

Abstract
The polymeric implant material poly(lactide-co-glycolide) (PLGA) degrades by a process of bulk degradation, which allows it to be used for the controlled release of therapeutic molecules from implants and microspheres. The temporal characterization of PLGA microsphere degradation has been limited by the need to destructively monitor the samples at each time point. In this study, a noninvasive imaging technology, optical coherence tomography (OCT), was utilized to characterize the in situ degradation of PLGA microspheres suspended within photo-crosslinked hyaluronic acid (HA) hydrogels. Microspheres with differing degradation rates were loaded with bovine serum albumin (BSA) as a marker protein, and temporal release of protein was correlated with morphological changes observed during 3-D OCT imaging. As proof-of-principle, a microsphere-loaded hydrogel scaffold was implanted in a modified rat calvarial critical size defect model and imaged using OCT. This animal model presents the opportunity to monitor microsphere degradation over time in living animals.
Index Terms—: Optical coherence tomography (OCT)
I. Introduction
OPTICAL coherence tomography (OCT) has been used since the early 1990s to image samples of biological relevance [1]. Its superb spatial resolution (<10 μm) and moderate penetration depth in biological tissue (1–2 mm) offer advantages over other high-resolution noninvasive optical imaging techniques [2]. OCT utilizes low coherence light to perform optical ranging, and both 2-D and 3-D imaging can then be performed noninvasively and in real time. OCT has primarily been applied to distinguish normal from pathologic structures both in vivo and ex vivo, in tissues such as esophagus [3], artery [4], cartilage [5], etc.; however, OCT has only recently become an attractive technique to monitor and evaluate therapies, particularly when using a tissue engineering or regenerative medicine approach. For example, OCT has been used to examine the bone/cartilage interface after chondrocyte implantation into a cartilage defect [6], image bone tissue engineering constructs in vitro [7], [8], and measure the response of tendon cells to different in vitro culture conditions [9]. In the present study, the use of OCT is further extended to characterize temporal changes within the scaffold materials themselves. These morphological changes in the scaffolds are linked to their potential to promote healing, particularly through variations in the release rate of protein therapeutics, which is controlled by the degradation of protein-loaded poly(lactide-co-glycolide) (PLGA) microspheres.
The PLGA microsphere and hyaluronic acid (HA) hydrogel composite system presented here is being developed for the delivery of bioactive molecules for bone repair applications. Local and controlled delivery is essential for the presentation of molecules that induce cellular proliferation, migration, and/or differentiation in a wound site because it minimizes the chance of side effects at remote locations and reduces the overall dose needed. PLGA has been used for over 40 years as a degradable implant material [10]. Hydration of polymeric implants leads to cleavage of the PLGA chains into low molecular weight fragments that can eventually be dissolved and cleared. The ratio of lactide to glycolide, enantiomeric form, crystallinity, and initial molecular weight of the polymer all affect the degradation rate. In vitro studies by Vert and coworkers have shown that heterogeneous degradation occurs in PLGA devices, with a faster degradation rate in the core than at the surface due to an autocatalytic effect from degradation products trapped within the core [11]–[16]. The degradation of PLGA microspheres has also been well characterized in vivo [17]–[19]. However, all of these studies used destructive end point microscopy techniques to monitor morphological changes. The ability of OCT to non-destructively image microsphere morphology in situ could provide a means of monitoring microsphere degradation and directly tie morphological changes to therapeutic protein release, which would be especially beneficial for comparisons between the in vitro and in vivo performance of such drug delivery systems.
This study seeks to develop a meaningful model for noninvasive in vivo evaluation of drug delivery systems for bone repair. The first goal was to use OCT to measure changes in implant morphology in an in vitro protein release assay, which would determine if quantifiable metrics to evaluate implant performance could be obtained. While measuring the rate of protein release in vitro is standard, directly measuring local protein concentration in vivo is difficult due to diffusion from the site and background contributions from endogenous proteins. The hypothesis of this research is that measurements obtained from OCT images, such as particle size, morphology, or overall number, could provide a novel way to characterize implant degradation, which relates to protein release. These measurements of microsphere morphology could eventually be carried out in vivo. Therefore, the second goal of the study was to demonstrate that a rat calvarial critical size defect model could be modified in such a way that noninvasive imaging using OCT could be performed, that appropriate imaging parameters could be identified, and that implant morphology could be seen in the resulting images. A critical size defect will not heal on its own [20], and the model is the gold standard used to evaluate treatments for bone regeneration [21]. This model further has the advantage of being in a nonweight-bearing location. While these measurements could eventually be carried out in vivo in the calvarial defect model, the in vitro results also have a broader impact for the characterization of drug delivery systems targeting a range of tissue types.
II. Materials and Methods
A. Materials
All reagents were obtained from Sigma-Aldrich (St. Louis, MO) unless otherwise noted.
B. Preparation of Glycidyl Methacrylate Modified Hyaluronic Acid (HA-GMA)
HA (220 000 Da, Lifecore, Chaska, MN) was modified with glycidyl methacrylate (GMA) to conjugate reactive vinyl groups, similar to previously published techniques [22]. An aqueous solution of HA at pH 9 was reacted with a 29-fold molar excess of GMA for seven days at room temperature. The HA-GMA was purified by 2 × precipitation in tetrahydrofuran (Fisher Scientific, Hampton, NH), redissolved in DI water, and lyophilized. The degree of substitution (number of methacrylate groups per 100 disaccharide units) was determined by 1H-NMR using an Avance DRX-499 (Bruker, Billerica, MA).
