

Abstract
Duodenopancreatic neuroendocrine neoplasia (DP-NEN) is in approximately 10% of cases of multiple endocrine neoplasia type 1 (MEN1). We encountered a case in which the onset of NEN led to suspicion and diagnosis of MEN1. Although genetic testing showed MEN1 variant of uncertain significance (VUS), we considered it pathological from the clinical course, promoting the provision of genetic counseling and screening for relatives. MEN1 has a variety of clinical manifestations, and DP-NENs are the second-most common manifestation after primary hyperparathyroidism (pHPT). It is important to assume that MEN1 is an underlying cause of NEN.
Keywords: Multiple endocrine neoplasia type 1, Genetic counseling, Duodenopancreatic neuroendocrine neoplasia
Introduction
Multiple endocrine neoplasia (MEN) is a disease that causes tumors and hyperplasia in multiple endocrine organs and is classified according to the combination of disease types into type I (MEN1), type II (MEN2A and MEN2B), and familial medullary thyroid carcinoma (FMTC) [1]. Although all are inherited in the form of autosomal manifestations, the causative genes are different in each case, and there are significant differences in their relationship with the clinical picture. In MEN2, penetrance, disease type, and severity vary depending on the mutation codon of the RET gene, the causative gene, and there is a correlation between mutation type and expression type [2]. In contrast, MEN1, the causative gene of MEN1, is a tumor-suppressor gene encoding the menin protein and is characterized by a lack of any association between the site of mutation and the clinical presentation, requiring individualized and careful management of both affected and unaffected carriers.
In the present study, we conducted a series of genetic counseling sessions for a family of patients diagnosed with MEN1 with MEN1 variant of unknown significance (VUS), following the onset of a pancreatic neuroendocrine tumor.
We herein report this family case and provide a review of the pertinent literature.
Case report
A 66-year-old woman (Fig. 1, II-5) was diagnosed clinically with MEN1 when she visited her previous physician with a chief complaint of abdominal pain, which was treated conservatively with a diagnosis of multiple duodenal ulcers, and was found to have parathyroid adenoma and pituitary adenoma in addition to a tumor of the pancreatic head. Because of diabetes mellitus, a functional pancreatic tumor was suspected, but glucagonoma was ruled out upon closer examination. Adrenal origin diabetes was also ruled out. We then considered the possibility that the pancreatic head tumor was a gastrinoma, which is extremely common with MEN1 and it can also cause duodenal ulcers. The corrected serum calcium and intact parathyroid hormone (iPTH) levels were elevated, 10.6 mg/dl and 108.5 pg/ml, respectively, suggesting primary hyperparathyroidism (pHPT).
Fig. 1.

Family tree
Pancreaticoduodenectomy was performed, and the macroscopic appearance of the pancreatic head tumor was a milky-white, substantial mass, and the tumor cells were homogeneous with nucleus segregation and were histopathologically positive for chromogranin A and gastrin (Fig. 2). Therefore, it was diagnosed as a low-grade NEN and considered likely to be a gastrinoma. Since the patient also had pHPT, MEN1 was suspected, so brain magnetic resonance imaging (MRI) was performed, revealing pituitary adenoma (Fig. 3). Serum prolactin (PRL) level was elevated, at 52.91 ng/ml, and the patient was diagnosed with prolactinoma, based on the findings of additional endocrine tests. Notably a markedly decreased bone density was also found. Based on these findings, MEN1 which is a hereditary disorder was suspected. The patient wished to undergo a genetic test and genetic counseling was conducted in the presence of her children.
Fig. 2.
