

Abstract
Hepatitis C virus (HCV) is a plus-stranded RNA virus that often chronically infects liver hepatocytes and causes liver cirrhosis and cancer. These viruses replicate their genomes employing error-prone replicases. Thereby, they routinely generate a large ‘cloud’ of RNA genomes (quasispecies) which—by trial and error—comprehensively explore the sequence space available for functional RNA genomes that maintain the ability for efficient replication and immune escape. In this context, it is important to identify which RNA secondary structures in the sequence space of the HCV genome are conserved, likely due to functional requirements. Here, we provide the first genome-wide multiple sequence alignment (MSA) with the prediction of RNA secondary structures throughout all representative full-length HCV genomes. We selected 57 representative genomes by clustering all complete HCV genomes from the BV-BRC database based on k-mer distributions and dimension reduction and adding RefSeq sequences. We include annotations of previously recognized features for easy comparison to other studies. Our results indicate that mainly the core coding region, the C-terminal NS5A region, and the NS5B region contain secondary structure elements that are conserved beyond coding sequence requirements, indicating functionality on the RNA level. In contrast, the genome regions in between contain less highly conserved structures. The results provide a complete description of all conserved RNA secondary structures and make clear that functionally important RNA secondary structures are present in certain HCV genome regions but are largely absent from other regions. Full-genome alignments of all branches of Hepacivirus C are provided in the supplement.
Keywords: Hepatitis C virus, Full-genome alignment, RNA secondary structure prediction
Subject terms: Computational biology and bioinformatics, Evolutionary biology, Coevolution, Evolutionary genetics
Introduction
Hepatitis C virus (HCV) is a major public health concern that affects an estimated 71 million people globally1 causing liver cirrhosis and hepatocellular carcinoma. The virus is a member of the species Hepacivirus C and the Flaviviridae family, which also includes Dengue, Zika, and yellow fever viruses. HCV is primarily transmitted through blood-to-blood contact, with injection drug use being the most common mode of transmission. It can also be transmitted through unsafe medical procedures, blood transfusions, and from mother to child during childbirth2–4.
HCV is a single-stranded positive-sense RNA virus. The HCV genome is approximately 9.6 kb in length and organized into a single open reading frame (ORF) flanked by two untranslated regions (UTRs) at the and ends. The ORF encodes a single polyprotein precursor that is co- and post-translationally cleaved by host and viral proteases into structural (C, E1, E2, p7) and non-structural proteins (NS2, NS3, NS4A, NS4B, NS5A, NS5B), which play critical roles in viral replication and pathogenesis5–7. The HCV genome is known to form several RNA secondary structures8–16. The UTR contains four structural domains (I–IV). Three of those (II–IV) form a highly structured RNA region called the internal ribosome entry site (IRES), which is responsible for controlling the translation of the viral polyprotein. The IRES allows HCV to circumvent the host cell’s cap-dependent translation initiation mechanism, which is commonly used by cellular mRNAs. The region downstream of SL I including alternative SL II structures17 contains binding sites for microRNA-122 (miR-122) involved in translation, replication, and RNA stability10,18–21. The UTR contains several RNA structures that are involved in regulating viral RNA replication and translation, including a hypervariable region (HVR), a poly-U/UC tract, and a highly conserved RNA secondary structure called X-tail. The X-tail is a conserved RNA stem-loop structure that serves as a binding site for host proteins and is involved in viral replication and translation10,14. Apart from the structural motifs located in the UTRs, HCV showcases RNA secondary structures within its coding region, such as the cis-replication element8–11,16,22,23.
The HCV genome is highly variable due to the error-prone nature of its NS5B replicase24,25 and generates a cloud of ‘quasispecies’ that covers a huge sequence space that allows the virus to adapt to changing host environments and escape the immune system26, with eight genotypes and 93 subtypes identified to date, and sequence diversity of approximately 30% between genotypes27,28, see Fig. S1. Its genome contains several complex RNA structures that are critical for viral replication and pathogenesis. Thus, understanding the structure and function of these RNA elements is crucial for the development of effective treatments for HCV infection.
Previous studies have predicted conserved RNA secondary structures in important parts of the HCV genome by different approaches. These studies either analyzed mainly the and UTRs as well as the end of the NS5B coding region10,16, or they focused on certain HCV genome hotspot regions in the coding regions using covariance analysis with genotype 2 sequences and extending this to other genotypes11. Another study confined the prediction of RNA secondary structures in a full-length genome to the isolates JFH-1 (genotype 2a), H77c (genotype 1a), and Con1 (genotype 1b)9. The above studies provided important information on conserved RNA secondary structures and their functions in certain HCV RNA genome regions. However, up to the present, a complete survey of all conserved RNA secondary structures in the full length HCV across all genotypes is missing. Therefore, we filtered and manually refined a set of 2549 HCV full-length genome sequences that fully represent the phylogenetic diversity of all known HCV isolates. From this set, 57 HCV RNA genome sequences were selected that represent the complete phylogenetic tree’s sequence space of HCV genomes.
The in silico calculation of full-genome alignments for viral sequences, coupled with the prediction of RNA secondary structures, presents a multifaceted challenge in computational biology. Viral genomes exhibit high genetic diversity, characterized by rapid mutation rates and the presence of insertions and deletions. The complexity for the construction of a multiple sequence-secondary structure alignment (MSSSA) lays at and is therefore, not computationally feasible. For current construction of MSSSAs, the genomes have to be divided into smaller subsequences for a reliable prediction. Addressing these challenges is pivotal, as such alignments provide crucial insights into the molecular evolution of viruses and their structural-functional relationships. In this context, the development of robust computational methodologies is essential to advance our understanding of viral biology and host-virus interactions.
In this study, we present a full-genome alignment of HCV coupled with RNA secondary structure annotation. The alignment was generated using a semi-automated approach and underwent curation led by experts in the field of HCV and its associated structural elements. Beyond established structures, our study reveals previously unrecognized RNA secondary structures, predicted through computational methods, that exhibit conservation across HCV genomes. Moreover, the results of our alignment—using sequences covering the complete phylogenetic sequence space of HCV isolates—suggest that our predictions likely cover virtually all possible conserved RNA secondary structures.
Material and methods
Data
We downloaded 2,606 HCV genomes (June 01, 2023) from the BV-BRC database29. To ensure the quality of the data, we filtered the genome status ‘complete’ and excluded the host group ‘lab’. Notably, despite the genome completeness filter, the majority of entries of this data set (80.5%) contain incomplete genomes, lacking the UTRs. Among these, 20 genomes were excluded from the analysis due to their sequences containing 10% or more ‘N’s. After identifying duplicated genomes, the data set was refined to a total of 2549 genomes. Both the original data set and the pre-filtered data set are included in the Supplementary Files F1 and F2 in Fasta (.fasta) format.
