

Abstract
Hematopoietic stem cells (HSC) are primarily dormant in a cell-cycle quiescence state to preserve their self-renewal capacity and long-term maintenance. How HSC maintain the balance between activation and quiescence remains largely unknown. Herein, we found that phosphatase, Mg2+/Mn2+ dependent 1B (Ppm1b) is required for the expansion of phenotypic HSC in vitro. By using a conditional knockout mouse model in which Ppm1b was specifically depleted in hematopoietic cells, we demonstrated that loss of Ppm1b impaired the HSC homeostasis and hematopoietic reconstitution. Ppm1b deficiency mice also exhibited B-cell leukocytopenia, which is due to the compromised commitment and proliferation of B-biased lymphoid progenitor cells from common lymphoid progenitors. With the aid of a small molecular inhibitor, we confirmed the roles of Ppm1b in adult hematopoiesis that phenocopied the effects with loss of Ppm1b. Furthermore, transcriptome profiling of Ppm1b-deficient HSC revealed the disruptive quiescence of HSC. Mechanistically, Ppm1b interacted with b-catenin and mediated its dephosphorylation. Loss of Ppm1b led to the decrease in the active b-catenin (non-phosphorylated) that interrupted the Wnt/b-catenin signaling in HSC, which consequently suppressed HSC expansion. Together, our study identified an indispensable role for Ppm1b in regulating HSC homeostasis via the Wnt/b-catenin pathway.
Introduction
Hematopoietic stem cells (HSC) have the potential for both self-renewal and multipotent differentiation into all blood cell lineages during the lifespan.1 They are primarily dormant in a cell-cycle quiescence state to preserve their self-renewal capacity and long-term maintenance, which is responsible for maintaining and rebuilding hematopoiesis.2,3 Functional HSC homeostasis is tightly regulated by multiple factors including cell cycle-associated factors and cell metabolism. Intrinsic programs, cell-autonomous and extrinsic signals from the microenvironment comprise a complicated network, which preserves the balance between activation and quiescence in HSC.4,5
The reversible phosphorylation of protein regulated by kinase and phosphatase is one of the main post-translational modifications that is involved in an extremely wide range of intracellular processes including metabolism, cell death, cell proliferation and differentiation.6,7 Recent studies have demonstrated that protein kinase plays a crucial role in the functional regulation of HSC, such as receptor tyrosine kinases (FLT3,8 c-KIT,9 and TIE210), glycogen synthase kinase 3b,11 sphingosine kinase 2,12 and Janus kinase 2.13 Despite the equally important biological significance, our understanding of protein phosphatases in hematopoietic regeneration still lags behind that of kinases due to their complicated active centers and lower substrate specificity. Emerging evidence of phosphatases in controlling HSC homeostasis and functions has been gradually elucidated in recent years. Src homology 2 domain-containing phosphatase 2 (Shp2) plays a critical role in controlling the survival and maintenance of HSC.14 Ptpn21 has been reported to control HSC homeostasis.15 STS1/STS2 functions in normal and stress hematopoiesis via the regulation of HSC fitness.16
The protein phosphatase 2C (PP2C) family is a member of the metal-dependent protein phosphatases (PPM) of serine/ threonine phosphatases. PP2C family members are known to be negative regulators of cell stress response pathways, and involved in cell cycle control via the dephosphorylation of cyclin-dependent kinases. PP2C family members such as PPM1K and PPM1D, have been demonstrated to play functional roles in maintaining the stemness of HSC.17,18 Protein phosphatase, Mg2+/Mn2+ dependent 1B (Ppm1b, also known as PP2Cβ), is involved in the regulation of multiple signaling pathways by targeting substrate protein dephosphorylation,19,20 including MAPK signaling, TGF-β signaling, TNF signaling, AMPK signaling, and cell cycle. Ppm1b showed high expression in embryonic stem cells,21 and its deficiency led to early pre-implantation lethality.22 Our previous study suggested that Ppm1b may participate in the regulation of hematopoietic processes such as B-/T-cell receptor signaling, FOXO signaling, and the pathogenesis of leukemia.23 It had consistently been reported that Ppm1b regulates terminal erythropoiesis by interacting with an erythroid Kruppel-like factor.24 However, the role of Ppm1b in hematopoiesis is still largely unknown.
In this study, we generated and characterized a conditional knockout (KO) mouse model in which Ppm1b was specifically depleted in hematopoietic cells, and demonstrated that loss of Ppm1b impaired the normal HSC pool and B-cell development. Furthermore, Ppm1b regulates the functional expansion of HSC via Wnt/β-catenin signaling.
Methods
Mice
Ppm1b floxed mice (Ppm1bfl/fl) were generated by inserting loxp sites flanking exon 2, which, when deleted, results in a frame-shift and forms a premature stop codon in the reading frame. Vav-cre mice were purchased from the Jackson Laboratory. The animals were kept in individually ventilated cages with filtered germ-free air, and maintained with sterilized water and irradiated food. The Institutional Animal Care and Use Committees at Shandong University (#19026) approved all animal experiments in our study.
Flow cytometric assays
Single-cell suspensions of tissues, cultured cells, or peripheral blood (PB) were prepared and stained as previously described.25,26 Detailed information for antibodies is provided in the Online Supplementary Appendix. Flow cytometric analysis was performed with an ACEA NovoCyte flow analyzer, and the results were further analyzed with FlowJo and NovoExpress software.
In vitro hematopoietic stem cell expansion
The purified lineage-negative cells were cultured in StemSpan expansion medium (StemCell) supplemented with 10 ng/mL mouse TPO (Peprotech) and 10 ng/mL mouse SCF (Peprotech) for different time periods as indicated in the figure legends (Figures 1-7).
Bone marrow transplantation
Non-competitive and competitive bone marrow transplantation (BMT) was performed as described previously.27 Briefly, for non-competitive BMT, 2x106 BM mononuclear cells (BMMC) (CD45.2) from Ppm1bCKO or Ppm1bfl/fl mice were injected retro-orbitally into lethally irradiated wild-type (WT) recipient mice (CD45.1). For competitive BMT, 1x106 BMMC cells from Ppm1bCKO or Ppm1bfl/fl mice (CD45.2) mixed with an equal number of WT CD45.1 BMMC were injected retro-orbitally into lethally-irradiated CD45.1 recipient mice. For serial competitive BMT, 2x106 donor BMMC from primary transplant recipient mice were transplanted into lethally irradiated CD45.1 congenic recipients. PB was analyzed every four weeks by flow cytometry to assess engraftment.
