

Abstract
The mechanistic basis for the metastasis of Ewing sarcomas remains poorly understood, as these tumors harbor few mutations beyond the chromosomal translocation that initiates the disease. Instead, the epigenome of Ewing sarcoma (EWS) cells reflects the regulatory state of genes associated with the DNA binding activity of the fusion oncoproteins EWSR1::FLI1 or EWSR1::ERG. In this study, we examined the EWSR1::FLI1/ERG’s repression of transcription factor genes, concentrating on those that exhibit a broader range of expression in tumors than in EWS cell lines. Focusing on one of these target genes, ETS1, we detected EWSR1::FLI1 binding and an H3K27me3 repressive mark at this locus. Depletion of EWSR1::FLI1 results in ETS1’s binding of promoter regions, substantially altering the transcriptome of EWS cells, including the upregulation of the gene encoding TENSIN3 (TNS3), a focal adhesion protein. EWS cell lines expressing ETS1 (CRISPRa) exhibited increased TNS3 expression and enhanced movement compared to control cells. Visualization of control EWS cells showed a distributed vinculin signal and a network-like organization of F-actin; in contrast, ETS1-activated EWS cells showed an accumulation of vinculin and F-actin towards the plasma membrane. Interestingly, the phenotype of ETS1-activated EWS cell lines depleted of TNS3 resembled the phenotype of the control cells. Critically, these findings have clinical relevance as TNS3 expression in EWS tumors positively correlates with that of ETS1.
Keywords: EWSR1::FLI1, ETS1, Ewing sarcoma, Transcriptional repression, TENSIN3, TNS3
Introduction
Since its discovery in the 1920s, the treatment of Ewing sarcoma (EWS), an aggressive bone and soft tissue sarcoma affecting children and young adults, has improved dramatically, and the current five-year survival rate for patients with localized disease is between 65 and 75% (1). However, patients with metastatic disease at diagnosis, typically pulmonary and osseous metastases, have five-year survival rates of about 50% and less than 30%, respectively (1). Highlighting the urgent need for the development of treatment strategies targeting EWS metastatic disease, an epidemiological study of over 800 EWS and EWS-like tumor cases identified that 35% of patients had detectable metastatic disease at presentation (2). However, advancing such therapies will require an enhanced understanding of the biological basis for the propensity of primary EWS tumor cells to disseminate and the processes that govern the generation of metastatic lesions.
In most solid malignancies, one determinant of the phenotypic plasticity required for metastasis is the genetic complexity of the primary tumor (3). In contrast, in EWS, a single oncogenic mutation drives tumor development. In most cases of EWS, the primary genetic event involves either a t(11:22) or t(21:22) translocation (4,5). These translocations result in the expression of either the EWSR1::FLI1 or EWSR1::ERG fusion oncoproteins that function as aberrant transcription factors due to the presence of the FLI1- or ERG-DNA binding domains of the ETS family of proteins (reviewed in (6)). Importantly, Ewing sarcomas harbor few additional mutations, and thus, there is a need to determine the alternative mechanisms by which EWS cells attain the phenotypic features required for metastasis.
The EWSR1::FLI1/ERG fusion oncoproteins function as activators and repressors of gene expression (7). Several recent studies have suggested that genes typically repressed by EWSR1::FLI1/ERG fusion proteins may become activated due to variable fusion protein levels or external stimuli and that such changes in gene expression may contribute to the development of metastatic EWS (8–11). Repressed EWSR1::FLI1/ERG gene targets include many encoding proteins involved in cell-cell and cell-extracellular interactions, specifically proteins involved in actin cytoskeletal organization, extracellular matrix (ECM)-receptor interactions, and focal adhesion formation – processes associated with cell migration and invasion (9). Altered expression of repressed EWSR1::FLI1/ERG gene targets because of variable activity of the fusion protein within a tumor, effects of the microenvironment on tumor cell signaling, or other factors could, thus, result in the phenotypic changes associated with processes that promote the dissemination of tumor cells and consequently metastasis (reviewed in (12)).
Current limitations to the study of EWS metastasis include lack of statistically powered clinical datasets that match samples from primary tumor and metastases and the paucity of in-vivo models of EWS metastasis. In this study, we have harnessed alternative resources, specifically tractable EWS cell lines and tumor gene expression profiles, to evaluate the hypothesis that repressed regulators of cell differentiation can contribute to EWS cell dissemination if activated, even transiently, independent of the mechanism by which this occurs. Utilizing epigenomic and transcriptomic approaches, we have identified ETS1 as a repressed gene target of EWSR1::FLI1. Using ectopic expression of ETS1 and CRISPR activation (CRISPRa) of the endogenous gene, we demonstrate that ETS1’s regulation of gene expression is distinct from that of EWSR1::FLI1 and that its expression induces many transcriptomic changes, including the increased expression of the focal adhesion associated protein TENSIN3 (TNS3) and enhanced cell movement and migration. Critically, using multiple datasets, we observe a positive correlation between ETS1 and TNS3 RNA levels in EWS tumor samples, suggesting that EWS tumor cells expressing ETS1 have the potential to exhibit a phenotype that promotes cell movement.
Materials and Methods
Cell lines and reagents.
Supplementary Table S1 details the cell lines, siRNAs and PCR primers, plasmids, and other critical reagents used in this study. Cells were cultured in RPM1–1640 or DMEM media (Thermo Fisher Scientific, Waltham, MA) supplemented with 10% FBS and Plasmocin Prophylactic (Invivogen, San Diego, CA) and grown at 37°C, 5% CO2. We confirmed the identity of cell lines using short tandem repeat (STR) analysis (ATCC; see data availability), and we monitored for mycoplasma contamination using the MycoAlert Plus system (Lonza, Walkersville, MD).
RNAi, qRT-PCR analysis, and RNA sequencing.
Cells were reverse transfected using 20 nM siRNA, and total RNA was isolated 48 hours (hrs) post-transfection and cDNA synthesized as previously described (13–15). Quantitative Real-Time PCR (qRT-PCR) was performed using PowerUp SYBR Green Master Mix (A25778; Thermo Fisher Scientific) on an ABI StepOne Plus Real-Time PCR system (Applied Biosystems, Foster City, CA) or a CFX384 (Bio-Rad, Hercules, CA). Fold-change gene expression was calculated by the ΔΔCT method and normalized to NACA mRNA levels. Samples were generated from three independent experiments unless stated otherwise. For paired-end RNA sequencing (RNA-seq), RNA was extracted (Maxwell 16 LEV simplyRNA purification kit, Promega, Madison, WI), and libraries were prepared and sequenced using standard protocols and a Nextseq 2000 instrument (Illumina, San Diego, CA). The CCR Collaborative Bioinformatics Resource (CCBR) RNA-seq pipeline was used for data analysis https://bioinformatics.ccr.cancer.gov/ccbr/pipelines-software/ccbr-pipeliner/ (see Supplementary Materials and Methods for additional details).
Generation of plasmids and stable cell lines.
To generate the ETS1-pLOC construct, a synthetic human ETS1 cDNA (NM_001143820) with custom flanking restriction enzyme sites (GENEWIZ, South Plainfield, NJ) was cloned into the pLOC vector (OHS5832; Dharmacon/Horizon Discovery, Cambridge, UK). Lentivirus was produced in HEK-293T cells using either the Trans-Lentiviral ORF Packaging Kit (Dharmacon/Horizon Discovery) or Lipofectamine 3000 (Thermo Fisher Scientific). Viral supernatants were collected 72 hrs post-transfection, concentrated (PEG Virus Precipitation Kit, Abcam, Waltham, MA), and added dropwise to cells in the presence of media supplemented with 8 μg/ml polybrene. Transduced cells were selected using puromycin (2 μg/ml, Thermo Fisher Scientific). For the generation of CRISPR lines, cells were transduced with virus expressing dCas9-VP64 (Addgene, Watertown, MA), selected using blasticidin (6 – 10 μg/ml, Thermo Fisher Scientific), and subjected to single-cell sorting. Plasmids expressing ETS1 sgRNAs (see Supplementary Table S1) were co-electroporated into the dCas9-VP64-expressing cells using the Amaxa™ Cell Line Nucleofector™ Kit R (Lonza, VCA-1001) and further subjected to puromycin selection and single-cell cloning. CRISPR-activation efficiency was assessed by qPCR and/or immunoblotting.
Chromatin Immunoprecipitation (ChIP) and CUT&RUN analysis.
ChIP assays were performed using the SimpleChIP plus Enzymatic Chromatin IP kit (9005S; Cell Signaling Technology, Danvers, MA) following the manufacturer’s protocol. For quantitative ChIP-PCR-based analysis, the ChIP-enriched DNA was analyzed using the primers listed in Supplementary Table S1. For ChIP-sequencing (ChIP-seq) and Cleavage Under Targets and Release Using Nuclease (CUT&RUN), two independent replicates of each sample were prepared except for the ChIP-seq analysis of H3K27ac, which was prepared in triplicate. Drosophila and Saccharomyces cerevisiae spike-ins were used to normalize ChIP-seq and CUT&RUN assays, respectively. See Supplementary Materials and Methods for detailed descriptions of the ChIP-seq and CUT&RUN protocols (the latter carried out as previously described (16)) and the analytical pipelines used to examine these datasets (https://bioinformatics.ccr.cancer.gov/ccbr/pipelines-software/ccbr-pipeliner/; https://github.com/CCBR/CARLISLE).
GGAA motif quantification.
Quantification of the GGAA composition of EWSR1::FLI1 and ETS1 peaks was performed as previously described (17) with modifications. Briefly, the genomic sequence within a range of 500 bp around the summit of EWSR1::FLI1 or ETS1 peaks called by MACS2 was extracted (hg38). The number of (GGAA)n or (TTCC)n repeats (from 1–4 or >4 consecutive GGAA motifs without any gap) in each 500 bp range counted.
Immunoblotting and Immunofluorescence.
Whole-cell lysates were prepared using cell extraction buffer (62 mM Tris-HCl, pH 8.0, 2% SDS, 10% Glycerol) and sonicated. Protein concentrations were determined using a BCA assay (Thermo Fisher Scientific). 20 – 30 μg of each protein sample was analyzed following standard immunoblotting protocols (see Supplementary Materials and Methods) using antibodies at the dilutions detailed in Supplementary Table S1, added sequentially. For immunofluorescence (IF) analysis, SK-N-MC and ES-5838 (2 × 105) cells were plated in each well of a 6-well plate on round coverslips and grown for 72 to 96 hrs. Cells grown on the coverslips were processed using standard procedures (see Supplementary Materials and Methods) and analyzed using antibodies at the dilutions detailed in Supplementary Table S1 and the DAPI stain to define nuclei. For confocal microscopy, two independent samples were prepared as described above and imaged using a region of interest (ROI) of 526 × 526 pixels with at least four nuclei in each plane of view. Ten planes of view were imaged for each sample across both replicates. Super-resolution microscopy was performed on independent samples and imaged using an ROI of 2000 × 2000 pixels to attain single-cell resolution.
