

Abstract
Idiopathic multicentric Castleman disease (iMCD) is a rare hematologic disorder with heterogeneous presentations ranging from moderate constitutional symptoms to life-threatening multiorgan system involvement. There are vastly different clinical subtypes, with some patients demonstrating thrombocytopenia, anasarca, fever/elevated C-reactive protein, reticulin fibrosis/renal failure, and organomegaly (TAFRO) and others having milder/more moderate symptoms with potential for severe disease (not otherwise specified, NOS). Due to its rarity and heterogeneity, the natural history and long-term burden of iMCD are poorly understood. We investigated real-world medical data from ACCELERATE, a large natural history registry of patients with Castleman disease, to better characterize the long-term disease burden experienced by these patients. We found that iMCD-TAFRO patients face a significant hospitalization burden, requiring more time in the hospital than iMCD-NOS patients during the year surrounding diagnosis (median [interquartile range]: 36 [18-61] days vs. 0 [0-4] days; P<0.001). In addition, we found life-sustaining interventions, such as mechanical ventilation (17%) and dialysis (27%), were required among iMCD patients, predominantly those with iMCD-TAFRO. iMCD-NOS patients, however, spent a significantly greater proportion of time following disease onset in a state of disease flare (median 52.3% vs. 18.9%; P=0.004). Lastly, we observed severe iMCD-related morbidities, such as acute renal failure, sepsis and pneumonia, among others, arising after iMCD diagnosis, impairing the patients’ quality of life. These data demonstrate a substantial disease burden experienced by iMCD patients and emphasize the importance of ongoing research into iMCD to aid disease control.
Introduction
Idiopathic multicentric Castleman disease (iMCD) is a rare lymphoproliferative disorder characterized by widespread lymphadenopathy and a systemic inflammatory syndrome. While the pathophysiology of the disease remains poorly understood, existing data have implicated interleukin 6 and the mTOR and JAK-STAT pathways.1-5 As with other inflammatory disorders, iMCD is characterized by constitutional symptoms and systemic inflammation that can lead to multiorgan failure or death.6 Several different clinical subtypes of iMCD have been identified. Patients with the most severe subtype who demonstrate thrombocytopenia, anasarca, fever/elevated C-reactive protein, reticulin fibrosis/renal failure, and organomegaly are classified as having the TAFRO subtype.7 Other patients who do not meet TAFRO criteria are said to have iMCD not otherwise specified (NOS) and typically present with a milder clinical phenotype.7 A subset of NOS patients have thrombocytosis and hyper-gammaglobulinemia and have historically been described as having idiopathic plasmacytic lymphadenopathy.8 A recent epidemiological study based on USA insurance claims found that approximately 1,000 patients were diagnosed with iMCD each year.9 Making a diagnosis of iMCD is challenging because it is based on non-specific clinical features and characteristic lymph node histopathology. Underdiagnosis is likely as diagnostic criteria were not developed until 2017, and there is no known diagnostic serum biomarker.10 Challenges with diagnosis and limited understanding of the disease burden may contribute to poor outcomes. Data from the USA before 2012 suggested 5- and 10-year mortality rates of 35% and 60% for iMCD patients whereas more recent data from electronic medical records suggest 25% mortality at 5 years.11,12 Three-year survival estimates based on a recent large cohort of patients in China were 65.7% for TAFRO patients, 87.2% for NOS patients without the characteristics of idiopathic plasmacytic lymphadenopathy, and 98.5% for NOS patients with the features of idiopathic plasmacytic lymphadenopathy.13 In the USA and Europe, siltuximab, a monoclonal antibody directed against interleukin 6, is the only approved treatment and first-line recommended therapy.14,15 Tocilizumab, a monoclonal antibody directed against the interleukin 6 receptor, is recommended when siltuximab is not available and is approved for use in iMCD in Japan.16 For patients with severe disease who do not respond to first-line treatment with interleukin 6 blockade, multiagent cytotoxic chemotherapy is recommended.15
Due to its rarity and heterogeneous nature, the natural history and long-term burden of iMCD are not well understood. In light of the limited understanding of the burden of disease and the significant consequences of underestimating the risks of iMCD,17 we sought to investigate the burden of iMCD on patients and the healthcare system. ACCELERATE (NCT02817997), a longitudinal natural history study of Castleman disease, is ideally positioned to characterize the natural history and burden associated with iMCD. Herein, we present data from ACCELERATE to demonstrate that iMCD patients face a high burden of disease with severe multisystem organ involvement, the onset of life-threatening iMCD-related morbidities, long periods of hospitalizations during which interventions are frequently required, and extensive periods of time spent with flares of active disease.
Methods
Study population
Patients with a pathology report suggestive of Castleman disease were invited to enroll into ACCELERATE beginning in October 2016.18 Enrollment is open to patients in the USA and globally. Given that the registry website is written in English and records from institutions in English-speaking countries are more feasibly obtained, there is a bias towards enrollment of English-speaking patients. After enrollment, all available medical data from the time of symptom onset to the time of analysis were collected from each patient’s treating institution(s). Data were reviewed and abstracted into the study database, and each case underwent a rigorous, systematic review by three hematopathologists and four hematologist-oncologists to confirm the accuracy of the iMCD diagnosis. While it is impossible to know the exact number of patients in this study who have been investigated previously, it is possible that some patients have been included in prior case reports/small series.