C. Preparation of Protein-Loaded PLGA Microspheres
PLGA microspheres were fabricated using the ProLease technique [23]. Briefly, BSA was micronized by spray freeze-drying a solution of 10 mg/mL BSA and 2 mg/mL trehalose in 5 mM sodium succinate (pH 5). The micronized BSA/trehalose was suspended by ultrasonication at 11 mg/mL in a methylene chloride (Fisher) solution containing 100 mg/mL PLGA (inherent viscosity (I.V.) = 0.17 dL/g or 0.39 dL/g, Absorbable Polymers International, Pelham, AL) and was then sprayed through an ultrasonic nozzle to produce atomized droplets. These droplets were immediately collected in a liquid nitrogen bath to freeze the particles, and then the particles were allowed to enter a cold (−80°C) ethanol (AAPER, Shelbyville, KY) bath where the methylene chloride was extracted over several days to harden the microspheres. The microspheres were collected by filtration and dried. Protein content was determined by dissolving the microspheres in methylene chloride, centrifuging the insoluble protein, and dissolving the protein pellet in DI water. The amount of protein was quantified using a bicinchonic acid (BCA) assay (Pierce, Rockford, IL).
D. Preparation of Cross-Linked HA-GMA Hydrogels
Hydrogels containing microspheres were formed by mixing equal volumes of a microsphere suspension (200 mg/mL in DI water) and an HA-GMA solution (40 mg/mL in DI water). The HA-GMA/microsphere suspension was mixed with a photo-initiator at a volume ratio of 10:1 (polymer:initiator). The photo-initiator, 2-hydroxy-1-[4-(hydroxyethoxy)phenyl]-2-methyl-1-propanone (Irgacure 2959—Ciba, Tarrytown, NY), was dissolved at a concentration of 20 mg/mL in methanol. Cross-linking was initiated by exposure to UV radiation (365 nm) for 30 min. To measure protein release and microsphere morphology, 100 μL hydrogels were fabricated in wells of a 96-well plate. HA hydrogels without microspheres were formed from a 20 mg/mL HA-GMA solution.
E. Protein Release From PLGA Microspheres
Microspheres were suspended at a 10 mg/mL in DI water and placed at 37 °C on a rotating shaker. Triplicate samples were prepared for each molecular weight of PLGA. At selected time points, the microspheres were pelleted by centrifugation, the release buffer was removed and replaced with fresh DI water, and the microspheres were resuspended. Protein in the release buffer was quantified using a BCA assay.
F. Protein Release From HA Hydrogels
Lyophilized HA hydrogels were rehydrated in a solution of BSA (100 μg BSA in 100 μL DI water) or DI water. Hydrogels were allowed to swell for 2 h at room temperature and then placed at 37 °C in 12-well plates with 3 mL DI water as the release buffer (triplicate samples). At selected time points, the release buffer was completely removed and replaced with fresh DI water, and protein concentration was quantified using a Coomassie Plus assay (Pierce).
G. Protein Release From PLGA Microspheres in HA Hydrogels
Hydrogels with or without PLGA microspheres were placed in Snapwell inserts (Corning, Acton, MA) immediately after formation, and the inserts were glued to 6-well plates to prevent motion. The well was filled with 3 mL DI water as a release buffer, and an additional 400 μL DI water was added to the insert to equilibrate and swell the hydrogel. The samples (three replicates per condition) were placed at 37 °C. Additional DI water was added to the wells at intermediate time points to compensate for evaporation. Further, a humidified chamber and plate sealers were used to reduce evaporation. The release buffer was completely removed and replaced after imaging at the selected time points. Protein concentration in the release buffer was quantified using a Coomassie Plus assay. Calculations of the amount of protein released were based on the actual amount of buffer retrieved from each well at each time point.
H. In Vitro OCT Imaging
For each imaging session, each 6-well plate was attached to the imaging stage in a fixed position and orientation. The starting x-y coordinates for each well were recorded during the first imaging session and used as reference for subsequent imaging sessions. Registration of the laser’s incident position was confirmed with reference ink marks on the imaging stage as well as on each plate. OCT imaging was performed on three hydrogels for each condition with a light source having a center wavelength of 825 nm and a spectral bandwidth of 150 nm. The axial resolution was about 2.8 μm, and the power on the sample was approximately 3 mW. A region of 1000μm × 50μm × 600μm–800μm was imaged for each hydrogel. 3-D scan series consisted of multiple 2-D cross-sectional scans (1 mm wide) with 2.5 μm between each adjacent 2-D scan. The acquisition rate was approximately 100 A-scans per second, corresponding to ca. 0.1 Hz (frames/second) with each frame consisting of 1000 A-scans. For each 2-D image, the beam focus was stepwise tracked in the z-direction ten times in 40 μm increments, starting from the top surface of the hydrogels, and images within the focal zones were digitally combined to create in-focus cross-sectional images.
I. In Vivo OCT Imaging
A 25 μL hydrogel with 0.75 mg PLGA microspheres was implanted in a modified rat calvarial critical size defect model and imaged using OCT. The surgical protocol was approved by the University of Washington (UW) Institutional Animal Care and Use Committee (IACUC) and was conducted under aseptic conditions using sterile materials. Briefly, a 5 mm circular defect was created in the parietal bones of an anesthetized adult male Sprague Dawley rat (Charles River Laboratories, Wilmington, MA), and the hydrogel scaffold was placed in the defect. The defect region was covered with a custom-designed “window chamber,” which was affixed to the skull with miniature screws (Small Parts, Miami Lakes, FL) and Jet dental acrylic (Lang Dental, Wheeling, IL). OCT imaging was performed on the anesthetized animal using the above in vitro imaging parameters. The animal was then sacrificed, and OCT imaging was repeated post mortem.
J. Image Processing and Analysis
Raw OCT intensity values were used to generate grayscale TIFF images for quantification. Multiple images with different focal regions were processed in MATLAB (The Math Works, Natick, MA) to create composite in-focus images. The z-dimension was scaled by the index of refraction of the hydrogel (i.e., ca. 1.33). The 3-D image stacks were also resectioned and reconstructed using MATLAB. Contrast enhancement using a linear mapping to 8-bit grayscale with background subtraction was performed on the images for display. The MetaMorph Imaging System (Molecular Devices Corporation, Sunnyvale, CA) was used to measure microsphere diameters and to quantify the number of particles per volume. A region that was 1000μm × 50μm × 200μm in the 3-D image stack was analyzed for each of the triplicate hydrogels per condition at each time point. To determine the diameter of the microspheres, the circumference of an individual microsphere was traced in each cross-sectional image, and the greatest width was selected as the overall microsphere diameter. Incompletely sectioned microspheres that did not reach a maximum width in the image sequence were not measured or counted.