Finding representative genomes
We performed clustering of the pre-filtered data set (2549 genomes) based on k-mers to select sequences representing the data set. After calculating the k-mer profiles of the input sequences, we performed a dimension reduction by principal component analysis (PCA) followed by clustering using HDBSCAN v0.8.2730. HDBSCAN resulted in 36 representative genomes which were selected for further analysis, as this method provided comprehensive coverage of the genome information space. For comparison, we clustered sequences with five algorithms: cd-hit-est31,32, MMSeqs233, sumaclust34, vclust35, and HDBSCAN30 (see Table S1). The workflow is implemented in ViralClust36. Despite all filters applied, partial genomes are present in the data set, and thus, the cluster representatives calculated by HDBSCAN did too. We removed six sequences manually from the set of representative genomes because they were too short (less than 1000 nt: MK468966, MK468983, MK469005, MK468990, and OM896954), or were not related to a functional polyprotein (EU862828). However, it was necessary to manually enlarge our set of representative genomes to fully display the entire spectrum of HCV samples. Utilizing the phylogenetic tree of the pre-filtered data set, see Fig. S1 and Supplementary File F3, we added 20 genomes representing outliers or subtrees not covered by the clustering results (black squares in Fig. S1). Additionally, for comparison to known strains, we added the NCBI RefSeq genomes of HCV (NC_038882, NC_004102, NC_009823, NC_009824, NC_009825, NC_009826, NC_009827, NC_030791) to our final set of representative genomes37. We removed one sequence (OM896952) because of high redundancy with NC_009824. Finally, a total of 57 representative genomes covering all eight genotypes of HCV were selected, see supplementary file F4. About 50% of the representative HCV genomes (27) contain the UTRs (see supplementary information subsection “Genome completeness of representative genomes”). The genome length of the selected genomes ranges from 9036 nt (MN164872.1) to 9711 nt (NC_009823.1).
Multiple sequence alignment and RNA secondary structure prediction
The 57 representative genomes served as input for alignment construction. We computed an initial multiple sequence alignment (MSA) using MAFFT v7.52038 to identify highly conserved regions, which served as ‘anchors’ for further steps. Anchors are defined as segments in the MSA, requiring a minimum length of 10 nucleotides, and exhibiting an average Shannon entropy value lower than 0.1. Subsequently, we focused our analysis on the subregions between these anchors, utilizing LocARNA v2.0.039. The subregions were then merged into one MSA, followed by an RNA secondary structure prediction of the full-genome alignment with a window-based approach. These steps are implemented in VeGETA using Python v3.7.1240,41. Additionally, we intensively examined and slightly curated the alignment manually. Finally, we added the annotation of conserved RNA secondary structures described in the literature as well as novel ones. Based on the nucleotide alignment, we constructed the protein alignment of the representative genomes.
Results and discussion
In this study, we present a comprehensive analysis of all conserved RNA secondary structures that occur in the complete sequence space represented by the phylogenetic tree of all HCV isolates (see Fig. S1). Thus, the results are supposed to provide a complete description of RNA secondary structures that may be advantageous for the viral life cycle.
It is known that functionally important RNA secondary structures not only occur in the untranslated regions of mRNAs but also in the coding regions42. A variety of molecular mechanisms can be envisioned to be employed by RNA secondary structure elements. RNA secondary structure elements in the protein-coding region can be used for influencing the translational outcome of a given RNA43, for example by inducing a ribosome frameshift or termination reinitiation. Specific RNA elements can be used for packaging selectively one RNA while another longer RNA species is excluded from packaging by translational inactivation44. A translating ribosome may also displace proteins from an RNA secondary structure element of the RNA and by that induce a kind of ‘burn after reading’ degradation of the RNA45. Thus, it is important to identify those RNA secondary structures that have been selected for their function from the available sequence space produced by the error-prone replicases of RNA plus strand viruses.
Basic statistics of the full-genome alignments
Our nucleotide-based alignment contains 57 representative HCV genomes including all eight genotypes. The alignment spans 9831 residues and 23 061 gaps, averaging approximately 405 gaps per sequence. Approximately 8.5% of the sequences within our alignment contain non-ACGU characters, highlighting sequence variations that may have functional significance. Half of the sequences (28/57) exhibit a full UTR; four sequences lack the UTR entirely; 12 sequences are deficient in both stem-loops I (SL I) and II (SL II) in the UTR, and 13 sequences lack only SL I. For the UTR, only 11 sequences display a complete X-tail structure; three sequences show partial X-tail formations, all others lack the UTR. The seed sequence (ACACUCC) of the first miR-122 binding site of the UTR directly downstream of the SL I is present in all 32 sequences covering that region; the second miR-122 binding site (CACUCC) directly downstream is present in 37/40 sequences, and one other sequence contains CGCUCC which would also allow miR-122 binding by G-U base pairing. In the UTR, the miR-122 seed sequence ACACUCC is contained in 41/44 sequences, in contrast, three of the sequences in genotypes 6 and 8 do not contain this site.
We added additional information to our nucleotide alignment: (1) gene annotations (shown as annotation line #=GC Annotation in the stk file); (2) the F/ARFP frameshift (notated with ‘f’ in the Annotation line in the stk file); (3) the RNA secondary structures (including pseudoknots) documented in the literature, along with alternative structural configurations (see Table 1); (4) incorporated in-silico predicted novel RNA secondary structures, providing a comprehensive view of potential conformations within the HCV genome.
Table 1.
Conserved RNA secondary structures (SS) in HCV genomes, along with their corresponding Rfam model IDs (if available)46,47.