Limiting dilution assays
Different doses (1x104, 2x104, 4x104, 8x104) of BMMC from Ppm1bCKO or Ppm1bfl/fl mice (CD45.2) plus 2x105 competitor BMMC (CD45.1) were transplanted retro-orbitally into lethally irradiated WT recipient mice (CD45.1). Sixteen weeks after transplantation, the frequency of donor-derived cells (CD45.2) was evaluated by flow cytometry. ELDA (bioinf. wehi.edu.au/software/elda/) and L-Calc software (StemCell) were used to analyze HSC frequency.
Proximity ligation assay
Proximity ligation assay (PLA) was performed using the DuoLink reagents as previously described.28 Briefly, LSK cells were sorted by an Aria III flow cytometer and plated in 8-mm glass coverslips. Cells were fixed with 4% paraformaldehyde and permeabilized with 0.5% Triton X-100 separately, followed by incubating with blocking solution. Cells were then incubated with Ppm1b and β-catenin overnight at 4°C. After incubation with PLA probes, probe ligation, and amplification separately, cells were visualized using an Axio Observer fluorescent A1 microscope.
Results
Ppm1b inhibition led to the compromised proliferation of phenotypic hematopoietic stem cells
By qRT-PCR, we confirmed that Ppm1b is highly expressed in LSK (Lin-Sca-1+c-Kit+) cells that represent relatively enriched HSC, compared to that of hematopoietic progenitor cell (Lin-Sca-1-c-Kit+, LSK) and lineage-negative BM cells (Online Supplementary Figure S1A). To explore the role of Ppm1b in HSC function, we first treated the HSC isolated from mouse BM with Ppm1b inhibitor HN252 which we developed in our previous study.23 The cells in the LSK fraction exhibited a dose-dependent decrease after 48 hours (hr) of culture with HN252, accompanied by the reduced LSK cells (Figure 1A, Online Supplementary Figure S1B). HN252 treatment also led to a significant increase in apoptotic cells in the LSK fraction (Online Supplementary Figure S1C), but the overall apoptosis rate was relatively low (<5%), suggesting that cell death is not the main cause of impaired HSC expansion induced by HN252.
We then examined the proliferation of HSC via CFSE staining, in which cell division was tracked via the changes in intracellular fluorescence.29,30 Interestingly, the relative mean fluorescence intensity (MFI) of CFSE in LSK was 2-fold higher than that of the control group after 48 hr of culture with HN252, indicating that Ppm1b inhibition led to the proliferative suppression of HSC (Figure 1B). Compared to the freshly isolated LSK from BM (uncultured), the frequency of HSC at the G0-phase (Ki67-) was obviously decreased, which represents quiescent cells, while active cycling cells (Ki67+) were dramatically increased. This is consistent with previous findings that HSC undergoes active cell cycling stimulated by supplemented hematopoietic cytokines in in vitro culture.31,32 Notably, HN252 treatment led to a significant increase in G0 cells coupled with the reduced G1 and G2/M-phase frequency in LSK, suggesting that Ppm1b is required for HSC to switch from quiescence to an active state (Figure 1C, D).
Figure 1.

Ppm1b inhibition led to the repressed proliferation of phenotypic hematopoietic stem cells. (A) Lineage negative cells were cultured with HN252 at multiple concentrations (0, 5, 10, and 20 μM) for 48 hours (hr). The relative proliferation ratio of Lin-Sca-1+c-Kit+ (LSK) cells was quantified by flow cytometry, and normalized to the uncultured cells. (B) CFSE labeled lineage negative cells were cultured with HN252 (10 µM) or vehicle (control) for 48 hr, and the mean fluorescence intensity (MFI) of CFSE in LSK cells was quantified by flow cytometry. (C) Representative cell cycle profile of LSK cells after in vitro culture with control or HN252 (10 μM) for 48 hr. Cell cycle was analyzed using Hoechst 33342 and Ki67 staining by flow cytometry. (D) Quantification of cells in indicated phases in (C). (E and F) Lineage negative cells were transduced with retroviruses encoding Ppm1b shRNA (shP1B-1 or shP1B-2) and cultured for 48 hr; the cell number (E) and apoptosis (F) were analyzed by flow cytometry. P values were determined by one-way ANOVA with Tukey’s multiple comparisons test (A) or by unpaired two-tailed Student t test (B-F). Data are presented as mean ± Standard Deviation from 3 independent experiments. See also Online Supplementary Figure S1.
In line with the findings of HN252 treatment, Ppm1b knockdown significantly inhibited the proliferation of LSK in in vitro culture, accompanied by a remarkable increase in cell death in LSK (Figure 1E, F). These data indicate that Ppm1b plays functional roles in the proliferation of HSC.
Ppm1b deficiency mice exhibited B-cell leukocytopenia
To explore the role of Ppm1b in the hematopoietic system, we generated hematopoietic-specific Ppm1b KO mice (conditional KO, CKO) by crossing mice bearing Ppm1b allele with loxP-flanked exon 2 with Vav-Cre transgenic mice (Online Supplementary Figure S2A). The ablation efficiency of Ppm1b was confirmed in mononuclear cells of BM and PB by qRT-PCR (Online Supplementary Figure S2B). CKO mice exhibited a dominant defect in white blood cells (WBC) that was mainly due to the reduction of lymphocytes in PB, whereas red blood cells (RBC) and platelets (PLT) were unaffected (Figure 2A, Online Supplementary Figure S2C). Interestingly, the low lymphocyte attributed to the significantly reduced B cells (B220+) but not T cells (CD3+) in PB from CKO mice compared to that of control mice (Online Supplementary Figure S2D). Similar defective B lymphocytes were also observed in CKO BM (Figure 2B). We next examined the B-cell compartment in the BM according to the well-established surface markers (Online Supplementary Figure S2E), which is characterized by pre-pro B cell, pro-B cell, pre-B cell, immature B cell, and mature B cell.33 Loss of Ppm1b resulted in a remarkable decrease in cell number in all subpopulations of B lymphocytes compared to that of WT controls, although there was no difference in their relative frequencies between the two groups of mice (Online Supplementary Figure S2E-I). Furthermore, common lymphoid progenitor (CLP) cells that commit into B-lineage progenitor cells also exhibited a decrease in CKO mice (Figure 2C, D). However, loss of Ppm1b did not disrupt the T lymphogenesis that also starts with the commitment of CLP, evidenced by the equal thymus and subpopulations of T lymphocytes (Online Supplementary Figure S2J-L). These findings suggest that Ppm1b is required for the commitment and proliferation of B-biased lymphoid progenitor cells from CLP, which is further supported by the markedly reduced colony number and size from CKO lineage-negative BM cells supplemented with IL-7 (Figure 2E, F, Online Supplementary Figure S2M).