Outcome definitions
Diease severity at diagnosis was classified according to the iMCD treatment guidelines and at least two criteria had to be met for a diagnosis of severe iMCD: an Eastern Cooperative Oncology Group performance status ≥2 or hospitalization, fluid retention, hemoglobin ≤8.0 g/dL, pulmonary involvement/interstitial pneumonitis with dyspnea, or stage IV renal dysfunction within 90 days of the diagnosis made by lymph node biopsy.15
To ensure standardization, we defined a new flare as the onset of at least two new iMCD disease symptoms or the clinical worsening of previously stable signs or symptoms. Flares also required at least one laboratory finding from the iMCD diagnostic minor criteria that was previously normal to become abnormal. A flare ended when a patient achieved at least 50% reduction of symptoms (minor criteria from the iMCD diagnostic criteria).10
The Systematized Nomenclature of Medicine - Clinical Terms (SNOMED-CT) ontology was used to categorize iMCD-related morbidities and comorbidities.
Patient-reported outcomes
Patients enrolled in ACCELERATE were given outcome surveys every 3 months throughout the duration of their enrollment. Data were analyzed from patients who completed the survey at least once. Response data from EuroQoL five-dimension, five-level (EQ-5D-5L), a validated quality-of-life (QOL) questionnaire, and from the MCD symptom score, a validated MCD symptom survey developed for the phase II siltuximab clinical trial, were analyzed for each patient’s most recent survey completion date.14 The EQ-5D-5L captures health on the day of the survey, as well as ease with each of mobility, self-care, usual activities, pain/discomfort, and anxiety/depression. The MCD symptom survey captures 16 symptoms on a scale scored from 1 to 6 with 1 being the least severe (did not experience) and 6 being the most severe (very severe). The sum of the responses yields the total MCD symptom score. The 16 symptoms assessed included cough, shortness of breath, loss of appetite, fatigue, lack of energy, feeling weak, sores or rash on skin (skin lesions), itching, numbness or tingling, pain, fever, swollen lymph nodes, swelling or edema in other body areas, night sweats, and excessive daytime sweating.
Statistical analyses
A χ2 test was used to compare proportions, and Wilcoxon rank sum was used to test for differences between continuous data. To compare hospitalization rates between iMCD patients and the general population, a matched case-control analysis was conducted using public data from the 2018 National Health Information Survey (NHIS), a cross-sectional household survey and the principal source of information on the health of the non-institutionalized population in the USA.19,20 For each case, four controls were matched for age (within 1 year), sex, and race.19,20 Kruskal-Wallis followed by Wilcoxon rank sum testing with the Bonferroni correction were used to compare controls with iMCD subtypes. Spearman rank correlation test was used to determine the correlation between QOL score and MCD symptom score. Analyses were performed using R v 4.04.
Ethical approval
The ACCELERATE natural history registry has received ethical approval from the University of Pennsylvania Institutional Review Board, with the most recent approval being given on March 9, 2023 (protocol: 824758).
Results
Patients with idiopathic multicentric Castleman disease present with severe clinical and laboratory abnormalities
Of the 136 patients suspected to have iMCD who were reviewed, the ACCELERATE expert panel confirmed that 102 (75%) met both clinical and pathological criteria for iMCD.10 The cohort with a confirmed diagnosis had a slight predominance of males (n=58, 56.9%), a median (interquartile range [IQR]) age of 35.3 (22.2-47.5) years, and 65% were identified as White (Table 1). The median (IQR) follow-up time after iMCD diagnosis was 3.4 (1.3-6.1) years for the full cohort, 3.3 (1.2-5.3) years for TAFRO patients, and 3.6 (1.6-8.1) years for iMCD-NOS patients. Eight patients were dead at the time of data collection (Online Supplementary Table S1). Sixty-one (60%) patients met TAFRO criteria7 and 41 (40%) were classified as having iMCD-NOS. TAFRO patients tended to be more commonly male (TAFRO 62% compared to NOS 49%) and younger (mean [standard deviation]: 33.0 [17.4] vs. 40.1 [13.9] years) than NOS patients. First, we investigated disease severity at diagnosis and across age ranges. Of the 100 patients with sufficient information to determine disease severity at diagnosis, 77 (77%) initially presented with severe disease. Notably, we found iMCD in individuals of all ages, with the youngest patient in the cohort diagnosed at 1.8 years and the oldest at 74.4 years. We examined severity by age range visually and observed that the majority of patients <30 or >60 years of age presented with severe disease (Figure 1A). When we examined this trend statistically, we found a significant difference among the groups (P=5.3x10-4). Patients <30 and patients >60 years old were more likely to have severe disease (94.6% and 90.0%, respectively) as compared to patients between 30 and 60 years old (62.2%) (Online Supplementary Table S2). All but one patient >60 years old presented with severe disease and all but two of the patients <30 years presented with severe disease, while a large proportion of patients in the 30- to 60-year-old age group presented with mild-moderate disease, which likely affected this result (Figure 1A).
Table 1.
Characteristics of the cohort of patients with idiopathic multicentric Castleman disease.