III. Results
A. Scaffold Fabrication
The presence of acrylate peaks at 5.6 and 6.1 ppm in the HA-GMA 1H-NMR spectrum compared to the unmodified HA 1H-NMR spectrum confirmed the functionalization of the HA (data not shown). The degree of substitution (number of methacrylate groups per 100 disaccharide units), which indicates the amount of potential cross-links that can be formed, was determined to be 17. It was calculated by taking ratios of the acrylate peaks, which arise from the methacrylate group only, and the methyl peak at 1.9 ppm, which includes protons from the methacrylate group and backbone methyl group. Hydrogels formed at 20 mg/mL HA-GMA exhibited a swelling ratio of 88, which also indicates the extent of cross-linking. The swelling ratio was calculated as Ws/Wd where Ws is the wet weight of the hydrogel after equilibration in excess DI water and Wd is the dry weight of the hydrogel. The hydrogels were molded to swell to completely fill the Snapwell inserts after equilibrating in DI water so that OCT imaging could be performed over nearly the same region at different time points. The hydrogel matrix remained intact for over six weeks during the study. By 10 weeks in DI water, the hydrogels began to exhibit dimensional instability and were partially collapsed or completely disintegrated so the imaging study was terminated.
B. Protein Release
Two different molecular weights of PLGA were used to control the protein release rate. As seen in Fig. 1(A), microspheres fabricated from a lower molecular weight PLGA (I.V. = 0.17 dL/g; molecular weight ≈ 12 kDa) degraded faster and released protein faster than microspheres fabricated from a higher molecular weight PLGA (I.V. = 0.39 dL/g; molecular weight ≈ 40 kDa). Both batches had a total protein content of 66 μg BSA per mg microspheres. The 12 kDa PLGA microspheres had a higher initial burst (74% of protein content) compared to the 40 kDa PLGA microspheres (9% of protein content). After the initial burst, the release rates of BSA were about the same for the first three weeks for both 12 kDa and 40 kDa PLGA microspheres (0.6 μg BSA/mg microspheres/day). After three weeks, release from the 12 kDa PLGA microspheres slowed to 0.2 μg BSA/mg microspheres/day. By six weeks the total protein released reached a plateau, which was approximately 100% of the initial protein content. Release from the 40 kDa PLGA microspheres remained at 0.6 μg BSA/mg microspheres/day for six weeks before slowing to 0.2 μg BSA/mg microspheres/day, and then continued at this slower rate for at least 70 days.
Fig. 1.

BSA release from constructs. (A) Cumulative BSA release from microspheres made with different molecular weights of PLGA. (B) Release of BSA from HA hydrogels loaded with 100 μg BSA or no protein as control. (C) Release of BSA from 12 kDa or 40 kDa PLGA microspheres suspended within HA hydrogels or hydrogels without microspheres or protein as control. Results are shown as mean and standard error from triplicate samples.
The release profile of BSA directly loaded in the HA hydrogels is shown in Fig. 1(B). A Bradford-based, Coomassie Plus assay was used to quantify protein release because components that leached from the HA hydrogels interfered with the BCA assay. Release of protein from the HA hydrogels was slower than expected. Release is influenced by electrostatic interactions between the protein and HA, which are in turn affected by the pI of the protein and the pH of the release buffer. While the pI of BSA is low, suggesting rapid release, the pH of the DI water used as the release buffer was also low (ca. 5.5) and acted to slow release. When the microspheres were suspended within HA hydrogels and placed in the Snapwell insert, the measured release of protein [Fig. 1(C)] was slowed compared to the microspheres alone due to diffusion of protein through the hydrogel, electrostatic interactions between BSA and HA, and limitation of release to only the bottom surface of the cylindrical hydrogel through the Snapwell membrane. However, the faster release of protein from the 12 kDa PLGA microspheres relative to the 40 kDa PLGA microspheres was again observed. Both formulations had an initial lag before beginning to release protein—14 days for the 12 kDa PLGA microspheres within the HA hydrogels and 21 days for the 40 kDa PLGA microspheres within the HA hydrogels. After this lag, the 12 kDA PLGA microspheres within the HA hydrogels released protein at a rate of 0.3 μg/mg microspheres/day for six weeks and then slowed to 0.05 μg/mg microspheres/day. The 40 kDa PLGA microspheres within the HA hydrogels released protein steadily at a rate of 0.1 μg/mg microspheres/day after the initial lag for the duration of the measurement period.
C. OCT Imaging and Initial Microsphere Morphology
OCT images of control and microsphere-loaded HA hydrogels were successfully obtained. Composite in-focus cross-sectional images were digitally prepared in a way similar to ultrasound C-mode imaging. As seen in Fig. 2, OCT images of control HA hydrogels without microspheres confirmed that the hydrogels were optically transparent. Besides a dark line due to the reflection from the surface of the hydrogel (i.e., the air-hydrogel interface), no structures were visible in these images, even with contrast enhancement. OCT images of HA hydrogels with 12 kDa or 40 kDa PLGA microspheres showed the presence of particulates embedded within the hydrogel (Fig. 2). At early time points, these particles were solid with a graded intensity. The reduced intensity at the bottom end of each particle is consistent with loss of the incident light’s power deeper within the nontransparent material. Particles located further down within the hydrogels, beneath other particles, also appeared lower in intensity.
Fig. 2.

Temporal sequence of high resolution OCT images of degrading PLGA microspheres within HA hydrogels during the release study. Approximately the same region of the hydrogel was imaged at each time point by fixing the plate in the same location on the stage and aligning the beam to reference points on the stage and plate. Images show a small region selected directly beneath the top surface of the hydrogel, which was visible as a dark line running horizontally across the image. (A, D, G, J) Control HA hydrogels. (B, E, H, K) HA hydrogels with 12 kDa PLGA microspheres. (C, F, I, L) HA hydrogels with 40 kDa PLGA microspheres. (A)–(C) Day 1. (D)–(F) Day 7. (G)–(I) Day 21. (J)–(L) Day 42. The scale bar represents 200 μm.