| RNA SS | Genomic region | Alignment position | JFH-1 position | Rfam v12 | Rfam v14.10 | ||
|---|---|---|---|---|---|---|---|
| S | E | S | E | ||||
| SL I | UTR | 13 | 28 | 5 | 19 | RF00061 | RF00061 |
| SL II | UTR | 52 | 126 | 43 | 117 | RF00061 | RF00061 |
| SL III | UTR | 133 | 334 | 124 | 322 | RF00061 | RF00061 |
| SL IV | UTR/C | 346 | 360 | 334 | 348 | RF00061 | RF00061 |
| SL V | C | 400 | 435 | 388 | 423 | RF00620 | RF00620 |
| SL VI | C | 439 | 519 | 427 | 507 | RF00620 | RF00620 |
| SL 562 | C | 574 | 595 | 562 | 582 | ||
| SL 588 | C | 601 | 678 | 588 | 665 | RF04220 | |
| SL 669 | C | 684 | 761 | 671 | 748 | RF04221 | |
| J 750 | C | 762 | 839 | 749 | 826 | RF04219 | |
| SL 833 | C | 846 | 869 | 833 | 856 | RF00*** | |
| SL 1412 | E1 | 1430 | 1467 | 1414 | 1451 | RF04305 | |
| SL 1850 | E1 | 1881 | 2013 | 1850 | 1982 | ||
| SL 2313 | E1 | 2356 | 2433 | 2313 | 2390 | ||
| SL 2531 | E2 | 2 574 | 2591 | 2531 | 2548 | RF04308 | |
| SL 2549 | E2/p7 | 2592 | 2636 | 2549 | 2592 | RF04308 | |
| SL 3308 | NS2/NS3 | 3352 | 3644 | 3308 | 3600 | ||
| SL 3844 | NS3 | 3888 | 3952 | 3844 | 3908 | ||
| SL 4005 | NS3 | 4049 | 4099 | 4005 | 4056 | ||
| SL 4214 | NS3 | 4258 | 4223 | 4214 | 4279 | ||
| SL 4527 | NS3 | 4571 | 4591 | 4527 | 4547 | ||
| SL 4621 | NS3 | 4665 | 4725 | 4621 | 4681 | ||
| SL 4691 | NS3 | 4735 | 4788 | 4691 | 4744 | ||
| SL 5016 | NS3 | 5060 | 5125 | 5016 | 5081 | ||
| SL 5128 | NS3 | 5172 | 5249 | 5128 | 5205 | ||
| SL 5357 | NS4A | 5401 | 5448 | 5357 | 5404 | ||
| SL 5647 | NS4B | 5691 | 5777 | 5647 | 5733 | ||
| SL 6027 | NS4B | 6071 | 6197 | 6027 | 6153 | ||
| SL 6270 | NS5A | 6314 | 6410 | 6270 | 6366 | ||
| SL 6371 | NS5A | 6415 | 6446 | 6371 | 6402 | ||
| SL 6530 | NS5A | 6574 | 6689 | 6530 | 6645 | ||
| SL 7516 | NS5A | 7584 | 7603 | 7516 | 7535 | ||
| SL 7536 | NS5A | 7604 | 7702 | 7536 | 7634 | ||
| SL 7816 | NS5B | 7893 | 7905 | 7816 | 7828 | ||
| J 7880 | NS5B | 7959 | 8073 | 7882 | 7996 | RF00*** | |
| SL 8001 | NS5B | 8077 | 8126 | 8000 | 8049 | RF04306 | |
| SL 8075 | NS5B | 8152 | 8174 | 8075 | 8097 | ||
| SL 8299 | NS5B | 8376 | 8396 | 8299 | 8334 | ||
| SL 8670 | NS5B | 8722 | 8803 | 8645 | 8726 | RF04307 | |
| 5BSL1 | NS5B | 9194 | 9231 | 9117 | 9154 | RF04218 | |
| 5BSL2 | NS5B | 9276 | 9335 | 9198 | 9257 | RF00468 | RF00468 |
| 5BSL3.1 | NS5B | 9361 | 9406 | 9283 | 9328 | RF00260 | RF00260 |
| 5BSL3.2 | NS5B | 9410 | 9455 | 9332 | 9377 | RF00260 | RF00260 |
| 5BSL3.3 | NS5B | 9467 | 9497 | 9389 | 9419 | RF00260 | RF00260 |
| SL IV | NS5B | 9505 | 9533 | 9427 | 9452 | RF00469 | RF00260 |
| X-tail | UTR | 9727 | 9826 | 9579 | 9678 | RF00481 | RF00481 |
We updated and merged six Rfam models of v12 into five Rfam models (v14.10); confirmed the conservation of a total of 16 previously predicted RNA secondary structures throughout the phylogenetic tree (available in Rfam v14.10); and added further 23 novel conserved RNA families into Rfam (bold font). indicates that the existing Rfam model was updated with this publication. We named novel structures according to their position in the genome JFH-1 (S—Start; E—End). ***Rfam models will follow in the near future.
The protein alignment of the 57 representative genomes encompasses a total of 3017 residues, with an average of 37 gaps (2136 gaps in total). A minor proportion of not characterized amino acid characters (0.0015%) accounting for a total of 262 occurrences, can be found in the alignment. These ‘X’s are based on ‘N’s in the sequences downloaded from NCBI. We provide the nucleotide, protein, and combined alignments in the supplementary material with several formats, such as Stockholm (stk), ClustalW (aln), and Fasta (fasta) (see Files F5 and Files F7), which can be conveniently visualized using tools such as ClustalX48,49, Jalview50 or Emacs RALEE mode51. We have visualized the possible color codes available in Emacs RALEE mode in Fig. S5 based on the examples SL V and SL VI.
Alignment confirms previously annotated RNA secondary structures
The full-genome alignment of HCV genomes reveals the presence of well-characterized RNA secondary structures, see Table 1, that are consistent with the existing literature9–11,18. All structures, including the long-range interactions and the genome circularization presented by Fricke et al.10, are annotated in our alignment. Therefore, the above alignment is validated by its prediction of these structures which had been demonstrated to be functional cis-elements involved in the regulation of HCV RNA translation and replication. These known structural elements include the highly conserved IRES (see Figs. S2 and S3), which plays a crucial role in HCV translation initiation10,18, as well as stem-loop structures within the core coding region (see Fig. S6). Additionally, the interaction of the sequence in the left base of SL VI in the core coding region with the single-stranded region between SL I and SL II in the UTR was shown; this interaction had been demonstrated to act inhibitory on translation52–54, while this inhibition can be relieved by microRNA-122 binding to the region between SL I and SL II55,56 (see S4). In the NS5B coding region (see Fig. 1 and Figs. S9–S13) the cis-replication element (CRE, composed of 5BSL3.1, 5BSL3.2, 5BSL3.3) and the highly conserved X-tail in the UTR (both conformations) (see Fig. 2 and Fig. S14) were identified. Moreover, we predicted several conserved RNA secondary structures in the coding region of HCV genomes (see Fig. 1, Figs. S6, S9, S10, S11, S12, and S13) in agreement with the literature9–11. The identification of these known structures in the core coding region57,58 and in the NS5B region9,11 not only validates the accuracy of our alignment but also underscores their functional importance in HCV biology: their conservation across different HCV genotypes further highlights their critical roles in viral replication, translation, and infection.
Figure 1.

Known RNA secondary structures in the downstream NS5B coding region including the cis-replication element (CRE, 5BSL3.2) which are relevant for replication control. (A) RNA secondary structures are colored by the number of base pair types illustrating the extent of covariations in double-stranded regions. The SL IV (or 5BSL3.4) contains the NS5B stop codon in the apical loop. The structure was visualized using R2DT59. The nucleotide sequence shows the most informative sequence (IUPAC code) calculated by RNAalifold60 based on the alignment. Lowercase letters indicate gaps in the alignment column. (B) RNA secondary structure dot-bracket annotation (SS), structure consensus (StrucC), sequence consensus (SeqC), and the fraction of nucleotides used from synonymous codons (SynCo). Thereby, a low value indicates that only a few nucleotide(s) out of all nucleotides possible for synonymous codons are actually used by the different HCV isolates, indicating a high degree of primary sequence conservation which goes beyond the requirements of the coding sequence. This provides evidence that a conserved functional RNA element may overlap with the coding sequence. Alignments shown in Figs. S12 and S13.
