

Abstract
Visualizing activity patterns of distinct cell types during complex behaviors is essential to understand complex neural networks. It remains challenging to excite multiple fluorophores simultaneously so that different types of neurons can be imaged. In this Letter, we report a multicolor fiber-optic two-photon endomicroscopy platform in which two pulses from a Ti:sapphire laser and an optical parametric oscillator were synchronized and delivered through a single customized double-clad fiber to excite multiple chromophores. A third virtual wavelength could also be generated by spatial-temporal overlapping of the two pulses. The performance of the fiber-optic multicolor two-photon endomicroscope was demonstrated by in vivo imaging of a mouse cerebral cortex with “Brainbow” labeling.
Two-photon fluorescence microscopy (TPFM) is an enabling tool in biomedical applications, especially for neuroscience studies [1–3]. One important extension of TPFM to explore brain function is multicolor capability. A key goal of neuroscience is to understand how interactions between different cell types enable complex behaviors. The distinct patterns of activity exhibited by neurons and nonneuronal cells in the brain can be assessed using different fluorescent indicators [4]. However, one major challenge of multicolor two-photon imaging on live animals, particularly freely walking animals, is the bulky size of the bench-top TPFM. The animals have to be head-constrained [5] during a real-time experiment; the constraint and physical stress will affect neural activities and preclude many behavior studies, such as social interaction. Fiber-optic two-photon endomicroscopy allows the animal to be released from fixation. The technique provides submicron resolution with imaging quality approaching a bench-top microscope while maintaining an extremely compact size and light weight [6–10], making it ideal for in vivo imaging in freely walking animals [11–14]. Here we report the first miniature fiber-optic probe capable of performing two-photon excited multicolor imaging.
There are some basic approaches to performing multicolor two-photon imaging for more than two spectrally distinct chromophores. The first approach is to use a single wavelength to excite all fluorophores [15]. It is convenient and low in cost. However, a single wavelength limits our choice of target fluorophores, and it is hard to achieve efficient excitation simultaneously for every component. The second approach is to use multiple wavelengths during imaging, which includes sequential imaging with a single tunable laser [16] or imaging with a multiple laser source combination. However, the sequential imaging mode dramatically sacrifices imaging speed, and multiple laser combinations introduce much cost. The third method is to use two coherent laser pulses derived from an optical parametric oscillator (OPO). By temporal and spatial overlapping, we can synchronize the two coherent pulses and generate the third virtual wavelength [17]. These three wavelengths, which cover a big range from 750 to 1200 nm, are suitable for many fluorescent proteins and calcium indicators that are commonly used in neuroscience studies. This method shows more benefits in practice, and we introduced it into the fiber-optic endomicroscopy system to achieve multicolor imaging.
However, fiber-optic endomicroscopy is very different from the bench-top system, and there are some extra design considerations for implementing multicolor two-photon fluorescence imaging modality. (1) The first is dispersion compensation in which the femtosecond laser pulses are subject to temporal broadening when propagating through the DCF core, leading to loss of peak intensity. To obtain sufficient two-photon excitation efficiency, it is necessary to optimize separate dispersion compensation for each individual wavelength. (2) The second is fiber coupling efficiency in which the two input beams need to be coupled into the DCF core with high efficiency. (3) The third is femtosecond pulse synchronization in which the third virtual wavelength can work properly only when the two real excitation pulses are temporally overlapped at the focus.