Cell proliferation and trans-well chemotaxis migration assays.
For proliferation assays, SK-N-MC and ES-5838 (4 × 104) cells per well were plated in a 24-well plate and placed in an IncucyteS3 incubator (Sartorius, Essen BioScience, Inc. Ann Arbor, MI). Nine images per well were taken every 6 hrs for seven days. Images were analyzed using the Incucyte 2020B GUI Analysis Software. For trans-well chemotaxis migration assays, 2.5 ×103 cells/well were plated in FBS-free media in the upper chamber of a 96-well Incucyte ClearView cell migration plate (8 μm pore size; #4582 Sartorius). Cells were incubated at 37°C for 45 minutes to allow cells to settle on the membrane before adding RPMI-1640 media supplemented with 10% FBS to the bottom chamber. The cells were placed in an IncucyteS3 incubator at 37°C with images of top/bottom chambers taken every 6 hrs for at least 72 hrs. To quantify the migratory phenotype of cells we normalized the total cell area (μm2) imaged within the bottom chamber at each time point to that of the cell area imaged within the top chamber of the control dCas9-VP64 modified EWS cells at the initial time point. To assess the chemotaxis migration of siRNA-transfected cells, we transfected 4 ×104 cells using 20 nM siRNA and 0.7 μl RNAiMax per well of a 24 well-format ultra-low attachment plate (Corning 3473) and incubated the cells for 24 hrs. The transfected cells were pelleted at low speed, and 2.5 ×103 cells/well were plated in FBS-free media in the upper chamber of a 96-well Incucyte ClearView cell migration plate, and the migratory phenotype of cells were assessed as described.
Cell motility assay and tracking analysis.
SK-N-MC and ES-5838 (1.8 × 103 cells/well) were plated in a 96-well ImageLock microplate (#4379 Sartorius) and incubated at 37°C overnight to allow cells to settle. Following transfer to an IncucyteS3 incubator (Sartorius), cells were imaged every 2 hrs for 100 hrs. Two phase-contrast images per well were captured at each time point at 20X magnification. Images up to 96 hrs across representative wells were compiled into a stack in Fiji (18). For each stack, an ROI of 800 × 800 pixels was defined, and cells within this ROI were segmented using Otsu thresholds in Fiji. The distance traveled was measured using the TrackMate plugin in Fiji. Only cells with a mean quality >100 across all time points with a minimum duration of ten occurrences in 96 hrs were assessed. Distance traveled per segmented cells over 96 hrs in each representative well was obtained from TrackMate analysis, exported to Prism, and plotted as shown. To assess the range of travel over 96 hrs, we used the minimum and maximum values traveled by cells within a sampled population and determined the range by subtracting these values. Phase contrast movies (two frames per second) were generated using Fiji. Confluency data from all the wells from each sample was analyzed as previously described.
External data sets.
Normalized RNA-seq data (TMM-RPKM - trimmed mean of M values-reads per kb per million mapped reads) for 79 EWS tumors and 42 EWS cell lines were supplied by Dr Javed Khan, Genetics Branch, CCR. These data are deposited in dbGAP submissions phs001928.v1.p1, phs000768, and phs001052 and combined in https://oncogenomics.ccr.cancer.gov/production/public/viewProjectDetails/24421 and described in Brohl et al., 2021 (19). Supplementary Table S1 includes a list of the 42 EWS cell lines within this dataset. The R2 Genomics Analysis and Visualization Platform (http://r2.amc.nl) (20) was used to evaluate deposited EWS tumor expression profiles assessed using microarray-based platforms. We used the data reported from the following cohorts: “Gene Expression Profiling of Ewing Sarcoma Tumors Reveals the Prognostic Importance of Tumor-Stromal Interactions: A Report from the Children’s Oncology Group” GSE63157 (85 tumors) (21); “ Expression profiling of Ewing sarcoma samples” GSE34620 (117 tumors) (22); “Overcoming resistance to conventional drugs in Ewing’s sarcoma and identification of molecular predictors of outcome” GSE12102 (37 tumors) (23); “Expression profiling of Ewing sarcoma samples” GSE142162 79 samples (24). The Kaplan Meier analysis performed using the R2 Genomics employed the median expression value for each gene to separate the data into two groups (high and low expression). A Chi-squared test was used to assess whether either high or low expression is significantly associated with a lower overall survival probability or not.
Statistical and graphical analyses.
Calculations were performed in Excel (Microsoft), and data were exported to Prism 8.0.0 for Mac (GraphPad, La Jolla, CA) for statistical analyses. Unless stated otherwise, results are shown as mean ± standard error of the mean (SEM). A p-value of <0.05 was considered significant, though, for some analyses, more stringent criteria were applied. Correlation analysis and generation of the lollipop plot were performed in RStudio using ggcorrplot and ggplot2 packages. The cor, corr_p.mat, and p.adjust arguments were used to calculate correlation, correlation significance level (p-value), and correlation significance level correction for multiple comparison (adjusted p-value), respectively. Standard R functions and packages (ggplot2, dplyr, ggrepel, and VennDiagram) were used to perform other analyses and to generate plots.
Data availability.
The data and resources generated are available upon reasonable request by contacting the corresponding author. The RNA-seq, ChIP-seq, and CUT&RUN datasets reported here are available in the Gene Expression Omnibus at GSE243184. Images of source immunoblots, STR fingerprinting information for all cell lines, and additional cell images (confocal and super-resolution microscopy) are available at FigShare, DOI:10.6084/m9.figshare.c.7114060.
Results
EWSR1::FLI1 represses the expression of multiple transcriptional factor genes.
To identify transcription factor genes repressed by the EWSR1::FLI1 fusion oncoprotein, we silenced the fusion oncogene in TC-32, TC-71, and A673 EWS cell lines (Supplementary Fig. S1A) and identified the differentially expressed genes (DEGs) (±1.5-fold change; FDR <0.05) (Supplementary Table S2). The DEGs observed in all three EWS cell lines showed enrichment for genes regulating the organization of the extracellular matrix, cell adhesion, and migration as showing an increased expression following EWSR1::FLI1 depletion (1107 genes) and enrichment for genes that function in the cell cycle, showing decreased expression (1111 genes) (Supplementary Fig. S1B, Supplementary Table S2). We next examined a curated list of genes encoding proteins with bona-fide DNA binding activity (25) and determined that 128 of the 2218 genes encode transcription factors (Supplementary Table S3). Of these 128 genes, 61 exhibited decreased expression, including HOXD13, NKX2–2, NR0B1, and SOX2, previously linked to Ewing sarcoma biology (Supplementary Fig. S1C) (26–28). Of the upregulated genes after EWSR1::FLI1 silencing, 67 encoded transcription factors, including established regulators of cell differentiation and developmental processes, such as SNAI2, ETS1, JUNB, and RUNX2 (Fig. 1A).
Figure 1: The EWSR1::FLI1/ERG fusion oncoproteins repress the expression of multiple transcription factor genes.

(A) Differential expression of transcription factor genes that demonstrated a significant increase in expression following the silencing of EWSR1::FLI1 (48 hrs post-siRNA transfection) in TC-32, TC-71, and A673 EWS cells (three biological replicates; fold change >1.5, FDR < 0.05). (B) Comparative expression of selected transcription factor genes in EWS tumor samples (n=79, blue circles) and EWS cell lines (n=42, orange circles) (19) (p-values determined using an unpaired t-test with Welch’s correction). (C) Immunoblots of whole-cell lysates prepared from TC-32 were transfected with the indicated siRNA (48 hrs post-transfection) and analyzed using the antibodies against the indicated proteins. (D, E) EWSR1::FLI1 binding, and H3K9me3, H3K27me3, and H3K27ac CUT&RUN-seq signals were detected in unmodified TC-32 cells and H3K27ac ChIP-seq signals in control and siFLI1-transfected TC-32 cells (48 hrs post-transfection) at (D) the RUNX2 locus and (E) the SNAI2 locus. The black arrows highlight the increase in the H3K27ac marks following the silencing of EWSR1::FLI1. Each track shows read-depth data normalized using the appropriate spike-in controls. (F, G) Kaplan-Meier curves (overall survival probability) of (F) SNAI2 and (G) ETS1 mRNA levels in EWS tumors (n=85) reported previously (21) (microarray-based analysis) and analyzed and plotted using the R2 Genomics Visualization platform (20). Median values of SNAI2 and ETS1 expression defined the classification of low and high gene expression groups.
To assess the potential clinical relevance of EWSR1::FLI1’s regulation of the transcription factor genes that exhibit an increase in expression following the depletion of the fusion oncoprotein, we examined normalized RNA-seq data from EWS tumors (n=79) and cell lines (n=42) (19). The distribution of gene expression in tumors relative to cell lines was higher (p<0.01) for 37 of the 67 transcription factor genes that exhibited an increase in expression following the silencing of EWSR1::FLI1 in EWS cell lines (Fig. 1B, Supplementary Fig. S1D). Based on these results, such genes may exhibit more variable expression in EWS tumors than cell line data suggest. To gain additional evidence that EWSR1::FLI1 represses the expression of specific transcription factors and extend our analysis to the EWSR1::ERG fusion protein, we examined the expression of the nine genes in two additional EWS cell lines, observing a significant increase in the expression of seven in SK-N-MC cells following silencing of EWSR1::FLI1 and five in TC-106 cells post-silencing of EWSR1::ERG, including ETS1 and SNAI2 (Supplementary Fig. S1E). Analysis of the proteins encoded by four of these genes, ETS1, SNAI2, JUNB, and RUNX2, confirmed that depletion of EWSR1::FLI1 in TC-32 and SK-N-MC cells increases their expression (Fig. 1C, Supplementary Fig. S1F). These results emphasize EWSR1::FLI1/ERG’s repression of multiple transcription factors but also suggest that the degree of repression and the resulting phenotypic effects may vary in tumors.
EWSR1::FLI1 represses the expression of ETS1 and other regulators of cell differentiation.
