

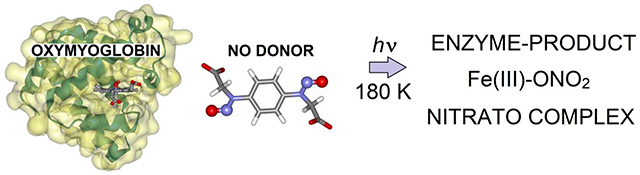
Abstract
The NO dioxygenation reaction catalyzed by heme-containing globin proteins is a crucial aerobic detoxification pathway. Accordingly, the second order reaction of NO with oxymyoglobin and oxyhemoglobin has been the focus of a large number of kinetic and spectroscopic studies. Stopped-flow and rapid-freeze-quench (RFQ) measurements have provided evidence for the formation of a Fe(III)-nitrato complex with millisecond lifetime prior to release of the nitrate product, but the temporal resolution of these techniques is insufficient for the characterization of precursor species. Most mechanistic models assume the formation of an initial Fe(III)-peroxynitrite species prior to homolytic cleavage of the O-O bond and recombination of the resulting NO2 and Fe(IV)=O species. Here we report vibrational spectroscopy measurements for the reaction of oxymyoglobin with a photolabile caged NO donor at cryogenic temperatures. We show that this approach offers efficient formation and trapping of the Fe(III)-nitrato, enzyme-product, complex at 180 K. Resonance Raman spectra of the Fe(III)-nitrato complex trapped via RFQ in the liquid phase and photolabile NO release at cryogenic temperatures are indistinguishable, demonstrating the complementarity of these approaches. Caged NO is released by irradiation <180 K but diffusion into the heme pocket is fully inhibited. Our data provide no evidence for Fe(III)-peroxynitrite of Fe(IV)=O species, supporting low activation energies for the NO to nitrate conversion at the oxymyoglobin reaction site. Photorelease of NO at cryogenic temperatures allows monitoring of the reaction by transmittance FTIR which provides valuable quantitative information and promising prospects for the detection of protein sidechain reorganization events in NO-reacting metalloenzymes.
Keywords: Nitric Oxide, Caged compounds, Heme Proteins, Cryogenic Spectroscopy, Raman, FTIR
Graphical Abstract
INTRODUCTION
Nitric oxide (NO) is a stable radical in anaerobic aqueous solutions, but it readily reacts with other radical species and transition metals leading to its broad toxicity and its critical role in the mammalian immune response [1–3]. In macrophages, NO can accumulate to micromolar concentrations and reacts with superoxide (O2−), produced by NADPH oxidase, in a diffusion-controlled fashion to generate peroxynitrite (ONOO−):
| (equation 1) |
Peroxynitrite is highly reactive and versatile with a near neutral pKa of 6.8 and cis and trans isomers of comparable energies [4,5]. While it can decay via isomerization to nitrate (NO3−) in aqueous solution, it also reacts with a wide range of biomolecules, including metalloenzymes, to produce additional reactive nitrogen species (RNS) and deleterious molecular defects. Pathogenic microorganisms depend on metal-containing superoxide dismutases and low molecular weight thiol antioxidants to scavenge reactive oxygen species (ROS), and on NO-sensing proteins to control and regulate the gene expression of NO-detoxifying enzymes and repair proteins. Flavohemoglobins and other heme-containing proteins such as truncated hemoglobins are the primary NO-detoxifying enzymes in aerobic conditions. These enzymes catalyze NO dioxygenase activity that uses O2 to efficiently convert NO to NO3− [6,7]:
| (equation 2) |
In this reaction, a second-order radical combination of NO with the heme iron(III)-superoxo complex forms a putative iron(III)-peroxynitrite intermediate. This intermediate is proposed to subsequently isomerize to NO3− via homolytic O-O bond cleavage and radical recombination of the resulting NO2 and ferryl heme Fe(IV)=O species (Scheme 1) [7–9].
Scheme 1.
