

ABSTRACT
Beech leaf disease (BLD) is a newly emerging disease in North America that affects American beech (Fagus grandifolia). It is increasingly recognized that BLD is caused by a subspecies of the anguinid nematode Litylenchus crenatae subsp. mccannii (hereafter L. crenatae), which is likely native to East Asia. How nematode infestation of leaves affects the leaf microbiome and whether changes in the microbiome could contribute to BLD symptoms remain uncertain. In this study, we examined bacterial and fungal communities associated with the leaves of F. grandifolia across nine sites in Ohio and Pennsylvania that were either symptomatic or asymptomatic for BLD and used qPCR to measure relative nematode infestation levels. We found significantly higher levels of infestation at sites visibly symptomatic for BLD. Low levels of nematode infestation were also observed at asymptomatic sites, which suggests that nematodes can be present without visible symptoms evident. Bacterial and fungal communities were significantly affected by sampling site and symptomology, but only fungal communities were affected by nematode presence alone. We found many significant indicators of both bacteria and fungi related to symptoms of BLD, with taxa generally occurring in both asymptomatic and symptomatic leaves, suggesting that microbes are not responsible for BLD but could act as opportunistic pathogens. Of particular interest was the fungal genus Erysiphe, which is common in the Fagaceae and is reported to overwinter in buds—a strategy consistent with L. crenatae. The specific role microbes play in opportunistic infection of leaves affected by L. crenatae will require additional study.
IMPORTANCE
Beech leaf disease (BLD) is an emerging threat to American beech (Fagus grandifolia) and has spread quickly throughout the northeastern United States and into southern Canada. This disease leads to disfigurement of leaves and is marked by characteristic dark, interveinal banding, followed by leaf curling and drop in more advanced stages. BLD tends to especially affect understory leaves, which can lead to substantial thinning of the forest understory where F. grandifolia is a dominant tree species. Understanding the cause of BLD is necessary to employ management strategies that protect F. grandifolia and the forests where it is a foundation tree species. Current research has confirmed that the foliar nematode Litylenchus crenatae subsp. mccannii is required for BLD, but whether other organisms are involved is currently unknown. Here, we present a study that investigated leaf-associated fungi and bacteria of F. grandifolia to understand more about how microorganisms may contribute to BLD.
KEYWORDS: bacteria, beech leaf disease, Fagus grandifolia, fungi, leaf nematodes, Litylenchus crenatae subsp. mccannii, plant microbiome
INTRODUCTION
Beech leaf disease (BLD) is an emerging threat to trees in the genus Fagus and especially American beech (Fagus grandifolia), which is a dominant tree species in many temperate forests of eastern North America (1, 2). First observed in Lake County, Ohio, USA, in 2012 (3), BLD has now spread throughout much of the northern United States, from Ohio to Maine, and extends into southern Ontario (Fig. S1). BLD also can affect European (Fagus sylvatica), Chinese (Fagus engleriana), and Oriental beech (Fagus orientalis). Initially, BLD symptoms consist of interveinal darkening and thickening of leaf tissue, often followed by curling of leaf tissue (4, 5). New leaves emerge from buds in spring already showing signs of BLD, suggesting that damage may occur within buds over winter (6). Severely affected leaves can senesce by late summer, and understory leaves and branches are often most impacted by BLD. This can lead to substantial thinning and mortality of understory trees in many forests (7), which suggests that BLD has the potential to affect the survival and regeneration of beech in many forests (8).
Recent work has shown that the nematode Litylenchus crenatae subsp. mccannii (hereafter L. crenatae) is required for the development of BLD (4). However, whether symptoms of BLD are due only to the presence of the nematode or whether bacteria or fungi are involved in BLD is uncertain. In previous work conducted in a 16-year-old American beech plantation, we showed that although bacterial and fungal communities were unaffected by BLD on buds, bacterial communities were significantly affected by BLD on leaves, indicating that the nematode could be altering leaf bacterial communities (5). A limited sequencing approach found that the bacterial genus Wolbachia was a significant indicator of leaves symptomatic for BLD (5). Wolbachia is often found as an intracellular parasite of arthropods (9, 10) and has also been found as a component of the microbiome of some nematodes (10). The bacterial genus Methylocystis was also a significant indicator of symptomatic leaves, and the bacterial genus Mucilaginibacter was found only on leaves symptomatic for BLD. None of these genera are known to cause plant disease; however, they could be members of the nematode microbiome.
Recent analysis of the leaf microbiome using high-throughput sequencing on forest trees at three locations (two in Ohio and one in Pennsylvania) found no significant effect of BLD symptoms on leaf-associated bacterial communities (11). However, indicator species analysis confirmed the bacterial genera Wolbachia, Erwinia, Paenibacillus, and Pseudomonas were only found on symptomatic leaves, and one fungal genus, Paraphaeosphaeria, was found to be an indicator of leaves symptomatic for BLD (11). Fungal communities were significantly different among sample locations, however, and these three locations varied in their disease progression (11). Taken together, the results from this study (11) show that large community shifts with BLD symptoms may not be apparent, but specific bacterial and fungal taxa could be members of the nematode microbiome and could be involved in the development and progression of BLD. To date, studies of the phyllosphere microbiome in the presence of BLD have been limited in the number of locations studied (one location in reference five and three locations in reference 11). Therefore, there is a need for greater analysis and study of bacterial and fungal communities associated with BLD at regional scales to identify key components of the nematode microbiome that could facilitate nematode feeding or behavior and to understand the possible role of microbes in BLD symptom development and progression.
In this study, we used high-throughput sequencing to examine bacterial and fungal communities associated with leaves of F. grandifolia that were symptomatic or asymptomatic for BLD across nine sites in Ohio and Pennsylvania. Selected sites were either symptomatic or asymptomatic for BLD based on visual symptoms and were confirmed to have L. crenatae presence or absence through polymerase chain reaction (PCR). In addition, we used qPCR to determine the potential population size of L. crenatae in F. grandifolia leaves at these sites. We hypothesized that (i) nematode population size would be consistently higher in sites with symptomatic leaves, (ii) BLD symptomatic leaves would be associated with specific bacterial and/or fungal taxa across sampled sites, which would suggest an important role of phyllosphere bacteria or fungi in BLD, and (iii) L. crenatae presence would alter phyllosphere bacteria and fungi irrespective of the presence of BLD symptoms. Our overall goal was to advance our understanding of the relationship between BLD symptoms, nematode distribution, and microbial communities in plant leaves and buds.
RESULTS
Nematode presence and quantification in leaves and buds
Pictures of typical BLD symptoms are shown in Fig. 1. We detected the presence of the nematode L. crenatae in leaf tissue at all nine sites of our study region, although five of our nine sampling locations did not contain beech trees with noticeable symptoms of BLD at the time of sampling (Fig. 2). The detection at symptomatic sites was confirmed, as all positive PCR amplifications that were sent for sequencing were identified as L. crenatae. At asymptomatic sites, we list positive detection only if the PCR amplification was confirmed to be L. crenatae through sequencing. Two of the asymptomatic sites displayed no infestation of buds by L. crenatae, whereas one site (ANF south) had very low levels of bud infestation. At symptomatic sites, nearly 100% of leaf and bud tissue revealed positive infestation by L. crenatae. Asymptomatic sites had lower levels of nematode detection, which ranged from 0%–100% depending on the tissue examined and site (Fig. 2).
Fig 1.

Pictures of F. grandifolia leaves that are symptomatic for beech leaf disease with the characteristic interveinal darkening of the leaf tissue (indicated by arrows). Left photograph: Photograph of beech leaves affected by BLD within a mixed mesophytic forest in Ohio, June 2023. Right photograph: Close-up of leaves affected by BLD in an Ohio forest, September 2022.
Fig 2.
