

Abstract
Background
Inherited retinal dystrophies (IRD) are one of the main causes of incurable blindness worldwide. IRD are caused by mutations in genes that encode essential proteins for the retina, leading to photoreceptor degeneration and loss of visual function. IRD generates an enormous global financial burden due to the lack of understanding of a significant part of its pathophysiology, molecular diagnosis, and the near absence of non-palliative treatment options. Patient-derived induced pluripotent stem cells (iPSC) for IRD seem to be an excellent option for addressing these questions, serving as exceptional tools for in-depth studies of IRD pathophysiology and testing new therapeutic approaches.
Methods
From a cohort of 8 patients with PROM1-related IRD, we identified 3 patients carrying the same variant (c.1354dupT) but expressing three different IRD phenotypes: Cone and rod dystrophy (CORD), Retinitis pigmentosa (RP), and Stargardt disease type 4 (STGD4). These three target patients, along with one healthy relative from each, underwent comprehensive ophthalmic examinations and their genetic panel study was expanded through clinical exome sequencing (CES). Subsequently, non-integrative patient-derived iPSC were generated and fully characterized. Correction of the c.1354dupT mutation was performed using CRISPR/Cas9, and the genetic restoration of the PROM1 gene was confirmed through flow cytometry and western blotting in the patient-derived iPSC lines.
Results
CES revealed that 2 target patients with the c.1354dupT mutation presented monoallelic variants in genes associated with the complement system or photoreceptor differentiation and peroxisome biogenesis disorders, respectively. The pluripotency and functionality of the patient-derived iPSC lines were confirmed, and the correction of the target mutation fully restored the capability of encoding Prominin-1 (CD133) in the genetically repaired patient-derived iPSC lines.
Conclusions
The c.1354dupT mutation in the PROM1 gene is associated to three distinct AR phenotypes of IRD. This pleotropic effect might be related to the influence of monoallelic variants in other genes associated with retinal dystrophies. However, further evidence needs to be provided. Future experiments should include gene-edited patient-derived iPSC due to its potential as disease modelling tools to elucidate this matter in question.
Graphical Abstract

Supplementary Information
The online version contains supplementary material available at 10.1186/s13287-024-03804-2.
Keywords: iPSC, Retinal diseases, PROM1 gene, CD133, Retinitis pigmentosa, Cone-rod dystrophy, Stargardt’s type 4 disease
Background
Inherited retinal dystrophies (IRD) are a group of rare Mendelian inheritance diseases caused by mutations in genes that encode essential proteins for various retinal cell processes, usually leading to visual loss due to progressive photoreceptor degeneration [1, 2]. IRD are the main cause of visual impairment in the working age group of 16 to 65 years and the second most common reason among children, affecting their quality of life and imposing an enormous financial burden globally [3, 4].
Progressive IRD can be clinically subclassified as Retinitis Pigmentosa (RP), which is the most prevalent form and primarily affects rods; Progressive Cone Dystrophy (PCD), primarily affecting cones, although it frequently evolves to Cone-Rod Dystrophy (CORD); and Macular Dystrophy (MD), affecting both rods and cones in the macular region. Inheritance patterns include autosomal dominant (AD), autosomal recessive (AR), and X-linked (XL) Inheritance [5–7].
Various clinical presentations of inherited retinal diseases (IRD) may arise from distinct variants within the same gene [8–11], while specific mutations in a single gene can result in different IRD phenotypes [12–16]. Due to the significant phenotypic and genotypic heterogeneity, molecular diagnosis based on Next Generation Sequencing (NGS) methodologies is deemed essential for IRD diagnosis [17, 18]. NGS techniques, including targeted panels or Clinical Exome Sequencing (CES) have shown considerable utility in accurately determining genotype–phenotype correlations in IRD patients [18–21].
PROM1-related retinopathies can manifest as pan-retinal phenotypes, as in Retinitis Pigmentosa type 41 (RP type 41, OMIM 268000) and Cone-Rod Dystrophy (CORD, OMIM 604116 or 120,970); or as macular dystrophies such as Stargardt's disease type 4 (STGD4, OMIM 248200) [22, 23]. Additionally, some specific single mutations in the PROM1 gene have been associated with both AD and AR forms of the disease, commonly AD forms tend to be of later onset and AR earlier [8, 23, 24]. Even within the same family, it has been reported substantial differences in the severity and extension of retinal degeneration. This clinical heterogeneity may be due to the influence of modifier genes, epigenetic and environmental factors [25].
PROM1 encodes the 5-domains transmembrane glycoprotein Prominin-1 (CD133) [26]. Historically recognised as stem cell marker, plays a crucial role in fundamental cellular processes, including self-renewal, metabolism or differentiation [27]. In the retina, prominin-1 is expressed at the base of the photoreceptors’ outer segments (OS). It is fundamental for the morphogenesis and structure of the OS membranes due to its interaction with the protocadherin-21, actin filaments, and myosin II, and it also plays an important role in the regulation of calcium-dependent chlorine channels [28, 29]. Likewise, it has significant implication in the regulation of autophagy in the retinal pigmented epithelium (RPE), and its deficiency increases apoptosis in retinal glial cells [30, 31].
There is currently no treatment available for IRD, apart from the unique additive therapy for biallelic mutations in the RPE65 gene (Luxturna, voretigene neparvovec-rzyl) established in 2017 [32]. However, gene therapy for IRD is an active area of research, and some clinical trials have shown encouraging results, such as those conducted on RPGR or CNGA3 genes [33, 34]. Other gene therapy clinical trials have been paused due to various issues that need to be addressed, as seen in the case of CHM gene [35]. The challenge lies in the limitation of specific gene therapy, making it difficult to correct the more than 300 genes associated with IRD [36]. To date, there are not potential therapeutic alternatives for patients in advanced stages of retinal degeneration other than stem cell therapy and optogenetics [37]. In this regard, patient-derived induced pluripotent stem cells (iPSC) appear to be a promising option for addressing these challenges.
iPSC are obtained by reprogramming somatic cells through the introduction of a set of pluripotency transcription factors, originally: Oct3/4, Sox2, c-Myc, and Klf4 [38]. They provide an accessible and abundant source of cells potentially capable of differentiating into retinal cells such as RPE and photoreceptors, without the ethical concerns associated to Embryonic Stem Cells (ESC) [39]. Patient-derived iPSC are valuable tools for in vitro disease modelling (disease-in-a-dish) and drug screening for retinal diseases [40]. In addition, iPSC with corrected mutations using CRISPR/Cas9 editing can be used to study gene function or generate cell therapy products as an alternative treatment to replace the degenerated retina in advanced disease [41–44]. At present, only two PROM1-related iPSC lines, both associated with the same mutation c.619G > T (p.E207X), have been reported [45].
Upon analysing a cohort of eight PROM1-related IRD patients we identified the pleiotropic effect of the IRD variant c.1354dupT (p.Tyr452Leufs*13) in homozygosity with AR inheritance. This mutation was linked to CORD, RP, and STGD4 in three different patients from our series. Therefore, the aim of this study was to elucidate the multi-phenotypic effect of the target mutation by expanding the patient’s genetic panel through CES, obtaining patient-derived iPSC, and genetically repairing those iPSC to study this mutation in vitro as an initial step towards designing new treatment options for these patients.
Materials and methods
Aim, design, and setting
To elucidate the pleiotropic effects of the IRD variant c.1354dupT (p.Tyr452Leufs*13) located in exon 13 of the PROM1 gene, by identifying associated modifier genes, and to create gene-edited patient-derived iPSC. The study was structured as a transversal study and experimental research. It was carried out at the Institute for Applied Ophthalmobiology (IOBA) and at the Institute of Biomedicine and Molecular Genetics (IBGM), both affiliated with the University of Valladolid.
Patient selection and sampling
A series of eight patients previously diagnosed with PROM1-related IRD was revised (Table 1). They underwent clinical and genetic characterization, and three presenting distinct phenotypes associated with the homozygous c.1354dupT (p.Tyr452Leufs*13) mutation were initially selected for the experimental study. Furthermore, a first-degree relative of each of these target patients was included.
Table 1.
Genetic background of identified patients carrying PROM1-related retinopathies
| Patient | Phenotype | Gender | Age at inclusion (years) | Ethnicity | Allele 1 | Allele 2 | Zygosity | Inheritance mode | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Exon/Intron | Nucleotide | Protein | Exon/Intron | Nucleotide | Protein | |||||||
| IRD 1 | CORD | F | 47 | Caucasic | Exon 13 | c.1354dupT | p.Tyr452Leufs*13 | Exon 13 | c.1354dupT | p.Tyr452Leufs*13 | Homozygosity | AR |
| IRD 2 | RP | F | 54 | Caucasic | Exon 13 | c.1354dupT | p.Tyr452Leufs*13 | Exon 13 | c.1354dupT | p.Tyr452Leufs*13 | Homozygosity | AR |
| IRD 3 | STGD4 | M | 20 | Caucasic | Exon 13 | c.1354dupT | p.Tyr452Leufs*13 | Exon 13 | c.1354dupT | p.Tyr452Leufs*13 | Homozygosity | AR |
| IRD 4 | CORD | F | 32 | Caucasic | Exon 5 | c.615 T > A | p.Tyr205* | Intron 20–21? | c.2240G > A | p.Trp747* | Compound heterozygosity | AR |
| IRD 5 | CORD | M | 41 | Caucasic | Exon 8 | c.869delG | p.Ser290IleFs*2 | Exon 8 | c.869delG | p.Ser290IleFs*2 | Homozygosity | AR |
| IRD 6 | STGD 4 | M | 41 | African | Exon 10 | c.1117C > T | p.Arg373Cys | – | – | – | Heterozygosity | AD |
| IRD 7 | STGD 4 | M | 37 | Latin | Exon 10 | c.1117C > T | p.Arg373Cys | – | – | – | Heterozygosity | AD |
| IRD 8 | CORD | F | 64 | Caucasic | Intron 17 | c.1984-1G > T | r.(spl?) | Intron 21 | c.2327A > T | p.Asp776Val | Compound heterozygosity | AR |
IRD: inherited retinal dystrophy; CORD: cone-rod dystrophy; STGD4: Stargardt disease type 4; RP: retinitis pigmentosa; AD: autosomal dominant; AR: autosomal recessive, F: female; M: male
Peripheral blood samples were collected from the patients and their respective relatives for CES. Dermal biopsy samples were obtained from the three target patients and were used to generate iPSC.
Ophthalmological examination
To confirm the patient’s phenotype and establish their pedigree patterns, a comprehensive anamnesis and ophthalmological examination were performed. Visual acuity (VA) was tested using an Early Treatment Diabetic Retinopathy Study panel and recorded as the logarithmic of minimum angle of resolution (logMAR) scale. Retinography and fundus autofluorescence (FAF) were obtained with the TRC 50DX Retinograph (TOPCON Europe Medical BV, Rotterdam, the Netherlands). Sweep-source optical coherence tomography (OCT) was gathered using the PLEX® Elite 9000 OCT (Carl Zeiss AC). Visual field (VF) testing was performed when fixation was possible with the Humphrey Field Analyzer (Carl Zeiss Meditec, Dublin, CA). A full field electroretinogram (ERG) was also performed according to the regularly updated standards of the International Society for Clinical Electrophysiology of Vision (ISCEV) [46].
Clinical exome sequencing
To investigate the potential influence of other modifying genes that could account for the varied phenotypic expressions associated with the target mutation, we conducted clinical CES of three target patients and three healthy relatives. DNA was extracted from blood cells, the Roche exome capture library was prepared, and extensive CES was carried out on a NovaSeq 6000 S4 (Illumina, CA, USA) with 90 × coverage. Bioinformatic analysis of the exons and adjacent intronic regions was performed on a total of 998 genes linked to retinal diseases (Supplementary file 1). We concentrated on genes associated with IRD [47], Age-related Macular Disease (AMD) [48], and genes coding for the phenotype HP:000047, indicative of abnormal retinal morphology [49]. Subsequently, in-silico predictors were applied as filters, and the findings were validated by Sanger sequencing using the ABI Prism 3130xl genetic analyzer (Thermo Fisher, MA, USA). Only variants classified as pathogenic or likely pathogenic were considered for further analysis.
