

ABSTRACT
We previously reported the Marimo cell line, which was established from the bone marrow cells of a patient with essential thrombocythemia (ET) at the last stage after transformation to acute myeloid leukemia (AML). This cell line is widely used for the biological analysis of ET because it harbors CALR mutation. However, genetic processes during disease progression in the original patient were not analyzed. We sequentially analyzed the genetic status in the original patient samples during disease progression. The ET clone had already acquired CALR and MPL mutations, and TP53 and NRAS mutations affected the disease progression from ET to AML in this patient. Particularly, the variant allele frequency of the NRAS mutation increased along with the disease progression after transformation, and the NRAS-mutated clone selectively proliferated in vitro, resulting in the establishment of the Marimo cell line. Although CALR and MPL mutations co-existed, MPL was not expressed in Marimo cells or any clinical samples. Furthermore, mitogen-activated protein kinase (MAPK) but not the JAK2-STAT pathway was activated. These results collectively indicate that MAPK activation is mainly associated with the proliferation ability of Marimo cells.
Key Words: essential thrombocythemia, transformation, clonal evolution, CALR, Marimo cell line
INTRODUCTION
Essential thrombocythemia (ET) is a chronic myeloproliferative neoplasm (MPN) characterized by megakaryocyte hyperplasia, thrombocytosis, thrombotic and hemorrhagic complications, and potential transformation into myelofibrosis and acute myeloid leukemia (AML). Most ET patients have a mutation in one of the JAK2, CALR, or MPL gene, but these mutations do not reportedly affect survival of patients with ET.1 It has been demonstrated that several additional genetic alterations, which are involved in epigenetic regulation, cell-growth signaling, and RNA splicing machinery, cooperate with JAK2, CALR, or MPL mutation in the development of secondary AML from ET.2,3 The order of mutation acquisition also influences the clonal evolution of MPNs4; however, the genetic process of leukemic transformation from MPNs varies, and is not fully understood.5
The Marimo cell line was established from bone marrow (BM) cells of a female patient with ET at the last stage after transformation to AML in our laboratory,6 and CALR mutation in Marimo was subsequently demonstrated by another group.7 Fortunately, serial clinical samples of the patient from whom Marimo was established were preserved with informed consent in our laboratory. In this study, we analyzed genetic alterations in the original patient samples at the stages of ET, transformation to AML, and disease progression after intensive chemotherapy for AML, and demonstrated the clonal evolution process from the ET state to establishment of the Marimo cell line.
CASE PRESENTATION
The clinical history of the patient was reported previously6 and is briefly shown in Fig. 1A. The patient was 58 years old at diagnosis and intermittently treated with ranimustine and busulfan to control the platelet count. BM aspiration analysis in the late stage of the ET phase showed an abnormal karyotype, 46,XX,der(15)t(1;15)(q23;p12~13), with few blasts (0.5%, Point 1). Ten years after diagnosis, she developed AML (Point 2) and was treated with daunorubicin and cytarabine. On day 15 after chemotherapy, she received further chemotherapy because of an increase in the peripheral blast count. Although peripheral blasts disappeared after two courses of chemotherapy, severe pancytopenia and BM hypoplasia were prolonged. After three months, the peripheral and BM blast counts increased again (Point 3), and Marimo was established by in vitro culture using BM cells at this point. She received carboplatin, cytarabine, vindesine and cyclophosphamide as a third-line of regimen, and died of renal and respiratory failure on the ninth day of the chemotherapy.
Fig. 1.

Mutated genes and MPL expression in clinical samples and Marimo cells
Fig. 1A: Clinical course of the patient from whom the Marimo cell line was established. Ind#1, daunorubicin, N4-behenoyl-1-beta-D-arabinofuranosylcytosine, mercaptopurine, and prednisolone; Ind#2, mitoxantrone, etoposide, N4-behenoyl-1-beta-Darabinofuranosylcytosine, and prednisolone; Ind#3, carboplatin, cytarabine, vindesine, cyclophosphamide, and prednisolone.
Fig. 1B: Mutations in clinical samples (Point 1 [ET] and Points 2–3 [AML]) and Marimo cells.
Fig. 1C: A model for the development of Marimo cells from ET and AML clones.