Loss of Ppm1b led to the suppression of phenotypic hematopoietic stem cell expansion in vivo
Ppm1bCKO mice exhibited a significantly diminished frequency and absolute number of HSC (lineage-Sca1+c-Kit+, LSK) in BM (Figure 3A, Online Supplementary Figure S3A), while the committed HPC (lineage-Sca1-c-Kit+, LK) were comparable between these two groups of mice (Online Supplementary Figure S3B). To further explore the impact of Ppm1b deficiency on the HSC, we examined its subpopulations according to the different identifications.8,34 Interestingly, Ppm1bCKO mice exhibited the obviously decreased long-term HSC (CD34-CD135-LSK, LT-HSC) and short-term HSC (CD34+CD135-LSK, ST-HSC) (Figure 3B, Online Supplementary Figure S3C). A similar reduction was also observed in SLAM-HSC (CD150+CD48-LSK) representing the quiescent HSC in Ppm1bCKO mice (Online Supplementary Figure S3D).35 To further determine the number of functional HSC of Ppm1bCKO mice in the BM, we performed limiting dilution assays and found that the frequency of competitive repopulation units (CRU) in Ppm1bCKO mice was significantly lower than that of control mice (Figure 3C). Indeed, Ppm1bCKO HSC showed comparable homing activity to WT ones (Online Supplementary Figure S3E).
Moreover, Ppm1b deletion had an undetectable effect on the HSC survival (data not shown). Cell cycle analysis by Ki67 staining demonstrated that Ppm1b-deficient HSC showed more quiescence than that of WT control (Figure 3D). This is confirmed by the low cell division of ex vivo cultured Ppm1b-deficient LSK indicated by the relatively high CFSE staining (Online Supplementary Figure S3F). These findings indicate that loss of Ppm1b impaired the active proliferation of HSC.
To confirm the regulatory role of Ppm1b in functional HSC, we challenged CKO mice with 5-fluorouracil (5-FU) that eliminates cycling cells and triggers the hematopoietic repopulation.36 All the Ppm1bCKO mice died at 20 days of continuous 5-FU injection, whereas 40% of WT mice survived after 3 rounds of administration (Figure 3E), indicating that Ppm1b is required for the stressed reconstitution of HSC. Consistently, the frequencies of HSC at the G0 and G1 phase in CKO mice also showed a more dramatic decrease after 5-FU administration compared with the steady state (Online Supplementary Figure S3G), suggesting that Ppm1b deletion impaired the transition between G0 to G1 phase under stress hematopoiesis.
To further assess the function of Ppm1b-deficient HSC, we performed competitive serial transplantation assays (Online Supplementary Figure S3H). Ppm1bCKO-derived cells showed a successive reduction in the primary recipients and subsequent recipients (Online Supplementary Figure S3 I), indicating the impaired reconstitution of CKO HSC. Notably, the frequencies of LSK and SLAM-LSK derived from Ppm1bCKO BM were also significantly reduced in primary and subsequent recipients (Figure 3F, Online Supplementary Figure S3J). In addition, B cells derived from Ppm1bCKO BM also showed a progressive decrease in the recipients (Online Supplementary Figure S3K, L), which is consistent with the defect of B cells in primary CKO mice.
Ppm1b regulated the hematopoietic stem cell pool and B-cell differentiation in adult hematopoiesis
Given that the defects of Ppm1bCKO mice may be due to a consequence of embryo stage, we analyzed whether Ppm1b also play roles in adult hematopoiesis. To this end, WT mice were pretreated with Ppm1b inhibitor HN252 and then challenged with a sublethal dose of 5-FU (Figure 4A). Compared to control group mice, HN252 treatment led to a greater decrease and subsequently slower recovery of WBC and lymphocytes but not PLT and RBC in PB (Figure 4B, Online Supplementary Figure S4A). HN252-treated mice showed markedly reduced B220+ cells in PB and BM after 12 days of 5-FU injection when the blood cells had completely recovered in control mice (Figure 4C, D). Moreover, it is notable that the proportion of LSK cells was also significantly reduced in HN252-treated mice BM upon the challenge of 5-FU (Figure 4E). These findings phenocopied the defect of the HSC pool and B-cell differentiation in Ppm1bCKO mice. To avoid the off-target effect of HN252, we performed experiments to confirm the specificity of HN252 on Ppm1b. There were no significant changes in WBC and lymphocytes in PB observed in Ppm1b deficient mice with or without HN252 treatment after 5-FU injection (Online Supplementary Figure S4B), as well as the cell number of B220+ cells and the proportion of LSK cells in the BM (Online Supplementary Figure S4C, D). In addition, HN252 treatment showed no notable effect on the relative mean fluorescence intensity of CFSE in Ppm1b-deficient LSK (Online Supplementary Figure S4E). These results indicate that HN252 affects the HSC pool and B-cell differentiation in adult hematopoiesis mainly via targeting Ppm1b.
Figure 2.

Ppm1b deficiency mice exhibited B-cell leukocytopenia. (A) Absolute number of white blood cells (WBC) and lymphocytes in peripheral blood from Ppm1bCKO and Ppm1bfl/fl mice at the age of 8 weeks (N=14). (B) Statistical analysis of the percentage of B cells in bone marrow (BM) from indicated mice (N=6). (C) Representative flow cytometric profiles of the strategy for common lymphoid progenitors (CLP), Lin-Sca-1lowc-KitlowCD127+ cells. (D) Quantification of the cell number of CLP cells in BM from indicated mice (N=4). (E) Representative profiles of the colonies after 7 days of culture from indicated mice. (F) Quantification of the colony number in (I) (N=3). All P values were determined by unpaired two-tailed Student t test. See also Online Supplementary Figure S2.
Figure 3.

Loss of Ppm1b leads to the suppression of phenotypic hematopoietic stem cell expansion in vivo. (A) Statistical analysis of the percentage (left) and number (right) of Lin-Sca-1+c-Kit+ (LSK) cells in 8-week-old Ppm1bCKO and Ppm1bfl/fl mice. (A) N=4. (B) Statistical analysis of the percentage of LT-HSC, ST-HSC and MPP cells (all gated from LSK) (N=4). (C) Limiting dilution transplantation assays were performed by transplanting different doses of bone marrow mononuclear cells (BMMC) from indicated mice. (Left) The number of functional hematopoietic stem cell (HSC) in bone marrow (BM) of Ppm1bCKO and Ppm1bfl/fl mice was measured by flow cytometry after 16 weeks of transplantation. Recipients with <2% donor-derived cells in BM were defined as non-respondent (N=8). (Right) Fraction of HSC in BM of Ppm1bCKO and Ppm1bfl/fl mice by Poisson statistical analysis of the data using L-Calc software. Solid lines indicate the best-fit linear model for each dataset. Dashed lines for 95% Confidence Intervals; χ2 test=4.39; P=0.0361. (D) Cell cycle analysis of LSK cells from indicated mice by flow cytometry using Hoechst 33342 and Ki67 staining (N=4). (E) Kaplan-Meier survival curves of Ppm1bCKO and Ppm1bfl/fl mice treated thrice with 5-FU (150 mg/kg) (N=12). (F) Percentage of donor-derived LSK cells in the BM of the primary and secondary recipient mice (N=6). Two-tailed unpaired Student t test was used to generate the P values. The significance for survival analyses was calculated by logrank (Mantel-Cox) test. See also Online Supplementary Figure S3. BMT: bone marrow transplantation; CRU: competitive repopulating units.