Figure 2.

The two alternative structures of the highly conserved X region of the UTR. (A) The conformation with the three stem-loops SL 1, 2, and 3. In this form, the apical loop of SL 2 can make a long-range interaction (LRI) with the apical loop of the CRE/5BSL3.2 (‘kissing loop’ interaction). Among the selected sequences, only 11 isolates had a complete UTR sequence (please see the additional alignment of only X sequences in Fig. S14 and F9). (B) The conformation with SL 2 and 3 restructured to form the DLS that is speculated to be involved in HCV RNA genome dimerization61–63. (C) Additional sequence and RNA secondary structure features of the consensus of the 11 isolates, with two alternative dot-bracket outputs. As in parts of the UTR, the strong conservation of the primary sequence indicates that both overlapping structures shown in (A) and (B) may be functionally important, thereby limiting the extent of possible covariations in the RNA secondary structure regions of each conformation.
The CRE/5BSL3.2 and the 5BSL3.3 are important for HCV replication22,23. These functional aspects are reflected by the high conservation of the 5BSL3.2 and 3.3 (see Fig. 1 and Fig. S13), not only in their RNA secondary structure regions but also in their single-stranded loops and bulges, which are conserved beyond coding sequence requirements, since only selected nucleotides are actually used from those possible in synonymous codons (see Fig. 1 and Fig. S13). Likely, in the early phase after HCV infection, the CRE apical loop can interact with the SL 2 of the X region by forming a ‘kissing loop’ interaction64, and this interaction may stabilize its SL 2, SL 3 conformation65,66. At a time when sufficient amounts of NS5B replicase have been translated, NS5B can bind the CRE/5BSL3.2 and 5BSL3.323,67, thereby disabling the CRE—SL 2 interaction and allowing refolding of the SL 3 and SL 2 in the X region to form the overlapping dimerization linkage sequence (DLS)61,62. Binding of NS5B to the CRE then is supposed to be involved in starting RNA minus strand synthesis at the HCV RNA end, whereas the role of the DLS and its putative role in dimerization of the full-length HCV genome in this process is not yet fully understood. These functional aspects in turn validate our alignment approach for identifying functionally important RNA secondary structures.
Similar constraints may apply to the region including SL II and the preceding sequence between SLs I and II is highly conserved in the primary sequence due to two alternative conformations that fulfill different tasks in the viral life cycle68. The classical conformation of SL II allows binding of two complexes of miR-122 with Argonaute (AGO) protein to the single-stranded region upstream of SL II and has roles in HCV genome replication19, promoting translation20 and stabilization of the genome against nucleolytic degradation21. The alternative conformation SL II17, however, appears to have a role in HCV assembly68. We also confirmed the SL IIId and its conserved alternative form SL IIId* which showed up previously10 in the IRES (see Figs. S2 and S3). This alternative SL IIId* is predicted to be slightly more stable than the classical SL IIId. We can only speculate if this alternative SL IIId* represents a structure that may be important in the IRES when not bound to ribosomes (an additional discussion of this aspect can be found in the Supplementary Materials Subsection ‘Alignment confirms previously predicted RNA secondary structures—Additional Information’).
Improvements to Rfam virus families
We improved the models in the Rfam database46,47 (see Table 1) to ensure comprehensive coverage of the entire phylogenetic clade of the HCV sequences. In total, we identified 39 conserved structural regions across the HCV genomes, of which 23 were novel. We updated the six Rfam families from release 12 to nine families in release 14.10, of which all are validated by the literature. The HCV Rfam families were reviewed for covariance support with R-scape69. Only a small number of base pairs (1–3) exhibited covariance support in each family, and this consistency was observed across families of both non-coding and coding sequences/regions. The remaining 30 structured regions will be used to create additional Rfam families in future releases. In the following, we compare the five models to the previously well-described models from Rfam v12: (1) We reduced the IRES model (RF00061) from 79 to 51 sequences, spanning 356 nucleotide positions in the alignment (previously 413). The new model includes now SL I (from 30 HCV genomes) and SL II (covered by 41 genomes), which was absent due to sequencing problems of the very genome end in previous times. Importantly, the new model includes now SL IV from 51 HCV genomes, which is located at the transition from UTR to core gene, containing the start codon of the polyprotein. (2) The SL V and SL VI model (RF00620) was updated from 36 sequences to 56 (MK548369 excluded because of non-ACGU characters) and now spans an alignment length of 136 positions. The previous model contained 153 alignment positions, indicating a major reduction of gaps in the novel alignment. SL V was reduced by one base pair and SL VI by three base pairs. (3) The 5BSL2 model (RF00468) has now been reduced from 110 to 57 genomes. This measurement allows us to not compose a bias towards closely related, highly over-represented sequences. Our alignment expands the stem-loop by four base pairs. (4) The CRE model (RF00260) is now represented by all 57 selected genomes (previously 52). Only 5BSL3.2 of the CRE has been included in the old model, therefore the new model spans now 183 nucleotide positions (instead of 51 nucleotides) including the complete CRE (5BSL3.1, 5BSL3.2, and 5BSL3.3) and SL IV. (5) The model of SL IV (RF00469), located at the transition from NS5B gene to UTR, containing the stop codon of the polyprotein, comprised 110 sequences. This structure is now naturally merged into model RF00260. (6) Lastly, the X-tail model (RF00481) now contains only 11 sequences, the old model contained 22 HCV genomes. However, the old model only contained sequences of genotype 1–3. Therefore, although the total number of sequences has been reduced, the variety of the X-tail has been enlarged by our new model and is now spanning genotypes 1–6. We expanded SL I by one base pair. In total, we were able to predict 16 structural elements in the entire phylogenetic tree of HCV that had been previously confirmed10,11,16, see Figs. 1, 2, Figs. S2, S6, S7 and S9. We added these and the 23 novel HCV RNA secondary structures to Rfam v14, see Fig. 3 and Fig. S15. An information page for all HCV models in Rfam is provided at the following link: https://rfam.org/viruses/hcv.
Figure 3.