To investigate EWSR1::FLI1’s repression of specific genes, including those encoding transcription factors, we assessed its associations with distinct epigenetic states (Supplementary Table S4). Using samples generated from TC-32 cells, we employed CUT&RUN to determine baseline EWSR1::FLI1 binding sites (two independent experiments, two biological replicates each) indicated as A and B, respectively – Supplementary Figs. S2A, S2B) and mapped histone modifications associated with transcriptional activation, H3K27ac (Supplementary Fig. S2A), and repression, H3K9me3 and H3K27me3 (Supplementary Fig. S2B). Complementing these analyses, we conducted ChIP-seq analyses of H3K27ac in the presence and absence of EWSR1::FLI1 (Supplementary Fig. S2C). The EWSR1::FLI1 CUT&RUN analyses identified over 25,000 binding sites, which overall exhibited enrichment for the ETS consensus sequence motif (Supplementary Fig. S2D) and the occupancy of multimeric GGAA sequences (≥ 4 GGAA repeats) previously reported for the fusion oncoprotein (Supplementary Fig. S2E) (29). Also consistent with published results (7,30), we observed the predominate binding of EWSR1::FLI1 to genomic sites present in distal regions of genes and their H3K27ac peaks (Supplementary Fig. S2F). Examining an exemplar EWSR1::FLI1 repressed gene PHLDA1, we observed evidence of EWSR1::FLI1 binding, most consistently, across replicates at the 3’ of the gene and an increase in gene activation in the absence of EWSR1::FLI1 as indicated by the detection of H3K27ac (Supplementary Fig. S2G). We also examined the repressive marks and observed H3K27me3 across the gene body, suggesting that at this locus, EWSR1::FLI1 binding results in the recruitment of polycomb-repressive marks.
Next, focusing on the >60 transcription factor genes that exhibited upregulated expression following depletion of EWSR1::FLI1 (Fig. 1A), we interrogated the binding of EWSR1::FLI1, H3K9me3, and H3K27me3 modifications at these loci (Supplementary Table S5). Using standard peak calling (MACS2 narrow peaks, p-value <0.01 in both CUT&RUN datasets) and annotation (Homer) tools, 61 loci showed evidence of EWSR1::FLI1 binding in the proximity of the gene body, albeit one of which exhibited a range of expression in the EWS tumor samples when compared to cell lines. Next, we examined the annotations of repressive marks (MACS2 broad peaks, p-value <0.01; Homer), which assigned H3K9me3 at 23 of the 60 transcription factor genes (~38%), H3K27me3 at nine genes (~15%), and both H3K9me3 and H3K27me3 in the proximity of seven genes (~12%). The remaining loci showed no annotated marks. As examples, the RUNX2 locus (Fig. 1D) showed evidence of H3K9me3 modifications in its proximity, and the SNAI2 locus (Fig. 1E) showed evidence of H3K9me3 and H3K27me3 modifications, with examination of the relative location of these marks showing H3K27me3 as the closest peak. At these loci, we observed similar distributions of the H3K27ac signals whether detected by CUT&RUN or ChIP-seq; however, due to the technical differences between these methods, the magnitude of their signals differs. Nevertheless, consistent with our analysis of their gene expression following the depletion of EWSR1::FLI1, we observed an increase in the H3K27ac signals present at each locus following the silencing of EWSR1::FLI1.
To obtain additional evidence that alterations in the transcriptional regulators our studies delineated as repressed by EWSR1::FLI1 in EWS cell lines are relevant to disease, we interrogated one of the few EWS tumor gene expression datasets (21) associated with clinical outcomes using the R2 Genomics Visualization platform (20). Using the R2 platform, we assessed the expression levels available for 57 of the 67 transcription factor genes that exhibit increased expression following silencing of EWSR1::FLI1, comparing the overall survival probability associated with tumors expressing either above or below the median expression levels of a given gene (Supplementary Table S5). This analysis indicated a trend towards greater than the median level of gene expression of 38 genes as associated with a poorer overall survival probability, including SNAI2 (Fig. 1F), with this comparison reaching statistical significance for two genes, CREB3L2 and ETS1 (Fig. 1G). CREB3L2 is a member of the CREB3 family of transcription factors that regulates multiple process, including, the unfolded protein response (31), while ETS1 is the founding member of the ETS family of transcription factors. The trend towards higher ETS1 expression and poorer EWS overall survival, as well as detection of its expression in some EWS tumors is intriguing, because during embryonic development, ETS1 regulates the movement of several early progenitor sub-types including endothelial and neural crest cells, suggesting that the expression of ETS1 in an EWS cell it could induce a transcriptional program favoring cell motility and migration (32–37). We thus selected to examine the regulation of ETS1 expression by EWSR1::FLI1 in further detail.
ETS1 is located adjacent to FLI1 on chromosome 11, and the alignment of RNA-seq data from EWSR1::FLI1 expressing EWS cell lines illustrates a striking juxtaposition of the effects of silencing the fusion oncogene and the expression of ETS1. As expected, we detect a decrease in the reads mapping to the 3’ FLI1 exons following the silencing of EWSR1::FLI1, whereas there is an increase in ETS1 expression (Fig. 2A). Using TC-32-derived samples, we observed EWSR1::FLI1’s binding of multiple sites within ETS1 (Fig. 2B), an observation we confirmed and extended to SK-N-MC-derived samples using ChIP-qPCR (Fig. 2C, Supplementary Fig. S2H). Analysis of the H3K27me3 and H3K9me3 marks showed a distribution of H3K27me3 at a region adjacent to ETS1 but not the latter mark (Fig. 2B). H3K27me3 ChIP-qPCR validated this finding and showed evidence for the same mark upstream of the ETS1 locus in SK-N-MC cells (Fig. 2D, Supplementary Fig. S2I). Furthermore, we detected an increase in the H3K4me3 mark (Supplementary Table S6, Supplementary Fig. S2J) at the ETS1 TSS and H3K27ac following silencing of the fusion oncogene (Fig. 2E), confirming transcriptional activity at this locus in the absence of EWSR1::FLI1. Based on these results, we hypothesized that variabilities in EWSR1::FLI1 transcriptional activity may explain, at least in some cases, the range of ETS1 mRNA levels observed in EWS tumors (Fig. 1B). To investigate the consequences of ETS1 expression in EWS cells, we next assessed its binding in EWS cells depleted of EWSR1::FLI1.
Figure 2: ETS1 is a repressed gene target of EWSR1::FLI1.

(A) RNA-seq read depth of control and EWSR1::FLI1-silenced TC-32, A673, and TC-71 cells (48 hrs post-transfection) focusing on the ETS1 and FLI1 loci. For clarity, different scales show the read depth at the ETS1 locus in TC-71 cells. The black arrows indicate the changes in read depth at the ETS1 locus and the 3’ FLI1 exons present in the fusion transcript (B) EWSR1::FLI1 binding, H3K27me3, and H3K9me3 modifications at the ETS1 and FLI1 loci of TC-32 cells. (C) ChIP-qPCR analysis (mean ± SEM of three replicates) of EWSR1::FLI1 binding at the indicated locations of the ETS1 locus in TC-32 and SK-N-MC cells. See Supplementary Fig. S2H for sequence information (D) ChIP-qPCR analysis (mean ± SEM of three replicates) of H3K27me3 deposition at the indicated locations of the ETS1 locus in TC-32 and SK-N-MC cells. See Supplementary Fig. S2I for sequence information. (E) The ETS1 locus showing H3K4me3 and H3K27ac modifications detected in control (siNeg) and siFLI1-transfected TC-32 cells (48 hrs post-transfection). The black arrows highlight the increase in the H3K4me3 and H3K27ac signals following the silencing of EWSR1::FLI1. (B, E) Each track shows read-depth data normalized using the appropriate spike-in controls.
ETS1’s regulation of gene expression is distinct from that of EWSR1::FLI1.
Following the depletion of EWSR1::FLI1, ChIP-seq analysis defined 2635 ETS1 binding sites in TC-32 cells (Fig. 3A, Supplementary Table S7). In contrast to EWSR1::FLI1, global ETS1 chromatin occupancy in EWSR1::FLI1-silenced cells predominantly mapped to promoter regions (~84%) (Fig. 3B). While ETS1 bound regions displayed enrichment for the canonical ETS DNA binding motifs (Fig. 3C), ETS’s binding at GGAA motifs was predominately at single and interspersed GGAAs (Fig. 3D). This is in contrast with EWSR1::FLI1’s binding at GGAA microsatellites and its aberrant function (7,38,39), and suggests that ETS1 genomic occupancy is distinct from that of EWSR1::FLI1.
Figure 3: ETS1 DNA binding is distinct from that of EWSR1::FLI1.