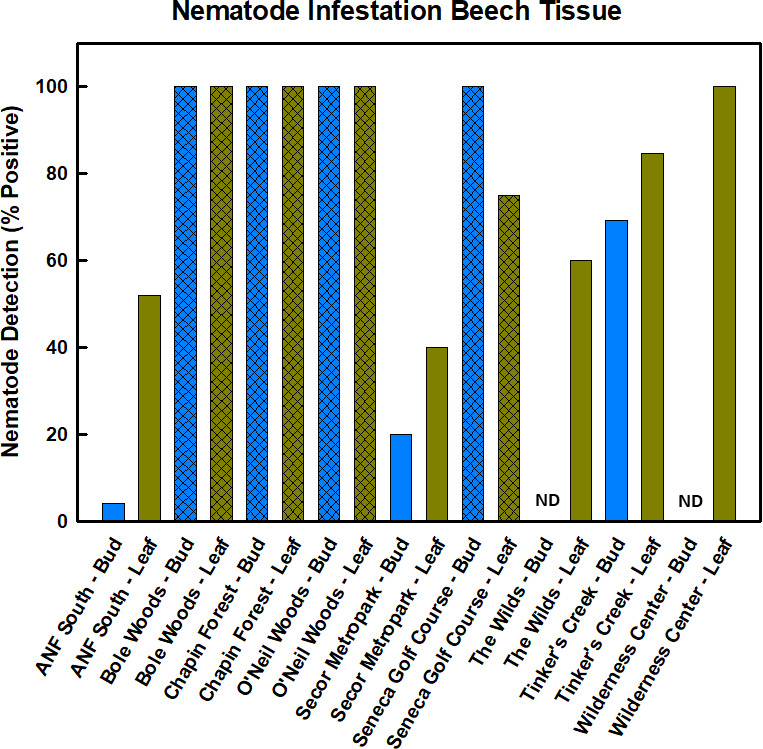
Detection of nematode infestation of leaf and bud tissue across all nine sites. Presence was determined from positive PCR amplification using general nematode primers. All positive PCR products were sequenced to confirm identity as L. crenatae. Percent of samples positive for L. crenatae is shown for each site; hashed bars are symptomatic sites. ND equals not detected.
qPCR was less sensitive than standard PCR in detecting the presence of the nematode (Fig. 3). Many samples that revealed nematode presence with standard PCR did not contain nematode copy numbers sufficient to distinguish the sample from background using qPCR. The melt temperature for nematode-positive qPCR amplicons was 82.5°C, and non-template controls and other samples deemed “not detected” for qPCR had melting temperatures routinely 10°C lower than pure nematode extracts. Wide variability in quantification among replicate samples from the same site was common. For example, we quantified nematode infestation from four samples collected at O’Neil Woods with copy numbers per sample ranging from 1.37 × 106 to 2.04 × 109 with a mean of 5.13 × 108 ± 5.09 × 108. This high level of variability was typical for many of the sites examined using qPCR. Nonetheless, samples from symptomatic sites had nematode copy numbers 2–3 orders of magnitude greater than asymptomatic sites (Fig. 3).
Fig 3.
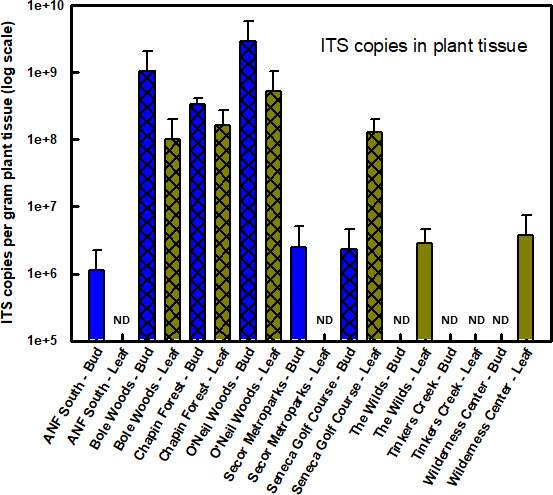
Relative differences in nematode population size across sites and samples as determined by qPCR estimates of ITS copy numbers. Cross-hatching indicates site with visible symptoms of beech leaf disease. ND equals not detected.
Molecular analysis of bacterial communities of leaf tissue
We detected a total of 3,361 bacterial zOTUs in the leaf microbiome. Nonmetric multidimensional scaling (NMDS) ordination of the bacterial community produced a 2-dimensional solution with a stress value of 0.14 and showed separation of the sites in ordination space (Fig. 4). There were significant overall regional differences in the bacterial microbiome between sites [Table 1, permutational multivariate analysis of variance (PERMANOVA) F = 11.3 and P value = 0.0002], which may be influenced by significant dispersion between sites [Table 1, permutational multivariate analysis of dispersion (PERMDISP) F = 10.586 and P value = 0.001]. Nematode presence did not significantly affect bacterial communities across the region (Table 1), although a significant interaction between site and nematode presence was observed (Table 1, F = 1.7, P = 0.004). This site by nematode interaction was observed on the NMDS graph, where bacterial microbiome samples from the same site separated in ordination space from one another based on nematode presence, but the direction of separation differed between sites (Fig. 4). PERMANOVA also found significant differences in bacterial communities between symptomatic and asymptomatic samples (Table 1, F = 8.0, P value = 0.0002), indicating differences in the leaf bacterial microbiome with BLD symptoms. This was driven by a change in leaf community structure at the symptomatic sites of O’Neil Woods, Bole Woods, and Seneca Golf Course, as these sites shifted in ordination space compared with the other sites (Fig. 4). In addition, the symptomatic sites had greater variability in bacterial community structure, as indicated by the significant dispersion (Table 1) and the higher distance to centroid measurements for the symptomatic sites compared with the asymptomatic sites (Fig. S2).
Fig 4.

Two-dimensional NMDS plot showing bacterial community structure differences between asymptomatic and symptomatic leaf tissues, as well as the presence or absence of L. crenatae (stress = 0.14). Each point is a mean of the axis scores (plus or minus standard error) for the samples that differed in nematode detection at each site. Some sites had differing nematode detection (ANF South, Secor Metropark, Seneca Golf Course, Tinker’s Creek, and The Wilds), and some sites had L. crenatae detected in all samples (Bole Woods, Chapin Forest, O’Neil Woods, and the Wilderness Center).
TABLE 1.
PERMANOVA and PERMDISP analyses of bacterial and fungal communities testing the interactive effects of site and nematode presence and the main effect of BLD symptoma
| Source | df | SS | R2 | F | P |
|---|---|---|---|---|---|
| Bacterial community PERMANOVA | |||||
| Model: Distance~Site*Nematode_Presence | |||||
| Site | 8 | 13.346 | 0.360 | 11.3102 | 0.0002 |
| Nematode presence | 1 | 0.213 | 0.00575 | 1.4464 | 0.1026 |
| Site × nematode presence | 4 | 0.982 | 0.0265 | 1.6651 | 0.004 |
| Residuals | 153 | 22.568 | 0.608 | ||
| Total | 166 | 37.11 | 1 | ||
| Dispersion - site | 8 | 0.709 | 10.586 | 0.001 | |
| Dispersion - nematode | 1 | 0.0144 | 1.364 | 0.251 | |
| Model: Distance ~ symptom | |||||
| Symptom BLD | 1 | 1.725 | 0.0465 | 8.0441 | 0.0002 |
| Residual | 165 | 35.385 | 0.953 | ||
| Total | 166 | 37.110 | 1 | ||
| Dispersion - symptom | 1 | 0.179 | 18.820 | 0.001 | |
| Fungal community PERMANOVA | |||||
| Model: Distance~Site*Nematode_Presence | |||||
| Site | 8 | 18.771 | 0.448 | 16.260 | 0.0002 |
| Nematode presence | 1 | 0.308 | 0.00736 | 2.138 | 0.0182 |
| Site × nematode presence | 3 | 0.581 | 0.0139 | 1.342 | 0.0826 |
| Residuals | 154 | 22.222 | 0.531 | ||
| Total | 166 | 41.882 | 1 | ||
| Dispersion - site | 8 | 0.3464 | 10.428 | 0.001 | |
| Dispersion - nematode | 1 | 0.0343 | 9.216 | 0.01 | |
| Model: Distance~symptom | |||||
| Symptom BLD | 1 | 2.921 | 0.0697 | 12.370 | 0.0002 |
| Residual | 165 | 38.961 | 0.930 | ||
| Total | 166 | 41.882 | 1 | ||
| Dispersion - symptom | 1 | 0.0668 | 20.308 | 0.001 | |
PERMANOVA analyses were run with the code adonis2. The dispersion statistics reported here were permutation tests on the distances to centroid between groups (the nine sites, nematode presence vs. absence, or symptomatic vs. asymptomatic tissue), which were calculated with the code betadisper. (See Materials and Methods for details.)