Our multicolor fiber-optic two-photon setup is shown in Fig. 1. A tunable multiwavelength laser system (Chameleon Compact OPO pumped by Chameleon Ultra II, Coherent) provided two coherent femtosecond 80 MHz pulse trains for the pump (830 nm) and Stokes (1050 nm) lines. Two sets of half-wave plates (HWP1: WPH10M-808, HWP2: WPH10M-1064, Thorlabs) and polarization beam splitters (PBS1: CM1-PBS252, PBS2: CM1-PBS253, Thorlabs) combinations were used for power control. Different from a bench-top microscopy system, the first consideration is dispersion compensation. Here two sets of transmission grading pairs (GP1: WP-900/930–25.4; GP2: WP-500/1030–25 × 35, Wasatch Photonics) were introduced to compensate for the material dispersion in the fiber for each pulse train independently. For each grating pair, the separation was controlled by a line stage, and the entire setup was further mounted on a rotation stage to turn the input angle for optimal dispersion compensation [6]. After passing through grading pairs, the pre-chirped pulse trains were combined via a dichroic mirror (DM1: DMLP900, Thorlabs) and coupled into a single double-clad fiber (DCF) through the coupling lens (CL) [PLN 20× Objective, Olympus]. The epifluorescence signal collected by the micro-objective in the endomicroscope was delivered/transmitted through the core and inner clad of the same DCF to the proximal end of the probe and directed by a dichroic mirror (DM2: FF685-Di02, Semrock) to a photomultiplier tube (H10771P-40, Hamamatsu) for detection. The endomicroscope design is similar to what was reported previously [10], where the use of an achromatic micro-objective lens and a customized DCF helped improve the signal-to-background ratio [10,18,19]. 2D excitation beam scanning was performed by the piezoelectric (PZT) scanner. Amplitude-modulated sine and cosine waveforms with a frequency close to the mechanical resonant frequency of the fiber-optic cantilever (~1.2 kHz) were applied to the two orthogonal pairs of electrodes to produce an open-close spiral scanning pattern. The imaging frame rate is ~1.5 Hz, and the size of the field of view (FOV) is ~120 μm in diameter.
Fig. 1.
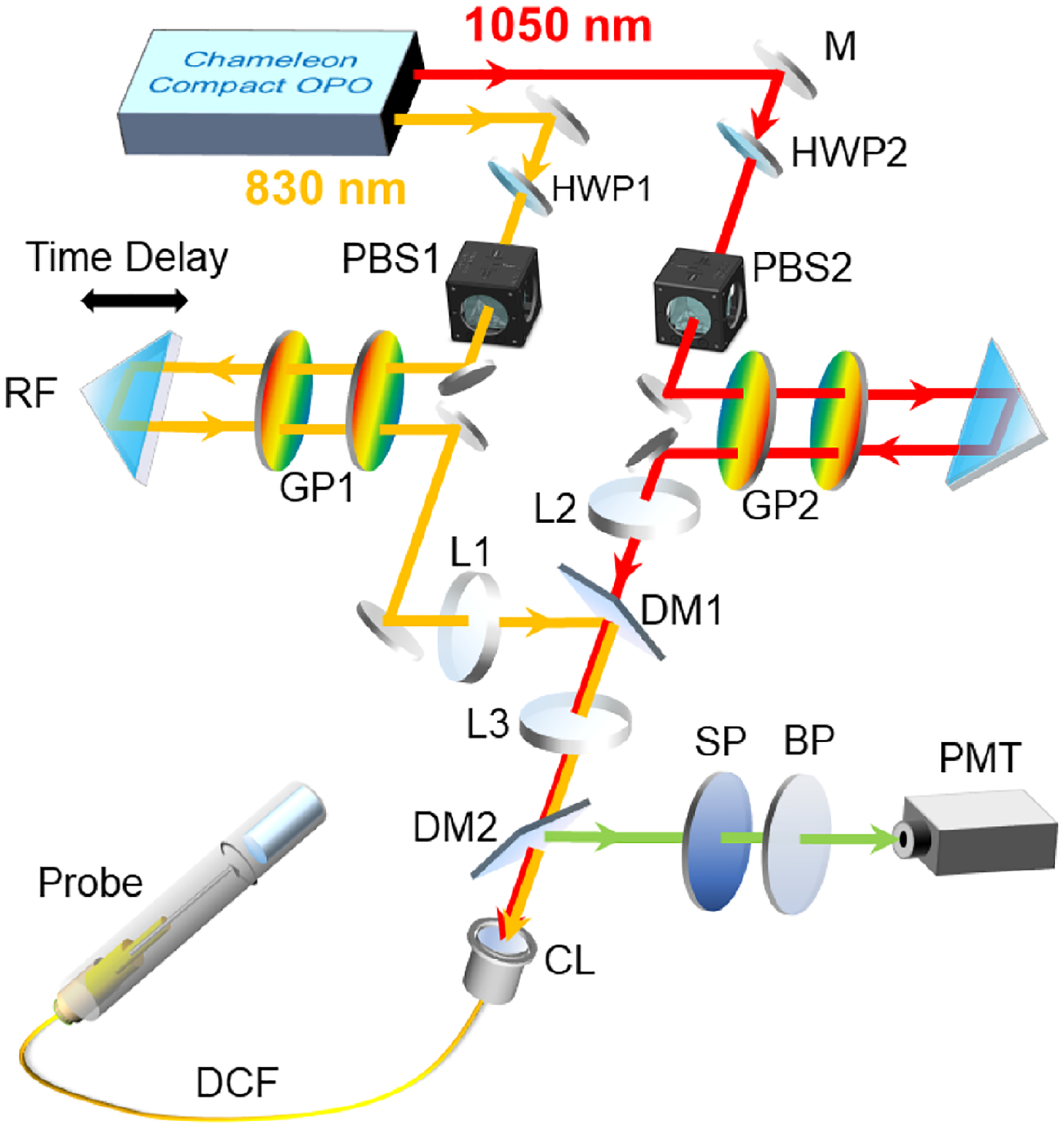