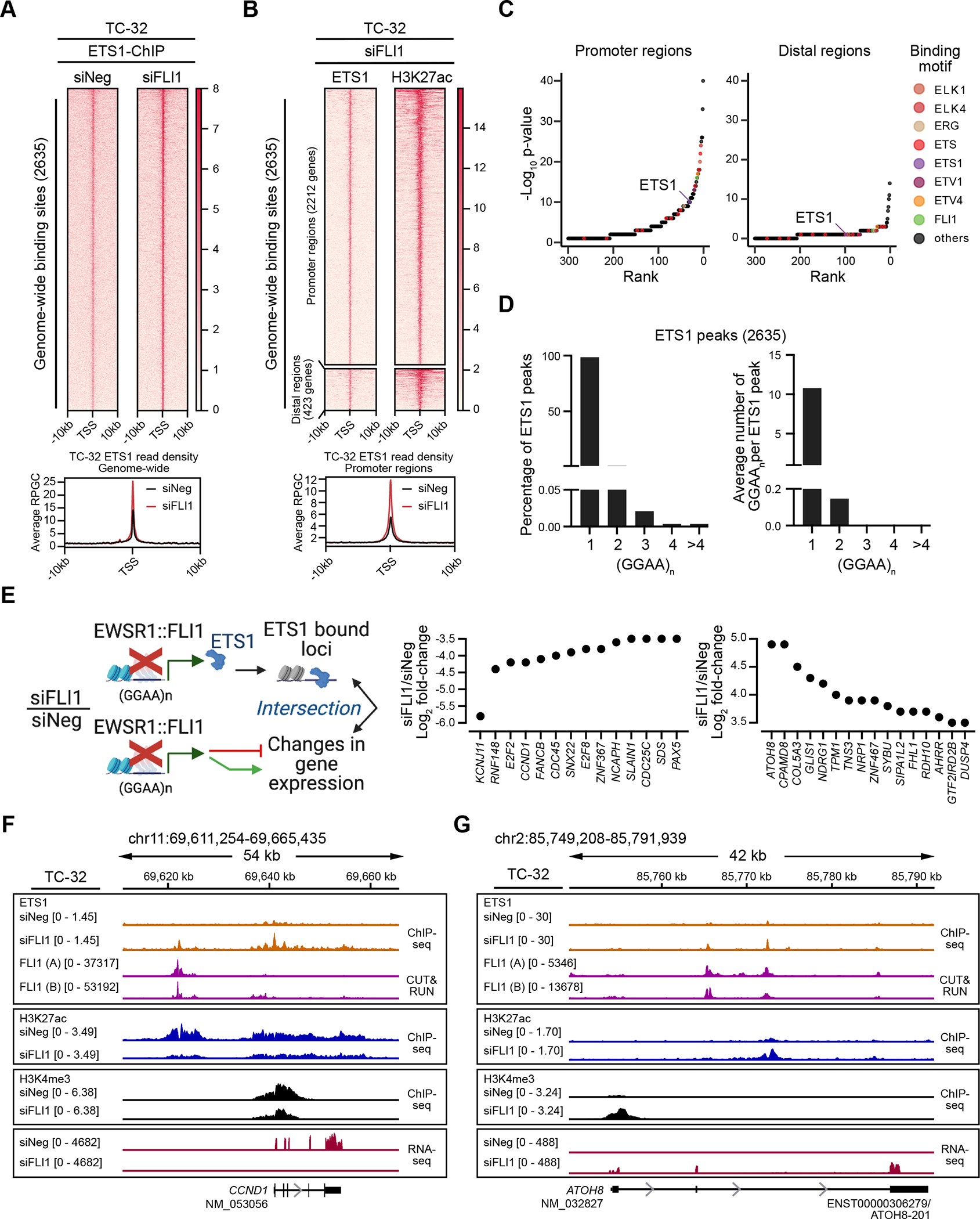
(A) Genome-wide heatmaps of ETS1 ChIP-seq peak-centered signals and read densities in control (siNeg) and EWSR1::FLI1-silenced (siFLI1) TC-32 cells (48 hrs post-transfection) showing 10kb windows (two biological replicates). Rankings of the bound regions are based on the siFLI1 signal. (B) Cluster heatmaps of ChIP-seq peak-centered signals for ETS1 and H3K27ac in TC-32 siFLI1-transfected cells (48 hrs post-transfection) showing 10kb windows. Rankings of the cluster regions are based on the ETS1 signals, with the gene promoter regions defined as −3kb and +1kb from TSS and the distal regions as sites existing outside promoter regions. Below the heatmap is a summary of the ETS1 read densities at promoter regions in control (siNeg) and EWSR1::FLI1-silenced (siFLI1) TC-32 cells. (C) Sequence motifs enriched in ETS1/H3K27ac promoter and ETS1/H3K27ac distal regions as defined in (B). (D) The percentage of peaks with at least one (GGAA)n (left) and the average number of (GGAA)n per peak (right) in ETS1 binding sites based on consecutive GGAA repeat sequence motifs of 1, 2, 3, 4, or over 4. (E) Schematic illustrating the intersectional analysis performed (left) and the changes in expression of ETS1-bound genes that are protein-encoding and exhibit changes in expression following depletion of EWSR1::FLI1 (greater or less than ±3.5 Log2-fold) (right). (F) The CCND1 locus of TC-32 cells assayed under the indicated conditions showing ETS1 and EWSR1::FLI1 binding, H3K9me3, H3K27me3, H3K27ac, and H3K4me3 modifications, and gene expression. (G) The ATOH8 locus of TC-32 cells assayed under the indicated conditions showing ETS1 and EWSR1::FLI1 binding, H3K9me3, H3K27me3, H3K27ac, and H3K4me3 modifications, and gene expression. The schematic in E was generated using BioRender. (F, G) Each of the ChIP-seq and CUT&RUN tracks shows read-depth data normalized using the appropriate spike-in controls except the ETS1-ChIP seq tracks, which were scaled by library size.
To identify putative downstream targets regulated by ETS1 in the absence of EWSR1::FLI1, we intersected ETS1 occupancy and significantly deregulated genes from the RNA-Seq analysis of EWSR1::FLI1-silenced TC-32 cells (Fig. 1 and Supplementary Fig. 1). This analysis defined 958 genes as bound by ETS1, including the genes shown in Fig. 3E, which illustrates ETS1’s association with genes that exhibit decreased or increased expression following the silencing of EWSR1::FLI1. As an example of ETS1’s association with an activated EWSR1::FLI1 gene target, we examined its binding at the CCND1 locus (Fig. 3F). Many studies have noted the high expression of CCND1, a critical regulator of the cell cycle, in EWS cells and its down-regulation following depletion of EWSR1::FLI1 (40–42). Our analysis confirmed the decrease in CCND1 RNA levels in EWSR1::FLI1-silenced TC-32 cells and a reduction in the H3K4me3 and H3K27ac marks at this locus. Interestingly, under control conditions, we detected EWSR1::FLI1 binding upstream of CCND1 with no evidence of ETS1 binding. However, following the depletion of EWSR1::FLI1, we observed a less pronounced ETS1 peak in the same vicinity as EWSR1::FLI1 and a prominent ETS1 peak at the CCND1 promoter. ETS1, like other ETS proteins, can repress and activate gene expression, leading us to speculate that at the CCND1 locus, ETS1 may function as a transcriptional repressor or a weaker activator of CCND1 in the absence of EWSR1::FLI1. However, further studies will need to assess this suggestion. In contrast, ETS1 may promote the expression of ATOH8 (Fig. 3G), which of those genes bound by ETS1 exhibited the greatest increase in expression following the silencing of EWSR1::FLI1 (Fig. 3E). ATOH8 encodes a transcription factor recently implicated in the regulation of chondrogenic differentiation (43), cellular plasticity (44), and the differentiation and maintenance of skeletal muscle (45). Gene expression analysis and H3K4me3 and H3K27ac epigenetic marks all indicate that EWSR1::FLI1 suppresses the expression of ATOH8 as each mark increases following depletion of the fusion oncoprotein. Consistent with ETS1 contributing to promoting ATOH8 expression, we observed ETS1 binding at two regions within this locus. However, in this case, these peaks coincided with EWSR1::FLI1 peaks, suggesting that ETS1 and EWSR1::FLI1 proteins may compete for binding at some sites.
ETS1 regulates gene targets repressed by EWSR1::FLI1.
Our previous experiment interrogated ETS1 function in the absence of EWSR1::FLI1, but to better recapitulate the scenario in tumors expressing both ETS1 and EWSR1::FLI1, we ectopically expressed ETS1 in TC-32 cells. We generated two single-cell clones, one that expressed ETS1 at levels comparable to EWSR1::FLI1-silenced cells (TC-32-ETS1-High) and one expressing lower levels of ETS1 (TC-32-ETS1-Low) (Fig. 4A, Supplementary Fig. S3A), neither of which exhibited changes in EWSR1::FLI1 expression (Supplementary Fig. S3B). Transcriptomic analysis of the TC-32-ETS1-High and ETS1-Low cells versus control cells identified over 5000 genes as exhibiting significantly altered expression in both clones. (Supplementary Fig. S3C, Supplementary Table S8). We identified a common set of 1886 downregulated and 1874 upregulated genes in TC-32-ETS1-High and ETS1-Low cells (Supplementary Fig. S3D; Supplementary Table S8); however, because the ETS1 protein levels in the TC-32 ETS1-High cells recapitulated most closely those seen upon EWSR1::FLI1 depletion (Fig. 4A), we continued our analysis using results generated from this clone, beginning with the identification of genes bound and regulated by ETS1. Intersecting the TC-32 (siFLI1/siNeg) ETS1 ChIP-seq and the ETS1-High mRNA expression datasets, we defined 522 genes as direct targets of ETS1 (Supplementary Table S8), including those detailed in Fig. 4B, and which included ATOH8 (Supplementary Fig. S3E). Interestingly, of the 265 genes positively regulated by ETS1, 103 exhibited an increase in expression following depletion of EWSR1::FLI1 in TC-32 cells (Fig. 4C; Supplementary Table S8), suggesting that ETS1 could activate the expression of these genes in tumors if expressed.
Figure 4: ETS1-regulated genes in EWS cell lines and tumors.

(A) Immunoblot-based assessment of ETS1 following its ectopic expression (two independent single-cell clones) or upon the depletion of EWSR1::FLI1 in TC-32 cells (48 hrs post-siRNA transfection). (B) Model of the intersection of the transcriptome-wide results of ectopic ETS1 expression in TC-32 cells and the detection of ETS1-binding following the depletion of EWSR1::FLI1 in TC-32 cells (left) and results of this analysis focusing on protein-encoding genes that exhibited ±2-fold-change in expression (FDR <0.05) or higher following the ectopic expression of ETS1 (right). (C) A model depicting the analysis of the expression of ETS1-positively regulated genes and genes that exhibit an increase in expression following the depletion of EWSR1::FLI1 (48 hrs post-siRNA transfection) and the results of this analysis highlighting selected genes. (D) A model depicting the correlation analysis of ETS1 expression and the expression of ETS1-positively regulated genes in EWS tumors and the results of this analysis using the gene expression profiles of 79 EWS tumors (19) focused on the most significantly correlated genes (FDR <0.01). (E) Correlation of the expression of ETS1 and COL5A3 in EWS tumor samples (n=79) (19). (F) The COL5A3 locus of TC-32 cells assayed. under the indicated conditions showing H3K27ac and H3K4me3 modifications, mRNA, H3K9me3 and H3K27me3 modifications, and ETS1 and EWSR1::FLI1 binding. The black box indicates ETS1’s binding at the 5’ end of the COL5A3 locus. Each of the ChIP-seq and CUT&RUN tracks shows read-depth data normalized using the appropriate spike-in controls except the ETS1-ChIP seq tracks, which were scaled by library size. (G) ChIP-qPCR analysis (mean ± SEM of three replicates) of ETS1 binding at the 5’ end of the COL5A3 locus in TC-32 cells. Schematics in B, C, and D were generated using BioRender.
Next, we considered the hypothesis that ETS1 expression and the expression of some of the ETS1-regulated genes our cell line studies defined should correlate in EWS tumor samples. Focusing on the 265 genes positively regulated by ETS1, we assessed the correlation of their mRNA levels with that of ETS1 using the normalized RNA-seq data from 79 EWS tumors (19) (Fig. 4D). We observed highly significant correlation between the expression of ETS1 and 36 of these genes, including several involved in cell differentiation or cell-cell or cell-matrix interactions, such as COL5A3, which encodes one of the type V collagens that forms a component of the ECM (46). The positive correlation between ETS1 and COL5A3 expression reflects that 16 of the 20 tumors expressing the highest levels of ETS1 (top quartile) also express median or higher levels of COL5A3 (Fig. 4E). Interestingly, three published gene expression datasets (microarray) also show positive correlations of ETS1 and COL5A3 expression in EWS tumor samples (R>0.7; Supplementary Fig. S3F). Examining the transcription factor binding and epigenetic marks at the COL5A3 locus and its expression, we observed changes in the H3K27ac and H3K4me3 marks present at the 5’ end of the locus and an increase in its mRNA levels, which the ectopic expression of ETS1 recapitulated. Under unperturbed conditions, analysis of the annotated repressive marks revealed H3K9me3 deposition in the vicinity of COL5A3. Examining ETS1’s putative regulation of COL5A3 expression, we noted two ETS1 bound sites in the proximity of COL5A3, one downstream of its 3’ end that overlaps with an EWSR1::FLI1 binding site and one close to the TSS (Fig. 4F), which we confirmed as bound by ETS1 using ChIP-qPCR (Fig. 4G). These findings are consistent with our hypothesis that ETS1’s transcriptional activity can promote the expression of genes categorized as repressed by EWSR1::FLI1 and that the activation of one or more of these genes has the potential to induce phenotypic changes.