We next investigated the clinical and laboratory abnormalities present at diagnosis (Online Supplementary Table S3). Specifically, we focused on the degree of anemia and hypoalbuminemia as markers of disease activity at diagnosis in this cohort. Across all patients, the median (IQR) hemoglobin concentration was 7.8 (6.6-10.5) g/dL and the median (IQR) albumin level was 2.2 (1.8-2.9) g/dL; patients in both TAFRO and NOS subgroups had median levels below the lower limits of normal. Eighty-seven (85.3%) patients had anemia and 81 (79.4%) had hypoalbuminemia at diagnosis (Figure 1B). We also determined the proportions of patients overall and by subtype who had clinical abnormalities (Figure 1C, Online Supplementary Table S3). A large majority of patients presented with fluid retention (84%), splenomegaly (72%), and/or hepatomegaly (60%). While TAFRO patients accounted for most patients with
Figure 1.

Patients with idiopathic multicentric Castleman disease demonstrate severe disease at diagnosis. (A) A large proportion of patients with idiopathic multicentric Castleman disease (iMCD) across all ages had severe disease. Notably, 95% of patients under the age of 30 years presented with severe disease at diagnosis. (B) Within the cohort of 102 iMCD patients, 87 (85.3%) had anemia and 81 (79.4%) had hypoalbuminemia at diagnosis. *Lower limit of normal for hemoglobin in males (12.5 g/dL). †Lower limit of normal for hemoglobin in females (11.5 g/dL). ‡Lower limit of normal for albumin (3.5 g/dL). (C) A large majority of patients presented with clinical symptoms, which ranged from mild to severe. Patients with both clinical subtypes of iMCD (TAFRO and NOS) demonstrated clinical abnormalities. TAFRO: thrombocytopenia, anasarca, fever/elevated C-reactive protein, reticulin fibrosis/renal failure, and organomegaly; NOS: not otherwise specified. these clinical signs, we found a substantial subset of NOS patients with the same severe clinical features. Among the 102 patients, 77 (76%) presented with severe disease at diagnosis, including 57 (93%) TAFRO patients and 20 (51%) NOS patients (Table 1). These data demonstrate that iMCD patients of any age and subtype can have severe and burdensome disease.
Idiopathic multicentric Castleman disease patients with TAFRO face significant hospitalization burden
We examined the number of hospitalized days due to iMCD symptoms as another measure of disease burden. In the year surrounding diagnosis (6 months preceding and after the diagnostic biopsy), iMCD patients were hospitalized for a median (IQR) of 18 (0.5-38.0) days. When we stratified by days hospitalized prior to and following diagnosis, we found that patients were hospitalized a median (IQR) of 5 (0-13.8) days in the 6 months preceding their diagnosis (Figure 2A). In the 6 months following diagnosis, patients were hospitalized a median (IQR) of 7.5 (0-25.0) days. Eleven patients (10.8%) spent more than 60 days in hospital in the year surrounding their diagnosis, highlighting the morbidity of this disease.
Next, we examined hospitalizations by subtype of iMCD to evaluate any differences in burden of hospitalizations. We found that TAFRO patients spent significantly more days in hospital than did NOS patients. The median (IQR) days in hospital in the year surrounding diagnosis was 36 (18-61) for patients with TAFRO and 0 (0-4) for NOS patients (W=153.5, P=1.3x10-13) (Figure 2B). In order to understand both subtypes in the context of a general population and given that most of our cohort were from the USA, we accessed the NHIS 2018 survey data20 and identified a control population of 408 individuals matched for age (±1 year), sex, and race. In a 12-month period, control patients were hospitalized for a median (IQR) of 0 (0-0) days. The number of days hospitalized differed between the general population and both TAFRO (W=47.5; P<2.2x10-16) and NOS (W=5,638; P=8.3x10-13) patients. This demonstrates that compared to the general population, patients with iMCD require substantially greater use of the healthcare system and frequently need to be admitted to hospital.
Figure 2.

Patients with idiopathic multicentric Castleman disease face a high burden of hospitalizations. (A) Patients with idiopathic multicentric Castleman disease (iMCD) are hospitalized a large proportion of time in the 6 months leading up to their diagnosis and the 6 months following diagnosis (year around diagnosis). The graph depicts each patient on the Y axis with the time hospitalized prior to diagnosis and the number of days hospitalized after diagnosis. Red indicates the time that iMCD patients with TAFRO (thrombocytopenia, anasarca, fever/elevated C-reactive protein, reticulin fibrosis/renal failure, and organomegaly) spent in hospital, and blue indicates the time that patients with iMCD not otherwise specified (NOS) were hospitalized. In the 6 months prior to diagnosis, patients were hospitalized a mean (standard deviation [SD]) of 10.6 (17.5) days and a median (interquartile range [IQR]) of 5 (0-13.8) days, while in the year following diagnosis, patients were hospitalized a mean (SD) of 18.9 (28.2) days and a median (IQR) of 7.5 (0-25.0) days. (B) A sample of the USA population from the 2018 National Health Information Survey (NHIS), a proxy for the general population, spent significantly less time hospitalized in a 12-month period (median [IQR]: 0 [0-0], mean [SD]: 0.24 [1.3] days) than either TAFRO patients (median [IQR]: 35 [18-61], mean [SD]: 46.0 [42.0] days; P<2.2x10-16) or NOS patients (median [IQR]: 0 [0-4], mean [SD]:4.9 [9.0] days; P=8.3x10-13) spent hospitalized in the year around diagnosis. TAFRO patients were hospitalized significantly more days than NOS patients within the year of diagnosis (P=1.3x10-13). ***P<0.001.
