

Abstract

We previously reported trisubstituted pyrimidine lead compounds, namely, ARN22089 and ARN25062, which block the interaction between CDC42 with its specific downstream effector, a PAK protein. This interaction is crucial for the progression of multiple tumor types. Such inhibitors showed anticancer efficacy in vivo. Here, we describe a second class of CDC42 inhibitors with favorable drug-like properties. Out of the 25 compounds here reported, compound 15 (ARN25499) stands out as the best lead compound with an improved pharmacokinetic profile, increased bioavailability, and efficacy in an in vivo PDX tumor mouse model. Our results indicate that these CDC42 inhibitors represent a promising chemical class toward the discovery of anticancer drugs, with ARN25499 as an additional lead candidate for preclinical development.
Introduction
CDC42 GTPases (CDC42, RHOJ, RHOQ) have emerged as appealing targets for the rational design of anticancer drugs as they are overexpressed in multiple cancers.1−4 Functionally, they play a role in multiple pathways required for tumor progression including cell migration, angiogenesis, and resistance to targeted therapies.3,5,6 In addition to its role in cancer development, CDC42 influences cardiovascular physiology, immune system function, nervous system function, and bone remodeling.7 CDC42 GTPases are in their active conformation when bound to GTP and switch to their “off” conformation when they hydrolyze GTP to GDP. Also, CDC42 protein activity is finely regulated by (i) guanine nucleotide exchange factors (GEFs), which exchange GDP for GTP to turn the protein “on”, (ii) GTPase activating proteins (GAPs), which help switch between “on” and “off” conformations, and (iii) guanine nucleotide dissociation inhibitors (GDIs), which keep GTPases in their “on” state.8,9 When the protein binds GTP, it can then activate downstream effectors, leading to altered cytoskeleton organization, polarity, adhesion, and migration, as well as cell proliferation.10
On these bases, several approaches have been taken to specifically target CDC42.11−14 These include (i) inhibiting CDC42-regulator and effector interactions, (ii) direct inhibition of effector kinases, and (iii) covalent irreversible inhibition of GEF-catalyzed nucleotide exchange.11,13 Several studies disclosed new small molecules that target CDC42–GEF interfaces such as ZCL278,15 ZCL367,16 CASIN,17 AZA197,18 and NSC23766.19 However, these approaches have failed to make it to the clinic because of lack of specificity and because of off-target toxicity secondary to the promiscuous activity of GEFs toward multiple Rho family members.20,21
We recently developed a new class of CDC42 GTPase interaction inhibitors.22,23 This class was rationally designed to specifically inhibit the interaction between CDC42 and its downstream effectors, differing from the other small molecules that interfere with the CDC42-GEF interaction, thus avoiding potential hematologic side effects.22 In particular, these inhibitors feature a triazine/pyrimidine core functionalized with several 6-membered heteroaryl groups, aniline, and completely saturated piperidine moieties (Scaffold A, Figure 1). In addition, our structure–activity-relationship (SAR) study allowed the identification of the best functionalities on a pyrimidine core that were also retained on the symmetric triazine core, as well as the identification of the influence of different types of masked and free saturated heterocycles groups. Importantly, compounds ARN22089 and ARN25062 (Figure 1) are drug-like CDC42/RHOJ pyrimidine derivatives with a significant ability to inhibit tumors in patient-derived xenografts (PDX) in vivo.23 On the other hand, compound 1 (Figure 1, which was originally named compound 29 in ref (23)) resulted in a promising triazine inhibitor with one-digit micromolar activity in four cancer cell lines (SKMel3, WM3248, A375, SW480). However, it is characterized by low solubility (Sk = 1 μM).23 These results prompted us to further investigate the SAR of these new CDC42 interaction inhibitors.
Figure 1.

Representation of new chemical scaffolds B–D explored starting from initial hits.22,23
Here, we present 25 additional derivatives that expand this new class of CDC42/RHOJ inhibitors, enhancing their drug-like profile. As described in Figure 1, we used compound 1,23 ARN22089,22 and ARN2506223 as our starting point for further derivatization of their core scaffold, generating scaffolds of types B–D (Figure 1), which show good potency in five cancer cell lines, and improved metabolic stability on mouse microsomes. Through the resulting SAR, we identified and characterized, in vitro and in vivo, compound 15 (ARN25499), which has (i) excellent kinetic and thermodynamic solubility, (ii) an improved in vivo bioavailability compared to previous leads, and (iii) a significant inhibition of tumor growth in a PDX mouse model.
Results and Discussion
Exploring the Structure of the New CDC42/RHOJ Inhibitors
To briefly recapitulate our previous drug discovery program on targeting CDC42/RHOJ, we mention that we have recently discovered a new class of trisubstituted pyrimidines (scaffold A, Figure 1, X = CH) as drug-like CDC42/RHOJ inhibitors.23 Scaffold A features 6-membered (hetero)aryl groups on position 6 of the core, several differently functionalized anilines on position 4, and piperidines on carbon 2. Additionally, we have preliminarily evaluated two triazine counterparts of this pyrimidine class (scaffold A, Figure 1, X = N), showing promising activity in our cell-based assay.23 This effort allowed us to identify ARN22089 and ARN25062 (Figure 1) as pyrimidine derivatives, and the triazine ARN24928 (Figure 1). All these lead compounds showed favorable PK and in vivo efficacy.23 This data prompted us to expand this class of triazine inhibitors and to complete the SAR on trisubstituted pyrimidines. Specifically, we previously found that the free 4-piperidine moiety represented the best moiety in terms of potency and synthetic feasibility, thus we maintained it in the majority of the derivatives. Moreover to improve the kinetic solubility of the triazine 1 (Sk = 1 μM, Figure 1)23 and the metabolic stability in mouse microsomes of selected pyrimidines (ARN22089–27 min, ARN25062–45 min),23 here we mainly explored: (i) the effect of different heteroaryl cycles of scaffold B; (ii) the inversion of piperidine/aniline positions of scaffold C, and (iii) the embedding of a 3′-nitrogen of the aniline moiety in a bicyclic group, as in scaffold D. We assessed the antiproliferative activity of all compounds in four melanoma cancer cell lines that have a BRAFV600E mutation (SKM28, SKMel3, WM3248, A375), and a colon cancer line (SW480) with a KRASG12 V mutation (Tables 1–3). These transformed cells have active RHOJ/CDC42 activity that is known to activate downstream Raf-MEK-ERK and PI3K-Akt pathways.24,25
Table 1. Modification at R1 of Triazine Class.

All IC50 values have a R2 > 0.90.
Compounds initially reported by Brindani et al.23
ND = not determined.
The compound features a completely saturated piperidin-4-yl substituent.
The compound features a partially saturated 1,2,3,6-tetrahydropyridin-4-yl substituent.
Table 3. Modification at R1 and R2 of “Classic” Pyrimidine Class.

All IC50 values have a R2 > 0.90.
Compounds initially reported by Jahid et al.22
Exploration of Heterocycles on the Triazine Core
In our previous work, we observed that the m-trifluoromethylaniline moiety improves the potency of pyrimidine derivatives.23 Surprisingly, the same modification on a triazine core as in compound 1 dropped the kinetic solubility to 1 μM but maintained a similar potency compared to the related pyrimidine counterpart.23 Thus, we explored different heteroaryl substituents R1 on the triazine core, maintaining the m-trifluoromethylaniline. The aim was to improve the solubility and explore the contribution of this substituent to the activity (Table 1). While the substitution of the phenyl group with a pyrimidine group in derivative 2 generally decreased the activity, especially in SKM28 (IC50 = 55.5 μM, Table 1, entry 2), compared to compound 1, the introduction of a bicyclic heteroaromatic group such as isoquinolyl-, N-methylindolyl-, indazolyl-, benzofuran-3-yl- in 3–6 restored the activity in the one digit micromolar range in all cancer cell lines (Table 1, entries 3–6). Indeed, among 3–6, the indazolyl group in compound 5 gave the best effect in potency, showing an IC50 in the range of 2.6–3.9 μM in all five cancer cell lines. We then investigated the role of smaller 5-membered heterocycles. Derivative 7 with N-methylpyrazolyl group maintained the activity in SKM28 but decreased the activity in other cancer cell lines, displaying the IC50 of ∼3–8 fold higher (Table 1, entry 7) compared to 1. The shifting of the N-methyl group in the imidazolyl substituent of 8 displayed a negative impact on the antiproliferative activity of 1, annihilating the effect on A375 and decreasing the activity of ∼10-fold on WM3248 (Table 1, entry 8). Instead, the presence of free nitrogen of the pyrrolyl substituent of compound 9 restored the activity in all cancer cell lines (Table 1, entry 9). We then evaluated the effect of the geometry of this substituent by introducing a completely saturated pyrrolidinyl group in derivative 10, which resulted in a complete loss of activity compared to its aromatic counterpart 9 (Table 1, entries 9–10). Finally, the introduction of a sulfur heteroatom in the thiophenyl substituent of 11 exhibited one-digit micromolar activity on all cancer cell lines (Table 1, entry 11). Notably, this analogue features the partially saturated 1,2,3,6-tetrahydropyridin-4-yl instead of the typical piperidine moiety. This was due to synthetic reasons (vide infra), as it is characterized by a slight instability in DMSO, probably due to the susceptibility of thiophene to oxidative processes.
Overall, these data suggested that key features for the activity of this class of CDC42/RHOJ inhibitors are their electronic structure in terms of the molecular size, their conformational freedom, and their number and exact position of the heteroatom in the heterocycle moiety. Specifically, analogues 3–6 pointed out that a heteroaryl bicycle is generally preferable to a monocycle, and derivative 11 suggested a good tolerance of a different conformation of piperidine ring induced by the intracyclic double C–C constraint.
Exploration of “Inverted” Trisubstituted Pyrimidine
The initial goal was to exchange the piperidine and the aniline moieties of ARN22089 and ARN25062 to evaluate the impact of the different orientations of these substituents on the activity. Therefore, we introduced meta-N,N-dimethylamino- and meta-trifluoroaniline moieties on position 2 and the piperidine on position 4 of the pyrimidine core, maintaining a phenyl substituent on position 6. Thus, we produced the analogues 12 and 13, which have comparable antiproliferative activity in all cell lines (Table 2, entries 1, 2). Generally, these new inhibitors have been proven equally potent to the original ARN22089 and ARN25062.23 This result paved the way toward the development of a second type of CDC42/RHOJ inhibitors, where variously substituted anilines were inserted on the C2 of the pyrimidine cycle (Table 2). While the introduction of a meta-trifluoromethoxy phenyl in compound 14, or two substituents as in 15 and 16, were generally well tolerated, the presence of free hydroxyl or carboxylic functional groups on the aniline of 17 and 18 annihilated the activity in the cells (Table 2, entries 3–7). We then investigated the impact of naked or substituted pyridines as in analogues 19–23 to evaluate the role of the basic functionality in this portion of the chemical structure. We found out that the naked 4-piperidine in derivative 19 decreased the potency of 2–3 folds compared to ARN25062 (Table 2, entry 8). The insertion of vicinal trifluoromethyl-, difluoromethoxyl-, or methoxyl group in the pyridine ring of compounds 20–22 restored the activity, probably due both to an alteration of the basic strength of the pyridine site and to additional interactions established by fluorine atoms or a methoxy group with the target (Table 2, entries 9–11). A similar, although less relevant, effect was observed with the regioisomer of compound 22, where the pyridine nitrogen is in meta-position and the methoxyl group is in para position of the aniline substituent (Table 2, entry 12 compound 23).
Table 2. Modification at R1 Aniline Substituents of “Inverted” Pyrimidine Class.

Activity of two reference compounds: ARN22089 (IC50–SKM28 = 24.8 μM,–SKMel3 = 4.2 μM,–WM3248 = 4.5 μM,–A375 = 4.9 μM,–SW480 = 8.6 μM), ARN25062 (IC50–SKM28 = 6.1 μM,–SKMel3 = 4.6 μM,–WM3248 = 9.3 μM,–A375 = 5.1 μM,–SW480 = 5.9 μM).
All IC50 values have a R2 > 0.90.
Compound as hydrobromide salt.
NA = not active.
Evaluation of Our New Analogues of ARN22089
The investigation of new triazines 2–11 highlighted a positive contribution of bicyclic heteroaryl substituents and a tolerance of partially saturated piperidine moieties. We further proved these results through the compounds 24–26, close analogs of our previous lead compound ARN22089 (Table 3). Derivative 24 maintained the activity of ARN22089, evidencing that a different saturation and conformation of the piperidine do not significantly affect potency (Table 3, entry 2). Moreover, we embedded the meta N-methyl amino group of the aniline moiety in an indole group, both in methylated and free forms, generating compounds 25 and 26 (Table 3, entries 3,4). These analogues inhibit the cell viability for all cell lines, confirming a positive effect of a bulkier bicyclic aniline moiety.
Evaluation of Druglike Properties of Our New Leads
After this initial evaluation, we assessed the drug-like profile of this novel RHOJ/CDC42 inhibitors by studying the kinetic solubility, plasma, and phase-I microsomal stability of 18 selected compounds. This was done to define the key chemical elements for a drug-like profile (Table 4). Notably, the first disclosed triazine 1 was very stable in plasma and mouse microsomes (>120 and >60 min, Table 4), but poorly soluble (Sk = 1 μM). Among the selected triazine derivatives 3–6, 9, and 11, only compound 9 showed an excellent kinetic solubility of 237 ± 11 μM (Table 4, entry 8), suggesting that the pyrrolyl ring positively impacts the solubility in water. All other triazine inhibitors have a kinetic solubility <12 μM (Table 4, entries 4–7, 9). On the contrary, the good plasma and microsomal stability of triazine 1 was maintained.
Table 4. Kinetic and Thermodynamic Solubility, Aggregation by NMR, Plasma, and Microsomal Stability in Mouse of Selected Compounds.
| entry | compound | kinetic solubility (Sk)(μM) | thermodynamic solubility (S ThD) (μM)b | aggregation by NMR (50 μM)b | T1/2 plasma (min)b | T1/2 microsomal (min)b |
|---|---|---|---|---|---|---|
| 1 | ARN22089a | 250 | ND | no | 71 | 27 |
| 2 | ARN25062a | 168 | ND | no | >120 | 45 |
| 3 | 1a | 1 | ND | ND | >120 | >60 |
| 4 | 3 | 6 ± 1 | ND | ND | >120 | >60 |
| 5 | 4 | <1 | ND | ND | >120 | >60 |
| 6 | 5 | 1 | ND | ND | >120 | >60 |
| 7 | 6 | 12 | ND | ND | >120 | >60 |
| 8 | 9 | 237 ± 11 | ND | no | >120 | >60 |
| 9 | 11 | <1 | ND | ND | >120 | >60 |
| 10 | 12 | 13 ± 4 | ND | ND | 60 | >60 |
| 11 | 13 | <1 | ND | ND | >120 | >60 |
| 12 | 14 | <1 | ND | ND | >120 | >60 |
| 13 | 15 (ARN25499) | >250 | 371 ± 5 | no | >120 | >60 |
| 14 | 15 HCl | 244 ± 2 | 341 ± 38 | ND | ND | ND |
| 15 | 16 | 245 ± 4 | ND | no | 89 | >60 |
| 16 | 20 (ARN25375) | 222 ± 2 | 106 ± 3 | no | >120 | >60 |
| 17 | 20 HCl | 244 ± 2 | 1 ± 0 | ND | ND | ND |
| 18 | 21 | <2 | ND | ND | >120 | >60 |
| 19 | 22 | 238 ± 7 | ND | no | >120 | >60 |
| 20 | 23 | 247 ± 6 | ND | no | >120 | >60 |
| 21 | 24 | <1 | ND | ND | >120 | 37 |
| 22 | 25 | 239 ± 5 | ND | yes | >120 | >60 |
| 23 | 26 | 68 | ND | ND | >120 | >60 |
Compounds initially reported by Brindani et al.23
ND = not detected.
From the first class of trisubstituted pyrimidines, we previously selected ARN22089 and ARN25062 as the most promising compounds for further in vivo studies. Notably, both compounds showed high kinetic solubility (250 and 168 μM, Table 4), and ARN25062 exhibited ameliorated plasma and microsomal stability (>120 and 45 min, Table 4) compared to ARN22089 (71 and 27 min, Table 4). Here, our goal was to improve the microsomal stability through an expansion of the initial SAR study for the “inverted” trisubstituted pyrimidine class (Table 2). Among the selected nine compounds 12–16 and 20–23 (Table 4, entries 10–13, 15, 16, 17–20), derivatives 15, 16, 20, 22, 23 exhibited excellent kinetic solubility (222 to >250 μM range). Indeed, the shifting of the N-methylamino aniline from C4 into C2 of the pyridine core as in compound 12 drastically dropped solubility. However, it ameliorated microsomal stability compared to our previous lead ARN22089 (Table 4, entry 10 vs entry 1). The same trend was observed for the solubility and microsomal stability parameters, in the case of the meta-trifluoromethylaniline moiety that was shifted from the C4 of the previous candidate ARN25062 to the C2 of the analogue 13 (Figure 1, Table 4, entry 11 vs entry 2). No improvement was observed when the trifluoromethoxy group of 14 replaced the trifluoromethyl group of 13 (Table 4, entry 12). On the other hand, when the meta-trifluoromethyl group was inserted in a pyridin-4-amine as in 20, the solubility was completely restored to 222 ± 2 μM value (Table 4, entry 16), suggesting a positive contribution of an additional basic site of the pyridine. Notably, the pyridine analogues 22–23 exhibited excellent kinetic solubility (Table 4, entries 19, 20), except for compound 21 with the 2-difluoromethylpyridine group (Table 4, entry 18). On the other hand, the disubstituted phenyl rings of compounds 15 and 16 improved sensibly the solubility (Table 4, entries 13, 15). Generally, all derivatives of this second class of trisubstituted pyrimidines possess excellent plasma and improved metabolic stability. Specifically, only compounds 12 and 16 showed a moderate decrease in plasma stability.
Moreover, the closest structural analogues 24–26 of ARN22089 were further analyzed (Table 4, entries 21–23). The presence of the double C–C bond in 24 annihilated the solubility, ameliorated the plasma stability, and approximately maintained the same metabolic stability. The solubility was partially recovered through the insertion of the free indole group in 26 with Sk of 68 μM and completely restored for N-methylindole in 25 with Sk 239 ± 5 μM (Table 4, entries 22–23).
The solubility and the aggregation state of selected compounds 9, 15, 16, 20, 22, 23, and 25 were further evaluated by NMR analysis under experimental conditions employed in microscale thermophoresis (MST) and NMR (Tris buffer– vide infra) according to the SPAM filter approach to avoid false-positive results.26 Since the starting compounds (ARN22089 and ARN25062) showed aggregation at 100 μM, these six compounds were tested in the binding assay buffer at the maximum concentration of 50 μM, in the presence of an internal reference (4-trifluoromethyl benzoic acid, 200 μM). The seven selected compounds were soluble in Tris buffer at least up to 50 μM (data not shown) in line with kinetics solubility data. Indeed compound 25 showed aggregation at 50 μM differently from other compounds, thus it was excluded from further binding evaluation studies (vide infra).
Notably, derivatives 15 (ARN25499) and 20 (ARN25375) resulted in being the best compounds in terms of potency in all cell lines, and kinetic solubility, also showing a strong improvement in microsomal stability. To preliminary evaluate the pH-dependent solubility of these two compounds, we assessed both the kinetic and thermodynamic solubility for both the neutral and the hydrochloride forms of 15 (ARN25499) and 20 (ARN25375) (Table 4, entries 13, 14, 16, 17). The kinetic solubility was maintained for the corresponding hydrochloride forms for both derivatives (Table 4, entries 14, 17). The same behavior has been observed for the thermodynamic solubility of analogue 15, which is >300 μM for the neutral and salt form (Table 4, entries 13, 14). On the contrary, the hydrochloride form of 20 exhibited a thermodynamic solubility 100-fold lower than the related neutral form (Table 4, entries 16, 17), suggesting a significant pH-dependent behavior for the trifluoromethylpyridine analogue 20.
Microscale Thermophoresis (MST) and NMR for CDC42-Binding Validation
To investigate the binding of the most promising compounds to the target, we have employed microscale thermophoresis (MST) and NMR techniques. Among the previously selected 18 new inhibitors, we further selected the best compounds in terms of potency, drug-like profile, and aggregation by NMR, namely, compounds 9, 15, 16, 20, 22, 23.
We first employed microscale thermophoresis (MST) to retrieve information on the ability of the six selected compounds to engage our target.27 Wild-type His-CDC42 (Ile4-Pro182) was used as a target macromolecule. The protein was activated or inactivated through loading of GppNHp or GDP, respectively. Loading efficiency was evaluated by measuring the protein intact mass with ESI MS+.22 Only protein samples with loading efficiency higher than 90% were used in MST experiments. Target proteins were Red-NHS labeled prior to use. Changes in their normalized fluorescence intensity (ΔFnorm [‰]= Fhot/Fcold) were recorded. ΔFnorm at 2.5 s (t-jump) and 15 s (thermophoresis signal) were evaluated. Compounds were tested for binding at a concentration of 50 μM in the presence of 0.5% v/v DMSO. Assays were set up in the Tris-HCl buffer. As shown in Figure 2, a change in the fluorescence signal was detected for compound 15 irrespectively of the CDC42 activation state, displaying good confidence in binding and signal-to-noise ratio higher than 12, both for the active and inactive state of CDC42 (Figure 2, Table S1, Supporting Information). While compounds 9 and 16 led to a fluorescence change only in the presence of the GDP-inactivated CDC42, compound 20 showed binding for both GDP- and GppNHp-CDC42, with a slightly lower confidence in binding than the other compounds (Figure 2, Table S1, Supporting Information). On the other hand, compounds 22, and 23 did not exhibit binding by MST for both active and inactive states of CDC42 (Figure 2, Table S1, Supporting Information). In all the binding events, both the T-jump fluorescence intensity and the thermophoresis signal were modified, except for compound 20 which displayed only a change in the T-jump initial fluorescence intensity.
Figure 2.

(A–E) Example of MST traces of selected compounds 22 (A) and 15 (B). In panel A, there was no change in fluorescence and no binding events. In panel B a good change in fluorescence was displayed, highlighting a potential binding event. (C) Graph displays the difference in normalized fluorescence (ΔFnorm [‰]= Fhot/Fcold) between protein:compound sample and a protein-only sample. Compounds (C) were tested at 50 μM toward the activated (loaded with GppNHp) or inactivated (loaded with GDP) His-Cdc42. A single blue asterisk indicates a signal-to-noise ratio above 5, while double red asterisks indicate a signal-to-noise ratio above 12. ΔFnorm at 2.5 s (t-jump) and 15 s (thermophoresis signal) were reported. (D,E) example of binding check by 19F T2 filter NMR experiments [compound 9-(E)], and by WaterLOGSY [Compound 16-(F)]. Compounds were tested in the absence of protein and in the presence of GppNHp (black) or GDP (gray), and in the presence of activated (loaded with GppNHp) His-CDC42 (red) or inactivated (loaded with GDP) His-CDC42 (blue). The arrows indicate where a difference in the compound NMR signal is observed after the addition of the protein, highlighting a binding event.
The binding of 6 compounds was also evaluated by NMR experiments,28 testing them at 50 μM in the absence and in the presence of both His-CDC42 (2 μM) by 19F T2 filters29,30 (fluorinated compounds) and WaterLOGSY (Water-Ligand Observation with Gradient SpectroscopY)31 experiments. In 19F T2 Filter experiments, the binding event induces a line-broadening of the 19F NMR signal of the binding molecule in the presence of the protein, which results in a decrease of its intensity compared to the signals recorded in the absence of protein (as an example see Figure 2D). On the other end, in WaterLOGSY experiments, the binding event is identified by 1H molecule signal change from negative, in the absence of protein, to less negative or to positive in the presence of protein due to the transfer of magnetization from bulk water to the compound interacting with the macromolecule (examples see Figure 2E).
In the NMR experiments, all 6 compounds bind to the activated form of His-CDC42 and only compound 22 does not bind to the inactivated form (Table S1, Supporting Information), whereas only compounds 9, 15, 16, and 20 bind to the protein in the MST experiments. These results were expected since we know that NMR is highly sensitive even to very weak binders, therefore it is quite common that binding events identified by NMR are not detected by other biophysical techniques.32
Molecular Modeling for Binding Validation
Based on overall data, we further performed molecular modeling studies of compounds 15 (ARN25499) and 20 (ARN25375) to validate their binding toward the active state of CDC42. We utilized the drug-binding pocket previously identified at the CDC42-PAK protein–protein interface for our computational investigations. Correspondingly, we conducted molecular docking analysis of the new derivative compounds, 15 (ARN25499) and 20 (ARN25375), on the GTP-bound active configuration of CDC42.22,23
Our computational results indicate that both compounds 15 (ARN25499) and 20 (ARN25375) fit inside the effector pocket of CDC42 (see Figure 3A, B), matching with the binding mode earlier proposed for compounds belonging to the previously identified class of CDC42/RHOJ inhibitors (e.g., ARN22089, ARN24928, and ARN25062).22,23 As previously described, the pocket possesses a hydrophobic nature that is well-suited for accommodating the phenyl group on C6 of both 15 (ARN25499) and 20 (ARN25375), serving as an anchor point for the binding to the allosteric pocket.23
Figure 3.