Schematic of a multicolor fiber-optic two-photon endomicroscopy platform. M, mirror; HWP, half-wave plate; PBS, polarization beam splitter; RF, retroreflector; GP, grating pair; DM, dichroic mirror; CL, coupling lens; SP, short-pass filter; BP, bandpass filter; DCF, double-clad fiber. The two-photon endomicroscope includes a PZT scanner, a DCF cantilever, a customized achromatic micro-objective (~300 μm working distance in water) and multiple mechanical adapters. The entire assembled probe weighed ~1 g with an outer diameter of ~2.8 mm.
The second challenge is how to achieve high coupling efficiency for both beams. Different wavelengths lead to different effective focal lengths of CL. The effective focal length is defined as feff = πωD/4λ. Here ω is the mode field diameter of the core of the DCF, and D is the 1/e2 diameter of the collimated beam incident on the lens. To make sure the fiber CL shares the same effective focal length for both input beams, we chose three doublet lens, L1 (AC254–250-B, Thorlabs), L2 (AC254–200-B, Thorlabs), and L3 (AC254–75-B, Thorlabs), to manually control the input beam sizes. Here the focal lengths satisfied fL2/fL1 ≈ λ1/λ2, and the fine-tuning was performed by changing the position of L1. After alignment, we could achieve over 50% coupling efficiency at the end of the imaging probe for both wavelengths.
The third challenge is how to generate the third virtual wavelength. Here the virtual wavelength results from the two-color two-photon absorption mechanism. Different from the traditional two-photon absorption, where the chromophore simultaneously absorbs two photons of the same wavelength, in this case, the chromophore is excited via simultaneous absorption of one photon from each beam, as shown in Fig. 2(a). Because the energy carried by a photon is hc/λ, synchronizing pulses with center wavelengths at λ1 and λ2 produce an equivalent wavelength at λ3 that satisfies the relation 2/λ3 = 1/λ1+ 1/λ2. This virtual wavelength depends on the combination of λ1 and λ2, and its equivalent intensity depends on the output power of the two input beams and synchronization of the pulses. Pulse synchronization requires both spatial and temporal overlapping. In our case, the two pulse trains are confined into the narrow fiber core (~5 μm in diameter), which means the first requirement has been met, so the dominant factor of synchronization is temporal overlapping. Here we controlled the time delay by tuning the time delay introduced by the retroreflector in the grating pair-based dispersion compensation module of the 830 nm beam path, as shown in Fig. 1.
Fig. 2.
