

Abstract
The patterns of low-risk myelodysplastic syndrome (MDS) progression and the clinical and molecular features of those patterns have not been well described. We divided our low-risk (LR) MDS patients (N=1,914) into 4 cohorts: 1) patients who remained LR-MDS (LR-LR; N=1,300; 68%), 2) patients who progressed from LR to high-risk (HR) MDS (LR-HR) without transformation into acute myeloid leukemia (AML) (N=317; 16.5%), 3) patients who progressed from LR to HR MDS and then AML (LR-HR-AML; N=124; 6.5%), and 4) patients who progressed from LR MDS directly to AML (LR-AML; N=173; 9%). Risk factors for progression included: male gender, low absolute neutrophil count (ANC), low platelet count, high bone marrow (BM) blasts, ferritin >1000 mcg/L, albumin <3.5 g/dL, multi-lineage dysplasia (MLD), and lack of ring sideroblasts. Among patients with marked BM fibrosis (N=49), 18% progressed directly to AML. Somatic mutations (SM) associated with an increased risk of direct or indirect AML progression included SRSF2 and NRAS. SM in IDH1, IDH2 and NPM1 were more common in patients with direct AML transformation. SM associated with progression to higher risk disease only, without AML transformation, were ASXL1, TP53, RUNX1, and CBL. SF3B1 mutation was associated with less progression. About 171 patients (13.1% of all LR-LR patients) died within two years of diagnosis of LR-MDS without disease progression. Among the 61 cases with documented cause of death, 18 patients (29.5%) died from cytopenia and MDS-related complications. Identifying patterns of disease progression of LR MDS patients and their predictive factors will be crucial to be able to tailor therapy accordingly.
Introduction
Myelodysplastic syndromes (MDS) are heterogeneous stem cell neoplasms that primarily affect the elderly population. The approach to treatment of MDS includes management of symptoms or cytopenias, and treatments to alter the natural history of the disease in order to prevent progression to acute myeloid leukemia (AML).
Management is largely risk-adapted based on various prognostic scoring systems. The International Prognostic Scoring System (IPSS) was introduced in 1997 and was an important standard to assess prognosis in patients with MDS at initial diagnosis.1 The IPSS divides patients into 4 risk categories: low, intermediate-1, intermediate-2, and high risk.1 Bone marrow (BM) blasts, karyotype and number of cytopenias are the only 3 variables comprising the IPSS. The Revised International Prognostic Scoring System (IPSS-R) was introduced in 2012. This added a refined cytogenetic classification, degree of cytopenia, and improved BM blast percentage as categories, and introduced 5 subgroups of MDS: very low (VL) risk, low risk (LR), intermediate, high, and very high risk.2 Some other tools that have been developed and widely used for risk stratification include the Global MD Anderson Model, the lower risk MD Anderson model (LR-MDAS), and the WHO-based prognostic scoring system (WPSS).3-5
In low-risk disease (LR MDS), the aim of therapy is to improve cytopenias in patients with anemia, neutropenia and/ or thrombocytopenia in order to improve quality of life and prevent complications from cytopenias in an attempt to prolong overall survival (OS). Cytopenia-related complications and exacerbation of co-existing comorbidities significantly affect the disease burden, and affect quality of life and OS in low-risk disease.6 ,7 The LR-MDAS model suggested that almost a third of the LR MDS patients stratified by the IPSS progress or die within a short time after diagnosis. It incorporated age, hemoglobin, severity of thrombocytopenia, karyotype, and BM blasts ≥4% as variables, and divided patients into 3 groups, upstaging 25% of LR MDS patients into the higher risk disease category.
Historically, 30-40% of MDS patients progress to AML; however, unfortunately, the majority of patients are thought to succumb from disease complications, namely cytopenia or an interplay of cytopenia with comorbidities. The patterns of LR MDS disease progression and causes of mortality are not well delineated in literature. Disease progression can be in the form of further BM failure associated with more severe cytopenias or increased myeloblasts with transformation to higher risk disease and/or AML. The molecular phenotypes associated with different patterns of disease progression and clonal evolution have not been well studied. Hence, we undertook this study to better delineate factors that can help identify patients with LR MDS at risk for progression and/or death early in their disease course who may benefit from early intervention rather than the “wait and watch” approach.
