

Abstract
To improve the efficacy of organic solar cells (OSCs), novel small acceptor molecules (CTD1–CTD7) were designed by modification at the terminal acceptors of reference compound CTR. The optoelectronic properties of the investigated compounds (CTD1–CTD7) were accomplished by employing density functional theory (DFT) in combination with time-dependent density functional theory (TD-DFT). The M06 functional along with a 6-311G(d,p) basis set was utilized for calculating various parameters such as: frontier molecular orbitals (FMO), absorption maxima (λmax), binding energy (Eb), transition density matrix (TDM), density of states (DOS), and open circuit voltage (Voc) of entitled chromophores. A red shift in the absorption spectra of all designed chromophores (CTD1–CTD7) was observed as compared to CTR, accompanied by low excitation energy. Particularly, CTD4 was characterized by the highest λmax value of 685.791 nm and the lowest transition energy value of 1.801 eV which might be ascribed to the robust electron-withdrawing end-capped acceptor group. The observed reduced binding energy (Eb) was linked to an elevated rate of exciton dissociation and substantial charge transfer from central core in HOMO towards terminal acceptors in LUMO. These results were further supported by the outcomes from TDM and DOS analyses. Among all entitled chromophores, CTD4 exhibited bathochromic shift (685.791 nm), minimum HOMO/LUMO band gap of 2.347 eV with greater CT. Thus, it can be concluded that by employing molecular engineering with efficient acceptor moieties, the efficiency of photovoltaic materials could be improved.
Keywords: NF-OSCs, Photovoltaic response, Chrysene derivatives, Open circuit voltage, Binding energy
Subject terms: Chemistry, Materials science, Optics and photonics
Introduction
Over the last decade, significant advancements have been achieved in organic solar cells (OSCs) on account of enhanced insights into device optimization, molecular design, and operational mechanisms1. Organic solar cells consist of organic semiconductors acting as electron donors and acceptors2. Despite the substantial development of donor materials over three decades, a limited number of acceptors have historically demonstrated the ability to achieve high power conversion efficiencies3. Non-fullerene acceptor (NFA) molecules have emerged as an auspicious and competent category of compounds, characterized by high efficiencies, low band gaps, tunable energy levels, and strong UV–Vis absorption4. The development of NFAs mark a transformative leap in cell efficiencies, with the notable achievement of reaching the significant milestone of 20%5,6. Recently, modifying the chemical structure has proven to be a successful strategy for adjusting the photovoltaic characteristics of non-fullerene compounds. This modification involves altering the side chains, end-capped acceptor units, and the core molecule7,8. Functionalizing the end groups (EG) of central donor core with non-fullerene acceptors (NFA) demonstrated to be an effective approach for boosting the performance of organic solar cells (OSCs)7–9. This, in turn, plays a substantial role in shaping the power conversion efficiency (PCE) and various other performance metrics of NFA-based OSCs (NF-OSCs)9. The A–π–A architecture, consisting of central electron-donating π-spacer unit combined with two electron-deficient end-capped acceptor groups on both sides is considered as an effective approach in NF–OSCs to improve intramolecular charge transfer (ICT)10. A novel electron-donating core, formed by condensing chrysene with two thiophenes via two dihydrobenzene rings, was synthesized by Lu and his coworkers. Utilizing this core along with two electron-accepting end groups of 1,1-dicyanomethylene-3-indanone, a new Z-shaped fused-ring electron acceptor was developed and synthesized. This chrysene-based compound exhibited strong absorption within the 500–850 nm range, a bandgap measuring 1.50 eV, and a charge mobility of 2.5 × 10−4 cm2 V−1 s−1 11. In another report, chrysene based compound with PCE value upto 24.7% on blending with poly (3-hexylthiophene) (P3HT) and phenyl-C61-butyric acid methyl ester (PCBM) polymers was stated12. Moreover, in a previous study, a chrysene-core based derivative with Voc value of 1.20 V has been reported13.
Encouraged by findings, we developed A–π–A type seven novel chrysene-based, non-fullerene, fused-ring electron acceptor materials (CTD1–CTD7). The designing of reference compound CTR has been carried out by the insertion of highly efficient benzothiophene end-capped acceptor at the peripherals of a recently synthesized parent molecule CTIC-4F24. The DFT study of compounds containing chrysene core fused with benzodithiophene terminal acceptors has not been reported yet. Introducing the electron-proficient benzothiophene unit into the end-group raises the energy levels of the lowest unoccupied molecular orbitals (LUMO) in non-fullerene acceptors (NFAs), resulting in increased Voc outputs. Moreover, it encourages antiparallel π–π stacking of the end-groups, thereby facilitating efficient electron transport14. The reference compound was further fabricated to design CTD1–CTD7 derivatives by introducing various electrophilic groups in to the benzothiophene acceptors. The newly designed molecules CTD1–CTD7 possess unique characteristics attributable to the presence of diverse groups and atoms in their terminal acceptor regions. Hence, employing various electrophilic groups leads to the formation of more efficient OSC molecules from chrysene core.
To assess the photovoltaic and optoelectronic properties of these materials, DFT and TD-DFT calculations have been employed. Analyses including frontier molecular orbital (FMO), density of states (DOS), transition density matrix (TDM), binding energy (Eb), and open circuit voltage (Voc) have been conducted to elucidate the electronic, photophysical, photovoltaic, and charge transfer characteristics of the reference molecule CTR and its designed compounds (CTD1–CTD7). Noticeably, all the designed derivatives showed good Voc values, revealing their potential for paramount current generation capacity. Thus, it is highly anticipated that these newly designed compounds (CTD1–CTD7) will play a remarkable role in advancing the development of highly efficient organic solar cell (OSC) materials.
