

Abstract
Background
Lymnaeid snails of the genus Austropeplea are an important vector of the liver fluke (Fasciola hepatica), contributing to livestock production losses in Australia and New Zealand. However, the species status within Austropeplea is ambiguous due to heavy reliance on morphological analysis and a relative lack of genetic data. This study aimed to characterise the mitochondrial genome of A. cf. brazieri, an intermediate host of liver fluke in eastern Victoria.
Methods
The mitochondrial genome was assembled and annotated from a combination of second- and third-generation sequencing data. For comparative purposes, we performed phylogenetic analyses of the concatenated nucleotide sequences of the mitochondrial protein-coding genes, cytochrome c oxidase subunit 1 and 16S genes.
Results
The assembled mt genome was 13,757 base pairs and comprised 37 genes, including 13 protein-coding genes, 22 transfer RNA genes and 2 ribosomal RNA genes. The mt genome length, gene order and nucleotide compositions were similar to related species of lymnaeids. Phylogenetic analyses of the mt nucleotide sequences placed A. cf. brazieri within the same clade as Orientogalba ollula with strong statistical supports. Phylogenies of the cox1 and 16S mt sequences were constructed due to the wide availability of these sequences representing the lymnaeid taxa. As expected in both these phylogenies, A. cf. brazieri clustered with other Austropeplea sequences, but the nodal supports were low.
Conclusions
The representative mt genome of A. cf. brazieri should provide a useful resource for future molecular, epidemiology and parasitological studies of this socio-economically important lymnaeid species.
Graphical abstract

Supplementary Information
The online version contains supplementary material available at 10.1186/s13071-024-06358-7.
Keywords: Austropeplea , Australia, Fasciola hepatica, Snail, Intermediate host, Mitochondrial genome
Background
Freshwater gastropods of the family Lymnaeidae, known commonly as pond snails, are a diverse group with a worldwide distribution. Many lymnaeid taxa are vectors of socio-economically significant parasitic trematodes (flukes) [1]. In Australia, where livestock production is impacted by fascioliasis, a disease caused by the liver fluke Fasciola hepatica [2–4], lymnaeid snails of the genus Austropeplea are considered the most important native intermediate hosts of this parasitic trematode [5, 6].
Austropeplea is a semi-amphibious lymnaeid, occurring in freshwater habitats, such as ponds, streams and wetlands in south-eastern Australia and in New Zealand [7]. Currently, this genus is proposed to comprise at least four species and two subgenera [7], although the number of species still requires verification. In the past, 23 Australian and New Zealand lymnaeid species–group names were synonymised as Austropeplea tomentosa on the basis that their morphological variation related to phenotypic plasticity, induced by environmental factors [8]. Recently, however, it was proposed that at least three species of Austropeplea are endemic to south-eastern Australia (A. brazieri and A. subaquatilis), including Tasmania (A. huonensis) [7] and that A. tomentosa is exclusive to New Zealand on the basis of combined morphological and molecular investigations [9]. However, current molecular systematic studies of the Lymnaeidae conducted to date have utilised DNA sequence data only for a very small number of genetic markers in nuclear DNA (particularly the internal transcribed spacers of rDNA) and in mitochondrial (mt) DNA (cox1 and 16S genes) [10–14]. Thus, conclusions regarding species status and phylogenetic position within the family Lymnaeidae are likely restricting. Nonetheless, these studies have underpinned the next step, which is to use genomic data sets to ‘barcode’ species or taxa (the majority of which are presently defined using morphological data) for robust analyses of relationships among them.
In the present study, we characterised the first complete mitochondrial genome of a key representative of the Lymnaeidae. The focus here is on a taxon we refer to as Austropeplea cf. brazieri, which is a fluke pond snail that is distributed in south-eastern Australia [7] and inferred to be the predominant intermediate host of liver fluke affecting livestock production in this region [5]. The designation of this taxon indicates its indeterminate species status. The mt genome provided in this study will serve as a reference mt genome for future taxonomic, phylogenetic and ecological work on key snail vectors of parasitic trematodes.