Induced pluripotent stem cells generation
Fibroblast isolation
A dermal biopsy was performed at the patient’s cranial gluteal area, previously disinfected with 10% povidone iodine and rinsed with saline solution, under local anesthesia injected subcutaneously (1% lidocaine), using a 3-mm biopsy punch (Stiefel, Middlesex, UK). The biopsies were immersed in IMDM medium supplemented with 10% fetal bovine serum (FBS), 3% penicillin/streptomycin (P/S), 3X Amphotericin B and 0.1 µM 2-Mercaptoetanol (Gibco, Invitrogen, Paisley, UK. Cat# 11,510,596, 26,140,079, 11,548,876, 15,290,018, 31,350,010). Biopsies were then cut into small pieces (small as possible, approximately 0.5 × 0.5 mm) and cultured in collagen-coated (Sigma-Aldrich, Saint Louis, USA. Cat# C3867-1VL) 100-mm culture dishes (Thermo Fisher, MA, USA. Cat# A30907) using “fibroblast medium”, composed by IMDM supplemented with 10% FBS, 1X P/S and 1X Amphotericin B and 0.1 µM 2-Mercaptoetanol (Gibco, Invitrogen, Paisley, UK. Cat# 11,510,596, 26,140,079, 11,548,876, 15,290,018, 31,350,010) under standard conditions (37Cº, 5% CO2 atmosphere). The fibroblast medium was refreshed every two days. Biopsies pieces were maintained in culture until fibroblasts outgrowth was observed, approximately at day 10 to 14 (Fig. 1).
Fig. 1.

Generation of patient-derived iPSC from IRD1 patient. At D0, dermal biopsy is cut into small pieces, seeded in 100-mm culture dishes, and maintained with IMDM medium. At D14, outgrowth of fibroblast is noticeable from biopsy pieces. At D28, passage 2 patient-derived fibroblast are reprogrammed using the episomal vectors through electroporation. At D38, three days after electroporation, IMDM medium is changed to mTSR-E7 medium. At D56, three weeks after electroporation, iPSC colonies have emerged between fibroblasts. iPSC colonies are picked up manually and seeded in new culture dishes using the mTeSR plus medium. At D70, passage 2 iPSC colonies displaying compact colonies with distinct borders, well-defined edges, and large nuclei. Days after electroporation*
Non-integrative fibroblast reprogramming
The patient’s fibroblasts were reprogrammed using The Epi5 Episomal iPSC Reprogramming Kit (Thermo Fisher, MA, USA. Cat# A15960) containing the reprogramming vectors pCE-hOCT3/4 (OCT4 gene), pCE-hSK (Sox2 and KlF4 genes), pCE-hUL (L-Myc and Lin28 genes) in Tube A, and the pCE-mP53DD (mp53DD gene), and pCXB-EBNA1 (EBNA1 gene) in Tube B. Passage 2 fibroblast (150,000 cells) were electroporated with 1 µL of Tubes A and B, using the 100 µL pipette tip of the Neon Transfection System (Thermo Fisher, MA, USA. Cat# MPK5000) under the following pulse conditions: 1400 V; 20 ms; two pulses. The electroporated fibroblast were then seeded in Matrigel-coated (23 µg/cm2; Corning Life Sciences, NY, USA. Cat# 11,593,620) 6-well plates (Thermo Fisher Scientific, MA, USA. Cat# 11,337,694) using fibroblast medium, being changed every 24 h until day 3. Subsequently, the medium was replaced by the TeSR™-E7™ Medium (Stem Cells Technologies, Cambridge, UK. Cat# 05914) until iPSC colonies emerged, approximately 21 days. The iPSC clones were manually picked using a 22Gx2″ hypodermic needle (Terumo, Madrid, Spain), the Leica LED2500-TL3000 ERGO microscope (Thermo Fisher, MA, USA), and transferred with a p200 Pipette (Gilson PIPETMAN; Thermo Fisher Scientific, MA, USA. Cat# 1,232,613) to a Matrigel-coated (23 µg/cm2; Corning Life Sciences, NY, USA. Cat# 11,593,620) 6-well plate with mTeSR™ Plus medium supplemented with 10 µM Rock inhibitor Y-27632 (Stem Cells Technologies, Cambridge, UK. Cat# 100–0276, 72,304). iPSC were passaged as clumps using 0.5 mM EDTA (Thermo Fisher, MA, USA. Cat# 10,135,423) every 5–7 days, and frozen using freezing medium composed by 90% FBS (Thermo Fisher, MA, USA. Cat# 26,140,079) and 10% DMSO (Sigma-Aldrich, Saint Louis, USA. Cat# 34,869) (Fig. 1).
Induced pluripotent stem cells (iPSC) characterization
Alkaline phosphatase staining
To demonstrate the characteristic upregulation of alkaline phosphatase (AP) activity in the generated iPSC lines, we performed the AP blue membrane substrate solution assay (Sigma-Aldrich, Saint Louis, USA. Cat# AB0300). On day 5, a single 6-well plate (Thermo Fisher Scientific, MA, USA. Cat# 11,337,694) was fixed with 4% paraformaldehyde (Panreac Quimica, Barcelona, Spain) for 1 min. The plate was then washed with phosphatase-buffered saline (PBS) prewarmed to 64 °C (Thermo Fisher, MA, USA. Cat# 10,010,023) and incubated for 20 min at the same temperature. Afterwards, 1.5 ml of a 1:1 mixture of kit solutions A and B was added, and the plate was incubated in the dark at room temperature (RT) for 10 min. Direct micrographs were obtained using a Nikon Eclipse TS100 inverted microscope (Nikon Instruments, NY, USA).
Immunocytochemical characterization
Immunocytochemical characterization was performed as previously described [50]. Briefly, iPSC lines were grown on Matrigel-coated (23 µg/cm2; Corning Life Sciences, NY, USA. Cat# 11,593,620) μ-Slide 8-well culture plates (Ibidi, München, Germany. Cat# 80,826) and fixed with 10% formalin solution (Sigma-Aldrich, Saint Louis, USA. Cat# HT501128). The plates were rinsed with Tris-buffered saline (TBS; Sigma-Aldrich, Saint Louis, USA. Cat# T5912) and blocked with a mixture of TBS, 0,3% Triton X-100 and 3% donkey serum, (Sigma-Aldrich, Saint Louis, USA Cat# X100, D9663) for 60 min. Primary antibodies associated with pluripotency markers (OCT4, SSEA3, SOX2, SSEA4, TRA1-60, NANOG, TRA-1–81) and germ layers markers (β-III Tubulin, α-1 Fetoprotein, α- Smooth Muscle Actin) were applied, followed by incubation with their respective species-specific secondary antibody. Nuclei were visualized by immunostaining with DAPI (4',6-diamidino-2-phenylindole, Invitrogen, CA, USA. Cat# 10,184,322). The specific conditions for the primary and secondary antibodies are detailed in Supplementary file 2.
All immunocytochemical analyses were carried out in triplicate for each experimental condition. Controls, were primary and/or secondary antibodies were omitted, were processed concurrently. Immunofluorescence micrographs were captured using a LEICA TCS SP8 LIGHTNING confocal microscope (Leica Microsystems, Hesse, Germany) and analysed with LEICA LAS AF software (Leica Microsystems, Hesse, Germany).
Expression of pluripotency factors and silencing of reprogramming vectors
To assess the expression levels of endogenous pluripotency factors, we conducted a gene-specific primer-based quantitative reverse transcription PCR (qRT-PCR) for the genes SOX2, OCT3/4, KLF4, LMYC, LIN28. On the other hand, to probe the absence of reprogramming vectors, we performed a dual assay: a copy number qPCR, using genomic DNA (gDNA) and targeting a region common to all reprogramming vectors (within the EBNA1 gene), and a vector-specific primer-based qRT-PCR, using complementary DNA (cDNA), for each exogenous reprogramming factor delivered by the reprogramming vectors.
For qRT-PCR assays, RNA was extracted from the patient-derived iPSC lines at passage 6 using the Trizol Reagent (Invitrogen, CA, USA. Cat# 15,596,026) according to the manufacturer’s protocol. The purity and concentration of the RNA were determined using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, MA, USA). cDNA synthesis was performed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, CA, USA. Cat# 4,368,814), following the manufacturer’s guidelines.
For EBNA1 copy number qPCR, gDNA extraction was performed using ethanol precipitation [51]. A standard curve was generated from serial dilutions of a vector contained in the Epi5 Episomal iPSC Reprogramming Kit (Thermo Fisher, MA, USA. Cat# A15960), pCE-hOCT3/4 (Addgene #41,813). The number of copies of EBNA1 in each dilution was determined based on the length of the pCE-hOCT3/4 plasmid.
All qPCRs were carried out with SYBR Green PCR Master Mix (Applied Biosystems, CA, USA. Cat# 4,309,155) on the LightCycler 480 Instrument II System (Roche, Switzerland). The cycling conditions were as follows: an initial denaturation at 95 °C for 10 min, followed by 45 cycles of 95 °C for 15 s, 60 °C for 30 s, and 72 °C for 45 s, with a final extension at 72 °C for 5 min and a subsequent melting curve analysis. GAPDH served as the housekeeping gene for normalizing mRNA expression levels. The threshold cycle was determined for each reaction, and gene expression levels were quantified using the 2−ΔΔCt method [51]. All qPCR assays were conducted in triplicate for each experimental condition.
Untransfected fibroblasts from IRD1 patient served as the negative control for the expression of pluripotency factors essay; while fibroblasts from IRD1, transfected with the same episomal vectors as the iPSC lines, 72 h post-transfection, served as positive control. Primers used in qRT-PCR essays for endogenous pluripotency factors (gene primers) and exogenous reprogramming vectors (plasmid primers) are listed in Supplementary file 3.
In vitro differentiation into three germinal layers
To assess the differentiation potential of patient-derived iPSC lines into cells from the three germ layers, three-dimensional iPSC aggregates known as embryoid bodies (EBs) were specifically induced to differentiate into cell types characteristic of each layer. iPSC were cultured to 80% confluence on Matrigel-coated (23 µg/cm2; Corning Life Sciences, NY, USA. Cat# 11,593,620) 60 mm culture plates (Thermo Fisher, MA, USA). They were then dissociated with 0,5 mM EDTA (Thermo Fisher, MA, USA. Cat# 10,135,423), resuspended in mTeSR™ Plus medium (Stem Cells Technologies, Cambridge, UK. Cat# 100–0276) and seeded into 96-well V-bottom ultra-low attachment plates (Sigma-Aldrich, Saint Louis, USA. Cat# 10,462,012), which were centrifuged to facilitate EB formation. The EBs were cultured in ultra-low attachment 60-mm plates (Corning Life Sciences, NY, USA. Cat# 3261) for 3 days, subsequently transferred to Matrigel coated (23 µg/cm2; Corning Life Sciences, NY, USA. Cat# 11,593,620) μ-Slide 8 well culture plates (Ibidi, München, Germany. Cat# 80,826) and maintained for two to three weeks with three distinct differentiation media: endoderm medium (DMEM supplemented with 20% FBS, 2 mM Glutamax™, 100 μM non-essential amino acids, 100 μM 2-Mercaptoetanol and 1X P/S, [Gibco, Invitrogen, Paisley, UK. Cat# 13,345,364, 26,140,079, 35,050,061, 11,140,050, 11,528,926, 11548876]), mesoderm medium (endoderm medium supplemented with 100 μM ascorbic acid [Sigma-Aldrich, Saint Louis, USA. Cat# V-038]), and ectoderm medium (50% DMEM F12, 50% neurobasal medium, 2 mM Glutamax™, 1X N2 supplement, 1X B27 supplement and 1X P/S [Gibco, Invitrogen, Paisley, UK. 11,320,033, 21,103,049, 35,050,061, 17,502,048, 17,504,044, 11548876]).