Fig. 1D: Western blot analysis of MPL and CALR in AML cells (Point 3), Marimo and the erythroblastic leukemia cell line HEL.
Fig. 1E: RT-PCR analysis of MPL mRNA levels in Marimo, the megakaryoblastic leukemia cell line CMK86, HEL, and acute myeloid leukemia cell line HL60.
AML: acute myeloid leukemia
BM: bone marrow
ET: essential thrombocythemia
Ind: induction therapy
IFN-α: interferon alpha
Plt: platelet
WBC: white blood cell
BM mononuclear cells were obtained at Points 2 and 3 and cryopreserved until use. At Point 1, only high-molecular-weight DNA was preserved. We obtained informed consent from the patient to use sequential samples for banking and molecular analysis, and approval was obtained from the ethics committee of Nagoya University School of Medicine.
Target sequencing of 54 genes, which are frequently identified in the presence of myeloid malignancies, was performed using the TruSight Myeloid Sequencing Panel according to the manufacturer’s instructions (Illumina, San Diego, CA, USA). Sequence variation annotation was performed using known polymorphism databases, followed by mutation characterization, as previously reported.8,9 Each predicted variant sequence was confirmed by Sanger sequencing. In Marimo cells, CALR (c.1099_1159del; p.L367Tfs*43), MPL (c.1514G>A; p.S505N), TP53 (c.404G>A; p.C135Y), and NRAS (c.181C>A; p.Q61K) mutations were identified, and the variant allele frequency (VAF) was 56.0%, 47.7%, 99.7%, and 54.3%, respectively. CALR and MPL mutations were observed throughout the ET (Point 1) and AML (Points 2 and 3) phases with almost the same allele frequencies as Marimo (Fig. 1B, C). TP53 mutation was also identified at a low VAF in the ET phase (Point 1, VAF: 3.0%), and the VAF increased to 92.8% after leukemic transformation (Point 2). Furthermore, NRAS mutation emerged at a low VAF (3.2%) at leukemic transformation (Point 2), and the VAF increased to 25.4% after progression of AML (Point 3) (Fig. 1B, C).
Since MPL (p.S505N) mutation was identified at almost the same VAF as that of CALR (p.L367Tfs*43) mutation throughout the course of the disease, we examined the expression of MPL in clinical samples and Marimo cells. Western blotting was performed for analysis of the protein expression with anti-MPL, anti-JAK2, anti-phospho-STAT5 (Tyr694), anti-phospho-AKT (Ser473), anti-AKT, anti-phospho-p44/42 mitogen-activated protein kinase (MAPK) (Thr202/Tyr204), anti-MAPK, anti-β-actin (Cell Signaling Technology, Beverly, MA, USA), anti-STAT5 (BD Transduction Laboratories, San Jose, CA, USA) and anti-CALR (Merck Millipore Corporation, Billerica, MA, USA) antibodies. The expression level of MPL transcripts was examined by RT-PCR using the following primers: forward primer, 5´-TTCTGGATCCACCAGGCTGT-3´ and reverse primer, 5´-CACAGGGCATGCCTCAGTCT-3´. RNA-seq was also performed on the AML sample (Point 3) and Marimo with a HiSeq2500 sequencing system (Illumina). Sequencing reads were aligned and counted for each gene with GenomonExpression (https://github.com/Genomon-Project/GenomonExpression) for expression analysis. Fusion genes were analyzed with Genomon-fusion. Western blot analysis did not detect MPL protein in any clinical samples or Marimo cells (Fig. 1D). In addition, mRNA of MPL was not detected in clinical samples or Marimo cells by either RT-PCR (Fig. 1E) or RNA-seq. RNA-seq analysis did not identify a chimeric transcript in the clinical sample at Point 3 or Marimo cells.