ETS1-mediated activation promotes EWS cell migration.
To investigate the phenotypic consequence of increased ETS1 expression in EWS cells, we activated endogenous ETS1 expression in SK-N-MC (EWSR1::FLI1) and ES-5838 (EWSR1::ERG) cell lines using the dCas9-VP64 CRISPRa modification and three gRNA targeting ETS1. We chose to employ dCas9-VP64 transcriptional activation to increase expression of the endogenous gene and we used the SK-N-MC and ES-5838 cell lines to extend our analysis to additional EWS cells. Of note, the SK-N-MC and ES-5838 cell lines originated from tumor cells found at metastatic sites (SK-N-MC—retro-orbital and ES-5838—pleural effusion). However, SK-N-MC and ES-5838 cells express comparable levels of their respective fusion oncoproteins as EWS cell lines derived at other stages of disease and sites (Supplementary Fig. S4A) and express little (SK-N-MC) to no (ES-5838) detectable ETS1 (Supplementary Figs. S4B, S4C). Following confirmation of ETS1 activation in SK-N-MC and ES-5838 cells (Supplementary Figs. S4B, S4C) and determination that the expression of their respective EWSR1-fusion oncoproteins remained unaffected (Supplementary Fig. S4D), we analyzed their growth and migratory phenotypes. Cell proliferation analysis revealed that endogenous activation of ETS1 did not significantly increase cell growth compared to control cells (unmodified and dCas9-VP64) (Supplementary Fig. S4E); however, trans-well chemotaxis assays revealed a significant increase in the migratory phenotype of the ETS1-CRISPR-activated EWS cells compared to control cells (Fig. 5A). To further assess the effect of activating ETS1 on the phenotype of SK-N-MC and ES-5838 cells, we next used longitudinal imaging and cell tracking analysis to capture the attachment and movement of cells over 96 hrs. We confirmed minimal differences in the proliferative rate of the control and ETS1-activated cell lines (Supplementary Fig. S4F), but cell tracking detected evidence of differences in their movement (Fig. 5B – 5D, Videos 1 – 4). Specifically, we observed some cells within the population of the ETS1-activated cell lines showed a greater range of movement over a defined area than control cells (Figs. 5C, 5D), suggesting that ETS1 has the potential to enhance the movement of EWS cells.
Figure 5: ETS1 promotes the movement of EWS cells.

(A) Trans-well chemotaxis migration assays showing the total cell area (μm2) present in the bottom chamber normalized to the total cell area present in the top chamber of the dCas9-VP64 modified EWS cells at the initial time point. (Controls: unmodified and dCas9-VP64 – SK-N-MC 16 replicates each; ES-5838, 32 replicates and CRISPRa-ETS1 EWS cells – SK-N-MC, 24 replicates each, ES-5838, 32 replicates; data plotted as mean ± SEM). The results shown represent three independent experiments, and the p-value reports student t-tests performed at each time point. (B) Images of the movement of the indicated cells over 96 hrs. See also Videos 1 – 4. (C) The quantification of cell movement plotted as total distance traveled over 96 hrs (eight ROIs). The first data set in each graph corresponds to the analysis of the ROIs shown in Fig. 5B, and the Y axis indicates the number of cells tracked per ROI. The horizontal bars indicate the median value for each group of tracked cells. (D) The range of cell movement was determined for eight ROIs per the indicated cell line shown in Fig. 5C. P-values were determined using an unpaired t-test with Welch’s correction. (B-D) Cell movement was determined using images taken every 2 hrs and generated by assessing nuclei (segmented using Otsu thresholds in Fiji) within an ROI of 800 × 800 pixels (one ROI per eight independent wells per cell line) (TrackMate, Fiji plug-in). Assessed nuclei had a mean quality of greater than 100 across all time points with a minimum duration of ten occurrences in 96 hrs as determined by the TrackMate, Fiji plug-in. (E) Representative single channel and merged images of vinculin (green) and DAPI (blue) in the indicated cells. Arrows indicate characteristics related to the descriptive statements below these images. (F) Representative single channel and merged images of phalloidin for F-actin (green) and DAPI (blue) in the indicated cells. Arrows indicate characteristics related to the descriptive statements included below the zoomed inset. (E, F) Cell images, scale bar = 5 μm, zoomed inset, scale bar = 1 μm. Images shown represent 30 images across two independent replicates.
Cell movement involves the formation of focal adhesions and the reorganization of the cytoskeleton. Focal adhesions consist of macromolecular protein complexes that regulate cellular responses to cues from the ECM by controlling mechanical force and downstream signaling cascades that alter the organization of vinculin and F-actin, among other proteins. Using IF and super-resolution microscopy, we compared the distribution of vinculin, a marker of focal adhesions, and phalloidin (47,48), a phallotoxin that is selective for F-actin, a marker of cytoskeletal organization, in the EWS control cells (unmodified and dCas9-VP64) and the cells in which we had activated ETS1 and observed several differences (Figs. 5E, 5F; Supplementary Figs. S4G, S4H). Specifically, in control SK-N-MC and ES-5838 cells, we observed low vinculin IF signals distributed throughout cytoplasm (Fig. 5E, Supplementary Fig. S4G), but following the activation of ETS1, we observed enhanced vinculin IF signals that accumulated towards the plasma membrane (Fig. 5E). Consistent with alterations in the distribution of vinculin, we also observed changes in the organization of F-actin. Specifically, under control conditions, F-actin in SK-N-MC and ES-5838 cells exhibited a network-like organization (Fig. 5F, Supplementary Fig. S4H), but a redistribution of F-actin towards the plasma membrane following the activation of ETS1 (Fig. 5F). These findings suggest ETS1 regulates the expression of one or more proteins that function in mediating the changes in the configuration of the cytoskeleton and potentially cell movement.
ETS1 regulates the expression of the focal adhesion protein TENSIN3.
Examination of the functions associated with the positively ETS1-regulated genes (Fig. 4B), whose expression in tumors also correlated with ETS1 (Fig. 4D), led us to next consider TNS3 as a possible candidate for contributing to the phenotype of the ETS1-activated EWS cells compared with control cells. TNS3 encodes TENSIN3, a focal adhesion protein recently defined as a regulator of oligodendrocyte differentiation during murine development and the osteogenic differentiation of bone marrow stromal cells (49,50). Not previously studied in EWS, we noted that 18 of 20 EWS tumors (RNA-seq) expressing the highest levels of ETS1 (top quartile) also express median or higher levels of TNS3 (Fig. 6A), and multiple EWS tumor gene expression profiles (microarray-based), show that the expression of ETS1 and TNS3 positively correlate (Fig. 6B, Supplementary Fig. S4I). Also, high TNS3 expression shows a trend towards an association with a reduced overall survival probability (Fig. 6C). Under unperturbed conditions, in TC-32 cells, we observed two H3K9me3 peaks in proximity to the 5’ end of TNS3, but minimal H3K27me3, H3K4me3, and H3K27ac signals, and no mRNA expression (Fig. 6D; upper tracks). Consistent with EWSR1::FLI1’s mediation of this repressive state, we detected EWSR1::FLI1 binding at multiple sites on the TNS3 gene body (Fig. 6D; middle purple tracks). In contrast, following the depletion of EWSR1::FLI1, we observed an increase in the TNS3 mRNA levels, which ETS1 ectopic expression, in the presence of EWSR1::FLI1, recapitulated (Fig. 6D; middle red tracks). Furthermore, in the absence of EWSR1::FLI1, we observed an increase in the H3K4me3 mark at the TSS of TNS3, a concomitant increase in H3K27ac across the locus, and ETS1 binding at a site between exons 1 and 2 of TNS3 (Fig. 6D; lower orange tracks, indicated by a black box). We also confirmed TNS3’s upregulation in the CRISPRa-ETS1 EWS cell lines using qRT-PCR (Fig. 6E), immunoblotting (Fig. 6F), and IF (Fig. 6G and Supplementary Figs. S4J, S4K).
Figure 6: ETS1 regulates the expression of TENSIN3.

(A) The expression of ETS1 and TNS3 in EWS tumor samples (n=79). (B) The expression of ETS1 and TNS3 mRNA levels (microarray-based analysis) in EWS tumors (n=85) reported previously (21), analyzed and plotted using the R2 Genomics Visualization platform (20). (C) Kaplan-Meier curves (overall survival probability) of TNS3 mRNA levels (microarray-based analysis) in EWS tumors (n=85) reported previously (21) and analyzed and plotted using the R2 Genomics Visualization platform (20). The median expression of TNS3 was used to define low and high gene expression groups. (D) The TNS3 locus of TC-32 cells assayed under control conditions showing H3K9me3, H3K27me3, H3K4me3, and H3K27ac modifications, and mRNA expression and EWSR1::FLI1 binding; H3K4me3 and H3K27ac modifications following the silencing of EWSR1::FLI1; TNS3 mRNA levels following the depletion of EWSR1::FLI1 or the ectopic expression of ETS1; and ETS1 binding in the presence or absence of EWSR1::FLI1. The black box highlights ETS1 binding at the TNS3 locus. Each of the ChIP-seq and CUT&RUN tracks shows read-depth data normalized using the appropriate spike-in controls except the ETS1-ChIP seq tracks, which were scaled by library size. (E) The expression of TNS3 assessed by qRT-PCR analysis (mean ± SEM of three replicates) in the indicated unmodified and modified EWS cell lines. Results are shown as. (F) Immunoblots assessing TNS3 protein expression following the activation of ETS1 in SK-N-MC and ES-5838 cells. (G) Confocal images showing the single-channel and merged TNS3 (magenta, IF) and DAPI stain (blue) signals observed in unmodified and modified SK-N-MC or ES-58368 cells as indicated. Scale bar = 25 μm. The images represent 30 images across two independent replicates.