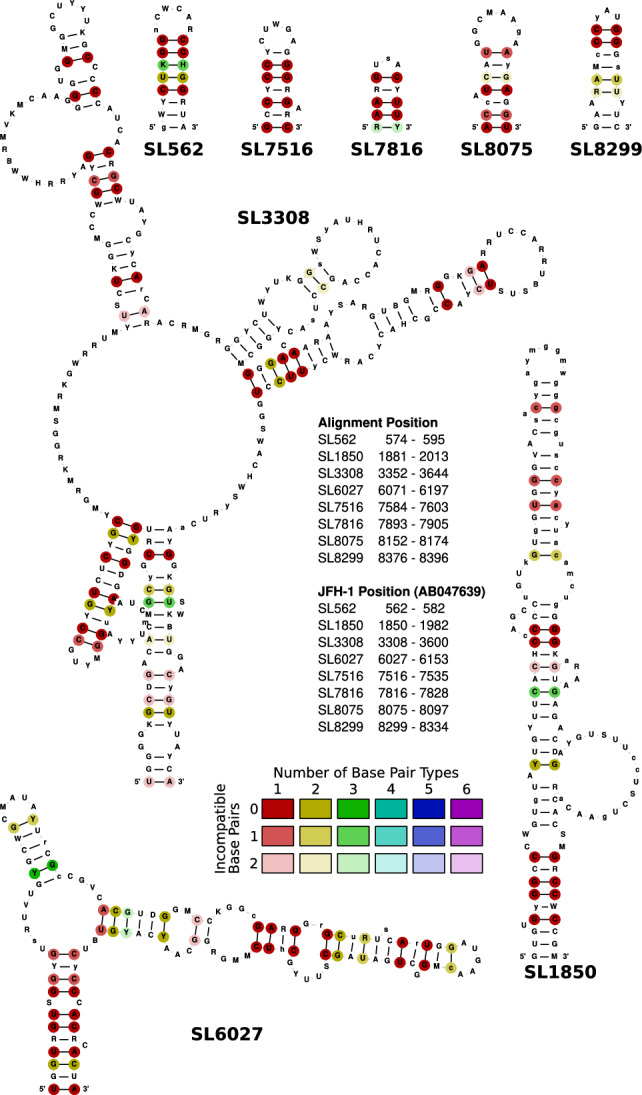
Eight novel selected conserved RNA secondary structure candidates from coding regions of the HCV alignment. RNA secondary structures are colored as in Fig. 1A.
New RNA secondary structure element candidates
Our analysis revealed several novel RNA secondary structures in the coding region using our in silico prediction method (see Fig. 3 and Fig. S15). We selected novel RNA secondary structures based on their (1) conservation at the sequence and structural level in the representative sequences regarding compensatory mutations and (2) the use of synonymous codons especially in the hairpin region. We predicted 23 novel candidates to likely be functional elements due to their RNA secondary structure conservation which shows compensatory mutations despite being placed in the coding region, including the restricted use of synonymous codons (Fig. 3 and Fig. S15). SL 562 (see Fig. 3) is one example for a novel predicted short hairpin, which shows compensatory mutations despite being placed in the coding region of the core protein. SL 562 is represented in all 57 sequences highly conserved and structured.
At alignment pos. 7582 (SL 7516, see Fig. 3; and JFH-1 pos. 7516), an RNA secondary structure element with a seven base pair stem and a five nucleotide loop is predicted to be conserved that shows good conservation in the StructConsensus, Consensus as well as in the low number of exchanges in synonymous codons, indicating conservation beyond coding sequence requirements (see Fig. 3 and Fig. S16). In these terms, this newly predicted element is better conserved than the J 7880 element which was shown to be functional in early HCV replication9, suggesting that this new element may have functional importance, even though the degree of its conservation does not fully reach that of the CRE. Similarly, newly predicted RNA secondary structures at positions 7893 (SL 7816, 7816 in JFH-1), pos. 8152 (SL 8075, 8075 in JFH-1), and pos. 8376 (SL 8299, 8299 in JFH-1) are well conserved and are good candidates for functional RNA elements (see Fig. 3 and Fig. S16). In contrast, some presumable RNA structures in the E1/E2 region, located at alignment positions 1430, 2574, and 2592 in the alignment (SL 1412, SL 2531, and SL 2549; JFH-1 pos. 1414, 2531, and 2549; see Figs. S7 and S8), are not well enough conserved in terms of StructConsensus (StrucC), Consensus sequence (SeqC) and the limited use of synonymous codons (SynCo) to suggest a possible function.
The above predicted well-conserved structures represent previously unrecognized RNA elements conserved in the representative HCV genomes (see Fig. 3 and Figs. S15–S17), highlighting the complexity and diversity of RNA secondary structures within this viral species. The discovery of these novel structures opens up new avenues for understanding their potential functional roles in HCV replication, translation, and pathogenesis. Further investigations are warranted to experimentally validate and explore the functional significance of these newly identified RNA secondary structures in the context of HCV biology.
A detailed sequence and RNA secondary structure comparison reveals hints into incongruent evolution
Consensus structures are defined by base pairs that are conserved despite substitutions in the underlying sequence. In other words, base pairs that structurally correspond to each other are usually formed by pairs of nucleotides that are homologous according to their position in the sequence context. This is not always the case, however, as demonstrated by the example of the 5BSL2 stem-loop structures from three representative HCV isolates, Fig. 4. From a coarse-grained perspective, there is a consensus comprising three helical substructures. A more detailed analysis of the individual stem-loop structures, however, not only shows the expected overall conservation of the structure but also surprising differences. In addition to the expected variation, e.g., of the presence or absence of base pairs at the ends of individual helices or variations in loop sizes, we observe that the innermost helix and the hairpin loop are not formed by homologous nucleotides. Instead, a well-conserved stretch of five nucleotides forming the helix in the consensus (green) is shifted by one position in MW689971 and three positions in AY232740 and NC_009827, each. As a consequence, the terminal hairpin is conserved as a structural feature, but its individual base pairs are formed by different, non-homologous sequence positions. A similar situation is visible in the middle (red) and outer (blue) stem. The nucleotides forming the middle stem are shifted by four nucleotides relative to the consensus.
Figure 4.

5BSL2 as an example of incongruent evolution. The consensus RNA secondary structure differs from the structures into which individual sequences would fold, with sequence and structure shifted relative to each other. A helix of five nucleotides (green) in the consensus is shifted by one position in MW689971 and three positions in AY232740 and three positions in NC_009827. The nucleotides that form the middle (red) stem are shifted by four nucleotides compared to the consensus.
The conservation of secondary structures realized by non-homologous base pairs was termed incongruent evolution70 in contrast to the more familiar and much more frequent congruent case. Incongruent evolution can be understood as divergence of sequence alignment and structure alignment. Starting from a sequence alignment, such as the one shown on the bottom of Fig. 4, this leads to an apparently poor conservation of the structure, indicated by the white base pairs in the consensus structure. On the other hand, focusing on the structure (shown here by helices in corresponding positions) results in mismatches (indicated by the colored intervals). It has been shown in Ref.71 that incongruences of sequence and structure can be explained mechanistically, e.g. by flexible structural intermediates. Selection pressures that act independently, i.e., in different functional contexts, to preserve sequence and secondary structure are particularly conducive to incongruent patterns42,72,73 such as the ones in the 5BSL2 stem-loop structure.