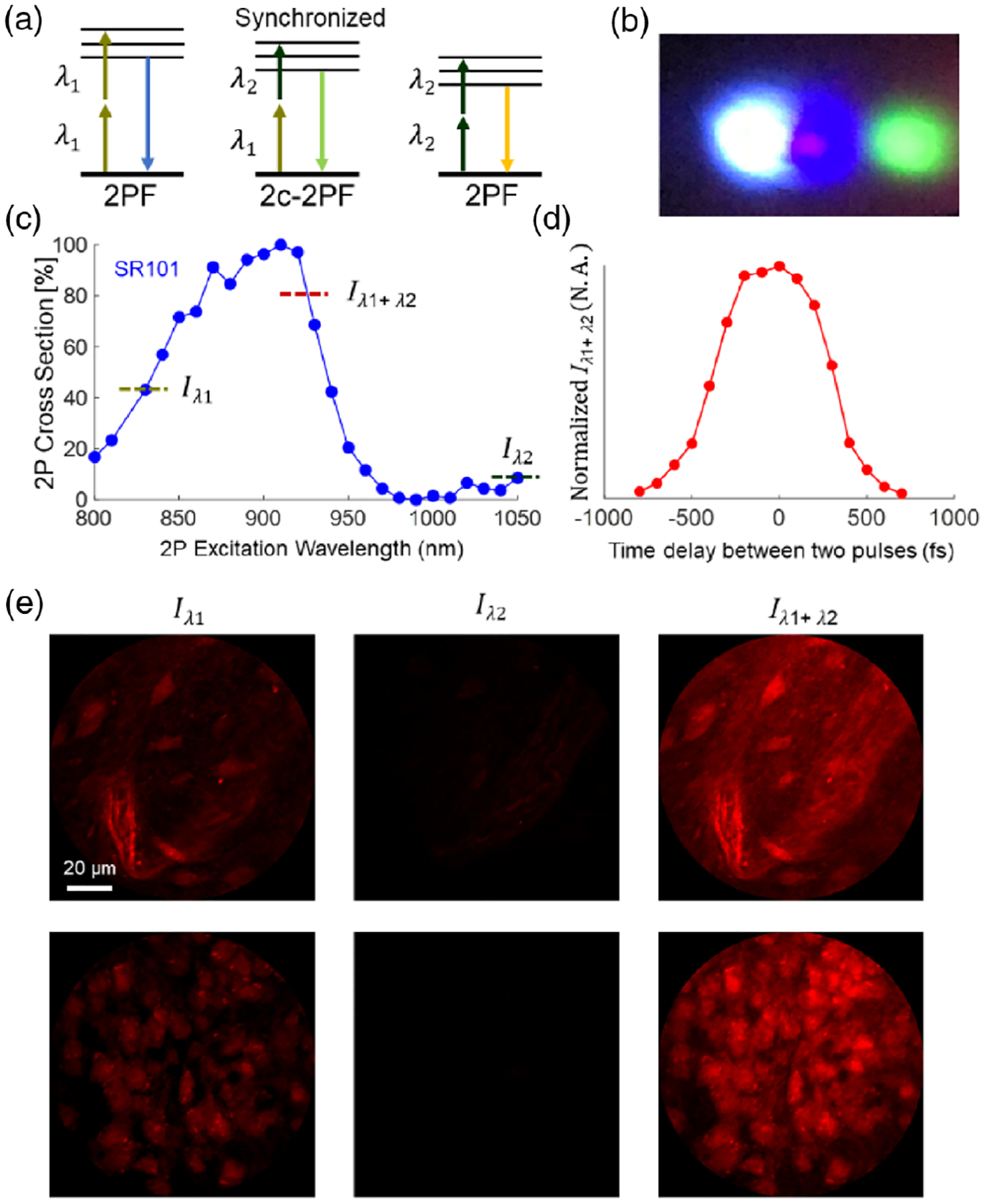
Validation of the third virtual wavelength. (a) Schematic of one-color two-photon absorption and two-color two-photon absorption. (b) SFG in BBO crystal after successful pulse synchronization. Three colors (from left to right): 415, 463.5, and 525 nm, corresponding to λ1/2, λ3/2, and λ2/2, respectively. (c) Normalized two-photon action cross section of SR101. (d) Fluorescence intensity at the virtual excitation Iλ1+λ2 versus the time delay between the two constituent laser pulses. (e) ex vivo imaging of a brain tissue slice (staining with SR101). Left, λ1 = 830 nm only; middle, λ2 = 1050 nm only; right, λ1 and λ2 simultaneously. (The time delay is optimized.)