TNS3 contributes to the transduction of signals between the extracellular environment and the cytoskeleton by binding focal adhesion complex components, including vinculin, via another cytoskeletal protein, TALIN (51). We thus hypothesized that ETS1’s upregulation of TNS3 contributes to the cytoskeletal reorganization observed in the CRISPRa-ETS1 EWS cells compared to control cells. To evaluate this hypothesis, we used confocal and super-resolution microscopy to visualize TNS3 and vinculin in the CRISPRa-ETS1 EWS cells to image multiple and single cells, respectively (Supplementary Fig. S5 and Figs. 7A, 7B). The confocal images of multiple control SK-N-MC and ES-5838 cells (Supplementary Fig. S5A and S5B, respectively) showed minimal expression of TNS3 and low vinculin IF signals in all cells, but following the activation of ETS1, most cells exhibit increased TNS3 expression and vinculin accumulation at the plasma membrane. Super-resolution microscopy of single cells confirmed the overlap of the TNS3 and vinculin signals at specific regions of the plasma membrane, particularly membrane projections in the CRISPRa-ETS1 EWS cells (Figs. 7A, 7B). We also examined TNS3’s localization in the context of F-actin organization using confocal microscopy to capture images of multiple cells (Supplementary Figs. S6A, S6B) and super-resolution microscopy of single cell (Supplementary Figs. S6C, S6D). As before, we observed that F-actin has a network-like organization in the control cells, which distributes toward the plasma membrane upon activation of ETS1. We also observed evidence of TNS3 in the proximity of F-actin, but in this case, we saw minimal overlaps in their signals, suggesting that if TNS3 contributes to cytoskeletal reorganization it is via its adhesion-associated functions (51). To assess this hypothesis, we validated two siRNAs targeting TNS3 (Supplementary Fig. S7A) and employed one (siTNS3.2) to evaluate if silencing of TNS3 in the CRISPRa-ETS1 cells alters their phenotype (Figs. 7C, 7D; Supplementary Figs. S7B, S7C). We observed that a reduction in TNS3 expression in both the CRISPRa-ETS1 cell lines resulted in less regional accumulation of vinculin at the plasma membrane and a reversal of the F-actin phenotype from one of accumulation at the plasma membrane to a more network-like organization. Critically, we observed a partial rescue of the migratory phenotype, with no changes in the proliferative rate, upon TNS3 depletion relative to control in ES-5838 cells (Fig. 7E). These observations indicate that ETS1-expressing EWS tumor cells have the potential to exhibit TNS3-dependent changes in their cytoskeleton that are consistent with the promotion of cell movement (Fig. 7F), a critical aspect of the metastatic process.
Figure 7: EWS cells expressing ETS1 exhibit TENSIN3-dependent changes in cytoskeletal organization.

(A) Super-resolution single channel and merged images of TNS3 (magenta, IF), vinculin (green, IF), and DAPI (blue) in the indicated modified SK-N-MC cells, including the indicated inset. See Supplementary Figure S5A for complementary multi-cell images. (B) Super-resolution single channel and merged images of TNS3 (magenta, IF), vinculin (green, IF), and DAPI (blue) in the indicated modified ES-5838 cells, including the indicated inset. See Supplementary Figure S5B for complementary multi-cell images. (C) Super-resolution single channel and merged images of TNS3 single channel and merged images (including the indicated inset) of TNS3 (magenta, IF), vinculin (green, IF), and DAPI (blue) in ES-5838-CRISPRa-ETS1 cells transfected with either a control siRNA (siNeg) (upper images) or a siRNA targeting TNS3 (lower images) (48 hrs post-transfection). (D) Super-resolution single channel and merged images (including the indicated inset) of TNS3 (magenta, IF), phalloidin for F-actin (green, IF), and DAPI (blue) in the ES-5838-CRISPRa-ETS1 cells transfected with control siRNA (siNeg) (upper images) or a siRNA targeting TNS3 (lower images). Images represent 30 images across three independent replicates. Scale bar = 5 μm, insets, scale bar = 1 μm. (E) Cell confluency and trans-well chemotaxis migration assays of the indicated modified ES-5838 EWS cell lines transfected with the indicated siRNAs. Cell confluency - each data point indicates the mean ± SEM of 12 wells; cell migration - each data point indicates the mean ± SEM of 48 wells. The p-value reports student t-tests performed at each time point. (F) A model of ETS’s regulation of TNS3 and the contribution of TNS3 to the cytoskeletal phenotype and movement of EWS cells. Schematic generated using BioRender.
Discussion
Many studies have interrogated the epigenetic mechanisms and genes that the EWSR1::FLI1/ERG fusion proteins activate as these are critical to the initiation and promotion of tumorigenesis (reviewed in (6,52), but less characterized is the effect of the altered expression of genes associated with the repressive function of these fusion oncoproteins. However, a convergence of recent reports has demonstrated that the epigenomes of EWS cells in vivo are more heterogeneous and responsive to external cues than earlier studies had suggested (10,28,53–56). These findings have highlighted the importance of investigating the genes categorized as repressed by EWSR1::FLI1/ERG because of their potential to contribute to processes linked to metastasis if expressed. Here, focusing on transcription factor genes repressed by EWSR1::FLI1 (Fig. 1), we demonstrated the fusion protein’s repression of the ETS transcription factor, ETS1, via the deposition of the polycomb repressive complex-2 (PRC2)-associated H3K27me3 histone modification (Fig. 2). Interestingly, while ETS1 and EWSR1::FLI1 both bind the same ETS DNA consensus sequence, we showed that ETS1 and EWSR1::FLI1 occupy distinct regions on chromatin, with the former predominantly binding within promoter regions, and the latter, distal regions (Fig. 3). We also demonstrated that ETS1 could function as a transcriptional regulator even in the presence of EWSR1::FLI1, altering the expression of hundreds of genes, including the genes encoding the fibrillar collagen COL5A3 and the focal adhesion protein TNS3 (Fig. 4), suggesting that ETS1 could exert substantial phenotypic effects if expressed in tumor cells. Consistent with this concept, we observed that the activation of endogenous ETS1 in two EWS cell lines promotes alterations in their movement (Fig. 5) and TNS3-dependent reorganization of the cytoskeleton (Figs. 6 and 7). Importantly, interrogation of multiple tumor gene expression profiles showed evidence for elevated ETS1 mRNA levels in a subset of Ewing sarcomas and the positive correlation of ETS1 and TNS3 expression. Furthermore, a dataset that enables analysis of gene expression and clinical outcomes indicated that higher ETS1 expression is associated with a trend towards poorer overall survival probability (Fig. 1G), as is the expression of TNS3. (Fig. 6C).
ETS1 is a critical regulator of normal embryonic development (57,58). However, it exhibits an entirely different expression pattern in adult tissues, specifically immune cell sub-types, including B cells, T cells, and NK cells (59–62). In the developing embryo, ETS1 contributes to the regulation of the mobility and invasive properties of several progenitor cell types, including endothelial cells that contribute to angiogenesis (32,33,63) and the diverse cell types that arise from neural crest cells (34–37,64–66). Interestingly, neural crest cells are among the few cells that tolerate EWSR1::FLI1 expression (67). Studies of mice, chick, and Xenopus embryos have noted Ets1 expression in pre-migratory and migrating neural crest and neural crest-derived cells, including cardiac and cranial neural crest cells (36,37,64–66). The expression of Ets1 in cardiac neural crest cells appears particularly important as the analysis of Ets1−/− mice on some genetic backgrounds demonstrates nearly complete perinatal lethality due to the failure of cardiac neural crest cells to migrate appropriately, resulting in heart malformation (36,66). In chick embryos, studies have also defined ETS1’s contributions to the gene regulatory network that defines migratory cranial neural crest cells, which give rise to cartilage and bone, regulating the expression of at least four markers of this cell type, RXRG, LTK, COL9A3, LMO4 (65). Interestingly, we noted ETS1 binding at the LMO4 and COL9A3 loci following the silencing of EWSR1::FLI1 and their upregulation following the ectopic expression of ETS1 in TC-32 EWS cells (Supplementary Table S8). Considering the invasive and migratory characteristics of the progenitor cells that express ETS1, we hypothesized that its expression in an EWS cell could promote the movement of such a cell away from the site of primary tumor growth. Supporting this hypothesis, we observed that the ETS1-CRISPRa EWS cell lines exhibited enhanced cell movement, migration, and cytoskeletal reorganization compared to control cells (Fig. 5).
The cytoskeletal reorganization that regulates cell movement requires the function of many proteins, including members of the TENSIN protein family (TNS1/2/3), which function as focal adhesion adaptor proteins (reviewed in (68–70). In this study, we noted in the ETS1-activated EWS cells the accumulation of vinculin towards the plasma membrane (Figs. 5E and 7A, 7B) and overlaps in the vinculin and TNS3 IF signals at membrane projections not seen in control cells (Figs. 7A, 7B, Supplementary Figs. S5). Examination of the distribution of F-actin showed a network-like organization in control cells that showed greater presence at the leading edge of cells following activation of TNS3 (Supplementary Fig. S6). Both the change in the distribution in vinculin and F-actin are consistent with the enhanced formation of focal adhesions and cell movement. Critically, the depletion of TNS3 reversed these changes in the phenotype of ETS1-activated cells to that of cells expressing little or no ETS1 (Figs. 7C, 7D, Supplementary Fig. S7).
However, we note that one limitation of this study was our inability to quantify these changes in signal distribution and future studies will need to confirm these findings using additional markers of focal adhesions and the cytoskeleton.
We present several lines of evidence that ETS1 regulates the expression of TNS3 and that this finding has clinical relevance. First, we observed that ETS1 binds the TNS3 promoter (Fig. 6D) and second, that the ectopic expression of ETS1 and activation of endogenous ETS1 expression increases TNS3 expression (Figs. 4B, 6E, 6F). Importantly, interrogation of tumor gene expression profiles from five independent datasets showed a positive correlation of ETS1 and TNS3 expression (Figs. 6A and 6B, and Supplementary Fig. S4I) and association of high TNS3 expression with a poorer overall survival probability (Fig. 6C). Building on these observations, critical next steps will include further assessment of phenotypic consequences of TNS3 expression in EWS cells and determination of whether elevated expression of ETS1, TNS3, or both contribute to the risk of metastasis, including the performance of in vivo xenograft studies. It will also be critical to demonstrate that the expression of ETS1 and TNS3 detected in bulk RNA-seq data derives predominantly from tumor cells rather than other cell types. The increasing availability of single cell RNA sequencing data from tumor samples, including Ewing sarcomas (53,55) associated with clinical outcomes, should soon facilitate such analysis.