We hypothesized that initiating a course of treatment for iMCD would result in a reduction in the time spent in hospital. To test this hypothesis, we first compared the proportion of patients hospitalized at the initiation of the first iMCD treatment regimen, excluding corticosteroid monotherapy, to the proportion hospitalized 4 weeks after initiating a treatment regimen. Four weeks after initiation was selected to allow sufficient time for the respective treatment regimen to have an effect. Among the 99 patients who received treatment with at least one regimen, 49 (49.5%) were hospitalized at the time of starting the treatment regimen. After 4 weeks of treatment, the number hospitalized had been reduced to 20 (20.2%) patients (X=17.4; P=3.0x10-5). We stratified the patients by type of treatment regimen, considering the regimens most frequently administered as first-line therapy, including siltuximab ± corticosteroids, tocilizumab ± corticosteroids, rituximab ± corticosteroids, chemotherapy-based regimens, and immunomodulator(s) ± corticosteroids. Corticosteroid monotherapy was not included in this analysis given that this approach is not recommended and patients often receive a short course while awaiting a correct diagnosis. We found that among the 32 patients treated with siltuximab ± corticosteroids as first-line therapy, 11 (34.3%) were hospitalized at the time of regimen initiation, and two (6.3%) were hospitalized 4 weeks after starting the treatment regimen (P=0.01). While fewer patients were hospitalized 4 weeks after starting treatment with each of the other regimens, we did not find any other statistically significant differences, although small sample sizes limit interpretation (Online Supplementary Table S4). Overall, these data demonstrate the clear burden of hospitalizations early in the course of iMCD and the importance of administering targeted therapy at the time of diagnosis.
Patients with idiopathic multicentric Castleman disease often require urgent interventions and demonstrate multisystem organ involvement
Considering the degree of hospitalization burden, we next investigated the distribution of organ system involvement among iMCD patients ± 365 days from diagnosis (Online Supplementary Methods). Cytopenias were the most common laboratory abnormalities, experienced by 97.1% (n=99) of the cohort. This most frequent abnormality was anemia, which was present in 87 (85.3%) patients. We also found large proportions of patients experiencing symptoms or laboratory abnormalities related to other organ systems, such as respiratory (n=90, 88.2%), gastrointestinal (n=86, 84.3%), hepatic (n=76, 74.5%), neurological (n=68, 66.7%), renal (n=63, 61.8%), cardiovascular (n=63, 61.8%), and ocular (n=27, 26.5%) systems (Figure 3A, Online Supplementary Tables S5 and S6).
Next, we looked at life-sustaining interventions related to organ system involvement. We found that 27 (26.5%) patients required dialysis and 17 (16.7%) patients required a ventilator during at least one iMCD-related hospitalization. Additionally, 47 (46.0%) patients required paracentesis, 42 (41.1%) patients received a red blood cell transfusion, and 22 (21.6%) patients received a platelet transfusion (Figure 3B). These interventions, including all platelet transfusions, were mostly required for TAFRO patients.
We also quantified the proportion of time patients spent in a state of flare. We found that NOS patients spent a significantly greater proportion of time in flare from presentation until last known follow-up (median [IQR]: 52.3% [21.0-99.6]) compared to TAFRO patients (18.9% [10.8-52.5], W=1,673; P=0.004) (Figure 3C, D). Of the eight deceased patients in this cohort, six had TAFRO and five of these TAFRO patients died within 2 years of diagnosis. These data suggest that while TAFRO patients experience a greater degree of organ failure and more hospital interventions, NOS patients experience a longer continuation of milder, chronic symptoms.
Patients with idiopathic multicentric Castleman disease develop severe disease-related morbidities and comorbidities following diagnosis
We next sought to identify and quantify the morbidities and comorbidities occurring both before and after the diagnosis of iMCD. Prior to the iMCD diagnosis, we found that the most common conditions diagnosed among the full cohort were hypertension (n=26, 25.5%), obesity (n=23, 22.5%), asthma (n=21, 20.6%), gastroesophageal reflux disease (n=14, 13.7%), and depression (n=11, 10.8%) (Figure 4A). Stratification by iMCD subtype did not reveal apparent differences in conditions identified before the diagnosis of iMCD (Online Supplementary Figure S1).
Following the iMCD diagnosis, 48% (n=49) of the cohort developed acute renal failure, 15.7% (n=16) developed chronic kidney disease, 13.7% (n=14) developed iron deficiency anemia, 10.7% (n=11) developed pneumonia, 6.9% (n=7) developed sepsis, and 6.9% (n=7) developed thrombotic microangiopathy (Figure 4B). Acute renal failure was the most common iMCD-related morbidity diagnosed following disease onset in both subtypes (Online Supplementary Figure S2). Sepsis and thrombotic microangiopathy were, however, primarily diagnosed in TAFRO patients. We also stratified the top three morbidities arising for the full cohort following iMCD onset by organ systems, revealing other serious conditions such as atypical hemolytic uremic syndrome (n=5, 4.9%), acute cholecystitis (n=5, 4.9%), and congestive heart failure (n=4, 3.9%) (Online Supplementary Table S7). Few malignancies were identified following diagnosis; the most common malignancy was papillary thyroid carcinoma, which occurred in two patients 1.3 and 3.6 years after iMCD diagnosis (Online Supplementary Table S8).