Methods
Patient selection
We identified 1,914 patients with an established diagnosis of very low- and low-risk MDS as per the IPSS-R from the Moffitt Cancer Center MDS database. Laboratory values and prognostic scores were determined at the time of diagnosis or referral prior to treatment. The study was approved by the Institutional Review Board. We divided our patients into 4 cohorts based on the pattern of their disease progression: 1) patients who remained in the low-risk MDS category throughout their period of follow-up (LR-LR); 2) patients who progressed from LR to HR MDS (LR-HR) without AML transformation; 3) patients who progressed from LR to HR MDS and then AML (LR-HR-AML); and 4) patients who directly progressed from LR MDS to AML (LR-AML).
Characterization of mutational profile
Next generation sequencing (NGS) was performed on peripheral blood or BM as a part of standard work up during patients’ initial presentation. The NGS panels targeted up to 406 genes across multiple platforms: FoundationOne Heme, Genoptix Myeloid Molecular Profile, Genoptix NexCourse Complete, Illumina TruSight Myeloid-54, and the Moffitt 98-gene Myeloid Action Panel.
Definitions of survival
Overall survival (OS) was measured from time of diagnosis until death or censored at time of last patient follow-up. For patients that progressed to AML, leukemia-free survival (LFS) was calculated from the time of diagnosis to development of AML.
Statistical methods
Categorical variables were compared using Fisher’s exact and χ2 tests, and quantitative data were compared using the Mann-Whitney U test. Time-to-event analyses were assessed using the Kaplan-Meier method. The log-rank test was used to compare OS between groups. Statistical analyses were performed using IBM SPSS statistics version 26.
Results
We divided the patients with low-risk and very low-risk MDS into the 4 cohorts described above. The majority of the patients, 68% (N=1,300) remained in LR-MDS without progressing to high-risk MDS or AML, 16.5% patients (N=317) progressed from low-risk to high-risk MDS (LR-HR) without AML transformation, 6.5% patients (N=124) progressed from low-risk to high-risk MDS and then AML (LR-HR-AML), and 9% patients (N=173) progressed from low-risk MDS directly to AML (LR-AML). At a median follow-up of 99 months, the median OS for the LR-LR, LR-HR, LR-HR-AML, and LR-AML groups was 80.7, 61.2, 48.3, and 42.8 months, respectively (P<0.0001) (Figure 1). The median time to progression to AML in the LR-HR-AML and LR-AML groups was 29 months, and the median time to progress from LR to HR-MDS was 22 months.
Figure 1.
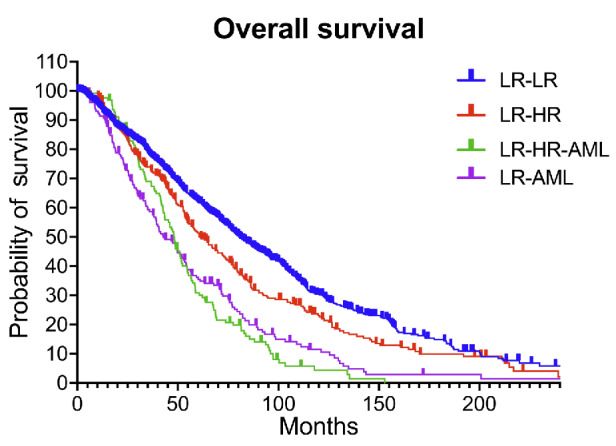
Overall survival of the 4 cohorts. At a median follow up of 99 months, the median overall survival (OS) for the patients who remained in the low-risk (LR) myelodysplastic syndromes (MDS) category throughout their period of follow-up (LR-LR), patients who progressed from LR to high-risk (HR) MDS without acute myeloid leukemia (AML) transformation (LR-HR), patients who progressed from LR to HR MDS and then AML (LR-HR-AML), and patients who directly progressed from LR MDS to AML (LR-AML) was 80.7, 61.2, 48.3, and 42.8 months, respectively.