The overall bacterial microbiome of both asymptomatic and symptomatic leaves was dominated by taxa in the Proteobacteria, Bacteroidetes, and Actinobacteria (Fig. 5). We used indicator species analysis, which in our study used both zOTU abundance and occurrence values, to determine whether some taxa are significantly associated with certain groups or types of samples. Indicator species analysis found 637 significant bacterial indicators of symptomatic leaves and 31 significant bacterial indicators of asymptomatic leaves (q value < 0.05; Supplemental data S1). For asymptomatic leaves, 18 of the 31 significant indicators could be identified to a taxonomic level lower than phylum and only seven could be identified to as genus (Fig. S3). These significant indicators largely matched with Alphaproteobacteria, but there were varied orders, families, and genera within this class (Table S2). The significant indicators of symptomatic leaves included 118 different taxa (using the lowest taxonomic identity; see Materials and Methods). The significant symptomatic indicators that were most frequently found across sites had close identity to Kineococcus, Amnibacterium, Curtobacterium, Actinoplanes, Abditibacteriaceae, and Methylobacterium (Fig. 6). The majority of significant indicator taxa of symptomatic leaves were also found on asymptomatic leaves (although typically at a lower frequency across samples; Fig. 6). For example, the significant zOTU that matched with Kineococcus was found in 99% of symptomatic leaves and 94% of asymptomatic leaves (Fig. 6). The relative abundance of Kineococcus was slightly lower at asymptomatic sites, but not generally different between symptomatic and asymptomatic leaf tissues (on average, the relative abundance was 0.0079 in symptomatic tissue and 0.0071 in asymptomatic tissue; see Fig. 5).
Fig 5.

Leaf bacterial community taxonomic distribution from asymptomatic and symptomatic sites. The bars show the mean relative abundance at each site of the top 15 bacterial genera found with Illumina sequence analysis.
Fig 6.

The lowest taxonomic identity for significant bacterial indicators of symptomatic leaf tissue. The frequency that the taxa were found in leaf samples at symptomatic or asymptomatic sites is shown. If more than one zOTU had the same lowest taxonomic identity, the frequencies shown here are an average.
Some significant indicators matched with taxa that were found in greater relative abundance in symptomatic tissue, particularly Nocardioides, Mucilaginibacter, Chryseobacterium, Pedobacter, and Novosphingobium. Of the 637 significant bacterial zOTUs for symptomatic sites, 11 zOTUs matched with the genus Nocardioides, 24 zOTUs matched with the genus Mucilaginibacter, seven zOTUs matched with the genus Chryseobacterium, 12 zOTUs matched with the genus Pedobacter, and three matched with the genus Novosphingobium (Table S3). All five of these genera matched with zOTUs that were significant with Wald tests and showed significantly greater abundance in symptomatic leaf tissue [i.e., positive log2-fold change (LFC) values; Table S4]. In addition, these genera had visually higher relative sequence abundance at symptomatic sites compared with asymptomatic sites, but the increase was driven by certain sites (Fig. 4). For example, Nocardioides relative abundance was highest at O’Neil Woods, Mucilaginibacter increases with symptom were driven by O’Neil Woods and Seneca Golf Course, and Bole Woods harbored the highest relative abundances of Chryseobacterium and Novosphingobium (Fig. 5).
Mucilaginibacter is the only genus that has been previously suggested as a potential indicator of BLD symptoms. The other genera suggested as indicators of BLD symptoms were either in low relative abundance (i.e., not in the top 15 most abundant genera of the current study and, thus, not included in Fig. 5; Pseudomonas, Erwinia, and Paenibacillus) or not seen in our analysis (Methylocystis and Wolbachia). It should be noted that both Pseudomonas (nine zOTUs) and Erwinia (three zOTUs) were found among the significant indicator taxa of symptomatic tissue (Fig. 6) and had positive LFC values with Wald tests (four significant Pseudomonas zOTUs and one significant Erwinia zOTU; Table S4). Other genera included in the top 15 most abundant were Methylobacterium, Sphingomonas, and Massilia (Fig. 5). All three of these genera matched with zOTUs with significant positive LFC values (14, 34, and 13 zOTUs, respectively; Table S4); however, all were in similar relative abundance between symptomatic and asymptomatic sites (Fig. 5). Overall, our analysis found many significant indicators for symptomatic leaves (637 zOTUs) spanning a large number of taxa (118 different taxa, 57 of which were at the genus level; Fig. 6) and a large number of zOTUs with significantly increased abundance in symptomatic tissue (274 zOTUs with positive LFC with Wald tests; Table S4).
Molecular analysis of fungal communities of leaf tissue
A total of 3,418 fungal zOTUs were detected on the leaves of beech trees across the region. NMDS ordination of the fungal community structure produced a three-dimensional solution with a stress value of 0.12 (Fig. 7). PERMANOVA found significant differences between sites (Table 1, F = 16.3, P value = 0.0002), indicating the presence of regional differences in the fungal microbiome. This was evident by the separation in ordination space between the nine sites (Fig. 7). Nematode presence also significantly affected the fungal communities (Table 1, F = 2.1, P value < 0.02), which could be seen with shifts in ordination space for the four sites where L. crenatae detection varied. This shift in community structure with nematode presence was found even at ANF South that was asymptomatic for BLD at the time of our sampling (Fig. 7). [Note that the sites with varied nematode detection (Fig. 7) differed from those with bacterial community results (Fig. 4) because the one Tinker’s Creek sample with no nematode detection had too few fungal sequences and was, thus removed from the pipeline.] There was no significant interaction between site and nematode presence for fungi. Significant differences in fungal communities were also seen between symptomatic and asymptomatic sites overall (Table 1, F = 12.4, P value = 0.0002), which was largely driven by separation of the asymptomatic sites of ANF South, Tinker’s Creek, and The Wilds. Site, nematode presence, and BLD symptom all showed significant differences with PERMDISP where there was greater variability in the fungal microbiome at asymptomatic sites and in the absence of L. crenatae (Fig. S4). This suggests a potential homogenization of the fungal leaf microbiome with BLD incidence.
Fig 7.

Two-dimensional NMDS plot showing fungal community structure differences between asymptomatic and symptomatic leaf tissue, as well as the presence or absence of L. crenatae (stress = 0.12). Each point is a mean of the axis scores (plus or minus standard error) for the samples that differed in nematode detection at each site. Some sites had differing nematode detection (ANF South, Secor Metropark, Seneca Golf Course, and The Wilds), and some sites had L. crenatae detected in all samples (Bole Woods, Chapin Forest, O’Neil Woods, Tinker’s Creek, and the Wilderness Center).
The fungal genera comprising the leaf microbiome were largely consistent between symptomatic and asymptomatic sites and found in similar relative abundance, on average (Fig. 8; Table S5 and S6). An exception was the genus Erysiphe, which includes the species that causes powdery mildew on plants. This genus, on average, was found in higher relative sequence abundance at symptomatic sites compared with asymptomatic sites (Fig. 8). At the zOTU level, however, not many significant indicators matched with Erysiphe. Only three of the 613 significant indicators for symptomatic tissue were identified as Erysiphe (Table S6). None of these three significant indicators were found ubiquitously in the symptomatic tissue samples (Fig. 9); they were found in 5.5%, 23.3%, and 72.6% of symptomatic samples (Table S6). The zOTU that was found in 72.6% of symptomatic samples was zOTU2, which matched to the Erysiphe genus, but did not have high confidence to any species with SINTAX. This same zOTU was the only significant Erysiphe zOTU with the Wald tests and had a positive LFC value, indicating it had greater abundance at symptomatic sites (Table S7). These results suggest that the high relative abundance of Erysiphe overall was driven by one zOTU.