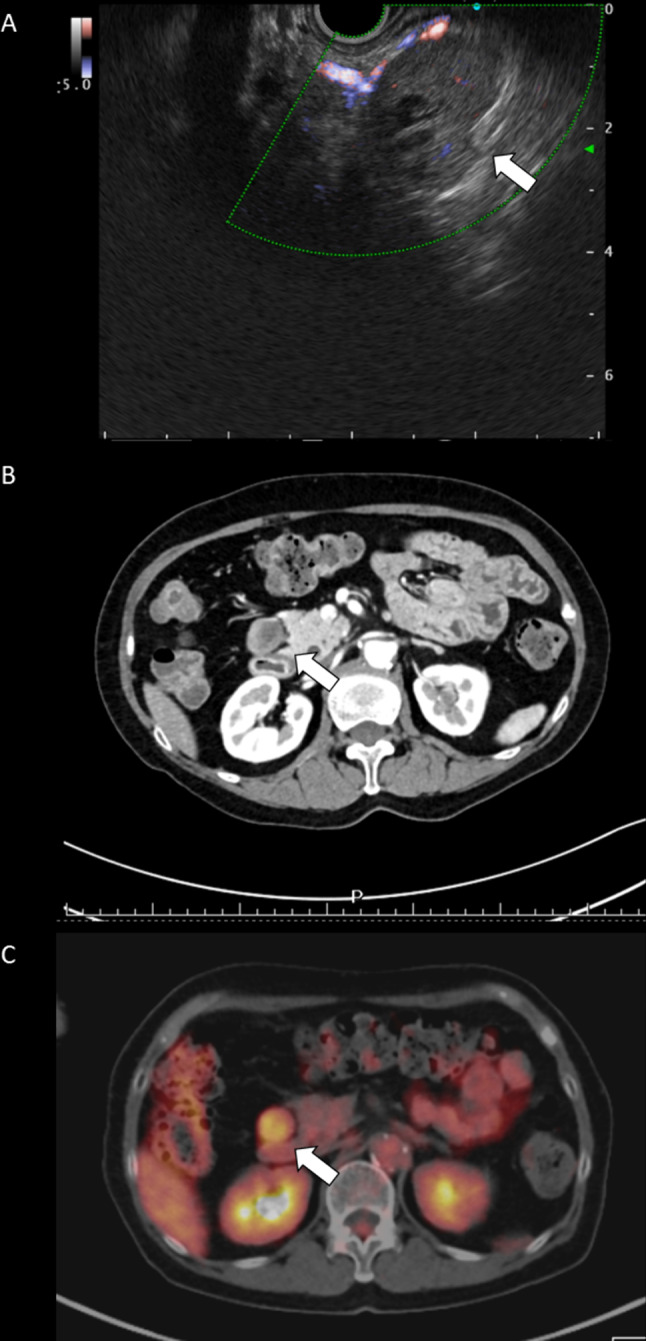
Preoperative examination. A Endoscopic ultrasonography findings showing the tumor of pancreas head to be well-circumscribed, hypoechoic with partially cystic change and hypovascular Doppler signaling. B Enhanced abdominal computed tomography (CT) findings showing an oval and poor-enhanced mass in front of the right kidney. C Planar and single photon emission computed tomography (SPECT) & maximum intensity projection (MIP) findings showing abnormal uptake in the pancreas head
Fig. 3.

A Macroscopic appearance of the specimen of the pancreas head, showing milky white and oval mass in the circle area. B–D Microscopic appearance of the tumor, B Hematoxylin–Eosin stain (Low-power field), showing homogeneous tumor cells with unevenly distributed nuclei. C-D Immunohistochemical findings, showing the tumor cells were C positive for chromogranin A, and D positive for gastrin
The patient’s family history was consistent with MEN1. She had an older brother (Fig. 1, II-2) who had died from thymic neuroendocrine tumor in his 70s and her mother (Fig. 1, I-2) had suffered from thymic disease and diabetes. Her 41-year-old son (Fig. 1, III-1) had been diagnosed with diabetes in his 20 s, and her 39-year-old daughter (Fig. 1, III-3) had been healthy at birth. Both of these children of the patient strongly requested genetic test be performed on their mother, which was granted. A MEN1 variant of unknown significance (VUS), (NM_130799.2): c.514G > T (p. Asp172Tyr), was detected. Another genetic counseling session was conducted, including an explanation of the variants. Although this variant was classified as a VUS in American College of Medical Genetics and Genomics (ACMG) Classification, we considered it a pathogenic variant, referred to as this variant by some manuscripts and databases; in ClinVar, it was referred to as either pathogenic or likely pathogenic (variation ID: 428,032), dbSNP: rs1114167494, The human genetic mutation database (HGMD®): CM981258, and the variant observed in this case was deemed to be a rare variant that had not been detected in ExAC (Database Commons), 1000 Genomes, or gnomAD. Screening for MEN1-related diseases was performed in both children, and pHPT was found in the daughter, who was thought to be healthy. Both son and daughter underwent genetic testing as well, and both showed the same variants as their mother. Based on these results, a third genetic counseling session was held, including the daughter's husband, to discuss future measures for the originator’s siblings and grandchildren. The siblings were offered another genetic counseling session at a later date, and genetic tests for the 7-year-old and 5-year-old grandchildren were planned.