Table 1 shows baseline characteristics of the 4 cohorts. The median age at diagnosis of patients in the 4 cohorts was similar (70, 68, 69, and 70 years respectively; P=0.484). Clinical and laboratory risk factors for progression to higher risk or AML included male gender (P<0.0001), low absolute neutrophil count (ANC) (P=0.04), low platelet count (P<0.001), high BM blasts (P<0.001), ferritin >1000 mcg/L (P<0.0001), albumin <3.5 g/dL (P=0.012). Pathologic risk factors for progression included multi-lineage dysplasia (MLD) (P<0.001) and lack of ring sideroblasts (P<0.0001). Patients with any del5q abnormality had a higher rate of progression to HR-MDS (P=0.001). Among patients with grade 2-3 BM fibrosis (N=49), 18% progressed directly to AML (P=0.009). There was no difference in rates of progression when patients were stratified by the lower risk MD Anderson model (LR-MDAS). Table 1 summarizes treatment patterns among the cohorts. As expected, more patients in the LR-AML (53.8%) and LR-HR-AML (87.9%) cohorts were treated with hypomethylating agents, while more patients in the LR-LR (24.6%) and LR-HR (30%) cohorts were treated with lenalidomide. As expected, the rate of allogeneic stem cell transplantation (alloSCT) was significantly lower in patients that remained low-risk compared to the other 3 cohorts that progressed to high-risk disease and/or AML: N=63 (4.8%) in the LR-LR cohort; N=53 (16.7%) in the LR-HR cohort; N=18 (14.5%) in the LR-HR-AML cohort; and N=27 (15.6%) in the LR-AML cohort (P<0.0001). Gene mutations associated with an increased risk of direct or indirect AML progression included SRSF2 (P<0.0001) and NRAS (P=0.003). Mutations in IDH1 (P=0.0001), IDH2 (P=0.029), and NPM1 (P<0.0001) were more common in patients with direct AML transformation from lower risk. Mutations associated with progression to both higher risk disease only, without AML and/or direct/indirect AML were A S X L 1 (P=0.009), TP53 (P<0.0001), RUNX1 (P<0.0001), and CBL (P=0.0074). The presence of an ETV6 mutation was associated with progression to HR-MDS but not AML (P<0.0001). PHF6 mutations were associated with progression to HR-MDS and then AML (P=0.0093). SF3B1 mutation was associated with less progression (P<0.0001) (Table 2, Figure 2).
Table 1.
Baseline, laboratory and pathologic characteristics of the four cohorts.

We divided the patients in the LR-LR cohort (i.e., those who remained low-risk; N=1,300) into 3 subgroups based on the number of patients that died in <2, 2-5, and >5 years, and found that about 171 (13.1%) patients of all LR-LR patients died <2 years after diagnosis of LR-MDS without disease progression to HR-MDS and/or AML. Among the 61 cases with documented cause of death, 18 patients (29.5%) died from cytopenia and MDS-related complications, including 2 from infection and 16 from anemia.
Discussion
To the best of our knowledge, this is the first and one of the largest cohorts to explore in detail the different patterns of low-risk MDS disease progression, including progressing to HR-MDS without AML transformation (16.5%), HR-MDS to AML (6.5%), and directly to AML (9%). The majority of LR-MDS patients do not actually progress during their life span. Our overall rate of progression to HR-MDS and/or AML (32%) was higher than the 8% reported by de Swart et al. in the VL and LR IPSS-R cohort in their study validating the IPSS-R in LR-MDS patients.8 We noted that male gender was one of the factors associated with disease progression. Similar to our observation, female gender has been shown to be associated with better survival.8
The LR-MDAS model was put forth by Garcia-Manero et al. in 2007 to identify patients with LR MDS who may benefit from early intervention.4 They included 856 patients with low or intermediate-1 risk disease based on the IPSS. They reported that low platelets, anemia, older age (≥ 60 years), high BM blasts (≥ 4%), and poor-risk cytogenetics were associated with worse survival. The LR-MDAS model was also validated by us in 1,288 LR-MDS patients using the IPSS.9 However, in our study, the LR-MDAS model was not able to predict for progression to HR-MDS or AML. This shows that other parameters, such as ferritin, albumin, pathologic characteristics like MLD, BM fibrosis, and, most importantly, mutational profile need to be incorporated to provide a better prognosis in LR MDS patients.