Short tandem repeat analysis
To verify the genetic concordance between the patient’s fibroblasts and their respective iPSC lines, DNA fingerprinting analysis was conducted. The amplification of short tandem repeat (STR) was performed through multiple PCR using the kit GenePrint® 10 System PCR Amplification Kit (Promega, Wisconsin, USA. Cat# B9510). STR analysis at 10 loci (CSF1PO, D13S317, D16S539, D21S11, D5S818, D7S820, TH01, TPOX, vWA, and Amelogenin for sex determination) were carried out in accordance with the ASN-0002 standard established by the American Tissue Culture Collection Standards Development Organization Workgroup for cell line authentication. The amplified samples were evaluated on a 3730 DNA Analyzer (Applied Biosystems. CA. USA) using the POP-7 polymer and the ILS600 size standard. The results were analysed using the GeneMapper® software (v4.1, Applied Biosystems).
Karyotyping
To assess the genomic integrity of the generated iPSC lines, a G-banded metaphase chromosome analysis with a resolution of 300–500 bands was conducted. The patient-derived iPSC colonies at 70% of confluence were incubated in KaryoMax colcemid (Thermo Fisher, MA, USA. Cat 15,212,012) at a final concentration of 0,1 μg/mL for 3 h to induce mitotic arrest during metaphase. The cells were then trypsinized, exposed to a prewarmed 37ºC hypotonic solution (KCl 75 mM) for 15 min and fixed with methanol/glacial acetic acid (3:1) solution. A total of 20 metaphases were analysed.
Target mutation sequencing
The target mutation (c.1354dupT) was sequenced in the iPSC lines. gDNA extraction was performed using ethanol precipitation [52]. The region of the PROM1 gene containing the target mutation was amplified by conventional PCR. Subsequently, the PCR-amplified fragments were sequenced using Capillary Electrophoresis Sanger Sequencing on an ABI 3730xl DNA analyser (Thermo Fisher, MA, USA). The sequencing primer PROM1seqFW is listed in Supplementary file 3.
Patient-derived iPSC genetic repair
gRNA designing
The gRNA were designed in silico using the CRISPOR web tool for genome editing [53], selected based on their proximity to the mutation and their specificity and efficiency scores. gRNAs were synthesized in vitro using the GeneArt™ Precision gRNA Synthesis Kit (Invitrogen, Paisley, UK. Cat# A29377) following manufacturer’s instructions. Four gRNAs (40RE, 47FW, 66FW, 77FW) and their corresponding repair oligonucleotides were generated.
The repair oligonucleotides (Alt-R HDR Donor Oligos) were engineered to include silent mutations, near the target mutation site, using the web tool Silent Mutator (https://molbiotools.com/silentmutator.php) [54] and the DNAStar Lasergene software (v.7.1, DNAstar). Two of these silent mutations were introduce to create a new restriction site for the Ssp1 enzyme (Thermo Fisher, MA, USA, Cat# ER0771), allowing the recognition of proper integration of the repair oligonucleotide. The other two silent mutations were designed to destroy the Protospacer Adjacent Motif (PAM) sequence, preventing the edited allele from being targeted repeatedly by the gRNA (Fig. 2).
Fig. 2.

Design of a gene editing strategy for repairing the c.1354dupT (p.Tyr452 Leufs*13) mutation in the exon 13 of the PROM1 gene. A In silico representation of the target sequence in a WT case. The amino acid affected by target mutation “Tyrosine” is highlighted in a green circle B In silico representation of a patient affect by the c.1354dupT (p.Tyr452 Leufs*13). The mutation is indicated by a black arrow and represented by a lowercase “t”. The Leucine generated due the mutation’s frameshift is highlighted in a red circle, and the consequent stop codon is indicated by a black arrow. The localization of the four designed gRNAs (40RE, 47RE, 66FW and 77FW) are framed in orange C In silico representation of the target sequence in a genetically repaired case. Blue arrows indicate silent mutations that introduce a restriction site for the Ssp1 and destroy the Protospacer Adjacent Motif (PAM) sequence to prevent the edited allele from being targeted again by the gRNA. The amino acid affected by target mutation “Tyrosine” is highlighted in a green circle. The WT reading frame is recovered
The predicted gene-editing efficiency of the designed four gRNA guides, were tested in vitro by editing the U2OS osteosarcoma cell line (ATCC HTB-96). The U2OS cells (1 × 106 cells) were transfected with each gRNA (1 µg), the corresponding repair oligonucleotide (15µL at 10 µM), and the Cas9-protein v2 TrueCut™ (5 µg; Invitrogen, Paisley, UK. Cat# A36498) using the 100 µL pipette tip of an Neon Transfection System (Thermo Fisher, MA, USA. Cat# MPK5000), under the following pulse conditions: 1230 V; 10 Ms; 4 pulses. After 24 h of culture, gDNA from each essay was extracted, and the region containing the target mutation was amplified by conventional PCR using the PROM1seq primer. The PCR products were purified using the Wizard® PCR Preps DNA Purification System (Promega, Wisconsin, USA. Cat# A7170) and digested with the Ssp1(Thermo Fisher, MA, USA, Cat# ER0771) enzyme for 3 h at 37 °C to ascertain the most effective CRISPR/Cas9 editing. Untransfected U2OS cells served as the wild type (WT) control.
Detection of gene-edited iPSC clones
As a screening method to identify successfully genetically repaired iPSC clones, a specific primer incorporating the silent mutations present in the selected repair oligonucleotide were designed (ALELO40RE primer) (Supplementary file 3). Due to the similarity between a WT and a gene edited sequence, amplified by the ALELO40RE primer, a gradient conventional PCR assay was conducted to determine the optimal annealing temperature to identified successfully genetically repaired iPSC clones.
To test this method, U2OS cells were transfected, under the previously described conditions, with the selected gRNA and repair oligonucleotide. After 24 h of culture, gDNA was extracted and a gradient conventional PCR was performed.
Patient-derived iPSC gene editing
After 5 days of culture, the iPSC lines derived from patients IRD1 and 2, were dissociated using Accutase (Stem Cells Technologies, Cambridge, UK. Cat# 07920) and resuspended in mTeSR™ Plus medium supplemented with CloneR™2 (Stem Cells Technologies, Cambridge, UK. Cat# 100–0276, 100–0691). A total of 1 × 106 cells were electroporated with the selected gRNA (1,5µL at 1µL/µg), the repair oligonucleotide (15 µL at 20 µM) and Cas9-protein v2 TrueCut™ (1,5µL at 5µL/µg, Thermo Fisher, MA, USA. Cat# A36498) using the Neon Transfection System (Thermo Fisher, MA, USA. Cat# MPK5000), with the following pulse conditions: 1200 V; 30 Ms; 1 pulse. The electroporated cells were plated at a density of 50 cells/cm2 on Matrigel coated (23 µg/cm2; Corning Life Sciences, NY, USA. Cat# 11,593,620) 6-well plates (Thermo Fisher Scientific, MA, USA. Cat# 11,337,694) using mTeSR™ Plus medium supplemented with CloneR™2 (Stem Cells Technologies, Cambridge, UK. Cat# 100–0276, 100–0691) for 24 h. The mTeSR™ Plus medium (Stem Cells Technologies, Cambridge, UK. Cat# 100–0276) was refreshed daily until the colonies reached a sufficient size for manual picking. iPSC clones were subsequently plated in 24-well plates (Thermo Fisher, MA, USA. Cat# 142,475), duplicated and cryopreserved.
The gDNA was extracted from 30 clones of each patient-derived iPSC lines and was amplified via conventional PCR using the PROM1seq primer (Supplementary file 3). The PCR products of the selected clones were purified with the Wizard® PCR Preps DNA Purification System (Promega, Wisconsin, USA. Cat# A7170) and digested with the Ssp1 enzyme (Thermo Fisher, MA, USA, Cat# ER0771) for 3 h at 37 °C. Proper integration of the repair oligonucleotide in the selected clones was then confirmed by Sanger sequencing using the PROM1seqFW primer (Supplementary file 3). The gene-edited clones were assessed for their ability to encode Prominin-1 through CD133 flow cytometry and western blotting.
Flow cytometry
The iPSC line derived from patient IRD1, its corresponding genetically repaired iPSC line, and the iPSC control line [FiPS] Ctrl1-Ep6F-5 were labelled with an anti-CD133 antibody conjugated to allophycocyanin (APC) (Supplementary file 2), following the manufacturer’s protocol. The flow cytometry assay was performed using the Gallios Flow Cytometer (Beckman Coulter, Indianapolis, USA) and the data were processed with Kaluza Analysis Software (Beckman Coulter, Indianapolis, USA). iPSC lines without staining served as negative controls for the assay.
Western blotting
Proteins obtained from the iPSC line derived from patient IRD2, its corresponding repaired iPSC line, and the control iPSC line [FiPS] Ctrl1-Ep6F-5 were extracted using RIPA buffer (25 mM Tris–HCl pH 7.4, 1 mM EDTA, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) with added protease inhibitors. Protein samples (20 µg each) were separated by SDS-PAGE technique and subsequently transferred to PVDF membranes. The proteins CD133 and β-actin (as a loading control) were immunodetected using specific antibodies (Supplementary file 2). Images were acquired using a GS-800 Calibrated Densitometer (Bio-Rad, Madrid, Spain) and analysed with Quantity One software (Bio-Rad, Madrid, Spain).
Results
Patient selection, ophthalmological examination and CES results
Eight patients with PROM1-related IRD were identified and studied at the IOBA’s clinical area. Their genetic background, clinical examination and functional and imaging characteristics are summarized in Tables 1, 2, and 3. Three of them presented retinopathy associated with the homozygous c.1354dupT (p.Tr452 Leufs*13) mutation in the PROM1 gene inherited in an autosomal recessive manner, and were selected for the generation of iPSC lines, as they exhibited three distinct phenotypes: IRD1 is a 47-year-old female with the CORD phenotype (Fig. 3A); IRD2 is a 54-year-old female with the RP phenotype (Fig. 3B); and IRD3 is a 20-year-old male with the STGD4 phenotype (Fig. 3C). Relatives of the target patients included: a 46-year-old female, who is the sister of IRD1; a 21-year-old female, who is the daughter of IRD2; and a 52-year-old female, who is the mother of IRD3. None of these relatives exhibited abnormalities upon ophthalmological examination. The results of the CES are presented in Table 4.
Table 2.
Clinical characteristics of the cohort of 8 patients with PROM1-related retinopathies
| Patient | Phenotype | Age of onset (years) | Symptoms | Refractive error | BCVA LogMAR/age first visit | BCVA LogMAR/age second visit | BCVA LogMAR/age last visit | Family history | Associated pathology |
|---|---|---|---|---|---|---|---|---|---|
| IRD 1 | CORD | 6 | Loss of AV followed by loss of VF | Hypermetropia | RE: 1.3/23 LE: 1.0 | RE: 3.0/34 LE: 1.3 | OD: 3.0/46 OD: 3.0 | Paternal grandfather, two paternal great-uncles affected. A cousin of his father's affected by RP (great-grandparents with relative consanguinity) | Nistagmus, pseudophakia |
| IRD 2 | RP | 5 | Diagnosed at the age of 5 upon revision following her brother's diagnosis. Simultaneous loss of VA and VF at 7 years of age, and nyctalopia at 19 years old. No visual rest at the age of 33 | Hypermetropia | RE:1.0/14 LE: 1.0 | RE: LP/45 LE: LP | OD: LP/54 OI: LP | 2 affected male brothers | Nistagmus, strabismus, pseudophakia |
| IRD 3 | STGD4 | 13 | Loss of AV and photophobia | Mild myopia | RE: 0.4/17 LE: 0.5 | RE: 0.3/18 LE: 0.4 | OD: 0.5/20 OI: 0.6 | No Ocular disease. Father Charcot-Marie-Tooth & patient asymptomatic have the PMMD2 gene duplicated. Consanguinity (grandfathers were cousins) | No |
| IRD 4 | CORD | 3 | Loss of AV followed by loss of VF, glare | Moderate myopia | NA | NA | OD: 3.0/41 OI: 3.0 | 1 affected male brother | Nistagmus |
| IRD 5 | CORD | 12 | Loss of AV and photophobia, followed by dyschromatopsia, glare and loss of VF | High myopia | RE: 0.4/12 LE: 0.4 | RE: 1.0/23 LE: 1.0 | OD: 3.0/32 OI: 3.0 | 3 affected brothers | Nistagmus |
| IRD 6 | STGD4 | 31 | Loss of AV and metamorphopsia | Mild myopia, high astigmatism | RE: 0.3/31 LE: 0.2 | RE: 0.10/32 LE: 0.10 | OD: 0.00/39 OI: 0.92 | Daughter possibly affected | No |
| IRD 7 | STGD4 | 25 | Loss of AV | Emmetropia | RE:0.1/30 LE: 0.17 | RE:0.5/31 LE: 0.17 | OD:0.1/37 OI: 0.3 | 1 affected male brother with more widespread disease. The father and one paternal uncle are also affected | Strabismus, cataract |
| IRD 8 | CORD | 13 | Loss of AV and photophobia | Mild hypermetropia. astigmatism | RE: LP/40 LE: LP | RE: LP/48 LE: LP | RE: NLP/64 LE: NLP | 2 cousins from the paternal family (male and female) affected RP. Relative consanguinity, as all are from a village with a population of fewer than 100 inhabitants | Nistagmus, cataract |
IRD: Inherited retinal dystrophy. BCVA: best corrected visual acuity. VF: visual field. RE: right eye. LE: left eye. LP: light perception. NLP: no light perception. CORD: cone – Rod Dystrophy. NA: not available. STGD4: Stargardt’s disease type 4. RP: retinitis pigmentosa
Table 3.