The trick to validate temporal overlapping out of the probe is to utilize the sum-frequency generation (SFG) in a beta barium borate (BBO) crystal. The output beams from the fiber tip (without micro-objective) were sent to a rotating BBO crystal, which was mounted on a stepper motor. Here the stepper motor was introduced to sweep the incident angle quickly for the phase-matching condition of the SFG. An 1800 lpmm transmission grating was placed right after the crystal to separate the SFG (2/(1/λ1 + 1/λ2)) from the two SHG (λ1/2 and λ2/2) signals for easy observation. Once we achieved the temporal overlapping by tuning the time delay, we could resolve three different colors on the screen, shown in Fig. 2(b). After successful synchronization of two input pulses, our multicolor system enables three two-photon excitation wavelengths at 830, 927, and 1050 nm. The lateral resolutions at these three wavelengths are ~0.8, ~0.9, and ~1.0 μm, respectively, and the axial resolutions are ~4.5, ~5.0, and ~5.7 μm, respectively. The resolution was estimated by the full width at half-maximum of the point-spread functions when imaging across 0.2 μm diameter fluorescent beads.
Next, we carried out ex vivo imaging to estimate the performance after the generation of the third wavelength. Here we chose sulforhodamine 101 (SR101) as the testing dye. For ex vivo imaging experiments, a wild-type mouse (C57BL/6J, 20-week-old) was deeply euthanized by isoflurane and decapitated. The entire brain was extracted and placed in ice-cold phosphate-buffered saline solution. A brain slice (~2×2× 0.5 mm, located around the somatosensory cortex) was carefully resected from the rest of the brain tissue with a razor blade. Dye loading was achieved by soaking the slice in a dark chamber containing SR101 (500 nM) for 15 min at 36°C and then rinsing it for 15 min in saline solution before imaging. During imaging, we acquired data under three excitation conditions: turn on λ1 only (output power was ~30 mW), turn on λ2 only (output power was ~30 mW), and turn on λ1 and λ2 simultaneously (the total output power was ~60 mW). According to the two-photon excitation cross section of SR101 [20] shown in Fig. 2(c), the virtual wavelength at 927 nm has a much larger cross section than the 830 or 1050 nm constituents; therefore, two-wavelength excitation of SR101 shows much better efficiency than each single-wavelength excitation. Furthermore, two-wavelength excitation can be modulated by tuning the time delay between the two pulses [17], as shown in Fig. 2(d). Representative images obtained under three excitation conditions from an identical FOV are compared in Fig. 2(e). Compared with single-wavelength excitation, this two-synchronized-wavelength approach provided much higher excitation efficiency for SR101. The third extra virtual wavelength, combined with the two real input wavelengths, enables a semi-broadband excitation source for two-photon imaging.
Another consideration is the intrinsic background noise. The proper pulse synchronization will introduce some other nonlinear processes, which might interfere with the fluorescence signals. Here the major component is the parasitic background due to nonlinear four-wave mixing (FWM) occurring in the fiber core. In our case, because the longest detection wavelength of our endomicroscopy system is cut off at 609 nm (for red fluorescent protein [RFP] fluorescence signals), which is far from the FWM signals (686 and 1429 nm), the background noise will not affect the imaging results.