Methods
Sample collection
Specimens of A. cf. brazieri were collected from a roadside irrigation channel in Werribee South, Victoria, Australia (latitude −37.944706, longitude 144.698857) and maintained in aquaria within a designated laboratory in the Department of Veterinary Biosciences, The University of Melbourne, Victoria, Australia. Individual snails were de-shelled, thoroughly washed in phosphate-buffered saline (PBS, pH 7.0), snap frozen in liquid nitrogen and stored at − 80 °C prior to DNA isolation.
DNA isolation, library construction and sequencing
The Nanobind Tissue Kit (PacBio, Menlo Park, CA, USA) was used to isolate high molecular weight genomic DNA from a single adult A. cf. brazieri. The quality of isolated DNA was evaluated using an Agilent 4200 TapeStation system (Thermo Fisher Scientific Waltham, MA, USA) and Genomic DNA ScreenTape (Thermo Fisher Scientific). The Ligation Sequencing Kit (SQK-LSK109; Oxford Nanopore Technologies) was used to construct a genomic DNA library following the manufacturer’s protocol. The library was then sequenced using a MinION sequencer (Oxford Nanopore Technologies). The flow cell used to sequence the library was washed using the Flow Cell Wash Kit (EXP-WSH003; Oxford Nanopore Technologies) and re-used to re-sequence the same DNA library. Base calling from raw FAST5 reads was done using the program Guppy v.5 (Oxford Nanopore Technologies) and saved in the FASTQ format [15].
Assembly and isolation of the mitochondrial genome
De novo assembly of the long-read sequences was performed using FLYE v.2.6 [16] with the -nano-raw option, and errors were corrected using medaka_consensus in the Medaka package v.0.10.0 (https://github.com/nanoporetech/medaka). The long-read sequence data were then mapped back to the assembled mitochondrial (mt) genome using Minimap2 v.2.0 [17], and mosdepth [18] was used to estimate genome coverage.
Initial annotation of tRNA, rRNA and protein-encoding gene regions was performed on the MITOS webserver [19] using the invertebrates mt genetic code (https://www.ncbi.nlm.nih.gov/Taxonomy/Utils/wprintgc.cgi; translation_table 5). Protein-coding genes were further curated in the program Geneious v.11.1.5 [20] using open reading frames (ORF) and published lymnaeid mt genomes as a guide (Table 1). The complete mt genome sequence was deposited in the GenBank database under accession no. PP100270 (Fig. 1). Raw sequence data are available from Sequence Read Archive (SRA) under accession no. SAMN39324652 with NCBI BioProject accession no. PRJNA1088272.
Table 1.
Mitochondrial genome sequences of snail species or strains used in the present study, with GenBank accession numbers and references listed
| GenBank accession number | Species − ‘strain’ | Length (bp) | G + C content (%) | References |
|---|---|---|---|---|
| PP100270 | Austropeplea cf. brazieri | 13,757 | 26.71 | Present study |
| KP098538 | Radix sp. “MOTU3” | 13,963 | 28.73 | [21] |
| KP098539 | Radix sp.—‘MOTU5’ | 13,832 | 25.98 | [21] |
| KP098541 | Radix balthica | 13,983 | 28.68 | [21] |
| MT862404 | Peregriana peregra | 14,023 | 28.44 | Direct submission |
| MT862422 | Galba truncatula | 13,855 | 26.06 | Direct submission |
| MT947902 | Bulinus truncatus | 13,767 | 24.29 | Direct submission |
| MW221941 | Lymnaea stagnalis | 13,834 | 28.13 | Direct submission |
| NC005439 | Biomphalaria glabrata | 13,670 | 25.37 | [22] |
| NC018536 | Orientogalba ollula | 13,768 | 27.32 | [23] |
| NC026538 | Radix auricularia | 13,745 | 29.31 | [21] |
| NC042905 | Pseudosuccinea columella | 13,757 | 26.66 | Direct submission |
| NC054237a | Radix plicatula | 13,751 | 29.69 | [24] |
aThe name of the organism listed under this GenBank accession number is Ampullaceana lagotis, but its referenced publication [24] clearly states that this mitochondrial sequence belongs to Radix plicatula; therefore, we have listed it in this study as such
Fig. 1.
