. Identification of a Novel Mutation in TNFAIP3 in a Family With Poly-Autoimmunity. Front. Immunol. 2022;13 doi: 10.3389/fimmu.2022.804401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chapoval S.P., Vadasz Z., Chapoval A.I., Toubi E. Semaphorins 4A and 4D in chronic inflammatory diseases. Inflamm. Res. 2017;66:111–117. doi: 10.1007/s00011-016-0983-5. [DOI] [PubMed] [Google Scholar]
- 61.Das T., Chen Z., Hendriks R.W., Kool M. A20/Tumor Necrosis Factor α-Induced Protein 3 in Immune Cells Controls Development of Autoinflammation and Autoimmunity: Lessons from Mouse Models. Front. Immunol. 2018;9:104. doi: 10.3389/fimmu.2018.00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kong X., Zheng Z., Song G., Zhang Z., Liu H., Kang J., Sun G., Sun G., Huang T., Li X., et al. Over-Expression of GUSB Leads to Primary Resistance of Anti-PD1 Therapy in Hepatocellular Carcinoma. Front. Immunol. 2022;13 doi: 10.3389/fimmu.2022.876048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhao K., Yi Y., Ma Z., Zhang W. INHBA is a Prognostic Biomarker and Correlated With Immune Cell Infiltration in Cervical Cancer. Front. Genet. 2021;12 doi: 10.3389/fgene.2021.705512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hum D.W., Bell A.W., Rozen R., MacKenzie R.E. Primary structure of a human trifunctional enzyme. Isolation of a cDNA encoding methylenetetrahydrofolate dehydrogenase-methenyltetrahydrofolate cyclohydrolase-formyltetrahydrofolate synthetase. J. Biol. Chem. 1988;263:15946–15950. [PubMed] [Google Scholar]
- 65.Moruzzi S., Guarini P., Udali S., Ruzzenente A., Guglielmi A., Conci S., Pattini P., Martinelli N., Olivieri O., Tammen S.A., et al. One-carbon genetic variants and the role of MTHFD1 1958G>A in liver and colon cancer risk according to global DNA methylation. PLoS One. 2017;12 doi: 10.1371/journal.pone.0185792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Duijvesz D., Burnum-Johnson K.E., Gritsenko M.A., Hoogland A.M., Vredenbregt-van den Berg M.S., Willemsen R., Luider T., Paša-Tolić L., Jenster G. Proteomic profiling of exosomes leads to the identification of novel biomarkers for prostate cancer. PLoS One. 2013;8 doi: 10.1371/journal.pone.0082589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hemani G., Zheng J., Elsworth B., Wade K.H., Haberland V., Baird D., Laurin C., Burgess S., Bowden J., Langdon R., et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife. 2018;7 doi: 10.7554/eLife.34408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Giambartolomei C., Vukcevic D., Schadt E.E., Franke L., Hingorani A.D., Wallace C., Plagnol V. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014;10 doi: 10.1371/journal.pgen.1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yu G., Wang L.G., Han Y., He Q.Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A.R., Bender D., Maller J., Sklar P., de Bakker P.I.W., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Warde-Farley D., Donaldson S.L., Comes O., Zuberi K., Badrawi R., Chao P., Franz M., Grouios C., Kazi F., Lopes C.T., et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010;38:W214–W220. doi: 10.1093/nar/gkq537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhu Z., Zhang F., Hu H., Bakshi A., Robinson M.R., Powell J.E., Montgomery G.W., Goddard M.E., Wray N.R., Visscher P.M., Yang J. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 2016;48:481–487. doi: 10.1038/ng.3538. [DOI] [PubMed] [Google Scholar]
- 73.Willer C.J., Li Y., Abecasis G.R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Skrivankova V.W., Richmond R.C., Woolf B.A.R., Yarmolinsky J., Davies N.M., Swanson S.A., VanderWeele T.J., Higgins J.P.T., Timpson N.J., Dimou N., et al. Strengthening the Reporting of Observational Studies in Epidemiology Using Mendelian Randomization: The STROBE-MR Statement. JAMA. 2021;326:1614–1621. doi: 10.1001/jama.2021.18236. [DOI] [PubMed] [Google Scholar]
- 75.Suhre K., Arnold M., Bhagwat A.M., Cotton R.J., Engelke R., Raffler J., Sarwath H., Thareja G., Wahl A., DeLisle R.K., et al. Connecting genetic risk to disease end points through the human blood plasma proteome. Nat. Commun. 2017;8 doi: 10.1038/ncomms14357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nikpay M., Goel A., Won H.H., Hall L.M., Willenborg C., Kanoni S., Saleheen D., Kyriakou T., Nelson C.P., Hopewell J.C., et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat. Genet. 2015;47:1121–1130. doi: 10.1038/ng.3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Brion M.J.A., Shakhbazov K., Visscher P.M. Calculating statistical power in Mendelian randomization studies. Int. J. Epidemiol. 2013;42:1497–1501. doi: 10.1093/ije/dyt179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Foley C.N., Staley J.R., Breen P.G., Sun B.B., Kirk P.D.W., Burgess S., Howson J.M.M. A fast and efficient colocalization algorithm for identifying shared genetic risk factors across multiple traits. Nat. Commun. 2021;12:764. doi: 10.1038/s41467-020-20885-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pierce B.L., Tong L., Argos M., Demanelis K., Jasmine F., Rakibuz-Zaman M., Sarwar G., Islam M.T., Shahriar H., Islam T., et al. Co-occurring expression and methylation QTLs allow detection of common causal variants and shared biological mechanisms. Nat. Commun. 2018;9:804. doi: 10.1038/s41467-018-03209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu B., Gloudemans M.J., Rao A.S., Ingelsson E., Montgomery S.B. Abundant associations with gene expression complicate GWAS follow-up. Nat. Genet. 2019;51:768–769. doi: 10.1038/s41588-019-0404-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Murphy A.E., Schilder B.M., Skene N.G. MungeSumstats: a Bioconductor package for the standardization and quality control of many GWAS summary statistics. Bioinformatics. 2021;37:4593–4596. doi: 10.1093/bioinformatics/btab665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yuan S., Xu F., Li X., Chen J., Zheng J., Mantzoros C.S., Larsson S.C. Plasma proteins and onset of type 2 diabetes and diabetic complications: Proteome-wide Mendelian randomization and colocalization analyses. Cell Rep. Med. 2023;4 doi: 10.1016/j.xcrm.2023.101174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Papadimitriou N., Dimou N., Tsilidis K.K., Banbury B., Martin R.M., Lewis S.J., Kazmi N., Robinson T.M., Albanes D., Aleksandrova K., et al. Physical activity and risks of breast and colorectal cancer: a Mendelian randomisation analysis. Nat. Commun. 2020;11:597. doi: 10.1038/s41467-020-14389-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sun J., Zhao J., Jiang F., Wang L., Xiao Q., Han F., Chen J., Yuan S., Wei J., Larsson S.C., et al. Identification of novel protein biomarkers and drug targets for colorectal cancer by integrating human plasma proteome with genome. Genome Med. 2023;15:75. doi: 10.1186/s13073-023-01229-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
This paper analyzes existing, publicly available data. These access URLs for the datasets are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.