Model structure of compound 15 (ARN25499, panel A) and 20 (ARN25375, panel B) compounds bound to the allosteric drug-binding pocket of CDC42 is reported. The structure of CDC42 is represented as a white surface while the identified drug-binding pocket is shown in both stick and transparent blue surface. 15-ARN25499 and 20-ARN25375 are reported as yellow and green sticks, respectively.
To further evaluate the binding configurations, we conducted equilibrium molecular dynamics (MD) simulations of both 15 (ARN25499) and 20 (ARN25375) in complex with CDC42 (Figure 4). Importantly, the binding pose of both 15 (ARN25499) and 20 (ARN25375) in the CDC42-ligand complexes was maintained during 500 ns-long MD simulations (RMSD = 3.40 ± 0.79 Å and 4.25 ± 0.45 for 15 (ARN25499) and 20 (ARN25375), respectively (Figure 4A, B, right panels). Indeed, as shown in Figure 4 (left panels), 15 (ARN25499) and 20 (ARN25375) tightly bind the target pocket throughout the simulation time.
Figure 4.

Molecular dynamics (MD) simulations of the protein–ligand complexes. The structural representation of CDC42 in a complex with compound 15 (ARN25499, panel A) or compound 20 (ARN25375, panel B) is reported on the left. CDC42 is represented as a cartoon, while the binding pocket is highlighted as both a stick and a transparent blue surface. Multiple MD snapshots of the 15 (ARN25499, yellow) and 20 (ARN25375, green) binding poses are shown as sticks. On the right, the RMSD over time for the CDC42-ligand complexes is reported with the RMSD running averages highlighted in bold.
In Vivo Pharmacokinetics of the Selected Follow-Up/Backup Leads
Based on the overall results and druglike profile of our compounds, the neutral form of compounds 15 (ARN25499) and 20 (ARN25375) were selected as candidates for in vivo pharmacokinetics (PK) studies, given further experiments to assess their in vivo efficacy in animal models (mouse specie) of cancer and also compare the PK profile to the previous lead compound (ARN22089).23
We tested two different routes of administration: (i) intravenous (I.V.) injection at a concentration of 3 mg/kg (n = 3 animals for each time point), and (ii) oral (P.O.) treatment at a dose of 10 mg/kg (n = 3 animals for each time point, Figure 5). During the PK studies, via either I.V. or P.O. administration, ARN25499 and ARN25375 were well tolerated by all animals, and no treatment-related toxicological signs were observed. While the I.V. profiles of the two inhibitors are comparable, the P.O. profiles exhibited significantly different behaviors consistently with the thermodynamic solubility data of hydrochloride and neutral forms. Indeed, ARN25499 and ARN25375 reached a Cmax of 320 and 643 ng/mL in 5 min after I.V. administration, respectively, followed by a protracted elimination phase. Both compounds were still detectable after 4 h at a concentration of 74 and 84 ng/mL, respectively. After oral administration (10 mg/kg), both compounds achieved the maximum concentration in 2 h, with Cmax values of 140 and 99 ng/mL for ARN25499 and ARN25375, respectively. Notably, the elimination phase of ARN25499 was slower than ARN25375-. ARN25499 and ARN25375 were still detectable after 8 h (7 ng/mL) and 4 h (40 ng/mL). Compound ARN25499 showed good exposure with a longer half-life of 257 min (I.V.), and 168 min (P.O.) compared to ARN25375, which showed a half-life of 115 min (I.V.) and 91 min (P.O). In summary, these data indicate that both compounds are well tolerated after a single injection. Remarkably, while ARN25375 exhibited a favorable pharmacokinetic profile of only I.V., with a bioavailability of 8% following its pH-dependent thermodynamic solubility (Table 4), ARN25499 possesses favorable pharmacokinetic profiles of both I.V. and P.O. with bioavailability in mouse of 22% (Figure 5).
Figure 5.

Mouse PK profiles of ARN25499 (15) and ARN25375 (20) following intravenous (I.V.) and oral (P.O.) administration at 3 and 10 mg/kg, respectively, and the corresponding observed and calculated PK parameters.
In Vivo Efficacy and In Vitro Activity of Selected Follow-Up/Backup Leads
We next test the efficacy of ARN25499 since it has favorable a PK profile, overall. We used the NOD scid gamma (NSG) model and inoculated a chunk of melanoma patient-derived xenograft (PDX) tumor on either side of the mouse. When the tumors reached a measurable size range, we treated the mice with 10 mg/kg ARN25499 via I.V. daily, for 2 weeks. Drug treatment inhibited the growth of tumors as compared to vehicle (Figure 6A), and ARN25499 treated tumors had a significantly smaller volume as compared with vehicle-treated tumors after 2 weeks of treatment (Figure 6B). Finally, a two-week treatment with the compound showed no adverse effect on the animals and no change in body weight (Figure 6C). Next, we compared the pathway engagement of ARN25499 and ARN25375 with ARN22089 in vitro in WM3248 cells. Our previous reverse phase protein analysis of ARN22089 treated cells indicated that drug treatment inhibited the accumulation of pERK and pS6.22 Following a 6-h incubation period, ARN25499 inhibited the accumulation of pERK and pS6 to a greater extent than ARN22089 (Figure 6D).
Figure 6.

ARN25499 slows the growth of melanoma PDX tumors in NSG mice. (A) Line plot showing the growth curves of the vehicle and ARN24599 treated tumors in mean ± SEM. A small chunk of tumors was inoculated on either side under the back skin of NSG mice. When tumors reached the initial size of about 150–200 mm3, mice were injected via tail vein with 10 mg/kg ARN25499 or vehicle (9 tumors per group) daily for 2 weeks. The tumor and weight of the mouse were measured every other day with a caliper and weight scale. (B) Scatterplot showing the blue (vehicle) and red (ARN25499) dots representing tumor volume in a millimeter cube at the end of the 2-week treatment. GraphPad Prism 9 was used to generate plot and statistical analysis using 2 way ANOVA (A) and unpaired two-tailed t test (B), ***,**p-value ≤0.0001, 0.0047, respectively. (C) The line plot (mean ± SD) shows no significant difference in weights between vehicle and ARN25499 treated mice for the 2-week treatment. (D) WM3248 cells were treated with 10 μM of ARN22089, ARN25375, and ARN25499 for 6 h. The accumulation of pS6 and pERK were measured by immunoblotting. Relative densitometry of pERK and pS6 as compared to unphosphorylated forms of the protein were determined and are reported below each lane. A representative blot of three independent biologic replicates is shown.
Chemistry
The triazine analogues 2–11 were obtained through a 4–5 steps synthetic route as depicted in Scheme 1. As previously developed, the order of the steps was crucial to successfully introduce the desired substituents.23 First, the nucleophilic aromatic substitution (SNAr) of m-trifluoromethylaniline on cyanuric chloride 27 allowed the preparation of intermediate 28 with 89% excellent yield, which underwent two sequential Suzuki couplings. Specifically, the first Suzuki employed (1-(tert-butoxycarbonyl)-1,2,3,6-tetrahydropyridin-4-yl)boronic acid under classic conditions of Pd(dppf)Cl2·DCM and aqueous K2CO3 in 1,4-dioxane dry at 80 °C to afford common intermediate 30 with 55% yield. Compound 30 represented a useful building block for the product diversification for this kind of scaffold, since the second Suzuki coupling with a wide range of boronic acids or esters 31a–i under slightly stronger conditions produced trisubstituted triazines 32a–i functionalized with several five-, six-membered, mono- and biheterocycles with 23–79% range yield. Indeed, compound 32e was accessed in a one-pot fashion from monosubstituted triazine 28 with a 45% yield. Then the reduction and Boc removal transformations got access to final products 2–10. Generally, the reduction was conducted in the presence of HCOONH4, Pd(OH)2/C, MeOH, or EtOH under reflux, affording precursors 33a–g and 33h’ with 25–92% range yield. Notably these conditions have been proven to be too harsh for the generation of the desired derivative 33h with aromatic pyrrole moiety due to the over-reduction of substrate 32h to 33h’, where the pyrrole ring was reduced to a completely saturated pyrrolidine ring with a very good 86% yield. Thus, we successfully applied milder conditions of reduction in the presence of Et3SiH, Pd/C in EtOH at room temperature to obtain precursor 33h starting from 32h with 60% yield. The double C–C bond of 32 turned out to be strongly dependent on the electronic features of the inserted heterocycle in conjugation with the triazine core.33 This hypothesis was further supported by substrate 32i, whose double C–C bond of 1,2,3,6-tetrahydropyridine has not been reduced both with classic conditions under prolonged time (5 days), neither under different conditions (i.e., NiCl2·H2O, NaBH4 in MeOH; or NaBH4 in the presence of TFA or AcOH in THF at 50 °C). Thus, compound 32i was directly subjected to BOC deprotection to afford final compound 11 with a 39% yield.
Scheme 1. Synthetic Route toward Trisubstituted Triazines 2–11.
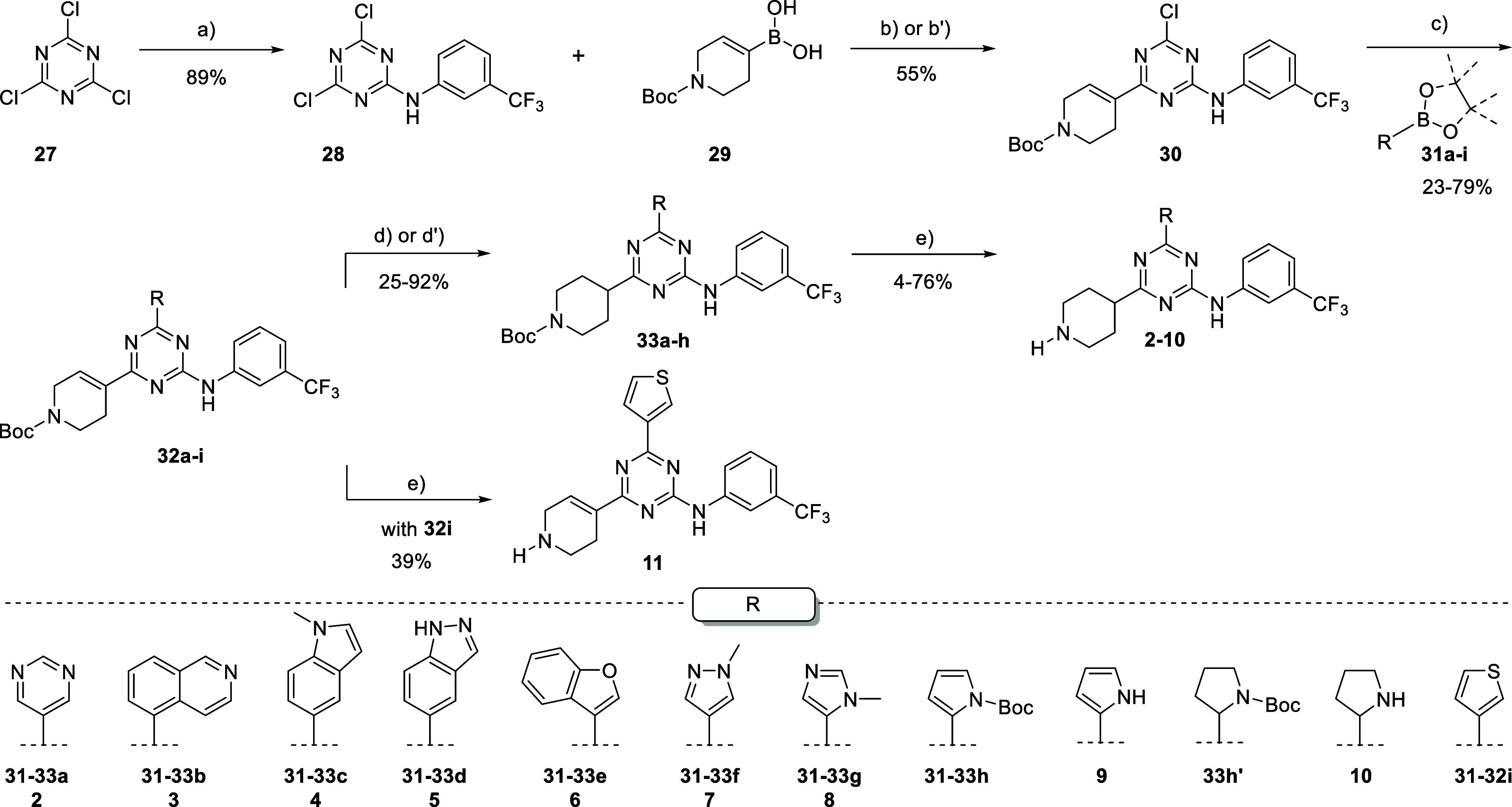
Reagents and conditions: (a) cyanuric chloride, 3-(trifluoromethyl)aniline, DIPEA, 0 °C; (b) (1-(tert-Butoxycarbonyl)-1,2,3,6-tetrahydropyridin-4-yl)boronic acid, Pd(dppf)Cl2·DCM, K2CO3 2M, 80 °C 1,4-dioxane; (b’) from 29 to 32e: (1-(tert-butoxycarbonyl)-1,2,3,6-tetrahydropyridin-4-yl)boronic acid, Pd(dppf)Cl2·DCM, K2CO3 2M, 80 °C; then Pd(dppf)Cl2·DCM, benzofuran-3-ylboronic acid, 120 °C; (c) Het-B(OH)2, Pd(dppf)Cl2·DCM, K2CO3 2M, 100 °C; (d) HCOONH4,Pd(OH)2/C, MeOH or EtOH, reflux; (d’) from 32h to 33h: Et3SiH, Pd/C, EtOH, rt; and (e) from compound 32j to 11, and from 33a–h to 2–10: HCl 4 M 1,4-dioxane solution, 1,4-dioxane, 0 °C to rt.
As shown in Scheme 2, we were able to develop a straightforward and efficient 5-step synthetic route for the obtainment of “inverted” trisubstituted pyrimidine 12–16, and 19–23. Since this analogue features the aniline moiety on C2, we performed the one-pot synthesis of intermediate 37 through two sequential Suzuki coupling with phenyl boronic acid 35 and boronic ester 36 in the presence of PdCl2(dppf)·DCM and aqueous K2CO3. For this strategy, we exploited the well-known reactivity order of each position of the pyrimidine halides 34, which follows the general order C4(6) > C2. We drove the insertion of each boronic partner through the temperature regulation: 60 °C for the first phenyl group, and 110 °C for the second tetrahydropyridinyl moiety. Thus, we nicely obtained intermediate 37 with a 55% yield just through one step starting from commercially available 34. This compound represented again our divergent point for the introduction of different aniline substituents on C2 through the Buchwald–Hartwig reaction. Thus, the reaction of 37 with aniline 38a–j in the presence of Pd(OAc)2, (±)-BINAP or Xantphos, and Cs2CO3 at 120 °C afforded intermediates 39a–j with 60–85% range yield. This transformation occurred together with the isomerization of the double C–C bond of tetrahydropyridine producing a mixture of regioisomers, but this did not affect our synthetic plane, since the next step involved the reduction of the aforementioned double bond for the obtainment of precursors 40a–j. The final Boc removal with HCl in dioxane afforded final products 12–16 and 19–23 (Scheme 2).
Scheme 2. Synthetic Route toward “Inverted” Trisubstituted Pyrimidine 12–16, 19–23.
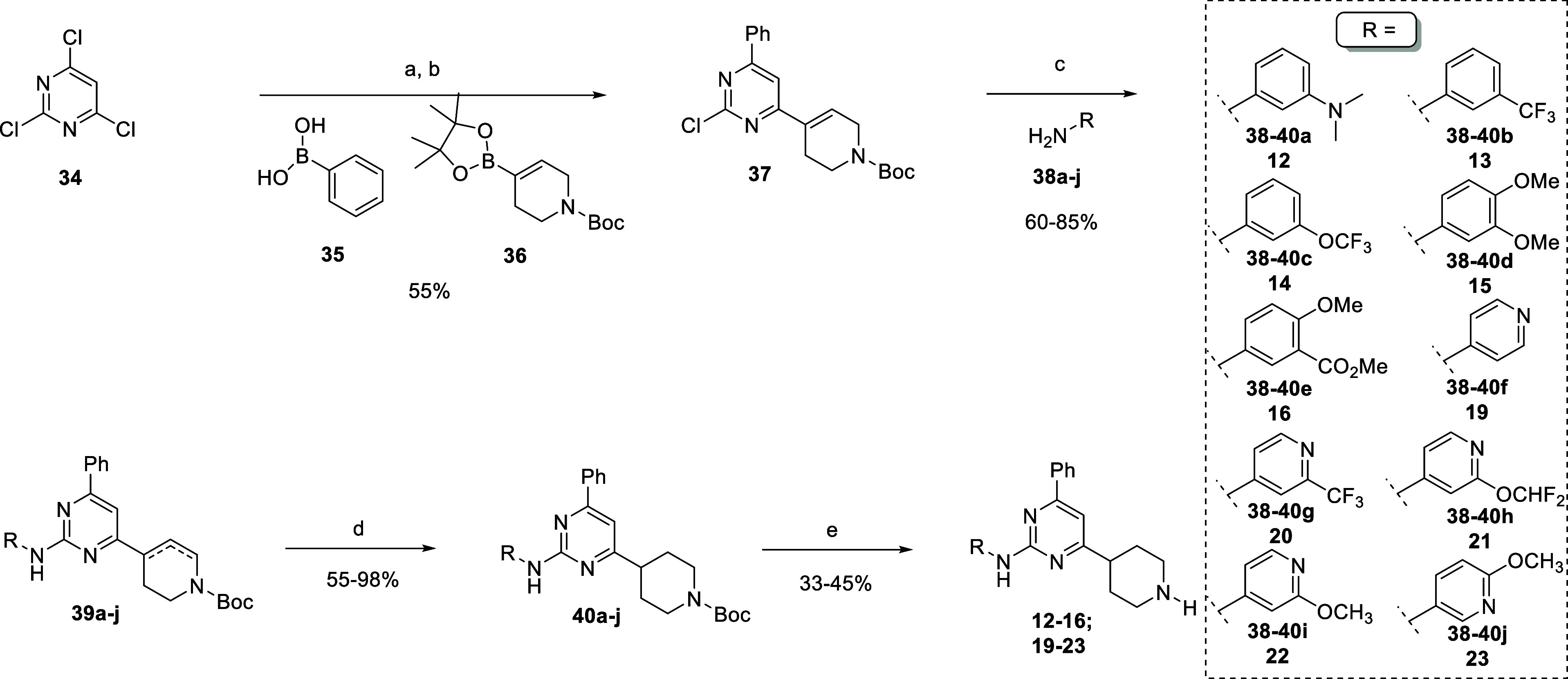
Reagents and conditions: (a) phenylboronic acid 35, PdCl2(dppf)·DCM, K2CO3 (2M)aq, 1,4-dioxane dry, 60 °C, then (b) compound 36, PdCl2(dppf)·DCM, 110 °C; (c) compounds 38a–j, Pd(OAc)2, (±)-BINAP or Xantphos, Cs2CO3, 1,4-dioxane dry, 120 °C; (d) Pd(OH)2/C, NH4COOH, MeOH dry, 80 °C; and (e) HCl (4 M in 1,4-dioxane), 1,4-dioxane dry.
On the other hand, the unmasking of catechol and salicylic groups in 17 and 18 was easily achieved through the treatment of intermediate 40d–e with BBr3 in DCM, respectively (Scheme 3). The synthesis of the products 24–26 was carried out following the synthesis strategy previously developed (Scheme 4).23 Compound 24 was obtained through 3 synthetic steps starting from dichloro-phenyl pyrimidine 41. As previously described, SNAr of 41 with aniline 38a, followed by the Suzuki coupling with 36 afforded intermediate 43a. Thus, the double C–C bond of tetrahydropyridine was maintained in the final product 24 by direct Boc deprotection of 43a with HCl in dioxane (56% yield). Instead, the synthesis of the derivatives 25 and 26 required the additional double C–C reduction step. Thus, SNAr with N-Methyl- and N-Boc-aniline 38k–l afforded intermediates 42b–c, respectively. The next Sukuki coupling with 36, double C–C reduction with Pd(OH)2/C and ammonium formate and Boc deprotection with HCl in dioxane furnished final 25 and 26 with 33 and 16% yields after three steps starting from 36, respectively.
Scheme 3. Synthesis of Derivatives 17, 18.

Reagents and conditions: (a) BBr3 (1 M in DCM), DCM dry.
Scheme 4. Synthesis of “Classic” Tri-Substituted Pyrimidine 24–26.
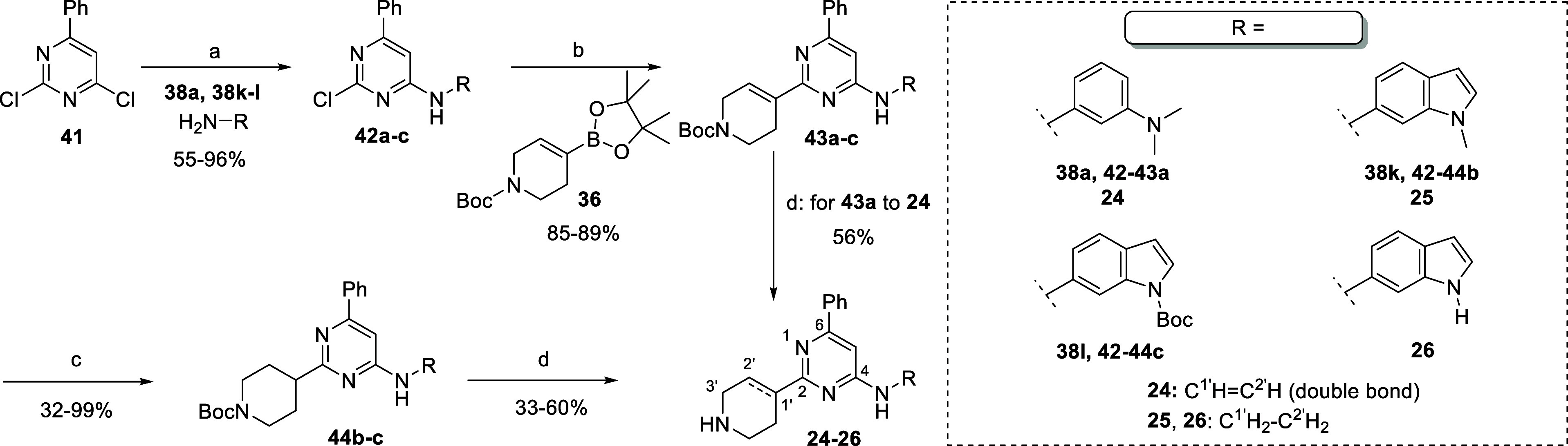
Reagents and conditions: (a) LiHMDS (1 M in THF), aniline 38a, 38k–l, THF dry; (b) compound 36, Pd(Cl2)(dppf)·DCM, K2CO3 (2M)aq, 1,4-dioxane dry; (c) Pd(OH)2/C, NH4COOH, MeOH dry, 80 °C; and (d) HCl (4 M in 1,4-dioxane dry), 1,4-dioxane dry.
Conclusions
Based on our previous results on trisubstituted pyrimidines as a promising class of CDC42-PAK interaction inhibitors, we have here reported the design, synthesis, and extensive characterization of a new series of pyrimidine/triazine derivatives to block CDC42. The resulting SAR elucidated the key structural features that enhanced the antiproliferative activity against five cancer cell lines and the overall drug-like profile. Generally, the introduction of bicyclic heterocycles such as isoquinolyl-, indolyl-, indazolyl- and benzofuranyl- favorably impact the compounds’ activity, as proven by the set of triazines (1–11, Table 1) and classic pyrimidine derivatives (25–26, Table 3). On the other hand, the shifting of the position of the piperidine moiety as in derivative 12 (Table 2) is well tolerated. This modification prompted the development of a new series of trisubstituted pyrimidines, such as 12–23, which unveil the favorable effect of methoxy and fluorinated groups (Table 3). Taken together, our new data on the inhibitory activity, drug-likeness profile (Table 4), in vitro binding data at the target supported also by computational studies, and the in vivo favorable pharmacokinetic profile, indicate the novel derivative 15 (ARN25499) as the most drug-like candidate of this novel chemical series of CDC42 inhibitors. Indeed, this compound showed improved in vitro metabolic stability compared to our previous leads ARN22089 and ARN25062,23 and optimal kinetic and thermodynamic solubility in both neutral and salt forms. Importantly, ARN25499 shows also a slightly improved in vivo oral bioavailability and a notable efficacy to inhibit tumor growth in a PDX tumor mouse model. Clearly, since substituted pyrimidine represents a privileged chemical scaffold extensively characterized for its multiple biological properties,34−38 additional target selectivity and off-target activity tests will be performed to move into advanced preclinical studies of ARN25499, as similarly already performed for the previous lead ARN22089.22
In conclusion, these promising findings elevate this compound as an additional lead drug candidate of our program on CDC42 inhibitors, which are now ready for further preclinical characterization and in vivo efficacy studies in other cancer models.