Future studies will also need to investigate if the expression of ETS1 and the other transcription factor genes data suggests some EWS tumor cells express at higher levels than EWS cell lines is a result of heterogenous EWSR1::FLI1/ERG expression and/or due to the transduction of external stimuli. The mechanisms by which EWSR1::FLI1 represses gene expression in cell lines and to what extent this recapitulates such processes in tumors are less well-defined than its recruitment of factors that activate gene expression, but previous studies have shown that in contrast to gene activation, the repression of genes by EWSR1::FLI1 occurs at non-repeat canonical ETS binding sites (7). Here, we identified H3K27me3 and/or H3K9me3 marks adjacent to several transcription factor genes repressed by EWSR1::FLI1/ERG (Supplementary Table S4), though not all, suggesting that EWSR1-fusion proteins can mediate gene repression through additional mechanisms. H3K27me3 repression is facultative and allows repressed genes to be accessible by transcription factor binding in a different cellular state, including during development (reviewed in (71,72). Interestingly, we observed H3K27me3 marks adjacent to ETS1 and SNAI2, both of which exhibited increased expression following depletion of EWSR1::FLI1 (Fig. 1E), confirming that at these loci, fusion oncoprotein activity does not mediate the formation of an irreversible heterochromatic state. It is noteworthy that a recent comprehensive study of 18 EWS cell lines that examined their expression profiles using high-density microarrays under control conditions and following the shRNA-mediated targeting of EWSR1::FLI1 or EWSR1::ERG also reported the upregulation of SNAI2 in 16 of 18 cell lines (73). However, a further comparison of our observations and the datasets reported by Orth and coworkers detected only slight increases in ETS1 expression in A673 and TC-32 cells and no change in ETS1 expression in TC-71, SK-N-MC, and TC-106 cells, among others. These differences may reflect the relative sensitivity of the assays used; nevertheless, we have reproducibly detected changes in ETS1 expression by RNA-seq and/or qPCR and protein analysis following siRNA-mediated targeting of EWSR1-FLI1/ERG fusion transcripts in multiple EWS cell lines (Fig. 1) and found evidence that at least some EWS tumors express ETS1. Critically, we show that even in the presence of EWSR1::FLI1, the expression of ETS1 could have profound effects on the phenotype of EWS cells, including promoting cell movement, an essential aspect of the metastatic process.
Supplementary Material
Implications:
ETS1’s transcriptional regulation of the gene encoding the focal adhesion protein TENSIN3 in Ewing sarcoma cells promotes cell movement, a critical step in the evolution of metastasis.
Acknowledgments
The Intramural Research Program of the National Cancer Institute (NCI), Center for Cancer Research (CCR), National Institutes of Health supported this study: ZIA BC 011704 supports N.J.C; ZIC BC 010858 supports the CCR Confocal Microscopy Core Facility; and the generation of CRISPR-Cas9 reagents included the support of Federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261201500003I. This project was funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, Department of Health and Human Services, under Contract No. 75N91019D00024. This work utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov). We thank Raj Chari and the Genome Modification Core, Laboratory Animal Sciences Program at the Frederick National Lab for Cancer Research for assistance with the generation of CRISPR-Cas9 reagents, and the CCR Flow Cytometry and CCR Sequencing Cores for technical assistance, and the CCR Microscopy Core, particularly Michael Kruhlak and Andy Tran for technical support and advice. We thank Javed Khan, Jun S. Wei, and Young Song for access to Ewing sarcoma RNA-seq data (Oncogenomics Section, Genetics Branch). We thank Carla Neckles and Alison Cross, former members of the Functional Genetics Section, Genetics Branch, for early discussion and technical assistance related to this work, and Sanjit Mukherjee, Bob Walker, and Fan Yang (Molecular Genetics Section, Genetics Branch) for advice and guidance. We also thank Christine Heske, Pediatric Oncology Branch, CCR, for useful discussion.
The content of this publication does not necessarily reflect the views or policies of the U.S. Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Financial support:
Funding from the Intramural Research Program of the Center for Cancer Research, NCI, NIH supported N.J. Caplen (ZIA BC 011704). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Conflict of Interest: The authors declare no potential conflicts of interest.
References
- 1.Gaspar N, Hawkins DS, Dirksen U, Lewis IJ, Ferrari S, Le Deley MC, et al. Ewing Sarcoma: Current Management and Future Approaches Through Collaboration. J Clin Oncol 2015;33:3036–46 [DOI] [PubMed] [Google Scholar]
- 2.Ramkumar DB, Ramkumar N, Miller BJ, Henderson ER. Risk factors for detectable metastatic disease at presentation in Ewing sarcoma - An analysis of the SEER registry. Cancer Epidemiol 2018;57:134–9 [DOI] [PubMed] [Google Scholar]
- 3.Rogiers A, Lobon I, Spain L, Turajlic S. The Genetic Evolution of Metastasis. Cancer Res 2022;82:1849–57 [DOI] [PubMed] [Google Scholar]
- 4.Delattre O, Zucman J, Plougastel B, Desmaze C, Melot T, Peter M, et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature 1992;359:162–5 [DOI] [PubMed] [Google Scholar]
- 5.Sorensen PH, Lessnick SL, Lopez-Terrada D, Liu XF, Triche TJ, Denny CT. A second Ewing’s sarcoma translocation, t(21;22), fuses the EWS gene to another ETS-family transcription factor, ERG. Nat Genet 1994;6:146–51 [DOI] [PubMed] [Google Scholar]
- 6.Grunewald TGP, Cidre-Aranaz F, Surdez D, Tomazou EM, de Alava E, Kovar H, et al. Ewing sarcoma. Nat Rev Dis Primers 2018;4:5. [DOI] [PubMed] [Google Scholar]
- 7.Riggi N, Knoechel B, Gillespie SM, Rheinbay E, Boulay G, Suva ML, et al. EWS-FLI1 utilizes divergent chromatin remodeling mechanisms to directly activate or repress enhancer elements in Ewing sarcoma. Cancer Cell 2014;26:668–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chaturvedi A, Hoffman LM, Welm AL, Lessnick SL, Beckerle MC. The EWS/FLI Oncogene Drives Changes in Cellular Morphology, Adhesion, and Migration in Ewing Sarcoma. Genes Cancer 2012;3:102–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chaturvedi A, Hoffman LM, Jensen CC, Lin YC, Grossmann AH, Randall RL, et al. Molecular dissection of the mechanism by which EWS/FLI expression compromises actin cytoskeletal integrity and cell adhesion in Ewing sarcoma. Mol Biol Cell 2014;25:2695–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Franzetti GA, Laud-Duval K, van der Ent W, Brisac A, Irondelle M, Aubert S, et al. Cell-to-cell heterogeneity of EWSR1-FLI1 activity determines proliferation/migration choices in Ewing sarcoma cells. Oncogene 2017;36:3505–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keskin T, Rucci B, Cornaz-Buros S, Martin P, Fusco C, Broye L, et al. A live single-cell reporter assay links intratumor heterogeneity to metastatic proclivity in Ewing sarcoma. Sci Adv 2021;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Apfelbaum AA, Wrenn ED, Lawlor ER. The importance of fusion protein activity in Ewing sarcoma and the cell intrinsic and extrinsic factors that regulate it: A review. Front Oncol 2022;12:1044707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grohar PJ, Kim S, Rangel Rivera GO, Sen N, Haddock S, Harlow ML, et al. Functional Genomic Screening Reveals Splicing of the EWS-FLI1 Fusion Transcript as a Vulnerability in Ewing Sarcoma. Cell Rep 2016;14:598–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neckles C, Boer RE, Aboreden N, Cross AM, Walker RL, Kim BH, et al. HNRNPH1-dependent splicing of a fusion oncogene reveals a targetable RNA G-quadruplex interaction. RNA 2019;25:1731–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vo T, Brownmiller T, Hall K, Jones TL, Choudhari S, Grammatikakis I, et al. HNRNPH1 destabilizes the G-quadruplex structures formed by G-rich RNA sequences that regulate the alternative splicing of an oncogenic fusion transcript. Nucleic Acids Res 2022;50:6474–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu DY, Ellegast JM, Ross KN, Malone CF, Lin S, Mabe NW, et al. The ETS transcription factor ETV6 constrains the transcriptional activity of EWS-FLI to promote Ewing sarcoma. Nat Cell Biol 2023;25:285–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao Y, He XY, Wu XS, Huang YH, Toneyan S, Ha T, et al. ETV6 dependency in Ewing sarcoma by antagonism of EWS-FLI1-mediated enhancer activation. Nat Cell Biol 2023;25:298–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 2012;9:676–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brohl AS, Sindiri S, Wei JS, Milewski D, Chou HC, Song YK, et al. Immuno-transcriptomic profiling of extracranial pediatric solid malignancies. Cell Rep 2021;37:110047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.R2: Genomics Analysis and Visualization Platform <http://r2.amc.nl>.