Fig 8.

Leaf fungal community taxonomic distribution from asymptomatic and symptomatic sites. The bars show the mean relative abundance at each site of the top 15 fungal genera found with Illumina sequence analysis.
Fig 9.

The lowest taxonomic identity for significant fungal indicators of symptomatic leaf tissue. The frequency that the taxa were found in leaf samples at symptomatic or asymptomatic sites is shown. If more than one zOTU had the same lowest taxonomic identity, the frequencies shown here are an average.
A genus that was encountered more frequently in the indicator species analysis and Wald tests and is known to contain a number of pathogenic species was Exobasidium. Nineteen of the 311 significant fungal indicators of asymptomatic leaves and 28 of the 613 significant fungal indicators of symptomatic leaves matched with Exobasidium (Table S5 and S6), as did 43 of the 504 significant Wald Tests zOTUs (Table S7). Exobasidium indicators of symptomatic tissue were more frequently encountered in symptomatic sites (Fig. 9), whereas Exobasidium indicators of asymptomatic tissue were more frequently encountered in asymptomatic sites (Fig. 10), as expected. Similar to the indicator species analysis (ISA) results that found indicators of both symptomatic and asymptomatic tissues, the significant Wald tests included both positive and negative LFC values for Exobasidium zOTUs (Table S7). Furthermore, Exobasidium overall was in similar relative abundance between asymptomatic and symptomatic sites (Fig. 8).
Fig 10.

The lowest taxonomic identity for significant fungal indicators of asymptomatic leaf tissue. The frequency that the taxa were found in leaf samples at asymptomatic or symptomatic sites is shown. If more than one zOTU had the same lowest taxonomic identity, the frequencies shown here are an average.
Other potentially pathogenic genera include Uwebraunia, Taphrina, and Colletotrichum. There was one significant Uwebraunia zOTU and one significant Taphrina zOTU of symptomatic tissue (Table S6); however, these genera were in low abundance overall and not in the top 15 fungal genera (Fig. 8). Collectotrichum also had relatively low sequence abundance compared with the other genera (Fig. 8) and was only found as a significant indicator of asymptomatic leaves (and had only negative LFC values with the Wald tests; Tables S4 and S7). The genus Paraphaeosphaeria was previously linked to BLD symptoms but was not matched with any zOTU in the current study. Similar to the bacterial sequence data, our results overall show a large number of fungal taxa associated with both asymptomatic and symptomatic sites (Tables S5 to S7) that spanned a large number of taxa (Fig. 9 and 10).
DISCUSSION
Beech leaf disease was first noted in Lake County Ohio in 2012, and since its initial detection, it has spread through the northern United States and into the Canadian province of Ontario. It has previously been determined that the anguinid nematode L. crenatae subsp. mccannii is the causative agent of BLD (4, 12); however, the role of other microbial taxa in BLD causation or symptomology cannot be excluded. We previously found evidence that some bacterial genera, notably Mucilaginibacter and Wolbachia, were significantly associated with leaf or bud tissue symptomatic for BLD (5). The goal of the current study was to expand on this and explore whether microbial taxa are consistently associated with BLD symptoms across multiple regions, which would suggest a functional role for those taxa in BLD causation or symptomatology. We observed significant variability in the microbiome of beech leaves impacted by symptoms of beech leaf disease across our study region during the early stages of infestation and before the disease had spread substantially out of Ohio. Our samples were collected in October and November of 2018, and as of November 2019, BLD had been found in just four U.S. states and Ontario (13). Both nematode infestation and fungal and bacterial community structure were associated with leaf symptoms, despite variability between sites. Our results suggest that BLD impacts on the leaf microbiome may be site -specific and determined in part by the level of L. crenatae infestation.
The PCR approach with general nematode primers was able to successfully detect L. crenatae in beech leaf tissue and revealed large differences in both degree of infestation and potential population size between symptomatic and asymptomatic sites. We have noted in previous work that primers TW81 and 5.8SM5 can produce non-specific amplification under some circumstances, such as when infestation of tissue is low (5). All positive PCR amplifications in this study were confirmed to be L. crenatae through direct sequencing; thus, the lack of specificity of these primers under some conditions and their use as a general nematode primer does not impact our results. Given the putative role of L. crenatae as the ultimate cause of BLD (4, 12), it is not surprising that we found much higher nematode gene copy numbers in samples from symptomatic sites. Symptomatic sites typically had gene copy numbers 100–1000× higher than asymptomatic sites, which is consistent with L. crenatae being the causative agent of BLD. However, the presence of L. crenatae at asymptomatic sites suggests that the nematode can occur, at least in low numbers, in the absence of typical BLD symptoms (e.g., dark interveinal banding). Our PCR approach revealed infestation at the asymptomatic locations of Allegheny National Forest (ANF South) and the Wilderness Center, although gene copy numbers at these sites were low (see Fig. 2). Although these sites were asymptomatic at the time the samples were collected for the current study, BLD symptoms were noted in the surrounding areas and within the same county for the Wilderness Center. It is likely that although both sites were asymptomatic, a low level of nematode infestation was present at the sampling time, and our results are picking up that low level of infestation. The number of nematodes needed to induce symptoms of BLD is uncertain and will require additional study. It should be noted that we also found high variability in nematode population size with qPCR. This could be the result of our sampling protocol and the potential spatially patchy occurrence of nematodes on the leaves. One might assume that nematode density is higher where dark banding occurs, and we did not exclusively sample dark -banded areas. Rather we sampled along both sides of the leaf mid-vein, which may or may not have included dark banded areas on symptomatic leaves. Although this could result in a more even perspective of total leaf nematode loads, it would also potentially reflect the high underlying variability in nematode numbers within the leaf. Recent work suggests that nematode -feeding behavior changes leaf morphology and development, but these changes do not occur in the absence of the nematode even within the same leaf (12). Our inclusion of non-banded areas of the leaf in our sampling likely increased the variability in nematode population levels we detected.
We found significant effects of site on leaf bacterial communities as well as a site by nematode interaction. At most sites with different nematode presence, detection of L. crenatae caused a divergence of the bacterial community. This was the case for both symptomatic sites (e.g., Seneca Golf Course) and asymptomatic sites (e,g, ANF), where the samples with nematodes detected separated in ordination space from the samples where nematodes were not detected. When leaf symptoms were examined alone, there was a significant effect on leaf bacterial community structure. Similarly, we found significant site, nematode, and symptom effects on leaf fungal communities; however, we did not see any significant site by nematode interaction, indicating that the nematode effects on fungal communities were not affected by the site itself. This is consistent with the findings of Ewing et al. (11) where significant differences in phyllosphere fungal communities were found with BLD symptoms. The bacterial results in our current study are also in agreement with previous work where we found BLD symptoms in leaves to affect bacterial community structure (5). However, both the Ewing et al. (11) and Burke et al. (5) studies only saw limited effects of BLD symptoms on the phyllosphere microbiome and the taxonomic group affected differed between the two studies (fungi in 11 and bacteria in 5). Both Ewing et al. (11) and Burke et al. (5) were conducted on a limited number of sites (three and one, respectively). At a regional scale, the current study found directional shifts in both leaf bacterial and fungal communities with BLD symptoms and also evidence that the presence of L. crenatae, even in the absence of symptoms, can alter the phyllosphere microbiome.
The reason why BLD symptoms and L. crenatae could affect the phyllosphere community is currently unknown. However, it was recently shown that nematode -feeding behavior within buds increases plant cell division and leads to increased cell layers and hypertrophy of plant cells within the nematode-infested portions of the mature leaves after bud break (12). This hypertrophy of the leaf cells and increased leaf cell layers leads to the interveinal banding that is typical of BLD symptoms (12). It is known that resource availability and host-microbe interactions are strong drivers of survival and community structure for the leaf microbiome (14–16). It seems likely that the altered number of cell layers could directly impact the ability of phyllosphere microbes to acquire carbon and nutrients, as well as to interact with their host; however, additional studies are needed to discern the mechanism for leaf microbiome changes with BLD. One goal of this study was to explore whether phyllosphere microbes were affected by symptoms alone or only by the presence of L. crenatae. Our study suggests that L. crenatae can alter the phyllosphere microbiome in the absence of symptoms. Future studies should explore the relationship between nematode population size and phyllosphere alteration and explicitly examine local leaf “hot spots” of nematode density (e.g., dark banded regions where nematode presence is high) and areas along the same leaf where nematode are in low abundance or absent. This could shed light on the nature of opportunistic infections of bacteria or fungi in the presence of leaf damage by the nematode.