Transcriptome profiling of Ppm1b-deficient hematopoietic stem cells reveals altered stem cell property
To explore the basis of abnormal hematopoiesis upon Ppm1b deficiency, we performed RNA sequencing analysis with purified lineage negative cells from Ppm1bCKO and the corresponding mice (Online Supplementary Figure S5A). Enriched gene sets were notably related to B-cell activation and immunoglobulin production (Online Supplementary Figure S5B). Given that transcription factors are crucial in controlling B-cell development,37 we next examined the expression of the multiple transcriptional regulators in B220+ BM cells. Compared to B220+ cells from the corresponding mice, Ppm1bCKO B cells showed significant downregulation of these transcription factors (Online Supplementary Figure S5C), in which flow cytometry further confirmed Forkhead Box O1 (FOXO1) protein levels to be reduced (Online Supplementary Figure S5D).
Figure 4.

Ppm1b regulates hematopoietic stem cell pool and B-cell differentiation in stressed hematopoiesis. (A) Experimental design for HN252-treated mice in response to 5-FU-induced emergency hematopoiesis. Long blue arrow indicates HN252 or Vehicle injection every 2 days. Black arrow indicates the time point for 5-FU injection. Red arrows indicates the bleeding for blood assays. (B) Quantification of white blood cells (WBC) and lymphocytes in peripheral blood at indicated time points during HN252 injection (N=6). (C) The mice were treated with HN252 as in (A), and the ratio of B cells in peripheral blood was analyzed by flow cytometry (N=6). (D and E) The number of B cells (D) and the percentage of Lin-Sca-1+c-Kit+ (LSK) cells (E) from indicated mice in (C) were measured by flow cytometry (N=6). Data are presented as mean ± Standard Deviation. All P values were determined by unpaired two-tailed Student t test. See also Online Supplementary Figure S4. BM: bone marrow; GO: gene ontology; PBMC: peripheral blood mononuclear cells.
Moreover, KEGG enrichment analysis revealed that Wnt signaling pathway was substantially enriched in the Ppm1bCKO group (Figure 5A, Online Supplementary Table S1). Several Wnt signaling-related genes were down-regulated upon the deletion of Ppm1b, including Fzd1, Jun, Camk2b, Lrp5, Ccnd1, and Gpc4 (Online Supplementary Figure S6A).
The reduced pool of HSC in Ppm1bCKO mice prompted us to investigate the basis of impaired HSC. We then performed single-cell RNA sequencing with sorted LSK cells from Ppm1bCKO mice and their corresponding controls. A total of 3,651 CKO and 3,713 WT cells were divided into 24 clusters with unsupervised clustering, which was visualized with uniform manifold approximation and projection (UMAP) (Online Supplementary Figure S6A, B). Four subpopulations referring to the commitment gene markers were identified, including myeloid-biased (pMy), lymphoid-biased (pLy), megakaryocytic-biased (pMk), and erythroid-biased (pEry) clusters (Figure 5B). The remaining cell populations, characterized by the maintenance of stemness and lacking a specific propensity for lineage differentiation according to enrichment analysis, were annotated as “non-primed”. This cluster was further confirmed by the elevated expression of Hlf, a master transcription factor predominantly expressed in an HSC subpopulation with extensive self-renewal capacity (Figure 5C).38 Consistent with the findings in flow cytometric analysis, the relative frequency of the “non-primed” cluster was also reduced in the Ppm1bCKO group (Online Supplementary Figure S6C).
Figure 5.

Transcriptome profiling of Ppm1b-deficient hematopoietic stem cells reveals altered stem cell properties. (A) KEGG pathway analysis indicating altered signaling pathways including Wnt signaling between Ppm1bCKO and Ppm1bfl/fl LSK cells (P<0.05). Numbers shown in each bar indicate the number of enriched proteins in relative terms. (B) A uniform manifold approximation and projection (UMAP) plot of 7,364 Lin-Sca-1+c-Kit+ (LSK) cells (3,651 for pm1bfl/fl, 3,713 for Ppm1bCKO) categorized into 5 clusters based on distinct differentiation potentials. (C) UMAP plots illustrating principal identifiers (expression of marker genes) of various LSK sub-populations. (D) Volcano plot showing differential expressed genes (DEG) between Ppm1bCKO and Ppm1bfl/fl within each cluster (P.adjust<0.01). (E). Gene ontology (GO) analysis of down-regulated DEG between Ppm1bCKO and Ppm1bfl/fl in the non-primed cluster (P<0.05). See also Online Supplementary Figures S5 and S6.
Several genes associated with HSC homeostasis were identified in the non-primed cluster between CKO and WT groups (Figure 5D, Online Supplementary Table S2). Nf4a1 has been reported to restrict HSC proliferation in part through activation of a C/EBPα-driven antiproliferative network by directly binding to a hematopoietic-specific Cebpa enhancer, a member of the Nr4a subfamily of nuclear receptors.39 Fos serves as a gatekeeper in the cell cycle progression of dormant HSC, whose sustained expression inhibits HSC entry into the cell cycle.40 These two genes showed obvious upregulation in CKO HSC compared to that of the corresponding controls (Figure 5D). On the other hand, it has been demonstrated that down-regulated Txnip is required for the hematopoietic reconstitution and HSC self-renewal capacity.41 In line with the previous report that low FLT3 expression led to a blockage at G0 phase in HSC,42 CKO HSC showed reduced FLT3 expression and enhanced quiescence. Furthermore, GO enrichments with these differentially expressed genes revealed that the physiological activity was down-regulated in CKO HSC, such as cytokine-mediated signaling, mRNA metabolism, and translation, which phenocopies the quiescent status (Figure 5E). These transcriptomic changes further confirmed the impact of Ppm1b on HSC expansion.