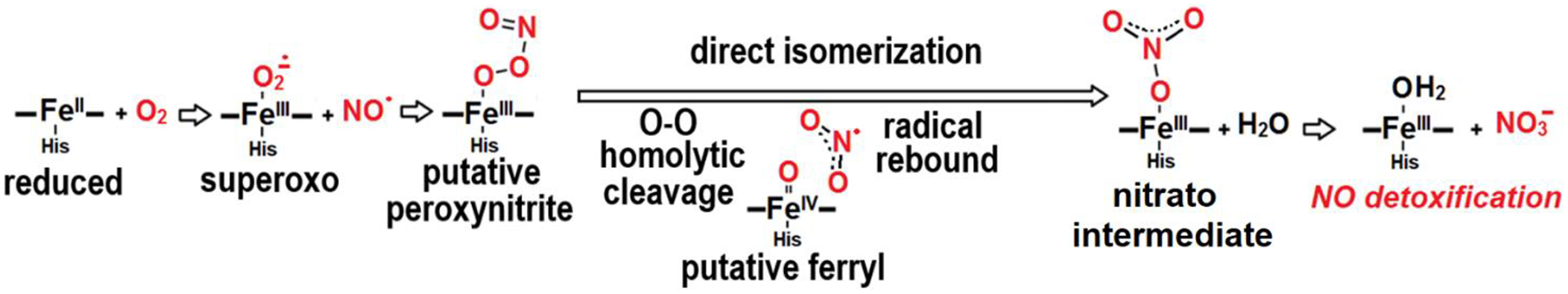
Proposed mechanisms for the NO dioxygenase reaction in hemoproteins
Monitoring this NO reaction with oxymyoglobin (oxyMb) using resonance Raman spectroscopy along with a rapid-freeze-quench approach (RFQ-RR), we showed that a millisecond intermediate, previously characterized by UV-vis stopped-flow and RFQ-EPR as a high-spin ferric species [10–12], corresponds to a transient Fe(III)-nitrato complex prior to NO3− release [13]. Accordingly, recent theoretical modeling of the NO deoxygenation reaction in Mycobacterium tuberculosis truncated hemoglobin predicts very low activation energy barriers throughout this reaction with predicted lifetimes for putative Fe(III)-peroxynitrite and Fe(IV)=O intermediates in the picosecond timescale [14,15].
Recently, Tosha, Shiro and coworkers used N,N’-bis-(carboxymethyl)-N,N’-dinitroso-p-phenylenediamine (Scheme 2), a photolabile caged NO initially designed by Fujimori and coworkers [16], to access intermediate species of the enzymatic reaction in various metalloenyzmes [17–19].
Scheme 2.

Structure and photoreaction of N,N’-bis-(carboxymethyl)-N,N’-dinitroso-p-phenylenediamine
In one of these studies, the NO donor is irradiated at cryogenic temperatures to produce a trapped non-heme iron-nitrosyl intermediate in the cytochrome c-dependent NO reductase of Pseudomonas aeruginosa (P.a. cNOR) [19]. Accordingly, we decided to test this approach to probe the NO deoxygenation reaction in oxyMb. In contrast to time-resolved spectroscopy and RFQ techniques, the approach used here cannot generate kinetic information, but it can allow the accumulation of intermediate species if their decay routes are inhibited at lower temperatures than their formation routes. Using low-temperature UV-vis, RR, and FTIR spectroscopies, we show that photorelease and migration of NO at cryogenic temperature forms the same Fe(III)-nitrato complex previously characterized via a RFQ approach [13]. A significant advantage of the cryogenic NO photorelease over the RFQ approach is its amenability to FTIR spectroscopy. In the experiments described herein, the cryogenic light-induced FTIR difference spectra simultaneously identify the presence of the dominant Fe(III)-nitrato complex and of minor ferrous- and ferric-nitrosyl contaminants. The FTIR data also detect the production of nitrous oxide (N2O) in experiments where the NO donor is used alone, providing indirect evidence for the sub-stoichiometric release of HNO from the photolabile NO donor.
MATERIAL AND METHODS
The N,N’-bis-(carboxymethyl)-N,N’-dinitroso-p-phenylenediamine NO donor was synthesized following published protocols [16,20]. p-phenylenediamine, ethyl bromoacetate, sodium acetate and sodium nitrite, were used as received from Sigma-Aldrich. Sodium nitrite (15N) was used as received from Cambridge Isotope Laboratories.
Lyophilized horse heart myoglobin was purchased from Sigma. The protein was brought into a glovebox with a controlled atmosphere of less than 1 ppm O2 (Omni-Lab System; Vacuum Atmospheres Co.) and dissolved in 100 mM phosphate buffer pH 7.5 to a concentration of ~ 3 mM before reduction by addition of 10 mM sodium dithionite before washing the excess reductant and dithionite oxidation product with a PD10 column or desalting spin column (Zebra, Pierce).
Complete heme reduction and dithionite removal was confirmed by UV-vis spectroscopy with a Cary 50 spectrometer and the deoxyMb concentration was determined and adjusted to the desired concentration by dilution or further concentrated with microcon filtrating device (10 kDa cutoff, Millipore). DeoxyMb solutions were then transferred to sealed serum bottles and exposed to 16O2 (Airgas) or 18O2 (99% 18O; ICON Stable Isotopes) or simply exposed to air outside the glovebox. Formation of oxyMb was confirmed by UV-vis spectroscopy before addition of 2 equiv NO donor.
Low-temperature UV-vis and FTIR photolysis experiments were conducted on thin films prepared with myoglobin solutions with 1-to-2 equiv NO donor sandwiched between two CaF2 windows and a 15-μm Mylar spacer. FTIR cells were mounted to a closed-cycle cryostat system (Displex, Advanced Research Systems) equipped with CaF2 windows before placing the cryostat inside the sample compartment of a Cary-50 UV-vis or Bruker Vertex 80 FTIR spectrometer and kept in the dark until reaching the desired cryogenic temperature. The temperature of the sample was monitored and controlled with a Cryo-Con 32 unit (Cryogenic Control Systems). The FTIR spectrometer was equipped with a liquid N2-cooled MCT detector and with constant purging of the instrument with compressed air depleted of water vapor and CO2 (purge gas generator, Puregas LLC). Multiple sets of 2000-scan accumulations at 4-cm−1 resolution were collected and compared before and after sample illuminations. Photolysis was performed by illumination of the sample directly in the FTIR sample chamber using the 355-nm laser emission of a frequency-triple YAG laser (Surelite SLII-10, Continuum) at 10 Hz repetition rate. The 600-mW laser beam was defocused to insure illuminate of the entirety of the IR film inside the cryostat. The photolysis process was efficient and required ~ 1-minute illumination to maximize the UV-vis and FTIR differential signals at 180 K.