Additional functional and imaging characteristics of the identified patients with PROM1-related retinopathy
| Patient | Fundoscopy | FAF Pattern | VF | ERG | OCT | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patched /mottled posterior pole HºAF | HerAF ring | Preserved fovea | Periphery HºAF | Central scotoma | Concentric retraction | Cone-rod pattern | Rod-Cone pattern | Subnormal scotopic and photopic | Abolished scotopic and photopic | ONL disruption in the macula with preserved fovea | ONL disruption in the macula without preserved fovea | Panretinal ONL disruption | SR deposits | Herrefl foci | ||
| IRD 1 | Macular atrophy, pigment in bone spicules, papillary pallor, vascular attenuation | Yes | – | – | Yes | Abolished | – | – | – | Yes | – | Yes | Yes | Yes | Yes | |
| IRD 2 | Pigment in bone spicules, papillary pallor, macular atrophy, vascular attenuation | Yes | – | – | – | Abolished | – | – | – | Yes | – | Yes | Yes | Yes | – | |
| IRD 3 | Bull’s eye maculopathy | – | Yes | – | – | Yes | – | Only subnormal photopic | Yes | – | – | Yes | Yes | |||
| IRD 4 | Macular atrophy, mild pigmentation in bone spicules & atrophic plaques in the inferior periphery | Yes | – | – | – | NA | NA | NA | NA | NA | Yes | Yes | – | – | Yes | Yes |
| IRD 5 | Macular atrophy, pigment in bone spicules & atrophic plaques in the mid–periphery, papillary pallor, vascular attenuation | Yes | – | – | – | Abolished | – | – | – | Yes | – | Yes | Yes | Yes | – | |
| IRD 6 | Macular atrophy without flecks | Yes | – | – | – | Yes | – | Only subnormal photopic | – | Yes | – | Yes | Yes | |||
| IRD 7 | Bull’s eye maculopathy, optic nerve pallor, macular atrophy, vascular attenuation, whitish mottling | Yes | Yes | – | Yes | Abolished | NA | NA | NA | NA | Yes | – | Yes | Yes | – | |
| IRD 8 | Pigment in bone spicules and atrophic plaques in the periphery, macular atrophy, vascular attenuation | Yes | – | – | – | Abolished | – | – | – | Yes | – | Yes | Yes/ Choroid absent | Yes | Yes | |
Abbreviations. FAF: Fundus autofluorescence. VF: visual field. ERG: Electroretinogram. OCT: optical coherence tomography. SR: subretinal. Herrefl: hyperreflective. NA: not available. HºAF: hypoautofluorescence. HerAF: hyperautofluorescence. ONL: outer nuclear layer. IRD: Inherited retinal dystrophy
Fig. 3.

Phenotypic Variability of the c.1354dupT (p.Tr452 Leufs*13) Mutation in the PROM1 Gene in Three Target Patients. A Patient IRD1 is a 47-year-old female displaying a CORD phenotype. Retinography reveals pigment in bone spicules, papillary pallor, macular atrophy, and vascular attenuation. Fundus Autofluorescence (FAF) displays patchy/mottled hypoautofluorescent patterns. Optical Coherence Tomotraphy (OCT) presents panretinal outer retinal layers (ORL) disruption with foveal involvement. B Patient IRD2 is a 54-year-old female exhibiting an RP phenotype. Retinography with bone spicules pigmentation, papillary pallor, macular atrophy, and vascular attenuation. FAF showing patchy/mottled hypoautofluorescent patterns. OCT reveals panretinal ORL disruption including the fovea. C Patient IRD3, a 20-year-old male, exhibits an STGD4 phenotype. Retinography displays Bull’s eye Maculopathy. FAF shows a hypoautofluorescent ring around the fovea. OCT exhibits ORL disruption of the previously described ring with preserved fovea, mainly affecting the RPE line but with photoreceptors still present in the macular area. D The pedigree analysis reveals an autosomal recessive (AR) inheritance pattern for all three target patients. IRD1’s paternal grandfather and two paternal great-uncles are affected; additionally, a cousin of his father received an RP diagnosis. That family resides in a small village in Cantabria and exhibits relatively high consanguinity. IRD2 has two affected male brothers and a non-affected son (her husband has congenital achromatopsia, highlighted in pink). IRD3 has no reported family history of ocular disease but few members present Charcot-Marie-Tooth (CMT) disease, highlighted in pink. The parents of IRD2 are second cousins, indicating consanguinity; IRD3’s father has CMT disease due to a duplication of the PMP22 gene mutation in homozygosis, and IRD3 was heterozygous for this mutation as well, albeit asymptomatic
Table 4.
Clinical exome sequencing results in patients with IRD related to the homozygous c.1354dupT mutation in the PROM1 gene and their healthy relatives
| Subject | Phenotype | Gene | RefSeq | Exon | Mutation | Classification | Genotype |
|---|---|---|---|---|---|---|---|
| IRD1 | CORD | PROM1 | NM_006017.3 | 13 | c.1354dupT (p.Tr452 Leufs*13) | Pathogenic | Homozygosity |
| C2 | NM_000063.6 | 6 | c.841_849 + 19del | Probably pathogenic [67] | Heterozygosity | ||
| IRD1’s sister | Control | PROM1 | NM_006017.3 | 13 | c.1354dupT (p.Tr452 Leufs*13) | Pathogenic | Heterozygosity |
| POMT1 | NM_001077365.2 | 20 | c.2097C > A (p.Tyr699*) | Pathogenic | Heterozygosity | ||
| IRD2 | RP | PROM1 | NM_006017.3 | 13 | c.1354dupT (p.Tr452 Leufs*13) | Pathogenic | Homozygosity |
| IRD2’s daughter | Control | PROM1 | NM_006017.3 | 13 | c.1354dupT (p.Tr452 Leufs*13) | Pathogenic | Heterozygosity |
| CNGB3 | NM_019098.5 | 10 | c.1148del (p.Thr383fs) | Probably pathogenic | Heterozygosity | ||
| HYDIN | NM_001270974.2 | 67 | c.11461C > T (p.Arg3821*) | Probably pathogenic | Heterozygosity | ||
| IRD3 | STGD4 | PROM1 | NM_006017.3 | 13 | c.1354dupT (p.Tr452 Leufs*13) | Pathogenic | Homozygosity |
| SAMD11 | NM_001385641.1 | 14 | c.2377C > T (p.Arg793*) | Probably pathogenic [65] | Heterozygosity | ||
| PEX6 | NM_000287.4 | 8 | c.1802G > A (p.Arg601Gln) | Probably pathogenic [66] | Heterozygosity | ||
| IRD3’s mother | Control | PROM1 | NM_006017.3 | 13 | c.1354dupT (p.Tyr452fs) | Pathogenic | Heterozygosity |
| PEX6 | NM_000287.4 | 8 | c.1802G > A (p.Arg601Gln) | Probably pathogenic | Heterozygosity |
The sequencing data supporting the results reported in this table are suitable as supplementary material in Supplementary files 6, 7, 8, 9, 10, 11
iPSC generation and characterization
Generation of Patient’s iPSC Lines. A primary fibroblast cell line was isolated from the dermal biopsy of each patient. The fibroblast lines: [CORD]-hFb, [RP]-hFb, and [STGD4]-hFb, were generated from the corresponding patient. The cell line populations showed the typical fibroblast-like morphology at phase contrast microscopy examination. Patient fibroblasts lines were reprogrammed into three iPSC lines, one for each phenotype. The generated iPSC lines: [CORD]-FiPSC1-Ep5F-2, [RP]-FiPSC1-Ep5F-10, [STGD4]-FiPSC1-Ep5F-8; displayed compact colonies with distinct borders, well-defined edges, and large nuclei.
AP staining and Pluripotency Immunocytochemistry. The generated iPSC lines tested positive for AP activity, exhibiting distinct blue staining (Fig. 4A). Furthermore, all of them displayed immunoreactivity for the nuclear markers (OCT4, Nanog, Sox2) and cell membrane markers (SSEA-3, SSEA-4, Tra 1–60, Tra 1–80) associated with pluripotency (Fig. 4B; Supplementary file 4A).
Fig. 4.

Characterization of the generated patient-derived iPSC lines. Alkaline phosphatase (AP) reactivity analysis. A Positive AP test results for all generated iPSC lines: [CORD]-FiPSC1-Ep5F-2, [RP]-FiPSC1-Ep5F-10, [STGD4]-FiPSC1-Ep5F-8. 50 µm scale. B Pluripotency analysis. Immunoreactivity of [RP]-FiPSC1-Ep5F-10 for pluripotency markers OCT4, SSEA3, SOX2, SSEA4, TRA1-60, NANOG, and TRA-1–81. 25 µm scale. C Molecular analysis of all generated iPSC lines. Relative mRNA expression of endogenous reprogramming factors (SOX2, OCT3/4, KLF4, LMYC, LIN28) apparently higher in the patient’s iPSC lines in comparison to unstransfected fibroblast. Above, the number of copies of the EBNA1 gene evidently lower in the generated iPSC lines than in 72 h post-transfection fibroblast, electroporated in the same reprogramming conditions. D Analysis of the functional pluripotency of embryoid bodies (EB) derived from [RP]-FiPSC1-Ep5F-10. Immunoreactivity to ectoderm (β-III Tubulin), mesoderm (α-SMA) and endoderm (α-1 Fetoprotein) markers, along with phase contrast micrography of EB. 50 µm and 200 µm scale. E Identity analysis of the generated patient-derived iPSC lines. Correspondence between the patient’s fibroblast lines: [RP]-hFb, [STGD4]-hFb, [CRD]-hFb, and their respective patient-derived iPSC line for the Short Tandem Repetition (STR) loci: CSF1PO, D13S317, D16S539, D21S11, D5S818, D7S820, TH01, TPOX y vWA, and Amelogenin. F Cytogenetic integrity of the generated iPSC lines. G-banded metaphase analysis of the patient-derived iPSC lines
Molecular Analysis of the Reprogramming Factors. The relative mRNA expression of the endogenous reprograming factors (SOX2, OCT3/4, KLF4, LMYC, LIN28) were apparently higher in the patient’s iPSC lines compared to the control. Besides, the number of copies of the EBNA1 gene was lower in the generated iPSC lines than in the control. Furthermore, the relative mRNA expression of the exogenous reprogramming vectors was lower in the iPSC lines than in the control (data available on request) (Fig. 4C).
In Vitro Differentiation. The cell populations that emerged from the EB of each patient-derived iPSC line expressed immunoreactivity for specific markers of each germinal layer. The cytoplasm of cell populations induced to undergo endodermal differentiation were immunoreactive to α-1 Fetoprotein. Cell populations exposed to mesodermal differentiation showed a dense filamentous pattern of immunoreactivity to α- Smooth Muscle Actin (α-SMA). Meanwhile, cell populations treated with ectodermal differentiation medium expressed a thin filamentous pattern of β-III Tubulin immunoreactivity in their cytoplasm (Fig. 4D; Supplementary file 4B).