Experimental Section
Chemistry
Chemistry General Considerations
All the commercially available reagents and solvents were used as purchased from vendors without further purification. Dry solvents were purchased from Sigma-Aldrich. Automated column chromatography purifications were done using a Teledyne ISCO apparatus (CombiFlash Rf) with prepacked silica gel columns of different sizes (from 4 g up to 24 g) and mixtures of increasing polarity of cyclohexane and ethyl acetate (EtOAc) or dichloromethane (DCM) and methanol (MeOH). NMR data were collected on 400 or 600 MHz (1H) and 100 or 150 MHz (13C). Spectra were acquired at 300 K, using deuterated dimethyl sulfoxide (DMSO-d6) or deuterated chloroform (CDCl3) as solvents. For 1H NMR, data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, dd = double of doublets, t = triplet, q = quartet, m = multiplet), coupling constants (Hz), and integration. UPLC-MS analyses were run on a Waters ACQUITY UPLC-MS instrument consisting of a single quadrupole detector (SQD) mass spectrometer equipped with an electrospray ionization interface (ESI) and a photodiode array detector (PDA) from Waters Inc. (Milford, MA, USA). PDA range was 210–400 nm. The analyses were performed on an ACQUITY UPLC BEH C18 column (50 × 2.1 mmID, particle size 1.7 μm) with a VanGuard BEH C18 precolumn (5 × 2.1 mmID, particle size 1.7 μm) (for method 1, 2 and 3) and an ACQUITY UPLC HSS T3 (50 × 2.1 mmID, particle size 1.8 μM) with a VanGuard HSS T3 precolumn (5 × 2.1 mmID, particle size 1.8 μM) (for method 4). The mobile phase was 10 mM NH4OAc in H2O at pH 5 adjusted with AcOH (A) and 10 mM NH4OAc in MeCN-H2O (95:5) at pH 5 (B). Electrospray ionization in positive and negative modes was applied in the mass scan range of 100–500 Da. Depending on the analysis method used, a different gradient increasing the proportion of mobile phase B was applied. For analysis method 1, the mobile phase B proportion increased from 5 to 95% in 2.5 min. For analysis method 2, the mobile phase B proportion increased from 50 to 100% in 2.5 min. For analysis method 3, the mobile phase B proportion increased from 70 to 100% in 2.5 min. The analysis method 4, the mobile phase B portion increased from 0 to 50% in 2.5 min. The QC analysis was performed starting from a 10 mM stock solution of the test compound in DMSO-d6 and further diluted 20-fold with MeCN-H2O (1:1) for analysis. The analyses were performed on a Waters ACQUITY UPLC-MS system consisting of a single quadrupole detector mass spectrometer as described above. The analyses were run on an ACQUITY UPLC BEH C18 column (100 × 2.1 mmID, particle size 1.7 μm) with a VanGuard BEH C18 precolumn (5 × 2.1 mmID, particle size 1.7 μm). The mobile phase was 10 mM NH4OAc in H2O at pH 5 adjusted with AcOH (A) and 10 mM NH4OAc in MeCN-H2O (95:5) at pH 5 (B) with 0.5 mL/min as flow rate. A linear gradient was applied: 0–0.2 min: 10%B, 0.2–6.2 min: 10–90%B, 6.2–6.3 min: 90–100%B, 6.3–7.0 min: 100%B (QC method). High-resolution mass spectrometry (HRMS) for accurate mass measurements was performed on a Sciex TripleTOF High-resolution LC-MS using a Waters UPLC ACQUITY chromatographic system (from Waters Inc., Milford, MA, USA) coupled to a TripleTOF 5600+ Mass Spectrometer (from Sciex, Warrington, UK) equipped with a NanoSpray III Ion source. The analyses were run on an ACQUITY UPLC BEH C18 column (50 × 2.1 mmID, particle size 1.7 μm), using H2O + 0.1% HCOOH (A) and MeCN + 0.1% HCOOH as mobile phase. All final compounds displayed ≥95% purity as determined by NMR and UPLC-MS (UV at 215 nm) analysis unless otherwise indicated. Compounds 27, 31a–i, 34–36, 38a–j were purchased from Sigma-Aldrich or Fluorochem and used as such without further purification. Intermediates 28–30, 41, 42–43a were synthesized as previously reported.22,23
General Procedure 1: SNAr
Method A
To a DCM solution (0.5M) of cyanuric chloride (1.3 equiv), corresponding aniline (1.0 equiv) was dropwise added at 0 °C, followed by DIPEA (1.1 equiv). After 10 min, H2O was slowly added at the same temperature to quench the excess reagent, and the resulting mixture was left to stir for 10–15 min. The organic layer was separated and dried with Na2SO4, the solvent was evaporated, and the resulting crude was purified by silica gel chromatography.
Method B
A solution (0.15 M) of 6-phenyl-2,4-dichloropyrimidine 42 (1.00 equiv) with a suitable aniline of type 38 (1.00 mmol) in THF was cooled to −60 °C. To this solution was added dropwise LiHMDS (1.0 M in THF, 2.5 equiv). After the complete conversion of the starting material, water was added, and the mixture was extracted with EtOAc. The combined organic layers were washed with brine, dried (Na2SO4), and concentrated in vacuo. Final normal phase chromatographic purification (Cyclohexane/EtOAc) provided the desired products.
General Procedure 2: Suzuki Cross-Coupling
To a degassed 1,4-dioxane solution (0.13 M) of heteroaryl halide (1.0 equiv), suitable boronic acid (1.2 equiv), Pd(dppf)Cl2·DCM (0.05 equiv) and K2CO3 (2 M)aq (2.0 equiv) were added. The resulting mixture was sparged with Argon for a further 10 min and heated at different temperatures in dependence of the substrate: 80 °C for compound 28, 100 °C for compound 30, and 120 °C for compound of type 42. Then, H2O and EtOAc were added and separated; aqueous layers were extracted two times with EtOAc and collected organic layers were dried over Na2SO4. The solvent was evaporated and the resulting crude was purified by silica gel chromatography.
General Procedure 3: Buchwald Reaction for the Obtainment of Compounds 39a–j
A mixture of compound 37 (1 equiv), proper aniline 38a–j (1.2 equiv), Pd(OAc)2 (5 mol %), (±)-BINAP or Xantphos (5 mol %), Cs2CO3 (1.5 equiv) in 1,4-dioxane dry (0.15M) stirred at 120 °C until complete consumption of starting material. After that, water and brine were added, the aqueous layer was extracted with EtOAc, and collected organic layers were dried over Na2SO4, filtered, and concentrated under a vacuum. Intermediate 5a-j were purified by silica.
General Procedure 4: Double C–C Reduction
Method A
Under N2 atmosphere, a suspension of intermediate 32a–i, 39a–j, 43b–c (1 equiv), ammonium formate (6 equiv), Pd(OH)2/C (20% of starting material weight) in MeOH dry (0.04 M solution) was stirred at reflux temperature until reaction completion. The catalyst was filtered off through a Celite coarse patch, and the resulting filtrate concentrated to dryness at low pressure. Final chromatographic normal phase purification (cyclohexane/EtOAc) afforded pure desired products.
Method B
Reduction with Et3SiH. Under the N2 atmosphere, Et3SiH (10 equiv) was dropwise added to a mixture of substrate 32i (1.0 equiv) and Pd/C (20% w/w of starting material weight) in EtOH (0.5 M). When no intermediate was detected by UPLC or TLC, the catalyst was filtered off through a Celite coarse patch, and the resulting filtrate concentrated to dryness at low pressure. The resulting crude was purified by silica gel chromatography.
General Procedure 5: Boc Removal
Under the N2 atmosphere, HCl (4 M in dioxane) (10 equiv) was added to a solution of intermediate of type 6, 16, 17 (1 equiv) in 1,4-dioxane dry (0.06 M). After completion of reaction, NaOH (2 M) aq was added until pH = 7, an aqueous layer was extracted with EtOAc, and collected organic layers were dried over Na2SO4, filtered, and concentrated under vacuum. Final purification by alumina (DCM/MeOH or DCM/MeOH·NH3) afforded the pure desired compound of type 7 and 18.
General Procedure 6: Methyl and Boc Removal
Under the N2 atmosphere, BBr3 (1 M in DCM) (6 equiv) was added to a solution of intermediate of type 6 (1 equiv) in CHCl3 dry (0.06 M). After completion of the reaction, MeOH was added, and the mixture was concentrated under vacuum. The crude was purified by trituration with EtOAc, obtaining pure products.
4-(Piperidin-4-yl)-6-(pyrimidin-5-yl)-N-(3-(trifluoromethyl)phenyl)-1,3,5-triazin-2-amine (2)
Compound 2 was synthesized following general procedure 5 using compound 33a (50 mg, 0.10 mmol), HCl 4 M 1,4-dioxane solution (0.25 mL, 1.0 mmol), and 1,4-dioxane (1.7 mL). After 24 h, no conversion occurred; thus, HCl 4 M (0.28 mL) was added, and the reaction mixture was stirred overnight. After a further 24 h, HCl 4 M (0.28 mL) was again added, and a standard workup was performed after 24 h. Purification by silica (elution by a gradient from 100/0 to 90/10 DCM/MeOH(NH3 1N)) afforded a white solid as the product (25 mg, 62% yield). UPLC-MS: tR = 0.52 min (method 1). MS (ESI) m/z: 402.0 [M + H]+, calcd for C19H19F3N7 [M + H]+: 402.4. HRMS (ESI) m/z: 402.1649 calcd for C19H19F3N7 [M + H]+; found m/z: 402.1660. 1H NMR (600 MHz, DMSO-d6) δ 10.74 (s, 1H), 9.58 (s, 2H), 9.41 (s, 1H), 8.57–8.29 (m, 1H), 8.00 (s, 1H), 7.63 (s, 1H), 7.44 (d, J = 7.7 Hz, 1H), 3.04 (dt, J = 12.3, 3.4 Hz, 2H), 2.81 (s, 1H), 2.61 (td, J = 12.0, 2.5 Hz, 2H), 2.06–1.86 (m, 2H), 1.79–1.65 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 182.0 (Cq), 167.5 (Cq), 164.0 (Cq), 160.8 (CH), 156.5 (CH, 2C), 139.6 (Cq), 130.0 (CH), 129.4 (Cq), 129.3 (q2, JCF = 31.7 Hz, Cq), 124.2 (q1, JCF = 272.8 Hz), 123.8 (CH), 119.4 (CH), 116.6 (CH), 45.8 (CH2, 2C), 44.9 (CH), 30.8 (CH2, 2C). 19F NMR (565 MHz, DMSO-d6) δ−60.32.
4-(Isoquinolin-5-yl)-6-(piperidin-4-yl)-N-(3-(trifluoromethyl)phenyl)-1,3,5-triazin-2-amine (3)
Compound 3 was synthesized following general procedure 5 using compound 33b (68 mg, 0.123 mmol), HCl 4 M 1,4-dioxane solution (0.31 mL, 1.23 mmol), 1,4-dioxane (2.0 mL). After 16 h, a standard workup has been performed. Purification by silica (elution by a gradient from 100/0 to 90/10 DCM/MeOH(NH3 1N)) afforded a yellowish unclean solid that was subjected to trituration with petroleum ether/EtOAc/MeOH 85:13:2. A pure white solid was collected by trituration (27 mg, 49% yield). UPLC-MS: tR = 1.91 min (method 1). MS (ESI) m/z: 451.0 [M + H]+, calcd for C24H22F3N6 [M + H]+: 451.5. HRMS (ESI) m/z: 451.1853 calcd for C24H22F3N6 [M + H]+; found m/z: 451.1861. 1H NMR (600 MHz, DMSO-d6) δ 10.68 (bs, 1H), 9.43 (s, 1H), 8.89 (bs, 1H), 8.58 (d, J = 6.7 Hz, 2H), 8.52–8.34 (m, 2H), 8.02 (bs, 1H), 7.86 (t, J = 7.7 Hz, 1H), 7.60 (t, J = 7.9 Hz, 1H), 7.42 (d, J = 7.7 Hz, 1H), 3.09 (dd, J = 12.2, 3.4 Hz, 2H), 2.87 (t, J = 12.2 Hz, 1H), 2.67 (td, J = 12.1, 2.5 Hz, 2H), 2.12–1.95 (m, 2H), 1.80 (dd, J = 12.1, 4.0 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 181.8 (Cq), 172.3 (Cq), 164.5 (Cq), 153.7 (CH), 144.4 (CH, 2C), 140.4 (Cq), 134.2 (CH), 133.5 (Cq), 132.7 (Cq), 132.3 (CH), 130.3 (CH), 129.9 (q2, JCF = 31.2 Hz, Cq), 129.2 (Cq), 127.2 (CH), 124.7 (q1, JCF = 272.8 Hz, Cq), 124.2 (CH), 119.7 (CH), 119.0 (CH), 117.0 (CH), 46.0 (CH2, 2C), 45.1 (CH), 30.9 (CH2, 2C). 19F NMR (565 MHz, DMSO-d6) δ−60.28.
Ethyl-(1H-indol-5-yl)-6-(piperidin-4-yl)-N-(3-(trifluoromethyl)phenyl)-1,3,5-triazin-2-amine (4)
Compound 4 was synthesized following general procedure 5 using compound 33c (100 mg, 0.18 mmol), HCl 4 M 1,4-dioxane solution (0.45 mL, 1.8 mmol), and 1,4-dioxane (3.0 mL). After 3.5 h, a standard workup has been performed. Purification by silica (elution by a gradient from 95/5 to 80/20 DCM/MeOH (1N NH3)) afforded not pure compound that has been subjected to trituration with petroleum ether giving the pure product as a white solid (40 mg, 49% yield). UPLC-MS: tR = 1.18 min (method 2). MS (ESI) m/z: 453.0 [M + H]+, calcd for C24H24F3N6 [M + H]+: 453.2. HRMS (ESI) m/z: 453.2009 calcd for C24H24F3N6 [M + H]+; found m/z: 453.2018. 1H NMR (400 MHz, DMSO-d6) δ 10.41 (s, 1H), 8.75 (d, J = 1.6 Hz, 1H), 8.61 (s, 1H), 8.28 (dd, J = 8.8, 1.7 Hz, 1H), 7.98 (d, J = 8.3 Hz, 1H), 7.66–7.54 (m, 2H), 7.43 (d, J = 3.1 Hz, 1H), 7.40 (d, J = 7.8 Hz, 1H), 6.60 (d, J = 3.1 Hz, 1H), 3.85 (s, 3H), 3.14–3.02 (m, 2H), 2.78 (t, J = 11.6 Hz, 1H), 2.72–2.57 (m, 2H), 2.06–1.89 (m, 2H), 1.85–1.68 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 181.1 (Cq), 171.4 (Cq), 164.1 (Cq), 140.3 (Cq), 138.8 (Cq), 131.3 (CH), 129.8 (CH), 129.4 (q2, JCF = 31.2 Hz, Cq), 127.3 (q1, JCF = 214.4 Hz, Cq), 125.2 (Cq), 123.4 (CH), 121.8 (CH), 121.3 (CH), 118.7 (CH), 116.2 (CH), 109.7 (CH), 102.0 (CH), 45.8 (CH2, 2C), 45.0 (CH), 32.7 (CH3), 30.8 (CH2, 2C). 19F NMR (565 MHz, DMSO-d6) δ −61.26.
4-(1H-Indazol-5-yl)-6-(piperidin-4-yl)-N-(3-(trifluoromethyl)phenyl)-1,3,5-triazin-2-amine (Compound 5)
Compound 5 was synthesized following general procedure 5 using compound 33d (27 mg, 0.05 mmol), HCl 4 M 1,4-dioxane solution (0.125 mL, 0.5 mmol), and 1,4-dioxane dry (0.08 mL). After 6 h, a standard workup has been performed. Purification by silica (elution by a gradient from 99/1 to 85/15 DCM/MeOH(NH3 1N)) afforded a white solid as the product (11 mg, 50% yield). UPLC-MS: tR = 0.76 min (method 2). MS (ESI) m/z: 440.0 [M + H]+, calcd for C22H21F3N7 [M + H]+: 439.5. HRMS (ESI) m/z: 440.1805 calcd for C22H21F3N7 [M + H]+; found m/z: 440.1814. 1H NMR (400 MHz, DMSO-d6) δ 13.37 (s, 1H), 10.49 (s, 1H), 8.93 (s, 1H), 8.50 (s, 1H), 8.43 (dd, J = 8.8, 1.6 Hz, 1H), 8.28 (s, 1H), 8.03 (d, J = 8.1 Hz, 1H), 7.67 (d, J = 8.9 Hz, 1H), 7.62 (t, J = 8.1 Hz, 1H), 7.42 (d, J = 7.7 Hz, 1H), 3.15–3.06 (m, 2H), 2.88–2.78 (m, 1H), 2.74–2.63 (m, 2H), 2.05–1.97 (m, 2H), 1.79 (dd, J = 12.0, 3.9 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 181.1 (Cq), 170.7 (Cq), 164.2 (Cq), 141.8 (Cq), 140.1 (Cq), 135.2 (CH), 129.8 (CH), 129.4 (q2, JCF = 31.4 Hz, Cq), 128.1 (Cq), 125.7 (CH), 124.3 (q1, JCF = 272.3 Hz, Cq), 123.6 (CH), 123.1 (Cq), 122.3 (CH), 118.9 (CH), 116.4 (CH), 110.3 (CH), 45.4 (CH2, 2C), 44.3 (CH), 30.2 (CH2, 2C). 19F NMR (565 MHz, DMSO-d6) δ−60.28.
4-(Benzofuran-3-yl)-6-(piperidin-4-yl)-N-(3-(trifluoromethyl)phenyl)-1,3,5-triazin-2-amine (Compound 6)
Compound 6 was synthesized following general procedure 5 using 33e (102 mg, 0.19 mmol), HCl 4 M 1,4-dioxane solution (0.48 mL, 1.90 mmol), 1,4-dioxane (3.2 mL). After 7 h, HCl 4 M (0.24 mL) was added again and the reaction mixture was stirred overnight. Further, HCl 4 M (0.24 mL) has been added and after 16 h standard workup has been performed. The crude product was purified by neutral alumina (elution by gradient DCM/EtOH 100:0 to 90:10) giving the pure product as a white solid (3.5 mg, 4% yield). UPLC-MS: tR = 1.27 min (method 2). MS (ESI) m/z: 440.0 [M + H]+, calcd for C23H21F3N5O [M + H]+: 440.5. 1H NMR (400 MHz, DMSO-d6) δ 10.50 (s, 1H), 8.85 (s, 1H), 8.34 (d, J = 54.9 Hz, 2H), 8.03 (d, J = 8.2 Hz, 1H), 7.72 (d, J = 8.1 Hz, 1H), 7.63 (t, J = 8.0 Hz, 1H), 7.51–7.32 (m, 3H), 3.10–3.02 (m, 2H), 2.84–2.73 (m, 1H), 2.68–2.56 (m, 2H), 2.05–1.91 (m, 2H), 1.81–1.67 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 181.3 (Cq), 167.8 (Cq), 163.9 (Cq), 155.5 (Cq), 150.7 (CH), 139.9 (Cq), 129.9 (CH), 129.4 (q2, JCF = 31.2 Hz, Cq), 125.2 (CH), 124.6 (Cq), 124.3 (q1, JCF = 273.2 Hz, Cq), 123.9 (CH, 2C), 122.7 (CH), 119.8 (Cq), 119.2 (CH), 116.5 (CH), 111.8 (CH), 45.9 (CH2, 2C), 44.9 (CH), 30.9 (CH2, 2C). 19F NMR (565 MHz, DMSO-d6) δ−60.1.
4-(1-Methyl-1H-pyrazol-4-yl)-6-(piperidin-4-yl)-N-(3-(trifluoromethyl)phenyl)-1,3,5-triazin-2-amine (Compound 7)
Compound 7 was synthesized following general procedure 5 using compound 33f (75 mg, 0.14 mmol), HCl, and 4 M 1,4-dioxane solution (0.35 mL, 1.4 mmol), 1,4-dioxane (2.3 mL). After 16 h, a standard workup has been performed. Purification by silica (elution by a gradient from 100/0 to 90/10 DCM/MeOH(1N NH3)) afforded the pure product as a white solid (43 mg, 76% yield). UPLC-MS: tR = 1.76 min (method 1). MS (ESI) m/z: 404.0 [M + H]+, calcd for C19H21F3N7 [M + H]+: 403.4. HRMS (ESI) m/z: 404.1805 calcd for C19H21F3N7 [M + H]+; found m/z: 404.1815. 1H NMR (600 MHz, DMSO-d6) δ 10.37 (s, 1H), 8.42 (s, 1H), 8.41 (s, 1H), 8.07–7.96 (m, 2H), 7.58 (t, J = 8.0 Hz, 1H), 7.38 (d, J = 6.0 Hz, 1H), 3.93 (s, 3H), 3.04 (dt, J = 12.3, 3.4 Hz, 2H), 2.72–2.67 (m, 1H), 2.60 (td, J = 12.1, 2.5 Hz, 2H), 1.97–1.85 (m, 2H), 1.69 (qd, J = 12.1, 3.9 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 181.2 (Cq), 167.1 (Cq), 163.9 (Cq), 140.2 (Cq), 139.3 (CH), 133.1 (CH), 129.8 (CH), 129.3 (q2, JCF = 31.3 Hz, Cq), 124.3 (q1, JCF = 272.4 Hz, Cq), 123.4 (CH), 120.8 (Cq), 118.7 (q3, JCF = 4.0 Hz, CH), 116.1 (q3, JCF = 4.2 Hz, CH), 45.7 (CH2), 44.7 (CH), 38.6 (CH3), 30.6 (CH2). 19F NMR (565 MHz, DMSO-d6) δ−60.31.
4-(1-Methyl-1H-imidazol-5-yl)-6-(piperidin-4-yl)-N-(3-(trifluoromethyl)phenyl)-1,3,5-triazin-2-amine (Compound 8)
Compound 8 was synthesized following general procedure 5 using compound 33g (83 mg, 0.17 mmol), HCl, and 4 M 1,4-dioxane solution (0.42 mL, 1.7 mmol) in 1,4-dioxane (2.8 mL). After 2 days, a standard workup has been performed. Purification by silica (elution by a gradient from 98/2 to 90/10 DCM/MeOH(NH3 1N)) afforded unclean compound, that was subjected to trituration with pentane/EtOAc 9/1 furnishing a white solid as the product (48 mg, 72% yield). UPLC-MS: tR = 1.65 min (method 2). MS (ESI) m/z: 404.0 [M + H]+, calcd for C19H21F3N7 [M + H]+: 404.4. HRMS (ESI) m/z: 404.1805 calcd for C19H21F3N7 [M + H]+; found m/z: 404.1815. 1H NMR (600 MHz, DMSO-d6) δ 10.41 (s, 1H), 8.38 (s, 1H), 7.93 (s, 1H), 7.91 (s, 1H), 7.85 (s, 1H), 7.59 (t, J = 8.0 Hz, 1H), 7.40 (d, J = 7.7 Hz, 1H), 4.04 (s, 3H), 3.14–2.97 (m, 2H), 2.73 (t, J = 11.4 Hz, 1H), 2.63 (t, J = 12.1 Hz, 2H), 2.01–1.89 (m, 2H), 1.70 (qd, J = 12.2, 3.9 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 180.9 (Cq), 164.7 (Cq), 163.6 (Cq), 143.8 (CH), 139.9 (Cq), 136.1 (CH), 129.8 (CH), 129.3 (q2, JCF = 31.7 Hz, Cq), 129.0 (Cq), 124.2 (q1, JCF = 271.8 Hz), 123.8 (CH), 119.1 (CH), 116.4 (CH), 45.6 (CH2, 2C), 44.5 (CH), 35.2 (CH3), 30.4 (CH2, 2C). 19F NMR (565 MHz, DMSO-d6) δ−60.28.