In the study by Greenberg et al. that introduced the IPSS-R, some other factors were reported that predicted survival, but their impact on the prognostic score was low. Among them were variables including performance status, serum ferritin, and lactate dehydrogenase.2 In the past, serum ferritin had been linked to worse survival, but not to progression.
Table 2.
Mutational profile of the 4 cohorts.

In a study on 107 LR MDS patients, Kawabata et al. showed that the 3-year OS for the high ferritin group (ferritin >210 ng/mL) was significantly shorter than the low ferritin group (66% vs. 79%), while the rates of leukemic progression were similar between the 2 groups.10 In contrast, we noted higher ferritin in patients that progressed to HR-MDS, HR-AML, and directly to AML, but this may reflect the severity of the anemia as the disease progresses, and is most likely the result of progression rather than its cause. Considering high ferritin levels, iron chelation therapy (ICT) has been suggested to improve EFS and OS.11,12 In our study, the LR-HR cohort had the most patients that were treated with ICT, probably because this group included patients that could tolerate ICT, while the other two groups, LR-HR-AML and LR-AML, were patients in a poorer medical condition, with progression to AML, and so a smaller number of patients were treated with ICT.
Mutations that were associated with increased risk of progression to AML in our study were SRSF2 and NRAS. Mutations associated with progression to higher risk disease only, without AML and/or direct/indirect AML, were ASXL1, TP53, RUNX1, and CBL. The presence of an ETV6 mutation was associated with progression to HR-MDS, but not to AML. Similar to our study, Bejar et al., in their study validating LR-MDAS in lower risk patients, reported that ASXL1, U2AF1, SRSF2, RUNX1, NRAS, and CBL were over-represented in the highest-risk LR-PSS category.13 In their study, ASXL1, RUNX1, EZH2, SRSF2, U2AF1, and NRAS mutations were also associated with shorter OS.13 We also found that IDH1, IDH2, and NPM1 were more common in patients with direct AML transformation. Similar to our study, Bejar et al. reported that SF3B1 mutation was significantly under-represented in the higher risk LR-MDS category and showed a non-significant trend toward longer OS.13 Recently, the molecular IPSS (IPSS-M) was introduced, which combines mutations with other hematologic and cytogenetic parameters.14 Using the IPSS-M, Bernard et al. were able to re-stratify 46% patients. We validated the IPSS-M at our center and found that 45% of patients were re-stratified, including approximately 75% of patients that the IPSS-R classified as very-low (VL) who were upstaged (the majority to IPSS-M low and ML/MH).15
In our study, at a median follow up of 99 months, the median OS for the LR-LR, LR-HR, LR-HR-AML, and LR-AML groups was 80.7, 61.2, 48.3, and 42.8 months, respectively. In the study by Greenberg et al. on the IPSS-R, the OS for the VL and LR IPSS-R categories was 8.8 and 5.3 years, which is comparable to the OS of our LR-LR (6.7 years) and LR-HR (5.1 years) cohorts.2 The median time to progression to AML in our LR-HR-AML and LR-AML groups was 29 months, and the median time to progress from LR to HR-MDS was 22 months. Greenberg et al. reported the time for 25% of patients to develop AML (AML/25%) to be around 10.8 years for the LR group and not reached for the VL risk group.2
Figure 2.

Algorithm showing patterns of progression and of low-risk myelodysplastic syndromes. IPSS-R: Revised International Prognostic Scoring System; LR-LR: patients who remained in the low-risk (LR) myelodysplastic syndromes (MDS) category throughout their period of follow-up; LR-HR: patients who progressed from LR to high-risk (HR) MDS without acute myeloid leukemia (AML) transformation; LR-HR-AML: patients who progressed from LR to HR MDS and then AML; LR-AML: patients who directly progressed from LR MDS to AML.