Computational procedure
The computational investigation involving density functional theory (DFT) and time-dependent density functional theory (TD-DFT) were carried out using the Gaussian 0915 software package, with visualization of results facilitated by GaussView 6.016. The M06 17 coupled with the 6-311G(d,p) basis set was employed to perform all analyses. At first geometrical optimization was accomplished to get true minima structures. By utilizing these optimized structures, various photovoltaic and optoelectronic parameters such as frontier molecular orbitals (FMOs), transition density matrix (TDM), global reactivity descriptors (GRDs), density of state graphs (DOS), binding energy (Eb), and open circuit voltage (Voc) were computed at the entitled functional. Multiple software like Avogadro18, Multiwfn19, PyMOlyze20, GaussSum21, Origin22 and Chemcraft23 were used to interpret data in tabular and and different graphs (Fig. 1).
Figure 1.

Optimized structures of CTR tailored chromophores CTD1–CTD7.
Results and discussion
Inspiring from the research of Zhao et al.24, we have designed a series of small organic molecular non-fullerene acceptors (NFAs) from parent compound CTIC-4F categorized as CTR and CTD1–CTD7 with A–π–A architecture, incorporating a 4,4,12,12-tetramethyl-4,12-dihydrodibenzo[4,5:10,11]tetraceno[1,2-b:7,8-b']dithiophene π-spacer moiety. The terminal acceptor group of the parent molecule was substituted with benzothiophene-based acceptor to design the reference chromophore (CTR), as depicted in Fig. 2.
Figure 2.

Designing of reference molecule (CTR) from parent chromophore.
The peripheral electron accepting units of CTR were systematically substituted with diverse electron-deficient groups with the objective of formulating efficient non-fullerene OSCs. Through the substitution of terminal groups like –F, –Cl, –CN, –NO2, –CF3, –SO3H, and –CH3COO, seven distinct derivatives CTD1, CTD2, CTD3, CTD4, CTD5, CTD6 and CTD7 were designed, respectively (Fig. 3). The IUPAC names and 2D-structures of these derivatives are arranged in Table S21, while the optimized structures of designed derivatives are presented in Fig. 1. The Cartesian coordinates of investigated compounds are portrayed in Tables S22–S29.
Figure 3.

Schematic representation of designing of derivatives (CTD1–CTD7) from reference (CTR).
The primary objective of this work is to design NFAs with enhanced photovoltaic properties. In this study, our focus is on designing new NFAs that exhibit significantly improved photovoltaic characteristics, including a narrow optical bandgap, low excitation energy and increased absorption. The theoretically simulated parameters aim to facilitate the development of molecules with superior properties for solar cell applications.
Electronic studies
Investigating the FMOs provides valuable insights into a molecule's reactivity, helping establish the active site through orbital distribution25. The determination of effective charge transfer across donor and acceptor components in photovoltaic devices heavily relies on assessing the energy levels of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO)26–29. Analyzing the HOMO–LUMO relationship is essential for exploring quantum chemical properties. This study aids in predicting reactive sites within π-electron systems and offers explanations for various reactions within conjugated frameworks30. The energy band gap in frontier molecular orbitals (FMOs) plays a crucial role in molecular modeling, influencing dynamic structural stability, charge generation, electrical conductivity, chemical reactivity, short-circuit current (Jsc), power conversion efficiency (PCE) and open-circuit voltage (Voc)31,32. A smaller band gap (Eg) in FMOs facilitates the flow of charge from donor’s HOMO and acceptor’s LUMO, facilitating current generation33. FMO analysis on reference compound CTR and designed derivatives CTD1–CTD7 has been conducted, utilizing the M06 level of theory in conjunction with the 6-311G(d,p) basis set. Figure 2 illustrates the alignment of FMOs of CTR and CTD1–CTD7. Table 1 represents energies of HOMO–LUMO and their respective energy gaps.
Table 1.
Energies of frontier molecular orbitals of the CTR and CTD1–CTD7.
| Compound | HOMO | LUMO | Eg |
|---|---|---|---|
| CTR | − 5.772 | − 3.279 | 2.493 |
| CTD1 | − 5.816 | − 3.348 | 2.468 |
| CTD2 | − 5.833 | − 3.377 | 2.456 |
| CTD3 | − 5.938 | − 3.534 | 2.404 |
| CTD4 | − 5.950 | − 3.578 | 2.327 |
| CTD5 | − 5.885 | − 3.443 | 2.442 |
| CTD6 | − 5.946 | − 3.546 | 2.400 |
| CTD7 | − 5.834 | − 3.383 | 2.451 |
Band gap = ELUMO − EHOMO, Units in eV.
Table 1 illustrates the HOMO and LUMO energies for reference molecule CTR as − 5.772/− 3.279 eV corresponding energy gap of 2.493 eV. In comparison, all derivatives (CTD1–CTD7) exhibited HOMO/LUMO energies of − 5.816/− 3.348, − 5.833/− 3.377, − 5.938/− 3.534, − 5.950/− 3.578, − 5.885/− 3.443, − 5.946/− 3.546, and − 5.834/− 3.383 eV, along with energy gaps of 2.468, 2.456, 2.404, 2.327, 2.442, 2.400, and 2.451 eV, respectively. The decreasing energy gap observed in these derivatives can be ascribed to the successive substitution of robust electron-withdrawing moieties, increasing acceptor strength and enhancing the charge transference as compared to CTR. It is worth noting that the intramolecular charge transfer (ICT) of a compound is inversely proportional to energy band gap. A minor energy gap indicates better transfer of charge and vice versa34. General decreasing sequence of band gap is given as: CTR > CTD1 > CTD2 > CTD7 > CTD5 > CTD3 > CTD6 > CTD4 with Eg values 2.493, 2.468, 2.456, 2.451, 2.442, 2.404, 2.400, and 2.327 eV, respectively. Thus, CTD4 chromophore represented an effective charge mobility across terminal acceptors through the chrysene donor by showing the lowest energy gap between the molecular orbitals, making it an efficient candidate for applications in photovoltaic devices.