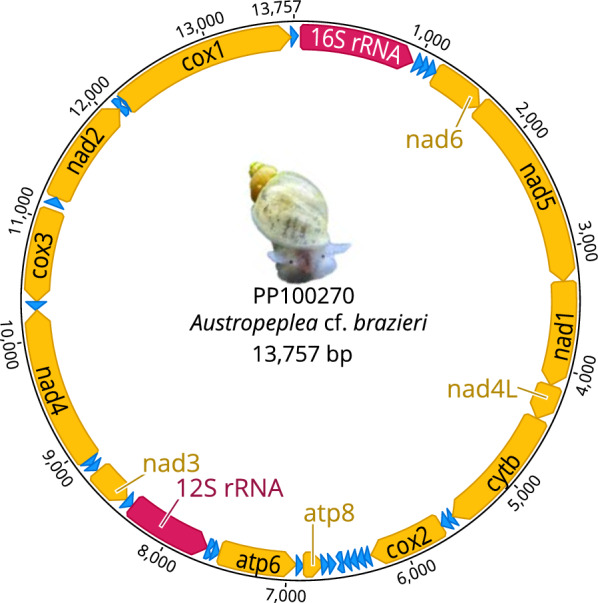
Reference mitochondrial genome of Austropeplea cf. brazieri (GenBank accession no. PP100270). The direction of gene transcription is shown with an arrow. Long (16S) and short (12S) ribosomal RNA subunits are shown in red and protein-encoding genes are shown in yellow
Whole mt genome and single loci comparative analyses
The complete mt genome of A. cf. brazieri was compared with the available reference mt genomes of other lymnaeids from the NCBI database (Table 1). We used the key words ‘Lymnaeidae + mitochondrial + genome’ and ‘Lymnaeidae + mitochondrion + genome’ in the ‘Nucleotide’ database (20 June 2023). The Bulinus truncatus and Biomphalaria glabrata (family Planorbidae) were used as outgroups. The comparison was performed with progressiveMauve v.2.4.0 [25] using the settings -hmm-identity = 0.95 and -island-gap-size = 10. Mitochondrial protein-coding genes were subsequently extracted and aligned as separate nucleotide coding sequences or inferred amino acid sequences using MUSCLE v.3.7 [26] alignment tool. The optimal nucleotide substitution model for each aligned sequence was determined using ModelTest-NG v.0.1.6 [27]. The aligned sequences were then subjected to phylogenetic analysis using Bayesian inference (BI) or maximum likelihood (ML) methods employing Monte Carlo Markov chain analysis in the program MrBayes v.3.2.2 [28] and IQ-tree v.2.2.2.7 [29], respectively. For the BI analysis, posterior probabilities (PP) were calculated using the optimal nucleotide substitution model (cox1 and 16S rRNA: GTR + I + G), generating 2,000,000 trees and sampling every 200th tree until potential scale reduction factors for each parameter approached 1. The initial 25% of trees were discarded as burn-in, and the others were used to construct a majority rule tree. Maximum likelihood trees and bootstrap (BS) supports were inferred using the optimal nucleotide substitution models and using the option ‘-B 10000 -bnni -minsup 0.5 -bi 1000’. The initial 10% of the 10,000 trees were discarded as burn-in, and the others were used to construct a majority rule tree. Phylogenetic trees were rendered and annotated using ggtree v.1.10.5 [30] in R v.4.3.1 (http://www.R-project.org/).
Due to the wide availability of mt cox1 and 16S rRNA sequence data for the family Lymnaeidae, we performed phylogenetic analyses of A. cf. brazieri and other lymnaeids based on these individual genes. Mitochondrial cox1 and 16S rRNA sequence data were downloaded from NCBI nucleotide sequence database (7 June 2023; Supplementary Table S1), with B. truncatus (gene ID 70630849) and Bi. glabrata (gene ID 2746309) (family Planorbidae) as outgroups. For the cox1 dataset, the key words ‘Lymnaeidae cox1’, ‘Lymnaeidae coi’, ‘Lymnaeidae cytochrome c oxidase subunit I’ and ‘Lymnaeidae cytochrome c oxidase subunit 1’ were used (Supplementary Table S1). For the 16S rRNA dataset, the key word ‘Lymnaeidae 16S’ was used. For each dataset, identical nucleotide sequences were removed using CD-HIT-EST v.4.6 [31]. Sequences incorrectly placed in the Lymnaeidae family or containing too few nucleotide sequences were also removed. The remaining sequences were aligned using MUSCLE. The optimal nucleotide substitution model for aligned sequences was then assessed using the program ModelTest-NG v.0.1.6 [27]. The aligned sequences were concatenated and then subjected to phylogenetic analysis using BI or ML methods as described above.