The particles were oval-shaped with a greater height than width. This lengthening of the particles in the z-dimension results from the mismatch in index of refraction between the PLGA microspheres and the hydrated HA hydrogel. When converting the raw OCT data to TIFF image format, the images were digitally rescaled in the z-dimension using the index of refraction of the HA hydrogel, which was experimentally measured at 1.33 and is similar to that of water. Assuming the particles to be truly spherical, the index of refraction of the PLGA microspheres could be calculated by taking the ratio of the height to width of the particles within the OCT images. The average ratio of height-to-width was measured on the image slice through the center of each microsphere and was calculated to be 1.25 for the 12 kDa PLGA microspheres and 1.14 for the 40 kDa PLGA microspheres at Day 1. Correcting for the original scaling factor (n = 1.33), this would result in a measured index of refraction of 1.51–1.67 for PLGA. This refractive index is consistent with that reported for other polymers, such as polystyrene (n = 1.57–1.61) [24] or a different polyester (n = 1.54) [25]. Additionally, resectioning of the 3-D scan data in the XY (horizontal) plane, which should not be affected by the index of refraction, confirmed that these particles initially appeared spherical (Fig. 3).
Fig. 3.

Representation of 3-D scan data in the 3-D coordinate system, showing high resolution OCT images in each plane. (A) 12 kDa PLGA microspheres in HA hydrogel at Day 1. (B) 40 kDa PLGA microspheres in HA hydrogel at Day 1.
The 3-D scan data were also used to obtain a measure of the overall diameter for each microsphere. The width of each slice through an individual microsphere was measured in each cross-sectional image, and the greatest width was selected as the overall microsphere diameter. The average microsphere diameter was thus determined to be 26±10 μm for the 12 kDa PLGA microspheres and 30±12 μm for the 40 kDa PLGA microspheres at Day 1. Since the microspheres were fabricated using identical conditions, it was expected that they would be similar in size prior to degradation.
D. Temporal Characterization of Morphological Changes
Images from a 1000 μm × 50 μm × 200 μm region of each of the triplicate hydrogels with either 12 kDa or 40 kDa PLGA microspheres were analyzed at each time point. Only particles that were greater than 50% contained within the region (i.e., reached maximum diameter) were included in the analysis for particle counts as well as refractive index and diameter measurements. Qualitative assessment of the OCT images showed morphological changes of the microspheres with time, which was particularly apparent in the 12 kDa PLGA samples (Fig. 2). No changes occurred in the control samples, which remained completely free of any structures for the six weeks of imaging. By 10 weeks, all hydrogels had begun to degrade, and all images had a reduced height between the top surface of the hydrogel and the supporting membrane or had no visible hydrogel. The ratio of particle height-to-width remained fairly constant throughout the study [Fig. 4(A)], which indicates that the calculated refractive index is not changing with time. Measurement of microsphere diameter, as described above, showed that both samples had a trend of decreasing particle size with time. As seen in Fig. 4(B), the average particle size of the microspheres remained approximately the same or slightly increased for two weeks before beginning to decrease. In images of hydrogels with the 40 kDa PLGA microspheres, the particle cross sections also predominantly remained solid throughout the course of the experiment [Fig. 4(D)]. The images of hydrogels with the 12 kDa PLGA microspheres initially contained particles that looked similar to those in the 40 kDa PLGA samples. With contrast enhancement, the background in the 12 kDa PLGA microsphere images was more uneven, with the appearance of smaller particulates (not included in the measurements) that persisted for the duration of the study. At later time points, some of the 12 kDa PLGA microsphere cross sections developed a ring-like appearance [example seen in Fig. 2(H)]. These hollow structures, which increased in number with time [Fig. 4(D)], could indicate that the core of the 12 kDa PLGA microspheres had degraded and dissolved away, leaving just the outer shell.
Fig. 4.
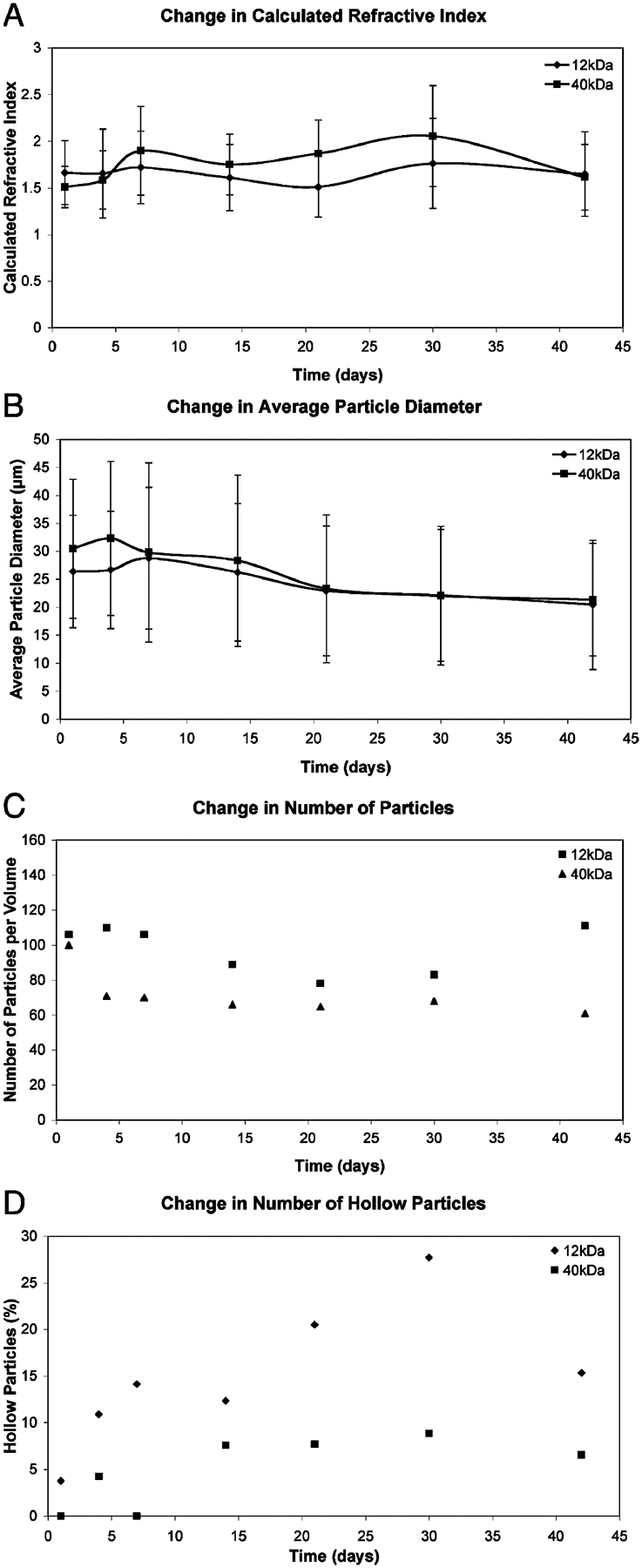
Morphological characterization of low and high molecular weight PLGA microspheres suspended within HA hydrogels as a function of time. Measurements taken for particles within a 1000 μm×50 μm×200 μm region in the 3-D OCT image stack from one hydrogel as representative of the three replicates. (A) Calculated refractive index ± standard deviation. (B) Average particle diameter ± standard deviation. (C) Total particle count per measured volume. (D) Hollow particle count per measured volume.