- 21.Volchenboum SL, Andrade J, Huang L, Barkauskas DA, Krailo M, Womer RB, et al. Gene Expression Profiling of Ewing Sarcoma Tumors Reveals the Prognostic Importance of Tumor-Stromal Interactions: A Report from the Children’s Oncology Group. J Pathol Clin Res 2015;1:83–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Postel-Vinay S, Veron AS, Tirode F, Pierron G, Reynaud S, Kovar H, et al. Common variants near TARDBP and EGR2 are associated with susceptibility to Ewing sarcoma. Nat Genet 2012;44:323–7 [DOI] [PubMed] [Google Scholar]
- 23.Scotlandi K, Remondini D, Castellani G, Manara MC, Nardi F, Cantiani L, et al. Overcoming resistance to conventional drugs in Ewing sarcoma and identification of molecular predictors of outcome. J Clin Oncol 2009;27:2209–16 [DOI] [PubMed] [Google Scholar]
- 24.Surdez D, Zaidi S, Grossetete S, Laud-Duval K, Ferre AS, Mous L, et al. STAG2 mutations alter CTCF-anchored loop extrusion, reduce cis-regulatory interactions and EWSR1-FLI1 activity in Ewing sarcoma. Cancer Cell 2021;39:810–26 e9 [DOI] [PubMed] [Google Scholar]
- 25.Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, et al. The Human Transcription Factors. Cell 2018;175:598–9 [DOI] [PubMed] [Google Scholar]
- 26.Kinsey M, Smith R, Iyer AK, McCabe ER, Lessnick SL. EWS/FLI and its downstream target NR0B1 interact directly to modulate transcription and oncogenesis in Ewing’s sarcoma. Cancer Res 2009;69:9047–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi X, Zheng Y, Jiang L, Zhou B, Yang W, Li L, et al. EWS-FLI1 regulates and cooperates with core regulatory circuitry in Ewing sarcoma. Nucleic Acids Res 2020;48:11434–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Apfelbaum AA, Wu F, Hawkins AG, Magnuson B, Jimenez JA, Taylor SD, et al. EWS::FLI1 and HOXD13 Control Tumor Cell Plasticity in Ewing Sarcoma. Clin Cancer Res 2022;28:4466–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gangwal K, Sankar S, Hollenhorst PC, Kinsey M, Haroldsen SC, Shah AA, et al. Microsatellites as EWS/FLI response elements in Ewing’s sarcoma. Proc Natl Acad Sci U S A 2008;105:10149–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tomazou EM, Sheffield NC, Schmidl C, Schuster M, Schonegger A, Datlinger P, et al. Epigenome mapping reveals distinct modes of gene regulation and widespread enhancer reprogramming by the oncogenic fusion protein EWS-FLI1. Cell Rep 2015;10:1082–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sampieri L, Di Giusto P, Alvarez C. CREB3 Transcription Factors: ER-Golgi Stress Transducers as Hubs for Cellular Homeostasis. Front Cell Dev Biol 2019;7:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wernert N, Raes MB, Lassalle P, Dehouck MP, Gosselin B, Vandenbunder B, Stehelin D. c-ets1 proto-oncogene is a transcription factor expressed in endothelial cells during tumor vascularization and other forms of angiogenesis in humans. Am J Pathol 1992;140:119–27 [PMC free article] [PubMed] [Google Scholar]
- 33.Wei G, Srinivasan R, Cantemir-Stone CZ, Sharma SM, Santhanam R, Weinstein M, et al. Ets1 and Ets2 are required for endothelial cell survival during embryonic angiogenesis. Blood 2009;114:1123–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tahtakran SA, Selleck MA. Ets-1 expression is associated with cranial neural crest migration and vasculogenesis in the chick embryo. Gene Expr Patterns 2003;3:455–8 [DOI] [PubMed] [Google Scholar]
- 35.Theveneau E, Duband JL, Altabef M. Ets-1 confers cranial features on neural crest delamination. PLoS One 2007;2:e1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gao Z, Kim GH, Mackinnon AC, Flagg AE, Bassett B, Earley JU, Svensson EC. Ets1 is required for proper migration and differentiation of the cardiac neural crest. Development 2010;137:1543–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barembaum M, Bronner ME. Identification and dissection of a key enhancer mediating cranial neural crest specific expression of transcription factor, Ets-1. Dev Biol 2013;382:567–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boulay G, Sandoval GJ, Riggi N, Iyer S, Buisson R, Naigles B, et al. Cancer-Specific Retargeting of BAF Complexes by a Prion-like Domain. Cell 2017;171:163–78 e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boulay G, Volorio A, Iyer S, Broye LC, Stamenkovic I, Riggi N, Rivera MN. Epigenome editing of microsatellite repeats defines tumor-specific enhancer functions and dependencies. Genes Dev 2018;32:1008–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kovar H, Jug G, Aryee DN, Zoubek A, Ambros P, Gruber B, et al. Among genes involved in the RB dependent cell cycle regulatory cascade, the p16 tumor suppressor gene is frequently lost in the Ewing family of tumors. Oncogene 1997;15:2225–32 [DOI] [PubMed] [Google Scholar]
- 41.Sanchez G, Bittencourt D, Laud K, Barbier J, Delattre O, Auboeuf D, Dutertre M. Alteration of cyclin D1 transcript elongation by a mutated transcription factor up-regulates the oncogenic D1b splice isoform in cancer. Proc Natl Acad Sci U S A 2008;105:6004–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kennedy AL, Vallurupalli M, Chen L, Crompton B, Cowley G, Vazquez F, et al. Functional, chemical genomic, and super-enhancer screening identify sensitivity to cyclin D1/CDK4 pathway inhibition in Ewing sarcoma. Oncotarget 2015;6:30178–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takacs R, Vago J, Poliska S, Pushparaj PN, Ducza L, Kovacs P, et al. The temporal transcriptomic signature of cartilage formation. Nucleic Acids Res 2023;51:3590–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huyghe A, Furlan G, Schroeder J, Cascales E, Trajkova A, Ruel M, et al. Comparative roadmaps of reprogramming and oncogenic transformation identify Bcl11b and Atoh8 as broad regulators of cellular plasticity. Nat Cell Biol 2022;24:1350–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Divvela SSK, Offei EB, Suerland F, Revuelta Garcia D, Kwiatkowski J, Balakrishnan-Renuka A, et al. Atonal homolog 8/Math6 regulates differentiation and maintenance of skeletal muscle. Front Cell Dev Biol 2022;10:950414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mak KM, Png CY, Lee DJ. Type V Collagen in Health, Disease, and Fibrosis. Anat Rec (Hoboken) 2016;299:613–29 [DOI] [PubMed] [Google Scholar]
- 47.Faulstich H, Zobeley S, Rinnerthaler G, Small JV. Fluorescent phallotoxins as probes for filamentous actin. J Muscle Res Cell Motil 1988;9:370–83 [DOI] [PubMed] [Google Scholar]
- 48.Szczesna D, Lehrer SS. The binding of fluorescent phallotoxins to actin in myofibrils. J Muscle Res Cell Motil 1993;14:594–7 [DOI] [PubMed] [Google Scholar]
- 49.Merour E, Hmidan H, Marie C, Helou PH, Lu H, Potel A, et al. Transient regulation of focal adhesion via Tensin3 is required for nascent oligodendrocyte differentiation. Elife 2022;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang S, van de Peppel J, Koedam M, van Leeuwen J, van der Eerden BCJ. Tensin-3 is involved in osteogenic versus adipogenic fate of human bone marrow stromal cells. Cell Mol Life Sci 2023;80:277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Atherton P, Konstantinou R, Neo SP, Wang E, Balloi E, Ptushkina M, et al. Tensin3 interaction with talin drives the formation of fibronectin-associated fibrillar adhesions. J Cell Biol 2022;221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kovar H Blocking the road, stopping the engine or killing the driver? Advances in targeting EWS/FLI-1 fusion in Ewing sarcoma as novel therapy. Expert Opin Ther Targets 2014;18:1315–28 [DOI] [PubMed] [Google Scholar]
- 53.Aynaud MM, Mirabeau O, Gruel N, Grossetete S, Boeva V, Durand S, et al. Transcriptional Programs Define Intratumoral Heterogeneity of Ewing Sarcoma at Single-Cell Resolution. Cell Rep 2020;30:1767–79 e6 [DOI] [PubMed] [Google Scholar]
- 54.Keskin T, Bakaric A, Waszyk P, Boulay G, Torsello M, Cornaz-Buros S, et al. LIN28B Underlies the Pathogenesis of a Subclass of Ewing Sarcoma LIN28B Control of EWS-FLI1 Stability. Cell Rep 2020;30:4567–83 e5 [DOI] [PubMed] [Google Scholar]
- 55.Khoogar R, Li F, Chen Y, Ignatius M, Lawlor ER, Kitagawa K, et al. Single-cell RNA profiling identifies diverse cellular responses to EWSR1/FLI1 downregulation in Ewing sarcoma cells. Cell Oncol (Dordr) 2022;45:19–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Katschnig AM, Kauer MO, Schwentner R, Tomazou EM, Mutz CN, Linder M, et al. EWS-FLI1 perturbs MRTFB/YAP-1/TEAD target gene regulation inhibiting cytoskeletal autoregulatory feedback in Ewing sarcoma. Oncogene 2017;36:5995–6005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kola I, Brookes S, Green AR, Garber R, Tymms M, Papas TS, Seth A. The Ets1 transcription factor is widely expressed during murine embryo development and is associated with mesodermal cells involved in morphogenetic processes such as organ formation. Proc Natl Acad Sci U S A 1993;90:7588–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Maroulakou IG, Papas TS, Green JE. Differential expression of ets-1 and ets-2 proto-oncogenes during murine embryogenesis. Oncogene 1994;9:1551–65 [PubMed] [Google Scholar]
- 59.Muthusamy N, Barton K, Leiden JM. Defective activation and survival of T cells lacking the Ets-1 transcription factor. Nature 1995;377:639–42 [DOI] [PubMed] [Google Scholar]
- 60.Bories JC, Willerford DM, Grevin D, Davidson L, Camus A, Martin P, et al. Increased T-cell apoptosis and terminal B-cell differentiation induced by inactivation of the Ets-1 proto-oncogene. Nature 1995;377:635–8 [DOI] [PubMed] [Google Scholar]
- 61.Mouly E, Chemin K, Nguyen HV, Chopin M, Mesnard L, Leite-de-Moraes M, et al. The Ets-1 transcription factor controls the development and function of natural regulatory T cells. J Exp Med 2010;207:2113–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Barton K, Muthusamy N, Fischer C, Ting CN, Walunas TL, Lanier LL, Leiden JM. The Ets-1 transcription factor is required for the development of natural killer cells in mice. Immunity 1998;9:555–63 [DOI] [PubMed] [Google Scholar]
- 63.Casie Chetty S, Sumanas S. Ets1 functions partially redundantly with Etv2 to promote embryonic vasculogenesis and angiogenesis in zebrafish. Dev Biol 2020;465:11–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ye M, Coldren C, Liang X, Mattina T, Goldmuntz E, Benson DW, et al. Deletion of ETS-1, a gene in the Jacobsen syndrome critical region, causes ventricular septal defects and abnormal ventricular morphology in mice. Hum Mol Genet 2010;19:648–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Simoes-Costa MS, McKeown SJ, Tan-Cabugao J, Sauka-Spengler T, Bronner ME. Dynamic and differential regulation of stem cell factor FoxD3 in the neural crest is Encrypted in the genome. PLoS Genet 2012;8:e1003142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lin L, Pinto A, Wang L, Fukatsu K, Yin Y, Bamforth SD, et al. ETS1 loss in mice impairs cardiac outflow tract septation via a cell migration defect autonomous to the neural crest. Hum Mol Genet 2022;31:4217–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.von Levetzow C, Jiang X, Gwye Y, von Levetzow G, Hung L, Cooper A, et al. Modeling initiation of Ewing sarcoma in human neural crest cells. PLoS One 2011;6:e19305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liao YC, Lo SH. Tensins - emerging insights into their domain functions, biological roles and disease relevance. J Cell Sci 2021;134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Blangy A Tensins are versatile regulators of Rho GTPase signalling and cell adhesion. Biol Cell 2017;109:115–26 [DOI] [PubMed] [Google Scholar]
- 70.Mainsiouw L, Ryan ME, Hafizi S, Fleming JC. The molecular and clinical role of Tensin 1/2/3 in cancer. J Cell Mol Med 2023;27:1763–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McCarthy RL, Zhang J, Zaret KS. Diverse heterochromatin states restricting cell identity and reprogramming. Trends Biochem Sci 2023;48:513–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Macrae TA, Fothergill-Robinson J, Ramalho-Santos M. Regulation, functions and transmission of bivalent chromatin during mammalian development. Nat Rev Mol Cell Biol 2023;24:6–26 [DOI] [PubMed] [Google Scholar]
- 73.Orth MF, Surdez D, Faehling T, Ehlers AC, Marchetto A, Grossetete S, et al. Systematic multi-omics cell line profiling uncovers principles of Ewing sarcoma fusion oncogene-mediated gene regulation. Cell Rep 2022;41:111761. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data and resources generated are available upon reasonable request by contacting the corresponding author. The RNA-seq, ChIP-seq, and CUT&RUN datasets reported here are available in the Gene Expression Omnibus at GSE243184. Images of source immunoblots, STR fingerprinting information for all cell lines, and additional cell images (confocal and super-resolution microscopy) are available at FigShare, DOI:10.6084/m9.figshare.c.7114060.