Comparison of nucleotide diversity patterns between the aligned mt protein-coding regions of A. cf. brazieri and protein-coding regions of the reference mt genome of Orientogalba ollula (= Galba pervia, GenBank accession no. NC018536) was performed using a sliding window analysis (Fig. 2). A sliding window analysis of nucleotide diversity (steps of 10 bp over 200-bp windows) was performed for each pairwise-alignment of concatenated genes using the PopGenome package [32] in R. For each comparison, nucleotide diversity values were plotted using the R package ggplot2 [33].
Fig. 2.

Sliding window analyses of the tRNA and concatenated protein-coding nucleotide sequences of Austropeplea cf. brazieri and Orientogalba ollula (Galba pervia, GenBank accession no. NC018536) mitochondrial genomes. Gene boundaries are indicated by vertical dotted lines. The horizontal dotted line indicates the average nucleotide diversity across both mitochondrial genomes
Results and discussion
The average depth of coverage of long-reads mapped to the mt genome of A. cf. brazieri was 463.09 (standard deviation = 30.56). For the short reads, the average depth of coverage was 958.85 (standard deviation 95.52). The mt genome of A. cf. brazieri (GenBank accession number PP100270) is circular and spans 13,757 base pairs (bp) in length (equivalent to 13.8 kb) which falls within range (13.6–14.1 kb) of heterobranch gastropod mt genomes sequenced so far [34]. We identified 37 genes which included 13 protein-coding genes, 22 transfer RNA genes and 2 ribosomal RNA genes (Fig. 1; Table 2). In most instances, start and stop codons were consistent with those of the mt genomes of most molluscs [34] and other invertebrates characterised to date [35].
Table 2.
Mitochondrial genes of Austropeplea cf. brazieri and their locations, GC contents, lengths, start/stop codons and direction of the protein-coding gene transcription
| Gene designations | Location (start/end) | Length (bp) | Start/stop codons | Transcription direction |
|---|---|---|---|---|
| rrnL | 1/986 | 962 | NA | Forward |
| tRNA-L1(tag) | 988/1051 | 64 | NA | Forward |
| tRNA-P(tgg) | 1045/1107 | 63 | NA | Forward |
| tRNA-A(tgc) | 1107/1170 | 64 | NA | Forward |
| nad6 | 1171/1629 | 459 | ATA/TAA | Forward |
| nad5 | 1631/3277 | 1647 | ATA/TAG | Forward |
| nad1 | 3279/4154 | 876 | ATT/TAA | Forward |
| nad4L | 4155/4452 | 298 | TTG/TAA | Forward |
| cytb | 4453/5535 | 1083 | ATT/TAA | Forward |
| tRNA-D(gtc) | 5536/5587 | 53 | NA | Forward |
| tRNA-F(gaa) | 5588/5650 | 62 | NA | Forward |
| cox2 | 5651/6294 | 643 | TTG/TAA | Forward |
| tRNA-Y(gta) | 6296/6345 | 50 | NA | Forward |
| tRNA-W(tca) | 6346/6405 | 60 | NA | Forward |
| tRNA-C(gca) | 6410/6468 | 59 | NA | Forward |
| tRNA-G(tcc) | 6470/6522 | 53 | NA | Forward |
| tRNA-H(gtg) | 6525/6582 | 58 | NA | Forward |
| tRNA-Q(ttg) | 6649/6591 | 59 | NA | Reverse |
| tRNA-L2(taa) | 6701/6650 | 53 | NA | Reverse |
| atp8 | 6853/6702 | 165 | ATC/TAA | Reverse |
| tRNA-N(gtt) | 6917/6854 | 64 | NA | Reverse |
| ATP6 | 7557/6917 | 641 | TTG/TAA | Reverse |
| tRNA-R(tcg) | 7620/7558 | 63 | NA | Reverse |
| tRNA-E(gaa) | 7672/7621 | 52 | NA | Reverse |
| rrnS | 8388/7673 | 717 | NA | Reverse |
| tRNA-M(cat) | 8457/8389 | 69 | NA | Reverse |
| nad3 | 8797/8460 | 340 | ATA/TAA | Reverse |
| tRNA-S2(tga) | 8866/8804 | 63 | NA | Reverse |
| tRNA-S1(gct) | 8863/8917 | 55 | NA | Forward |
| nad4 | 8918/10,243 | 1326 | ATT/TAG | Forward |
| tRNA-T(tgt) | 10,311/10,244 | 68 | NA | Reverse |
| cox3 | 11,092/10,313 | 780 | ATG/TAA | Reverse |
| tRNA-I(gat) | 11,133/11,197 | 65 | NA | Forward |
| nad2 | 11,198/12,121 | 907 | ATT/TAG | Forward |
| tRNA-K(ttt) | 12,102/12,180 | 79 | NA | Forward |