In addition, in the study reporting the LR-MDAS model, 90% of patients died without transforming to AML.4 When we looked at patients that remained in the LR-LR cohort, we found that 13.1% of patients died within two years of diagnosis, with 29% of the patients dying from cytopenias and MDS-related complications without progressing to HR disease or to AML. Greenberg et al. also described that 27% and 40% of patients belonging to the VL and the LR categories died, with 87% and 83% of patients not transforming to AML.2 In another similar study on outcomes of 531 patients with LR MDS by the Connect Myeloid Disease Registry by Komrokji et al., 54% of the 144 patients who had a follow-up BM autopsy available had a 5% increase in BM blasts. The overall rate of disease progression was about 15% in the Connect LR-MDS cohort. About 213 patients died from the 531 LR MDS patients, and 56.3% deaths were attributed to disease-related complications, followed by 14.6% related to cardiovascular disease (CVD).16 In another recent study on 2,396 LR MDS patients by Madry et al., infection (17.8%) and CVD (9.8%) were reported as the main causes of death other than disease progression. They also noted that the relative survival of patients with LR MDS decreases from 94.3% at the first year to 59.6% at five years, hence implying that >40% of patients with LR-MDS die, either directly or indirectly, from MDS-related causes within five years of diagnosis.17 This further affirms our belief that there is a need to better prognosticate these LR-MDS patients that might need earlier intervention in certain cases. Allogeneic stem cell transplantation (alloSCT) is the only cure for MDS. AlloSCT is generally recommended at the time of disease progression and is avoided for LR disease due to early transplant-related mortality, in contrast to the long OS for patients with LR MDS.18-20 If we can identify the patients with LR-MDS who die early, i.e., within two years, without progression clearly related to disease, we might be able to offer them an alloSCT, and significantly improve their survival and quality of life.
Despite being limited by the retrospective design, our study provides important conclusions among a large, predominantly treated cohort. We delineated and identified the clinical and molecular factors associated with different patterns of disease progression. For patients who remained in LR MDS, 13% died in <2 years, and for those with documented cause of death, one-third of deaths within two years were directly MDS-related. Other causes of mortality reported in LR MDS largely include cardiac events. The interplay of cytopenia and such comorbidities may be crucial, as cytopenia may exacerbate those conditions and indirectly contribute to mortality. In fact, several models incorporate comorbidities in MDS risk stratification, but do not detail the interplay between cytopenia and those concomitant diseases.21,22 There is increasing evidence that certain clonal hematopoietic events, such as TET-2 somatic mutation, are associated with other comorbidities such as atherosclerosis.23,24 The impact of treating cytopenias among patients with comorbidities, alleviating transfusion burden and/or achieving hematologic improvement should be further studied. Identifying those LR MDS patients with higher risk features of disease progression or disease-related death within two years will be crucial to tailor therapy accordingly.
Funding Statement
Funding: No funding was received for this work.
Data-sharing agreement
The MDS database related to this study is not available.
References
- 1.Greenberg P, Cox C, LeBeau MM, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997;89(6):2079-2088. [PubMed] [Google Scholar]
- 2.Greenberg PL, Tuechler H, Schanz J, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120(12):2454-2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kantarjian H, O’Brien S, Ravandi F, et al. Proposal for a new risk model in myelodysplastic syndrome that accounts for events not considered in the original International Prognostic Scoring System. Cancer. 2008;113(6):1351-1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garcia-Manero G, Shan J, Faderl S, et al. A prognostic score for patients with lower risk myelodysplastic syndrome. Leukemia. 2008;22(3):538-543. [DOI] [PubMed] [Google Scholar]
- 5.Malcovati L, Germing U, Kuendgen A, et al. Time-dependent prognostic scoring system for predicting survival and leukemic evolution in myelodysplastic syndromes. J Clin Oncol. 2007;25(23):3503-3510. [DOI] [PubMed] [Google Scholar]