DNA Fingerprinting, Karyotyping and Yarget Mutation Sequencing. Allelic variants for each analysed STR locus showed correspondence between the patient’s fibroblast lines and their corresponding iPSC lines (Fig. 4E). In addition, cytogenetic analysis did not detect any structural chromosomal abnormality in the analysed metaphases of the patient’s iPSC lines (Fig. 4F). Finally, the presence of the target mutation c.1354dupT was confirmed by Sanger sequencing in all patient’s iPSC lines.
Genetic repair of patient-derived iPSC lines
gRNA selection and detection of gene-edited iPSC clones
The gRNA40RE, 47RE, 77FW showed similar gene editing efficiency. The gRNA40RE (ACACGCCACACAGTAAGCCC) and the repair oligonucleotide 40RE (TCCAGGTGGCTGGGTGGCCTGGTCATCTGCTCTCTGCTGACCCTCATCGTAATATTTTACTACTTGGGCTTACTGTGTGGCGTGTGCGGCTATGACAGGCATGCCACCCCGACCAC) were selected (Fig. 5A). Additionally, it was determined that using the ALELO40RE primer at 56 °C of annealing temperature, it was possible to differentiate between a WT sequence from a gene edited sequence (Fig. 5B).
Fig. 5.
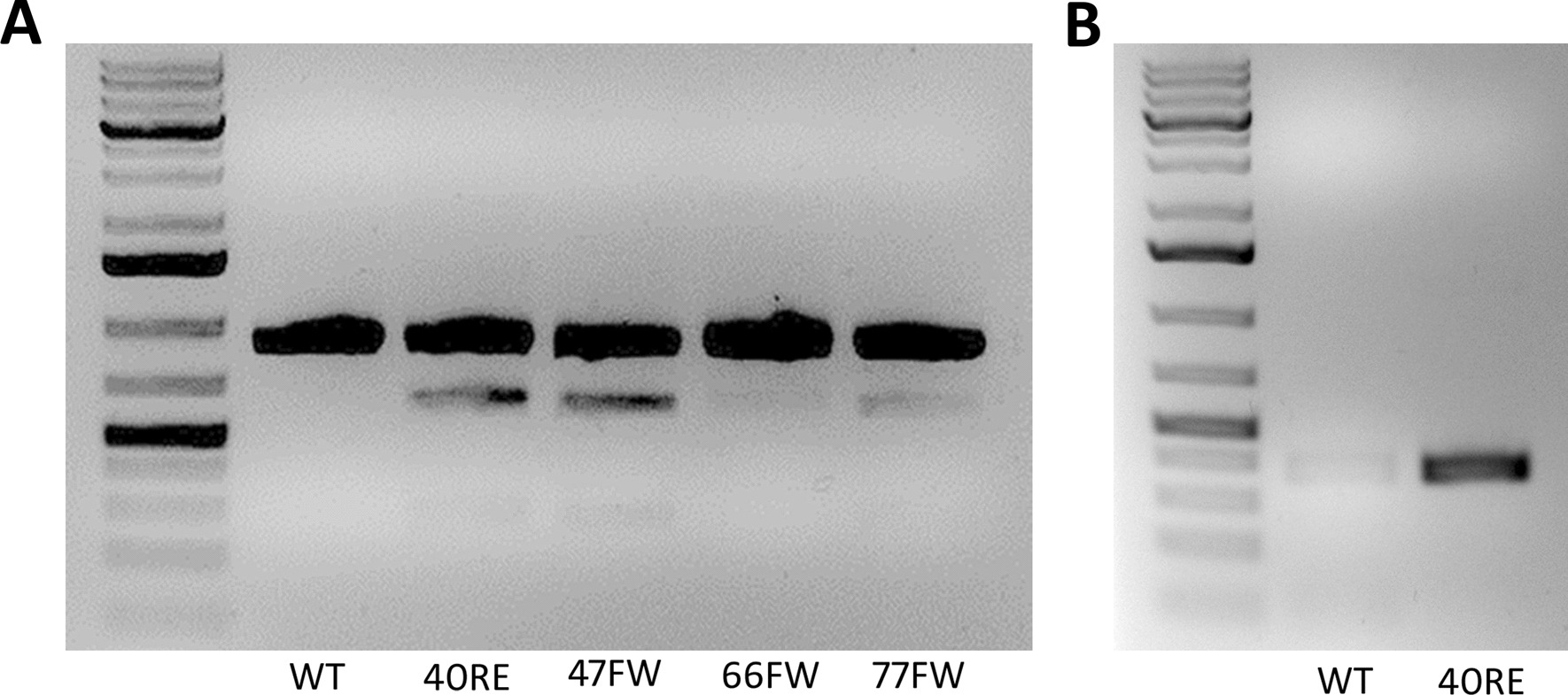
gRNA selection and detection of gene-edited iPSC clones (A) Comparison of gene editing efficiency among different gRNAs (40RE, 47RE, 66FW, 77FW). Ssp1 digestion of PCR products obtained from gDNA of U2OS cells edited with each gRNA and its respective repair oligonucleotide. Untransfected U2OS cells served wild type (WT) control (B) Screening test for detection of genetically repaired clones. A comparison of conventional PCR products using the ALELO40RE primer between untransfected U2OS cells (WT) and U2OS cells transfected with gRNA 40RE and the corresponding repair oligonucleotide 40RE (40RE)
The target mutation c.1354dupT (p.Tr452 Leufs*13) was corrected in the iPSC lines derived from patients IRD1 and 2, resulting in the generation of the gene-edited iPSC lines: [CORD]-FiPSC1-Ep5F-2-GC1 and [RP]-FiPSC1-Ep5F-10-GC1, respectively. Successful gene-edited was obtained in 10% of the clones selected from each patient. Proper integration of the repair oligonucleotide was confirmed by Sanger sequencing in both gene-edited iPSC lines (Fig. 6A).
Fig. 6.

Genetic restoration in the gene-edited iPSC lines derived from patient IRD1 and IRD2. A Comparison between sequencing electropherograms of a patient-derived iPSC line and its corresponding genetically repaired iPSC line. In the patient’s sequence the target mutation c.1354dupT is pointed by an arrow. In the repaired sequence the silent mutations (A > T) that generates the restriction site for the Ssp1 enzyme are pointed by arrows. B Flow cytometry histograms illustrating the relative number of CD133-positive cells in the iPSC control line iPSC control line [FiPS] Ctrl1-Ep6F-5, the iPSC line derived from patient IRD1 [CORD]-FiPSC1-Ep5F-2, and its corresponding genetically repaired iPSC line [CORD]-FiPSC1-Ep5F-2-GC1. C Western blotting results depicting CD133 protein expression in the iPSC control line [FiPS] Ctrl1-Ep6F-5, the iPSC line derived from patient IRD2 [RP]-FiPSC1-Ep5F-10, and its corresponding genetically repaired iPSC line [RP]-FiPSC1-Ep5F-10-GC1. Full-length gels are available in the figure at the Supplementary file 5
CD133 expression by the repaired iPSC lines
The quantification of the CD133-positive cell population in the flow cytometry assay revealed high CD133 expression in the iPSC control line [FiPS] Ctrl1-Ep6F-5, no expression in the iPSC line derived from patient IRD1, which was comparable to the non-stained negative control, and restoration of expression in the gene-edited iPSC line derived from patient IRD1 (Fig. 6B). In the western blotting assay, the CD133 was highly expressed in the iPSC control line, undetectable in the iPSC derived from patient IRD2, and its expression recovery was confirmed in the gene-edited iPSC line from patient IRD2 (Fig. 6C).
Discussion
I this study, we report a cohort of eight PROM1-related IRD patients, including three individuals harbouring the same mutation (c.1354dupT) but expressing different phenotypes (CORD, RP and STG4). We selected these three patients to elucidate the pleiotropic effect of the c.1354dupT in the PROM1 gene. To assess this matter in question, we expanded their genetic panel using CES and generated patient-derived iPSC to model the disease. Furthermore, we genetically repaired these iPSC as the first step towards designing therapeutic strategies. We plan to further study the disease by continuing our research in this direction.
Few cohort studies of PROM1-related retinopathies have been reported as it is a rare disease. In this regard, mutations in this gene account for 1 to 9.5% of AR CORD worldwide [8, 55, 56], 2% of AR RP and 4% AD RP in Spain [57]. In Asia, a cohort of 10 Japanese patients, considered as large cohort, were related to 3 variants, all exhibiting AD inheritance [58]; similarly, 10 Korean patients were all related to the same variant (p.Arg373Cys), also showing AD inheritance [25]. In Europe, the largest cohort included 25 Spanish patients, but only 7 of them underwent the main outcome measures FAF and OCT [22]; meanwhile, the second largest study included only 19 European patients [23]. In that sense, we report a non-negligible number of patients, including 5 different pathogenic PROM1 variants, heterogeneous phenotypical characteristics, both sexes between 20 and 64 years old, exhibiting AD and AR inheritance patterns, and main ophthalmology imaging (FAF and OCT) was performed in all of them.
Our target mutation c.1354dupT (p.Tyr452Leufs*) is a complete loss-of-function variant (LOF) resulting in a premature stop codon. Similar to our study, in the two largest European cohort studies [22, 23], the c.1354dupT mutation was the most prevalent variant, highlighting its importance in the European population. Furthermore, its previously unreported pleiotropic effect justified focusing the experimental part of this study on these patients.
The c.1354dupT mutation has been associated with early onset AR phenotypes of severe panretinal dystrophy and late onset moderate MD [23]; and AR forms of CORD and RP [22]. Additionally, we report an early-medium onset (13 years old) of an AR form of STGD4 in patient IRD3 (aged 20 years old), contrary to the typical late onset AD manifestation of STGD4 dystrophy with an evident bull’s eye maculopathy [24, 59]. Nevertheless, given the young age of this patient we cannot rule out that he will be evolving to CORD later in his live. Interestingly, this patient has also a duplication of the PMP22 gene, associated with Charcot-Marie-Tooth disease, although he remains asymptomatic.
Currently, it remains challenging to determinate whether PROM1-related dystrophies originate in the RPE or at the photoreceptors base due to its intricate role in the visual processes, participating in OS morphogenesis, RPE autophagy, and glial apoptosis [28–31]. At a clinical level, we could attempt to deduce it from the OCT and ERG results. We have observed in our series is that in older patients, and therefore with a more advanced condition, both RPE and photoreceptors are absent on OCT. However, interestingly, the two youngest patients, IRD 3 and IRD7, exhibit alteration of the RPE line with the photoreceptor line still present, as does IRD5 reviewed at 41 years of age, who presents a CORD phenotype and had the photoreceptor line preserved in the OCT. This leads us to suspect that at least in these patients, the disease primarily affects the RPE. However, IRD3 and IRD5 have photoreceptor function affected on ERG. Consequently, it would be impossible to definitively establish the primary affected cell type solely by observing these clinical features, since some IRDs affect the function of specific cell types without the disappearance of the corresponding retinal layer in histological studies or imaging tests until we are facing advanced stages of the disease.
Additionally, as supported by our study, most authors agree that early macular involvement is a common feature for PROM1-related IRD patients [60]. Furthermore, for PROM1-related STGD4 disease, it appears that pathological changes primarily affect the photoreceptors rather than the RPE. However, to deeply assess this matter, prospective studies conducting serial SD-OCT on a large cohort of PROM1-related IRD patients from the onset of symptoms and over several years would be necessary. This approach is outlined in the unique prospective cohort study of PROM1-associated retinal degeneration, examined with OCT but only for a 24-month duration [59].
Pleiotropic effects of IRD-related genes seem to be a common phenomenon in clinics, still poorly studied, and understood. The variant c.2539G > A, p.Glu847Lys in the HK1 gene has been associated to AD forms of MD, CORD and RP [12, 13]. Similarly, the c.629C > G, p.Pro210Arg variant in the PRPH2 gene with AD forms of macular dystrophy and RP [15, 16]. Additionally, similar to our target mutation, the c.1117C > T, p.Arg373Cys variant in the PROM1 gene, has been associated with AD forms of STGD4 [25, 61, 62], CORD [23–25] and RP [22, 23], displaying phenotypical variability even within the same family. Although presently unexplained, this phenomenon is likely related to modifier genes affecting penetrance, dominance, expressivity, and pleiotropy of the IRD-causing variant [63].