Quality of life is inversely correlated with symptoms in idiopathic multicentric Castleman disease
We questioned patients about their experiences living with iMCD by asking about QOL and the presence of ongoing iMCD-related symptoms. Fifty-nine (57.8%) patients responded. At a median (IQR) of 3.9 (2.3-8.0) years after diagnosis, iMCD patients reported a median (IQR) QOL score of 80 (71.5-90.0) in the EQ-5D-5L, on a scale of 0 to 100 with 0 being the worst health imaginable and 100 being the best health imaginable. This compares to a mean score of 80.4 in the general USA population.21 That same day, the patients reported a median MCD symptom score of 29.5 (IQR, 20-37; min-max: 16-96) with 16 being the best possible score (no iMCD symptoms) and 96 being the worst possible score. We investigated whether there was any relationship between a patient’s QOL score and number of days hospitalized prior to completing the survey, but did not found a correlation (R=0.038; P=0.8) (Online Supplementary Figure S3). We also looked for differences in subsequent QOL scores between patients who presented with severe disease and mild/moderate disease at baseline as well as between TAFRO and non-TAFRO patients and found no significant differences between groups (TAFRO vs. NOS: W=481, P=0.31; severe vs. mild/moderate at baseline: W=298.5, P=0.61). We determined the correlation between QOL score and MCD symptom score based on data collected a median (IQR) of 3.9 (2.3-8.0) years after diagnosis. QOL scores were inversely correlated with MCD symptom score (R=-0.69; P<0.001) (Figure 5). This negative correlation appeared to hold for patients with both NOS and TAFRO subtypes. This suggests that several years after diagnosis, patients reporting active iMCD symptoms may be having a decrease in their QOL from those symptoms.
Figure 3.

Patients with idiopathic multicentric Castleman disease experience a range of organ system involvement, require various hospital interventions, and demonstrate ongoing flares. (A) Organ system involvement in idiopathic multicentric Castleman disease (iMCD) ranked from most frequently observed to least frequently observed. Over 97% of the iMCD cohort experienced some hematopoietic dysfunction. Notably, both patients with TAFRO (thrombocytopenia, anasarca, fever/elevated C-reactive protein, reticulin fibrosis/renal failure, and organomegaly) and those with iMCD not otherwise specified (NOS) experienced significant organ system involvement and dysfunction. (B) The severity of organ dysfunction is reflected by the degree of healthcare intervention(s) required. Over one-quarter of patients (N=27 [26.5%]) required the use of a ventilator, and 17 (16.7%) patients required dialysis. Additionally, 47 (46.0%) patients required fluid removal (paracentesis), 42 (41.1%) patients received a red blood cell transfusion, and 22 (21.6%) patients received a platelet transfusion. (C) NOS patients spent a significantly greater proportion of time in flare from presentation until last known information (median [interquartile range]: 52.3% [21.0-99.6]) compared to TAFRO patients (18.9% [10.8-52.5], W=1673; P=0.004). (D) Each patient is represented by a vertical bar. The bar extends the length of follow-up from the start of the first flare. NOS patients are represented on the left, and TAFRO patients are represented on the right. The blue bars represent the proportion of time NOS patients spent in flare. The red bars represent the proportion of time TAFRO patients spent in flare. The designation ‘d’ indicates a deceased patient. RBC: red blood cells.
Figure 4.

Patients with idiopathic multicentric Castleman disease have numerous comorbid and morbid conditions contributing to the burden of the disease. (A) Prior to diagnosis, the most commonly diagnosed comorbidities among the full cohort of patients with idiopathic multicentric Castleman disease (iMCD) mirrored common comorbidities among the USA population and included hypertension (N=26, 25.5%), obesity (N=23, 22.5%), asthma (N=21, 20.6%), gastroesophageal reflux disease (N=14, 13.7%), and depression (N=11, 10.8%). (B) Following the diagnosis of iMCD, patients experienced an array of burdensome comorbidities and morbidities including acute renal failure (N=49, 48.0%), chronic kidney disease/chronic renal insufficiency (N=16, 15.7%) and iron deficiency anemia (N=11, 10.8%) among others. GERD: gastroesophageal reflux disease; ADHD: attention-deficit hyperactivity disorder; CKD/CRI: chronic kidney disease/chronic renal insufficiency; TMA: thrombotic microangiopathy; OSA: obstructive sleep apnea; NOS: not otherwise specified; TAFRO: thrombocytopenia, anasarca, fever/elevated C-reactive protein, reticulin fibrosis/ renal failure, and organomegaly.
Figure 5.