Conclusion
We presented the first comprehensive genome-wide multiple sequence alignment (MSA), incorporating computational predictions of RNA secondary structures across the entire HCV genomes (see F5–F9, and Table 1). Our selection of 57 representative genomes across the entire phylogenetic tree is based on clustering all complete HCV genomes from the BV-BRC with HDBSCAN using k-mer distributions and dimension reduction. We added manually the RefSeq sequences. The inclusion of annotations for previously identified features, such as genome annotations, secondary structures, pseudoknots, and alternative structures facilitates seamless comparisons with other research studies. By considering suboptimal structures during the predictions with LocARNA and RNAalifold, we can detect potential alternative conformations. Our in-depth analysis included conservation of the predicted RNA secondary structures, covariance in structured stem regions, and the use of nucleotides in synonymous codons. The latter output not only provides information about the overall conservation of a sequence but also information about the degree of conservation that extends beyond the requirements of the underlying amino acid sequence in coding regions. This information is important for complementing the overall degree of sequence conservation, in particular in single-stranded regions of the conserved RNA secondary structures like apical loops or bulges.
In the 5BSL2 stem-loop, we encountered examples of an incongruent mode of evolution, where sequence and structure are conserved, but the base pairs realizing the structure are not formed by homologous nucleotides. Such situations may be indicative of functionally independent selection pressures on sequence and structure72. As a consequence, part of the conserved structure is not detectable in a sequence-based alignment. Evolutionary incongruencies thus may reduce the sensitivity of consensus structure prediction methods. The local nature of the ‘shifts’ between sequence and structure, on the other hand, still makes it possible to detect larger structured elements, such as the 5BSL2 stem-loop, which contains a sufficient subset of congruent base pairs formed by homologous nucleotides.
All conserved RNA secondary structure models have been added to the Rfam database in version 14.10 or later. The alignment will serve as a standard for future work on HCV.
Supplementary Information
Author contributions
S.T. performed the clustering, alignment construction and refinement, and wrote the manuscript. K.L. implemented the clustering and initial alignment construction. N.O. integrated the RNA secondary structures in the Rfam database. B.S. integrated the RNA secondary structures in the Rfam database and revised the subsection about the Rfam models. P.F.S. wrote the subsection about incongruent evolution. M.N. evaluated the alignment and wrote the biological interpretation of the results. A.I.P. integrated the RNA secondary structures in the Rfam database. M.M. conceived the original research idea of full-genome alignment with RNA secondary structure annotation, designed the study, performed alignment construction and refinement, and wrote the manuscript. All authors read and approved the final version of the manuscript.
Funding
Open Access funding enabled and organized by Projekt DEAL. This work is funded by NFDI4Microbiota-NFDI 28/1-Project-ID 460129525, the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy-EXC 2051-Project-ID 390713860, the German state of Thuringia via the Thüringer Aufbaubank-Project-ID 2021 FGI 0009, the Thüringer Ministerium für Wirtschaft, Wissenschaft und Digitale Gesellschaft Grant “DigLeben”-Project-ID 5575/10-9 TMWBDG, the EU Horizon 2020 Grant “VIROINF”-Project-ID 955974, and FOR 5151 QuaLiPerF-TP4: MA5082/15-1, and the DFG-SFB 1021-Project-ID 197785619.
Data availability
The alignments are available in the supplementary information (see Files F5–F9): (1) the nucleotide alignment with additional annotations such as RNA secondary structures and genes (F5); (2) the protein alignment with gene annotation (F6); and (3) the nucleotide and protein alignment combined with additional annotations such as RNA secondary structures and genes (F7). The RNA secondary structure models are provided in the Rfam database46,47.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-62897-0.
References
- 1.Organization, W. H. Global progress report on HIV, viral hepatitis and sexually transmitted infections, 2021. Accountability for the global health sector strategies 2016–2021: Actions for impact (World Health Organization, 2021).
- 2.Mast EE, et al. Risk factors for perinatal transmission of hepatitis C virus (HCV) and the natural history of HCV infection acquired in infancy. J. Infect. Diseases. 2005;192:1880–1889. doi: 10.1086/497701. [DOI] [PubMed] [Google Scholar]
- 3.Prasad M, et al. Risk factors for perinatal transmission of Hepatitis C virus. Obstetr. Gynecol. 2023;142:449–456. doi: 10.1097/AOG.0000000000005306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roudot-Thoraval F. Epidemiology of hepatitis C virus infection. Clin. Res. Hepatol. Gastroenterol. 2021;45:101596. doi: 10.1016/j.clinre.2020.101596. [DOI] [PubMed] [Google Scholar]
- 5.Choo Q-L, et al. Isolation of a cDNA cLone derived from a blood-borne non-A, non-B viral Hepatitis genome. Science. 1989;244:359–362. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- 6.Kuo G, et al. An assay for circulating antibodies to a major etiologic virus of human non-A, non-B hepatitis. Science. 1989;244:362–364. doi: 10.1126/science.2496467. [DOI] [PubMed] [Google Scholar]