Figure 4 portrays surface diagrams of the FMOs, showcasing the spreading of electronic density clouds across molecules. For all the designed compounds (except CTD4), there is a significant concentration of charge density in the central part in HOMO, with a minor presence of electronic cloud observed over the end-capped acceptor groups in LUMO. For CTD4, the HOMO density is predominantly intense on the π-bridge, but the LUMO density is highly concentrated in the nitro group among electron-deficient end-capped groups. Consequently, compounds under analysis exhibited ICT from one terminal acceptor to the other across the π-core. HOMO − 1/LUMO + 1 and HOMO − 2/LUMO + 2 energies are detailed in Tables S1 and S2 respectively, and their corresponding FMO diagrams are presented in Fig. S1. Similar phenomena in terms of energies and charge transfer are observed among above level orbitals (HOMO − 1/LUMO + 1 and HOMO − 2/LUMO + 2). The GRDs of entitled chromophores are tabulated Table S3.
Figure 4.
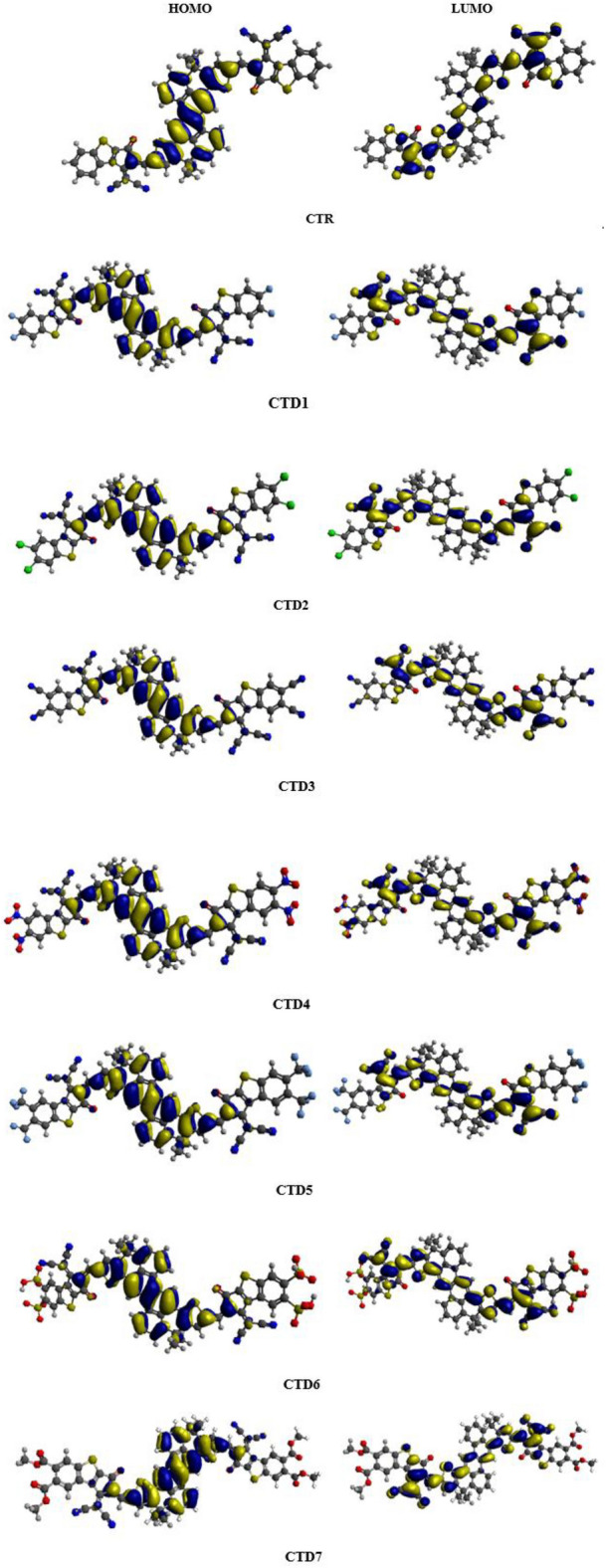
Energy Orbital diagrams of CTR and designed chromophores CTD1–CTD7.
Density of states (DOS)
DOS study is employed to determine the distinct contribution of each fragment within the molecular system35. This study reveals the distribution of charge, indicating that HOMO possesses a significant capacity to donate electrons, while LUMO exhibits capability to accept electrons36,37. The distribution of electronic charges varies with different acceptor units, as evidenced by the HOMO/LUMO percentage of density of states (DOS) presented in Table S4. In this context, the acceptor units show contributions of electronic charges as follows: 16.5%, 16.4%, 16.4%, 16.4%, 16.5%, 16.2%, 16.4%, and 16.3% to HOMO, while 65.1%, 66.2%, 65.9%, 62.5%, 63.5%, 63.8%, 61.3%, and 66.1% to LUMO for CTR and CTD1–CTD7, respectively. Similarly, the π-bridge demonstrates charge distribution as follows: 83.5%, 83.6%, 83.5%, 83.6%, 83.5%, 83.8%, 83.6%, and 83.7% to HOMO, whereas 34.9%, 33.8%, 34.1%, 37.5%, 36.5%, 36.2%, 38.7%, and 33.9% to LUMO for CTR and CTD1–CTD7, respectively. From DOS graphs, LUMO is indicated by positive values, HOMO is specified by negative values along the x-axis. The energy gap (Eg) expresses the distance between HOMOs and LUMOs (Fig. 5). CTD4 DOS spectrum suggested that the maximum positive charge density (HOMO) is located over the π-spacer at approximately − 10.5 eV, while LUMO is primarily located on the acceptor, with its largest peak occurring at 3.5 eV. Hence, DOS analysis revealed massive transfer of charges across terminal acceptors through the π-bridge in all studied chromophores (CTD1–CTD7).