The CES evaluation detected different associated monoallelic variants in two target patients that are recognised for being associated to AR phenotypes [64–67]; however, we cannot completely exclude nor confirm their phenotypic influence [68]. In IRD1, the monoallelic c.841_849 + 19del variant in the C2 gene is associated with complement component 2 deficiency through skipping of exon 6 during RNA splicing [67]. Many C2 variants has been associated with Age-related Macular Degeneration (AMD) [69]. However, the c.841_849 + 19del variant has not been associated with this pathology yet; nevertheless, due its early frameshift effect, it may cause similar microglia dysregulation as other AMD-related C2 variants [70].
Besides, the monoallelic c.2377C > T (p.Arg793*) variant in the SAMD11 gene found in IRD3 has been related to AR RP due to its interaction with the photoreceptor-specific transcription factor Cone-Rod homeobox (CRX) [65] and the Polycomb repressive complex 1 component (PRC1) [71], both implicated in the photoreceptor differentiation. Besides, the overexpression of the PCGF1 subunit of the PRC1 in cancer stem cells has been associated with an enhanced expression of CD133 [72]. Whereas, the monoallelic c.1802G > A (p.Arg601Gln) variant in the PEX6 gene has been associated with Peroxisome biogenesis disorders [66]. Additionally, CD133 has been associated with upregulation of Peroxisome proliferator-activated receptor alpha in cancer stem cells, which is essential for retinal lipid metabolism [73, 74]. Thus, it is plausible to correlate CD133 deficiency with a possible monoallelic alteration in photoreceptors differentiation and retinal peroxisome function in the IRD3 patient.
To clarify the possible influence of the mentioned monoallelic variants in the PROM1-related IRD phenotypes, it would be convenient to generate gene-edited patient-derived iPSC and murine models to study the associated variants independently. However, at present, none of these variants could be stated as modifier genes as none of them participate directly in the described Prominin-1 metabolic or function pathways [27–31, 75, 76]. On the other hand, other variants not related to retinal disease, and therefore not included in this study, may have some influence in this phenomenon. Further studies should include more comprehensive molecular diagnosis methods, such as whole exome or genome sequencing.
The generated patient-derived iPSC lines: [CORD]-FiPSC1-Ep5F-2, [RP]-FiPSC1-Ep5F-10, [STGD4]-FiPSC1-Ep5F-8, meet the characterization requirements the Carlos III Health Institute, the Spanish competent authority for iPSC banking and registration, in accordance with the guidelines established by the International Stem Cell Banking Initiative (ISCBI) [77]. In this sense, these iPSC lines can be considered as fully reprogrammed and functional iPSC. Although CD133 has been historically recognized as a stem cell marker, highly expressed in the cell membranes of undifferentiated cells such as ESC and iPSC [75, 78], our findings indicate that CD133 deficiency does not appear to affect reprogramming, pluripotency, or in vitro differentiation of iPSC. Furthermore, in contrast to CD133-KO ESC [76], cell proliferation, in terms of the number of passing days, did not appear to be affected when compared to the iPSC control line [FiPS] Ctrl1-Ep6F-5.
Although, PROM1 has been previously knocked out in RPE cells [30, 78] and murine models [79–81], the application of CRISPR/Cas9 genome editing on patient-derived iPSC to establish IRD-specific disease models enables researchers to study the distinct molecular effects associated with each IRD-related variant, extending beyond the mere absence of a protein as in KO murine models [82, 83]. In this regard, the next steps in our research horizon include obtaining gene-edited iPSC-derived RPE cells to evaluate this matter and to future cell therapy assays as they seem to be the most promising alternatives for replacement therapies [84].
To the best of our knowledge, this is the first report of a PROM1-related IRD mutation genetically repaired in patient-derived iPSC lines. After confirming the functional restoration of the generated genetic-repaired iPSC, they can be useful tools for preclinical cell therapy studies as an alternative treatment to replace degenerated retina in advanced diseases. Furthermore, the gene editing strategy could contribute to the design of future gene therapy studies to treat early stages of PROM1-related retinal dystrophy. Finally, we would like to highlight the relevance of having a better understanding of the pleiotropism phenomena of IRD-related genes and variants, as it may be a crucial milestone in the proper diagnosis, classification, and treatment of IRD.
Limitations of the study include the extensive phenotypic and genotypic heterogeneity present within IRD, which complicates the identification of correlations between associated variants and the patient phenotypic traits, even when employing massive NGS techniques such as CES. Future studies should consider expanding the patient cohort, encompassing a broader spectrum of mutations, and incorporating whole exome/genome sequencing to achieve a more comprehensive understanding. Furthermore, deeper characterization of a gene-edited iPSC derivatives, including RPE, photoreceptors, and retinal organoids, are essential to elucidate PROM1-related IRD phenotypic variability and disease mechanisms, and finally to explore the potential therapeutic utility of gene editing or cell therapy product development.
Conclusion
The c.1354dupT mutation in the PROM1 gene is associated to three distinct AR phenotypes of IRD. This pleotropic effect might be related to the influence of monoallelic variants in other genes associated with retinal dystrophies, but further evidence needs to be provided. Future experiments should include gene-edited patient-derived iPSC due to its potential as disease modelling tools to elucidate this matter in question. The generated patient-derived iPSC lines are fully reprogrammed and functional. Besides, the gene-editing strategy successfully restored the capability of the PROM1 gene to encode Prominin-1. After functional characterization, the gene-edited iPSC lines can be used as a basis for future development of gene and cell therapies for PROM1-related IRD patients.
Supplementary Information
Supplementary file 1: List of the 998 genes related to retinal diseases evaluated in the clinical exome sequencing analysis.
Supplementary file 2: Antibodies and their experimental conditions.
Supplementary file 3: Primer sequences for iPSC molecular characterization, sequencing, and gene editing.
Supplementary file 4: Pluripotency analysis. (A) Immunoreactivity of [CORD]-FiPSC1-Ep5F-2 and [STGD4]-FiPSC1-Ep5F-8 for the pluripotency markers OCT4, SSEA3, SOX2, SSEA4, TRA1-60, NANOG, and TRA-1-81. 25 µm scale. (B) Analysis of the functional pluripotency of the embryoid bodies (EBs) derived from [CORD]-FiPSC1-Ep5F-2 and [STGD4]-FiPSC1-Ep5F-8. Immunoreactivity to ectoderm (β-III Tubulin), mesoderm (α-SMA) and endoderm (α-1 Fetoprotein) markers. 25 µm scale.
Supplementary file 5: Full-length gels of Figure 4. (A) Full-length gels of Western blotting of CD133 protein expression in the iPSC control line [FiPS] Ctrl1-Ep6F-5, the iPSC line derived from patient IRD2 [RP]-FiPSC1-Ep5F-10, and its corresponding genetically repaired iPSC line [RP]-FiPSC1-Ep5F-10-GC1. (B). Full-length gels of Western blotting of β-actin (control).
Supplementary file 6: Sequencing data from IRD1.
Supplementary file 7: Sequencing data from IRD1’ sister.
Supplementary file 8: Sequencing data from IRD2.
Supplementary file 9: Sequencing data from IRD2’ daughter.
Supplementary file 10: Sequencing data from IRD3.
Supplementary file 11: Sequencing data from IRD3’ mother.
Acknowledgements
The authors would like to acknowledge the Institute of Biomedical Research of Bellvitge (IDIBELL) for their support in processing the cytogenetics and DNA fingerprinting studies. We also extend our gratitude to the Sanitary Research Institute La Fe for performing the Clinical Exome Sequencing analyses. Special thanks to Dra. Eva Richard from the Severo Ochoa Institute, Madrid, for generously sharing her iPSC characterization protocols with us. Lastly, we express our sincere appreciation to the patients and their relatives who participated in this study.
Abbreviations
- IRD
inherited retinal dystrophy
- iPSC
induced pluripotent stem cell
- RP
retinitis pigmentosa
- PCD
progressive cone dystrophy
- CORD
cone-rod dystrophy
- MD
macular dystrophy
- AD
autosomal dominant
- AR
autosomal recessive
- XL
X-linked
- NGS
next generation sequencing
- ES
exome sequencing
- STGD4
stargardt’s disease type 4
- OS
photoreceptor’s outer segments
- ESC
embryonic stem cells
- IOBA
institute for applied ophthalmobiology
- IBGM
institute of molecular biology and genetics
- CES
clinical exome sequencing
- BCVA
best corrected visual acuity
- FAF
fundus autofluorescence
- VF
visual field
- ERG
electroretinography
- OCT
optical coherence tomography
- AMD
age-related macular disease
- FBS
fetal bovine serum
- IMDM
Iscove′s modified Dulbecco′s medium
- EDTA
ethylenediamine tetraacetic acid
- AP
alkaline phosphatase
- PBS
phosphatase-buffered saline
- TBS
Tris-buffered saline
- DAPI
4',6-diamidino-2-fenilindol
- cDNA
complementary DNA
- mRNA
messenger RNA
- qRT-PCR
quantitative reverse transcription PCR
- STR
short tandem repeat
- α-SMA
α- Smooth Muscle Actin
Author contributions
KP-N, MAF, RMC-M, and IF-B designed the study. MAF, RC-M, and IF-B coordinated the study. KP-N, RMC-M, LAH-R, DG, YG-F, RP-V, MS, and MAF generated research data. KP-N, RMC-M, LAH-R, DG, JJT, MAF, and IF-B analysed and discussed the results. KP-N, RMC-M, MAF and IF-B wrote the manuscript. All authors reviewed the manuscript, contributed to the discussion, and approved the final version of the manuscript.
Funding
The study was funded by grant PID2020-118860RB-100 and PID2020-118517RB-I00 funded by MCIN/AEI/10.13039/501100011033. Programa Estratégico Instituto de Biología y Genética Molecular (IBGM), Junta de Castilla y León (CCVC8485). Consejería de Educación, Junta de Castilla y León (VA172P20). Centro en Red de Medicina Regenerativa y Terapia Celular de Castilla y León. Kevin Puertas-Neyra was funded by predoctoral contracts UVa2020 (co-funded by Santander Bank) and Junta de Castilla y Leon 2021.
Availability of data and materials
All data generated or analysed during this study are included within the article.
Declarations
Ethics approval and consent to participate
This study, originally approved under the title: “Estudio molecular del gen PROM1 en pacientes con Distrofias Hereditarias de la Retina y nuevas aproximaciones terapéuticas basadas en la edición genética (PROMedit)” [“Molecular Study of the PROM1 Gene in Patients with Hereditary Retinal Dystrophies and New Therapeutic Approaches Based on Gene Editing (PROMedit)”], received ethical approvals from multiple authorities. These approvals include the Research Committee of the Institute for Applied Ophthalmobiology (IOBA) from the University of Valladolid (registration code IOBA-2021–48, registered on November 9, 2021), the Ethics Committee for Medicament Research from East Valladolid (registration code PI 21–2490, registered on December 9, 2021) and the Guarantees Commission of the Carlos III Health Institute (ISCIII) on March 15, 2022 (without registration code). The study adhered to European and Spanish laws and regulations concerning the generation of human iPSC. The generated iPSC lines are registered in the Human Pluripotent Stem Cell Registry (hPSCreg®) under the codes: ESi125-A, ESi126-A and ESi127-A, and they will be deposited at the National Bank of Cell Lines (BNLC) of ISCIII. We received authorization from ISCIII to acquire the iPSC line [FiPS] Ctrl1-Ep6F-5 from BNLC, which was used as a control line. The study followed the principles outlined in the Helsinki Declaration of 1964 (last amended in 2013), data protection law 15/99 enacted on December 13th, and biomedical research law 14/2007 established on July 03. Informed consent was thoroughly explained to and signed by all participating individuals.
Consent for publication
Not applicable.
Competing interests
This research was partially presented at the German Stem Cell Network (GSCN) meeting 2022 and the European Association for Vision and Eye Research (EVER) meetings in 2022 and 2023. The authors declare no conflicts of interest.
Declaration of generative AI and AI-assisted technologies in the writing process
During the preparation of this work the authors used ChatGPT4 to improve readability and language of the manuscript. After using this tool, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Joint senior authors: Miguel A. de la Fuente and Ivan Fernandez-Bueno.