Loss of Ppm1b impaired the functional expansion of hematopoietic stem cells through Wnt/b-catenin pathway
Given that the phosphorylation of b-catenin functions as a downstream key node of Wnt signaling, and it has been previously identified as a potential subtract of PPM1B, we speculated that Ppm1b regulates HSC via Wnt/b-catenin signaling. Indeed, the phosphorylation of b-catenin was dramatically increased in Ppm1bCKO HSC compared to that of WT controls (Figure 6A). The target genes of Wnt/b-catenin signaling such as Cylin D1 and Myc, were also down-regulated upon Ppm1b deletion (Figure 6B). Proximity ligation assay in sorted LSK cells indicated that Ppm1b interacted with b-catenin (Figure 6C). Moreover, Ppm1b inhibitor HN252 treatment led to a considerable decrease in the active b-catenin (non-phosphorylated) in the LSK cells (Online Supplementary Figure S7A, B), which is further confirmed by the downregulation of its target genes (Online Supplementary Figure S7C). These data suggest that Ppm1b mediates the b-catenin activity via the dephosphorylation in HSC.
To confirm Wnt/b-catenin signaling is responsible for the impaired expansion of HSC with Ppm1b deficiency, we performed rescue experiments with a potent Wnt signaling activator BML-284 (BML)43 and found that BML could activate the expression of Wnt-targeted genes in Ppm1b deficient cells (Online Supplementary Figure S7D). Lineage negative BM cells were sorted from CKO and WT mice and labeled with CFSE, followed by in vitro culture with BML treatment. Compared to the high MFI of CFSE in Ppm1bCKO LSK cells after 48 hr ex vitro culture, BML treatment led to a significant reduction in CFSE staining (Figure 6D). This was further confirmed by the increase in LSK cells derived from Ppm1bCKO mice upon BML treatment (Online Supplementary Figure S7E), indicating that BML reversed the suppressed HSC expansion with Ppm1b deficiency.
To verify the functional roles of Ppm1b/Wnt/b-catenin axis in HSC expansion in vivo, we pretreated Ppm1bCKO and WT mice with BML for four days and then challenged them with a sublethal dose of 5-FU. Similar to the findings in ex vitro culture, the frequencies and cell number of HSC in Ppm1bCKO mice were remarkably increased by BML treatment, whereas the HSC in Ppm1bfl/fl mice remained unchanged (Figure 6E, Online Supplementary Figure S7F). Furthermore, BML treatment led to a significant decrease in LSK cells at G0 phases but a notable increase at the G2/S/M-phase in Ppm1bCKO mice, compared to that of the control group (Figure 6F). However, BML had no detectable impact on the cell cycle of LSK cells in WT control mice (Figure 6F). These data confirm that loss of Ppm1b impaired the HSC homeostasis via the blockage of Wnt/b-catenin signaling.
Discussion
In stable state conditions, most HSC are in a state of quiescence with only a small proportion undergoing self-renewal or differentiation.44,45 The balance between the quiescence and activation of HSC is tightly regulated to protect the HSC pool from exhaustion, which is also required for the capabilities of long-term hematopoiesis.46 In this study, we identified the critical role of Ppm1b in the regulation of HSC homeostasis and B-cell development in vitro and in vivo. Ppm1b inhibition suppressed the expansion of HSC due to the blockage of the cell cycle at G0 phase. Moreover, Ppm1b deficiency led to a reduction in the early B-cell progenitors committed from CLP, which consequently impaired B-cell differentiation (Figure 7). Altogether, our work demonstrated that Ppm1b emerges as a key regulator in normal hematopoiesis.
The canonical Wnt/b-catenin signaling has been documented to be crucial for the self-renewal and differentiation of HSC.47 However, its regulatory role in HSC remains a subject of debate.48 Weissman et al. found that constitutive activation of Wnt/b-catenin signaling led to the enhanced proliferation and repopulation of HSC via overexpression of active b-catenin.49 In line with this finding, Kincade et al. also confirmed that constitutively active b-catenin accelerates the expansion of HSPC in vitro.50 However, other research groups demonstrated that constitutive activation of b-catenin inhibited the repopulation and differentiation of HSC by using transgenic mice.51,52 These inconsistent results indicated that the role of Wnt/b-catenin signaling in HSC fate decisions is dependent on its degree. Our finding underscored the essential roles of Wnt/b-catenin signaling in HSC homeostasis. Loss of Ppm1b led to the decrease in active b-catenin (non-phosphorylated state) and its target genes’ expression in HSC, resulting in a blockage of self-renewal in Ppm1b-deficient HSC. This was efficiently reversed by activating Wnt signaling (Figure 7).
Figure 6.
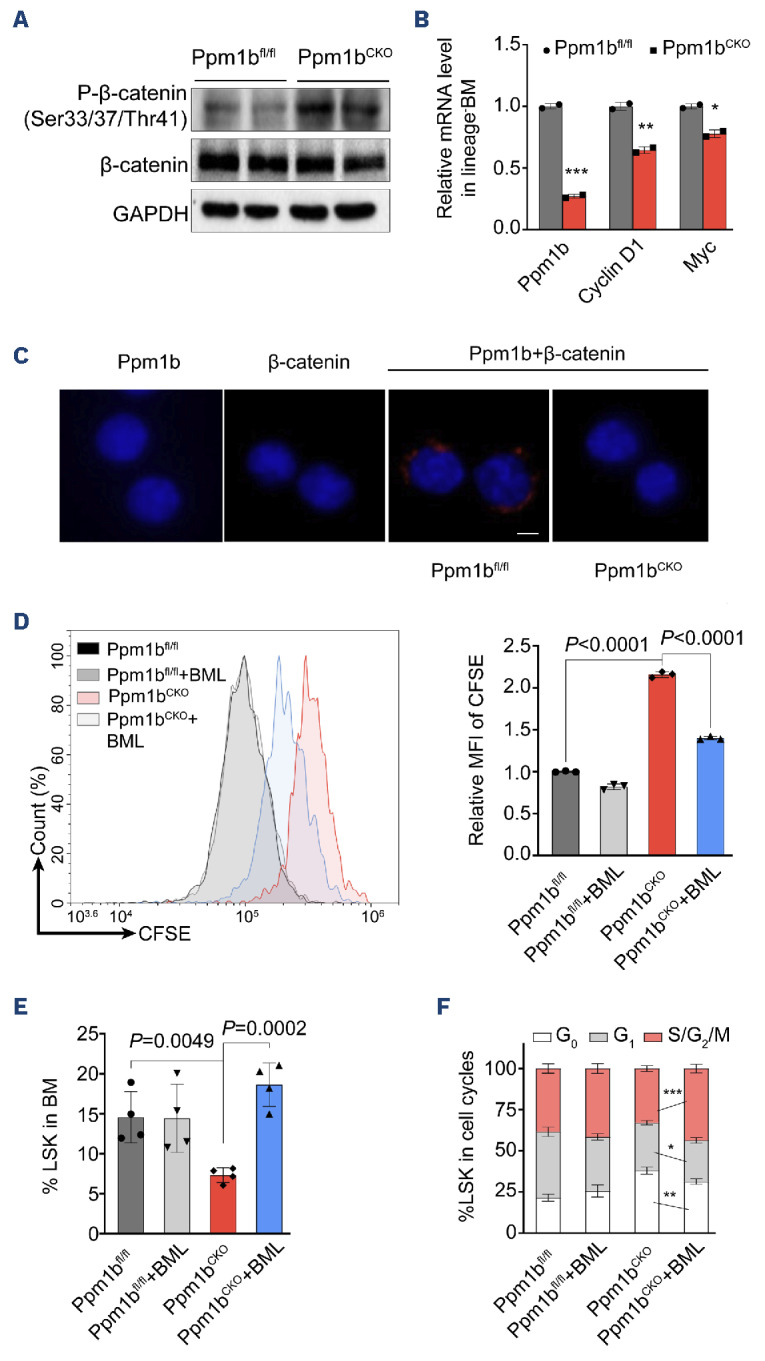