Figure 5.

Density of states (DOS) plots for compounds CTR and CTD1–CTD7.
Optical properties
Optical properties are essential for evaluating overall operational capabilities of organic solar cells (OSCs)38,39. The UV–Vis spectra of all the investigated molecules have been thoroughly examined using TD-DFT/M06/6-311G(d,p). Although, UV–Vis data were computed for six excited states (as presented in Tables S5–S20), only data pertaining to the first excited state are shown in Table 2. This selection is justified as the first excited state contains maximum values of wavelength of absorption (λmax), oscillator strength (fos), first excitation energy (E), and the highest percentage of intramolecular charge transfer (ICT).
Table 2.
The outcomes of UV–visible absorption spectroscopy for CTR and CTD1–CTD7 in both solvent and gaseous phases.
| Medium | Compounds | max (nm) | E(eV) | fos | MO contributions |
|---|---|---|---|---|---|
| Solvent | CTR | 651.109 | 1.904 | 2.005 | H → L (94%), H-1 → L + 1 (2%) |
| CTD1 | 657.497 | 1.886 | 1.979 | H → L (94%), H-1 → L + 1 (2%), H → L + 2 (2%) | |
| CTD2 | 662.061 | 1.873 | 2.081 | H → L (93%), H → L + 2 (2%) | |
| CTD3 | 678.510 | 1.827 | 2.228 | H → L (95%), | |
| CTD4 | 685.791 | 1.808 | 2.210 | H → L (95%), | |
| CTD5 | 666.295 | 1.861 | 2.094 | H → L (94%), H → L + 2 (2%) | |
| CTD6 | 680.223 | 1.823 | 2.243 | H → L (95%), | |
| CTD7 | 663.301 | 1.869 | 2.086 | H → L (94%), H → L + 2 (2%) | |
| Gaseous phase | CTR | 617.913 | 2.007 | 1.797 | H → L (96%) H-1 → L + 1 (2%) |
| CTD1 | 625.204 | 1.983 | 1.747 | H → L (96%) | |
| CTD2 | 628.627 | 1.972 | 1.882 | H → L (96%) | |
| CTD3 | 643.840 | 1.926 | 2.031 | H → L (97%) | |
| CTD4 | 647.606 | 1.915 | 2.027 | H → L (97%) | |
| CTD5 | 632.863 | 1.959 | 1.901 | H → L (96%) | |
| CTD6 | 644.643 | 1.923 | 2.092 | H → L (97%) | |
| CTD7 | 628.819 | 1.972 | 1.915 | H → L (96%) |
fos = oscillator strength, H = HOMO, L = LUMO, MO = molecular orbital
UV–Vis spectra for both the solution and gas phases are illustrated in Fig. 6. In the gaseous phase, the reference CTR and designed molecules CTD1–CTD7 exhibited a red shift in absorbance. But the values of maximum absorption in the chloroform solvent are shifted more bathochromically as a consequence of solvent effect. The optical properties of a compound are also substantially influenced by its internal morphology. The interaction of π–π conjugation between the rings of end groups is crucial in determining intermolecular electronic couplings and facilitating charge transport40,41. Moreover, as the electron-pulling capacity of the end groups increases, the intramolecular charge transfer (ICT) between the core and end groups strengthens, resulting in a red-shift in the absorption42. The CTR exhibited the λmax of 651.109 nm by showing oscillator strength and excitation energy and at 2.005 and 1.904 eV, respectively. Increase in absorption wavelength is observed as CTD1 (657.497 nm), CTD2 (662.061 nm), CTD7 (663.301 nm) and CTD5 (666.295 nm). This increase in wavelength can be attributed to the robust electrophilic nature of their end-capped acceptors. Moreover, a bathochromic shift is also observed in compounds CTD3 (678.510 nm) and CTD6 (680.223 nm). Interestingly, CTD4 showed the maximum results of λmax (685.791 nm) accompanying with minimum E (1.8808 eV) owing to the incorporation of powerful electron-pulling nitro groups in end-capped acceptor moieties. Decreasing trend of λmax in the gaseous phase for all compounds can be found as follows: CTD4 > CTD6 > CTD3 > CTD5 > CTD7 > CTD2 > CTD1 > CTR. The increase in absorption spectra is investigated as the electron withdrawing efficacy of moieties increased over the terminal acceptors. Remarkably, exactly similar trend of λmax in the solvent phase has been observed by proving CTD4 as excellent organic material for obtaining maximum efficiency of solar cell materials.
Figure 6.

UV–Vis absoption spectra of designed compounds (a) in solvent phase and (b) in gaseous phase.