Low temperature RR spectra (~110 K) were recorded in a backscattering geometry on frozen samples in EPR tubes maintained at 110 K in a liquid nitrogen Dewar and cold-finger sample holder. All RR spectra were collected using a backscattering geometry on a custom McPherson 2061/207 spectrograph (0.67 m with variable gratings) equipped with a Princeton Instruments liquid N2-cooled CCD detector (LN-1100PB) with a 407 nm excitation from a krypton laser (Innova 302, Coherent). A long-pass edge filter (RazorEdge, Semrock) was used to attenuate the Rayleigh scattering. Frequencies were calibrated relative to aspirin and are accurate to ±1 cm−1. The lack of photosensitivity of all samples was confirmed by comparing rapid acquisitions within a range of laser powers.
RESULTS AND DISCUSSION
Low-temperature UV-vis analysis of the reaction of deoxyMb and oxyMb with the caged NO donor
As a first step to determining the photolabile efficiency of our 355-nm laser excitation with the photolabile caged NO and diffusion efficiency of the photogenerated NO at cryogenic temperatures, we prepared a deoxyMb sample with 2 equiv of the NO donor and monitored UV-vis spectral changes after illumination at 77, 140, and 180 K. Traces obtained before and after illumination at 77 and 140 K are indistinguishable, indicating that the frozen solvation cage of deoxyMb prevents the photo-released NO to reach the heme Fe(II) ion center. In contrast, illumination for 1 minute at 180 K with the 355-nm laser source results in a complete blue shift of the Soret maximum from 438 to 423 nm with appearance of distinctive Q bands at 547 and 581 nm indicative of the formation myoglobin’s nitrosyl complex [21] (Figure 1, top traces). Using the same illumination conditions (same average power and duration) at 77 K before raising the sample temperature to 180 K produces only a partial conversion of deoxyMb to the nitrosyl complex, suggesting a less efficient photorelease of NO at 77 K, possibly as recombination of photolyzed NO with the caged radical (see Scheme 2) becomes more significant at 77 K.
Figure 1:

UV-vis spectra monitoring the cryogenic reaction of deoxyMb (top traces) and oxyMb (bottom traces) with the NO donor at 180 K. Black and red traces correspond to spectra obtained before and after the 355-nm illumination.
Reproducing these UV-vis experiments with oxyMb and 2 equiv of the caged NO reveals similar temperature dependence. Specifically, the Soret maximum of oxyMb at 77 K is observed at 419 nm and is unchanged after UV-illumination at 77 or 140 K. However, either warming these samples to 180 K, or with direct illumination of samples at 180 K, the Soret maximum change to 413 nm with appearance of a CT band around 635 nm indicative of the formation of high-spin ferric heme species (Figure 1, bottom traces). Further increase of the samples’ temperature to 210 and 220 K do not generate any significant UV-vis changes.
Resonance Raman analysis of the reaction of oxyMb with the caged NO donor
Low-temperature RR spectra of oxyMb show fully symmetric porphyrin modes v4, v3, v2 and v10 at 1378, 1508, 1586 and 1644 cm−1, respectively, that are typical of a 6-coordinate low-spin ferric heme species [22] (Figure 2A). While these RR modes are unchanged after UV irradiation at 110 K, annealing these irradiated samples at 190 K for 10 mins before returning to 110 K downshifts the v4, v3 and v2 modes to 1373, 1482 and 1566 cm−1, respectively, indicating conversion to a 6-coordinate high-spin ferric-heme species [23].