Loss of Ppm1b impaired the function of hematopoietic stem cells through Wnt/b-catenin pathway. (A) Western blot analysis of indicated proteins in purified lineage negative cells from Ppm1bCKO and Ppm1bfl/fl mice. (B) Quantification of mRNA expression of Ppm1b, cyclin D1 and Myc in cells from (A). 18S ribosomal RNA was used as an internal control. (C) Representative confocal images of proximity ligation assay between Ppm1b and b-catenin in Lin-Sca1+c-Kit+ (LSK) cells. Sorted LSK cells from 3 independent Ppm1bCKO and Ppm1bfl/fl mice were stained with indicated antibodies separately, and were visualized using a fluorescent microscope. Scale bar: 5 μM. (D) Lineage negative cells purified from indicated mice were labeled with CFSE, and then the cells were cultured with or without BML-284 (10 nM) for 48 hours (hr) in vitro. The fluorescence intensity of CFSE of LSK cells was quantified by flow cytometry. (E) Ppm-1bCKO and Ppm1bfl/fl mice were treated with BML-284 (5 mg/kg) or not for 4 days, followed by a single dose injection of 5-FU for another 7 days. The frequency of LSK cells was determined from indicated mice by flow cytometry (N=4). (F) Cell cycles of LSK cells in (E) were analyzed by flow cytometry using Hoechst 33342 and Ki67 staining (N=4). *P<0.05, **P<0.01, ***P<0.001. All P values were determined by unpaired two-tailed Student t test. Data are presented as mean ± Standard Deviation from 3 independent experiments. See also Online Supplementary Figure S7. BM: bone marrow; MFI: mean fluorescence intensity.
We demonstrated that Ppm1b interacted with β-catenin, and Ppm1b deletion led to the inhibition of Wnt/β-catenin signaling via the dephosphorylation in HSC. Although the role of Wnt/b-catenin signaling in B-cell development has been revealed by LEF1 KO mice that led to a reduction in B cells,53 we did not observe any changes in the phosphorylation of b-catenin and downstream target genes’ expression in Ppm1b-deficient B cells (data not shown). This is consistent with our findings that Ppm1b is required for the commitment and proliferation of B-biased lymphoid progenitor cells from CLP but not the consequent B-cell differentiation. Given that B-cell precursor acute lymphoblastic leukemia (BCP-ALL) is characterized by uncontrolled proliferation of early B-cell progenitors, our work identified Ppm1b as the potential target for the clinical treatment of BCP-ALL. Our data showed that loss of Ppm1b led to the defect in B-cell but not T-cell development. Indeed, lymphopoiesis is an intricate process that gives rise to all lymphocytes which starts with the commitment of HSC to CLP.54 Even though CLP generate T and B cells when properly stimulated,55 it seems that CLP are mainly progenitors for B cells, owing to the limited or negligible T-cell generation from CLP.56,57 By the expression of Ly6D, CLP can be defined as all-lymphoid progenitors (Ly6D-), which have the potential to generate T and natural killer cells, and B-cell biased lymphocyte progenitors (Ly6D+).58 Moreover, there are still CLP-independent pathways of innate T-cell development which are more efficient than CLP.59 Importantly, the changes in lymphocyte counts in the first two weeks after 5-FU were most likely not driven by differentiation from HSC but rather due to lymphocyte death and subsequent homeostatic proliferation, as well as differentiation from more committed progenitors (such as BM CLP or Flt3+ MPP) (Figure 4). Given that PLT and RBC in HN252-treated mice showed mild changes compared to that of the control group, a single 5-FU treatment is probably not sufficient to impair the function of HSC pretreated with the PPM1B inhibitor. These data would rather seem to point toward a CLP-intrinsic effect of PPM1B. However, these findings were not invalidated by the serial transplant or CRU data, suggesting that there might be two at least partially independent stages of PPM1B activity. Consequently, the impact of Ppm1b deletion on B-cell development may take place at the level of stem and progenitor cells, even in populations lacking an apparent inclination for lineage differentiation.
Figure 7.

Schematic diagram illustrates the regulatory role of Ppm1b in hematopoietic stem cell self-renewal and B-cell development. Ppm1b is required for hematopoietic stem cell (HSC) proliferation and early B-cell progenitor differentiation. Loss of Ppm1b led to the decrease of the active β-catenin (non-phosphorylated) that interrupted the Wnt/β-catenin signaling in HSC, which consequently suppressed HSC expansion. CLP: common lymphoid progenitors.
In summary, our study identified an essential role for Ppm1b in HSC homeostasis via Wnt/β-catenin signaling. Our findings may provide novel insights into the understanding of HSC self-renewal and B-cell development, and also offer clues for the pathogenesis of their related hematologic malignancies.
Supplementary Material
Acknowledgments
We thank the Translational Medicine Core Facility and Core Facility of the School of Basic Medical Sciences of Shandong University for consultation and instrument availability that supported this work. This work was supported by grants from the National Natural Science Foundation of China (81874294, 32300957), Taishan Scholars Program (TSQN201812015) and the key Program of Natural Science Foundation of Shandong Province (ZR2022LSW027), Qilu Young Scholars Program (to BZ), and the Multidisciplinary Research and Innovation Team of Young Scholars (2020QNQT007) of Shandong University. This work was also supported by the program for Innovative Research Team at University of the Ministry of Education of China (IRT_17R68), the academic promotion program of Shandong First Medical University (No.2019LJ003), the Natural Science Foundation of Shandong Province (ZR2023QH427), and the Innovation Team of Shandong Higher School Youth Innovation Technology Program (2022KJ197).