Finally, we illustrate our approach for live imaging through the “Brainbow”-labeled mouse cerebral cortex. The animal model (CAD-pac-CreER; Brainbow 2.1, Jackson) contains four fluorescence proteins: CFP, GFP, YFP, and RFP, and their peak two-photon excitation wavelengths are 830, 927, 960, and 1040, respectively. Our multicolor two-photon endomicroscopy system provides three different excitation wavelengths (830, 927, and 1050 nm), which enables high excitation efficiency for all four fluorophores simultaneously. The cranial window surgery was performed on a 10-week-old male mouse: a 4 mm wide craniotomy was drilled over the V1 cortex using a high-performance surgical drill. The surgical site was then closed using a cover glass (~4 mm diameter, 100 μm thickness) and vet glue. A one-gram titanium head restraining bar was attached to the head with dental cement for later fixation. After the mouse recovered from craniotomy surgery, we moved it to the endomicroscopy system for imaging. We anesthetized the mouse using 5% isoflurane and restrained the mouse by locking the head-restraining bar to a homemade platform. After the mouse head was fixed properly, the anesthesia setup was removed. The mouse woke up about 5 min post anesthesia, and it stayed awake through the entire imaging experiment. The multicolor imaging results of a Brainbow mouse are shown in Fig. 3. Benefiting from the semi-broadband excitation source, we can recognize several different anatomic structures (such as somas and blood vessels) simultaneously based on their distinct emission bands. The two-photon probe, manipulated via a three-axis translation stage, was gently placed against the cranial window surface. The focal plane was determined by fine-tuning the Z position to maximize the imaging signal-to-noise ratio. Once the focal plane was determined, the probe was moved laterally (in the X–Y plane) to search for different FOVs and collect images. All of the animal procedures were performed under the protocols approved by the Institutional Animal Care and Use Committee at the Johns Hopkins University.
Fig. 3.

In vivo imaging of a Brainbow-labeled mouse cortex using a multicolor two-photon endomicroscopy platform. BV, blood vessel; PP, parenchymal plaque; VP, vascular plaque. The three FOVs were obtained at the same focal plane (about 100 μm below the meninges). Scale bars: 20 μm.
Multicolor genetic labeling strategies open new avenues in the recording of neuronal connectivity among different components, and they have been widely used in the multiphoton microscopy system for ex vivo imaging of fixed tissue and in vivo imaging of head-fixed animals. However, the lack of appropriate imaging modality has hindered applications in freely behaving animals. Here we demonstrated the feasibility of multicolor imaging capability in a fiber-optic endomicroscopy system. We achieved simultaneous excitation of the multiple fluorophores with an optimized excitation efficiency due to broad-range, tunable excitation wavelengths. The approach can be used with many fluorophore combinations such as DAPI, GFP, mCherry for structural information and OGB1, GCaMP6 series, and jRCaMP series for functional information. This method shows great potential in many neuroscience applications. For example, social behavior studies involve the real-time recording of different types of neurons [21] and glial cells such as astrocytes [22]. In addition, this design principle can readily accommodate other fiber-optic imaging modalities such as micro-electro-mechanical systems (MEMS)-based miniature multiphoton microscopes [11,12,14]. Besides multicolor imaging capability, there are several technical innovations that can be integrated into our endomicroscopy system to further improve the imaging throughput such as z-scanning [23,24], higher imaging speed [13], and wider imaging FOV [25].
To conclude, we present a compact multicolor fiber-optic two-photon endomicroscopy platform. We achieve three-wavelength excitations by synchronizing the pulse trains from a femtosecond OPO and its pump. The ex vivo imaging demonstrates the benefits of the third virtual wavelength. Furthermore, we perform in vivo multicolor imaging of a mouse brain labeled with “Brainbow” transgenes. The results show that we can excite four different fluorescent proteins simultaneously with optimized excitation efficiency. The combination of multicolor imaging and fiber-optic endomicroscopy provides a promising tool for neuroscience studies.
Acknowledgment.
X. Li acknowledges the support of NIH and NSF Major Research Instrumentation (MRI). D. Bergles and X. Li acknowledge the support of Johns Hopkins Medicine.
Funding.
School of Medicine, Johns Hopkins University (Discovery Fund Synergy Award); National Science Foundation (CEBT1430030); National Institutes of Health (R01 CA153023).
Footnotes
Disclosures. The authors declare no conflicts of interest.