In the present case, the serum pro-GRP level decreased from 102.0 pg/ml to 67.3 pg/ml 3 months after the pancreaticoduodenectomy. An operation for pHPT was therefore planned due to the patient’s poor response to the non-invasive administered medical treatments. Regarding the presence of a prolactinoma, the patient was carefully observed and no enlargement of tumor nor any clinical changes have so far been identified.
Discussion
DP-NENs are the second-most common manifestation of MEN1. Approximately 80% of DP-NENs are estimated to be sporadic, but 74% of MEN1 patients have multiple tumors at the time of the diagnosis of DP-NENs [1]. Although MEN1 accounts for only 0.42% of gastrointestinal NENs, 16–25% of gastrinomas are reportedly complicated by MEN1 and most of gastrinomas with MEN1 occur in the duodenum [2, 3]. Therefore, when encountering these diseases, it is important to consider the possibility of MEN1. However, clinical experiences of MEN1 with germline variants are still limited in Japan, so accumulating more clinical experience as well as referencing previously published manuscripts and guidelines are expected to aid in our understanding of MEN1 organ manifestations. Furthermore, not only the early detection of NENs but also postoperative follow-up is very important for MEN1 patients. With respect to the follow-up period after surgery, there is no consensus. The development of associated tumors of MEN1 may occur at an older age, including the possibility of the recurrence of NEN, and it is considered necessary to continue periodic examinations throughout the patient’s life. NEN recurrence is seen in 16–20% of patients after partial resection or enucleation of the tumor [4, 5]. As we know, in order to treat neoplastic lesions such as NENs in MEN1, a more peculiar examination and a follow-up are needed than with sporadic lesions.
Because of the wide variety of departments that have the opportunity to encounter MEN1-related diseases, the diagnosis may be delayed, especially when the symptoms seem to be clinically specific. It is therefore up to the physician responsible for the initial consultation to anticipate the possibility of this disease, so it is important to share knowledge across multiple medical specialists. Furthermore, NENs in thymus as well as DP-NENs lead death. We, therefore, must concentrate our efforts on confirming correct genetic diagnosis when we encounter such diseases. The clinical significance of the genetic evaluation of MEN is remarkable. In Japan, genetic testing of RET for medullary thyroid carcinoma (MTC) received insurance coverage in 2016 for MEN2, which is characterized by MTC, parathyroid hyperplasia and pheochromocytoma, and testing of MEN1 of MEN1, which is characterized by pHPT, pancreatic and gastrointestinal neuroendocrine tumors, and pituitary tumors, was also included in 2020. A proper diagnosis of MEN is associated with not only an improvement in the patient’s prognosis but also the early detection and treatment of relatives, including younger generations. Therefore, a genetic diagnosis is very important to make in order to distinguish between these diseases and requires appropriate genetic counseling.
Explaining the concept of VUSs to patients is difficult. The amount of information that should be provided may be controversial. We attempt to provide genetic counseling to patients found to have VUSs by referring to clinical findings, their family history, and domestic and international databases. In the present case, the c.514G > T (p.Asp172Tyr) variant was found in MEN1 and reported as a VUS. Some previous reports found the same variant, including sporadic case of pHPT, pancreatic endocrine tumor, anterior pituitary tumor, and Zollinger–Ellison syndrome in 54 families with MEN1 reported by Giraud et al. in 1998 [6, 7]; 2 families and 1 other patient in 25 families with MEN1 reported by Poncin et al. in 1999 [8]; and 1 out of 324 MEN1 cases with pituitary lesions reported in a French–Belgian multicenter study in 2002 [9]. Therefore, in the present case, this variant seemed to be pathogenic, considering the clinical findings and their family history. In another interesting report by Fujii et al. in 1999, the same somatic variant was observed in a case of functional pancreatic NEN, insulinoma, although the presence of germline variant was not surveyed [10]. The accumulation of the further data concerning this variant is expected.