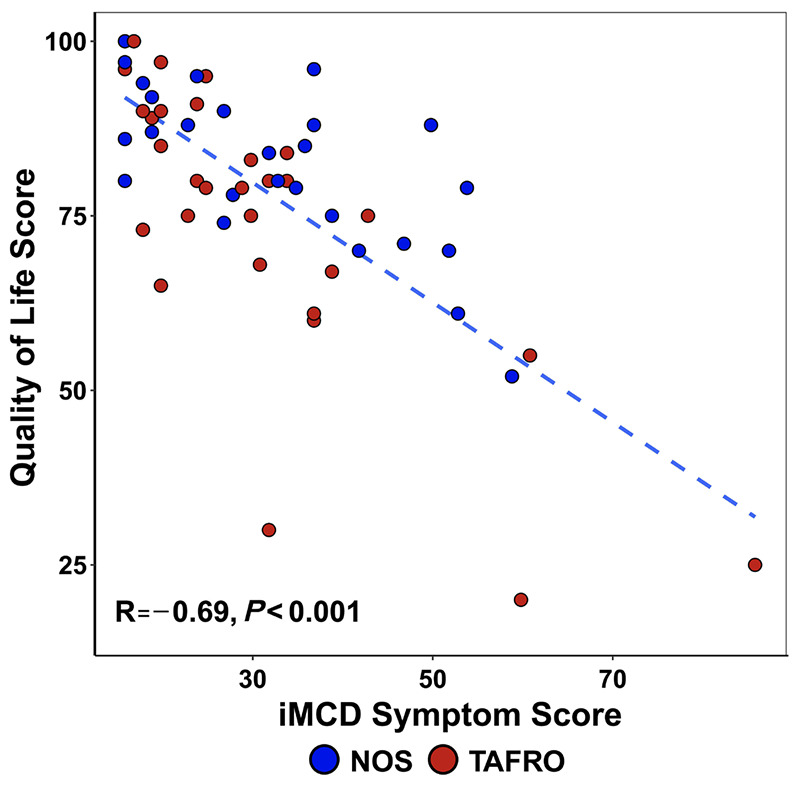
Quality of life of patients with idiopathic multicentric Castleman disease is inversely correlated with degree of symptoms. There is a correlation between quality-of-life score and idiopathic multicentric Castleman disease (iMCD) symptom score. Lower quality of life correlates with higher iMCD symptom score and higher quality of life correlates with lower iMCD symptom score (R=-0.69, P<0.001). NOS: not otherwise specified; TAFRO: thrombocytopenia, anasarca, fever/elevated C-reactive protein, reticulin fibrosis/renal failure, and organomegaly.
Discussion
iMCD is a rare, heterogeneous disorder, and its full impact on patients’ lives is still not well understood. Since the consensus diagnostic criteria for iMCD are relatively new, long-term epidemiological and medical data from patients with confirmed iMCD are sparse. The ACCELERATE natural history registry is ideally suited to better understand the burden of disease as the most extensive source of longitudinal clinical data (median 3.4 years of follow-up after diagnosis) for these patients. ACCELERATE also provides independent adjudication about the accuracy of diagnosis for each patient through a panel of clinicians and hematopathologists who are experienced in diagnosing and treating iMCD.18 We found that patients with iMCD require extensive use of the healthcare system and experience long-term effects in terms of iMCD-related morbidities and comorbidities, time in disease flare, and QOL.
There are distinct clinical subtypes of iMCD and, importantly, we found differences in the burden of disease between these subtypes. Patients with TAFRO required significantly longer stays in hospital and disproportionately required interventions such as dialysis, mechanical ventilation, and transfusions, illustrating the severe, life-threatening nature of this disease. NOS patients, however, spent a significantly greater proportion of time following diagnosis in a state of flare compared to TAFRO patients. These data reveal the challenges iMCD patients experience across the spectrum of clinical subtypes and severity. The patient-reported outcome data from our study indicate that patients with both subtypes continue to experience an impact on their QOL even years after diagnosis and that persistent iMCD-related symptoms lower QOL significantly. Notably, QOL scores reflect the state of patients who have thus far survived their disease and may, therefore, be biased towards a healthier subset of the disease population.
Although NOS patients spent significantly less time in hospital, we found that 51.3% met criteria for severe disease at presentation. This remains lower than the 93.4% of TAFRO patients who presented with severe disease. A recent study from China characterizing 418 iMCD-NOS patients applied the same definition of severity and found that only 87 (21%) patients met criteria for severe disease at presentation.13 The researchers also noted a lower 3-year overall survival rate among NOS patients with severe (76%) compared to mild/moderate (94%) disease and found that severity at diagnosis was associated with death. Notably, our study showed high proportions of patients in both younger (<30 years) and older (>60 years) age groups who presented with severe disease. In our study, patients under 30 years old were significantly more likely to present with severe disease, and nearly all patients <18 years presented with severe disease. This may be associated with the median age of the TAFRO subtype of patients being 31 years, compared to 39 years for patients with iMCD-NOS, and with the fact that 93% of TAFRO patients presented with severe disease. The relatively young age at diagnosis, burden of disease, and availability of disease-controlling therapies highlight the importance of rapid diagnosis and treatment.