It has been reported that Marimo is highly dependent on the MAPK signaling pathway, but not JAK/STAT caused by markedly reduced expression levels of JAK2 and STAT1/3/5 proteins.7 Therefore, we examined the expression and activation status of these molecules in clinical samples with or without the inhibitor of these signaling pathways: MEK inhibitor U0126 (Merck Millipore Corporation) or JAK inhibitor ruxolitinib (Selleck Chemicals, Houston, TX, USA). In this experiment, HEL, an erythroblastic leukemia cell line harboring JAK2-V617F mutation which causes constitutive activation of JAK-STAT pathway, and HL60, another AML cell line without JAK2-V617F mutation were used as controls.10,11 The phosphorylation of STAT5 has been reported to be inhibited by a pan JAK inhibitor in HEL.11 Expressions of JAK2 and STAT5 were not detected in clinical AML samples as Marimo cells. AKT was expressed in Marimo cells, but it was not phosphorylated. However, AKT was not expressed in clinical AML samples. MAPK was expressed in clinical samples as well as Marimo cells. Although MAPK was not activated in AML cells at AML transformation (Point 2), it was phosphorylated in AML cells in the chemo-resistant state (Point 3) as Marimo cells. Phosphorylation of MAPK in AML samples was reduced by MEK inhibitor U0126 treatment as Marimo cells (Fig. 2A). Consistent with these results, the MEK inhibitor U0126 inhibited the proliferation of Marimo cells, but the JAK inhibitor ruxolitinib did not (Fig. 2B). Gene set enrichment analysis with Hallmark gene sets revealed that Myc-target genes were significantly enriched in Marimo cells compared with clinical AML cells (Point 3) (Fig. 3A). Individual analysis also showed a significant enrichment in Myc-target genes sets in Marimo cells (Fig. 3B). Myc activation could be involved in the establishment of Marimo cells, which harbors the amplification of the c-Myc gene.6
Fig. 2.

Effect of MEK inhibition on AML and Marimo cells
Fig. 2A: Western blot analysis of MPL, CALR, JAK2, and signaling pathway proteins in AML cells, Marimo, and HEL treated with the MEK inhibitor U0126 (50 μM), JAK inhibitor ruxolitinib (1 μM), or DMSO for four hours.
Fig. 2B: The inhibitory effect of the MEK inhibitor on Marimo cell viability. Cell viability was measured using the MTT assay following the treatment with ruxolitinib (0.2–51.2 μM) or U0126 (0.1–400 μM) for 72 hours.
AML: acute myeloid leukemia
DMSO: dimethyl sulfoxide
MTT: 3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide
Ruxo: ruxolitinib
Fig. 3.
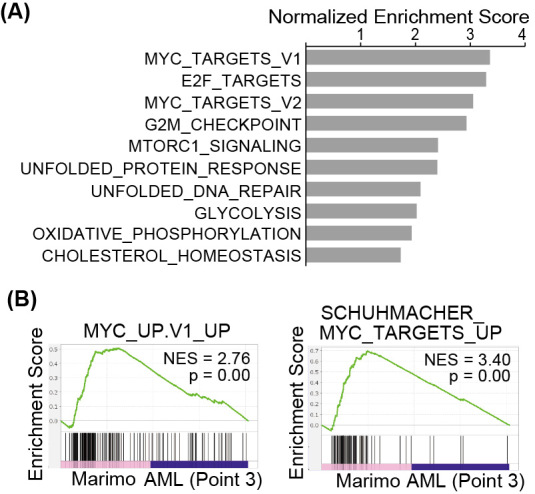
Gene set enrichment analysis in Marimo cells
Fig. 3A: Top-10 gene sets positively enriched in Marimo versus AML cells (Point 3), identified by Gene set enrichment analysis with Hallmark gene sets.
Fig. 3B: Enrichment plots of the indicated signatures.