| cox1 | 12,192/13,685 | 1493 | ATT/TAA | Forward |
| tRNA-V(tac) | 13,684/13,744 | 60 | NA | Forward |
The nucleotide composition within A. cf. brazieri mt genome was A + T biased (A = 36.9%, C = 11.9%, G = 12.8% and T = 38.5%). This A + T nucleotide composition bias has also been observed in other lymnaeid species sequenced to date, including Pseudosuccinea columella (73.3%) Orientogalba ollula (as Galba pervia) (72.69%), Radix auricularia (70.7%) and Radix plicutula (70.3%). The tRNAs (Table 2) were inferred to have a canonical structure. Two copies of a serine and a leucine tRNA were encoded, and all tRNAs were predicted to have DHU and TψC arms, except for tRNA-G(tcc) (without a TψC arm), tRNA-S1(gct) (without a DHU arm) and tRNA-S2(tga) (without a DHU arm).
The arrangement of genes within the mt genome of A. cf. brazieri are identical to those of O. ollula [23]. Future studies sequencing the complete mt genomes of additional taxa related to Austropeplea could potentially reveal alternative gene arrangements. The Heterobranchia, a group which Austropeplea belongs to, has been found to display the most variable gene arrangement among the Gastropoda [36].
Phylogenetic analyses (ML and BI) of the full-length mtDNA resulted in a phylogenetic tree with robust statistical supports (Fig. 3). There were good supports (BS/PP = 91/1) for the close relationship between A. cf. brazieri and O. ollula (as Galba pervia), originally described from eastern China [37]. This relationship, in addition to the clades comprising Radix and Ampullaceana, is consistent with the phylogeny based on the combined mt and nuclear sequence dataset [38]. However, with only 11 full-length lymnaeid mt genomes sequenced to date (including this study) out of around 175 described species, the inadequate representation of taxa within the current phylogenetic tree prevents any further interpretation.
Fig. 3.

Phylogenetic relationship of Austropeplea cf. brazieri with other representative lymnaeid snails. Biomphalaria glabrata and Bulinus truncatus (family Planorbidae) are the outgroups (Table 1). A phylogeny was inferred from concatenated nucleotide sequences derived from 12 mitochondrial protein-encoding genes using Bayesian inference (BI) and maximum likelihood (ML) analyses. Bootstrap (BS) support for the ML and posterior probability (PP) of the BI analyses are indicated at each node of the tree. The scale bar indicates phylogenetic distance in substitutions per site. The updated nomenclature is based on Supplementary Table S2
We also assessed the phylogenetic relationship between A. cf. brazieri with other lymnaeids using the mitochondrial cox1 and 16S genes due to the wide availability of these sequences within the Lymnaeidae (Figs. 4, 5, respectively). The phylogenetic tree comprising 83 cox1 sequences placed A. cf. brazieri in the same clade as A. tomentosa (GenBank AY227365) with strong support (BS/PP = 100/1). Since A. tomentosa is the only other species of Austropeplea with the cox1 gene sequenced, future studies containing additional cox1 sequences of this genus could determine the utility of this marker for resolving phylogenetic relationships within Austropeplea, although, at the genus level, the position of Austropeplea with other lymnaeid genera was ambiguous based on the cox1 gene. Similarly, the mt 16S phylogeny comprised 19 sequences (Fig. 5) and A. cf. brazieri fell within the Australian A. tomentosa group with low statistical support from both the ML and BI analyses. The lack of resolution within the current 16S phylogeny and the incongruences between this marker and other genetic regions [9] suggest that this topology may not accurately reflect the phylogenetic relationship of Austropeplea.