4-(Piperidin-4-yl)-6-(1H-pyrrol-2-yl)-N-(3-(trifluoromethyl)phenyl)-1,3,5-triazin-2-amine (Compound 9)
Compound 9 was synthesized following general procedure 5 using compound 33h (60 mg, 0.10 mmol), HCl 4 M 1,4-dioxane solution (0.25 mL, 1.0 mmol), and 1,4-dioxane (1.7 mL). After 2 days, a standard workup has been performed. Purification by silica (elution by a gradient from 99:1 to 90:10 DCM/MeOH(NH3 1N)) afforded a white solid as the product (15 mg, 38% yield). UPLC-MS: tR = 1.91 min (method 2). MS (ESI) m/z: 389.0 [M + H]+, calcd for C19H20F3N6 [M + H]+: 389.4. HRMS (ESI) m/z: 389.1696 calcd for C19H20F3N6 [M + H]+; found m/z: 389.1705. 1H NMR (600 MHz, DMSO-d6) δ 11.66 (s, 1H), 10.26 (s, 1H), 8.47 (s, 1H), 8.11–7.92 (m, 1H), 7.57 (t, J = 8.0 Hz, 1H), 7.36 (d, J = 7.7 Hz, 1H), 7.08–7.02 (m, 2H), 6.29–6.24 (m, 1H), 3.05 (dd, J = 12.1, 3.4 Hz, 2H), 2.74–2.67 (m, 1H), 2.62 (dd, J = 12.0, 2.5 Hz, 2H), 1.99–1.87 (m, 2H), 1.74 (dd, J = 12.2, 3.8 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 180.8 (Cq), 164.5 (Cq), 163.8 (Cq), 140.4 (Cq), 129.8 (CH), 129.3 (q2, JCF = 31.7 Hz, Cq), 128.7 (Cq), 124.3 (q1, JCF= 271.8 Hz, Cq), 124.2 (CH), 123.3 (CH), 118.5 (q3, JCF = 3.5 Hz, CH), 116.0 (CH), 113.7 (CH), 110.3 (CH), 45.7 (CH2, 2C), 44.7 (CH), 30.5 (CH2, 2C). 19F NMR (565 MHz, DMSO-d6) δ−60.3.
4-(Piperidin-4-yl)-6-(pyrrolidin-2-yl)-N-(3-(trifluoromethyl)phenyl)-1,3,5-triazin-2-amine (Compound 10)
Compound 10 was synthesized following general procedure 5 using compound 33h’ (123 mg, 0.21 mmol), HCl 4 M 1,4-dioxane solution (0.53 mL, 2.1 mmol), 1,4-dioxane (3.5 mL). After 16 h, no conversion occurred; thus, HCl 4 M (3.5 mL) was added and the reaction mixture was stirred over the weekend. Then, a standard workup has been performed. Purification by silica (elution by gradient 100/0 to 90/10 DCM/MeOH (1 N NH3)) afforded a yellowish unclean solid that was subjected to trituration with pentane (1.5 mL). The pure product was collected as a white solid after filtration (16 mg, 20% yield). UPLC-MS: tR = 1.68 min (method 1). MS (ESI) m/z: 393.0 [M + H]+, calcd for C19H24F3N6 [M + H]+: 393.4. 1H NMR (600 MHz, DMSO-d6) δ 10.46 (s, 1H), 8.40 (s, 1H), 7.92 (d, J = 8.3 Hz, 1H), 7.56 (t, J = 8.0 Hz, 1H), 7.37 (d, J = 7.7 Hz, 1H), 3.99 (s, 1H), 3.10–3.04 (m, 1H), 3.00 (dt, J = 12.3, 3.5 Hz, 2H), 2.86–2.79 (m, 1H), 2.72–2.63 (m, 1H), 2.56 (td, J = 12.1, 2.5 Hz, 2H), 2.16–2.09 (m, 1H), 1.90–1.78 (m, 3H), 1.76–1.69 (m, 2H), 1.63 (qd, J = 12.2, 4.0 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 181.2 (Cq), 173.0 (Cq), 163.8 (Cq), 140.0 (Cq), 129.8 (CH), 129.4 (q2, JCF = 31.7 Hz, Cq), 124.2 (q1, JCF = 272.8 Hz, Cq), 123.4 (CH), 118.9 (CH), 116.2 (CH), 62.9 (CH), 46.9 (CH2), 45.8 (CH2, 2C), 44.8 (CH), 32.0 (CH2), 30.9 (CH2, 2C), 25.8 (CH2). 19F NMR (565 MHz, DMSO-d6) δ−60.3.
4-(1,2,3,6-Tetrahydropyridin-4-yl)-6-(thiophen-3-yl)-N-(3-(trifluoromethyl)phenyl)-1,3,5-triazin-2-amine (Compound 11)
Compound 11 was synthesized following general procedure 5 using compound 32i (48 mg, 0.095 mmol), HCl 4 M 1,4-dioxane solution (0.25 mL, 0.10 mmol), and 1,4-dioxane (1.6 mL). After 16 h, no conversion occurred; thus, HCl 4 M (0.78 mL) was added and the reaction mixture was stirred over the weekend. Then, standard workup has been. Purification by silica (elution by a gradient from 99/1 to 96/4 DCM/MeOH(NH3 1N)) afforded unclean compound, that was subjected to trituration with pentane/EtOAc 1/1 furnishing a white solid as the product (15 mg, 39% yield). UPLC-MS: tR = 0.90 min (method 2). MS (ESI) m/z: 404.0 [M + H]+, calcd for C19H17F3N5S [M + H]+: 404.4. 1H NMR (400 MHz, DMSO-d6) δ 10.47 (s, 1H), 8.53 (dd, J = 3.0, 1.2 Hz, 1H), 8.50 (s, 1H), 7.98 (s, 1H), 7.83 (dd, J = 5.0, 1.2 Hz, 1H), 7.72 (t, J = 5.0, 3.1 Hz, 1H), 7.60 (t, J = 8.0 Hz, 1H), 7.53–7.46 (m, 1H), 7.40 (d, J = 7.7 Hz, 1H), 3.62–3.54 (m, 2H), 2.99 (t, J = 5.6 Hz, 2H), 2.60–2.53 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 171.6, 167.4, 164.8, 140.6, 140.6, 135.6, 134.1, 131.4, 130.3, 129.8 (q2, JCF = 31.7 Hz), 128.1, 127.3, 124.8 (q1, JCF = 271.8 Hz), 124.0, 119.4 (q3, JCF = 3.7 Hz, CH), 116.73 (q3, JCF = 4.5 Hz, CH), 45.0 (CH2), 42.3 (CH2), 24.7 (CH2). 19F NMR (565 MHz, DMSO-d6) δ−60.29.
N1,N1-Dimethyl-N3-(4-phenyl-6-(piperidin-4-yl)pyrimidin-2-yl)benzene-1,3-diamine (Compound 12)
Title compound was synthesized following the general procedure 5 using intermediate 40a (58 mg, 0.12 mmol) and HCl (4 M in dioxane) (0.32 mL) in 1,4-dioxane dry (2.1 mL). Purification by alumina (elution by a gradient from 100/0 to 98/2 DCM/MeOH·NH3 1N) afforded pure product 12 (15.5 mg, 33% yield). UPLC-MS: tR = 1.08 min (method 1). MS (ESI) m/z: 374.3 [M + H]+, calcd for C23H28N5 [M + H]+: 374.5. HRMS (ESI) m/z: 374.2339 calcd for C23H28N5 [M + H]+; found m/z: 374.2349. 1H NMR (400 MHz, DMSO-d6) δ 9.31 (s, 1H), 8.23–8.10 (m, 2H), 7.58 (s, 1H), 7.55–7.49 (m, 3H), 7.26 (s, 1H), 7.08 (t, J = 7.9 Hz, 1H), 7.06 (s, 1H), 6.35 (dt, J = 7.2, 2.3 Hz, 1H), 3.10–3.00 (m, 3H), 2.92 (s, 6H), 2.69 (tt, J = 11.8, 3.8 Hz, 1H), 2.59 (td, J = 12.0, 2.5 Hz, 2H), 1.83 (d, J = 12.1 Hz, 2H), 1.68 (qd, J = 12.2, 4.0 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 175.2 (Cq), 163.7 (Cq), 160.2 (Cq), 150.9 (Cq), 141.6 (Cq), 137.2 (Cq), 130.6 (CH), 128.7 (CH, 2C), 126.9 (CH, 2C), 107.4 (CH), 105.9 (CH), 105.1 (CH), 103.2 (CH), 46.2 (CH2, 2C), 44.4 (CH), 40.3 (CH3, 2C), 31.8 (CH2, 2C).
4-Phenyl-6-(piperidin-4-yl)-N-(3-(trifluoromethyl)phenyl)pyrimidin-2-amine (Compound 13)
The title compound was synthesized following the general procedure 5 previously described using intermediate 40b (45 mg, 0.09 mmol) and HCl (4 M in dioxane) (0.22 mL) in 1,4-dioxane dry (1.5 mL). Purification by alumina (elution by a gradient from 100/0 to 90/10 DCM/MeOH) afforded pure product 13 (16.2 mg, 45% yield). UPLC-MS: tR = 1.17 min (method 2). MS (ESI) m/z: 399.2 [M + H]+, calcd for C22H21F3N4 [M + H]+: 399.2. HRMS (ESI) m/z: 399.1791 calcd for C22H21F3N4 [M + H]+; found m/z: 399.1794. 1H NMR (400 MHz, DMSO-d6) δ 9.95 (s, 1H), 8.60 (bs, 1H), 8.18 (m, 2H), 7.96 (dd, J = 8.3, 1.4 Hz, 1H), 7.30–7.26 (m, 4H), 7.40 (s, 1H), 7.27 (d, J = 7.7 Hz, 1H), 3.06 (dt, J = 12.2, 3.2 Hz, 2H), 2.74 (tt, J = 11.9, 3.2 Hz, 1H), 2.60 (td, J = 12.0, 2.5 Hz, 2H), 1.85 (d, J = 12.5 Hz, 2H), 1.70 (qd, J = 12.2, 4.0 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 175.6 (Cq), 163.9 (Cq), 159.8 (Cq), 141.7 (Cq), 136.8 (Cq), 130.8 (CH), 129.5 (CH), 129.3 (q2, JCF = 30.9 Hz, Cq), 128.8 (CH, 2C), 126.9 (CH, 2C), 124.4 (q1, JCF = 271.8 Hz, Cq), 122.0 (CH), 117.1 (q3, JCF = 3.6 Hz, CH), 114.5 (q3, JCF = 4.3 Hz, CH), 106.3 (CH), 46.2 (CH2, 2C), 44.3 (CH), 31.8 (CH2, 2C). 19F NMR (376 MHz, DMSO-d6) δ – 61.3.
4-Phenyl-6-(piperidin-4-yl)-N-(3-(trifluoromethoxy)phenyl)pyrimidin-2-amine (Compound 14)
The title compound was synthesized following the general procedure 5 previously described using intermediate 40c (70 mg, 0.14 mmol) and HCl (4 M in dioxane) (0.34 mL) in 1,4-dioxane dry (2.3 mL). Purification by alumina (elution by a gradient from 100/0 to 95/5 DCM/MeOH) afforded pure product 14 (23.1 mg, 41% yield). UPLC-MS: tR = 2.41 min (method 1). MS (ESI) m/z: 415.0 [M + H]+, calcd for C22H22F3N4O [M + H]+: 415.2. HRMS (ESI) m/z: 415.1740 calcd for C22H22F3N4O [M + H]+; found m/z: 415.1747. 1H NMR (400 MHz, DMSO-d6) δ 9.91 (s, 1H), 8.22 (s, 1H), 8.17 (m, 2H), 7.70 (dd, J = 8.4, 2.0 Hz, 1H), 7.57–7.51 (m, 3H), 7.41 (t, J = 8.2 Hz, 1H), 7.38 (s, 1H), 6.86 (d, J = 8.0 Hz, 1H), 3.05 (dt, J = 12.0, 2.7 Hz, 2H), 2.72 (tt, J = 11.7, 3.7 Hz, 1H), 2.60 (td, J = 12.1, 2.5 Hz, 2H), 1.84 (d, J = 12.4 Hz, 2H), 1.69 (qd, J = 12.2, 3.9 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 175.6 (Cq), 164.0 (Cq), 159.7 (Cq), 148.7 (Cq), 142.7 (Cq), 136.9 (Cq), 130.8 (CH), 130.0 (CH), 128.8 (CH, 2C), 127.0 (CH, 2C), 120.6 (q1, JCF = 255.6 Hz, Cq), 117.2 (CH), 112.9 (CH), 110.4 (CH), 106.2 (CH), 46.2 (CH2, 2C), 44.3 (CH), 31.7 (CH2, 2C). 19F NMR (565 MHz, DMSO-d6) δ – 56.5.
N-(3,4-Dimethoxyphenyl)-4-phenyl-6-(piperidin-4-yl)pyrimidin-2-amine (Compound 15 – ARN25499)
The title compound was synthesized following the general procedure 5 using intermediate 40d (30 mg, 0.06 mmol) and HCl (4 M in dioxane) (0.15 mL) in 1,4-dioxane dry (1.0 mL). Purification by alumina (elution by a gradient from 100/0 to 95/5 DCM/MeOH) afforded pure compound 15 – ARN25499 (12.4 mg, 38% yield). UPLC-MS: tR = 1.79 min (method 1). MS (ESI) m/z: 391.0 [M + H]+, calcd for C23H27N4O2 [M + H]+: 391.2. HRMS (ESI) m/z: 391.2129 calcd for C23H27N4O2 [M + H]+; found m/z: 391.2146. 1H NMR (400 MHz, DMSO-d6) δ 9.35 (s, 1H), 8.21–8.11 (m, 2H), 7.78 (d, J = 2.4 Hz, 1H), 7.57–7.49 (m, 3H), 7.26 (dd, J = 8.6, 2.4 Hz, 1H), 7.25 (s, 1H), 6.90 (d, J = 8.7 Hz, 1H), 3.79 (s, 3H), 3.72 (s, 3H), 3.05 (d, J = 12.0 Hz, 2H), 2.70 (tt, J = 11.9, 4.4 Hz, 1H), 2.59 (td, J = 12.0, 2.5 Hz, 2H), 1.82 (d, J = 12.4 Hz, 2H), 1.70 (qd, J = 12.2, 4.0 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 175.2 (Cq), 163.7 (Cq), 160.1 (Cq), 148.5 (Cq), 143.4 (Cq), 137.2 (Cq), 134.8 (Cq), 130.6 (CH), 128.8 (CH, 2C), 126.9 (CH, 2C), 112.4 (CH), 110.4 (CH), 105.0 (CH), 104.3 (CH), 55.9 (CH3), 55.3 (CH3), 46.2 (CH2, 2C), 44.3 (CH), 31.8 (CH2, 2C).
Methyl 2-Methoxy-5-((4-phenyl-6-(piperidin-4-yl)pyrimidin-2-yl)amino)benzoate (Compound 16)
The title compound was synthesized following the general procedure 5 previously described using intermediate 40e (51.8 mg, 0.1 mmol) and HCl (4 M in dioxane) (0.25 mL) in 1,4-dioxane dry (1.7 mL). Purification by alumina (elution by a gradient from 100/0 to 95/5 DCM/MeOH) afforded pure compound 16 (17.6 mg, 42% yield). UPLC-MS: tR = 1.79 min (method 1). MS (ESI) m/z: 419.1 [M + H]+, calcd for C24H27N4O3 [M + H]+: 419.2. HRMS (ESI) m/z: 419.2078 calcd for C24H27N4O3 [M + H]+; found m/z: 419.2089. 1H NMR (400 MHz, DMSO-d6) δ 9.56 (s, 1H), 8.51 (d, J = 2.8 Hz, 1H), 8.18 (m, 2H), 7.85 (dd, J = 9.0, 2.8 Hz, 1H), 7.58–7.48 (m, 3H), 7.29 (s, 1H), 7.13 (d, J = 9.1 Hz, 1H), 3.82 (s, 3H), 3.80 (s, 3H), 3.05 (dt, J = 12.1, 3.2 Hz, 2H), 2.70 (tt, J = 11.8, 3.7 Hz, 1H), 2.59 (td, J = 12.1, 2.5 Hz, 2H), 1.83 (d, J = 12.8 Hz, 2H), 1.69 (qd, J = 12.2, 4.0 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 175.5 (Cq), 166.0 (Cq), 163.7 (Cq), 160.0 (Cq), 153.0 (Cq), 137.0 (Cq), 133.8 (Cq), 130.7 (CH), 128.8 (CH, 2C), 126.9 (CH, 2C), 123.9 (CH), 121.1 (CH), 119.4 (Cq), 113.2 (CH), 105.3 (CH), 56.1 (CH3), 51.8 (CH3), 46.2 (CH2, 2C), 44.3 (CH), 31.8 (CH2, 2C).
4-((4-Phenyl-6-(piperidin-4-yl)pyrimidin-2-yl)amino)benzene-1,2-diol Hydrobromide (Compound 17)
The title compound was synthesized following the general procedure 6 previously described using intermediate 40d (66 mg, 0.13 mmol) and BBr3 (1 M in DCM) (0.78 mL, 0.78 mmol) in CHCl3 dry (2.2 mL). Purification by trituration with EtOAc (1.5 mL) afforded pure compound 8a (16.6 mg, 34% yield). UPLC-MS: tR = 2.48 min (method 4). MS (ESI) m/z: 363.0 [M + H]+, calcd for C21H23N4O2 [M + H]+: 363.2. 1H NMR (400 MHz, DMSO-d6) δ 9.18 (s, 1H), 8.64 (d, J = 10.4 Hz, 1H), 8.37 (q, J = 10.5 Hz, 1H), 8.19–8.06 (m, 2H), 7.61–7.48 (m, 3H), 7.22 (d, J = 2.5 Hz, 1H), 7.21 (s, 1H), 7.08 (dd, J = 8.5, 2.5 Hz, 1H), 6.67 (d, J = 8.5 Hz, 1H), 3.41 (d, J = 12.9 Hz, 2H), 3.12–3.03 (m, 2H), 2.96 (tt, J = 11.4, 3.2 Hz, 1H), 2.13 (dd, J = 14.6, 3.7 Hz, 2H), 2.03–1.87 (m, 2H).
2-Hydroxy-5-((4-phenyl-6-(piperidin-4-yl)pyrimidin-2-yl)amino)benzoic Acid Hydrobromide (Compound 18)
The title compound was synthesized following the general procedure 6 previously described using intermediate 40e (31.1 mg, 0.06 mmol) and BBr3 (1 M in DCM) (0.36 mL, 0.36 mmol) in CHCl3 dry (1.0 mL). Purification by trituration with EtOAc (1.0 mL) afforded pure compound 18 (9.6 mg, 41% yield). UPLC-MS: tR = 2.37 min (method 4). MS (ESI) m/z: 391.0 [M + H]+, calcd for C22H23N4O3 [M + H]+: 391.4. HRMS (ESI) m/z: 391.1765 calcd for C22H23N4O3 [M + H]+; found m/z: 391.1771. 1H NMR (400 MHz, DMSO-d6) δ 10.87 (s, 1H), 9.57 (s, 1H), 8.74 (d, J = 11.2 Hz, 1H), 8.63 (s), 8.45 (q, J = 10.9 Hz, 1H), 8.22–8.20 (m, 2H), 7.81 (dd, J = 8.9, 2.8 Hz, 1H), 7.54–7.50 (m, 3H), 7.33 (s, 1H), 6.94 (d, J = 9.0 Hz, 1H), 3.40 (d, J = 12.4 Hz, 2H), 3.14–3.09 (m, 2H), 2.94 (tt, J = 11.4, 3.3 Hz, 1H), 2.18 (d, J = 13.8 Hz, 2H), 1.95 (qd, 12.4, 2.6 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 172.0 (Cq), 163.9 (Cq), 160.0 (Cq), 157.8 (Cq), 156.0 (Cq), 136.8 (Cq), 132.4 (Cq), 130.8 (CH), 128.8 (CH, 2C), 127.6 (CH), 127.0 (CH, 2C), 120.1 (CH), 116.9 (CH), 112.2 (Cq), 105.2 (CH), 43.0 (CH2, 2C), 40.2 (CH), 27.0 (CH2, 2C).
4-Phenyl-6-(piperidin-4-yl)-N-(pyridin-4-yl)pyrimidin-2-amine (Compound 19)
The title compound was synthesized following the general procedure 5 previously described using intermediate 40f (70 mg, 0.16 mmol) and HCl (4 M in dioxane) (0.40 mL) in 1,4-dioxane dry (2.7 mL). Purification by alumina (elution by gradient from 100/0 to 90/5 DCM/MeOH) afforded pure intermediate 19 (18.5 mg, 35% yield). UPLC-MS: tR = 1.88 min (method 1). MS (ESI) m/z: 332.1 [M + H]+, calcd for C20H22N5 [M + H]+: 332.4. HRMS (ESI) m/z: 332.1870 calcd for C20H22N5 [M + H]+; found m/z: 332.1878. 1H NMR (400 MHz, DMSO-d6) δ 10.04 (s, 1H), 8.42–8.32 (m, 2H), 8.23–8.15 (m, 2H), 7.89–7.82 (m, 2H), 7.63–7.50 (m, 3H), 7.44 (s, 1H), 3.06 (dt, J = 11.9, 3.2 Hz, 2H), 2.77 (tt, J = 11.7, 3.7 Hz, 1H), 2.61 (td, J = 12.1, 2.5 Hz, 2H), 1.84 (d, J = 12.4 Hz, 2H), 1.71 (qd, J = 11.0, 2.8 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 175.7 (Cq), 164.2 (Cq), 159.5 (Cq), 149.9 (CH, 2C), 147.4 (Cq), 136.7 (Cq), 130.9 (CH), 128.9 (CH, 2C), 127.1 (CH, 2C), 112.6 (CH, 2C), 107.0 (CH), 46.2 (CH2, 2C), 44.3 (CH), 31.8 (CH2, 2C).
4-Phenyl-6-(piperidin-4-yl)-N-(2-(trifluoromethyl)pyridin-4-yl)pyrimidin-2-amine (Compound 20 – ARN25375)
The title compound was synthesized following the general procedure 5 previously described using intermediate 40g (70 mg, 0.14 mmol) and HCl (4 M in dioxane) (0.35 mL) in 1,4-dioxane dry (2.3 mL). Purification by alumina (elution by a gradient from 100/0 to 96/4 DCM/MeOH) afforded pure compound 20 (25.1 mg, 45% yield). UPLC-MS: tR = 2.05 min (method 1). MS (ESI) m/z: 400.0 [M + H]+, calcd for C21H21F3N5 [M + H]+: 400.4. HRMS (ESI) m/z: 400.1744 calcd for C21H21F3N5 [M + H]+; found m/z: 400.1751. 1H NMR (400 MHz, DMSO-d6) δ 10.51 (s, 1H), 8.61 (d, J = 2.1 Hz, 1H), 8.54 (d, J = 5.6 Hz, 1H), 8.20 (dd, J = 6.7, 2.9 Hz, 2H), 7.98 (dd, J = 5.7, 2.1 Hz, 1H), 7.61–7.55 (m, 3H), 7.54 (s, 1H), 3.06 (dt, J = 12.1, 3.2 Hz, 2H), 2.79 (tt, J = 11.8, 3.7 Hz, 1H), 2.61 (td, J = 12.1, 2.5 Hz, 2H), 1.86 (d, J = 11.9 Hz, 2H), 1.71 (qd, J = 12.2, 4.0 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 175.9 (Cq), 164.2 (Cq), 159.3 (Cq), 150.5 (CH), 149.0 (Cq), 147.2 (q2, JCF = 32.9 Hz, Cq), 136.5 (Cq), 131.1 (CH), 128.9 (CH, 2C), 127.1 (CH, 2C), 121.9 (q1, JCF = 273.4 Hz, Cq), 114.7 (CH), 109.1 (q3, JCF = 3.4 Hz, CH), 107.8 (CH), 46.2 (CH2, 2C), 44.2 (CH), 31.8 (CH2, 2C). 19F NMR (376 MHz, DMSO-d6) δ – 66.9.
N-(2-(Difluoromethoxy)pyridin-4-yl)-4-phenyl-6-(piperidin-4-yl)pyrimidin-2-amine (Compound 21)
The title compound was synthesized following the general procedure 5 previously described using intermediate 40h (70 mg, 0.14 mmol) and HCl (4 M in dioxane) (0.35 mL) in 1,4-dioxane dry (2.3 mL). Purification by alumina (elution by a gradient from 100/0 to 96/4 DCM/MeOH) afforded pure compound 21 (25.1 mg, 45% yield). UPLC-MS: tR = 2.00 min (method 1). MS (ESI) m/z: 398.0 [M + H]+, calcd for C21H22F2N5O [M + H]+: 398.2. HRMS (ESI) m/z: 398.1787 calcd for C21H22F2N5O [M + H]+; found m/z: 398.1797. 1H NMR (400 MHz, DMSO-d6) δ 10.32 (s, 1H), 8.23–8.14 (m, 2H), 8.07 (d, J = 5.8 Hz, 1H), 7.68 (t, J = 73.4 Hz, 1H), 7.66 (dd, J = 5.8, 1.9 Hz, 1H), 7.64 (d, J = 1.8 Hz, 1H), 7.60–7.53 (m, 3H), 7.50 (s, 1H), 3.06 (dt, J = 12.1, 2.5 Hz, 2H), 2.68 (tt, J = 11.9, 3.6 Hz,1H), 2.61 (td, J = 12.1, 2.8 Hz, 2H), 1.85 (dd, J = 12.6, 2.4 Hz, 2H), 1.71 (qd, J = 12.2, 3.9 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 175.9 (Cq), 164.3 (Cq), 159.5 (Cq), 159.4 (Cq), 151.1 (Cq), 147.0 (CH), 136.6 (Cq), 131.0 (CH), 129.0 (CH, 2C), 127.1 (CH, 2C), 114.8 (t1, JCF = 254.1 Hz, CH), 110.4 (CH), 107.6 (CH), 97.8 (CH), 46.2 (CH2, 2C), 44.3 (CH), 31.8 (CH2, 2C). 19F NMR (376 MHz, DMSO-d6) δ – 86.5.