Greater the first molecular orbital (MO) contributions, higher the absorbance wavelength of compounds. The results clearly indicate that excitations originating from the first excited states, specifically HOMO → LUMO transitions, predominantly contribute (between 93 and 97%) to the absorption maxima observed in the designed compounds, regardless of whether they are in solvent or gas phases. It is noteworthy that molecules in which H → L transitions highly contribute tend to have higher λmax values. Therefore, CTD3, CTD4, and CTD6 holding the highest H → L contributions of 95% in solvent and 97% in gas, which account for the red-shift observed in the absorption spectra of these compounds.
Transition density matrix (TDM) and exciton binding energy (Eb)
The TDMs of designed compounds has also been examined using Multiwfn, as shown in Fig. 7. The transition density matrix provides insights into the collaboration of donor and acceptor moieties in the excited state, as well as information about location of electron–hole and electronic excitations43. It's noteworthy that hydrogen atoms were excluded from the analysis due to their minimal contribution in transitions.
Figure 7.

TDM graphs of designed compounds CTR and CTD1–CTD7.
TDM heatmaps elucidated the electronic excitation and nature of the transition in the first singlet excited state. Each molecule was divided into π-spacer (chrysene) and benzothiophene acceptor units with diverse electron-withdrawing groups (refer to Fig. 1). Almost, all the derivatives show a similar pattern of charge transference. The designed molecules (CTD1–CTD7) exhibited robust diagonal electron coherence in the transition density matrix (TDM) heatmaps. It is evident from the graphs that electron coherence effectively transferred from π-bridge towards the terminal acceptors (A), enabling the movement of electron density across the end-capped acceptors without entrapment. The results from TDM heat maps suggest a straightforward, smoother, and enhanced exciton dissociation in the excited state, offering potential advancements in solar cell technology.
The generation of the Frenkel exciton through the photoexcitation of small, conjugated molecules is a widely acknowledged phenomenon. In this phase, charge carriers are tightly bound by electrostatic forces, as opposed to existing as free charges. The successful dissociation of excitons leads to the creation of electrons and holes, a process made feasible by the presence of designed structures with low exciton binding energies44. The binding energy (Eb) is determined by subtracting energy required for optical transition (represented by ΔE) and the energy gap (Eg) between the HOMO and LUMO energy levels45. The equation used to calculate Eb is expressed as follows:
| 1 |
According to above equation, ΔE represents energy required by electron for ground state to the first singlet excitation, while Eg denotes the energy gap of HOMO and LUMO46. The exciton binding energy (Eb) calculations are provided in Table 3. Notably, the Eb value for the designed molecule CTD4 is the lowest (0.519 eV) among all the derivatives, making it an effective candidate to be used in new generation OSCs. The overall increasing trend of Eb for tailored chromophores is found as: CTD4 < CTD6 = CTD3 < CTD5 < CTD7 = CTD1 < CTD2 < CTR.
Table 3.
Calculated exciton binding energies for CTR and CTD1–CTD7 derivatives.
| Compounds | Eg | ΔE | Eb |
|---|---|---|---|
| CTR | 2.493 | 1.904 | 0.589 |
| CTD1 | 2.468 | 1.886 | 0.582 |
| CTD2 | 2.456 | 1.873 | 0.583 |
| CTD3 | 2.404 | 1.827 | 0.577 |
| CTD4 | 2.327 | 1.808 | 0.519 |
| CTD5 | 2.442 | 1.861 | 0.581 |
| CTD6 | 2.400 | 1.823 | 0.577 |
| CTD7 | 2.451 | 1.869 | 0.582 |
Molecular electrostatic potential (MEP)
MEPs are assessed to elucidate the quantitative representation of intermolecular charge distribution between the donor and acceptor regions of a molecular system47. MEP surfaces resemble a cloud with three main colors, each indicating a specific strength of the molecule's electrostatic potential. Red hues denote negative electrostatic potential, indicating electron-rich regions that could potentially serve as attack sites for electrophiles due to the intense electron density. Conversely, blue shades denote positive electrostatic potentials, signifying electron-deficient areas prone to nucleophilic attack and repulsive regions for protons due to the molecular nuclei's presence. Green-yellow shaded sites depict regions with a neutral electrostatic potential48,49. MEP graphs for all the studied molecules are illustrated in Fig. 8. Interestingly, a noticeable occurrence of red color is observed at the acceptor regions of all designed compounds CTD1–CTD7 compared to CTR molecule, indicating them as nucleophilic region. A distinct bluish cloud is particularly prominent in the central π-region, characterizing the π-spacer as an electrophilic site. Specifically, this observation is more prominent in the CTD3 and CTD4 compounds. This analysis indicates increased charge separation in investigated compounds, suggesting that these newly devised molecules could be effectively synthesized for OSCs.
Figure 8.

Molecular electrostatic potential diagrams of investigated molecules CTR and CTD1–CTD7.
Open circuit voltage (Voc)
Measuring open-circuit voltage (Voc) of the device is essential for evaluating the efficiency of an organic solar cell (OSC). The Voc represents the maximum voltage generated by a solar device when applied current is 0. Various factors such as: charge transfer, temperature of photovoltaic device, and light intensity significantly influence Voc50. When the highest voltage is achieved, HOMO of donor material couples with LUMO of acceptor substance. To enhance Voc values, the donor's HOMO energy state should be lower, and the acceptor's LUMO energy state should be higher51. In this study, the Voc of designed acceptor molecules CTR and CTD1–CTD7 was computed by combining these molecules with a J52Cl donor. According to available data, J52Cl is an efficient donor with HOMO/LUMO energies of − 5.299 and − 2.141 eV, respectively52. So, Eq. (2) has been utilized to provide a numerical estimate of Voc results of all studied chromophores.
| 2 |
Figure 9 elucidates the Voc calculations achieved through blending J52Cl (donor polymer) with variously designed acceptors CTD1–CTD7. Amongst all the studied chromophores CTR and CTD1–CTD7, the CTR depicted the highest value of open circuit voltage (1.72 V). On the other hand, compound CTD4 showed the lowest value (1.421 V) of Voc. Moreover, other entitled molecules represented intermediate results. Thus, the following trend in the open circuit voltage results is observed: CTR > CTD1 > CTD2 > CTD7 > CTD5 > CTD3 > CTD6 > CTD4. It is obvious from the results that all the derivatives exhibited maximum value of Voc, making them proficient OSCs material.