Figure 2:
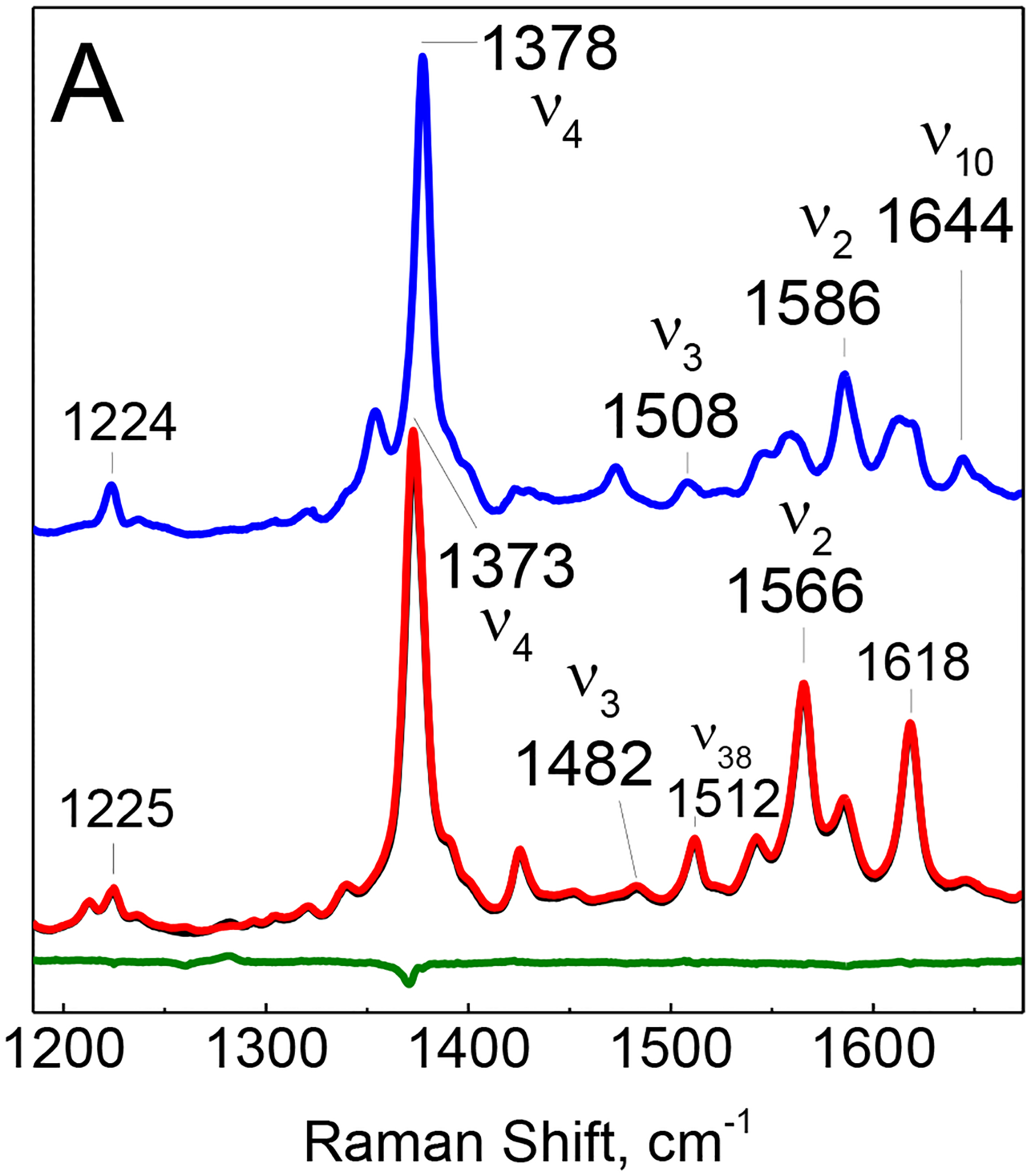

High- (A) and mid-frequency (B) regions of the RR spectra of oxyMb with unlabeled and 15N-labeled caged NO after illumination with the 355-nm emission from the YAG laser at 110 K. The blue trace corresponds to the RR spectrum of a sample maintained at 110 K while the black and red traces are spectra of illuminated oxyMb samples with unlabeled and 15N-labeled NO photo-donor incubated for 10 minutes at 190 K before data collection at 110 K. The green trace corresponds to the difference between the black and red traces.
Experiments performed with 15N-labeled caged NO identifies a single isotope sensitive RR band at 1282 cm−1 that downshifts to 1260 cm−1 with 15N (Figure 2B). These RR frequencies are exact match for the unlabeled and 15N-labeled vs(NO2) vibration of the iron(III)-nitrato complex previously trapped on the millisecond time using a rapid-freeze-quench approach on samples at 5 °C [13].
Low-temperature FTIR photolysis analysis of the reaction of oxyMb with caged NO
Low-temperature FTIR experiments were performed with 4 mM oxyMb and 1 equiv caged NO, collecting series of 2000-scan spectra before and after 355-nm illumination at 180 K. Light minus illuminated difference spectra produce numerous differential signals in the 1100 to 1950 cm−1 frequency window (Figure 3A). Control experiments with the caged NO alone identify differential signals associated with the photoconversion of the starting material to the quinone-imine byproduct after release of NO (Figure 3B). In these control experiments, the most prominent signals are negative bands corresponding to IR absorption features of the NO donor, indicating that the quinone-imine product is a much weaker IR absorbing species than the starting material. Labeling of the nitrosyl groups with 15N-atoms affects most severely two bands at 1416 and 1463 cm−1 with vibrational contributions from the NNO groups. As expected, the N-O stretching absorption for the photolyzed NO is too weak and broadened for detection in these FTIR difference spectra, but a distinctive positive band at 2229 cm−1 that downshifts to 2160 cm−1 with 15N-labeled caged NO is clearly observed in the high-frequency region (Figure 3B). These frequencies are 5 cm−1 higher than literature values for gas phase frequencies of the v3 mode of unlabeled and doubly labeled 15N15NO at 2224 and 2155 cm−1, respectively [24]. These IR absorptions are observed whether the samples are prepared in presence or absence of O2 and are indirect reporters of the photoproduction of nitroxyl (3NO− and 1HNO). Evaluating the concentration of N2O produced from the intensity of the v3 absorption bands and standard solutions of N2O suggest a yield of 0.10 ± 0.05 N2O per caged NO. This HNO yield at cryogenic temperatures is comparable to values reported earlier for the photochemistry of aqueous NONOate solutions at ambient temperatures [25,26]. As the HNO concentrations remain low relative to NO, HNO dimerization is expected to be outpaced by the reaction of HNO with NO to produce nitrite and N2O, which imply that we can assign the ~10% production of N2O to a ~10% photoproduction of HNO per NO donor.