Fig. 4.

Phylogenetic relationship of Austropeplea cf. brazieri with other representative lymnaeid snails inferred on the basis of an analysis of the aligned partial mitochondrial 16S rRNA gene sequences by Bayesian inference (BI) and maximum likelihood (ML) using Biomphalaria pfeifferi and Bulinus truncatus (family Planorbidae) outgroups. Bootstrap (BS) support for the ML and posterior probability (PP) of the BI analyses are indicated at each node of the tree. The partial mitochondrial 16S rRNA gene sequence of A. cf. brazieri sequenced here is denoted in bold type. The updated nomenclature is based on Supplementary Table S2. The scale bar indicates phylogenetic distance in substitutions per site. NSW New South Wales, TAS Tasmania, Vic Victoria
Fig. 5.

Phylogenetic relationship of Austropeplea cf. brazieri with other representative lymnaeid snails inferred on the basis of an analysis of the aligned partial cox1 mitochondrial gene sequences by Bayesian inference (BI) and maximum likelihood (ML) using Biomphalaria pfeifferi and Bulinus truncatus (family Planorbidae) as outgroups. Bootstrap (BS) support for the ML and posterior probability (PP) of the BI analyses are indicated at each node of the tree. The partial cox1 gene sequence of A. cf. brazieri sequenced here is denoted in bold type. The scale bar indicates phylogenetic distance (in substitutions per site)
Conclusions
Future studies to characterise the mt genomic sequences of the lymnaeids endemic to Australasia should allow some of the taxonomic ambiguities within this group to be addressed. Furthermore, such genomic datasets would provide greater insight into the phylogeny among the Australian lymnaeids and their relationships with other pond snails occurring worldwide. In conclusion, this study presented the first complete mt genome of A. cf. brazieri, which serves as a valuable resource for future molecular, epidemiological and ecological studies of this and related socio-economically important lymnaeid species.
Supplementary Information
Additional file 1: Table S1. Partial cox1 gene and 16S trees sequences used in the present study, with GenBank accession numbers, description of taxon and references listed; Supplementary Table S2. The updated nomenclature used in the phylogenetic tree of the current study and the previous species names corresponding to each of the GenBank accession numbers.
Acknowledgements
We acknowledge Ms Christine Andersen for collecting the snail used in this study.
Author contributions
N.D.Y. designed the study. C.G.G. collected the sample. N.D.Y., A.V.K. and C.E.F. performed the sequencing and data analyses. N.D.Y. drafted the first version of the manuscript. T.S., R.B.G., A.V.K. and N.D.Y. contributed to the writing and editing process. All authors have read and approved the final manuscript.
Funding
This study was supported by the Australian Research Council Discovery Project no. DP230100270.
Availability of data and materials
All relevant data are included in the article.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Aksenova OV, Bolotov IN, Khrebtova IS, Kondakov AV, Vinarski MV. Phylogeny and taxonomy of the family Lymnaeidae. In: Vinarski MV, Vazquez AO, editors. The Lymnaeidae: a handbook on their natural history and parasitological significance. Switzerland: Springer Nature; 1980. pp. 67–101. [Google Scholar]
- 2.Elliott TP, Kelley JM, Rawlin G, Spithill TW. High prevalence of fasciolosis and evaluation of drug efficacy against Fasciola hepatica in dairy cattle in the Maffra and Bairnsdale districts of Gippsland, Victoria, Australia. Vet Parasitol. 2015;209:117–124. doi: 10.1016/j.vetpar.2015.02.014. [DOI] [PubMed] [Google Scholar]
- 3.Lane J, Jubb T, Shephard R, Webb-Ware J, Fordyce G. Priority list of endemic diseases for the red meat industries. 2015. https://www.mla.com.au/research-and-development/reports/2023/B.AHE.0327-priority-list-of-endemic-diseases-for-the-red-meat-industry--2022-update/. Accessed 12 Dec 2023.