N-(2-Methoxypyridin-4-yl)-4-phenyl-6-(piperidin-4-yl)pyrimidin-2-amine (Compound 22)
The title compound was synthesized following the general procedure 5 previously described using intermediate 40i (70 mg, 0.15 mmol) and HCl (4 M in dioxane) (0.37 mL) in 1,4-dioxane dry (2.5 mL). Purification by alumina (elution by a gradient from 100/0 to 95/5 CHCl3/MeOH) afforded pure compound 22 (21.1 mg, 39% yield). UPLC-MS: tR = 1.70 min (method 1). MS (ESI) m/z: 362.1 [M + H]+, calcd for C21H24N5O [M + H]+: 362.2. HRMS (ESI) m/z: 362.1975 calcd for C21H24N5O [M + H]+; found m/z: 362.1992. 1H NMR (400 MHz, DMSO-d6) δ 10.01 (s, 1H), 8.17 (dd, J = 6.7, 2.8 Hz, 2H), 7.97 (d, J = 5.8 Hz, 1H), 7.58–7.54 (m, 3H), 7.50 (d, J = 1.8 Hz, 1H), 7.44 (s, 1H), 7.37 (dd, J = 5.8, 1.9 Hz, 1H), 3.83 (s, 3H), 3.08 (dt, J = 11.8, 2.8 Hz, 2H), 2.75 (tt, J = 11.8, 3.7 Hz, 1H), 2.60 (td, J = 12.1, 2.5 Hz, 2H), 1.84 (d, J = 12.4 Hz, 2H), 1.70 (qd, J = 12.2, 4.0 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 175.7 (Cq), 164.7 (Cq), 164.1 (Cq), 159.7 (Cq), 149.7 (Cq), 146.7 (CH), 136.8 (Cq), 130.9 (CH), 128.9 (CH, 2C), 127.0 (CH, 2C), 108.0 (CH), 106.9 (CH), 97.0 (CH), 53.0 (CH3), 46.2 (CH2, 2C), 44.4 (CH), 31.8 (CH2, 2C).
N-(6-Methoxypyridin-3-yl)-4-phenyl-6-(piperidin-4-yl)pyrimidin-2-amine (Compound 23)
Title compound was synthesized following the general procedure 5 previously described using intermediate 40j (44 mg, 0.09 mmol), HCl (4 M in dioxane) (0.24 mL) in 1,4-dioxane dry (1.5 mL). Purification by alumina (elution by gradient from 100/0 to 93/7 DCM/MeOH) afforded pure compound 23 (12.4 mg, 38% yield). UPLC-MS: tR = 1.79 min (method 1). MS (ESI) m/z: 362.0 [M + H]+, calcd for C21H24N5O [M + H]+: 362.5. HRMS (ESI) m/z: 362.1975 calcd for C21H24N5O [M + H]+; found m/z: 362.1985. 1H NMR (400 MHz, DMSO-d6) δ 9.48 (s, 1H), 8.68 (d, J = 2.7 Hz, 1H), 8.13 (dd, J = 6.7, 3.0 Hz, 2H), 8.07 (dd, J = 8.9, 2.8 Hz, 1H), 7.57–7.48 (m, 3H), 7.28 (s, 1H), 6.81 (d, J = 8.9 Hz, 1H), 3.83 (s, 3H), 3.07 (d, J = 11.8 Hz, 2H), 2.73 (tt, J = 11.9, 3.6 Hz, 1H), 2.62 (td, J = 12.2, 1.7 Hz, 2H), 1.83 (d, J = 12.0 Hz, 2H), 1.69 (qd, J = 12.3, 4.0 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 174.9 (Cq), 164.6 (Cq), 160.6 (Cq), 159.0 (Cq), 137.5 (CH), 137.4 (Cq), 132.2 (Cq), 131.8 (CH), 131.2 (CH), 129.3 (CH, 2C), 127.4 (CH, 2C), 110.2 (CH), 105.9 (CH), 53.5 (CH3), 45.1 (CH2, 2C), 43.1 (CH), 30.1 (CH2, 2C).
N1,N1-Dimethyl-N3-(6-phenyl-2-(1,2,3,6-tetrahydropyridin-4-yl)pyrimidin-4-yl)benzene-1,3-diamine (Compound 24)
The title compound was synthesized following the general procedure 5 previously described using intermediate 43a (60 mg, 0.13 mmol) and HCl (4 M in dioxane) (0.33 mL) in 1,4-dioxane dry (4.4 mL). Purification by alumina (elution by a gradient from 100/0 to 90/10 DCM/MeOH) afforded pure compound 24 (26.5 mg, 56% yield). UPLC-MS: tR = 2.06 min (method 1). MS (ESI) m/z: 372.3 [M + H]+, calcd for C23H26N5 [M + H]+: 372.2. 1H NMR (400 MHz, DMSO-d6) δ 9.42 (s, 1H), 8.07–8.05 (m, 2H), 7.57–7.45 (m, 3H), 7.40 (t, J = 2.3 Hz, 1H), 7.30–7.24 (m, 1H), 7.13 (t, J = 8.1 Hz, 1H), 7.09 (s, 1H), 6.95 (dd, J = 7.8, 1.9 Hz, 1H), 6.40 (dd, J = 8.2, 2.4 Hz, 1H), 3.46 (q, J = 3.2 Hz, 2H), 2.92 (bs, 8H), 2.58 (bs, 2H). 13C NMR (101 MHz, DMSO-d6) δ 163.9 (Cq), 161.1 (Cq), 160.7 (Cq), 150.9 (Cq), 141.0 (Cq), 137.5 (Cq), 135.2 (Cq), 131.8 (CH), 130.1 (CH), 129.0 (CH), 128.8 (CH, 2C), 126.3 (CH, 2C), 107.7 (CH), 106.7 (CH), 103.6 (CH), 99.8 (CH), 45.2 (CH2), 42.7 (CH2), 40.2 (CH3, 2C), 26.1 (CH2).
1-Methyl-N-(6-phenyl-2-(piperidin-4-yl)pyrimidin-4-yl)-1H-indol-6-amine (Compound 25)
The title compound was synthesized following the general procedure 5 previously described using intermediate 44b (50.0 mg, 0.10 mmol), HCl (4 M in dioxane) (0.25 mL) in 1,4-dioxane dry (1.7 mL). Purification by alumina (elution by a gradient from 100/0 to 95/5 DCM/MeOH) afforded pure compound 25 (22.9 mg, 60% yield). UPLC-MS: tR = 1.94 min (method 1). MS (ESI) m/z: 384.3 [M + H]+, calcd for C24H26N5 [M + H]+: 384.2. 1H NMR (DMSO-d6) δ 9.58 (bs, 1H), 8.27 (s, 1H), 8.02 (d, J = 6.9 Hz, 2H), 7.53–7.47 (m, 4H), 7.24–7.22 (m, 1H), 7.11–7.05 (m, 2H), 6.37 (d, J = 3.1 Hz, 1H), 3.77 (s, 3H), 3.07–3.04 (m, 2H), 2.82 (t, J = 11.8 Hz, 1H), 2.63 (t, J = 12.0 Hz, 2H), 2.00 (d, J = 13.0 Hz, 3H), 1.80 (q, J = 12.3 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 172.6 (Cq), 161.6 (Cq), 161.1 (Cq), 137.6 (Cq), 136.6 (Cq), 134.5 (Cq), 130.0 (CH), 129.1 (CH), 128.8 (CH, 2C), 126.5 (CH, 2C), 123.7 (Cq), 120.4 (CH), 113.3 (CH), 101.3 (CH), 100.3 (CH), 99.0 (CH), 46.1 (CHhh, 2C), 45.4 (CH), 32.4 (CH3), 31.9 (CH2, 2C).
N-(6-Phenyl-2-(piperidin-4-yl)pyrimidin-4-yl)-1H-indol-6-amine (Compound 26)
The title compound was synthesized following the general procedure 5 previously described using intermediate 44c (69.0 mg, 0.12 mmol) and HCl (4 M in dioxane) (0.30 mL) in 1,4-dioxane dry (2.0 mL). Purification by alumina (elution by a gradient from 100/0 to 95/5 DCM/MeOH) afforded pure compound 26 (14.6 mg, 33% yield). UPLC-MS: tR = 1.79 min (method 1). MS (ESI) m/z: 370.2 [M + H]+, calcd for C23H24N5 [M + H]+: 370.2. 1H NMR (400 MHz, DMSO-d6) δ 11.04 (s, 1H), 9.43 (s, 1H), 8.02–8.00 (m, 3H), 7.58–7.46 (m, 4H), 7.26 (t, J = 2.7 Hz, 1H), 7.13 (dd, J = 8.5, 1.9 Hz, 1H), 7.03 (s, 1H), 6.37 (t, J = 2.5 Hz, 1H), 3.07 (d, J = 12.0 Hz, 2H), 2.79 (t, J = 11.7 Hz, 1H), 2.68–2.58 (m, 2H), 1.94 (d, J = 12.7 Hz, 2H), 1.78 (qd, J = 12.2, 4.0 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 172.6 (Cq), 161.6 (Cq), 161.0 (Cq), 137.6 (Cq), 136.1 (Cq), 134.0 (Cq), 129.9 (CH), 128.7 (CH, 2C), 126.4 (CH, 2C), 124.7 (CH), 123.5 (Cq), 119.9 (CH), 113.5 (CH), 103.2 (CH), 101.0 (CH), 98.8 (CH), 46.1 (CH2, 2C), 45.5 (CH), 31.7 (CH2, 2C). Compound 26 displayed 93% as determined by UPLC-MS (UV at 215 nm) analysis (see Supporting Information).
tert-Butyl 4-(4-Chloro-6-((3-(trifluoromethyl)phenyl)amino)-1,3,5-triazin-2-yl)-3,6-dihydropyridine-1(2H)-carboxylate (Compound 30)
The protocol for the obtainment of intermediate 30 has been modified compared to that previously reported.23 Compound 30 was synthesized following general procedure 2 using intermediate 28 (1.0 g, 3.247 mmol), (1-(tert-butoxycarbonyl)-1,2,3,6-tetrahydropyridin-4-yl)boronic acid 29 (885 mg, 3.9 mmol), Pd(dppf)Cl2·DCM (132 mg, 0.162 mmol), K2CO3 (aq) 2 M (3.3 mL, 6.49 mmol) and 1,4-dioxane dry (20 mL). After 5 h, a standard workup has been performed. Purification by the gradient (elution by gradient 100/0 to 70/30 cyclohexane/EtOAc) gave the pure product as a white solid (816 mg, 55% yield). UPLC-MS and NMR analysis were consistent with those previously reported.23
tert-Butyl 4-(4-(Pyrimidin-5-yl)-6-((3-(trifluoromethyl)phenyl)amino)-1,3,5-triazin-2-yl)-3,6-dihydropyridine-1(2H)-carboxylate (Compound 32a)
Compound 32a was synthesized following general procedure 2 using compound 30 (200 mg, 0.44 mmol), pyrimidin-5-yl boronic acid 31a (65.3 mg, 0.53 mmol), Pd(dppf)Cl2·DCM (18.0 mg, 0.022 mmol), K2CO3 (2 M) aq (0.44 mL, 0.88 mmol) in 1, 4-dioxane dry (3.4 mL). After 1 h, a standard workup has been performed. Purification by silica (elution by gradient 90/10 to 60/40 cyclohexane/EtOAc) afforded the pure product as a white solid (174 mg, 79% yield). UPLC-MS: tR = 1.87 min (method 2). MS (ESI) m/z: 498.0 [M-H]−, calcd for C24H23F3N7O2 [M-H]−: 498.2. 1H NMR (400 MHz, DMSO-d6) δ 10.74 (s, 1H), 9.64 (s, 2H), 9.42 (s, 1H), 8.43 (s, 1H), 8.00 (s, 1H), 7.64 (t, J = 8.0 Hz, 1H), 7.57 (s, 1H), 7.45 (d, J = 7.8 Hz, 1H), 4.22–4.15 (m, 2H), 3.61–3.54 (m, 2H), 2.68–2.62 (m, 2H), 1.44 (s, 9H).
tert-Butyl 4-(4-(Isoquinolin-5-yl)-6-((3-(trifluoromethyl)phenyl)amino)-1,3,5-triazin-2-yl)-3,6-dihydropyridine-1(2H)-carboxylate (Compound 32b)
Compound 32b was synthesized following general procedure 2 using compound 30 (200 mg, 0.44 mmol), isoquinolin-5-yl boronic acid 31b (91 mg, 0.528 mmol), Pd(dppf)Cl2·DCM (18.0 mg, 0.022 mmol), K2CO3 (2 M) aq (0.44 mL, 0.876 mmol) and 1,4-dioxane dry (3.4 mL). After 3 h, a standard workup has been performed. Purification by silica (elution by a gradient from 70/30 to 40/60 cyclohexane/EtOAc) afforded the pure product as a yellowish solid (175 mg, 73% yield). UPLC-MS: tR = 2.21 min (method 2). MS (ESI) m/z: 549.0 [M + H]+, calcd for C29H28F3N6O2 [M + H]+: 549.2. 1H NMR (400 MHz, DMSO-d6) δ 10.67 (s, 1H), 9.43 (d, J = 1.0 Hz, 1H), 8.90 (s, 1H), 8.65–8.57 (m, 2H), 8.46 (s, 1H), 8.37 (d, J = 8.2 Hz, 1H), 8.02 (d, J = 7.8 Hz, 1H), 7.89–7.82 (m, 1H), 7.61 (t, J = 8.0 Hz, 1H), 7.49–7.39 (m, 2H), 4.23–4.14 (m, 2H), 3.62–3.54 (m, 2H), 2.72–2.65 (m, 2H), 1.44 (s, 9H).
tert-Butyl 4-(4-(1-Methyl-1H-indol-5-yl)-6-((3-(trifluoromethyl)phenyl)amino)-1,3,5-triazin-2-yl)-3,6-dihydropyridine-1(2H)-carboxylate (Compound 32c)
Compound 32c was synthesized following general procedure 6 using compound 30 (200 mg, 0.438 mmol), (1-methyl-1H-indol-5-yl)boronic acid 31c (92 mg, 0.526 mmol), Pd(dppf)Cl2·DCM (17.9 mg, 0.022 mmol), K2CO3(aq) 2 M (0.44 mL, 0.876 mmol) in 1,4 dioxane dry (3.4 mL). Purification by silica (elution by a gradient from 100/0 to 70/30 cyclohexane/EtOAc) afforded the pure product as a yellowish solid (180 mg, 74% yield). UPLC-MS: tR = 1.50 min (method 3). MS (ESI) m/z: 551.0 [M + H]+, calcd for C29H30F3N6O2 [M + H]+: 551.2. 1H NMR (400 MHz, CDCl3) δ 8.89 (d, J = 1.6 Hz, 1H), 8.48–8.38 (m, 2H), 7.70 (d, J = 7.8 Hz, 1H), 7.58–7.46 (m, 2H), 7.43–7.35 (m, 2H), 7.12 (d, J = 3.2 Hz, 1H), 6.63 (d, J = 3.3 Hz, 1H), 4.28–4.23 (m, 2H), 3.85 (s, 3H), 3.73–3.60 (m, 2H), 2.86–2.74 (m, 2H), 1.51 (s, 9H).
tert-Butyl 4-(4-(1H-Indazol-5-yl)-6-((3-(trifluoromethyl)phenyl)amino)-1,3,5-triazin-2-yl)-3,6-dihydropyridine-1(2H)-carboxylate (Compound 32d)
Compound 32d was synthesized following general procedure 2 using compound 30 (200 mg, 0.44 mmol), (1H-indazol-6-yl)boronic acid 31d (85.3 mg, 0.528 mmol), Pd(dppf)Cl2·DCM (18.0 mg, 0.022 mmol), and K2CO3 (2 M) aq (0.66 mL, 1.32 mmol) in 1,4-dioxane dry (3.4 mL). After 1.5 h, a standard workup has been performed. Purification by silica (elution by a gradient from 90/10 to 30/70 cyclohexane/EtOAc) afforded pure product that was washed with acetone. Filtration of solid furnished pure product as a white solid (55 mg, 23% yield). UPLC/MS: Rt = 1.97 min (method 2), [M–H]− = 536.0; [M–H] – Calculated for C27H25F3N7O2: 536.2. 1H NMR (400 MHz, DMSO-d6) δ 13.36 (s, 1H), 10.51 (s, 1H), 8.98 (s, 1H), 8.54–8.43 (m, 2H), 8.27 (s, 1H), 8.02 (d, J = 8.2 Hz, 1H), 7.71–7.59 (m, 2H), 7.51 (s, 1H), 7.42 (d, J = 7.8 Hz, 1H), 4.27–4.15 (m, 2H), 3.63–3.55 (m, 2H), 2.74–2.64 (m, 2H), 1.45 (s, 9H).
tert-Butyl 4-(4-(Benzofuran-3-yl)-6-((3-(trifluoromethyl)phenyl)amino)-1,3,5-triazin-2-yl)-3,6-dihydropyridine-1(2H)-carboxylate (Compound 32e)
To a degassed 1,4-dioxane solution (0.13 M, 5.0 mL) of compound 28 (4,6-dichloro-N-(3-(trifluoromethyl)phenyl)-1,3,5-triazin-2-amine) (240 mg, 0.777 mmol), (1-(tert-butoxycarbonyl)-1,2,3,6-tetrahydropyridin-4-yl)boronic acid 29 (211 mg, 0.932 mmol), Pd(dppf)Cl2·DCM (31.7 mg, 0.0388 mmol) and K2CO3(aq) 2 M (0.78 mL, 1.55 mmol) were added. The resulting mixture was sparged with argon for a further 10 min and heated up to 80 °C until consumption of starting material was detected by UPLC (5 h). Then, Pd(dppf)Cl2·DCM (31.7 mg, 0.0388 mmol) and new boronic acid benzofuran-3-ylboronic acid 31e (151 mg, 0.932 mmol) were added and reaction mixture was heated up to 120 °C until no intermediate was detected by UPLC. Then, H2O and EtOAc were added and separated; aqueous layers were extracted two times with EtOAc and collected organic layers were dried over Na2SO4. The solvent was evaporated and the resulting crude was purified by silica (elution by gradient 100/0 to 70/30 cyclohexane/EtOAc) giving pure product 32e as a white solid (189 mg, 45% yield for two steps). UPLC-MS: tR = 1.73 min (method 3). MS (ESI) m/z: 538.0 [M + H]+, calcd for C28H27F3N5O3 [M + H]+: 538.4. 1H NMR (400 MHz, CDCl3) δ 8.61 (s, 1H), 8.49–8.41 (m, 1H), 8.33 (s, 1H), 7.68 (d, J = 8.1 Hz, 1H), 7.60–7.55 (m, 1H), 7.54–7.46 (m, 2H), 7.43–7.37 (m, 4H), 4.28–4.23 (m, 2H), 3.72–3.64 (m, 2H), 2.77 (s, 2H), 1.52 (s, 9H).
tert-Butyl 4-(4-(1-Methyl-1H-pyrazol-4-yl)-6-((3-(trifluoromethyl)phenyl)amino)-1,3,5-triazin-2-yl)-3,6-dihydropyridine-1(2H)-carboxylate (Compound 32f)
Compound 32f was synthesized following general procedure 2 using compound 30 (200 mg, 0.44 mmol), (1-methyl-1H-pyrazol-4-yl)boronic acid (66.5 mg, 0.528 mmol), Pd(dppf)Cl2·DCM (18.0 mg, 0.022 mmol), K2CO3 (2 M) aq (0.44 mL, 0.876 mmol) in 1,4-dioxane dry (3.4 mL). After 1.5 h, a standard workup has been performed. Purification by silica (elution by a gradient from 90/10 to 40/60 cyclohexane/EtOAc) afforded the pure product as a white solid (143 mg, 65% yield). UPLC-MS: tR = 0.67 min (method 3). MS (ESI) m/z: 502.0 [M + H]+, calcd for C24H27F3N7O2 [M + H]+: 502.2. 1H NMR (400 MHz, CDCl3) δ 8.35 (s, 1H), 8.21 (s, 1H), 8.15 (s, 1H), 7.65 (d, J = 8.7 Hz, 1H), 7.51–7.39 (m, 3H), 7.36 (d, J = 7.5 Hz, 1H), 4.25–4.17 (m, 2H), 3.98 (s, 3H), 3.67–3.59 (m, 2H), 2.77–2.63 (m, 2H), 1.50 (s, 9H).
tert-Butyl 4-(4-(1-Methyl-1H-imidazol-5-yl)-6-((3-(trifluoromethyl)phenyl)amino)-1,3,5-triazin-2-yl)-3,6-dihydropyridine-1(2H)-carboxylate (Compound 32g)
Compound 32g was synthesized following general procedure 2 using compound 30 (100 mg, 0.22 mmol), (1-methyl-1H-imidazol-5-yl)boronic acid 31g (54.8 mg, 0.26 mmol), Pd(dppf)Cl2·DCM (9.0 mg, 0.011 mmol), K2CO3 (2 M) aq (0.22 mL, 0.44 mmol) in 1,4-dioxane dry (2.0 mL). After 1.5 h, the addition of H2O promotes the precipitation of a solid that has been filtered and washed with EtOAc, furnishing the desired product as a white solid (65 mg, 60% yield). UPLC-MS: tR = 1.61 min (method 2). MS (ESI) m/z: 502.0 [M + H]+, calcd for C24H27F3N7O2 [M + H]+: 502.2. 1H NMR (400 MHz, DMSO-d6) δ 10.43 (s, 1H), 8.41 (s, 1H), 8.01–7.84 (m, 4H), 7.60 (t, J = 8.0 Hz, 1H), 7.44–7.35 (m, 2H), 4.21–4.10 (m, 2H), 4.06 (s, 3H), 3.61–3.52 (m, 2H), 2.64–2.58 (m, 2H), 1.44 (s, 9H).
tert-Butyl 4-(4-(1-(tert-Butoxycarbonyl)-1H-pyrrol-2-yl)-6-((3-(trifluoromethyl)phenyl)amino)-1,3,5-triazin-2-yl)-3,6-dihydropyridine-1(2H)-carboxylate (Compound 32h)
Compound 32g was synthesized following general procedure 2 using compound 30 (200 mg, 0.44 mmol (1-(tert-butoxycarbonyl)-1H-pyrrol-2-yl)boronic acid 31h (111.4 mg, 0.53 mmol), Pd(dppf)Cl2·DCM (17.9 mg, 0.022 mmol), K2CO3 (2 M) aq (0.44 mL, 0.88 mmol) in 1,4-dioxane dry (3.5 mL). After 1.5 h, the addition of H2O promotes the precipitation of a solid that has been filtered and washed with AcOEt, furnishing the desired product as a white solid (170 mg, 65% yield). UPLC-MS: tR = 2.56 min (method 2). MS (ESI) m/z: 587.0 [M + H]+, calcd for C29H34F3N6O4 [M + H]+: 587.2. 1H NMR (400 MHz, DMSO-d6) δ 8.19 (s, 1H), 7.71 (s, 1H), 7.56–7.31 (m, 5H), 6.96 (bs, 1H), 6.28 (t, J = 3.3 Hz, 1H), 4.18 (bs, 2H), 3.61 (t, J = 5.0 Hz, 2H), 2.69 (s, 2H), 1.50 (s, 9H), 1.47 (s, 9H).
tert-Butyl 4-(4-(Thiophen-3-yl)-6-((3-(trifluoromethyl)phenyl)amino)-1,3,5-triazin-2-yl)-3,6-dihydropyridine-1(2H)-carboxylate (Compound 32i)
Compound 32i was synthesized following general procedure 2 using compound 30 (120 mg, 0.263 mmol), thiophen-3-ylboronic acid 31i (40 mg, 0.316 mmol), Pd(dppf)Cl2·DCM (10.6 mg, 0.013 mmol), and K2CO3 (2 M) aq (0.26 mL, 0.526 mmol) in 1,4-dioxane dry (2.0 mL). After 1.5 h, a standard workup has been performed. Purification by silica (elution by a gradient from 95/5 to 80/20 cyclohexane/EtOAc) afforded not a pure product that was washed with acetone. Filtration of solid furnished pure product as a white solid (99 mg, 70% yield). UPLC-MS: tR = 1.15 min (method 3). MS (ESI) m/z: 504.0 [M + H]+, calcd for C24H25F3N5O2S [M + H]+: 504.2. 1H NMR (400 MHz, DMSO-d6) δ 10.50 (s, 1H), 8.54 (d, J = 3.1 Hz, 1H), 8.46 (s, 1H), 8.01 (d, J = 8.3 Hz, 1H), 7.84 (d, J = 5.0 Hz, 1H), 7.72 (t, J = 5.0, 3.1 Hz, 1H), 7.60 (t, J = 8.0 Hz, 1H), 7.45 (s, 1H), 7.40 (d, J = 7.8 Hz, 1H), 4.19–4.14 (m, 2H), 3.60–3.52 (m, 2H), 2.66–2.62 (m, 2H), 1.44 (s, 9H).