Figure 3:

180-K “illuminated” minus “dark” FTIR photolysis difference spectra of oxyMb in presence of unlabeled (black) and 15N-labeled (red) caged NO donor (A), and of the caged NO alone (B).
Compared to the control FTIR difference spectra obtained with the caged NO alone, the data obtained with oxyMb show a positive band at 1283 cm−1 that downshifts to 1260 cm−1 with the 15N-labeled NO donor (Figure 3A). These frequencies match those observed by RR spectroscopy for the vs(NO2) of the nitratoMb complex. The assignment of these FTIR signals to the nitrato vs(NO2) mode is further supported by the splitting of this signal observed when oxyMb is prepared with 18O2 gas (see Figure S1). Isotope sensitive differential signals are also seen around 1500 cm−1 in a frequency range where a nitrato vas(NO2) mode would be expected [27]. Accordingly, a strong positive band at 1507 cm−1 is likely to correspond to the nitrato the vas(NO2) mode as it is only seen when both oxyMb and the unlabeled caged NO are present. Unfortunately, the expected vas(15NO2) counterpart to this unlabeled vas(NO2) mode is not clearly defined, even in the isotope-edited double difference spectrum as isotope sensitive modes from the caged NO also contribute differential signals in that same frequency range (Figure 4). Accidental degeneracy of nitrato group vibrations with porphyrin skeletal modes occurring between 1380 and 1460 cm−1 may also contribute to the complexity seen in this spectral range of the isotope-edited difference spectrum. FTIR signals from free nitrate in aqueous solution are broad [28] and are not detected in our experiments with oxyMb and the caged NO (see Figure S2).
Figure 4:

Isotope-edited double difference spectra of oxyMb with the caged NO donor (top black trace) and the caged NO alone (bottom blue trace). The traces are obtained by subtracting the “illuminated-15N” minus “dark-15N” difference spectrum from the “illuminated-14N” minus “dark-14N” spectrum.
Sharp differential signals in the 1600 to 1700 cm−1 region are only seen when both oxyMb and the NO donor are present (Figure 3A). However, these signals are unaffected by the 15N-labeleing of the NO donor and cancel out in the isotope-edited double difference spectrum (Figure 4). These signals must be associated with minor frequency shifts of protein vibrations upon conversion of oxyMb to nitratoMb.
The isotope-edited double difference spectrum of oxyMb also show a very weak positive signal at 1614 and 1924 cm−1 that downshift 28 and 38 cm−1 with 15N-labeled caged NO. These frequencies are characteristic of the v(N-O) mode of the ferrous- and ferric-nitrosyl complexes of Mb [29,30]. The concentration of these nitrosyl species can be estimated to ~ 0.1 mM based on the intensity of the v(N-O) bands, which presumably reflects ~ 2% metMb and deoxyMb contamination in our starting oxyMb solutions. The photoproduction of nitroxyl from the NO donor may also contribute to the formation of these ferric and ferrous-nitrosyl Mb contaminants. Indeed, the v3 signals from N2O seen with the NO donor alone and attributed to the formation of HNO are not seen in the spectra obtained with oxyMb (Figure 4).
Interestingly, the N2O signals observed in control experiments with the caged NO alone are absent of the FTIR different spectra obtained with oxyMb samples, implying that the sub-stoichiometric HNO produced by the NO donor is consumed by myoglobin before it can produce N2O. The product of oxyMb reaction with nitroxyl would be expected to lead to the formation of nitrito-metMb species, but anticipated concentration of this side product is too low to obtain direct evidence for this species in our FTIR spectra. Moreover, we cannot rule out metMb and deoxyMb contaminants in the oxyMb samples as possible source for the HNO consumption; both are known to react with HNO and produce ferrous-nitrosyl and ferrous-nitroxyl species [31,32].