References
- 1.Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. The Lancet. 2006;368(9549):1795–809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- 2.Traboulsi EI. Hope and major strides for genetic diseases of the eye. J Genet. 2009;88(4):395–7. doi: 10.1007/s12041-009-0060-8. [DOI] [PubMed] [Google Scholar]
- 3.Cross N, van Steen C, Zegaoui Y, Satherley A, Angelillo L. Retinitis pigmentosa: burden of disease and current unmet needs. Clin Ophthalmol. 2022;20(16):1993–2010. doi: 10.2147/OPTH.S365486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heath Jeffery RC, Mukhtar SA, McAllister IL, Morgan WH, Mackey DA, Chen FK. Inherited retinal diseases are the most common cause of blindness in the working-age population in Australia. Ophthalmic Genet. 2021;42(4):431–9. doi: 10.1080/13816810.2021.1913610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bocquet B, Lacroux A, Surget MO, Baudoin C, Marquette V, Manes G, et al. Relative frequencies of inherited retinal dystrophies and optic neuropathies in Southern France: assessment of 21-year data management. Ophthalmic Epidemiol. 2013;20(1):13–25. doi: 10.3109/09286586.2012.737890. [DOI] [PubMed] [Google Scholar]
- 6.Hamel C. Retinitis pigmentosa. Orphanet J Rare Dis. 2006;1(1):40. doi: 10.1186/1750-1172-1-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coco-Martin RM, Diego-Alonso M, Orduz-Montaña WA, Sanabria MR, Sanchez-Tocino H. Descriptive Study of a Cohort of 488 Patients with inherited retinal dystrophies. OPTH. 2021;15:1075–84. doi: 10.2147/OPTH.S293381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boulanger-Scemama E, El Shamieh S, Démontant V, Condroyer C, Antonio A, Michiels C, et al. Next-generation sequencing applied to a large French cone and cone-rod dystrophy cohort: mutation spectrum and new genotype-phenotype correlation. Orphanet J Rare Dis. 2015;24(10):85. doi: 10.1186/s13023-015-0300-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Allikmets R, Singh N, Sun H, Shroyer NF, Hutchinson A, Chidambaram A, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet. 1997;15(3):236–46. doi: 10.1038/ng0397-236. [DOI] [PubMed] [Google Scholar]
- 10.Martínez-Mir A, Paloma E, Allikmets R, Ayuso C, del Río T, Dean M, et al. Retinitis pigmentosa caused by a homozygous mutation in the Stargardt disease gene ABCR. Nat Genet. 1998;18(1):11–2. doi: 10.1038/ng0198-11. [DOI] [PubMed] [Google Scholar]
- 11.Maugeri A, Klevering BJ, Rohrschneider K, Blankenagel A, Brunner HG, Deutman AF, et al. Mutations in the ABCA4 (ABCR) gene are the major cause of autosomal recessive cone-rod dystrophy. Am J Hum Genet. 2000;67(4):960–6. doi: 10.1086/303079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yuan Z, Li B, Xu M, Chang EY, Li H, Yang L, et al. The phenotypic variability of HK1-associated retinal dystrophy. Sci Rep. 2017;7(1):7051. doi: 10.1038/s41598-017-07629-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sullivan LS, Koboldt DC, Bowne SJ, Lang S, Blanton SH, Cadena E, et al. A dominant mutation in hexokinase 1 (HK1) causes retinitis pigmentosa. Investig Ophthalmol Visual Sci. 2014;55(11):7147. doi: 10.1167/iovs.14-15419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang F, Wang Y, Zhang B, Zhao L, Lyubasyuk V, Wang K, et al. A missense mutation in HK1 leads to autosomal dominant retinitis pigmentosa. Investig Ophthalmol Vis Sci. 2014;55(11):7159–64. doi: 10.1167/iovs.14-15520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feist RM, White MF, Skalka H, Stone EM. Choroidal neovascularization in a patient with adult foveomacular dystrophy and a mutation in the retinal degeneration slow gene (Pro 210 Arg) Am J Ophthalmol. 1994;118(2):259–60. doi: 10.1016/S0002-9394(14)72913-7. [DOI] [PubMed] [Google Scholar]
- 16.Gorin MB, Jackson KE, Ferrell RE, Sheffield VC, Jacobson SG, Gass JD, et al. A peripherin/retinal degeneration slow mutation (Pro-210-Arg) associated with macular and peripheral retinal degeneration. Ophthalmology. 1995;102(2):246–55. doi: 10.1016/S0161-6420(95)31029-9. [DOI] [PubMed] [Google Scholar]
- 17.Huang XF, Huang F, Wu KC, Wu J, Chen J, Pang CP, et al. Genotype–phenotype correlation and mutation spectrum in a large cohort of patients with inherited retinal dystrophy revealed by next-generation sequencing. Genet Med. 2015;17(4):271–8. doi: 10.1038/gim.2014.138. [DOI] [PubMed] [Google Scholar]
- 18.Ganapathi M, Thomas-Wilson A, Buchovecky C, Dharmadhikari A, Barua S, Lee W, et al. Clinical exome sequencing for inherited retinal degenerations at a tertiary care center. Sci Rep. 2022;12(1):9358. doi: 10.1038/s41598-022-13026-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haer-Wigman L, van Zelst-Stams WA, Pfundt R, van den Born LI, Klaver CC, Verheij JB, et al. Diagnostic exome sequencing in 266 Dutch patients with visual impairment. Eur J Hum Genet. 2017;25(5):591–9. doi: 10.1038/ejhg.2017.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang L, Zhang J, Chen N, Wang L, Zhang F, Ma Z, et al. Application of whole exome and targeted panel sequencing in the clinical molecular diagnosis of 319 Chinese families with inherited retinal dystrophy and comparison study. Genes (Basel). 2018;9(7):360. doi: 10.3390/genes9070360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dockery A, Whelan L, Humphries P, Farrar GJ. Next-generation sequencing applications for inherited retinal diseases. Int J Mol Sci. 2021;22(11):5684. doi: 10.3390/ijms22115684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Del Pozo-Valero M, Martin-Merida I, Jimenez-Rolando B, Arteche A, Avila-Fernandez A, Blanco-Kelly F, et al. Expanded phenotypic spectrum of retinopathies associated with autosomal recessive and dominant mutations in PROM1. Am J Ophthalmol. 2019;207:204–14. doi: 10.1016/j.ajo.2019.05.014. [DOI] [PubMed] [Google Scholar]
- 23.Cehajic-Kapetanovic J, Birtel J, McClements ME, Shanks ME, Clouston P, Downes SM, et al. Clinical and molecular characterization of PROM1-related retinal degeneration. JAMA Netw Open. 2019;2(6):e195752. doi: 10.1001/jamanetworkopen.2019.5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Michaelides M, Gaillard MC, Escher P, Tiab L, Bedell M, Borruat FX, et al. The PROM1 mutation pR373C causes an autosomal dominant bull’s eye maculopathy associated with rod, rod-cone, and macular dystrophy. Invest Ophthalmol Vis Sci. 2010;51(9):4771–80. doi: 10.1167/iovs.09-4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hwang S, Kang SW, Jang JH, Kim SJ. Genetic and clinical characteristics of PROM1-related retinal degeneration in Korean. Sci Rep. 2023;13(1):21877. doi: 10.1038/s41598-023-49131-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maw MA. A frameshift mutation in prominin (mouse)-like 1 causes human retinal degeneration. Hum Mol Genet. 2001;9(1):27–34. doi: 10.1093/hmg/9.1.27. [DOI] [PubMed] [Google Scholar]
- 27.Li Z. CD133: a stem cell biomarker and beyond. Exp Hematol Oncol. 2013;2(1):17. doi: 10.1186/2162-3619-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hori A, Nishide K, Yasukuni Y, Haga K, Kakuta W, Ishikawa Y, et al. Prominin-1 modulates rho/ROCK-mediated membrane morphology and calcium-dependent intracellular chloride flux. Sci Rep. 2019;9(1):15911. doi: 10.1038/s41598-019-52040-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang Z, Chen Y, Lillo C, Chien J, Yu Z, Michaelides M, et al. Mutant prominin 1 found in patients with macular degeneration disrupts photoreceptor disk morphogenesis in mice. J Clin Invest. 2008;118(8):2908–16. doi: 10.1172/JCI35891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bhattacharya S, Yin J, Winborn CS, Zhang Q, Yue J, Chaum E. Prominin-1 is a novel regulator of autophagy in the human retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2017;58(4):2366–87. doi: 10.1167/iovs.16-21162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kobayashi Y, Watanabe S, Ong ALC, Shirai M, Yamashiro C, Ogata T, et al. Early manifestations and differential gene expression associated with photoreceptor degeneration in Prom1 -deficient retina. Disease Models & Mechanisms. 2021 Nov 1;14(11):dmm048962. [DOI] [PMC free article] [PubMed]
- 32.Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet. 2017;390(10097):849–60. doi: 10.1016/S0140-6736(17)31868-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cehajic-Kapetanovic J, Xue K, Martinez-Fernandez de la Camara C, Nanda A, Davies A, Wood LJ, et al. Initial results from a first-in-human gene therapy trial on X-linked retinitis pigmentosa caused by mutations in RPGR. Nat Med 2020;26(3): 354–359 [DOI] [PMC free article] [PubMed]
- 34.Fischer MD, Michalakis S, Wilhelm B, Zobor D, Muehlfriedel R, Kohl S, et al. Safety and vision outcomes of subretinal gene therapy targeting cone photoreceptors in achromatopsia: a nonrandomized controlled trial. JAMA Ophthalmol. 2020;138(6):643–51. doi: 10.1001/jamaophthalmol.2020.1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.MacDonald IM, Moen C, Duncan JL, Tsang SH, Cehajic-Kapetanovic J, Aleman TS. Perspectives on gene therapy: choroideremia represents a challenging model for the treatment of other inherited retinal degenerations. Transl Vis Sci Technol 9(3):17. [DOI] [PMC free article] [PubMed]
- 36.RetNet - Retinal Information Network [Internet]. [cited 2021 Jun 14]. Available from: https://sph.uth.edu/RETNET/
- 37.Scholl HPN, Strauss RW, Singh MS, Dalkara D, Roska B, Picaud S, et al. Emerging therapies for inherited retinal degeneration. Sci Transl Med 2016;8(368). [DOI] [PubMed]
- 38.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–72. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 39.Liang Y, Sun X, Duan C, Tang S, Chen J. Application of patient-derived induced pluripotent stem cells and organoids in inherited retinal diseases. Stem Cell Res Ther. 2023;27(14):340. doi: 10.1186/s13287-023-03564-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen FK, McLenachan S, Edel M, Da Cruz L, Coffey PJ, Mackey DA. iPS cells for modelling and treatment of retinal diseases. J Clin Med. 2014;3(4):1511–41. doi: 10.3390/jcm3041511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shi Y, Inoue H, Wu JC, Yamanaka S. Induced pluripotent stem cell technology: a decade of progress. Nat Rev Drug Discov. 2017;16(2):115–30. doi: 10.1038/nrd.2016.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coco-Martin RM, Pastor-Idoate S, Pastor JC. Cell replacement therapy for retinal and optic nerve diseases: cell sources, clinical trials and challenges. Pharmaceutics. 2021;13(6):865. doi: 10.3390/pharmaceutics13060865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maeda T, Mandai M, Sugita S, Kime C, Takahashi M. Strategies of pluripotent stem cell-based therapy for retinal degeneration: update and challenges. Trends Mol Med. 2022;28(5):388–404. doi: 10.1016/j.molmed.2022.03.001. [DOI] [PubMed] [Google Scholar]
- 44.Benati D, Leung A, Perdigao P, Toulis V, van der Spuy J, Recchia A. Induced pluripotent stem cells and genome-editing tools in determining gene function and therapy for inherited retinal disorders. Int J Mol Sci. 2022;23(23):15276. doi: 10.3390/ijms232315276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu P, Guo F, Xie B, Zhong X. Generation and characterization of two iPSC lines carrying heterozygous or homozygous nonsense mutation in PROM1 gene from a single family. Stem Cell Res. 2022;64:102913. doi: 10.1016/j.scr.2022.102913. [DOI] [PubMed] [Google Scholar]