Data-sharing statement
Supporting data are available in the Online Supplementary Appendix. RNA sequencing data that support the findings of this study have been deposited in NCBI SRA under accession number GSE23774. The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
References
- 1.Eaves CJ. Hematopoietic stem cells: concepts, definitions, and the new reality. Blood. 2015;125(17):2605-2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sieweke MH, Allen JE. Beyond stem cells: self-renewal of differentiated macrophages. Science. 2013;342(6161):1242974. [DOI] [PubMed] [Google Scholar]
- 3.de Haan G, Lazare SS. Aging of hematopoietic stem cells. Blood. 2018;131(5):479-487. [DOI] [PubMed] [Google Scholar]
- 4.Verovskaya EV, Dellorusso PV, Passegue E. Losing sense of self and surroundings: hematopoietic stem cell aging and leukemic transformation. Trends Mol Med. 2019;25(6):494-515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilkinson AC, Igarashi KJ, Nakauchi H. Haematopoietic stem cell self-renewal in vivo and ex vivo. Nat Rev Genet. 2020;21(9):541-554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Humphrey SJ, James DE, Mann M. Protein phosphorylation: a major switch mechanism for metabolic regulation. Trends Endocrinol Metab. 2015;26(12):676-687. [DOI] [PubMed] [Google Scholar]
- 7.Kamada R. Metal-dependent Ser/Thr protein phosphatase PPM family: evolution, structures, diseases and inhibitors. Pharmacol Ther. 2020;215:107622. [DOI] [PubMed] [Google Scholar]
- 8.Adolfsson J, Mansson R, Buza-Vidas N, et al. Identification of Flt3+ lympho-myeloid stem cells lacking erythro-megakaryocytic potential a revised road map for adult blood lineage commitment. Cell. 2005;121(2):295-306. [DOI] [PubMed] [Google Scholar]
- 9.Okada S, Nakauchi H, Nagayoshi K, Nishikawa S, Miura Y, Suda T. In vivo and in vitro stem cell function of c-kit- and Sca-1-positive murine hematopoietic cells. Blood. 1992;80(12):3044-3050. [PubMed] [Google Scholar]
- 10.Ito K, Turcotte R, Cui JH, et al. Self-renewal of a purified Tie2(+) hematopoietic stem cell population relies on mitochondrial clearance. Science. 2016;354(6316):1156-1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holmes T, O’Brien TA, Knight R, et al. Glycogen synthase kinase-3 beta inhibition preserves hematopoietic stem cell activity and inhibits leukemic cell growth. Stem Cells. 2008;26(5):1288-1297. [DOI] [PubMed] [Google Scholar]
- 12.Li CZ, Wu BH, Li YS, et al. Loss of sphingosine kinase 2 promotes the expansion of hematopoietic stem cells by improving their metabolic fitness. Blood. 2022;140(15):1686-1701. [DOI] [PubMed] [Google Scholar]
- 13.Zhang XF, Wang JF, Matczak E, Proper J, Groopman JE. Janus kinase 2 is involved in stromal cell-derived factor-1 alpha-induced tyrosine phosphorylation of focal adhesion proteins and migration of hematopoietic progenitor cells. Blood. 2001;97(11):3342-3348. [DOI] [PubMed] [Google Scholar]
- 14.Chan G, Cheung LS, Yang WT, et al. Essential role for Ptpn11 in survival of hematopoietic stem and progenitor cells. Blood. 2011;117(16):4253-4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ni F, Yu WM, Wang X, et al. Ptpn21 controls hematopoietic stem cell homeostasis and biomechanics. Cell Stem Cell. 2019;24(4):608-620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang J, Vakhrusheva O, Bandi SR, et al. The phosphatases STS1 and STS2 regulate hematopoietic stem and progenitor cell fitness. Stem Cell Reports. 2015;5(4):633-646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu XY, Zhang FF, Zhang YP, et al. PPM1K regulates hematopoiesis and leukemogenesis through CDC20-mediated ubiquitination of MEIS1 and p21. Cell Rep. 2018;23(5):1461-1475. [DOI] [PubMed] [Google Scholar]
- 18.Chen ZY, Yi WW, Morita YH, et al. Wip1 deficiency impairs haematopoietic stem cell function via p53 and mTORC1 pathways. Nat Commun. 2015;6:6808. [DOI] [PubMed] [Google Scholar]
- 19.Marley AE, Kline A, Crabtree G, Sullivan JE, Beri RK. The cloning expression and tissue distribution of human PP2C beta. FEBS Lett. 1998;431(1):121-124. [DOI] [PubMed] [Google Scholar]
- 20.Li ZY, Chen RY, Li YX, et al. A comprehensive overview of PPM1B: from biological functions to diseases. Eur J Pharmacol. 2023;947:175633. [DOI] [PubMed] [Google Scholar]
- 21.Skottman H, Mikkola M, Lundin K, et al. Gene expression signatures of seven individual human embryonic stem cell lines. Stem Cells. 2005;23(9):1343-1356. [DOI] [PubMed] [Google Scholar]
- 22.Sasaki M, Ohnishi M, Tashiro F, et al. Disruption of the mouse protein Ser/Thr phosphatase 2Cbeta gene leads to early preimplantation lethality. Mech Dev. 2007;124(6):489-499. [DOI] [PubMed] [Google Scholar]
- 23.Lu ZY, Xiao P, Zhou Y, et al. Identification of HN252 as a potent inhibitor of protein phosphatase PPM1B. J Cell Mol Med. 2020;24(22):13463-13471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yien YY, Bieker JJ. Functional Interactions between erythroid Kruppel-like factor (EKLF/KLF1) and protein phosphatase PPM1B/ PP2C beta. J Biol Chem. 2012;287(19):15193-15204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu ZY, Huang LX, Li YX, et al. Fine-tuning of cholesterol homeostasis controls erythroid differentiation. Adv Sci. 2022;9(2):2102669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu ZY, Xu GS, Li YX, Lu CH, Shen YM, Zhao BB. Discovery of N-arylcinnamamides as novel erythroblast enucleation inducers. Bioorg Chem. 2022;128:106105. [DOI] [PubMed] [Google Scholar]
- 27.Mei Y, Han X, Liu YJ, Yang J, Sumagin R, Ji P. Diaphanous-related formin mDia2 regulates beta2 integrins to control hematopoietic stem and progenitor cell engraftment. Nat Commun. 2020;11(1):3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blanchard EL, Loomis KH, Bhosle SM, et al. Proximity ligation assays for in situ detection of innate immune activation: focus on in vitro-transcribed mRNA. Mol Ther Nucleic Acids. 2019;14:52-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tario JD, Humphrey K, Bantly AD, Muirhead KA, Moore JS, Wallace PK. Optimized staining and proliferation modeling methods for cell division monitoring using cell tracking dyes. J Vis Exp. 2012;70:e4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Filby A, Begum J, Jalal M, Day W. Appraising the suitability of succinimidyl and lipophilic fluorescent dyes to track proliferation in non-quiescent cells by dye dilution. Methods. 2015;82:29-37. [DOI] [PubMed] [Google Scholar]