- 7.Takamizawa A, et al. Structure and organization of the hepatitis C virus genome isolated from human carriers. J. Virol. 1991;65:1105–1113. doi: 10.1128/jvi.65.3.1105-1113.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chu D, et al. Systematic analysis of enhancer and critical cis-acting RNA elements in the protein-encoding region of the hepatitis C virus genome. J. Virol. 2013;87:5678–5696. doi: 10.1128/JVI.00840-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mauger DM, et al. Functionally conserved architecture of hepatitis C virus RNA genomes. Proc. Natl. Acad. Sci. 2015;112:3692–3697. doi: 10.1073/pnas.1416266112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fricke M, et al. Conserved RNA secondary structures and long-range interactions in hepatitis C viruses. RNA. 2015;21:1219–1232. doi: 10.1261/rna.049338.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pirakitikulr N, Kohlway A, Lindenbach B, Pyle A. The coding region of the HCV genome contains a network of regulatory RNA structures. Mol. Cell. 2016;62:111–120. doi: 10.1016/j.molcel.2016.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown EA, Zhang H, Ping LH, Lemon SM. Secondary structure of the 5’ nontranslated regions of hepatitis C virus and pestivirus genomic RNAs. Nucleic Acids Res. 1992;20:5041–5045. doi: 10.1093/nar/20.19.5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tanaka T, Kato N, Cho MJ, Shimotohno K. A novel sequence found at the 3’ terminus of hepatitis C virus genome. Biochem. Biophys. Res. Commun. 1995;215:744–749. doi: 10.1006/bbrc.1995.2526. [DOI] [PubMed] [Google Scholar]
- 14.Blight KJ, Rice CM. Secondary structure determination of the conserved 98-base sequence at the 3’ terminus of hepatitis C virus genome RNA. J. Virol. 1997;71:7345–7352. doi: 10.1128/jvi.71.10.7345-7352.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Niepmann M, Shalamova LA, Gerresheim GK, Rossbach O. Signals involved in regulation of hepatitis C virus RNA genome translation and replication. Front. Microbiol. 2018;9:395. doi: 10.3389/fmicb.2018.00395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Romero-López C, Berzal-Herranz A. The role of the RNA–RNA interactome in the hepatitis C virus life cycle. Int. J. Mol. Sci. 2020;21:1479. doi: 10.3390/ijms21041479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schult P, et al. microRNA-122 amplifies hepatitis C virus translation by shaping the structure of the internal ribosomal entry site. Nat. Commun. 2018;9:2613. doi: 10.1038/s41467-018-05053-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsukiyama-Kohara K, Iizuka N, Kohara M, Nomoto A. Internal ribosome entry site within hepatitis C virus RNA. J. Virol. 1992;66:1476–1483. doi: 10.1128/jvi.66.3.1476-1483.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science (New York, NY) 2005;309:1577–1581. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- 20.Henke JI, et al. microRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J. 2008;27:3300–3310. doi: 10.1038/emboj.2008.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shimakami T, et al. Stabilization of hepatitis C virus RNA by an Ago2-miR-122 complex. Proc. Natl. Acad. Sci. USA. 2012;109:941–946. doi: 10.1073/pnas.1112263109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.You S, Stump DD, Branch AD, Rice CM. A cis-acting replication element in the sequence encoding the NS5B RNA-dependent RNA polymerase is required for hepatitis C virus RNA replication. J. Virol. 2004;78:1352–1366. doi: 10.1128/jvi.78.3.1352-1366.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee H, Shin H, Wimmer E, Paul AV. cis-acting RNA signals in the NS5B C-terminal coding sequence of the hepatitis C virus genome. J. Virol. 2004;78:10865–10877. doi: 10.1128/JVI.78.20.10865-10877.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsukiyama-Kohara K, Kohara M. Hepatitis C virus: Viral Quasispecies and genotypes. Int. J. Mol. Sci. 2017;19:23. doi: 10.3390/ijms19010023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Galli A, Bukh J. Mechanisms and consequences of genetic variation in hepatitis C virus (HCV) Curr. Topics Microbiol. Immunol. 2023;439:237–264. doi: 10.1007/978-3-031-15640-3_7. [DOI] [PubMed] [Google Scholar]
- 26.Domingo E, Perales C. Viral quasispecies. PLoS Genet. 2019;15:e1008271. doi: 10.1371/journal.pgen.1008271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith DB, et al. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: Updated criteria and genotype assignment web resource. Hepatology. 2014;59:318–327. doi: 10.1002/hep.26744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Borgia SM, et al. Identification of a novel hepatitis C virus genotype from Punjab, India: Expanding classification of hepatitis C virus into 8 genotypes. J. Infect. Diseases. 2018;218:1722–1729. doi: 10.1093/infdis/jiy401. [DOI] [PubMed] [Google Scholar]
- 29.BV-BRC. Introducing the Bacterial and Viral Bioinformatics Resource Center (BV-BRC): a resource combining PATRIC, IRD and ViPR. Nucleic Acids Res. 51, D678–D689. 10.1093/nar/gkac1003 (2023). [DOI] [PMC free article] [PubMed]
- 30.McInnes, L., Healy, J. & Astels, S. hdbscan: Hierarchical density based clustering. J. Open Source Softw. 10.21105/joss.00205 (2017).
- 31.Li W, Godzik A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 32.Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28:3150–3152. doi: 10.1093/bioinformatics/bts565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Steinegger M, Söding J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017;35:1026–1028. doi: 10.1038/nbt.3988. [DOI] [PubMed] [Google Scholar]
- 34.Mercier, C., Boyer, F., Bonin, A. & Coissac, E. SUMATRA and SUMACLUST: fast and exact comparison and clustering of sequences. Programs and Abstracts of the SeqBio 2013 Workshop (2013). https://git.metabarcoding.org/obitools/sumaclust/wikis/home/.
- 35.Rognes T, Flouri T, Nichols B, Quince C, Mahé F. VSEARCH: A versatile open source tool for metagenomics. PeerJ. 2016;4:e2584. doi: 10.7717/peerj.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lamkiewicz, K. & Marz, M. ViralClust—Find representative viruses for your dataset. 202x (in preparation), www.github.com/klamkiew/viralclust/.
- 37.Sayers EW, et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2022;50:D20–D26. doi: 10.1093/nar/gkab1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Katoh K, Misawa K, Kuma K-I, Miyata T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Will S, Reiche K, Hofacker IL, Stadler PF, Backofen R. Inferring non-coding RNA families and classes by means of genome-scale structure-based clustering. PLoS Comput. Biol. 2007;3:e65. doi: 10.1371/journal.pcbi.0030065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van Rossum, G. & Drake, F. L. Python 3 Reference Manual (CreateSpace, 2009).
- 41.Lamkiewicz, K. & Marz, M. VeGETA—Viral GEnome sTructure Alignments. 202x (in preparation). https://github.com/klamkiew/vegeta.