- 46.McCulloch DL, Marmor MF, Brigell MG, Hamilton R, Holder GE, Tzekov R, et al. ISCEV Standard for full-field clinical electroretinography (2015 update) Doc Ophthalmol. 2015;130(1):1–12. doi: 10.1007/s10633-014-9473-7. [DOI] [PubMed] [Google Scholar]
- 47.RetNet: Summaries [Internet]. [cited 2023 May 1]. Available from: https://web.sph.uth.edu/RetNet/sum-dis.htm#A-genes
- 48.Armento A, Ueffing M, Clark SJ. The complement system in age-related macular degeneration. Cell Mol Life Sci. 2021;78(10):4487–505. doi: 10.1007/s00018-021-03796-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Human Phenotype Ontology [Internet]. [cited 2023 Jun 2]. Available from: https://hpo.jax.org/app/browse/term/HP:0000479
- 50.Alonso-Barroso E, Brasil S, Briso-Montiano Á, Navarrete R, Pérez-Cerdá C, Ugarte M, et al. Generation and characterization of a human iPSC line from a patient with propionic acidemia due to defects in the PCCA gene. Stem Cell Res. 2017;23:173–7. doi: 10.1016/j.scr.2017.07.021. [DOI] [PubMed] [Google Scholar]
- 51.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 52.Green MR, Sambrook J. Precipitation of DNA with ethanol. Cold Spring Harb Protoc. 2016;2016(12):pdb.prot093377. [DOI] [PubMed]
- 53.Concordet JP, Haeussler M. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018;46(W1):W242–5. doi: 10.1093/nar/gky354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.MOLBIOTOOLS - Molecular Biology Online Apps [Internet]. [cited 2023 Sep 28]. https://molbiotools.com/
- 55.Littink KW, Koenekoop RK, van den Born LI, Collin RWJ, Moruz L, Veltman JA, et al. Homozygosity mapping in patients with cone-rod dystrophy: novel mutations and clinical characterizations. Invest Ophthalmol Vis Sci. 2010;51(11):5943–51. doi: 10.1167/iovs.10-5797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Birtel J, Eisenberger T, Gliem M, Müller PL, Herrmann P, Betz C, et al. Clinical and genetic characteristics of 251 consecutive patients with macular and cone/cone-rod dystrophy. Sci Rep. 2018;8(1):4824. doi: 10.1038/s41598-018-22096-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Perea-Romero I, Gordo G, Iancu IF, Del Pozo-Valero M, Almoguera B, Blanco-Kelly F, et al. Genetic landscape of 6089 inherited retinal dystrophies affected cases in Spain and their therapeutic and extended epidemiological implications. Sci Rep. 2021;11(1):1526. doi: 10.1038/s41598-021-81093-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fujinami K, Oishi A, Yang L, Arno G, Pontikos N, Yoshitake K, et al. Clinical and genetic characteristics of 10 Japanese patients with PROM1 -associated retinal disorder: A report of the phenotype spectrum and a literature review in the Japanese population. Am J Med Genet Pt C. 2020;184(3):656–74. doi: 10.1002/ajmg.c.31826. [DOI] [PubMed] [Google Scholar]
- 59.Großpötzl M, Riedl R, Schließleder G, Hu ZJ, Michaelides M, Sadda S, et al. Progression of PROM1-associated retinal degeneration as determined by spectral-domain optical coherence tomography Over a 24-month period. Am J Ophthalmol. 2024;259:109–16. doi: 10.1016/j.ajo.2023.11.010. [DOI] [PubMed] [Google Scholar]
- 60.Permanyer J, Navarro R, Friedman J, Pomares E, Castro-Navarro J, Marfany G, et al. Autosomal recessive retinitis pigmentosa with early macular affectation caused by premature truncation in PROM1. Invest Ophthalmol Vis Sci. 2010;51(5):2656–63. doi: 10.1167/iovs.09-4857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim JM, Lee C, Lee GI, Kim NKD, Ki CS, Park WY, et al. Identification of the PROM1 mutation p.R373C in a Korean patient with autosomal dominant stargardt-like macular dystrophy. Ann Lab Med. 2017;37(6):536–9. doi: 10.3343/alm.2017.37.6.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ricca AM, Han IC, Hoffmann J, Stone EM, Sohn EH. Macular atrophy and phenotypic variability in autosomal dominant stargardt-like macular dystrophy due to prom1 mutation. RETINA. 2023;43(7):1165. doi: 10.1097/IAE.0000000000003784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nadeau JH. Modifier genes in mice and humans. Nat Rev Genet. 2001;2(3):165–74. doi: 10.1038/35056009. [DOI] [PubMed] [Google Scholar]
- 64.Alsaif HS, Khan AO, Patel N, Alkuraya H, Hashem M, Abdulwahab F, et al. Congenital glaucoma and CYP1B1: an old story revisited. Hum Genet. 2019;138(8):1043–9. doi: 10.1007/s00439-018-1878-z. [DOI] [PubMed] [Google Scholar]
- 65.Corton M, Avila-Fernández A, Campello L, Sánchez M, Benavides B, López-Molina MI, et al. Identification of the photoreceptor transcriptional co-repressor SAMD11 as novel cause of autosomal recessive retinitis pigmentosa. Sci Rep. 2016;6(1):35370. doi: 10.1038/srep35370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yik WY, Steinberg SJ, Moser AB, Moser HW, Hacia JG. Identification of novel mutations and sequence variation in the Zellweger syndrome spectrum of peroxisome biogenesis disorders. Hum Mutat. 2009;30(3):E467–480. doi: 10.1002/humu.20932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Johnson CA, Densen P, Hurford RK, Colten HR, Wetsel RA. Type I human complement C2 deficiency A 28-base pair gene deletion causes skipping of exon 6 during RNA splicing. J Biol Chem. 1992;267(13):9347–53. doi: 10.1016/S0021-9258(19)50430-6. [DOI] [PubMed] [Google Scholar]
- 68.Heyne HO, Karjalainen J, Karczewski KJ, Lemmelä SM, Zhou W, Finn G, et al. Mono- and biallelic variant effects on disease at biobank scale. Nature. 2023;613(7944):519–25. doi: 10.1038/s41586-022-05420-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jakobsdottir J, Conley YP, Weeks DE, Ferrell RE, Gorin MB. C2 and CFB genes in age-related maculopathy and joint action with CFH and LOC387715 genes. PLoS One. 2008;3(5):e2199. doi: 10.1371/journal.pone.0002199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chu L, Bi C, Wang C, Zhou H. The relationship between complements and age-related macular degeneration and its pathogenesis. J Ophthalmol. 2024;2(2024):e6416773. doi: 10.1155/2024/6416773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kubo S, Yamamoto H, Kajimura N, Omori Y, Maeda Y, Chaya T, et al. Functional analysis of Samd11, a retinal photoreceptor PRC1 component, in establishing rod photoreceptor identity. Sci Rep. 2021;11(1):4180. doi: 10.1038/s41598-021-83781-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ji G, Zhou W, Du J, Zhou J, Wu D, Zhao M, et al. PCGF1 promotes epigenetic activation of stemness markers and colorectal cancer stem cell enrichment. Cell Death Dis. 2021;12(7):633. doi: 10.1038/s41419-021-03914-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Roy L, Bobbs A, Sattler R, Kurkewich JL, Dausinas PB, Nallathamby P, et al. CD133 promotes adhesion to the ovarian cancer metastatic niche. Cancer Growth Metastasis. 2018;9(11):1179064418767882. doi: 10.1177/1179064418767882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pearsall EA, Cheng R, Zhou K, Takahashi Y, Matlock HG, Vadvalkar SS, et al. PPARα is essential for retinal lipid metabolism and neuronal survival. BMC Biol. 2017;15(1):113. doi: 10.1186/s12915-017-0451-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Corbeil D, editor. Prominin-1 (CD133): new insights on stem & cancer stem cell biology. New York, NY: Springer New York; 2013 [cited 2021 Jun 9]. (Advances in Experimental Medicine and Biology; vol. 777). Available from: http://link.springer.com/10.1007/978-1-4614-5894-4
- 76.Wang H, Gong P, Li J, Fu Y, Zhou Z, Liu L. Role of CD133 in human embryonic stem cell proliferation and teratoma formation. Stem Cell Res Ther. 2020;27(11):208. doi: 10.1186/s13287-020-01729-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Crook JM, Hei D, Stacey G. The international stem cell banking initiative (ISCBI): raising standards to bank on. In Vitro Cell DevBiol-Animal. 2010;46(3):169–72. doi: 10.1007/s11626-010-9301-7. [DOI] [PubMed] [Google Scholar]
- 78.Bhattacharya S, Yin J, Huo W, Chaum E. Modeling of mitochondrial bioenergetics and autophagy impairment in MELAS-mutant iPSC-derived retinal pigment epithelial cells. Stem Cell Res Ther. 2022;13(1):260. doi: 10.1186/s13287-022-02937-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zacchigna S, Oh H, Wilsch-Bräuninger M, Missol-Kolka E, Jászai J, Jansen S, et al. Loss of the cholesterol-binding protein prominin-1/CD133 causes disk dysmorphogenesis and photoreceptor degeneration. J Neurosci. 2009;29(7):2297–308. doi: 10.1523/JNEUROSCI.2034-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dellett M, Sasai N, Nishide K, Becker S, Papadaki V, Limb GA, et al. Genetic background and light-dependent progression of photoreceptor cell degeneration in prominin-1 knockout mice. Invest Ophthalmol Vis Sci. 2015;56(1):164–76. doi: 10.1167/iovs.14-15479. [DOI] [PubMed] [Google Scholar]
- 81.Xiao YS, Liang J, Gao M, Sun JR, Liu Y, Chen JQ, et al. Deletion of prominin-1 in mice results in disrupted photoreceptor outer segment protein homeostasis. Int J Ophthalmol. 2021;14(9):1334–44. doi: 10.18240/ijo.2021.09.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Deng B. Mouse models and induced pluripotent stem cells in researching psychiatric disorders. Stem Cell Investig. 2017;17(4):62. doi: 10.21037/sci.2017.06.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pulman J, Sahel JA, Dalkara D. New editing tools for gene therapy in inherited retinal dystrophies. CRISPR J. 2022;5(3):377–88. doi: 10.1089/crispr.2021.0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Klymenko V, González Martínez OG, Zarbin M. Recent progress in retinal pigment epithelium cell-based therapy for retinal disease. Stem Cells Transl Med. 2024;13(4):317–31. doi: 10.1093/stcltm/szae004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary file 1: List of the 998 genes related to retinal diseases evaluated in the clinical exome sequencing analysis.
Supplementary file 2: Antibodies and their experimental conditions.
Supplementary file 3: Primer sequences for iPSC molecular characterization, sequencing, and gene editing.
Supplementary file 4: Pluripotency analysis. (A) Immunoreactivity of [CORD]-FiPSC1-Ep5F-2 and [STGD4]-FiPSC1-Ep5F-8 for the pluripotency markers OCT4, SSEA3, SOX2, SSEA4, TRA1-60, NANOG, and TRA-1-81. 25 µm scale. (B) Analysis of the functional pluripotency of the embryoid bodies (EBs) derived from [CORD]-FiPSC1-Ep5F-2 and [STGD4]-FiPSC1-Ep5F-8. Immunoreactivity to ectoderm (β-III Tubulin), mesoderm (α-SMA) and endoderm (α-1 Fetoprotein) markers. 25 µm scale.
Supplementary file 5: Full-length gels of Figure 4. (A) Full-length gels of Western blotting of CD133 protein expression in the iPSC control line [FiPS] Ctrl1-Ep6F-5, the iPSC line derived from patient IRD2 [RP]-FiPSC1-Ep5F-10, and its corresponding genetically repaired iPSC line [RP]-FiPSC1-Ep5F-10-GC1. (B). Full-length gels of Western blotting of β-actin (control).
Supplementary file 6: Sequencing data from IRD1.
Supplementary file 7: Sequencing data from IRD1’ sister.
Supplementary file 8: Sequencing data from IRD2.
Supplementary file 9: Sequencing data from IRD2’ daughter.
Supplementary file 10: Sequencing data from IRD3.
Supplementary file 11: Sequencing data from IRD3’ mother.
Data Availability Statement
All data generated or analysed during this study are included within the article.