Conclusion
We encountered a familial case of MEN1 with a VUS in MEN1. In present case, the patient had multiple neoplasms in addition to metabolic disorders due to this genetic disease. It is important to provide detailed genetic counseling to each individual client, and the identification of VUSs underscores the need for continuous research and collaboration to improve screening practices.
Acknowledgements
Written patient’s informed consent was obtained for publication of this report.
Abbreviations
- DP
Duodenopancreatic
- NEN
Neuroendocrine neoplasia
- MEN
Multiple endocrine neoplasia
- VUS
Variant of unknown significance
- pHPT
Primary hyperparathyroidism
- FMTC
Familial medullary thyroid carcinoma
- iPTH
Intact parathyroid hormone
- MRI
Magnetic resonance imaging
- PRL
Prolactin
- ACMG
American College of Medical Genetics and Genomics
- HGMD®
The human genetic mutation database
Author contributions
Y.T. carried out the genetic counselings, literature search and drafted the manuscript. T.N, T.M. and T.A. carried out the initial assessment and the management of the patient. A.A. supported to do diagnosis of the patient. K.I. evaluated histopathological features and contributed histological part. All authors read and approved the final manuscript.
Declarations
Conflict of interest
The authors fully declare any financial or other potential conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Sakurai A, Suzuki S, Kosugi S, MEN consortium of Japan et al. Multiple endocrine neoplasia type 1 in Japan: establishment and analysis of a multicentre database. Clin Endocrinol. 2012;76(4):533–539. doi: 10.1111/j.1365-2265.2011.04227.x. [DOI] [PubMed] [Google Scholar]
- 2.Ito T, Igarashi H, Nakamura K, et al. Epidemiological trends of pancreatic and gastrointestinal tumors in Japan: a nationwide surgery analysis. J Gastroenterol. 2015;50(1):58–64. doi: 10.1007/s00535-014-0934-2. [DOI] [PubMed] [Google Scholar]
- 3.Gibril F, Schmann M, Pace A, et al. Multiple endocrine neoplasia1 and Zollinger-Ellison syndrome: a prospective study of 107 cases and comparison with 1009 cases from the literature. Medicine. 2004;83(1):43–83. doi: 10.1097/01.md.0000112297.72510.32. [DOI] [PubMed] [Google Scholar]
- 4.Kouvaraki MA, Shapiro SE, Cote GJ, et al. Management of pancreatic endocrine tumors in multiple endocrine neoplasia type 1. World J Surg. 2006;30:643–653. doi: 10.1007/s00268-006-0360-y. [DOI] [PubMed] [Google Scholar]
- 5.Gauger PG, Doherty GM, Broome JT, et al. Completion pancreatectomy and duodenectomy for recurrent MEN-1 pancreaticoduodenal endocrine neoplasms. Surg. 2009;146(4):801–808. doi: 10.1016/j.surg.2009.06.038. [DOI] [PubMed] [Google Scholar]
- 6.Giraud S, Zhang XZ, Olga SS, et al. Germ-line mutation analysis in patients with multiple endocrine neoplasia type 1 and related disorders. Am J Hum Genet. 1998;63:455–467. doi: 10.1086/301953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wautot V, Vercherat C, Lespinasse J, et al. Germline mutation profile of MEN1 in multiple endocrine neoplasia type 1: search for correlation between phenotype and the functional domains of MEN1 protein. Hum Mutat. 2002;20:35–47. doi: 10.1002/humu.10092. [DOI] [PubMed] [Google Scholar]
- 8.Poncin J, Abs R, Velkeniers B, et al. Mutation analysis of the MEN1 gene in Belgian patients with multiple endocrine neoplasia type 1 and related disease. Hum Mutat. 1999;13:54–60. doi: 10.1002/(SICI)1098-1004(1999)13:1<54::AID-HUMU6>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 9.Vergès B, Boureille F, Goudet P, et al. Pituitary disease in MEN1 Type 1 (MEN1): data from the France-Belgium MEN1 multicenter study. J Clin Endocrino Metab. 2002;87:457–465. doi: 10.1210/jcem.87.2.8145. [DOI] [PubMed] [Google Scholar]
- 10.Fujii T, Kawai T, Saito K, et al. MEN1 gene mutations in sporadic neuroendocrine tumors of foregut derivation. Pathol Int. 1999;49:968–973. doi: 10.1046/j.1440-1827.1999.00971.x. [DOI] [PubMed] [Google Scholar]