E. In Vivo OCT Imaging
The surgical procedure for attachment of the “window chamber” over the critical size defect was successful. During live animal imaging, significant distortion of the image occurred due to breathing and heartbeat. Holding the rat’s head with ear bars in a modified stereotactic frame was able to significantly reduce (although not completely eliminate) this motion artifact. The rat was imaged again immediately after sacrifice, and representative post mortem images of a microsphere-loaded hydrogel in the defect are shown in Fig. 5. The microspheres are clearly visible in the hydrogel, which is approximately 0.9 mm thick. The cover slip, which is made of plastic, appears as a region of high contrast at the top of the image. Within the hydrogel scaffold region, particles similar to those seen in vitro could be observed. In images obtained by stepping the focal zone down through the implant [Fig. 5(B) and (C)], these particles were also shown to be solid with an oval cross section, again resulting from the refractive index mismatch between the PLGA microsphere and HA hydrogel. The particles were evenly distributed throughout the depth of the hydrogel. A darker line, about 100 μm thick, ran beneath the implant and can be clearly seen in Fig. 5(C). This structure is consistent in size and location with the dura mater, the thin membrane that runs between the parietal bones and the brain. This demonstrates that OCT was able to image through the full thickness of the implant within the defect.
Fig. 5.
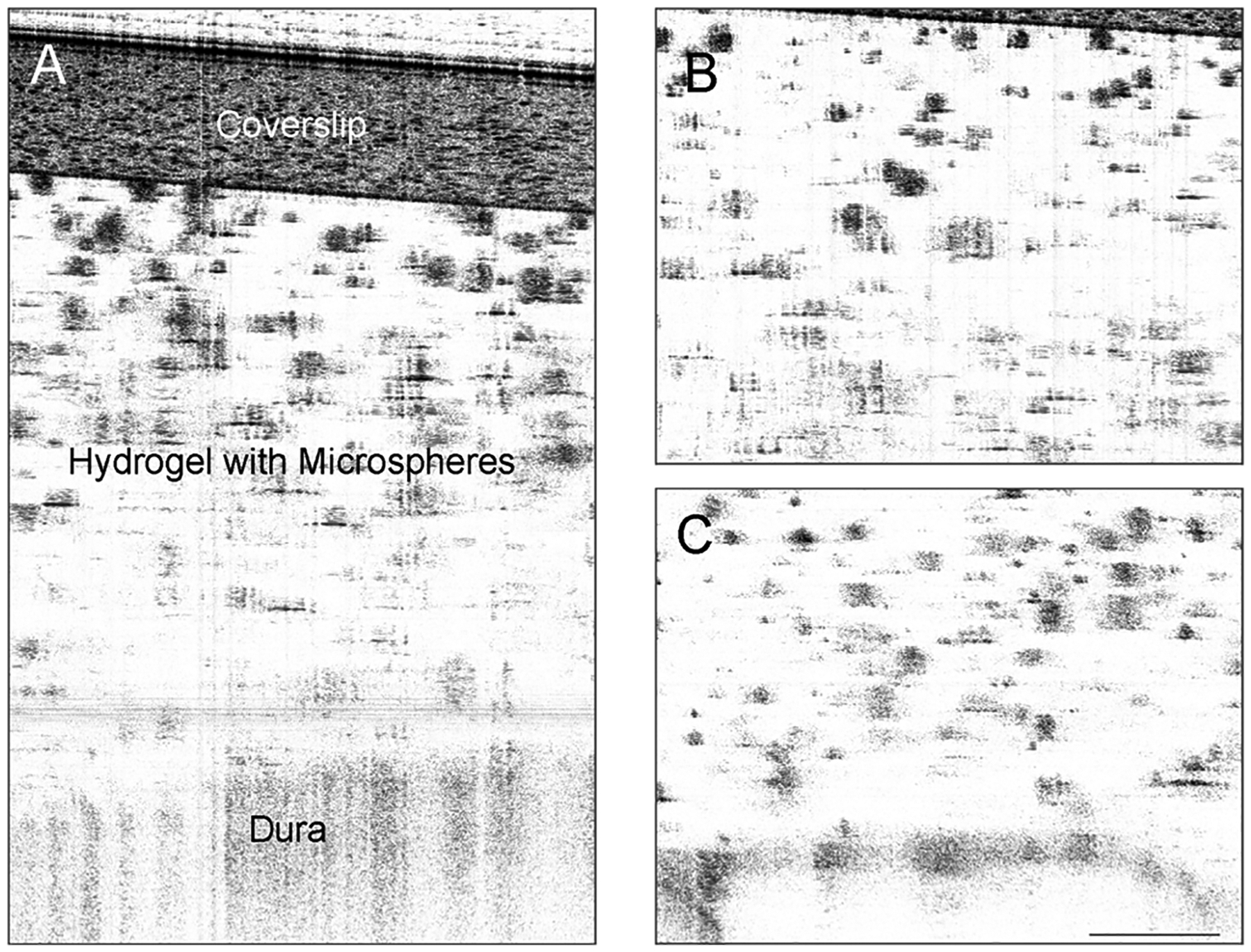
High resolution OCT images of scaffold in rat calvarial defect after sacrifice. OCT was able to penetrate through entire scaffold and image to the dura mater. Microspheres within the scaffold appear as dark shapes on a light background. (A) 2 mm depth scan showing plastic coverslip through dura. (B) In-focus image of top half of implant, showing part of the plastic coverslip at top. (C) In-focus image of bottom half of implant, showing dura at bottom. The scale bar represents 200 μm.