This study found that iMCD patients experienced a greater degree of hospitalization than the general population experiences in a given year, with this being primarily due to the increased rates of hospitalization among iMCD-TAFRO patients.20 Our observation that patients required extensive hospital care in the year surrounding diagnosis is consistent with the findings of a recent claims-based study.17 We found that 4 weeks after the first-administered treatment regimen, the proportion of hospitalized patients decreased significantly, including patients treated with siltuximab ± corticosteroids (34% vs. 6%), which is the current consensus first-line recommendation.15
We also found that the conditions most frequently diagnosed in our cohort prior to the diagnosis of iMCD were similar to those among the general USA population. A report from the Agency for Healthcare Research and Quality found that hypertension, diabetes, and chronic respiratory disease were among the most common conditions associated with inpatient stays and that depression was present in 10% of patients requiring admission to hospital,22 which is similar to the findings in our cohort prior to the diagnosis of iMCD. We did not identify a condition present in a large proportion of iMCD patients prior to the iMCD diagnosis that might suggest a predisposition or trigger to iMCD, which has an as-yet-unknown etiology. Following the diagnosis of iMCD, patients in both the TAFRO and NOS subtypes developed life-threatening morbidities, most commonly acute renal failure. Consistent with previous reports, we found a high degree of both renal and hepatic dysfunction.17 In this cohort, few patients developed a malignancy following the diagnosis of iMCD. In fact, only one patient developed a myeloid malignancy (myelodysplastic syndrome), which contrasts with a key finding from a systematic literature review and a claims-based study that found an increase in myeloid and solid malignancies.17,23 No patients developed diffuse large B-cell lymphoma despite previous reports of its association with MCD, though predominantly in human herpes virus 8-positive MCD.24-26 We found that our rate of malignancies was comparable to that in the control population in the claims-based study.17 This discrepancy may be due to insufficient follow-up time to detect subsequent malignancies or to more rigorous validation of the iMCD diagnosis in our cohort and recent formalization of exclusion criteria for iMCD. Specifically, iMCD diagnostic criteria preclude patients from being concurrently diagnosed with iMCD and a number of malignancies, given the overlapping histopathology and the fact that malignancies can cause reactive lymph node changes that resemble iMCD. It is, therefore, possible that some patients in the claims-based study who should have only been classified as having a malignancy were incorrectly classified as having both. Conversely, our strict inclusion criteria might have inadvertently excluded true iMCD patients who also developed malignancies.
This study has several limitations. First, 13 patients with the NOS subtype met the criteria for idiopathic plasmacytic lymphadenopathy (thrombocytosis and hypergammaglobulinemia) and may represent a distinct subtype from other iMCD-NOS patients.27 Due to the low number of patients and the similar, more moderate presentation, these patients were combined with other non-TAFRO patients in the NOS category.13 Second, our study included a larger proportion of TAFRO patients than NOS patients, so may not be representative of the whole iMCD population. To address this, we stratified our analyses between TAFRO and NOS subtypes. It is possible that the higher number of TAFRO patients in this study is a result of patients with more severe disease being more likely to seek resources online and thus enroll into the ACCELERATE registry. However, the study design for our registry is biased towards patients who survive and who are able to enroll themselves into a registry compared to patients who die shortly after diagnosis and would need a family member to enroll them.
Ultimately, our data demonstrate that an iMCD diagnosis imparts a long-term burden of disease on patients which results in high rates of hospitalization, the development of life-threatening iMCD-related morbidities and comorbidities requiring intensive interventions, and reduced QOL from active symptoms. These data demonstrate the importance of ongoing research into iMCD, the need to recognize and diagnose iMCD sooner, and the relevance of focused efforts to identify diagnostic and/or disease biomarkers to aid in disease management.
Supplementary Material
Acknowledgments
The authors thank all the patients and their families for their participation in the ACCELERATE registry. We also thank the Castleman Disease Collaborative Network (CDCN) and the ACCELERATE Registry team for their support. We thank the volunteers for the CDCN who have supported this research, including Mary Zuccato and Mileva Repasky. We also thank Shawnee Bernstein, Nathan Hersh, Gerard Hoeltzel, and Jeremy Zuckerberg for their contributions to this study. Finally, we thank Faizaan Ahkter, Erin Napier, Eric Haljasmaa, Katherine Floess, Mark-Avery Tamakloe, Victoria Powers, Alexander Gorzewski, Johnson Khor, Reece Williams, Jasira Ziglar, Amy Liu, Saishravan Shyamsundar, Criswell Lavery, and Bridget Austin.
Funding Statement
Funding: The ACCELERATE natural history registry has received funding from Janssen Pharmaceuticals (2016-2018), EUSA Pharma, LLC (USA), which has merged with Recordati Rare Diseases Inc. (2018-2022), and the U.S. Food & Drug Administration (R01FD007632) (2022-Present). DCF also receives funding from the National Heart, Lung, and Blood Institute (R01HL141408) (2018-present).
Data-sharing statement
All source data reported in this study are available by contacting the ACCELERATE team at accelerate@uphs.upenn.edu.