- 4.Kelley JM, Elliott TP, Beddoe T, Anderson G, Skuce P, Spithill TW. Current threat of Triclabendazole resistance in Fasciola hepatica. Trends Parasitol. 2016;32:458–469. doi: 10.1016/j.pt.2016.03.002. [DOI] [PubMed] [Google Scholar]
- 5.Boray J. Molluscs and parasitic diseases. Mollusca South Synth. 1998;5:65–70. [Google Scholar]
- 6.O'Shaughnessy J, Garcia-Campos A, McAloon CG, Fagan S, De Waal T, McElroy M, et al. Epidemiological investigation of a severe rumen fluke outbreak on an Irish dairy farm. Parasitology. 2018;145:948–952. doi: 10.1017/S0031182017002086. [DOI] [PubMed] [Google Scholar]
- 7.Ponder WF, Hallan A, Shea ME, Clark SA, Richards K, Klunzinger MW, et al. Australian freshwater molluscs revision 2. 2023. https://keys.lucidcentral.org/keys/v3/freshwater_molluscs/key/australian_freshwater_molluscs/Media/Html/entities/austropeplea.htm. Accessed 15 Nov 2023.
- 8.Boray JC, McMichael DF. The identity of the Australian lymnaeid snail host of Fasciola hepatica and its response to environment. Mar Freshw Res. 1961;12:150–163. doi: 10.1071/MF9610150. [DOI] [Google Scholar]
- 9.Puslednik L, Ponder WF, Dowton M, Davis AR. Examining the phylogeny of the Australasian Lymnaeidae (Heterobranchia: Pulmonata: Gastropoda) using mitochondrial, nuclear and morphological markers. Mol Phylogenet Evol. 2009;52:643–659. doi: 10.1016/j.ympev.2009.03.033. [DOI] [PubMed] [Google Scholar]
- 10.Bargues MD, Mas-Coma S. Reviewing lymnaeid vectors of fascioliasis by ribosomal DNA sequence analyses. J Helminthol. 2005;79:257–267. doi: 10.1079/JOH2005297. [DOI] [PubMed] [Google Scholar]
- 11.Dung BT, Doanh PN, The DT, Loan HT, Losson B, Caron Y. Morphological and molecular characterization of lymnaeid snails and their potential role in transmission of Fasciola spp. in Vietnam. Korean J Parasitol. 2013;51:657–662. doi: 10.3347/kjp.2013.51.6.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Standley CJ, Prepelitchi L, Pietrokovsky SM, Issia L, Stothard JR, Wisnivesky-Colli C. Molecular characterization of cryptic and sympatric lymnaeid species from the Galba/Fossaria group in Mendoza Province, Northern Patagonia, Argentina. Parasit Vectors. 2013;6:304. doi: 10.1186/1756-3305-6-304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vinarski MV, Aksenova OV, Bespalaya YV, Gofarov MY, Kondakov AV, Khrebtova IS, et al. How an ecological race is forming: morphological and genetic disparity among thermal and non-thermal populations of aquatic lymnaeid snails (Gastropoda: Lymnaeidae) Diversity. 2023;15:548. doi: 10.3390/d15040548. [DOI] [Google Scholar]
- 14.Correa AC, Escobar JS, Durand P, Renaud F, David P, Jarne P, et al. Bridging gaps in the molecular phylogeny of the Lymnaeidae (Gastropoda: Pulmonata), vectors of Fascioliasis. BMC Evol Biol. 2010;10:381. doi: 10.1186/1471-2148-10-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cock PJ, Fields CJ, Goto N, Heuer ML, Rice PM. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 2010;38:1767–1771. doi: 10.1093/nar/gkp1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kolmogorov M, Yuan J, Lin Y, Pevzner PA. Assembly of long, error-prone reads using repeat graphs. Nat Biotechnol. 2019;37:540–546. doi: 10.1038/s41587-019-0072-8. [DOI] [PubMed] [Google Scholar]
- 17.Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pedersen BS, Quinlan AR. Mosdepth: quick coverage calculation for genomes and exomes. Bioinformatics. 2018;34:867–868. doi: 10.1093/bioinformatics/btx699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, et al. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 2013;69:313–319. doi: 10.1016/j.ympev.2012.08.023. [DOI] [PubMed] [Google Scholar]