IV. Discussion and Conclusion
Degradable microspheres provide a means for controlled release of encapsulated proteins (or drugs) and allow therapeutic dosing to be maintained over an extended period of time from a single administration. Recently, techniques to embed microspheres within a scaffold matrix have been developed [26]–[29]. These composite scaffolds offer altered protein release profiles compared to microspheres alone, extended release of protein compared to protein directly loaded into the scaffold matrix, and better retention of the microspheres within an implant site. Characterization of composite scaffold performance, however, has been limited to traditional techniques used with degradable implants, namely in vitro protein release testing, monitoring of scaffold morphology by destructive examination such as SEM, and measuring of therapeutic endpoints in vivo. This study reports the use of the noninvasive imaging technique, OCT, to follow temporal changes in scaffold morphology in situ. These morphological changes were further compared to measurements of protein release from the scaffolds, which is indicative of their therapeutic potential. Whereas the focus of this correlation was an in vitro study, the potential to extend these results to in vivo monitoring of scaffold performance was demonstrated by the addition of a “window chamber” to the rat calvarial critical size defect model.
Protein release from the microspheres embedded within the hydrogels [Fig. 1(C)] was slower than protein release from the hydrogels alone [Fig. 1(B)], which is expected and further supported by other studies [28]. Compared to an aqueous suspension of microspheres [Fig. 1(A)], suspending the microspheres within the hydrogel matrix slowed the overall rate of protein release and reduced the initial burst release [Fig. 1(C)]. While the measured rate of protein release was artificially reduced by suspending the samples within a Snapwell insert, the observed changes in the release profile are supported by the results of others who have examined protein release from a microsphere-embedded hydrogel system [27]. The slower release of BSA from the HA hydrogels compared to that reported for similar hydrogel systems [28] is likely due to interactions between the BSA and HA that are affected by the pI of the protein and pH of the release buffer. Using a release buffer with a higher pH accelerated the release of BSA from HA hydrogels (data not shown).
Two different molecular weights of PLGA were selected that degrade at different rates, thus providing differing release kinetics with all other formulation parameters remaining the same. Since the 12 kDa PLGA microspheres released 100% of the protein loaded by five weeks when placed in aqueous suspension, it was expected that these microspheres would have undergone significant degradation during the course of the OCT imaging. Conversely, since the 40 kDa PLGA microspheres released only approximately 50% of the protein loaded after 10 weeks in suspension, it was expected that these microspheres might still be fairly intact. These expectations were met in the temporal sequence of OCT images (Fig. 2). The initial solid morphology of the particles in the OCT images may likely be due to the heterogeneous nature of the microspheres, which contain particulates of protein/trehalose within the PLGA matrix, potentially resulting in a refractive index mismatch throughout the particle. The initial slight increase in particle size for the microspheres could be a result of particle swelling upon hydration.
Quantitatively, the 12 kDa PLGA microspheres within the HA hydrogels showed a 23% decrease in average microsphere diameter over six weeks as well as an increase in the number of measurable particles per volume at the last time point [Fig. 4(C)]. This suggests that the 12 kDa PLGA microspheres are breaking down over the time course of the experiment. More importantly, the 12 kDa PLGA images also showed the presence of hollow particles, which increased in number at later time points [Fig. 4(D)]. There was also a smaller particulate seen in the background of the contrast-enhanced images that could be associated with smaller pieces of PLGA dissociating from the faster degrading 12 kDa PLGA microspheres. The hollow morphology indicates that there is no refractive index mismatch within the core, which would be expected once the polymer is degraded and cleared and protein is released. This hollow morphology is consistent with faster polymer degradation within the core of the microsphere followed by dissolution and clearing of the lower molecular weight fragments, which was also observed in SEM studies by Visscher et al. [17]–[19]. Whereas Grizzi et al. initially suggested that microspheres might fall below a critical dimensional limit and degrade homogeneously [16], additional studies by that group and others have shown that degradation at the surface of microspheres also proceeds more slowly [30], [31]. Further, Visscher et al. showed that there was no loss of microsphere integrity as the polymer initially degraded, which was characterized by molecular weight loss [19]. Erosion and disintegration into particulates only became apparent at later time points [19]. When measuring the particle diameters, the 40 kDa PLGA microspheres within the HA hydrogels also decreased in size; however, they remained fairly constant in number with time [Fig. 4(C)]. The particle cross sections remained solid and did not change much in appearance, suggesting that they had not yet reached the point of dissolution of small molecular weight fragments within the core.
The clear morphological differences observed between the two microsphere types as well as changes observed over time suggest that OCT may be a useful imaging technique for more sophisticated characterization of microsphere degradation. With careful registration, OCT could be used to track changes in the morphology of individual particles with time. This would offer advantages over techniques which monitor the degradation of the microspheres in bulk or that analyze different samples at each time point by destructive measurement methods. It would further provide the potential for analysis of the effect of particle size on degradation rate without the need to separate and group samples into particle size ranges before characterization and analysis. More importantly, OCT could be used to characterize microsphere degradation in vivo. By comparing morphological changes in vivo to those in vitro, it may indicate if the measured in vitro release profiles are actually predictive of those in vivo. This would give a more realistic measure of the controlled release properties and degradability of the microspheres in their true therapeutic setting.