References
- 1.Beck JT, Hsu SM, Wijdenes J, et al. Brief report: alleviation of systemic manifestations of Castleman’s disease by monoclonal anti-interleukin-6 antibody. N Engl J Med. 1994;330(9):602-605. [DOI] [PubMed] [Google Scholar]
- 2.Yoshizaki K, Matsuda T, Nishimoto N, et al. Pathogenic significance of interleukin-6 (IL-6/BSF-2) in Castleman’s disease. Blood. 1989;74(4):1360-1367. [PubMed] [Google Scholar]
- 3.Fajgenbaum DC, Langan RA, Japp AS, et al. Identifying and targeting pathogenic PI3K/AKT/mTOR signaling in IL-6-blockaderefractory idiopathic multicentric Castleman disease. J Clin Invest. 2019;129(10):4451-4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arenas DJ, Floess K, Kobrin D, et al. Increased mTOR activation in idiopathic multicentric Castleman disease. Blood. 2020;135(19):1673-1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pierson SK, Shenoy S, Oromendia AB, et al. Discovery and validation of a novel subgroup and therapeutic target in idiopathic multicentric Castleman disease. Blood Adv. 2021;5(17):3445-3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fajgenbaum DC, June CH. Cytokine storm. N Engl J Med. 2020;383(23):2255-2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nishimura Y, Fajgenbaum DC, Pierson SK, et al. Validated international definition of the thrombocytopenia, anasarca, fever, reticulin fibrosis, renal insufficiency, and organomegaly clinical subtype (TAFRO) of idiopathic multicentric Castleman disease. Am J Hematol. 2021;96(10):1241-1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takeuchi K. Idiopathic plasmacytic lymphadenopathy: a conceptual history along with a translation of the original Japanese article published in 1980. J Clin Exp Hematop. 2022;62(2):79-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mukherjee S, Martin R, Sande B, Paige JS, Fajgenbaum DC. Epidemiology and treatment patterns of idiopathic multicentric Castleman disease in the era of IL-6-directed therapy. Blood Adv. 2022;6(2):359-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fajgenbaum DC, Uldrick TS, Bagg A, et al. International, evidence-based consensus diagnostic criteria for HHV-8-negative/idiopathic multicentric Castleman disease. Blood. 2017;129(12):1646-1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dispenzieri A, Armitage JO, Loe MJ, et al. The clinical spectrum of Castleman’s disease. Am J Hematol. 2012;87(11):997-1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cohen AB, Swaminathan A, Wang XL, et al. Clinical characteristics, treatment patterns, and overall survival of real-world patients with idiopathic multicentric Castleman disease. J Clin Oncol. 2021;39(15 suppl):7048. [Google Scholar]
- 13.Zhang L, Dong YJ, Peng HL, et al. A national, multicenter, retrospective study of Castleman disease in China implementing CDCN criteria. Lancet Reg Health West Pac. 2023;34:100720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Rhee F, Wong RS, Munshi N, et al. Siltuximab for multicentric Castleman’s disease: a randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2014;15(9):966-974. [DOI] [PubMed] [Google Scholar]
- 15.van Rhee F, Voorhees P, Dispenzieri A, et al. International, evidence-based consensus treatment guidelines for idiopathic multicentric Castleman disease. Blood. 2018;132(20):2115-2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nishimoto N, Kanakura Y, Aozasa K, et al. Humanized anti-interleukin-6 receptor antibody treatment of multicentric Castleman disease. Blood. 2005;106(8):2627-2632. [DOI] [PubMed] [Google Scholar]
- 17.Mukherjee S, Kanhai K, Kauffman D, et al. Organ dysfunction, thrombotic events and malignancies in patients with idiopathic multicentric Castleman disease: a population-level US health claims analysis. Leukemia. 2022;36(10):2539-2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pierson SK, Khor JS, Ziglar J, et al. ACCELERATE: a patient-powered natural history study design enabling clinical and therapeutic discoveries in a rare disorder. Cell Rep Med. 2020;1(9):100158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.CDC. Hospital utilization (in non-Federal short-stay hospitals). 2018. https://www.cdc.gov/nchs/fastats/hospital.htm#print. Accessed June 2023. [Google Scholar]
- 20.NHIS. About the National Health Interview Survey. 2019. https://www.cdc.gov/nchs/nhis/about_nhis.htm. Accessed June 2023. [Google Scholar]
- 21.Jiang R, Janssen MFB, Pickard AS. US population norms for the EQ-5D-5L and comparison of norms from face-to-face and online samples. Qual Life Res. 2021;30(3):803-816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Owens PL, Liang L, Barrett ML, Fingar KR. Comorbidities associated with adult inpatient stays, 2019. Healthcare Cost and Utilization Project (HCUP) statistical briefs. Rockville (MD), 2006. [PubMed] [Google Scholar]
- 23.Liu AY, Nabel CS, Finkelman BS, et al. Idiopathic multicentric Castleman’s disease: a systematic literature review. Lancet Haematol. 2016;3(4):e163-175. [DOI] [PubMed] [Google Scholar]
- 24.Payera E, Zofia M, Zsofia S, et al. Diagnostic and therapeutic difficulties in diffuse large B-cell lymphoma arising from HHV8 positive Castleman disease. J Hematol. 2012;1(2):65-69. [Google Scholar]
- 25.Sukswai N, Lyapichev K, Medeiros LJ, Khoury JD. Unusual case of human herpesvirus 8-positive large B-cell lymphoma associated with Castleman disease. Clin Case Rep. 2019;7(3):587-588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Whooley M, Keirns D, Wu XX, Luker A, Silberstein PT, Hsia B. Diffuse large B-cell lymphoma in HHV8+ multicentric Castleman disease: a national analysis of demographic features. J Clin Oncol. 2023;41(16 suppl):e19535. [Google Scholar]
- 27.Nishikori A, Nishimura MF, Nishimura Y, et al. Idiopathic plasmacytic lymphadenopathy forms an independent subtype of idiopathic multicentric Castleman disease. Int J Mol Sci. 2022;23(18):10301. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All source data reported in this study are available by contacting the ACCELERATE team at accelerate@uphs.upenn.edu.