- 20.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feldmeyer B, Greshake B, Funke E, Ebersberger I, Pfenninger M. Positive selection in development and growth rate regulation genes involved in species divergence of the genus Radix. BMC Evol Biol. 2015;15:164. doi: 10.1186/s12862-015-0434-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DeJong RJ, Emery AM, Adema CM. The mitochondrial genome of Biomphalaria glabrata (Gastropoda: Basommatophora), intermediate host of Schistosoma mansoni. J Parasitol. 2004;90:991–997. doi: 10.1645/GE-284R. [DOI] [PubMed] [Google Scholar]
- 23.Liu GH, Wang SY, Huang WY, Zhao GH, Wei SJ, Song HQ, et al. The complete mitochondrial genome of Galba pervia (Gastropoda: Mollusca), an intermediate host snail of Fasciola spp. PLoS ONE. 2012;7:e42172. doi: 10.1371/journal.pone.0042172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qin D-M, Huang X-C, Yang L-M, Liu X-J, Wu R-W, Ouyang S, et al. Complete mitochondrial genome of the radicine pond snail Radix plicatula (Gastropoda: Lymnaeidae) Mitochondrial DNA Part B. 2019;4:2861–2862. doi: 10.1080/23802359.2019.1661300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Darling AE, Mau B, Perna NT. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE. 2010;5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Darriba D, Posada D, Kozlov AM, Stamatakis A, Morel B, Flouri T. ModelTest-NG: a new and scalable tool for the selection of DNA and protein evolutionary models. Mol Biol Evol. 2020;37:291–294. doi: 10.1093/molbev/msz189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ronquist F, Teslenko M, Van Der Mark P, Ayres DL, Darling A, Höhna S, et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, Von Haeseler A, et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol. 2020;37:1530–1534. doi: 10.1093/molbev/msaa015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu G, Smith DK, Zhu H, Guan Y, Lam TTY. ggtree: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol Evol. 2017;8:28–36. doi: 10.1111/2041-210X.12628. [DOI] [Google Scholar]
- 31.Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28:3150–3152. doi: 10.1093/bioinformatics/bts565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pfeifer B, Wittelsburger U, Ramos-Onsins SE, Lercher MJ. PopGenome: an efficient Swiss army knife for population genomic analyses in R. Mol Biol Evol. 2014;31:1929–1936. doi: 10.1093/molbev/msu136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wickham H. ggplot2: elegant graphics for data analysis. New York: Springer; 2009. [Google Scholar]
- 34.Ghiselli F, Gomes-dos-Santos A, Adema CM, Lopes-Lima M, Sharbrough J, Boore JL. Molluscan mitochondrial genomes break the rules. Philos Trans R Soc B Biol Sci. 2021;376:20200159. doi: 10.1098/rstb.2020.0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lavrov DV. Mitochondrial genomes in invertebrate animals. In: Bell E, editor. Molecular life sciences: an Encyclopedic reference. New York: Springer; 2021. pp. 1–8. [Google Scholar]
- 36.Sun S, Li Q, Kong L, Yu H. Multiple reversals of strand asymmetry in molluscs mitochondrial genomes, and consequences for phylogenetic inferences. Mol Phylogenet Evol. 2018;118:222–231. doi: 10.1016/j.ympev.2017.10.009. [DOI] [PubMed] [Google Scholar]
- 37.Vinarski MV, Aksenova OV, Bolotov IN. Taxonomic assessment of genetically-delineated species of radicine snails (Mollusca, Gastropoda, Lymnaeidae) Zoosyst Evo. 2020;96:577–608. doi: 10.3897/zse.96.52860. [DOI] [Google Scholar]
- 38.Aksenova OV, Bolotov IN, Gofarov MY, Kondakov AV, Vinarski MV, Bespalaya YV, et al. Species richness, molecular taxonomy and biogeography of the radicine pond snails (Gastropoda: Lymnaeidae) in the Old World. Sci Rep. 2018;8:11199. doi: 10.1038/s41598-018-29451-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. Partial cox1 gene and 16S trees sequences used in the present study, with GenBank accession numbers, description of taxon and references listed; Supplementary Table S2. The updated nomenclature used in the phylogenetic tree of the current study and the previous species names corresponding to each of the GenBank accession numbers.
Data Availability Statement
All relevant data are included in the article.