CONCLUSION
Using a photolabile caged NO donor at cryogenic temperatures, we have trapped a ferric-nitrato enzyme-product complex, for the NO dioxygenation reaction catalyzed by oxyMb. Below 180 K, the diffusion of the photolyzed NO into the heme distal pocket of Mb is inhibited, but above that temperature the radical reaction between NO and the superoxo group of oxyMb proceeds all the way to ferric-nitrate complex. The low-temperature UV-vis, RR, and FTIR data provide no evidence for the trapping of any of the putative early intermediate species such as ferric-peroxynitrite and Fe(IV)-oxo species. This failure to trap early intermediate species is consistent with theoretical modeling of this reaction and the prediction of near barrierless isomerization of Fe(III)-peroxynitrito to Fe(III)-nitrato species, but it clearly does not rule out the viability of these species as transient states in the NOD reaction of myoglobin. The 180-K thermal barrier for the initiation of the NO reaction in deoxyMb can be assumed to reflect collective motions of the protein matrix required for NO to gain access to near-attack position by the Fe(III)-superoxo group. Slightly higher thermal barriers (200 to 220 K) have been observed for non-geminate rebinding of CO to deoxyMb after photodissociation [33–35]. At ambient temperature, O2 access to the iron distal pocket is known to be gated by the distal histidine [36]; molecular dynamic simulations have proposed additional gated pathways across the surface of myoglobin [37,38], but experimental support for these alternative pathways remain limited. Tosha, Shiro and coworkers reported an equivalent 180-K thermal barrier for the reaction of NO with the detergent solubilized P.a. cNOR [19], but the gaping contrast in structures of these two proteins leave no doubts on the coincidental nature of these matching temperatures. Unlike Mb, P.a. cNOR is equipped with a large hydrophobic channel connecting the outer-hydrophobic surface of its twelve transmembrane α-helix bundle with the diiron active site anchored at its core [39].
Previous RFQ experiments with oxyMb and NO were performed in pH 9.0 buffer to allow for easy distinction between the high-spin nitrato complex and the low-spin hydroxy-metMb product [13]. It was unclear at the time if the alkaline conditions increase the lifetime of the nitrato complex and whether the same nitrato complex would form at neutral pH. The perfect match in the vs(NO2) frequencies seen in the RFQ samples and photogenerated complex indicates that the environment of the nitrato group is unaffected by the pH difference and that if pH 9.0 increase the lifetime of the nitrato complex it must be through changes at the egress route used by the nitrate group.
Our work here has shown how photoreleasing NO in frozen samples allows characterization of trapped reaction steps by low-temperature transmittance FTIR. In contrast to RR spectroscopy, transmittance FTIR provides direct access to quantitative information as well as evidence for protein sidechain conformational changes. Specifically, in the study discussed here, low-temperature FTIR spectra have revealed the presence of trace ferric- and ferrous-nitrosyl species, undetected in the RR data, and which we assigned to minor deoxyMb and metMb contaminants in the oxyMb starting material. It is conceivable that in some multistep reactions, this FTIR approach could afford information on conversion rates between intermediate species. Sharp derivative signals seen in the FTIR difference spectra associated with the conversion of oxyMb to the Fe(III)-nitrato complex that were unaffected by the labeling of the NO donor and absent from control experiments with the NO donor alone were indicators of perturbations at the porphyrin macrocycle and protein sidechains. Follow-up experiments with isotopic labeling of the protein and porphyrin substitutions would allow accurate assignments for these differential signals and lead to detailed reaction mechanism schemes.
Control FTIR experiments with the NO donor provide indirect evidence for the release of ~10 % nitroxyl upon UV irradiation. This small fraction of NO−/HNO production does not affect the promise of this photocryogenic approach to mechanistic studies of NO catalysis, but it will be important to keep this side-reaction in mind in future studies if multiple intermediate species are detected after photolysis.
Supplementary Material
Synopsis.
The graphic shows the structure of oxymyoglobin, the caged NO compound, and the result of uncaged at 180 K.
Highlights.
Nitric oxide readily diffuses into the active site of oxymyoglobin at 180 K.
The reaction of NO with oxymyoglobin at 180 K traps a Fe(III)-nitrato complex.
Transmittance FTIR data quantifies trace ferric- and ferrous-nitrosyl contaminants.