Environmental conditions are also known to be a strong driver of the leaf microbiome (14, 15). In beech trees specifically, it has been found that the location where individual European beech trees (F. sylvatica) were sourced affected the microbes (17). We found strong effects of site on the leaf microbiome in the current study, consistent with this. Given the interactive effects we found for site and nematode presence, it is possible that site conditions, such as underlying nutrient availability, also affect leaf resources and interact with nematode feeding.
Our indicator species analysis found many bacterial and fungal indicators of symptomatic leaves, with the vast majority of taxa that were significant indicators of symptomatic leaves also found on asymptomatic leaves. For bacteria, the genera that were in higher relative abundance at symptomatic sites tended to increase in just one of the four symptomatic sites. This suggests that the changes in the leaf microbiome we observed with BLD are likely the result of site-specific increases in opportunistic bacteria. Potentially, these bacteria are responding to changes in resources on the leaf due to nematode feeding damage or altered physiology (see above). Bacterial genera found in previous studies as indicators of symptomatic leaves that were also found as significant indicators in the current study are Mucilaginibacter (5), Pseudomonas, and Erwinia (11). However, these taxa were generally found in a low percentage of symptomatic leaf samples (less than 25% of symptomatic leaves). This further suggests that these taxa are acting opportunistically in symptomatic leaves and could be the result of changes in leaf damage or physiology by the nematode. If any of these genera were the cause of BLD, they would be found in symptomatic leaf tissue across the region, and this pattern is not evident in the results noted here.
On the contrary, one fungal genus, Erysiphe, was present in higher relative abundance across all symptomatic sites in the current study. Erysiphe is not suspected to be a cause of BLD, as it causes another plant disease, powdery mildew, which can affect beech (18, 19). However, perhaps Erisyphe is acting opportunistically on leaves that are already weakened by BLD. Fungi in the genus Erysiphe can overwinter in buds, and outbreaks of powdery mildew can be severe following insect defoliation events (19). Since there is strong evidence that L. crenatae causes damage within developing bud tissue (12), and the nematode likely overwinters within buds, coincident infection with Erysiphe in autumn could contribute to the development of some of the more advanced symptoms of BLD. However, this relationship will require additional study.
Other fungal taxa that could be acting as opportunistic pathogens or in response to them include the genera Aureobasidium and Taphrina. Both of these genera were significant indicators of symptomatic leaves in the current study and were also found to be indicators of symptomatic leaves in previous work (5). These fungal taxa were listed as among the abundant fungal taxa on European beech (F. sylvatica; 20). Taphrina was the most abundant fungal taxa on European beech leaves (20) and can cause leaf curl disease on Prunus persica (21). Aureobasidium includes the species Aureobasidium pullulans, which is known to be antagonistic to a number of pathogens (22). The genus Mycosphaerella was a significant indicator of asymptomatic leaves in the current study, although it has been shown to cause leaf spot disease on Japanese beech (23). Additional fungal taxa that were found as significant indicators of symptomatic leaves in the current study include the genus Uwebraunia, which causes leaf spots on eucalypts (24), and Colletotrichum, which causes leaf spots on apples (25). Overall, though, these fungal genera were all in low abundance in the current study.
Nonetheless, there is a still a possible role for the phyllosphere microorganisms that grow opportunistically on leaves containing L. crenatae. For example, Methylobacterium is a common resident of the leaf surface where it is known to assimilate methanol, which is a byproduct of pectin demethylation that occurs during cell wall metabolism (26). Increases in the abundance of Methylobacterium with nematode infestation and symptoms of BLD are consistent with the observation that nematode infestation dramatically increases cell division and alters leaf morphology (12), which likely increases methanol on leaf surfaces (26, 27). Such increases in leaf cell division with nematode feeding likely contribute to increases in the abundance of bacteria such as Methylobacterium dependent on the byproduct of cell wall synthesis.
Conclusion
Our regional examination of beech leaf disease microbial communities found significant differences in leaf infestation by L. crenatae subsp. mccannii, with higher levels of infestation at sites with visible signs of beech leaf disease (i.e., dark interveinal banding). We did however find low levels of nematode presence even at asymptomatic sites during sampling in 2018, suggesting that nematodes can be present at low levels prior to manifestation of visible symptoms of disease. Both bacterial and fungal communities were significantly affected by sampling site and symptomology, but only fungal communities were significantly affected by nematode presence alone. Although we found many significant indicators of both bacteria and fungi to symptoms of BLD, taxa generally occurred in both asymptomatic and symptomatic leaves, with indicator status being affected by relative changes in abundance between symptom types and largely driven by increased abundance at one site. This suggests that BLD is unlikely caused by bacterial or fungal pathogens, in accordance with previous studies but could act as opportunistic pathogens on nematode damaged tissue. The fungal genus Erysiphe was of particular interest as a possible opportunistic pathogen as it is commonly found on members of the Fagaceae and has been observed in previous studies to overwinter in buds, a behavioral pattern in common with L. crenatae. What role microbial taxa play in opportunistic infection of leaves affected by L. crenatae is unclear and will require additional study.
MATERIALS AND METHODS
Site description and field sampling
We examined nine sites for the incidence of beech leaf disease and phyllosphere microbial communities within 150 miles of Lake County, Ohio, the original focal point of the North American beech leaf disease outbreak. Our sites included four sites symptomatic for beech leaf disease based on visual assessment of typical characteristics (e.g., dark, interveinal banding; Fig. 1) and five sites that we considered asymptomatic for beech leaf disease. All sites consist of mixed, temperate deciduous forests containing American beech (F. grandifolia) (see Table S1 for detailed information about sites). Leaf and bud samples of F. grandifolia were collected between 22 October 2018 and 8 November 2018 from understory trees where leaf and bud material could be collected from the ground using hand shears. Buds had formed in the leaf axil of green but senescing leaf tissue, and leaf and associated buds were collected from each tree. Shears were sterilized and washed between clippings and four to five leaves, and associated buds were taken from each tree. Leaves and associated buds were packaged separately in plastic bags to eliminate cross-contamination. BLD symptoms are known to vary between leaves of the same individual tree; thus, each leaf and bud was accounted for as its own sample per site. Every leaf was also visually examined in the laboratory by the same two observers to determine whether characteristic banding was present on each leaf and avoid bias in determining the presence or absence of symptoms. The sampling collection totaled 183 leaves and associated buds from 37 trees from a total of nine sites.
DNA extraction
DNA from leaf and bud tissue was extracted using a bead beating, phenol-chloroform extraction protocol. In brief, three 0.5 cm leaf disks were removed from each leaf using a sterilized (autoclaved at 121°C for 30 min) cork corer. Leaf disks (on average 27 mg) were placed in a bead beating tube containing 300 mg of 400 µM sterile glass beads (VWR, West Chester, PA, USA) and 200 mg of 1 mm sterile glass beads (Chemglass, Vineland, NJ, USA). Bud tissue associated with each leaf was excised using a sterile scalpel, divided into 5 mm pieces, and placed within bead beating tubes as described above (on average, 31 mg of bud tissue wereas added to the tubes). Replicate leaf and bud samples were extracted individually. In addition, leaves and buds from the same plant were extracted separately to examine nematode presence in different plant tissues prior to plant autumn senescence. To each bead tube, we added 750 μL of 2% CTAB (cetyltrimethyl-ammonium bromide), and samples were bead beaten using a Precellys homogenizer (Bertin Technologies, France) for 80 seconds to lyse cells and release DNA. DNA was purified using phenol-chloroform extraction (28), followed by precipitation in 20% polyethylene glycol 8000 with 2.5 M NaCl. After precipitation, DNA was dried and suspended in 100 µL TE (Tris EDTA) buffer. DNA was stored in a 1.5 mL low retention microcentrifuge tube (Fisher Scientific) at −20°C until analysis.