REFERENCES
- 1.Denk W, Strickler JH, and Webb WW, Science 248, 73 (1990). [DOI] [PubMed] [Google Scholar]
- 2.Helmchen F and Denk W, Nat. Methods 2, 932 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Kerr JN and Denk W, Nat. Rev. Neurosci 9, 195 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Inoue M, Takeuchi A, Manita S, Horigane S-I, Sakamoto M, Kawakami R, Yamaguchi K, Otomo K, Yokoyama H, and Kim R, Cell 177, 1346 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Dombeck DA, Khabbaz AN, Collman F, Adelman TL, and Tank DW, Neuron 56, 43 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Myaing MT, MacDonald DJ, and Li X, Opt. Lett 31, 1076 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Liang W, Murari K, Zhang YY, Chen Y, Li M-J, and Li X, J. Biomed. Opt 17, 021108 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Y, Akins ML, Murari K, Xi J, Li M-J, Luby-Phelps K, Mahendroo M, and Li X, Proc. Natl. Acad. Sci. USA 109, 12878 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liang W, Hall G, and Li X, Opt. Express 26, 22877 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liang W, Hall G, Messerschmidt B, Li M-J, and Li X, Light Sci. Appl 6, e17082 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Piyawattanametha W, Cocker ED, Burns LD, Barretto RPJ, Jung JC, Ra H, Solgaard O, and Schnitzer MJ, Opt. Lett 34, 2309 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zong WJ, Wu RL, Li ML, Hu YH, Li YJ, Li JH, Rong H, Wu HT, Xu YY, Lu Y, Jia HB, Fan M, Zhou Z, Zhang YF, Wang AM, Chen LY, and Cheng HP, Nat. Methods 14, 713 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Park H-C, Guan H, Li A, Yue Y, Li M-J, Lu H, and Li X, Opt. Lett 45, 3605 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klioutchnikov A, Wallace DJ, Frosz MH, Zeltner R, Sawinski J, Pawlak V, Voit K-M, J. Russell P. St., and Kerr JN, Nat. Methods 17, 509 (2020). [DOI] [PubMed] [Google Scholar]
- 15.Entenberg D, Wyckoff J, Gligorijevic B, Roussos ET, Verkhusha VV, Pollard JW, and Condeelis J, Nat. Protoc 6, 1500 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dunn KW, Sandoval RM, Kelly KJ, Dagher PC, Tanner GA, Atkinson SJ, Bacallao RL, and Molitoris BA, Am. J. Physiol 283, C905 (2002). [DOI] [PubMed] [Google Scholar]
- 17.Mahou P, Zimmerley M, Loulier K, Matho KS, Labroille G, Morin X, Supatto W, Livet J, Débarre D, and Beaurepaire E, Nat. Methods 9, 815 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Wu Y, Xi J, Cobb MJ, and Li X, Opt. Lett 34, 953 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu Y and Li X, Biomed. Opt. Express 1, 1234 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mütze J, Iyer V, Macklin JJ, Colonell J, Karsh B, Petrášek Z, Schwille P, Looger LL, Lavis LD, and Harris TD, Biophys. J 102, 934 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Selimbeyoglu A, Kim CK, Inoue M, Lee SY, Hong AS, Kauvar I, Ramakrishnan C, Fenno LE, Davidson TJ, and Wright M, Sci. Transl. Med 9, eaah6733 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cao X, Li L-P, Wang Q, Wu Q, Hu H-H, Zhang M, Fang Y-Y, Zhang J, Li S-J, and Xiong W-C, Nat. Med 19, 773 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Li A, Liang W, Guan H, Gau Y-TA, Bergles DE, and Li X, Biomed. Opt. Express 8, 2519 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu Y, Zhang Y, Xi J, Li M-J, and Li X, J. Biomed. Opt 15, 060506 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liang W, Park H-C, Li K, Li A, Chen D, Guan H, Yue Y, Gau Y-TA, Bergles DE, Li M-J, and Li X, IEEE Trans. Med. Imaging 39, 3779 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]