- 31.Wilkinson AC, Ishida R, Nakauchi H, Yamazaki S. Long-term ex vivo expansion of mouse hematopoietic stem cells. Nat Protoc. 2020;15(2):628-648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilkinson AC, Nakauchi H. Stabilizing hematopoietic stem cells in vitro. Curr Opin Genet Dev. 2020;64:1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klein C, Zwick A, Kissel S, et al. Ptch2 loss drives myeloproliferation and myeloproliferative neoplasm progression. J Exp Med. 2016;213(2):273-290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang L, Bryder D, Adolfsson J, et al. Identification of Lin(-)Sca1(+) kit(+)CD34(+)Flt3- short-term hematopoietic stem cells capable of rapidly reconstituting and rescuing myeloablated transplant recipients. Blood. 2005;105(7):2717-2723. [DOI] [PubMed] [Google Scholar]
- 35.Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121(7):1109-1121. [DOI] [PubMed] [Google Scholar]
- 36.Takebe N, Zhao SC, Ural AU, et al. Retroviral transduction of human dihydropyrimidine dehydrogenase cDNA confers resistance to 5-fluorouracil in murine hematopoietic progenitor cells and human CD34(+)-enriched peripheral blood progenitor cells. Cancer Gene Ther. 2001;8(12):966-973. [DOI] [PubMed] [Google Scholar]
- 37.Nutt SL, Kee BL. The transcriptional regulation of B cell lineage commitment. Immunity. 2007;26(6):715-725. [DOI] [PubMed] [Google Scholar]
- 38.Lehnertz B, Chagraoui J, MacRae T, et al. HLF expression defines the human hematopoietic stem cell state. Blood. 2021;138(25):2642-2654. [DOI] [PubMed] [Google Scholar]
- 39.Freire PR, Conneely OM. NR4A1 and NR4A3 restrict HSC proliferation via reciprocal regulation of C/EBPα and inflammatory signaling. Blood. 2018;131(10):1081-1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Okada S, Fukuda T, Inada K, Tokuhisa T. Prolonged expression of c-fos suppresses cell cycle entry of dormant hematopoietic stem cells. Blood. 1999;93(3):816-825. [PubMed] [Google Scholar]
- 41.Jeong M, Piao ZH, Kim MS, et al. Thioredoxin-interacting protein regulates hematopoietic stem cell quiescence and mobilization under stress conditions. J Immunol. 2009;183(4):2495-2505. [DOI] [PubMed] [Google Scholar]
- 42.Gotze KS, Ramirez M, Tabor K, Small D, Matthews W, Civin CI. Flt3(high) and Flt3(low) CD34(+) progenitor cells isolated from human bone marrow are functionally distinct. Blood. 1998;91(6):1947-1958. [PubMed] [Google Scholar]
- 43.Liu J, Wu X, Mitchell B, Kintner C, Ding S, Schultz PG. A small-molecule agonist of the Wnt signaling pathway. Angew Chem Int Ed Engl. 2005;44(13):1987-1990. [DOI] [PubMed] [Google Scholar]
- 44.Pietras EM, Warr MR, Passegue E. Cell cycle regulation in hematopoietic stem cells. J Cell Biol. 2011;195(5):709-720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakamura-Ishizu A, Takizawa H, Suda T. The analysis, roles and regulation of quiescence in hematopoietic stem cells. Development. 2014;141(24):4656-4666. [DOI] [PubMed] [Google Scholar]
- 46.Orford KW, Scadden DT. Deconstructing stem cell self-renewal: genetic insights into cell-cycle regulation. Nat Rev Genet. 2008;9(2):115-128. [DOI] [PubMed] [Google Scholar]
- 47.Richter J, Traver D, Willert K. The role of Wnt signaling in hematopoietic stem cell development. Crit Rev Biochem Mol Biol. 2017;52(4):414-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carpenter KA, Thurlow KE, Craig SEL, Grainger S. Wnt regulation of hematopoietic stem cell development and disease. Curr Top Dev Biol. 2023;153:255-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reya T, Duncan AW, Ailles L, et al. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature. 2003;423(6938):409-414. [DOI] [PubMed] [Google Scholar]
- 50.Baba Y, Yokota T, Spits H, Garrett KP, Hayashi SI, Kincade PW. Constitutively active beta-catenin promotes expansion of multipotent hematopoietic progenitors in culture. J Immunol. 2006;177(4):2294-2303. [DOI] [PubMed] [Google Scholar]
- 51.Kirstetter P, Anderson K, Porse BT, Jacobsen SEW, Nerlov C. Activation of the canonical Wnt pathway leads to loss of hematopoietic stem cell repopulation and multilineage differentiation block. Nat Immunol. 2006;7(10):1048-1056. [DOI] [PubMed] [Google Scholar]
- 52.Scheller M, Huelsken J, Rosenbauer F, et al. Hematopoietic stem cell and multilineage defects generated by constitutive beta-catenin activation. Nat Immunol. 2006;7(10):1037-1047. [DOI] [PubMed] [Google Scholar]
- 53.Reya T, O’Riordan M, Okamura R, et al. Wnt signaling regulates B lymphocyte proliferation through a LEF-1 dependent mechanism. Immunity. 2000;13(1):15-24. [DOI] [PubMed] [Google Scholar]
- 54.Kondo M, Weissman IL, Akashi K. Identification of clonogenic common lymphoid progenitors in mouse bone marrow. Cell. 1997;91(5):661-672. [DOI] [PubMed] [Google Scholar]
- 55.Porritt HE, Rumfelt LL, Tabrizifard S, et al. Heterogeneity among DN1 prothymocytes reveals multiple progenitors with different capacities to generate T cell and non-T cell lineages. Immunity. 2004;20(6):735-745. [DOI] [PubMed] [Google Scholar]
- 56.Allman D, Sambandam A, Kim S, et al. Thymopoiesis independent of common lymphoid progenitors. Nat Immunol. 2003;4(2):168-174. [DOI] [PubMed] [Google Scholar]
- 57.Schwarz BA, Bhandoola A. Circulating hematopoietic progenitors with T lineage potential. Nat Immunol. 2004;5(9):953-960. [DOI] [PubMed] [Google Scholar]
- 58.Inlay MA, Bhattacharya D, Sahoo D, et al. Ly6d marks the earliest stage of B-cell specification and identifies the branchpoint between B-cell and T-cell development. Genes Dev. 2009;23(20):2376-2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ghaedi M, Steer CA, Martinez-Gonzalez I, Halim TYF, Abraham N, Takei F. Common-lymphoid-progenitor-independent pathways of innate and T lymphocyte development. Cell Rep. 2016;15(3):471-480. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Supporting data are available in the Online Supplementary Appendix. RNA sequencing data that support the findings of this study have been deposited in NCBI SRA under accession number GSE23774. The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.