Molecular analysis of nematode presence
The presence of L. crenatae was determined through PCR targeting the internal transcribed spacer (ITS1) region of rDNA with the forward primer TW81 (GTTTCCGTAGGTGAACCTGC) and the reverse primer 5.8SM5 (GGCGCAATGTGCATTCGA) (29–31). PCR reactions included 5× GoTaq buffer, 2 mM MgCl2, 0.8 mM deoxyribonucleotide triphosphates (dNTPs), 0.5 µg/µL bovine serum albumin (BSA), 2 units GoTaq DNA polymerase (Promega, Madison, WI, USA), 0.2 µM of forward and reverse primers, and 1 µL of DNA template in 25 µL total volume. PCR was completed on a PTC-2000 thermal cycler (Bio-Rad Laboratories, Inc. Hercules, CA) with the following conditions: an initial denaturation for 2 min at 94°C was followed by 35 cycles of denaturation for 30 seconds at 94°C, annealing for 45 seconds at 55°C, and extension for 3 min at 72°C, with a final extension of 10 min at 72°C (31). A subset of positive PCR reactions from symptomatic sites were sent for direct Sanger sequencing with the Big Dye Terminator Cycle Sequencing kit (Applied Biosystems) through the Biotechnology Resource Center at Cornell University. Similarly, all positive PCR products from asymptomatic sites were sent for direct sequencing to confirm the amplification of L. crenatae DNA.
qPCR estimation of nematode population size
Population size was estimated with qPCR on a subset of the leaf and bud samples. In total, 56 samples were used for qPCR including 38 leaf samples and 18 bud samples across all nine sites. Three technical replicate qPCR reactions were performed for each sample on a CFX Connect Real -Time System (Bio-Rad Laboratories, Inc., Hercules, CA). Each 20 µL qPCR reaction consisted of 1 µL of template DNA, 1X Supermix (Bio-Rad Laboratories, Inc., Hercules, CA), 0.4 µM of each primer (TW81 and 5.8SM5 as described above), and 0.5 mg/mL of bovine serum albumin. The thermal cycling conditions consisted of an initial denaturation of 95°C for 5 min, followed by 40 cycles of 95°C for 30 seconds, 55°C for 60 seconds, and 72°C for 60 seconds with plate reads after every 72°C step. qPCR specificity was determined using melting curves (65°C–95°C with reads every 0.5°C) and by running the amplicons on 2% agarose gels. Gene copy number was estimated by comparing the quantification cycle (Cq) in the samples with a standard curve using Bio-Rad CFX Maestro Software, version 4.1.2433.1219 (Bio-Rad Laboratories, Inc., Hercules, CA). Five-point standard curves (that ranged from 1 × 106 to 1 × 102 copies) were made with a transformed plasmid containing the ITS1 region of L. crenatae isolated from previous research (5) and quantified with a Nanodrop spectrophotometer. The Cq was determined manually for each run, such that the reaction efficiency and the r2 of the standard curve were optimized. The r2 for the standard curves ranged from 0.993 to 0.996, and the efficiencies of the runs ranged from 89.4% to 99.8%. All no-template controls (NTCs) were below the detection threshold. The Cq standard deviations for the technical replicates ranged from 0.021 to 0.75 cycles. Samples that had starting quantities lower than 100 copies or a melting temperature that deviated from the standard curve melting temperature by more than 2° were listed as “not detected.”
Molecular characterization of the leaf microbiome
The leaf microbiome was assessed with highthroughput sequencing on both bacterial and fungal communities. PCR amplification of the bacterial 16S rRNA gene was done with primers 799 f (AACMGGATTAGATACCCKG) and 1115 r (AGGGTTGCGCTCGTTG) (32, 33), and amplification of the fungal ITS2 gene was done with primers 58A2F (ATCGATGAAGAACGCAG) (34) and ITS4 (TCCTCCGCTTATTGATATGC) (35) with added Illumina overhang adapters (36). The bacterial primers used are optimal for plant leaf microbiome work as they avoid the amplification of plant plastids but allow for the amplification of leaf-associated bacteria. PCR was conducted in 25 µL reactions with 1 µL of leaf DNA, 0.2 µM of each primer, 2.0 mM MgCl, 0.2 mM of each dNTP, 0.5 µg/µL bovine serum albumin, and 1.0 unit of Fast Start Taq DNA Polymerase (Sigma Aldrich, Saint Louis, MO, USA) on a S1000 thermal cycler (Bio-Rad Laboratories, Inc. Hercules, CA). Thermocycling conditions involved an initial denaturation step at 95°C for 5 min, followed by 30 cycles for bacteria and 25 cycles for fungi of 95°C for 30 seconds, 60°C for 60 seconds, and 72°C for 60 seconds, and a final 5-min extension at 72°C. PCR products were quantified and sequenced as 2 × 250 bp reads on one flowcell of an Illumina Mi-Seq V3 sequencer (Illumina Inc., San Diego, Ca) at the Case Western Reserve University Genomics Core facility. PCR products with low yield were not included in the sequencing run.
High-throughput sequencing analysis
In total, our sequencing effort yielded 6,837,908 bacterial reads and 9,916,918 fungal reads that were processed in separate pipelines. Samples that contained less than 0.05% of the total number of sequences were not included in the respective pipelines; this removed 11 bacterial samples with less than 3,418 reads and 11 fungal samples with less than 4,958 reads and resulted in 167 leaf samples going through each pipeline. Sequences were processed in USEARCH, version 11.0.667 (37). First, forward and reverse sequence reads were merged with the fastq_mergepairs command, and then, control phiX reads were removed with the filter_phiX command. Reads were trimmed of PCR primers using Cut Adapt (v2.8; 38) where up to 15% mismatches were allowed during primer removal. Short reads (less than 250 bp for bacteria and 300 bp for fungi) with one or more sequence errors were removed with the fastq_filter command. The unoise3 command was used to create error-corrected and chimera-filtered sequence variants (i.e., zero radius OTUs or zOTUs) where zOTUs with fewer than eight sequence reads were removed (per the default settings). The merged reads from each leaf sample with control phiX and primers removed were then mapped to the zOTUs with the otutab command. Taxonomic assignments for the zOTUs were made with the SINTAX algorithm (39) by comparing against the RDP database for bacteria (using the rdp_16 s_v16 fasta file available at the usearch site; 40) and UNITE database for fungi (using a uchime/usearch/utax/sintax reference database downloaded from the UNITE site; the version used was 8.0, release date 2018–11-18; UNITE Community, 2019). For the 16S sequence reads, zOTUs that matched with chloroplasts (six zOTUs) or the domain Eukaryota (one zOTU) were searched for and removed. This resulted in 3,361 bacterial zOTUs and 3,418 fungal zOTUs.