The novel “window chamber” modification to the critical size defect model also provides an opportunity for repeated imaging of the healing defect over time in the same animal. In addition to changes in scaffold morphology, tissue regeneration can be observed, as has been seen with HA hydrogel scaffolds loaded directly with growth factors [32]. A major limitation to OCT images taken of live animals is the motion artifact due to breathing and heartbeat; however, this can be further minimized by a faster acquisition rate and by postprocessing of the images with a cross-correlation routine in addition to the use of stabilizing ear bars. These measurements were conducted with a benchtop ultrahigh-resolution OCT system that had a slow acquisition rate. Subsequently, a system with a much faster acquisition rate (e.g., 30 000 A-scans per second) was constructed, which would allow speed increases by more than a factor of 100–300. A secondary limitation observed in the in vivo images in this study is the distortion of some of the particles that are located beneath defects in the plastic cover slip. This can be reduced by using a glass cover slip, which will be optically transparent. A final limitation to the in vivo measurements is the penetration depth of OCT within tissue. The 800 nm source is based on a Ti:Sapphire laser, which has a broad spectrum bandwidth (i.e., 150 nm) and provides an ultrahigh resolution (i.e., ca. 2.5 μm). Although 800 nm light sources have a shallow imaging depth in tissue, they are very suitable for imaging the optically transparent hydrogels. The penetration depth of the source would be a concern when performing measurements in vivo after there has been tissue regrowth. However, for monitoring regrowth of the rat skull, which is generally less than 1 mm thick, an 800 nm light source should be sufficient. If needed, a broadband 1300 nm light source can be used, but it will come with a potential reduction in imaging resolution. In conclusion, this study demonstrates the potential of OCT for in situ temporal characterization of changes in scaffold morphology and develops an animal model that could be used to extend this characterization to measure in vivo scaffold performance.
Acknowledgment
The authors would like to thank M. Pak, M. Cobb, and D. MacDonald for assistance in image processing as well as Y. Chen and D. Thomas for assistance in image acquisition.
This work was supported in part by the National Institutes of Health (NIH) Health Bioengineered Allogeneic Tissue (BEAT) partnership under Grant 5 R24 HL64387 and in part by the National Science Foundation (NSF) under Grant EEC-9872882 and Grant EEC-9529161. The work of J. Patterson was supported by a NSF Graduate Fellowship. The work of X. D. Li was supported by a NSF Career Award.
Contributor Information
Jennifer Patterson, Department of Bioengineering, University of Washington, Seattle, WA 98195 USA.
Patrick S. Stayton, Department of Bioengineering, University of Washington, Seattle, WA 98195 USA
Xingde Li, Department of Bioengineering, University of Washington, Seattle, WA 98195 USA.
References
- [1].Huang D, Swanson EA, Lin CP, Schuman JS, Stinson WG, Chang W, Hee MR, Flotte T, Gregory K, Puliafito CA, and Fujimoto JG, “Optical coherence tomography,” Science, vol. 254, pp. 1178–1181, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Tadrous PJ, “Methods for imaging the structure and function of living tissues and cells: 1. Optical coherence tomography,” J. Pathol, vol. 191, pp. 115–119, 2000. [DOI] [PubMed] [Google Scholar]
- [3].Tearney GJ, Brezinski ME, Bouma BE, Boppart SA, Pitris C, Southern JF, and Fujimoto JG, “In vivo endoscopic optical biopsy with optical coherence tomography,” Science, vol. 276, pp. 2037–2039, 1997. [DOI] [PubMed] [Google Scholar]
- [4].Brezinski ME, Tearney GJ, Bouma BE, Izatt JA, Hee MR, Swanson EA, Southern JF, and Fujimoto JG, “Optical coherence tomography for optical biopsy. Properties and demonstration of vascular pathology,” Circulation, vol. 93, pp. 1206–1213, 1996. [DOI] [PubMed] [Google Scholar]
- [5].Li X, Martin S, Pitris C, Ghanta R, Stamper DL, Harman M, Fujimoto JG, and Brezinski ME, “High-resolution optical coherence tomographic imaging of osteoarthritic cartilage during open knee surgery,” Arthritis. Res. Therapy, vol. 7, pp. R318–R323, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Han CW, Chu CR, Adachi N, Usas A, Fu FH, Huard J, and Pan Y, “Analysis of rabbit articular cartilage repair after chondrocyte implantation using optical coherence tomography,” Osteoarthritis Cartilage, vol. 11, pp. 111–121, 2003. [DOI] [PubMed] [Google Scholar]
- [7].Tan W, Sendemir-Urkmez A, Fahrner LJ, Jamison R, Leckband D, and Boppart SA, “Structural and functional optical imaging of three-dimensional engineered tissue development,” Tissue Eng, vol. 10, pp. 1747–1756, 2004. [DOI] [PubMed] [Google Scholar]
- [8].Yang Y, Dubois A, Qin X-P, Li J, Haj AE, and Wang RK, “Investigation of optical coherence tomography as an imaging modality in tissue engineering,” Phys. Med. Biol, vol. 51, pp. 1649–1659, 2006. [DOI] [PubMed] [Google Scholar]
- [9].Bagnaninchi PO, Yang Y, Zghoul N, Maffulli N, Wang RK, and Haj AJE, “Chitosan microchannel scaffolds for tendon tissue engineering characterized using optical coherence tomography,” Tissue Eng, vol. 13, pp. 323–331, 2007. [DOI] [PubMed] [Google Scholar]