- 42.Fricke M, Gerst R, Ibrahim B, Niepmann M, Marz M. Global importance of RNA secondary structures in protein-coding sequences. Bioinformatics (Oxford, England) 2019;35:579–583. doi: 10.1093/bioinformatics/bty678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Firth AE, Brierley I. Non-canonical translation in RNA viruses. J. General Virol. 2012;93:1385–1409. doi: 10.1099/vir.0.042499-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nassal M, Junker-Niepmann M, Schaller H. Translational inactivation of RNA function: Discrimination against a subset of genomic transcripts during HBV nucleocapsid assembly. Cell. 1990;63:1357–1363. doi: 10.1016/0092-8674(90)90431-d. [DOI] [PubMed] [Google Scholar]
- 45.Chang T-C, et al. UNR, a new partner of poly(A)-binding protein, plays a key role in translationally coupled mRNA turnover mediated by the c-fos major coding-region determinant. Genes Develop. 2004;18:2010–2023. doi: 10.1101/gad.1219104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR. Rfam: An RNA family database. Nucleic Acids Res. 2003;31:439–441. doi: 10.1093/nar/gkg006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kalvari I, et al. Rfam 14: Expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 2020;49:D192–D200. doi: 10.1093/nar/gkaa1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Larkin MA, et al. Clustal W and Clustal X version 2.0. Bioinformatics (Oxford, England) 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 49.Jeanmougin F, Thompson JD, Gouy M, Higgins DG, Gibson TJ. Multiple sequence alignment with Clustal X. Trends Biochem. Sci. 1998;23:403–405. doi: 10.1016/s0968-0004(98)01285-7. [DOI] [PubMed] [Google Scholar]
- 50.Clamp M, Cuff J, Searle SM, Barton GJ. The Jalview Java alignment editor. Bioinformatics. 2004;20:426–427. doi: 10.1093/bioinformatics/btg430. [DOI] [PubMed] [Google Scholar]
- 51.Griffiths-Jones S. RALEE-RNA ALignment editor in Emacs. Bioinformatics (Oxford, England) 2005;21:257–259. doi: 10.1093/bioinformatics/bth489. [DOI] [PubMed] [Google Scholar]
- 52.Honda M, Rijnbrand R, Abell G, Kim D, Lemon SM. Natural variation in translational activities of the 5’ nontranslated RNAs of hepatitis C virus genotypes 1a and 1b: Evidence for a long-range RNA-RNA interaction outside of the internal ribosomal entry site. J. Virol. 1999;73:4941–4951. doi: 10.1128/JVI.73.6.4941-4951.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim YK, Lee SH, Kim CS, Seol SK, Jang SK. Long-range RNA-RNA interaction between the 5’ nontranslated region and the core-coding sequences of hepatitis C virus modulates the IRES-dependent translation. RNA (New York, NY) 2003;9:599–606. doi: 10.1261/rna.2185603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Beguiristain N, Robertson HD, Gómez J. RNase III cleavage demonstrates a long range RNA: RNA duplex element flanking the hepatitis C virus internal ribosome entry site. Nucleic Acids Res. 2005;33:5250–5261. doi: 10.1093/nar/gki822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Díaz-Toledano R, Ariza-Mateos A, Birk A, Martínez-García B, Gómez J. In vitro characterization of a miR-122-sensitive double-helical switch element in the 5’ region of hepatitis C virus RNA. Nucleic Acids Res. 2009;37:5498–5510. doi: 10.1093/nar/gkp553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goergen D, Niepmann M. Stimulation of Hepatitis C Virus RNA translation by microRNA-122 occurs under different conditions in vivo and in vitro. Virus Res. 2012;167:343–352. doi: 10.1016/j.virusres.2012.05.022. [DOI] [PubMed] [Google Scholar]
- 57.McMullan LK, et al. Evidence for a functional RNA element in the hepatitis C virus core gene. Proc. Natl. Acad. Sci. USA. 2007;104:2879–2884. doi: 10.1073/pnas.0611267104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vassilaki N, et al. Role of the hepatitis C virus core+1 open reading frame and core cis-acting RNA elements in viral RNA translation and replication. J. Virol. 2008;82:11503–11515. doi: 10.1128/JVI.01640-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sweeney BA, et al. R2DT is a framework for predicting and visualising RNA secondary structure using templates. Nat. Commun. 2021;12:3494. doi: 10.1038/s41467-021-23555-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lorenz R, et al. ViennaRNA package 2.0. Algorithms Mol. Biol. 2011;6:26. doi: 10.1186/1748-7188-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cristofari G, et al. The hepatitis C virus Core protein is a potent nucleic acid chaperone that directs dimerization of the viral (+) strand RNA in vitro. Nucleic Acids Res. 2004;32:2623–2631. doi: 10.1093/nar/gkh579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shetty S, Kim S, Shimakami T, Lemon SM, Mihailescu M-R. Hepatitis C virus genomic RNA dimerization is mediated via a kissing complex intermediate. RNA (New York, NY) 2010;16:913–925. doi: 10.1261/rna.1960410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Castillo-Martínez J, et al. Structure and function analysis of the essential 3’X domain of hepatitis C virus. RNA (New York, NY) 2020;26:186–198. doi: 10.1261/rna.073189.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Friebe P, Boudet J, Simorre J-P, Bartenschlager R. Kissing-loop interaction in the 3’ end of the hepatitis C virus genome essential for RNA replication. J. Virol. 2005;79:380–392. doi: 10.1128/JVI.79.1.380-392.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Romero-López C, Barroso-Deljesus A, García-Sacristán A, Briones C, Berzal-Herranz A. End-to-end crosstalk within the hepatitis C virus genome mediates the conformational switch of the 3’X-tail region. Nucleic Acids Res. 2014;42:567–582. doi: 10.1093/nar/gkt841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Castillo-Martínez, J., Fan, L., Szewczyk, M. P., Wang, Y.-X. & Gallego, J. The low-resolution structural models of hepatitis C virus RNA subdomain 5BSL3.2 and its distal complex with domain 3’X point to conserved regulatory mechanisms within the Flaviviridae family. Nucleic Acids Res. 50, 2287–2301. 10.1093/nar/gkac061 (2022). [DOI] [PMC free article] [PubMed]
- 67.Zhang J, et al. Inhibition of hepatitis C virus replication by pol III-directed overexpression of RNA decoys corresponding to stem-loop structures in the NS5B coding region. Virology. 2005;342:276–285. doi: 10.1016/j.virol.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 68.Rheault M, Cousineau SE, Fox DR, Abram QH, Sagan SM. Elucidating the distinct contributions of miR-122 in the HCV life cycle reveals insights into virion assembly. Nucleic Acids Res. 2023;51:2447–2463. doi: 10.1093/nar/gkad094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rivas E. RNA structure prediction using positive and negative evolutionary information. PLOS Comput. Biol. 2020;16:e1008387. doi: 10.1371/journal.pcbi.1008387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Waldl, M., Will, S., Wolfinger, M. T., Hofacker, I. L. & Stadler, P. F. Bi-alignments as Models of Incongruent Evolution of RNA Sequence and Secondary Structure. In Cazzaniga, P., Besozzi, D., Merelli, I. & Manzoni, L. (eds.) Computational Intelligence Methods for Bioinformatics and Biostatistics, vol. 12313, 159–170, 10.1007/978-3-030-63061-4_15 (Springer International Publishing, Cham, 2020). Series Title: Lecture Notes in Computer Science.
- 71.Stadler PF, Will S. Bi-alignments with affine gaps costs. Algorithms Mol. Biol. 2022;17:10. doi: 10.1186/s13015-022-00219-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nowick, K., Walter Costa, M. B., Höner Zu Siederdissen, C. & Stadler, P. F. Selection pressures on RNA sequences and structures. Evolut. Bioinform. Online. 15, 1176934319871919. 10.1177/1176934319871919 (2019). [DOI] [PMC free article] [PubMed]
- 73.Gerresheim GK, et al. Ribosome pausing at inefficient codons at the end of the replicase coding region is important for hepatitis C virus genome replication. Int. J. Mol. Sci. 2020;21:6955. doi: 10.3390/ijms21186955. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The alignments are available in the supplementary information (see Files F5–F9): (1) the nucleotide alignment with additional annotations such as RNA secondary structures and genes (F5); (2) the protein alignment with gene annotation (F6); and (3) the nucleotide and protein alignment combined with additional annotations such as RNA secondary structures and genes (F7). The RNA secondary structure models are provided in the Rfam database46,47.