Statistical analyses
All statistical analyses were conducted in R (version 4.2.1; 41) with bacterial and fungal communities analyzed separately and significance determined at α = 0.05. Raw sequence read counts were first normalized to account for differing sequence depth between samples (see 42) with the estimateSizeFactors function in the package DESeq2 (version 1.36.0; 43). Permutational multivariate analysis of variance (PERMANOVA), permutational multivariate analysis of dispersion (PERMDISP), and NMDS were all conducted in the vegan package (version 2.6–4; 44) on Bray-Curtis dissimilarity matrices calculated with the vegdist function and the option “method = bray.” PERMANOVA was conducted with the adonis2 function and used 4999 permutations. Two different PERMANOVA models were run for each community—one that investigated the interactive effects of site and nematode presence and another that compared symptomatic vs. asymptomatic sites. These two separate PERMANOVA models were necessary because sites were either symptomatic or asymptomatic. PERMDISP used the betadisper function followed by a permutation test with the function permutest and 999 permutations to test for multivariate dispersion. Differences in community composition between groups were visualized with NMDS, which used the function metamds. NMDS runs were made four times with dimensions ranging between one and four, and the final solution was selected after examining the stress values of each run and a scree plot. Axis scores of the resulting solution were averaged for each site × nematode presence in the package doBy (version 4.6.19), and NMDS graphs were made in the ggplot2 package (version 3.4.3). To identify any taxa that were associated with symptomatic or asymptomatic tissue, ISA was conducted in the indicspecies package (version 1.7.14; 45) with the multipatt function. The resulting P values were used to calculate q values with the package qvalue (version 2.28.0; 46), as this controls for the false discovery rate in large data sets, such as those generated with Illumina sequencing (47). Any indicators with both a P value and q value at 0.05 or lower were considered significant. To determine how often significant indicators were found in symptomatic or asymptomatic leaf tissue, the frequency that they were detected (i.e., the sequence number was above zero) in each sample was determined with the lapply function in the data.table package (version 1.14.8). Frequencies were averaged across all significant zOTUs that were identified as the same taxa (to the lowest taxonomic identification) with the doBy package and graphed in ggplot2. To further assess taxa associated with symptomatic or asymptomatic leaf tissue, Wald tests were run in the DESeq2 package with the function DESeq. The tests used the alpha = 0.05 option and had asymptomatic set as the baseline in the contrasts, which means that negative values indicate greater abundance in asymptomatic sites, whereas positive values indicate greater abundance in symptomatic sites. The LFC values were modified with the lfcShrink function in DESeq2 to control for variability. This modification used type = apeglm from the apeglm package (v. 1.18.0), as this method has been shown to have less bias than the default (48). LFC values were considered significantly different between symptoms if adjusted P-values, implemented by default in DESeq2 with the Benjamini-Hochberg false discovery rate, were below 0.05. Coupled with ISA and Wald tests, the phyloseq package (version 1.40.0; 49) was used to visualize the relative abundance of the top 15 bacterial and fungal genera and examine differences between symptomatic and asymptomatic sites. For this, the function transform_sample_counts was used to calculate the relative abundance on the raw reads (i.e., no normalization), and the function glom was used to agglomerate the relative abundance at the level of genus for the bacterial and fungal data separately. Bar graphs of the relative abundance were made in ggplot2.
ACKNOWLEDGMENTS
This work was funded by USDA, Forest Service Cooperative Agreement 18-CA-11420004-11, as well as funding from the Corning Institute for Education and Research.
Contributor Information
David J. Burke, Email: dburke@holdenarb.org.
Irina S. Druzhinina, Royal Botanic Gardens, Surrey, United Kingdom
DATA AVAILABILITY
Sequence reads are publicly available at the NCBI Sequence Read Archive under BioProject number PRJNA1066788.
SUPPLEMENTAL MATERIAL
The following material is available online at https://doi.org/10.1128/aem.00142-24.
Figures S1 to S4.
Tables S1 to S7.
ASM does not own the copyrights to Supplemental Material that may be linked to, or accessed through, an article. The authors have granted ASM a non-exclusive, world-wide license to publish the Supplemental Material files. Please contact the corresponding author directly for reuse.
REFERENCES
- 1. Cale JA, McNulty SA, Teale SA, Castello JD. 2013. The impact of beech thickets on northern hardwood forest biodiversity. Biol Invasions 15:699–706. doi: 10.1007/s10530-012-0319-5 [DOI] [Google Scholar]
- 2. Jensen PG, Demers CL, Mcnulty SA, Jakubas WJ, Humphries MM. 2012. Marten and fisher responses to fluctuations in prey populations and mast crops in the northern hardwood forest. J Wildl Manag 76:489–502. doi: 10.1002/jwmg.322 [DOI] [Google Scholar]
- 3. Ewing CJ, Hausman CE, Pogacnik J, Slot J, Bonello P. 2019. Beech leaf disease: an emerging forest epidemic. For Pathol 49:e12488. doi: 10.1111/efp.12488 [DOI] [Google Scholar]
- 4. Carta LK, Handoo ZA, Li S, Kantor M, Bauchan G, McCann D, Gabriel CK, Yu Q, Reed S, Koch J, Martin D, Burke DJ. 2020. Beech leaf disease symptoms caused by newly recognized nematode subspecies Litylenchus crenatae mccannii (Anguinata) described from Fagus grandifolia in North America. For Pathol 50:e12580. doi: 10.1111/efp.12580 [DOI] [Google Scholar]
- 5. Burke DJ, Hoke AJ, Koch J. 2020. The emergence of beech leaf disease in Ohio: probing the plant microbiome in search of the cause. For Pathol 50:e12579. doi: 10.1111/efp.12579 [DOI] [Google Scholar]
- 6. Reed SE, Greifenhagen S, Yu Q, Hoke A, Burke DJ, Carta LK, Handoo ZA, Kantor MR, Koch J. 2020. Foliar nematode, Litylenchus crenatae ssp. mccannii, population dynamics in leaves and buds of beech leaf disease‐affected trees in Canada and the US. For Path 50:e12599. doi: 10.1111/efp.12599 [DOI] [Google Scholar]
- 7. Martin DK, Volk D. 2021. Pest alert: beech leaf disease. R9–PR–001–21. USDA, Eastern Region State and Private Forestry [Google Scholar]
- 8. Reed SE, Volk D, Martin DKH, Hausman CE, Macy T, Tomon T, Cousins S. 2022. The distribution of beech leaf disease and the causal agents of beech bark disease (Cryptoccocus fagisuga, Neonectria faginata, N. ditissima) in forests surrounding Lake Erie and future implications. For Ecol Manage 503:119753. doi: 10.1016/j.foreco.2021.119753 [DOI] [Google Scholar]
- 9. Murfin KE, Dillman AR, Foster JM, Bulgheresi S, Slatko BE, Sternberg PW, Goodrich-Blair H. 2012. Nematode-bacterium symbioses - cooperation and conflict revealed in the 'omics' age. Biol Bull 223:85–102. doi: 10.1086/BBLv223n1p85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Werren JH, Baldo L, Clark ME. 2008. Wolbachia: master manipulators of invertebrate biology. Nat Rev Microbiol 6:741–751. doi: 10.1038/nrmicro1969 [DOI] [PubMed] [Google Scholar]
- 11. Ewing CJ, Slot J, Benítez MS, Rosa C, Malacrinò A, Bennett A, Bonello E. 2021. The foliar microbiome suggests that fungal and bacterial agents may be involved in the beech leaf disease pathosystem. Phytobiomes J 5:335–349. doi: 10.1094/PBIOMES-12-20-0088-R [DOI] [Google Scholar]
- 12. Vieira P, Kantor MR, Jansen A, Handoo ZA, Eisenback J 2. 2023. Cellular insights of beech leaf disease reveal abnormal ectopic cell division of symptomatic interveinal leaf areas. PLoS One 18:e0292588. doi: 10.1371/journal.pone.0292588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Martin DK, Volk D, Macy T, Pogacnik J. 2019. Forest Health pest alert: beech leaf disease. USDA, Eastern Region State and Private Forestry [Google Scholar]