- [10].Kronenthal RL, “Biodegradable polymers in medicine and surgery,” Polym. Sci. Technol, vol. 8, pp. 119–127, 1974. [Google Scholar]
- [11].Li SM, Garreau H, and Vert M, “Structure-property relationships in the case of the degradation of massive aliphatic poly-(alpha-hydroxy acids) in aqueous media, part 1: Poly (DL-lactic acid),” J. Mater. Sci. Mater. Med, vol. 1, pp. 123–130, 1990. [Google Scholar]
- [12].Li SM, Garreau H, and Vert M, “Structure-property relationships in the case of the degradation of massive aliphatic poly-(alpha-hydroxy acids) in aqueous media, part 2: Degradation of lactide-glycolide copolymers: PLA37.5GA25 and PLA75GA25,” J. Mater. Sci. Mater. Med, vol. 1, pp. 131–139, 1990. [Google Scholar]
- [13].Li SM, Garreau H, and Vert M, “Structure-property relationships in the case of the degradation of massive aliphatic poly-(alpha-hydroxy acids) in aqueous media, part 3: Influence of morphology of poly (l-lactic acid),” J. Mater. Sci. Mater. Med, vol. 1, pp. 198–206, 1990. [Google Scholar]
- [14].Therin M, Christel P, Li S, Garreau H, and Vert M, “In vivo degradation of massive poly (alpha-hydroxy acids): Validation of in vitro findings,” Biomaterials, vol. 13, pp. 594–600, 1992. [DOI] [PubMed] [Google Scholar]
- [15].Vert M, Mauduit J, and Li S, “Biodegradation of PLA/GA polymers: Increasing complexity,” Biomaterials, vol. 15, pp. 1209–1213, 1994. [DOI] [PubMed] [Google Scholar]
- [16].Grizzi I, Garreau H, Li S, and Vert M, “Hydrolytic degradation of devices based on poly (DL-lactic acid) size-dependence,” Biomaterials, vol. 16, pp. 305–311, 1995. [DOI] [PubMed] [Google Scholar]
- [17].Visscher GE, Pearson JE, Fong JW, Argentieri GJ, Robison RL, and Maulding HV, “Effect of particle size on the in vitro and in vivo degradation rates of poly (DL-lactide-co-glycolide) microcapsules,” J. Biomed. Mater. Res, vol. 22, pp. 733–746, 1988. [DOI] [PubMed] [Google Scholar]
- [18].Visscher GE, Robison MA, and Argentieri GJ, “Tissue response to biodegradable injectable microcapsules,” J. Biomater. Appl, vol. 2, pp. 118–131, 1987. [DOI] [PubMed] [Google Scholar]
- [19].Visscher GE, Robison RL, Maulding HV, Fong JW, Pearson JE, and Argentieri GJ, “Biodegradation of and tissue reaction to 50:50 poly (DL-lactide-co-glycolide) microcapsules,” J. Biomed. Mater. Res, vol. 19, pp. 349–365, 1985. [DOI] [PubMed] [Google Scholar]
- [20].Bosch C, Melsen B, and Vargervik K, “Importance of the critical-size bone defect in testing bone-regenerating materials,” J. Craniofac. Surg, vol. 9, pp. 310–316, 1998. [DOI] [PubMed] [Google Scholar]
- [21].Ray RD and Holloway JA, “Bone implants; preliminary report of an experimental study,” J. Bone. Joint. Surg. Am, vol. 39-A, pp. 1119–1128, 1957. [PubMed] [Google Scholar]
- [22].Leach J, Bivens K, Patrick C, and Schmidt C, “Photocrosslinked hyaluronic acid hydrogels: Natural, biodegradable tissue engineering scaffolds,” Biotechnol. Bioeng, vol. 82, pp. 578–589, 2003. [DOI] [PubMed] [Google Scholar]
- [23].Johnson OL, Jaworowicz W, Cleland JL, Bailey L, Charnis M, Duenas E, Wu C, Shepard D, Magil S, Last T, Jones AJ, and Putney SD, “The stabilization and encapsulation of human growth hormone into biodegradable microspheres,” Pharm. Res, vol. 14, pp. 730–735, 1997. [DOI] [PubMed] [Google Scholar]
- [24].Ma X, Lu JQ, Brock RS, Jacobs KM, Yang P, and Hu X-H, “Determination of complex refractive index of polystyrene microspheres from 370 to 1610 nm,” Phys. Med. Biol, vol. 48, pp. 4165–4172, 2003. [DOI] [PubMed] [Google Scholar]
- [25].Firbank M and Delpy DT, “A design for a stable and reproducible phantom for use in near infra-red imaging and spectroscopy,” Phys. Med. Biol, vol. 38, pp. 847–853, 1993. [DOI] [PubMed] [Google Scholar]
- [26].Hu Y, Hollinger JO, and Marra KG, “Controlled release from coated polymer microparticles embedded in tissue-engineered scaffolds,” J. Drug Target, vol. 9, pp. 431–438, 2001. [DOI] [PubMed] [Google Scholar]
- [27].DeFail AJ, Chu CR, Izzo N, and Marra KG, “Controlled release of bioactive TGF-beta 1 from microspheres embedded within biodegradable hydrogels,” Biomaterials, vol. 27, pp. 1579–1585, 2006. [DOI] [PubMed] [Google Scholar]
- [28].Leach JB and Schmidt CE, “Characterization of protein release from photocrosslinkable hyaluronic acid-polyethylene glycol hydrogel tissue engineering scaffolds,” Biomaterials, vol. 26, pp. 125–135, 2005. [DOI] [PubMed] [Google Scholar]
- [29].Richardson TP, Peters MC, Ennett AB, and Mooney DJ, “Polymeric system for dual growth factor delivery,” Nat. Biotechnol, vol. 19, pp. 1029–1034, 2001. [DOI] [PubMed] [Google Scholar]
- [30].Cleland JL, Duenas ET, Park A, Daugherty A, Kahn J, Kowalski J, and Cuthbertson A, “Development of poly-(D,L-lactide-coglycolide) microsphere formulations containing recombinant human vascular endothelial growth factor to promote local angiogenesis,” J. Control Release, vol. 72, pp. 13–24, 2001. [DOI] [PubMed] [Google Scholar]
- [31].Spenlehauer G, Vert M, Benoit JP, and Boddaert A, “In vitro and in vivo degradation of poly(D,L lactide/glycolide) type microspheres made by solvent evaporation method,” Biomaterials, vol. 10, pp. 557–563, 1989. [DOI] [PubMed] [Google Scholar]
- [32].Patterson J, Stayton PS, and Li X, “Optical coherence tomography for temporal characterization of bone regeneration induced by angiogenic and osteoinductive hydrogel scaffolds,” unpublished.