Figure 9.

Open circuit voltage diagrams for compounds CTR and CTD1–CTD7.
Conclusion
The present research employs theoretical calculations to explore the influence of structural modifications by substituting end-capped acceptor groups in OSCs featuring the chrysene unit.
By structural modeling of chrysene unit with benzothiophene acceptors, the photovoltaic and optoelectronic properties of organic chromophores have been tuned. The quantum chemical findings demonstrated proficient charge transfer from central π-conjugated bridge to end-capped acceptor moieties in all the molecules. Open circuit voltage was calculated by developing a complex between designed chromophore and donor polymer (HOMOdonor–LUMOacceptor) and interestingly, higher photovoltaic response was seen in derivatives. The designed molecule CTD4 demonstrated good properties, i.e. lowest Eb value (0.519 eV) and minimum band gap (2.327 eV) with maximum red-shifted absorption in both the solvent (685.791 nm) and gaseous phase (647.606 nm) amongst all CTD1–CTD7. Moreover, by relating the results of FMOs with GRPs, it is found that, CTD4 derived molecule is observed as softest (0.421 eV−1) molecule amongst all the studied derivatives portraying its maximum charge transferring properties. Thus, CTD4 can be considered as a remarkable material for future OSCs applications.
Supplementary Information
Acknowledgements
The authors thank the Researchers Supporting Project number (RSP2024R6), King Saud University, Riyadh, Saudi Arabia. The S.C.O. acknowledges the support from the doctoral research fund of the Affiliated Hospital of Southwest Medical University.
Author contributions
Iqra Shafiq: Conceptualization; methodology. Shehla Kousar: Data curation; formal analysis; writing original draft. Faiz Rasool: Conceptualization; resources. Tansir Ahamad: Conceptualization; methodology; software. Khurram Shahzad Munawar: Data curation; formal analysis; validation. Saifullah Bullo: Methodology; software; project administration. Suvash Chandra Ojha: Conceptualization; Article Write-up; Final Analysis.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Iqra Shafiq and Suvash Chandra Ojha.
Contributor Information
Saifullah Bullo, Email: saifullah.bullo@bnbwu.edu.pk.
Suvash Chandra Ojha, Email: suvash_ojha@swmu.edu.cn.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-65459-6.
References
- 1.Wang Y, et al. A halogenation strategy for over 12% efficiency nonfullerene organic solar cells. Adv. Energy Mater. 2018;8:1702870. doi: 10.1002/aenm.201702870. [DOI] [Google Scholar]
- 2.Shafiq I, et al. Exploration of photovoltaic behavior of benzodithiophene based non-fullerene chromophores: First theoretical framework for highly efficient photovoltaic parameters. J. Market. Res. 2023;24:1882–1896. [Google Scholar]
- 3.Zhao F, et al. Emerging approaches in enhancing the efficiency and stability in non-fullerene organic solar cells. Adv. Energy Mater. 2020;10:2002746. doi: 10.1002/aenm.202002746. [DOI] [Google Scholar]
- 4.Mehboob MY, Hussain R, Irshad Z, Adnan M. Role of acceptor guests in tuning optoelectronic properties of benzothiadiazole core based non-fullerene acceptors for high-performance bulk-heterojunction organic solar cells. J. Mol. Model. 2021;27:226. doi: 10.1007/s00894-021-04843-9. [DOI] [PubMed] [Google Scholar]
- 5.Armin A, et al. A history and perspective of non-fullerene electron acceptors for organic solar cells. Adv. Energy Mater. 2021;11:2003570. doi: 10.1002/aenm.202003570. [DOI] [Google Scholar]
- 6.Mehboob MY, Hussain R, Iqbal MMA, Irshad Z, Adnan M. First principle theoretical designing of W-shaped Dithienosilole-based acceptor materials having efficient photovoltaic properties for high-performance organic solar cells. J. Phys. Chem. Solids. 2021;157:110202. doi: 10.1016/j.jpcs.2021.110202. [DOI] [Google Scholar]
- 7.Asif Iqbal MM, Mehboob MY, Arshad M. Quinoxaline based unfused non-fullerene acceptor molecules with PTB7-Th donor polymer for high performance organic solar cell applications. J. Mol. Graph. Model. 2022;114:108181. doi: 10.1016/j.jmgm.2022.108181. [DOI] [PubMed] [Google Scholar]
- 8.Mehboob MY, Hussain R, Adnan M, Irshad Z. End-capped molecular engineering of S-shaped hepta-ring-containing fullerene-free acceptor molecules with remarkable photovoltaic characteristics for highly efficient organic solar cells. Energy Technol. 2021;9:2001090. doi: 10.1002/ente.202001090. [DOI] [Google Scholar]
- 9.Irfan A, Mahmood A. Designing of efficient acceptors for organic solar cells: Molecular modelling at DFT level. J. Clust. Sci. 2018;29:359–365. doi: 10.1007/s10876-018-1338-x. [DOI] [Google Scholar]
- 10.Shafiq I, et al. Exploration of photovoltaic behavior of benzodithiophene based non-fullerene chromophores: First theoretical framework for highly efficient photovoltaic parameters. J. Mater. Res. Technol. 2023;24:1882–1896. doi: 10.1016/j.jmrt.2023.03.077. [DOI] [Google Scholar]
- 11.Lu B, et al. Z-shaped fused-chrysene electron acceptors for organic photovoltaics. ACS Appl. Mater. Interfaces. 2019;11:33006–33011. doi: 10.1021/acsami.9b10834. [DOI] [PubMed] [Google Scholar]
- 12.Wang S, et al. A new dibenzo[g.p]chrysene derivative as an efficient anode buffer for inverted polymer solar cells. Organ. Electron. 2019;74:269–275. doi: 10.1016/j.orgel.2019.07.022. [DOI] [Google Scholar]
- 13.Structural, optical and photovoltaic properties of unfused Non-Fullerene acceptors for efficient solution processable organic solar cell (Estimated PCE greater than 12.4%): A DFT approach. J. Mol. Liquids. 341, 117428 (2021).