AML: acute myeloid leukemia
DISCUSSION
Sequential analysis of clinical samples revealed that the ET clone had already acquired CALR and MPL mutations, and TP53 and NRAS mutations were involved during the disease progression from ET to AML in this patient. Particularly, NRAS mutation provided the strong proliferation ability for the transformed AML cells. Therefore, NRAS-mutated clone selectively proliferated in vitro resulting in the establishment of the Marimo cell line. Notably, CALR and MPL mutations co-existed at the same VAFs from the chronic phase of ET in this patient. Although co-existence of these mutations was rarely reported in MPNs, the VAF of the MPL mutation was low in those patients.12-14 It has been reported that mutant CALR constitutively activates MPL, resulting in the activation of JAK2 and its downstream signal-transduction molecules such as STATs, MAPK, and AKT.15,16 However, the JAK2-STAT pathway was not activated in clinical samples or Marimo cells. Therefore, it was an interesting issue how mutant CALR and mutant MPL interacted and were involved in the pathophysiology of Marimo cells. MPL is expressed at various levels in AML cells and cell lines,17,18 whereas we did not detect the expression of MPL and mRNA of MPL in Marimo cells or any of the clinical samples. These results, therefore, suggest that the activation of MPL by mutant CALR could not occur, and the CALR mutation, but not MPL mutation, was the initiating event for ET development in this patient. Furthermore, it has been reported that CALR mutation activates MAPK signaling and is essential for Marimo cells using a novel method (Kinase Inhibitor Screen for Mapping Essential Targets).19 We also confirmed that the MAPK pathway was activated, but not the JAK2-STAT pathway in Marimo cells and the patient’s primary cells at AML transformation. Importantly, the NRAS mutation was not detected in the ET phase, and the VAF increased during disease progression, indicating that the NRAS mutation occurred somatically. These results indicate that MAPK activation was indeed involved in the proliferation ability of Marimo cells, but the activation was not solely dependent on the mutant CALR-mediating signals.
ACKNOWLEDGEMENTS
We would like to thank Ms Satomi Yamaji, Ms Manami Kira, and Ms Yukie Konishi for technical and secretarial assistance.
CONFLICT OF INTEREST
H.K. received research funding from FUJIFILM, Kyowa-Kirin, Bristol-Myers Squibb, Otsuka, Perseus Proteomics, Daiichi Sankyo, Abbvie, CURED, Astellas Pharma, Chugai, Zenyaku Kogyo, Nippon Shinyaku, Eisai, Takeda, Sumitomo Pharma, and Sanofi, and honoraria from Abbvie, Chugai, Astellas Pharma, and Novartis. The remaining authors declare no competing interests.
FUNDING
The present study was supported by Grants-in-Aid from the Practical Research for Innovative Cancer Control from the Japan Agency for Medical Research and Development, AMED (17ck0106251h and 20ck0106535h), the Project for Development of Innovative Research on Cancer Therapeutics (P-DIRECT) from AMED (19cm0106562h and 21cm0106581h), and the Japan Society for the Promotion of Science (JSPS) KAKENHI Grant Number JP 19K08835.
Abbreviations
- AML
acute myeloid leukemia
- BM
bone marrow
- ET
essential thrombocythemia
- MAPK
mitogen-activated protein kinase
- VAF
variant allele frequency
REFERENCES
- 1.Grinfeld J, Nangalia J, Baxter EJ, et al. Classification and Personalized Prognosis in Myeloproliferative Neoplasms. N Engl J Med. 2018;379(15):1416–1430. doi: 10.1056/NEJMoa1716614. [DOI] [PMC free article] [PubMed]
- 2.Rampal R, Ahn J, Abdel-Wahab O, et al. Genomic and functional analysis of leukemic transformation of myeloproliferative neoplasms. Proc Natl Acad Sci U S A. 2014;111(50):E5401–E5410. doi: 10.1073/pnas.1407792111. [DOI] [PMC free article] [PubMed]