ACKNOWLEDGEMENTS
This work was supported by National Institutes of Health Grant R01 GM147588 to P.M.L. and R35GM147515 to J.D.C. We also acknowledge Dr. Anthony Altomare for assistance in characterizing the NO donor by mass spectrometry.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
REFERENCES
- [1].Fang FC, and Vazquez-Torres A (2002) Nitric oxide production by human macrophages: there’s NO doubt about it, Am. J. Physiol. Lung Cell. Mol. Physiol 282, L941–943. [DOI] [PubMed] [Google Scholar]
- [2].Wink DA, Hines HB, Cheng RY, Switzer CH, Flores-Santana W, Vitek MP, Ridnour LA, and Colton CA (2011) Nitric oxide and redox mechanisms in the immune response, J. Leukoc. Biol 89, 873–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Toledo JC Jr., and Augusto O (2012) Connecting the chemical and biological properties of nitric oxide, Chem. Res. Toxicol 25, 975–989. [DOI] [PubMed] [Google Scholar]
- [4].Molina C, Kissner R, and Koppenol WH (2013) Decomposition kinetics of peroxynitrite: influence of pH and buffer, Dalton Trans. 42, 9898–9905. [DOI] [PubMed] [Google Scholar]
- [5].Goldstein S, and Merenyi G (2008) The chemistry of peroxynitrite: implications for biological activity, Methods Enzymol. 436, 49–61. [DOI] [PubMed] [Google Scholar]
- [6].Forrester MT, and Foster MW (2012) Protection from nitrosative stress: A central role for microbial flavohemoglobin, Free Radic. Biol. Med 52, 1620–1633. [DOI] [PubMed] [Google Scholar]
- [7].Gardner PR (2012) Hemoglobin: a nitric-oxide dioxygenase, Scientifica 2012, 683729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Su J, and Groves JT (2009) Direct detection of the oxygen rebound intermediates, ferryl Mb and NO2, in the reaction of metmyoglobin with peroxynitrite, J. Am. Chem. Soc 131, 12979–12988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lehnert N, Kim E, Dong HT, Harland JB, Hunt AP, Manickas EC, Oakley KM, Pham J, Reed GC, and Alfaro VS (2021) The biologically relevant coordination chemistry of iron and nitric oxide: electronic structure and reactivity, Chem. Rev 121, 14682–14905. [DOI] [PubMed] [Google Scholar]
- [10].Herold S, Exner M, and Nauser T (2001) Kinetic and mechanistic studies of the NO*-mediated oxidation of oxymyoglobin and oxyhemoglobin, Biochemistry 40, 3385–3395. [DOI] [PubMed] [Google Scholar]
- [11].Herold S (1999) Kinetic and spectroscopic characterization of an intermediate peroxynitrite complex in the nitrogen monoxide induced oxidation of oxyhemoglobin, FEBS Lett. 443, 81–84. [DOI] [PubMed] [Google Scholar]
- [12].Olson JS, Foley EW, Rogge C, Tsai AL, Doyle MP, and Lemon DD (2004) NO scavenging and the hypertensive effect of hemoglobin-based blood substitutes, Free Radic. Biol. Med 36, 685–697. [DOI] [PubMed] [Google Scholar]
- [13].Yukl ET, de Vries S, and Moënne-Loccoz P (2009) The millisecond intermediate in the reaction of nitric oxide with oxymyoglobin is an iron(III)-nitrato complex, not a peroxynitrite, J. Am. Chem. Soc 131, 7234–7235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Crespo A, Marti MA, Kalko SG, Morreale A, Orozco M, Gelpi JL, Luque FJ, and Estrin DA (2005) Theoretical study of the truncated hemoglobin HbN: exploring the molecular basis of the NO detoxification mechanism, J. Am. Chem. Soc 127, 4433–4444. [DOI] [PubMed] [Google Scholar]
- [15].Mishra S, and Meuwly M (2010) Atomistic simulation of NO dioxygenation in group I truncated hemoglobin, J. Am. Chem. Soc 132, 2968–2982. [DOI] [PubMed] [Google Scholar]
- [16].Namiki S, Arai T, and Fujimori K (1997) High-performance caged nitric oxide: a new molecular design, synthesis, and photochemical reaction, J. Am. Chem. Soc 119, 3840–3841. [Google Scholar]
- [17].Tosha T, Nomura T, Nishida T, Saeki N, Okubayashi K, Yamagiwa R, Sugahara M, Nakane T, Yamashita K, Hirata K, Ueno G, Kimura T, Hisano T, Muramoto K, Sawai H, Takeda H, Mizohata E, Yamashita A, Kanematsu Y, Takano Y, Nango E, Tanaka R, Nureki O, Shoji O, Ikemoto Y, Murakami H, Owada S, Tono K, Yabashi M, Yamamoto M, Ago H, Iwata S, Sugimoto H, Shiro Y, and Kubo M (2017) Capturing an initial intermediate during the P450nor enzymatic reaction using time-resolved XFEL crystallography and caged-substrate, Nat. Commun 8, 1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Nomura T, Kimura T, Kanematsu Y, Yamada D, Yamashita K, Hirata K, Ueno G, Murakami H, Hisano T, Yamagiwa R, Takeda H, Gopalasingam C, Kousaka R, Yanagisawa S, Shoji O, Kumasaka T, Yamamoto M, Takano Y, Sugimoto H, Tosha T, Kubo M, and Shiro Y (2021) Short-lived intermediate in N(2)O generation by P450 NO reductase captured by time-resolved IR spectroscopy and XFEL crystallography, Proc. Natl. Acad. Sci. U. S. A 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Takeda H, Shimba K, Horitani M, Kimura T, Nomura T, Kubo M, Shiro Y, and Tosha T (2023) Trapping of a mononitrosyl nonheme intermediate of nitric oxide reductase by cryo-photolysis of caged nitric oxide, J. Phys. Chem. B 127, 846–854. [DOI] [PubMed] [Google Scholar]