- 14.Chang S-L, et al. Isomerically pure benzothiophene-incorporated acceptor: Achieving improved Voc and Jsc of nonfullerene organic solar cells via end group manipulation. ACS Appl. Mater. Interfaces. 2019;11:33179–33187. doi: 10.1021/acsami.9b08462. [DOI] [PubMed] [Google Scholar]
- 15.Gaussian09, R. D. 01; Gaussian. Inc.: Wallingford, CT, USA (2009).
- 16.Dennington, R., Keith, T. A. & Millam, J. M. GaussView 6.0. 16. Semichem Inc.: Shawnee Mission, KS, USA 143–150 (2016).
- 17.Zhao Y, Truhlar DG. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Accounts. 2008;120:215–241. doi: 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]
- 18.Hanwell MD, et al. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012;4:17. doi: 10.1186/1758-2946-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu T, Chen F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012;33:580–592. doi: 10.1002/jcc.22885. [DOI] [PubMed] [Google Scholar]
- 20.Tenderholt, A. L. PyMOlyze, Version 1.1. h ttp. pymolyze. sourceforge. net.
- 21.O’boyle NM, Tenderholt AL, Langner KM. cclib: A library for package-independent computational chemistry algorithms. J. Comput. Chem. 2008;29:839–845. doi: 10.1002/jcc.20823. [DOI] [PubMed] [Google Scholar]
- 22.Stevenson KJ. Review of OriginPro 8.5. J. Am. Chem. Soc. 2011;133:5621–5621. doi: 10.1021/ja202216h. [DOI] [Google Scholar]
- 23.Zhurko, G. A. ChemCraft software, version 1.6. (2014).
- 24.Zhao J, et al. Synthesis and characterization of new nonfullerene electron acceptors with a chrysene core. Dyes Pigments. 2020;174:108012. doi: 10.1016/j.dyepig.2019.108012. [DOI] [Google Scholar]
- 25.Mehboob MY, Hussain R, Irshad Z, Adnan M. Designing of U-shaped acceptor molecules for indoor and outdoor organic solar cell applications. J. Phys. Organ. Chem. 2021;34:e4210. doi: 10.1002/poc.4210. [DOI] [Google Scholar]
- 26.Mehboob MY, et al. Designing of benzodithiophene core-based small molecular acceptors for efficient non-fullerene organic solar cells. Spectrochimica Acta Part A Mol. Biomol. Spectrosc. 2021;244:118873. doi: 10.1016/j.saa.2020.118873. [DOI] [PubMed] [Google Scholar]
- 27.Mehboob MY, et al. Quantum chemical design of near-infrared sensitive fused ring electron acceptors containing selenophene as π-bridge for high-performance organic solar cells. J. Phys. Organ. Chem. 2021;34:e4204. doi: 10.1002/poc.4204. [DOI] [Google Scholar]
- 28.Mehboob MY, et al. Designing N-phenylaniline-triazol configured donor materials with promising optoelectronic properties for high-efficiency solar cells. Comput. Theor. Chem. 2020;1186:112908. doi: 10.1016/j.comptc.2020.112908. [DOI] [Google Scholar]
- 29.Mehboob MY, et al. Designing of near-infrared sensitive asymmetric small molecular donors for high-efficiency organic solar cells. J. Theor. Comput. Chem. 2020;19:2050034. doi: 10.1142/S0219633620500340. [DOI] [Google Scholar]
- 30.Pathade VA, Waghchaure RH, Jagdale BS, Pawar TB, Pathade SS. Molecular structure, frontier molecular orbital and spectroscopic examination on dihydropyrimidinones: A comparative computational approach. J. Adv. Sci. Res. 2020;11:64–70. [Google Scholar]
- 31.Yaqoob, U. et al. (2021) Structural, optical and photovoltaic properties of unfused Non-Fullerene acceptors for efficient solution processable organic solar cell (Estimated PCE greater than 12.4%): A DFT approach. Journal of Molecular Liquids341, 117428.