tert-Butyl 4-(4-(Pyrimidin-5-yl)-6-((3-(trifluoromethyl)phenyl)amino)-1,3,5-triazin-2-yl)piperidine-1-carboxylate (Compound 33a)
Compound 33a was synthesized following general procedure 4-Method A using compound 32a (153 mg, 0.306 mmol), ammonium formate (113.6 mg, 1.8 mmol), Pd(OH)2/C (30.6 mg) and dry EtOH (5.0 mL) and THF (2.5 mL). After 1 h, a standard workup has been performed. Purification by silica (elution by gradient 80/20 to 50/50 cyclohexane/EtOAc) afforded the pure product as a white solid (51 mg, 32% yield). UPLC-MS: tR = 1.75 min (method 2). MS (ESI) m/z: 502.1 [M + H]+, calcd for C24H27F3N7O2 [M + H]+: 502.2. 1H NMR (400 MHz, DMSO-d6) δ 10.75 (s, 1H), 9.58 (s, 2H), 9.41 (s, 1H), 8.50–8.30 (m, 1H), 8.08–7.88 (m, 1H), 7.69–7.58 (m, 1H), 7.45 (d, J = 7.8 Hz, 1H), 4.03 (d, J = 13.0 Hz, 2H), 3.05–2.84 (m, 3H), 2.11–1.95 (m, 2H), 1.80–1.63 (m, 2H), 1.42 (s, 9H).
tert-Butyl 4-(4-(Isoquinolin-5-yl)-6-((3-(trifluoromethyl)phenyl)amino)-1,3,5-triazin-2-yl)piperidine-1-carboxylate (Compound 33b)
Compound 33b was synthesized following general procedure 4-Method A using compound 32b (147 mg, 0.26 mmol), ammonium formate (98.4 mg, 1.56 mmol), Pd(OH)2/C (30 mg) and dry EtOH (4.2 mL) and THF (2.1 mL). After 3.5 h, a standard workup has been performed. Purification by silica (elution by a gradient from 80/20 to 50/50 cyclohexane/EtOAc) afforded the pure product as a white foaming solid (63 mg, 43% yield). UPLC-MS: tR = 2.01 min (method 2). MS (ESI) m/z: 549.0 [M-H]−, calcd for C29H28F3N6O2 [M-H]−: 549.6. 1H NMR (400 MHz, DMSO-d6) δ 10.68 (s, 1H), 9.43 (s, 1H), 8.88 (s, 1H), 8.70–8.28 (m, 4H), 7.98 (d, J = 7.8 Hz, 1H), 7.86 (t, J = 7.8 Hz, 1H), 7.60 (t, J = 8.1 Hz, 1H), 7.42 (d, J = 7.8 Hz, 1H), 4.12–3.98 (m, 2H), 3.06–2.87 (m, 3H), 2.13–2.03 (m, 2H), 1.83–1.67 (m, 2H), 1.42 (s, 9H).
tert-Butyl 4-(4-(1-Methyl-1H-indol-5-yl)-6-((3-(trifluoromethyl)phenyl)amino)-1,3,5-triazin-2-yl)piperidine-1-carboxylate (Compound 33c)
Compound 33c was synthesized following general procedure 4-Method A using compound 32c (155 mg, 0.282 mmol), ammonium formate (106.8 mg, 1.69 mmol), Pd(OH)2/C (31 mg) and dry EtOH (4.0 mL) and THF (3.0 mL). Purification by silica (elution by gradient 95/5 to 70/30 cyclohexane/EtOAc) giving pure product 33c as a white foaming solid (105.0 mg, 67% yield). UPLC-MS: Rt = 1.42 min (method 3), [M + H]+ = 553.0; [M + H]+ Calculated for C29H32F3N6O2: 553.2. 1H NMR (400 MHz, DMSO-d6) δ 10.43 (s, 1H), 8.75 (d, J = 1.6 Hz, 1H), 8.61 (s, 1H), 8.28 (dd, J = 8.7, 1.7 Hz, 1H), 7.94 (d, J = 8.0 Hz, 1H), 7.65–7.54 (m, 2H), 7.47–7.37 (m, 2H), 6.60 (d, J = 3.1 Hz, 1H), 4.12–3.98 (m, 2H), 3.85 (s, 3H), 3.05–2.81 (m, 3H), 2.10–1.97 (m, 2H), 1.81–1.66 (m, 2H), 1.43 (s, 9H).
tert-Butyl 4-(4-(1H-Indazol-5-yl)-6-((3-(trifluoromethyl)phenyl)amino)-1,3,5-triazin-2-yl)piperidine-1-carboxylate (Compound 33d)
Compound 33d was synthesized following general procedure 4-Method A using compound 32d (110 mg, 0.205 mmol), ammonium formate (77.6 mg, 1.23 mmol), Pd(OH)2/C (22 mg), dry MeOH (3.4 mL), and THF (1.7 mL). After 2 h, a standard workup has been performed. Purification by silica (elution by a gradient from 95/5 to 50/50 cyclohexane/EtOAc) afforded the pure product as a white solid (28 mg, 25% yield). UPLC-MS: tR = 1.80 min (method 2). MS (ESI) m/z: 540.1 [M + H]+, calcd for C27H29F3N7O2 [M + H]+: 540.2. 1H NMR (400 MHz, DMSO-d6) δ 13.36 (s, 1H), 10.51 (s, 1H), 8.93 (s, 1H), 8.51 (s, 1H), 8.43 (d, J = 9.0 Hz, 1H), 8.27 (s, 1H), 7.99 (s, 1H), 7.71–7.56 (m, 2H), 7.42 (d, J = 7.8 Hz, 1H), 4.12–4.00 (m, 2H), 3.04–2.85 (m, 2H), 2.12–1.93 (m, 3H), 1.80–1.66 (m, 2H), 1.43 (s, 9H).
tert-Butyl 4-(4-(Benzofuran-3-yl)-6-((3-(trifluoromethyl)phenyl)amino)-1,3,5-triazin-2-yl)piperidine-1-carboxylate (Compound 33e)
Compound 33e was synthesized following general procedure 4-Method A using compound 32e (170 mg, 0.316 mmol), ammonium formate (100 mg, 1.90 mmol), Pd(OH)2/C (34 mg), and MeOH dry (3.2 mL). After 1.5 h, a standard workup was performed and the crude product was purified by silica (elution by gradient 90/10 to 85/15 cyclohexane/EtOAc) giving the pure product as a white foaming solid (108 mg, 63% yield). UPLC-MS: tR = 1.63 min (method 3). MS (ESI) m/z: 540.0 [M + H]+, calcd for C28H29F3N5O3 [M + H]+: 540.2. 1H NMR (400 MHz, CDCl3) δ 8.60 (s, 1H), 8.47–8.41 (m, 1H), 8.28 (s, 1H), 7.73–7.67 (m, 1H), 7.61–7.54 (m, 1H), 7.51 (t, J = 7.9 Hz, 1H), 7.43–7.33 (m, 4H), 4.24 (s, 2H), 3.01–2.83 (m, 3H), 2.20–2.04 (m, 2H), 1.89 (dd, J = 12.1, 4.3 Hz, 2H), 1.50 (s, 9H).
tert-Butyl 4-(4-(1-Methyl-1H-pyrazol-4-yl)-6-((3-(trifluoromethyl)phenyl)amino)-1,3,5-triazin-2-yl)piperidine-1-carboxylate (Compound 33f)
Compound 33f was synthesized following general procedure 4-Method A using compound 32f (132 mg, 0.263 mmol), ammonium formate (99.6 mg, 1.58 mmol), Pd(OH)2/C (26.4 mg) in EtOH dry (4.4 mL) and THF dry (2.2 mL). After 1 h, a standard workup has been performed. Purification by silica (elution by a gradient from 80/20 to 50/50 cyclohexane/EtOAc) afforded the pure product as a white solid (78 mg, 59% yield). UPLC-MS: tR = 1.68 min (method 3). MS (ESI) m/z: 504.0 [M + H]+, calcd for C24H29F3N7O2 [M + H]+: 504.2. 1H NMR (400 MHz, DMSO-d6) δ 10.39 (s, 1H), 8.48–8.37 (m, 2H), 8.04 (s, 1H), 8.04 (d, J = 0.7 Hz, 1H), 7.58 (t, J = 8.0 Hz, 1H), 7.37 (d, J = 7.4 Hz, 1H), 4.06–3.98 (m, 2H), 3.93 (s, 3H), 3.02–2.75 (m, 3H), 2.06–1.88 (m, 2H), 1.74–1.59 (m, 2H), 1.42 (s, 9H).
tert-Butyl 4-(4-(1-Methyl-1H-imidazol-5-yl)-6-((3-(trifluoromethyl)phenyl)amino)-1,3,5-triazin-2-yl)piperidine-1-carboxylate (Compound 33g)
Compound 33g was synthesized following general procedure 4-Method A using compound 32g (90 mg, 0.180 mmol), ammonium formate (68.1 mg, 1.08 mmol), Pd(OH)2/C (18 mg) in dry MeOH (3.0 mL) and THF (1.5 mL). After 5 h, a standard workup was performed giving pure product without any purification as a white solid (83 mg, 92% yield). UPLC-MS: tR = 1.47 min (method 2). MS (ESI) m/z: 504.0 [M + H]+, calcd for C24H29F3N7O2 [M + H]+: 504.2. 1H NMR (400 MHz, DMSO-d6) δ 10.41 (s, 1H), 8.39 (s, 1H), 7.93–7.89 (m, 2H), 7.86 (d, J = 1.1 Hz, 1H), 7.59 (t, J = 8.0 Hz, 1H), 7.40 (d, J = 7.7 Hz, 1H), 4.09–3.95 (m, 5H), 3.02–2.78 (m, 3H), 2.04–1.93 (m, 2H), 1.74–1.59 (m, 2H), 1.41 (s, 9H).
tert-Butyl 4-(4-(1-(tert-Butoxycarbonyl)-1H-pyrrol-2-yl)-6-((3-(trifluoromethyl)phenyl)amino)-1,3,5-triazin-2-yl)piperidine-1-carboxylate (Compound 33h)
Compound 33h was synthesized following general procedure 4-Method B using compound 32h (100 mg, 0.17 mmol), Et3SiH (0.27 mL, 1.7 mmol), Pd/C (20 mg) and dry EtOH (3.4 mL). After 3 h, further Et3SiH (0.27 mL, 1.7 mmol) was added at 0 °C, and the reaction mixture was left to stir overnight. After the addition of a further 5 equiv of reducing agent, a standard workup has been performed. Purification by silica (elution by gradient 95/5 to 80/20 cyclohexane/EtOAc) afforded the pure product as a white solid (60 mg, 60% yield). UPLC-MS: tR = 1.18 min (method 3). MS (ESI) m/z: 589.0 [M + H]+, calcd for C29H36F3N6O4 [M + H]+: 588.3. 1H NMR (400 MHz, DMSO-d6) δ 10.51 (s, 1H), 8.38 (s, 1H), 7.93 (d, J = 8.4 Hz, 1H), 7.57 (t, J = 8.0 Hz, 1H), 7.46 (t, J = 3.1, 1.7 Hz, 1H), 7.39 (d, J = 7.7 Hz, 1H), 6.90–6.85 (m, 1H), 6.35 (t, J = 3.3 Hz, 1H), 4.07–3.93 (m, 2H), 3.01–2.77 (m, 3H), 2.00–1.87 (m, 2H), 1.72–1.57 (m, 2H), 1.41 (s, 18H).
tert-Butyl 4-(4-(1-(tert-Butoxycarbonyl)pyrrolidin-2-yl)-6-((3-(trifluoromethyl)phenyl)amino)-1,3,5-triazin-2-yl)piperidine-1-carboxylate (Compound 33h’)
Compound 33h’ was synthesized following general procedure 4-Method A using compound 32h (145 mg, 0.247 mmol), ammonium formate (93.4 mg, 1.48 mmol), Pd(OH)2/C (29 mg), and dry EtOH (6.0 mL). After 1 h, a standard workup has been performed. Purification by silica (elution by gradient 90/10 to 60/40 cyclohexane/EtOAc) afforded the pure product as a white foaming solid (127 mg, 86% yield). UPLC-MS: tR = 2.25 min (method 2). MS (ESI) m/z: 593.0 [M + H]+, calcd for C29H40F3N6O4 [M + H]+: 592.3. 1H NMR (400 MHz, DMSO-d6) δ 10.47 (s, 1H), 8.36 (s, 1H), 7.91 (d, J = 8.4 Hz, 1H), 7.59–7.50 (m, 1H), 7.42–7.34 (m, 1H), 4.64–4.50 (m, 1H), 4.07–3.90 (m, 2H), 3.62–3.38 (m, 2H), 2.99–2.76 (m, 3H), 2.42–2.26 (m, 1H), 2.06–1.76 (m, 5H), 1.72–1.52 (m, 2H), 1.40 (s, 9H), 1.10 (s, 9H).
tert-Butyl 4-(2-Chloro-6-phenylpyrimidin-4-yl)-3,6-dihydropyridine-1(2H)-carboxylate (Compound 37)
K2CO3 (2 M) aq (2 mL), phenyl boronic acid 35 (256 mg, 2.1 mmol), and PdCl2(dppf)·DCM (163 mg, 0.2 mmol) were added to a solution of trichloropyrimidine 34 (366 mg, 2 mmol) in 1,4-dioxane dry (4 mL) under argon. The reaction mixture stirred at 65 °C for 2 h until complete consumption of starting material 34 (UPLC-MS of not isolated intermediate 2,4-dichloro-6-phenylpyrimidine: tR = 1.50 min (method 2). MS (ESI) m/z: 224.9 [M + H]+, calcd for C10H7Cl2N2 [M + H]+: 226.1). After that, boronic ester 36 (618 mg, 2 mmol) and a second addition of PdCl2(dppf)·DCM (82 mg, 0.1 mmol) was added and reaction mixture stirred at 90 °C for other 2h at 90 °C under argon until complete conversion of 2,4-dichloro-6-phenylpyrimidine intermediate. Then water (3 mL) was added and an aqueous layer was extracted with EtOAc (5 mL × 2). Collected organic layers were dried over Na2SO4, filtered, and concentrated under a vacuum. The desired product 37 was purified by silica eluting by a gradient from 100% cyclohexane to 85/15 cyclohexane/EtOAc, obtaining pure compound 3 (410 mg, 55%). UPLC-MS: tR = 2.20 min (method 2). MS (ESI) m/z: 372.1 [M + H]+, calcd for C20H23ClN3O2 [M + H]+: 372.9. 1H NMR (400 MHz, CDCl3) δ 8.12–8.02 (m, 2H), 7.59 (s, 1H), 7.56–7.47 (m, 3H), 7.05 (m, 1H), 4.20 (q, J = 3.0 Hz, 2H), 3.66 (t, J = 5.7 Hz, 2H), 2.64 (m, 2H), 1.49 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 167.7 (Cq), 167.2 (Cq), 161.7 (Cq), 154.8 (Cq), 135.8 (Cq), 133.2 (Cq), 131.8 (CH), 130.7 (CH), 129.2 (CH, 2C), 127.5 (CH, 2C), 109.7 (CH), 80.2 (Cq), 44.1 (CH2), 40.0 (CH2), 28.6 (CH2), 25.4 (CH3, 3C).
tert-Butyl 4-(2-((3-(Dimethylamino)phenyl)amino)-6-phenylpyrimidin-4-yl)-3,6-dihydropyridine-1(2H)-carboxylate (Compound 39a)
The title compound was synthesized following the general procedure 3 previously described using intermediate 37 (100 mg, 0.27 mmol), aniline 38a (44 mg, 0.32 mmol), Pd(OAc)2 (3.0 mg, 0.013 mmol), (±)-BINAP (8.4 mg, 0.013 mmol), Cs2CO3 (132.0 mg, 0.40 mmol) in 1,4 dioxane (1.8 mL). Purification by silica (elution by gradient from 95/5 to 80/20 cyclohexane/EtOAc) afforded pure intermediate 39a (89 mg, 70% yield). UPLC-MS: tR = 2.42 min (method 2). MS (ESI) m/z: 472.4 [M + H]+, calcd for C28H34N5O2 [M + H]+: 472.3. 1H NMR (400 MHz, CDCl3) δ 8.12–8.02 (m, 2H), 7.55–7.46 (m, 3H), 7.44 (s, 1H), 7.31 (bs, 1H), 7.22 (t, J = 7.9 Hz, 1H), 7.19 (s, 1H), 6.96 (s, 1H), 6.93 (s, 1H), 6.48 (d, J = 8.2 Hz, 1H), 4.18 (q, J = 3.0 Hz, 2H), 3.66 (t, J = 5.6 Hz, 2H), 2.69 (m, 2H), 1.51 (s, 9H).
tert-Butyl 4-(6-Phenyl-2-((3-(trifluoromethyl)phenyl)amino)pyrimidin-4-yl)- dihydropyridine-1(2H)-carboxylate (Compound 39b)
Title compound was synthesized following the general procedure 3 previously described using intermediate 37 (100 mg, 0.27 mmol), aniline 38b (0.04 mL, 0.32 mmol) in 1,4 dioxane (1.8 mL), Pd(OAc)2 (3.0 mg, 0.013 mmol), (±)-BINAP (8.4 mg, 0.013 mmol), and Cs2CO3 (132.0 mg, 0.40 mmol). Purification by silica (elution by gradient from 95/5 to 80/20 cyclohexane/EtOAc) afforded intermediate 39b (83 mg, 62% combined yield of the two isomers: an isomerization of the double bond occurred during the reaction). UPLC-MS: tR = 1.53 min (method 3). MS (ESI) m/z: 497.2 [M + H]+, calcd for C27H28F3N4O2 [M + H]+: 497.2. The mixture was used as such in the next step for the obtainment of intermediate 40b.
tert-Butyl 4-(6-Phenyl-2-((3-(trifluoromethoxy)phenyl)amino)pyrimidin-4-yl)- dihydropyridine-1(2H)-carboxylate (Compound 39c)
The title compound was synthesized following the general procedure 3 previously described using intermediate 37 (100 mg, 0.27 mmol), aniline 38c (0.04 mL, 0.32 mmol) in 1,4 dioxane (1.8 mL), Pd(OAc)2 (2.7 mg, 0.012 mmol), (±)-BINAP (7.4 mg, 0.012 mmol), and Cs2CO3 (131.0 mg, 0.40 mmol). Purification by silica (elution by gradient from 95/5 to 85/15 cyclohexane/EtOAc) afforded intermediate 39c (109 mg, 79% combined yield of the two isomers: an isomerization of the double bond occurred during the reaction). UPLC-MS: there are two main peaks related to the isomers α and β of 39c with double C–C bond shifted tR = 1.53 and 1.68 min (method 3). MS (ESI) m/z: 513.1 [M + H]+, calcd for C27H28F3N4O3 [M + H]+: 513.2. The mixture was used as such in the next step for the obtainment of intermediate 40c.
tert-Butyl 4-(2-((3,4-Dimethoxyphenyl)amino)-6-phenylpyrimidin-4-yl)-dihydropyridine-1(2H)-carboxylate (Compound 39d)
The title compound was synthesized following the general procedure 3 previously described using intermediate 37 (200 mg, 0.54 mmol), aniline 38d (99.0 mg, 0.64 mmol) in 1,4 dioxane (3.6 mL), Pd(OAc)2 (6.0 mg, 0.025 mmol), Xantphos (15.5 mg, 0.025 mmol), Cs2CO3 (263.0 mg, 0.81 mmol). Purification by silica (elution by a gradient from 80/20 to 75/25 cyclohexane/EtOAc) afforded intermediate 39d (197.9 mg, 75% combined yield of the two isomers: an isomerization of the double bond occurred during the reaction). UPLC-MS: tR = 1.97 min (method 2). MS (ESI) m/z: 489.1 [M + H]+, calcd for C28H33N4O4 [M + H]+: 489.2. The mixture was used as such in the next step for the obtainment of intermediate 40d.
tert-Butyl 4-(2-((4-Methoxy-3-(methoxycarbonyl)phenyl)amino)-6-phenylpyrimidin-4-yl)- dihydropyridine-1(2H)-carboxylate (Compound 39e)
The title compound was synthesized following the general procedure 3 previously described using intermediate 37 (200 mg, 0.54 mmol), aniline 38e (117.0 mg, 0.64 mmol) in 1,4 dioxane (3.6 mL), Pd(OAc)2 (6.0 mg, 0.025 mmol), Xantphos (15.5 mg, 0.025 mmol), and Cs2CO3 (263.0 mg, 0.81 mmol). Purification by silica (elution by a gradient from 100/0 to 75/25 cyclohexane/EtOAc) afforded intermediate 39e (236.4 mg, 85% combined yield of the two isomers: an isomerization of the double bond occurred during the reaction). UPLC-MS: tR = 1.92 min (method 2). MS (ESI) m/z: 517.1 [M + H]+, calcd for C29H33N4O5 [M + H]+: 517.2. The mixture was used as such in the next step for the obtainment of intermediate 40e.
tert-Butyl 4-(6-Phenyl-2-(pyridin-4-ylamino)pyrimidin-4-yl)-dihydropyridine-1(2H)-carboxylate (Compound 39f)
The title compound was synthesized following the general procedure 3 previously described using intermediate 37 (100 mg, 0.27 mmol), aniline 38f (30.4 mg, 0.32 mmol) in 1,4 dioxane (1.8 mL), Pd(OAc)2 (2.7 mg, 0.012 mmol), Xantphos (6.9 mg, 0.012 mmol), and Cs2CO3 (131.0 mg, 0.40 mmol). Purification by silica (elution by a gradient from 100/0 to 95/5 DCM/EtOH) afforded intermediate 39f (97.8 mg, 85% combined yield of the two isomers: an isomerization of the double bond occurred during the reaction). UPLC-MS: tR = 1.68 min (method 2). MS (ESI) m/z: 430.1 [M + H]+, calcd for C25H28N5O2 [M + H]+: 430.2. The mixture was used as such in the next step for the obtainment of intermediate 40f.
tert-Butyl 4-(6-Phenyl-2-((2-(trifluoromethyl)pyridin-4-yl)amino)pyrimidin-4-yl)- dihydropyridine-1(2H)-carboxylate (Compound 39g)
The title compound was synthesized following the general procedure 3 previously described using intermediate 37 (100 mg, 0.27 mmol), aniline 38g (52.3 mg, 0.32 mmol) in 1,4 dioxane (1.8 mL), Pd(OAc)2 (2.7 mg, 0.012 mmol), (±)-BINAP (7.4 mg, 0.012 mmol), and Cs2CO3 (131.0 mg, 0.40 mmol). Purification by silica (elution by a gradient from 100/0 to 75/25 cyclohexane/EtOAc) afforded pure intermediate 39g (85.6 mg, 80% combined yield of the two isomers: an isomerization of the double bond occurred during the reaction). UPLC-MS: tR = 2.30 min (method 2). MS (ESI) m/z: 498.1 [M + H]+, calcd for C26H27F3N5O2 [M + H]+: 498.2. The mixture was used as such in the next step for the obtainment of intermediate 40g.
tert-Butyl 4-(2-((2-(Difluoromethoxy)pyridin-4-yl)amino)-6-phenylpyrimidin-4-yl)- dihydropyridine-1(2H)-carboxylate (Compound 39h)
The title compound was synthesized following the general procedure 3 previously described using intermediate 37 (100 mg, 0.27 mmol), aniline 38h (51.7 mg, 0.32 mmol) in 1,4 dioxane (1.8 mL), Pd(OAc)2 (2.7 mg, 0.012 mmol), (±)-BINAP (7.4 mg, 0.012 mmol), Cs2CO3 (131.0 mg, 0.40 mmol). Purification by silica (elution by a gradient from 100/0 to 80/20 cyclohexane/EtOAc) afforded intermediate 39h (85.6 mg, 80% combined yield of the two isomers: an isomerization of the double bond occurred during the reaction). UPLC-MS: tR = 2.30 min (method 2). MS (ESI) m/z: 496.1 [M + H]+, calcd for C26H28F2N5O3 [M + H]+: 496.2. The mixture was used as such in the next step for the obtainment of intermediate 40h.
tert-Butyl 4-(2-((2-Methoxypyridin-4-yl)amino)-6-phenylpyrimidin-4-yl)-3,6-dihydropyridine-1(2H)-carboxylate (Compound 39i)
Title compound was synthesized following the general procedure 3 previously described using intermediate 37 (100 mg, 0.27 mmol), aniline 38i (40.0 mg, 0.32 mmol) in 1,4 dioxane (1.8 mL), Pd(OAc)2 (2.7 mg, 0.012 mmol), Xantphos (6.9 mg, 0.012 mmol), and Cs2CO3 (131.0 mg, 0.40 mmol). Purification by silica (elution by a gradient from 80/20 to 75/25 cyclohexane/EtOAc) afforded intermediate 39i (74.1 mg, 60% combined yield of the two isomers: an isomerization of the double bond occurred during the reaction). UPLC-MS: there are two main peaks related to the isomers tR = 1.95 min and 2.05 (method 2). MS (ESI) m/z: 460.1 [M + H]+, calcd for C26H30N5O3 [M + H]+: 460.2. The mixture was used as such in the next step for the obtainment of intermediate 40i.