- 14. Leveau JHJ. 2019. A brief from the leaf: latest research to inform our understanding of the phyllosphere microbiome. Curr Opin Microbiol 49:41–49. doi: 10.1016/j.mib.2019.10.002 [DOI] [PubMed] [Google Scholar]
- 15. Chaudhry V, Runge P, Sengupta P, Doehlemann G, Parker JE, Kemen E. 2021. Shaping the leaf microbiota: plant-microbe-microbe interactions. J Exp Bot 72:36–56. doi: 10.1093/jxb/eraa417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schäfer M, Pacheco AR, Künzler R, Bortfeld-Miller M, Field CM, Vayena E, Hatzimanikatis V, Vorholt JA. 2023. Metabolic interaction models recapitulate leaf microbiota ecology. Science 381:eadf5121. doi: 10.1126/science.adf5121 [DOI] [PubMed] [Google Scholar]
- 17. Unterseher M, Siddique AB, Brachmann A, Peršoh D. 2016. Diversity and composition of the leaf mycobiome of beech (Fagus sylvatica) are affected by local habitat conditions and leaf biochemistry. PLoS One 11:e0152878. doi: 10.1371/journal.pone.0152878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Takamatsu S, Sato Y, Mimuro G, Kom-un S. 2003. Erysiphe wadae: a new species of Erysiphe sect. Uncinula on Japanese beech. Mycoscience 44:165–171. doi: 10.1007/S10267-003-0100-9 [DOI] [Google Scholar]
- 19. Marçais B, Desprez-Loustau M-L. 2014. European oak powdery mildew: impact on trees, effects of environmental factors, and potential effects of climate change. Ann For Sci 71:633–642. doi: 10.1007/s13595-012-0252-x [DOI] [Google Scholar]
- 20. Cordier T, Robin C, Capdevielle X, Fabreguettes O, Desprez-Loustau M-L, Vacher C. 2012. The composition of phyllosphere fungal assemblages of European beech (Fagus sylvatica) varies significantly along an elevation gradient. New Phytol 196:510–519. doi: 10.1111/j.1469-8137.2012.04284.x [DOI] [PubMed] [Google Scholar]
- 21. Cissé OH, Almeida J, Fonseca A, Kumar AA, Salojärvi J, Overmyer K, Hauser PM, Pagni M. 2013. Genome sequencing of the plant pathogen Taphrina deformans, the causal agent of peach leaf curl. mBio 4:e00055-13. doi: 10.1128/mBio.00055-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schena L, Sialer MF, Gallitelli D. 2002. Molecular detection of strain L47 of Aureobasidium pullulans, a biocontrol agent of postharvest diseases. Plant Dis 86:54–60. doi: 10.1094/PDIS.2002.86.1.54 [DOI] [PubMed] [Google Scholar]
- 23. Kaneko R, Kakishima M. 2001. Mycosphaerella buna sp. nov. with a Pseudocercospora anamorph isolated from the leaves of Japanese beech. Mycoscience 42:59–66. doi: 10.1007/BF02463976 [DOI] [Google Scholar]
- 24. Aguín O, Sainz MJ, Ares A, Otero L, Pedro Mansilla J. 2013. Incidence, severity and causal fungal species of Mycosphaerella and Teratosphaeria diseases in Eucalyptus stands in Galica (NW Spain). For Ecol Manage 302:379–389. doi: 10.1016/j.foreco.2013.03.021 [DOI] [Google Scholar]
- 25. Moreira RR, Zielinski EC, Castellar C, Bergamin Filho A, May De Mio LL. 2021. Study of infection process of five species of Colletotrichum comparing symptoms of glomerella leaf spot and bitter rot in two apple cultivars. Eur J Plant Pathol 159:37–53. doi: 10.1007/s10658-020-02138-y [DOI] [Google Scholar]
- 26. Rastogi G, Coaker GL, Leveau JHJ. 2013. New insights into the structure and function of phyllosphere microbiota through high-throughput molecular approaches. FEMS Microbiol Lett 348:1–10. doi: 10.1111/1574-6968.12225 [DOI] [PubMed] [Google Scholar]
- 27. Turner TR, James EK, Poole PS. 2013. The plant microbiome. Genome Biol 14:209. doi: 10.1186/gb-2013-14-6-209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Burke DJ, Smemo KA, López-Gutiérrez JC, DeForest JL. 2012. Soil fungi influence the distribution of microbial functional groups that mediate forest greenhouse gas emissions. Soil Biol Biochem 53:112–119. doi: 10.1016/j.soilbio.2012.05.008 [DOI] [Google Scholar]
- 29. Maafi ZT, Subbotin S, Moens M. 2003. Molecular identification of cyst-forming nematodes (Heteroderidae) from Iran and a phylogeny based on the ITS sequences of rDNA. Nematol 5:99–111. doi: 10.1163/156854102765216731 [DOI] [Google Scholar]
- 30. Vovlas N, Subbotin SA, Troccoli A, Liébanas G, Castillo P. 2008. Molecular phylogeny of the genus Rotylenchus (Nematoda, Tylenchida) and description of a new species. Zool Scr 37:521–537. doi: 10.1111/j.1463-6409.2008.00337.x [DOI] [Google Scholar]
- 31. Esmaeili M, Heydari R, Ye W. 2017. Description of a new Anguinid nematode, Nothotylenchus phoenixae n. sp. (Nematoda: Anguinidae) associated with palm date trees and its phylogenetic relations within the family Anguinidae. J Nematol 49:268–275. doi: 10.21307/jofnem-2017-072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chelius MK, Triplett EW. 2001. The diversity of Archaea and bacteria in association with the roots of Zea mays L. Microb Ecol 41:252–263. doi: 10.1007/s002480000087 [DOI] [PubMed] [Google Scholar]
- 33. Redford AJ, Bowers RM, Knight R, Linhart Y, Fierer N. 2010. The ecology of the phyllosphere: geographic and phylogenetic variability in the distribution of bacteria on tree leaves. Environ Microbiol 12:2885–2893. doi: 10.1111/j.1462-2920.2010.02258.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Martin KJ, Rygiewicz PT. 2005. Fungal specific primers developed for analysis of the ITS region of environmental DNA extracts. BMC Microbiol 5:28. doi: 10.1186/1471-2180-5-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gardes M, White TJ, Fortin JA, Bruns TD, Taylor JW. 1991. Identification of indigenous and introduced symbiotic fungi in ectomycorrhizae by amplification of nuclear and mitochondrial ribosomal DNA. Can J Bot 69:180–190. doi: 10.1139/b91-026 [DOI] [Google Scholar]
- 36. Burke DJ, Carrino‐Kyker SR, Burns JH. 2019. Is it climate or chemistry? Soil fungal communities respond to soil nutrients in a multi-year high-resolution analysis. Ecosphere 10:e02896. doi: 10.1002/ecs2.2896 [DOI] [Google Scholar]
- 37. Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. doi: 10.1093/bioinformatics/btq461 [DOI] [PubMed] [Google Scholar]
- 38. Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17:10–12. doi: 10.14806/ej.17.1.200 [DOI] [Google Scholar]
- 39. Edgar RC. 2016. SINTAX: a simple non-Bayesian taxonomy classifier for 16S and ITS sequences. bioRxiv. doi: 10.1101/074161 [DOI]
- 40. Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, Brown CT, Porras-Alfaro A, Kuske CR, Tiedje JM. 2014. Ribosomal database project: data and tools for high throughput rRNA analysis. Nucleic Acids Res 42:D633–D642. doi: 10.1093/nar/gkt1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. R Development Core Team . 2015. R: a language and environment for statistical computing. R foundation for statistical computing, Vienna, Austria. http://www.r-project.org/. [Google Scholar]
- 42. McMurdie PJ, Holmes S. 2014. Waste not, want not: why rarefying microbiome data is inadmissible. PLoS Comput Biol 10:e1003531. doi: 10.1371/journal.pcbi.1003531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. doi: 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’Hara RB, Simpson GL, Sólymos P, Stevens MHH, Wagner H. 2013. Vegan: community ecology package. http://cran.r-project.org/package=vegan.
- 45. De Cáceres M, Legendre P. 2009. Associations between species and groups of sites: indices and statistical inference. Ecology 90:3566–3574. doi: 10.1890/08-1823.1 [DOI] [PubMed] [Google Scholar]
- 46. Storey JD, Bass AJ, Dabney A, Robinson D. 2022. qvalue: Q-value estimation for false discovery rate control. R package version 2.28.0
- 47. Storey JD, Tibshirani R. 2003. Statistical significance for genome wide studies. Proc Natl Acad Sci U S A 100:9440–9445. doi: 10.1073/pnas.1530509100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhu A, Ibrahim JG, Love MI. 2019. Heavy-tailed prior distributions for sequence count data: removing the noise and preserving large differences. Bioinformatics 35:2084–2092. doi: 10.1093/bioinformatics/bty895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. McMurdie PJ, Holmes S. 2013. Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1 to S4.
Tables S1 to S7.
Data Availability Statement
Sequence reads are publicly available at the NCBI Sequence Read Archive under BioProject number PRJNA1066788.