- [20].Handa NV, Li S, Gerbec JA, Sumitani N, Hawker CJ, and Klinger D (2016) Fully Aromatic High Performance Thermoset via Sydnone-Alkyne Cycloaddition, J Am Chem Soc 138, 6400–6403. [DOI] [PubMed] [Google Scholar]
- [21].Kharitonov VG B. J; Sharma VS (1996) Interactions of nitric oxide with heme prtoeins using UV-vis spectroscopy, In Methods in Nitric Oxide Research (Stamler M. F. a. J. S., Ed.), pp 39–45, John Wiley & Sons. [Google Scholar]
- [22].Ibrahim M, Denisov IG, Makris TM, Kincaid JR, and Sligar SG (2003) Resonance Raman spectroscopic studies of hydroperoxo-myoglobin at cryogenic temperatures, J. Am. Chem. Soc 125, 13714–13718. [DOI] [PubMed] [Google Scholar]
- [23].Spiro TG (1975) Resonance Raman spectroscopic studies of heme proteins, Biochim Biophys Acta 416, 169–189. [DOI] [PubMed] [Google Scholar]
- [24].Laane J, and Ohlsen JR (1980) Characterization of nitric oxides by vibrational spectroscopy, Prog. Inorg. Chem 27, 465–513. [Google Scholar]
- [25].Pavlos CM, Cohen AD, D’Sa RA, Sunoj RB, Wasylenko WA, Kapur P, Relyea HA, Kumar NA, Hadad CM, and Toscano JP (2003) Photochemistry of 1-(N,N-diethylamino)diazen-1-ium-1,2-diolate: an experimental and computational investigation, J. Am. Chem. Soc 125, 14934–14940. [DOI] [PubMed] [Google Scholar]
- [26].Lymar SV, and Shafirovich V (2007) Photoinduced release of nitroxyl and nitric oxide from diazeniumdiolates, J. Phys. Chem. B 111, 6861–6867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Nakamoto K (1997) Infrared and Raman spectroscopy of inorganic and coordination compounds, Vol. A-B, 5th ed., John Wiley and Sons, Inc., New York. [Google Scholar]
- [28].Hudson PK, Schwarz J, Baltrusaitis J, Gibson ER, and Grassian VH (2007) A spectroscopic study of atmospherically relevant concentrated aqueous nitrate solutions, J. Phys. Chem. A 111, 544–548. [DOI] [PubMed] [Google Scholar]
- [29].Miller LM, Pedraza AJ, and Chance MR (1997) Identification of conformational substates involved in nitric oxide binding to ferric and ferrous myoglobin through difference Fourier transform infrared spectroscopy (FTIR), Biochemistry 36, 12199–12207. [DOI] [PubMed] [Google Scholar]
- [30].Tsubaki M, and Yu NT (1982) Resonance Raman investigation of nitric oxide bonding in nitrosylhemoglobin A and -myoglobin: detection of bound N-O stretching and Fe-NO stretching vibrations from the hexacoordinated NO-heme complex, Biochemistry 21, 1140–1144. [DOI] [PubMed] [Google Scholar]
- [31].Lin R, and Farmer PJ (2000) The HNO adduct of myoglobin: synthesis and characterization, J. Am. Chem. Soc 122, 2393–2394. [Google Scholar]
- [32].Sulc F, Immoos CE, Pervitsky D, and Farmer PJ (2004) Efficient trapping of HNO by deoxymyoglobin, J. Am. Chem. Soc 126, 1096–1101. [DOI] [PubMed] [Google Scholar]
- [33].Austin RH, Beeson KW, Eisenstein L, Frauenfelder H, and Gunsalus IC (1975) Dynamics of ligand binding to myoglobin, Biochemistry 14, 5355–5373. [DOI] [PubMed] [Google Scholar]
- [34].Srajer V, Reinisch L, and Champion PM (1991) Investigation of laser-induced long-lived states of photolyzed MbCO, Biochemistry 30, 4886–4895. [DOI] [PubMed] [Google Scholar]
- [35].Alben JO, Beece D, Bowne SF, Doster W, Eisenstein L, Frauenfelder H, Good D, McDonald JD, Marden MC, Moh PP, Reinisch L, Reynolds AH, Shyamsunder E, and Yue KT (1982) Infrared spectroscopy of photodissociated carboxymyoglobin at low temperatures, Proc. Natl. Acad. Sci. U. S. A 79, 3744–3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Scott EE, Gibson QH, and Olson JS (2001) Mapping the pathways for O2 entry into and exit from myoglobin, J. Biol. Chem 276, 5177–5188. [DOI] [PubMed] [Google Scholar]
- [37].Elber R (2010) Ligand diffusion in globins: simulations versus experiment, Curr Opin Struct Biol 20, 162–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Yu TQ, Lapelosa M, Vanden-Eijnden E, and Abrams CF (2015) Full kinetics of CO entry, internal diffusion, and exit in myoglobin from transition-path theory simulations, J. Am. Chem. Soc 137, 3041–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hino T, Matsumoto Y, Nagano S, Sugimoto H, Fukumori Y, Murata T, Iwata S, and Shiro Y (2010) Structural basis of biological N2O generation by bacterial nitric oxide reductase, Science 330, 1666–1670. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.