- 6.Komrokji RS, Zhang L, Bennett JM. Myelodysplastic syndromes classification and risk stratification. Hematol Oncol Clin North Am. 2010;24(2):443-457. [DOI] [PubMed] [Google Scholar]
- 7.Volpe VO, Garcia-Manero G, Komrokji RS. Myelodysplastic syndromes: a new decade. Clin Lymphoma Myeloma Leuk. 2022;22(1):1-16. [DOI] [PubMed] [Google Scholar]
- 8.de Swart L, Smith A, Johnston TW, et al. Validation of the revised international prognostic scoring system (IPSS-R) in patients with lower-risk myelodysplastic syndromes: a report from the prospective European LeukaemiaNet MDS (EUMDS) registry. Br J Haematol. 2015;170(3):372-383. [DOI] [PubMed] [Google Scholar]
- 9.Komrokji R, Ramadan H, Al Ali N, et al. Validation of the Lower-Risk MD Anderson Prognostic Scoring System for Patients With Myelodysplastic Syndrome. Clin Lymphoma Myeloma Leuk. 2015;15 Suppl: S60-63. [DOI] [PubMed] [Google Scholar]
- 10.Kawabata H, Usuki K, Shindo-Ueda M, et al. Serum ferritin levels at diagnosis predict prognosis in patients with low blast count myelodysplastic syndromes. Int J Hematol. 2019;110(5):533-542. [DOI] [PubMed] [Google Scholar]
- 11.Angelucci E, Greenberg P, Izquierdo M, Garcia-Manero G. Iron chelation in transfusion-dependent patients with low- to intermediate-1-risk myelodysplastic syndromes. Ann Intern Med. 2020;173(7):595-596. [DOI] [PubMed] [Google Scholar]
- 12.Leitch HA, Parmar A, Wells RA, et al. Overall survival in lower IPSS risk MDS by receipt of iron chelation therapy, adjusting for patient-related factors and measuring from time of first red blood cell transfusion dependence: an MDS-CAN analysis. Br J Haematol. 2017;179(1):83-97. [DOI] [PubMed] [Google Scholar]
- 13.Bejar R, Stevenson KE, Caughey BA, et al. Validation of a prognostic model and the impact of mutations in patients with lower-risk myelodysplastic syndromes. J Clin Oncol. 2012;30(27):3376-3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bernard E, Tuechler H, Greenberg Peter L, et al. Molecular International Prognostic Scoring System for Myelodysplastic Syndromes. NEJM Evidence. 2022;1(7):EVIDoa2200008. [DOI] [PubMed] [Google Scholar]
- 15.Aguirre LE, Al Ali N, Ball S, et al. Validation of the Molecular International Prognostic Scoring System (IPSS-M) Risk Stratification Model for Myelodysplastic Syndromes. Blood. 2022;140(Suppl 1):1125-1127. [Google Scholar]
- 16.Komrokji RS, George TI, Erba HP, et al. Treatment patterns and outcomes of patients with lower-risk myelodysplastic syndromes in the connect ® myeloid disease registry. Blood. 2021;138(Suppl 1):3686. [Google Scholar]
- 17.Mądry K, Lis K, Fenaux P, et al. Cause of death and excess mortality in patients with lower-risk myelodysplastic syndromes (MDS): a report from the European MDS registry. Br J Haematol. 2023;200(4):451-461. [DOI] [PubMed] [Google Scholar]
- 18.Cutler CS, Lee SJ, Greenberg P, et al. A decision analysis of allogeneic bone marrow transplantation for the myelodysplastic syndromes: delayed transplantation for low-risk myelodysplasia is associated with improved outcome. Blood. 2004;104(2):579-585. [DOI] [PubMed] [Google Scholar]
- 19.Jain AG, Elmariah H. BMT for Myelodysplastic syndrome: when and where and how. Front Oncol. 2021;11:771614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Witte T, Bowen D, Robin M, et al. Allogeneic hematopoietic stem cell transplantation for MDS and CMML: recommendations from an international expert panel. Blood. 2017;129(13):1753-1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Della Porta MG, Malcovati L, Strupp C, et al. Risk stratification based on both disease status and extra-hematologic comorbidities in patients with myelodysplastic syndrome. Haematologica. 2011;96(3):441-449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Naqvi K, Garcia-Manero G, Sardesai S, et al. Association of comorbidities with overall survival in myelodysplastic syndrome: development of a prognostic model. J Clin Oncol. 2011;29(16):2240-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jaiswal S, Natarajan P, Silver AJ, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. 2017;377(2):111-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fuster JJ, MacLauchlan S, Zuriaga MA, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 2017;355(6327):842-847. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The MDS database related to this study is not available.