- 32.Mehboob MY, Adnan M, Hussain R, Irshad Z. Quantum chemical designing of banana-shaped acceptor materials with outstanding photovoltaic properties for high-performance non-fullerene organic solar cells. Synthetic Metals. 2021;277:116800. doi: 10.1016/j.synthmet.2021.116800. [DOI] [Google Scholar]
- 33.Bredas J-L. Mind the gap! Mater. Horizons. 2014;1:17–19. doi: 10.1039/C3MH00098B. [DOI] [Google Scholar]
- 34.Arshad MN, et al. Enhancing the photovoltaic properties via incorporation of selenophene units in organic chromophores with A2-π2-A1-π1-A2 configuration: A DFT-based exploration. Polymers. 2023;15:1508. doi: 10.3390/polym15061508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Taouali W, Alimi K, Sindhoo Nangraj A, Casida ME. Density-functional theory (DFT) and time-dependent DFT study of the chemical and physical origins of key photoproperties of end-group derivatives of a nonfullerene acceptor molecule for bulk heterojunction organic solar cells. J. Comput. Chem. 2023;44:2130–2148. doi: 10.1002/jcc.27186. [DOI] [PubMed] [Google Scholar]
- 36.Khalid M, et al. Photovoltaic response promoted via intramolecular charge transfer in pyrazoline-based small molecular acceptors: Efficient organic solar cells. Arab. J. Chem. 2023;16:105271. doi: 10.1016/j.arabjc.2023.105271. [DOI] [Google Scholar]
- 37.Shafiq I, et al. The impact of structural modifications into benzodithiophene compounds on electronic and optical properties for organic solar cells. Mater. Chem. Phys. 2023;308:128154. doi: 10.1016/j.matchemphys.2023.128154. [DOI] [Google Scholar]
- 38.Mehboob MY, et al. Efficient designing of half-moon-shaped chalcogen heterocycles as non-fullerene acceptors for organic solar cells. J. Mol. Model. 2022;28:125. doi: 10.1007/s00894-022-05116-9. [DOI] [PubMed] [Google Scholar]
- 39.Saeed MU, et al. Impact of end-group modifications and planarity on BDP-based non-fullerene acceptors for high-performance organic solar cells by using DFT approach. J. Mol. Model. 2022;28:397. doi: 10.1007/s00894-022-05382-7. [DOI] [PubMed] [Google Scholar]
- 40.Janjua MRSA. Deciphering the role of invited guest bridges in non-fullerene acceptor materials for high performance organic solar cells. Synthetic Metals. 2021;279:116865. doi: 10.1016/j.synthmet.2021.116865. [DOI] [Google Scholar]
- 41.Hussain R, et al. Molecular engineering of A-D–C–D–A configured small molecular acceptors (SMAs) with promising photovoltaic properties for high-efficiency fullerene-free organic solar cells. Optical Quant. Electron. 2020;52:1–20. doi: 10.1007/s11082-020-02482-7. [DOI] [Google Scholar]
- 42.Wang J, Zhan X. Fused-ring electron acceptors for photovoltaics and beyond. Acc. Chem. Res. 2021;54:132–143. doi: 10.1021/acs.accounts.0c00575. [DOI] [PubMed] [Google Scholar]
- 43.Nadeem Arshad M, et al. Donor moieties with D–π–a framing modulated electronic and nonlinear optical properties for non-fullerene-based chromophores. RSC Adv. 2022;12:4209–4223. doi: 10.1039/D1RA07183A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Asif T, Khera RA, Naveed A, Salim M, Iqbal J. Tuning the optoelectronic properties of naphthodithiophene (NDT) for designing of A-D-A type photovoltaic materials. Optik. 2021;247:167892. doi: 10.1016/j.ijleo.2021.167892. [DOI] [Google Scholar]
- 45.Khalid M, et al. Structural modeling of 1, 4 azaborine-based chromophores for organic solar cells using bracing units with benzothiophene-incorporated acceptors for exploration of photovoltaic behavior. J. Photochem. Photobiol. A Chem. 2023;445:115091. doi: 10.1016/j.jphotochem.2023.115091. [DOI] [Google Scholar]
- 46.Dvorak M, Wei S-H, Wu Z. Origin of the variation of exciton binding energy in semiconductors. Phys. Rev. Lett. 2013;110:016402. doi: 10.1103/PhysRevLett.110.016402. [DOI] [PubMed] [Google Scholar]
- 47.Sajid H, Ayub K, Gilani MA, Mahmood T. Donor-π-acceptor N-methyl-4,5-diazacarbazole based ultra-high performance organic solar cells: A density functional theory study. Energy Technol. 2023;11:2201164. doi: 10.1002/ente.202201164. [DOI] [Google Scholar]
- 48.Zubair H, et al. Effect of tailoring π-linkers with extended conjugation on the SJ-IC molecule for achieving high V OC and improved charge mobility towards enhanced photovoltaic applications. RSC Adv. 2023;13:26050–26068. doi: 10.1039/D3RA03317A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Adnan M, Mehboob MY, Hussain R, Irshad Z. Banana-shaped nonfullerene acceptor molecules for highly stable and efficient organic solar cells. Energy Fuels. 2021;35:11496–11506. doi: 10.1021/acs.energyfuels.1c01431. [DOI] [Google Scholar]
- 50.Aloufi FA, Halawani RF, Jamoussi B, Hajri AK, Zahi N. Quantum modification of indacenodithieno[3,2-b]thiophene-based non-fullerene acceptor molecules for organic solar cells of high efficiency. ACS Omega. 2023;8:21425–21437. doi: 10.1021/acsomega.2c07975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Khan MU, et al. First theoretical framework of Z-shaped acceptor materials with fused-chrysene core for high performance organic solar cells. Spectrochimica Acta Part A Mol. Biomol. Spectrosc. 2021;245:118938. doi: 10.1016/j.saa.2020.118938. [DOI] [PubMed] [Google Scholar]
- 52.Khalid M, et al. Exploration of efficient electron acceptors for organic solar cells: Rational design of indacenodithiophene based non-fullerene compounds. Sci. Rep. 2021;11:19931. doi: 10.1038/s41598-021-99254-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its supplementary information files.