tert-Butyl 4-(2-((6-Methoxypyridin-3-yl)amino)-6-phenylpyrimidin-4-yl)-dihydropyridine-1(2H)-carboxylate (Compound 39j)
The title compound was synthesized following the general procedure 3 previously described using intermediate 37 (100 mg, 0.27 mmol), aniline 38j (40.0 mg, 0.32 mmol) in 1,4-dioxane (1.8 mL), Pd(OAc)2 (2.7 mg, 0.012 mmol), Xantphos (6.9 mg, 0.012 mmol), and Cs2CO3 (131.0 mg, 0.40 mmol). Purification by silica (elution by a gradient from 80/20 to 75/25 cyclohexane/EtOAc) afforded intermediate 39j (82.3 mg, 66% combined yield of the two isomers: an isomerization of the double bond occurred during the reaction). UPLC-MS: there are two main peaks related to the isomers tR = 2.01 min and 2.05 (method 2). MS (ESI) m/z: 460.1 [M + H]+, calcd for C26H30N5O3 [M + H]+: 460.2.
tert-Butyl 4-(2-((3-(Dimethylamino)phenyl)amino)-6-phenylpyrimidin-4-yl)piperidine-1-carboxylate (Compound 40a)
The title compound was synthesized following the general procedure 4-Method A previously described using intermediate 39a (60 mg, 0.127 mmol), Pd(OH)2/C (12 mg), and NH4CO2H (48 mg, 0.76 mmol) in MeOH (3.2 mL). Purification by silica (elution by a gradient from 100/0 to 80/20 cyclohexane/EtOAc) afforded pure intermediate 40a (59 mg, 98% yield). UPLC-MS: tR = 2.30 min (method 2). MS (ESI) m/z: 474.3 [M + H]+, calcd for C28H36N5O2 [M + H]+: 474.3. 1H NMR (400 MHz, CDCl3) δ 8.12–8.02 (m, 2H), 7.52 (s, 1H), 7.51–7.43 (m, 3H), 7.20 (t, J = 8.1 Hz, 1H), 7.02 (s, 1H), 6.91 (d, J = 8.0 Hz, 1H), 6.48 (d, J = 8.3 Hz, 1H), 4.27 (bs, 2H), 2.93–2.70 (m, 3H), 1.97 (d, J = 13.0 Hz, 2H), 1.90–1.74 (qd, J = 12.8, 4.2 Hz, 2H), 1.49 (s, 9H).
tert-Butyl 4-(6-Phenyl-2-((3-(trifluoromethyl)phenyl)amino)pyrimidin-4-yl)piperidine-1-carboxylate (Compound 40b)
The title compound was synthesized following the general procedure 4-Method A previously described using intermediate 39b as an isomeric mixture (70 mg, 0.14 mmol), Pd(OH)2/C (14 mg), and NH4CO2H (53 mg, 0.84 mmol) in MeOH (3.5 mL). Purification by silica (elution by a gradient from 100/0 to 85/15 cyclohexane/EtOAc) afforded pure intermediate 40b (59.3 mg, 85% yield). UPLC-MS: tR = 1.49 min (method 3). MS (ESI) m/z: 499.2 [M + H]+, calcd for C27H30F3N4O2 [M + H]+: 499.6. 1H NMR (400 MHz, CDCl3) δ 8.39 (t, J = 2.0 Hz, 1H), 8.10–8.03 (m, 2H), 7.68 (dd, J = 8.2, 2.0 Hz, 1H), 7.54–7.47 (m, 3H), 7.44 (t, J = 8.0 Hz, 1H), 7.30–7.26 (m, 1H), 7.11 (s, 1H), 4.30 (bs, 2H), 2.90–2.84 (m, 2H), 2.80 (tt, J = 11.9, 3.8 Hz, 1H), 1.98 (d, J = 13.1 Hz, 2H), 1.80 (qd, J = 12.3, 4.4 Hz,2H), 1.49 (s, 9H).
tert-Butyl 4-(6-Phenyl-2-((3-(trifluoromethoxy)phenyl)amino)pyrimidin-4-yl)piperidine-1-carboxylate (Compound 40c)
The title compound was synthesized following the general procedure 4-Method A previously described using intermediate 39c as an isomeric mixture (110 mg, 0.21 mmol), Pd(OH)2/C (22 mg), NH4CO2H (79 mg, 1.26 mmol) in MeOH (5.2 mL). Purification by silica (elution by a gradient from 90/10 to 85/15 cyclohexane/EtOAc) afforded pure intermediate 40c (79.5 mg, 72% yield). UPLC-MS: tR = 1.50 min (method 3). MS (ESI) m/z: 515.1 [M + H]+, calcd for C27H30F3N4O3 [M + H]+: 515.6. 1H NMR (400 MHz, CDCl3) δ 8.11–8.01 (m, 3H), 7.58–7.44 (m, 3H), 7.37 (dt, J = 8.2, 1.5 Hz, 1H), 7.33 (t, J = 8.0 Hz, 1H), 7.09 (s, 1H), 6.86 (d, J = 7.9 Hz, 1H), 4.29 (bs, 2H), 2.91–2.83 (m, 2H), (tt, J = 11.9, 3.5 Hz, 1H), 1.98 (d, J = 13.1 Hz, 2H), 1.79 (qd, J = 12.5, 4.3 Hz, 2H), 1.49 (s, 9H).
tert-Butyl 4-(2-((3,4-Dimethoxyphenyl)amino)-6-phenylpyrimidin-4-yl)piperidine-1-carboxylate (Compound 40d)
The title compound was synthesized following the general procedure 4-Method A previously described using intermediate 39d as an isomeric mixture (197 mg, 0.40 mmol), Pd(OH)2/C (39.4 mg), NH4CO2H (151.4 mg, 2.5 mmol) in MeOH (10.0 mL). Purification by silica (elution by a gradient from 100/0 to 75/25 cyclohexane/EtOAc) afforded pure intermediate 40d (174.6 mg, 89% yield). UPLC-MS: tR = 1.91 min (method 2). MS (ESI) m/z: 491.1 [M + H]+, calcd for C28H35N4O4 [M + H]+: 491.3. 1H NMR (400 MHz, CDCl3) δ 8.10–8.00 (m, 2H), 7.85 (s, 1H), 7.64 (d, J = 2.5 Hz, 1H), 7.53–7.45 (m, 3H), 7.04 (dd, J = 8.5, 2.4 Hz, 1H), 7.01 (s, 1H), 6.86 (d, J = 8.7 Hz, 1H), 4.27 (bs, 2H), 3.92 (s, 3H), 3.88 (s, 3H), 2.89–2.81 (m, 2H), 2.79 (tt, J = 11.8, 3.9 Hz, 1H), 1.99 (d, J = 13.1 Hz, 2H), 1.80 (qd, J = 12.5, 4.4 Hz, 2H), 1.48 (s, 9H).
tert-Butyl 4-(2-((4-Methoxy-3-(methoxycarbonyl)phenyl)amino)-6-phenylpyrimidin-4-yl)piperidine-1-carboxylate (Compound 40e)
The title compound was synthesized following the general procedure 4-Method A previously described using intermediate 39e as an isomeric mixture (270 mg, 0.52 mmol), Pd(OH)2/C (54.0 mg), NH4CO2H (196.8 mg, 3.1 mmol) in MeOH (13.0 mL). Purification by silica (elution by a gradient from 100/0 to 75/25 DCM/EtOAc) afforded pure intermediate 6j (371.3 mg, 73% yield). UPLC-MS: tR = 1.88 min (method 2). MS (ESI) m/z: 519.1 [M + H]+, calcd for C29H35N4O5 [M + H]+: 519.2. 1H NMR (400 MHz, CDCl3) δ 8.36 (d, J = 2.9 Hz, 1H), 8.10–8.05 (m, 2H), 7.75 (dd, J = 8.9, 2.9 Hz, 1H), 7.53–7.48 (m, 3H), 7.05 (s, 1H), 7.00 (d, J = 9.0 Hz, 1H), 4.28 (s, 2H), 3.93 (s, 3H), 3.92 (s, 3H), 2.84 (m, 3H), 2.00 (d, J = 13.0 Hz, 2H), 1.80 (qd, J = 12.5, 4.3 Hz, 2H), 1.49 (s, 9H).
tert-Butyl 4-(6-Phenyl-2-(pyridin-4-ylamino)pyrimidin-4-yl)piperidine-1-carboxylate (Compound 40f)
The title compound was synthesized following the general procedure 4-Method B previously described using intermediate 39f as an isomeric mixture (100 mg, 0.23 mmol), Et3SiH (0.37 mL, 2.33 mmol), Pd/C (20 mg) in EtOH (3.8 mL). Purification by silica (elution by a gradient from 90/10 to 85/15 cyclohexane/EtOAc) afforded pure intermediate 40f (79.3 mg, 79% yield). UPLC-MS: tR = 1.34 min (method 2). MS (ESI) m/z: 432.1 [M + H]+, calcd for C25H30N5O2 [M + H]+: 432.5. 1H NMR (400 MHz, CDCl3) δ 8.43 (d, J = 6.4 Hz, 2H), 8.07–8.00 (m, 2H), 7.96 (d, J = 6.1 Hz, 2H), 7.51–7–50 (m, 4H), 7.22 (s, 1H), 4.26 (bs, 2H), 2.96–2.76 (m, 3H), 1.97 (d, J = 12.6 Hz, 2H), 1.79 (qd, J = 12.7, 4.4 Hz, 2H), 1.49 (s, 9H).
tert-Butyl 4-(6-Phenyl-2-((2-(trifluoromethyl)pyridin-4-yl)amino)pyrimidin-4-yl)piperidine-1-carboxylate (Compound 40g)
The title compound was synthesized following the general procedure 4-Method A previously described using intermediate 39g as an isomeric mixture (100 mg, 0.20 mmol), Pd(OH)2/C (20 mg), NH4CO2H (79 mg, 1.26 mmol) in MeOH (5.2 mL). Purification by silica (elution by a gradient from 100/0 to 85/15 cyclohexane/EtOAc) afforded pure intermediate 40g (64.9 mg, 65% yield). UPLC-MS: tR = 2.25 min (method 2). MS (ESI) m/z: 500.1 [M + H]+, calcd for C26H29F3N5O2 [M + H]+: 500.5. 1H NMR (400 MHz, CDCl3) δ 8.57 (d, J = 5.6 Hz, 1H), 8.40 (d, J = 2.2 Hz, 1H), 8.12–8.01 (m, 2H), 7.69 (dd, J = 5.6, 2.2 Hz, 2H), 7.57–7.48 (m, 3H), 7.21 (s, 1H), 4.30 (bs, 2H), 3.00–2.85 (m, 2H), 2.82 (tt, J = 11.9, 3.6 Hz, 1H), 1.98 (d, J = 12.9 Hz, 2H), 1.81 (qd, J = 12.5, 4.3 Hz, 3H), 1.50 (s, 9H).
tert-Butyl 4-(2-((2-(Difluoromethoxy)pyridin-4-yl)amino)-6-phenylpyrimidin-4-yl)piperidine-1-carboxylate (Compound 40h)
The title compound was synthesized following the general procedure 2-Method A previously described using intermediate 39h as an isomeric mixture (75 mg, 0.15 mmol), Pd(OH)2/C (15 mg), NH4CO2H (76.3 mg, 1.21 mmol) in MeOH (5.2 mL). Purification by silica (elution by a gradient from 100/0 to 80/20 cyclohexane/EtOAc) afforded pure intermediate 40h (63.4 mg, 85% yield). UPLC-MS: tR = 2.21 min (method 2). MS (ESI) m/z: 498.1 [M + H]+, calcd for C26H30F2N5O3 [M + H]+: 498.6. 1H NMR (400 MHz, CDCl3) δ 8.11–8.02 (m, 3H), 7.61–7.52 (m, 4H), 7.48 (t, J = 73.3 Hz, 1H), 7.33 (dd, J = 5.8, 1.9 Hz, 1H), 7.20 (s, 1H), 4.30 (bs, 2H), 2.93–2.87 (m, 3H), 2.02 (d, J = 12.6 Hz, 2H), 1.81 (qd, J = 12.4, 4.4 Hz, 2H), 1.49 (s, 9H).
tert-Butyl 4-(2-((2-Methoxypyridin-4-yl)amino)-6-phenylpyrimidin-4-yl)piperidine-1-carboxylate (Compound 40i)
The title compound was synthesized following the general procedure 4-Method A previously described using intermediate 39i as an isomeric mixture (80 mg, 0.17 mmol), Pd(OH)2/C (16 mg), NH4CO2H (64.4 mg, 1.02 mmol) in MeOH (5.5 mL). Purification by silica (elution by a gradient from 85/15 to 75/25 cyclohexane/EtOAc) afforded pure intermediate 40i (64.2 mg, 80% yield). UPLC-MS: tR = 1.95 min (method 2). MS (ESI) m/z: 462.1 [M + H]+, calcd for C26H32N5O3 [M + H]+: 462.2. 1H NMR (400 MHz, CDCl3) δ 8.04 (m, 3H), 7.64 (s, 1H), 7.55–7.49 (m, 3H), 7.47 (bs, 1H), 7.19 (dd, J = 6.0, 2.0 Hz, 1H), 7.15 (s, 1H), 4.29 (bs, 2H), 3.99 (s, 3H), 2.97–2.83 (m, 2H), 2.88 (tt, J = 11.8, 3.6 Hz, 1H), 1.97 (d, J = 12.5 Hz, 2H), 1.81 (qd, J = 12.5, 4.3 Hz, 2H), 1.49 (s, 9H).
tert-Butyl 4-(2-((6-Methoxypyridin-3-yl)amino)-6-phenylpyrimidin-4-yl)piperidine-1-carboxylate (Compound 40j)
The title compound was synthesized following the general procedure 4-Method A previously described using intermediate 39j as an isomeric mixture (80 mg, 0.17 mmol), Pd(OH)2/C (16 mg), NH4CO2H (64.4 mg, 1.02 mmol) in MeOH (5.5 mL). Purification by silica (elution by a gradient from 85/15 to 75/25 cyclohexane/EtOAc) afforded pure intermediate 40j (44.9 mg, 56% yield). UPLC-MS: tR = 1.91 min (method 2). MS (ESI) m/z: 462.1 [M + H]+, calcd for C26H32N5O3 [M + H]+: 462.6. 1H NMR (400 MHz, CDCl3) δ 8.46 (d, J = 2.7 Hz, 1H), 8.06–8.00 (m, 2H), 7.98 (dd, J = 8.9, 2.8 Hz, 1H), 7.52–7.46 (m, 4H), 7.03 (s, 1H), 6.78 (d, J = 9.0 Hz, 1H), 4.27 (s, 2H), 3.95 (s, 3H), 2.93–2.79 (m, 2H), 2.76 (tt, J = 11.9, 3.6 Hz, 1H), 1.97 (d, J = 13.0 Hz, 2H), 1.78 (qd, J = 12.5, 4.2 Hz, 2H), 1.49 (s, 9H).
N-(2-Chloro-6-phenylpyrimidin-4-yl)-1-methyl-1H-indol-6-amine (Compound 42b)
The title compound was synthesized following the general procedure 1 previously described using compound 41 (184 mg, 0.82 mmol), aniline 38k (120 mg, 0.82 mmol), LiHMDS (1.0 M in THF, 2.05 mL) in THF dry (5.5 mL). Purification by silica (elution by a gradient from 100/0 to 95/5 cyclohexane/EtOAc) afforded pure compound 42b (150.0 mg, 55%). UPLC-MS: tR = 2.52 min (method 1). MS (ESI) m/z: 335.2 [M + H]+, calcd for C19H16ClN4 [M + H]+: 335.1. 1H NMR (400 MHz, CDCl3) δ 7.91–7.83 (m, 2H), 7.67 (d, J = 8.3 Hz, 1H), 7.47–7.36 (m, 3H), 7.34 (d, J = 11.3 Hz, 2H), 7.13 (d, J = 3.1 Hz, 1H), 7.04 (dd, J = 8.3, 1.9 Hz, 1H), 6.88 (s, 1H), 6.54 (d, J = 3.1 Hz, 1H), 3.80 (s, 3H).
tert-Butyl 6-((2-Chloro-6-phenylpyrimidin-4-yl)amino)-1H-indole-1-carboxylate (Compound 42c)
The title compound was synthesized following the general procedure 1 previously described using compound 41 (100 mg, 0.82 mmol), aniline 38l (112.0 mg, 0.50 mmol), LiHMDS (1.0 M in THF, 2.05 mL) in THF dry (3.3 mL). Purification by silica (elution by a gradient from 100/0 to 95/5 cyclohexane/EtOAc) afforded pure compound 42c (202.0 mg, 96%). UPLC-MS: tR = 2.17 min (method 2). MS (ESI) m/z: 419.3 [M + H]+, calcd for C23H20ClN4O2 [M + H]+: 419.1. 1H NMR (400 MHz, CDCl3) δ 8.20 (s, 1H), 7.94–7.88 (m, 2H), 7.64 (d, J = 3.7 Hz, 1H), 7.60 (d, J = 8.3 Hz, 1H), 7.48–7.37 (m, 3H), 7.27 (s, 1H), 7.18 (dd, J = 8.3, 2.0 Hz, 1H), 7.04 (s, 1H), 6.60 (dd, J = 3.7, 0.8 Hz, 1H), 1.64 (s, 9H).
tert-Butyl 4-(4-((1-Methyl-1H-indol-6-yl)amino)-6-phenylpyrimidin-2-yl)-3,6-dihydropyridine-1(2H)-carboxylate (Compound 43b)
The title compound was synthesized following the general procedure 2 previously described using intermediate 41 (140.0 mg, 0.42 mmol) boronic ester 36 (155.8 mg, 0.50 mmol), Pd(Cl2)(dppf)·DCM (34.0 mg, 0.04 mmol), K2CO3 (2 M) aq (0.46 mL) in 1,4-dioxane dry (2.8 mL). Purification by silica (elution by a gradient from 100/0 to 75/25 cyclohexane/EtOAc) afforded pure intermediate 43b (175.0 mg, 86%). UPLC-MS: tR = 2.31 min (method 2). MS (ESI) m/z: 480.6 [M-H]−, calcd for C29H32N5O2 [M-H]−: 480.2. 1H NMR (400 MHz, CDCl3) δ 8.02–7.93 (m, 2H), 7.63 (d, J = 8.3 Hz, 1H), 7.53 (s, 1H), 7.46–7.40 (m, 3H), 7.23 (bs, 1H), 7.09 (d, J = 3.1 Hz, 1H), 7.04 (dd, J = 8.3, 1.9 Hz, 1H), 6.99 (s, 1H), 6.90 (s, 1H), 6.51 (dd, J = 3.1, 0.8 Hz, 1H), 4.20 (bs, 2H), 3.78 (s, 3H), 3.66 (bs, 2H), 2.84 (bs, 2H).
tert-Butyl 6-((2-(1-(tert-Butoxycarbonyl)-1,2,3,6-tetrahydropyridin-4-yl)-6-phenylpyrimidin-4-yl)amino)-1H-indole-1-carboxylate (Compound 43c)
The title compound was synthesized following the general procedure 2 previously described using intermediate 42c (190.0 mg, 0.45 mmol) boronic ester 36 (209.4 mg, 0.67 mmol), Pd(Cl2)(dppf)·DCM (36.8 mg, 0.05 mmol), K2CO3 (2 M) aq (0.68 mL) in 1,4 dioxane dry (5 mL). Purification by silica (elution by a gradient from 100/0 to 85/15 cyclohexane/EtOAc) afforded pure intermediate 43c (217.0 mg, 85%). UPLC-MS: tR = 2.31 min (method 2). MS (ESI) m/z: 566.4 [M-H]−, calcd for C33H36N5O4 [M-H]−: 566.3. 1H NMR (400 MHz, CDCl3) δ 8.50 (s, 1H), 8.06–8.0 (m, 2H), 7.58 (d, J = 3.7 Hz, 1H), 7.55 (d, J = 8.3 Hz, 1H), 7.49–7.40 (m, 3H), 7.34 (s, 1H), 7.18 (dd, J = 8.3, 2.0 Hz, 1H), 7.02 (s, 1H), 6.90 (s, 1H), 6.57 (d, J = 3.7 Hz, 1H), 4.22 (bs, 2H), 3.67 (t, J = 5.2 Hz, 2H), 2.85 (bs, 2H), 1.64 (s, 9H), 1.50 (s, 9H).
tert-Butyl 4-(4-((1-Methyl-1H-indol-6-yl)amino)-6-phenylpyrimidin-2-yl)piperidine-1-carboxylate (Compound 44b)
The title compound was synthesized following the general procedure 4-Method A previously described using intermediate 43b (85 mg, 0.18 mmol), Pd(OH)2/C (17.0 mg), NH4CO2H (68.1 mg, 1.1 mmol) in MeOH (4.5 mL). Purification by silica (elution by a gradient from 100/0 to 70/30 cyclohexane/EtOAc) afforded pure intermediate 44b (60.0 mg, 69% yield). UPLC-MS: tR = 2.23 min (method 2). MS (ESI) m/z: 484.5 [M + H]+, calcd for C29H34N5O2 [M + H]+: 484.6. 1H NMR (400 MHz, CDCl3) δ 7.97–7.87 (m, 2H), 7.63 (d, J = 8.4 Hz, 1H), 7.50 (s, 1H), 7.45–7.36 (m, 3H), 7.09 (dd, J = 3.1, 1.3 Hz, 1H), 7.03 (dd, J = 8.3, 1.9 Hz, 1H), 6.94 (bs, 1H), 6.87 (d, J = 1.2 Hz, 1H), 6.51 (dd, J = 3.1, 0.9 Hz, 1H), 4.23 (bs, 2H), 3.78 (s, 3H), 2.96–2.90 (m, 3H), 2.08 (bs, 2H), 1.93 (qd, J = 12.2, 3.9 Hz, 2H), 1.49 (s, 9H).
tert-Butyl 6-((2-(1-(tert-Butoxycarbonyl)piperidin-4-yl)-6-phenylpyrimidin-4-yl)amino)-1H-indole-1-carboxylate (Compound 44c)
The title compound was synthesized following the general procedure 4-Method A previously described using intermediate 43c (220.0 mg, 0.39 mmol), Pd(OH)2/C (44.0 mg), NH4CO2H (147.7 mg, 2.3 mmol) in MeOH (5.0 mL). Purification by silica (elution by a gradient from 100/0 to 75/25 cyclohexane/EtOAc) afforded pure intermediate 44c (69.2 mg, 32% yield). The yield has been affected by the formation of the over-reduced indoline derivative, which was separated by desired 44c by purification by silica. UPLC-MS: tR = 1.56 min (method 3). MS (ESI) m/z: 570.4 [M + H]+, calcd for C33H40N5O4 [M + H]+: 570.3. 1H NMR (400 MHz, CDCl3) δ 8.28 (s, 1H), 8.01–7.93 (m, 2H), 7.59 (d, J = 3.7 Hz, 1H), 7.57 (d, J = 8.3 Hz, 1H), 7.46–7.39 (m, 3H), 7.22 (dd, J = 8.3, 2.0 Hz, 1H), 7.09 (bs), 7.03 (s, 1H), 6.57 (d, J = 3.7 Hz, 1H), 4.23 (bs, 2H), 3.03–2.82 (m, 3H), 2.09 (d, J = 13.3 Hz, 2H), 1.92 (qd, J = 12.5, 3.9 Hz, 2H), 1.63 (s, 9H), 1.49 (s, 9H).
In Vitro and In Vivo Experiments
Cell Viability
Cell viability assay was performed as previously described.22 Cancer cell lines were cultured according to the method described in Jahid et al., 2022.22
In Vivo Efficacy
An in vivo efficacy test was performed as described.22,23 All animal experiments were approved by the UC Irvine Institutional Animal Care and Use Committee (IACUC) (AUP-23-116). NOD.Cg-Prkdcscid IL2rgtm1Wjl/SzJ (NSG) mice were purchased from The Jackson Laboratories (stock number 005557); both male and female mice were used in vehicle and inhibitor-treated groups (N = 6). After tumors reached the initial size range of 150–250 mm3, mice were administrated with 10 mg/kg ARN25499, or vehicle via tail vein injection for 2 weeks daily. Tumors were measured every other day with a caliper and tumor volume was calculated ((length × width × width)/2). At the end of 2 weeks, tumors were extracted and measured to determine volume (length × width × height). GraphPad Prism9 software was used to generate line and scatterplot graphs and determine significance using two-way ANOVA and unpaired two-tailed t-test. An equal number of males and females were used in the experiments.