- 3.Lasho TL, Mudireddy M, Finke CM, et al. Targeted next-generation sequencing in blast phase myeloproliferative neoplasms. Blood Adv. 2018;2(4):370–380. doi: 10.1182/bloodadvances.2018015875. [DOI] [PMC free article] [PubMed]
- 4.Ortmann CA, Kent DG, Nangalia J, et al. Effect of mutation order on myeloproliferative neoplasms. N Engl J Med. 2015;372(7):601–612. doi: 10.1056/NEJMoa1412098. [DOI] [PMC free article] [PubMed]
- 5.Dunbar AJ, Rampal RK, Levine R. Leukemia secondary to myeloproliferative neoplasms. Blood. 2020;136(1):61–70. doi: 10.1182/blood.2019000943. [DOI] [PMC free article] [PubMed]
- 6.Yoshida H, Kondo M, Ichihashi T, et al. A novel myeloid cell line, Marimo, derived from therapy-related acute myeloid leukemia during treatment of essential thrombocythemia: consistent chromosomal abnormalities and temporary C-MYC gene amplification. Cancer Genet Cytogenet. 1998;100(1):21–24. doi: 10.1016/s0165-4608(97)00017-4. [DOI] [PubMed]
- 7.Kollmann K, Nangalia J, Warsch W, et al. MARIMO cells harbor a CALR mutation but are not dependent on JAK2/STAT5 signaling. Leukemia. 2015;29(2):494–497. doi: 10.1038/leu.2014.285. [DOI] [PMC free article] [PubMed]
- 8.Nishiyama T, Ishikawa Y, Kawashima N, et al. Mutation analysis of therapy-related myeloid neoplasms. Cancer Genet. 2018;222–223:38–45. doi: 10.1016/j.cancergen.2018.02.006. [DOI] [PubMed]
- 9.Kawashima N, Ishikawa Y, Kim JH, et al. Comparison of clonal architecture between primary and immunodeficient mouse-engrafted acute myeloid leukemia cells. Nat Commun. 2022;13(1):1624. doi: 10.1038/s41467-022-29304-6. [DOI] [PMC free article] [PubMed]
- 10.Quentmeier H, MacLeod RA, Zaborski M, Drexler HG. JAK2 V617F tyrosine kinase mutation in cell lines derived from myeloproliferative disorders. Leukemia. 2006;20(3):471–476. doi: 10.1038/sj.leu.2404081. [DOI] [PubMed]
- 11.Walters DK, Goss VL, Stoffregen EP, et al. Phosphoproteomic analysis of AML cell lines identifies leukemic oncogenes. Leuk Res. 2006;30(9):1097–1104. doi: 10.1016/j.leukres.2006.01.001. [DOI] [PubMed]
- 12.Bernal M, Jiménez P, Puerta J, Ruíz-Cabello F, Jurado M. Co-mutated CALR and MPL driver genes in a patient with myeloproliferative neoplasm. Ann Hematol. 2017;96(8):1399–1401. doi: 10.1007/s00277-017-3023-9. [DOI] [PubMed]
- 13.Tashkandi H, Moore EM, Tomlinson B, Goebel T, Sadri N. Co-occurrence of type I CALR and two MPL mutations in patient with primary myelofibrosis. Ann Hematol. 2017;96(8):1417–1418. doi: 10.1007/s00277-017-3022-x. [DOI] [PubMed]
- 14.Partouche N, Conejero C, Barathon Q, et al. Emergence of MPLW515 mutation in a patient with CALR deletion: Evidence of secondary acquisition of MPL mutation in the CALR clone. Hematol Oncol. 2018;36(1):336–339. doi: 10.1002/hon.2431. [DOI] [PubMed]
- 15.Araki M, Yang Y, Masubuchi N, et al. Activation of the thrombopoietin receptor by mutant calreticulin in CALR-mutant myeloproliferative neoplasms. Blood. 2016;127(10):1307–1316. doi: 10.1182/blood-2015-09-671172. [DOI] [PubMed]
- 16.Rauch PJ, Ellgast JM, Widmer CC, et al. MPL expression on AML blasts predicts peripheral blood neutropenia and thrombocytopenia. Blood. 2016;128(18):2253–2257. doi: 10.1182/blood-2016-04-711986. [DOI] [PubMed]
- 17.Uhlén M, Fagerberg L, Hallström BM, et al. Proteomics.Tissue-based map of the human proteome. Science. 2015;347(6220):1260419. doi: 10.1126/science.1260419. [DOI] [PubMed]
- 18.Pronier E, Cifani P, Merlinsky TR, et al. Targeting the CALR interactome in myeloproliferative neoplasms. JCI Insight. 2018;3(22):e122703. doi: 10.1172/jci.insight.122703. [DOI] [PMC free article] [PubMed]
- 19.Kollmann K, Warsch W, Gonzalez-Arias C, et al. A novel signalling screen demonstrates that CALR mutations activate essential MAPK signalling and facilitate megakaryocyte differentiation. Leukemia. 2017;31(4):934–944. doi: 10.1038/leu.2016.280. [DOI] [PMC free article] [PubMed]