In Vitro Treatment and Western Blot Analysis
Immunoblotting
Melanoma cells (WM3248) were cultured as described22 and were seeded at 1.5 × 106 cells per 60.8 cm2 plate. Sixteen hrs. later, WM3248 cells were treated with 10 μM of ARN22089, ARN25375, or ARN25499 for 6 h. and lysed in Lysis buffer (Cytoskeleton, Inc.) containing a protease inhibitors cocktail. Lysates were then subjected to SDS-PAGE and transferred to PVDF membranes. The expression or phosphorylation of proteins was detected by Western blotting using the following primary Abs: pERK at 1:500, ERK at 1:6000, pS6 at 1:6000, S6 at 1:1000 for 16 h and appropriate HRP-secondary antibody for 2 h (all from Cell Signaling). ImageJ was used to perform densitometry. The experiments were performed in triplicate and representative results are shown.
Biophysical Methods
His-CDC42 Production and Purification
His-CDC42 wild-type (amino acids Ile4-Pro182) was expressed from the pET28a+ vector in E. coli BL21 (DE3) cells and purified, GppNHp or GDP-bound, as previously described.22,23
Binding Check of Hit Derivatives by Microscale Thermophoresis
MicroScale Thermophoresis experiments were performed according to the NanoTemper technologies protocols in a Monolith NT.115 Pico (Pico Red/Nano Blue - NanoTemper Technologies). His-CDC42 was RED – NHS labeled and used at a concentration of 10 nM. The compound concentration was 50 μM throughout all the experiments. DMSO was also constant across samples at 0.5% v/v. Solutions were prepared in 100 mM Trizma base (Sigma) pH 7.5, 40 mM NaCl, 0.05% v/v Tween 20, and incubated 5 min before loading on Premium Capillaries and analysis. Binding was detected at 24 °C, MST power high, and 20% LED power. The MST traces were recorded as follows: 3 s MST power off, 20 s MST power on, and 1 s MST power off. The difference in normalized fluorescence (ΔFnorm [‰] = Fhot/Fcold) between a protein:compound sample and a protein only sample at 1.5–2.5 s and 14.0–15.0 s is calculated and plotted through MO.Affinity analysis v2.3 (NanoTemper Technologies) and GraphPad Prism 8.0.0 (GraphPad Software, San Diego, California USA). Signal-to-noise ratio and response amplitude were used to evaluate the quality of the binding data according to NanoPedia instructions (NanoTemper Technologies). Only a signal-to-noise ratio of more than 5 and a response amplitude of more than 1.5 were considered acceptable, while a signal-to-noise of more than 12 was considered excellent.
NMR Confirmation of Target Engagement
The NMR experiments were performed as reported in Brindani et al. 202323 using a 5 mm CryoProbe QCI 1H/19F–13C/15N–D quadruple resonance, a shielded z-gradient coil, and an automatic sample changer SampleJet 600 MHz NMR system with temperature control. For all samples, a 1D 1H NMR experiment was recorded, and the water suppression was obtained using the standard NOESY (nuclear Overhauser effect spectroscopy) presat Bruker pulse sequence, with 64 k data points, a spectral width (sw) of 30 ppm, 64 scans, acquisition time (aq) of 1.835 s, a relaxation delay (d1) of 4 s and a mixing time of 10 ms. The WaterLOGSY experiments were achieved with a 7.5 ms long 180° Gaussian-shaped pulse, aq 0.852 s, mixing time of 1.7 s, relaxation delay of 2 s, and 1024 scans. 19F T2 filter experiments were recorded using a CPMG spin–echo scheme with a 35 ms time interval between the 180 pulses and different total lengths (140 and 240 ms, respectively), 32 scans, sw 40 ppm, aq 0.72s, and d1 5 s. The data were multiplied with an exponential window function with 1 Hz line broadening prior to Fourier transformation for 1H 1D, and 19F T2 filter experiments and 2 Hz line broadening for the WaterLOGSY experiments. The solubility of the compounds was evaluated both in PBS and Tris buffer by 1H 1D experiments and aggregation by WaterLOGSY, testing the compounds in the binding assays buffer at the theoretical concentrations of 10, 25, and 50 in the presence of 200 μM 4-trifluoromethyl benzoic acid (internal reference).
For the binding experiments, all the compounds were tested at 50 μM in 20 mM Trizma base (Sigma) pH 7.5, 40 mM NaCl, 5 mM MgCl2, 5 μM EDTA, 10% D2O (for the lock signal), and in the presence of 2 μM GppNHp or GDP and 2 μM His-CDC42 (loaded with GDP) or His-CDC42 (loaded with GppNHp). The total amount of DMSO-d6 in all samples was 1%. All fluorine chemical shifts were referred to the CFCl3signal in water.
Computational Methods
Molecular Docking
To predict and evaluate the interaction between CDC42 and ARN25499/ARN25375 compounds, we first performed molecular docking. As the receptor structure, we employed the CDC42 protein in complex with the CRIB domain of PAK6 (PDB code 2ODB, resolution of 2.4 Å) where we formerly identified a previously unappreciated allosteric pocket at the protein–protein interface.22 The structure was refined by using the Protein Preparation Wizard39 workflow implemented in Maestro Release 2021–3. Specifically, hydrogen atoms were added, and charges and protonation states were assigned titrating the protein at physiologic pH. The steric clashes were relieved by performing a small number of minimization steps until the RMSD of the non-hydrogen atoms reached 0.30 Å. The formerly identified pocket was used to center the grid. Precisely, the cubic grid box of 26 × 26 × 26 Å3 was centered on the previously identified hit compound of the CDC42-ligand complex.22 The compound was prepared using LigPrep software implemented in Maestro. First, we added hydrogens and generated ionization states at pH 7.4 ± 0.5. Then, we generated tautomers and all stereochemical isomers. Finally, we used Glide40−42 to perform the molecular docking, using Extra Precision and retaining a maximum of 20 poses. The best docking pose was chosen for further investigation through classical MD simulation.
Molecular Dynamics Simulations
Molecular dynamics (MD) simulations were performed on the protein–ligand complexes obtained from our docking calculations. The GTP substrate as well as the catalytic Mg2+ ion in the active site of the proteins were considered. The systems were hydrated with a 14 Å layer of TIP3P water molecules43 from the protein center. The coordinates of the water molecules at the catalytic center were taken from the PDB X-ray structure 2ODB. Sodium ions were added to neutralize the charge of the systems. The final models are enclosed in a box of ∼89 × 89 × 89 Å3, containing ∼18,800 water molecules, resulting in ∼59,000 atoms for each system. The AMBER-ff14SB force field44 was used for the parametrization of the protein. The parameters for the ligands ARN25499 and ARN25375 were determined via Hartree–Fock calculation, with 6-31G* basis set, convergence criterium SCF = Tight after structure optimization (DFT B3LYP functional; 6-31G* basis set). The Merz–Singh–Kollman scheme45 was used for the atomic charge assignment. The GTP and the Mg2+ were parametrized according to Meagher et al. and Allner et al. respectively.46,47 Joung-Chetham parameters were used for monovalent ions.48 All MD simulations were performed with Amber49 and all the systems were objects of the following equilibration protocol. To relax the water molecule and the ions, we performed an energy minimization imposing a harmonic potential of 300 kcal/mol Å2 on the backbone, the GTP, and the docked compounds. Then, two consecutive MD simulations in NVT and NPT ensembles (1 and 10 ns, respectively) were carried out, imposing the previous positional restraints. To relax the solute, two additional energy minimization steps were performed imposing positional restraints of 20 kcal/mol Å2 and without any restraints, respectively. Such minimized systems were heated up to 303 K with four consecutive MD simulations in NVT (∼0.1 ns, 100 K) and NPT ensembles (∼0.1 ns, 100 K; ∼0.1 ns, 200 K; ∼0.2 ns, 303 K), imposing the previous positional restraints of 20 kcal/mol Å2. We used the Andersen-like temperature-coupling scheme50 while pressure control was achieved with Monte Carlo barostat at a reference pressure of 1 atm. Long-range electrostatics were treated with the particle mesh Ewald method. We performed an additional MD simulation (∼1.5 ns) in the NPT ensemble at 303 K without any restraint to relax the system at such a temperature. Finally, multiple replicas of 500 ns were performed in the NPT ensemble for each system with an integration time step of 2 fs.
Acknowledgments
We thank the Italian Association for Cancer Research (AIRC) for financial support (IG 23679). We thank S. Venzano for the preparation of the plates with the compounds’ solution for the screening.
Glossary
Abbreviations
- DCM
dichloromethane
- DMF
N,N-dimethylformamide
- DMSO
dimethyl sulfoxide
- EtOAc
ethyl acetate
- MST
microscale thermophoresis
- THF
tetrahydrofuran
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.4c00855.
1H, 13C, 19F NMR spectral data and chromatography analysis of key compounds; MST and NMR analyses for binding evaluation; procedures for aqueous kinetic and thermodynamic solubility, in vitro metabolic stability, and in vitro plasmatic stability; and pharmacokinetic studies (PDF)
Molecular formula strings (CSV)
Author Contributions
# N.B. and L.M.V. contributed equally to this work.
The authors declare the following competing financial interest(s): A.K.G., M.D.V., N.B., L.M.V., A.M. are co-inventors on patents related to this work.
Supplementary Material
References
- Bray K.; Gillette M.; Young J.; Loughran E.; Hwang M.; Sears J. C.; Vargo-Gogola T. Cdc42 overexpression induces hyperbranching in the developing mammary gland by enhancing cell migration. Breast Cancer Res. 2013, 15 (5), R91. 10.1186/bcr3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy N. P.; Binti Ahmad Mokhtar A. M.; Mott H. R.; Owen D. Molecular subversion of Cdc42 signalling in cancer. Biochem. Soc. Trans. 2021, 49 (3), 1425–1442. 10.1042/BST20200557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv J.; Song Y. Could cell division cycle protein 42 be a target for lung cancer treatment?. Transl Cancer Res. 2019, 8 (1), 312–318. 10.21037/tcr.2019.01.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.; Craig B. T.; Romain C. V.; Qiao J.; Chung D. H. Silencing of CDC42 inhibits neuroblastoma cell proliferation and transformation. Cancer Lett. 2014, 355 (2), 210–216. 10.1016/j.canlet.2014.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez del Pulgar T.; Benitah S. A.; Valeron P. F.; Espina C.; Lacal J. C. Rho GTPase expression in tumourigenesis: evidence for a significant link. Bioessays 2005, 27 (6), 602–613. 10.1002/bies.20238. [DOI] [PubMed] [Google Scholar]
- Haga R. B.; Ridley A. J. Rho GTPases: Regulation and roles in cancer cell biology. Small GTPases 2016, 7 (4), 207–221. 10.1080/21541248.2016.1232583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melendez J.; Grogg M.; Zheng Y. Signaling role of Cdc42 in regulating mammalian physiology. J. Biol. Chem. 2011, 286 (4), 2375–2381. 10.1074/jbc.R110.200329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hercyk B. S.; Rich-Robinson J.; Mitoubsi A. S.; Harrell M. A.; Das M. E. A novel interplay between GEFs orchestrates Cdc42 activity during cell polarity and cytokinesis in fission yeast. J. Cell Sci. 2019, 132 (23), jcs236018 10.1242/jcs.236018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray J. L.; von Delft F.; Brennan P. E. Targeting the Small GTPase Superfamily through Their Regulatory Proteins. Angew. Chem., Int. Ed. Engl. 2020, 59 (16), 6342–6366. 10.1002/anie.201900585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotteret S.; Chernoff J. The evolutionary history of effectors downstream of Cdc42 and Rac. Genome Biol. 2002, 3 (2), REVIEWS0002 10.1186/gb-2002-3-2-reviews0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy N. P.; Mott H. R.; Owen D. Progress in the therapeutic inhibition of Cdc42 signalling. Biochem. Soc. Trans. 2021, 49 (3), 1443–1456. 10.1042/BST20210112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado M. D. M.; Dharmawardhane S. Targeting Rac and Cdc42 GTPases in Cancer. Cancer Res. 2018, 78 (12), 3101–3111. 10.1158/0008-5472.CAN-18-0619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado M. D. M.; Medina J. I.; Velazquez L.; Dharmawardhane S. Targeting Rac and Cdc42 GEFs in Metastatic Cancer. Front. Cell Dev. Biol. 2020, 8, 201. 10.3389/fcell.2020.00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X. H.; Lv L. C.; Duan J.; Wu Y. M.; He S. J.; Hu Z. Z.; Xiong L. X. Regulating Cdc42 and Its Signaling Pathways in Cancer: Small Molecules and MicroRNA as New Treatment Candidates. Molecules 2018, 23 (4), 787. 10.3390/molecules23040787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesland A.; Zhao Y.; Chen Y. H.; Wang L.; Zhou H.; Lu Q. Small molecule targeting Cdc42-intersectin interaction disrupts Golgi organization and suppresses cell motility. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (4), 1261–1266. 10.1073/pnas.1116051110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar B. J.; Zhao Y.; Zhou H.; Huo S.; Chen Y. H.; Lu Q. Inhibition of Cdc42-intersectin interaction by small molecule ZCL367 impedes cancer cell cycle progression, proliferation, migration, and tumor growth. Cancer Biol. Ther 2019, 20 (6), 740–749. 10.1080/15384047.2018.1564559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W.; Du W.; Shang X.; Wang L.; Evelyn C.; Florian M. C.; Ryan M. A.; Rayes A.; Zhao X.; Setchell K.; Meller J.; Guo F.; Nassar N.; Geiger H.; Pang Q.; Zheng Y. Rational identification of a Cdc42 inhibitor presents a new regimen for long-term hematopoietic stem cell mobilization. Leukemia 2019, 33 (3), 749–761. 10.1038/s41375-018-0251-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zins K.; Gunawardhana S.; Lucas T.; Abraham D.; Aharinejad S. Targeting Cdc42 with the small molecule drug AZA197 suppresses primary colon cancer growth and prolongs survival in a preclinical mouse xenograft model by downregulation of PAK1 activity. J. Transl. Med. 2013, 11, 295. 10.1186/1479-5876-11-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y.; Dickerson J. B.; Guo F.; Zheng J.; Zheng Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc. Natl. Acad. Sci. U. S. A. 2004, 101 (20), 7618–7623. 10.1073/pnas.0307512101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller P. M.; Rademacher J.; Bagshaw R. D.; Wortmann C.; Barth C.; van Unen J.; Alp K. M.; Giudice G.; Eccles R. L.; Heinrich L. E.; Pascual-Vargas P.; Sanchez-Castro M.; Brandenburg L.; Mbamalu G.; Tucholska M.; Spatt L.; Czajkowski M. T.; Welke R. W.; Zhang S.; Nguyen V.; Rrustemi T.; Trnka P.; Freitag K.; Larsen B.; Popp O.; Mertins P.; Gingras A. C.; Roth F. P.; Colwill K.; Bakal C.; Pertz O.; Pawson T.; Petsalaki E.; Rocks O. Systems analysis of RhoGEF and RhoGAP regulatory proteins reveals spatially organized RAC1 signalling from integrin adhesions. Nat. Cell Biol. 2020, 22 (4), 498–511. 10.1038/s41556-020-0488-x. [DOI] [PubMed] [Google Scholar]
- Dütting S.; Heidenreich J.; Cherpokova D.; Amin E.; Zhang S. C.; Ahmadian M. R.; Brakebusch C.; Nieswandt B. Critical off-target effects of the widely used Rac1 inhibitors NSC23766 and EHT1864 in mouse platelets. J. Thromb. Haemost. 2015, 13, 827–838. 10.1111/jth.12861. [DOI] [PubMed] [Google Scholar]
- Jahid S.; Ortega J. A.; Vuong L. M.; Acquistapace I. M.; Hachey S. J.; Flesher J. L.; La Serra M. A.; Brindani N.; La Sala G.; Manigrasso J.; Arencibia J. M.; Bertozzi S. M.; Summa M.; Bertorelli R.; Armirotti A.; Jin R.; Liu Z.; Chen C. F.; Edwards R.; Hughes C. C. W.; De Vivo M.; Ganesan A. K. Structure-based design of CDC42 effector interaction inhibitors for the treatment of cancer. Cell Rep 2022, 39 (4), 110760 10.1016/j.celrep.2022.110760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brindani N.; Vuong L. M.; Acquistapace I. M.; La Serra M. A.; Ortega J. A.; Veronesi M.; Bertozzi S. M.; Summa M.; Girotto S.; Bertorelli R.; Armirotti A.; Ganesan A. K.; De Vivo M. Design, Synthesis, In Vitro and In Vivo Characterization of CDC42 GTPase Interaction Inhibitors for the Treatment of Cancer. J. Med. Chem. 2023, 66 (8), 5981–6001. 10.1021/acs.jmedchem.3c00276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stengel K. R.; Zheng Y. Essential role of Cdc42 in Ras-induced transformation revealed by gene targeting. PLoS One 2012, 7 (6), e37317 10.1371/journal.pone.0037317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C. M.; Li H.; Gasman S.; Huang J.; Schiff R.; Chang E. C. Compartmentalized Ras proteins transform NIH 3T3 cells with different efficiencies. Mol. Cell. Biol. 2011, 31 (5), 983–997. 10.1128/MCB.00137-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalvit C.; Caronni D.; Mongelli N.; Veronesi M.; Vulpetti A. NMR-based quality control approach for the identification of false positives and false negatives in high throughput screening. Curr. Drug Discov Technol. 2006, 3 (2), 115–124. 10.2174/157016306778108875. [DOI] [PubMed] [Google Scholar]
- Seidel S. A.; Dijkman P. M.; Lea W. A.; van den Bogaart G.; Jerabek-Willemsen M.; Lazic A.; Joseph J. S.; Srinivasan P.; Baaske P.; Simeonov A.; Katritch I.; Melo F. A.; Ladbury J. E.; Schreiber G.; Watts A.; Braun D.; Duhr S. Microscale thermophoresis quantifies biomolecular interactions under previously challenging conditions. Methods 2013, 59 (3), 301–315. 10.1016/j.ymeth.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mureddu L. G.; Vuister G. W. Fragment-Based Drug Discovery by NMR. Where Are the Successes and Where can It Be Improved?. Front Mol. Biosci 2022, 9, 834453 10.3389/fmolb.2022.834453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalvit C.; Flocco M.; Veronesi M.; Stockman B. J. Fluorine-NMR competition binding experiments for high-throughput screening of large compound mixtures. Comb Chem. High Throughput Screen 2002, 5 (8), 605–611. 10.2174/1386207023329923. [DOI] [PubMed] [Google Scholar]
- Dalvit C.; Piotto M. (19) F NMR transverse and longitudinal relaxation filter experiments for screening: a theoretical and experimental analysis. Magn. Reson. Chem. 2017, 55 (2), 106–114. 10.1002/mrc.4500. [DOI] [PubMed] [Google Scholar]
- Dalvit C.; Pevarello P.; Tato M.; Veronesi M.; Vulpetti A.; Sundstrom M. Identification of compounds with binding affinity to proteins via magnetization transfer from bulk water. J. Biomol NMR 2000, 18 (1), 65–68. 10.1023/A:1008354229396. [DOI] [PubMed] [Google Scholar]
- Dalvit C. NMR methods in fragment screening: theory and a comparison with other biophysical techniques. Drug Discov Today 2009, 14 (21–22), 1051–1057. 10.1016/j.drudis.2009.07.013. [DOI] [PubMed] [Google Scholar]
- Troll T. Reduction Potentials of Substituted as-Triazines and s-Tetrazines in acetonitrile. Electrochim. Acta 1982, 27 (9), 1311–1314. 10.1016/0013-4686(82)80153-9. [DOI] [Google Scholar]
- Chiacchio M. A.; Iannazzo D.; Romeo R.; Giofre S. V.; Legnani L. Pyridine and Pyrimidine Derivatives as Privileged Scaffolds in Biologically Active Agents. Curr. Med. Chem. 2020, 26 (40), 7166–7195. 10.2174/0929867325666180904125400. [DOI] [PubMed] [Google Scholar]
- Sander K.; Kottke T.; Tanrikulu Y.; Proschak E.; Weizel L.; Schneider E. H.; Seifert R.; Schneider G.; Stark H. 2,4-Diaminopyrimidines as histamine H4 receptor ligands--Scaffold optimization and pharmacological characterization. Bioorg. Med. Chem. 2009, 17 (20), 7186–7196. 10.1016/j.bmc.2009.08.059. [DOI] [PubMed] [Google Scholar]
- Mowbray C. E.; Bell A. S.; Clarke N. P.; Collins M.; Jones R. M.; Lane C. A.; Liu W. L.; Newman S. D.; Paradowski M.; Schenck E. J.; Selby M. D.; Swain N. A.; Williams D. H. Challenges of drug discovery in novel target space. The discovery and evaluation of PF-3893787: a novel histamine H4 receptor antagonist. Bioorg. Med. Chem. Lett. 2011, 21 (21), 6596–6602. 10.1016/j.bmcl.2011.07.125. [DOI] [PubMed] [Google Scholar]
- Hammer S. G.; Gobleder S.; Naporra F.; Wittmann H. J.; Elz S.; Heinrich M. R.; Strasser A. 2,4-Diaminopyrimidines as dual ligands at the histamine H1 and H4 receptor-H1/H4-receptor selectivity. Bioorg. Med. Chem. Lett. 2016, 26 (2), 292–300. 10.1016/j.bmcl.2015.12.035. [DOI] [PubMed] [Google Scholar]
- Natarajan R.; Anthoni Samy H. N.; Sivaperuman A.; Subramani A. Structure-Activity Relationships of Pyrimidine Derivatives and their Biological Activity - A Review. Med. Chem. 2022, 19 (1), 10–30. 10.2174/1573406418666220509100356. [DOI] [PubMed] [Google Scholar]
- Sastry G. M.; Adzhigirey M.; Day T.; Annabhimoju R.; Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27 (3), 221–234. 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- Friesner R. A.; Murphy R. B.; Repasky M. P.; Frye L. L.; Greenwood J. R.; Halgren T. A.; Sanschagrin P. C.; Mainz D. T. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49 (21), 6177–6196. 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- Halgren T. A.; Murphy R. B.; Friesner R. A.; Beard H. S.; Frye L. L.; Pollard W. T.; Banks J. L. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47 (7), 1750–1759. 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; Shaw D. E.; Francis P.; Shenkin P. S. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47 (7), 1739–1749. 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79 (2), 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Maier J. A.; Martinez C.; Kasavajhala K.; Wickstrom L.; Hauser K. E.; Simmerling C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput 2015, 11 (8), 3696–3713. 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh U. C.; Kollman P. A. An approach to computing electrostatic charges for molecules. J. Comput. Chem. 1984, 5 (2), 129–145. 10.1002/jcc.540050204. [DOI] [Google Scholar]
- Meagher K. L.; Redman L. T.; Carlson H. A. Development of polyphosphate parameters for use with the AMBER force field. J. Comput. Chem. 2003, 24 (9), 1016–1025. 10.1002/jcc.10262. [DOI] [PubMed] [Google Scholar]
- Allner O.; Nilsson L.; Villa A. Magnesium Ion-Water Coordination and Exchange in Biomolecular Simulations. J. Chem. Theory Comput 2012, 8 (4), 1493–1502. 10.1021/ct3000734. [DOI] [PubMed] [Google Scholar]
- Joung I. S.; Cheatham T. E. Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J. Phys. Chem. B 2008, 112 (30), 9020–9041. 10.1021/jp8001614. [DOI] [PMC free article] [PubMed] [Google Scholar]; third,
- Case D. A.; Aktulga H. M.; Belfon K. A. A.; Ben-Shalom I.; Berryman J. T.; Brozell S. R.; Cerutti D. S.; Cheatham T. E.; Cisneros G. A.; Cruzeiro V. W. D.; Darden T. A.; Duke R. E.; Giambasu G.; Gilson M. K.; Gohlke H.; Goetz A. W.; Harris R.; Izadi S.; Izmailov S. A.; Kasavajhala K.; Kaymak M. C.; King E.; Kovalenko A.; Kurtzman T.; Lee T. S.; LeGrand S.; Li P.; Lin C.; Liu J.; Luchko T.; Luo R.; Machado M.; Man V.; Manathunga M.; Merz K. M.; Miao Y.; Mikhailovskii O.; Monard G.; Nguyen H.; O’Hearn K. A.; Onufriev A.; Pan F.; Pantano S.; Qi R.; Rahnamoun A.; Roe D. R.; Roitberg A.; Sagui C.; Schott-Verdugo S.; Shajan A.; Shen J.; Simmerling C. L.; Skrynnikov N.; Smith J.; Swails J. M.; Walker R. C.; Wang J.; Wang J.; Wei H.; Wolf R. M.; Wu X.; Xiong Y.; Xue Y.; York D. M.; Zhao S.; Kollman P. A.. Amber; University of California: San Francisco, 2022. [Google Scholar]
- Andrea T. A.; Swope W. C.; Andersen H. C. The role of long ranged forces in determining the structure and properties of liquid water. J. Chem. Phys. 1983, 79, 4576–4584. 10.1063/1.446373. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.