

Abstract
Metabolic reprogramming is an important cancer hallmark that plays a key role in cancer malignancies and therapy resistance. Cancer cells reprogram the metabolic pathways to generate not only energy and building blocks but also produce numerous key signaling metabolites to impact signaling and epigenetic/transcriptional regulation for cancer cell proliferation and survival. A deeper understanding of the mechanisms by which metabolic reprogramming is regulated in cancer may provide potential new strategies for cancer targeting. Recent studies suggest that deregulated transcription factors have been observed in various human cancers and significantly impact metabolism and signaling in cancer. In this review, we highlight the key transcription factors that are involved in metabolic control, dissect the crosstalk between signaling and transcription factors in metabolic reprogramming, and offer therapeutic strategies targeting deregulated transcription factors for cancer treatment.
1. Introduction
Cancer cells demand substantial energy for abnormal and uncontrolled growth. To meet the requirement of energy for their growth and proliferation, cancer cells often evolve to rewire the metabolic pathways [1]. The mechanisms of how cancer cells reshape metabolic pathways have begun to be revealed. Tremendous progress towards elucidating the metabolic pathways and the derived metabolites involved in metabolic reprogramming for cancer control by transcriptional regulation has been made in recent years [2]. Although there are still limited studies demonstrating the role of the transcription factors in regulating the metabolic pathways for cancer cell regulation, numerous transcription factors that regulate the expression of diverse target genes for metabolic reprogramming involved in cancer malignant features have been identified [2]. It is important to note that the genes that encode the metabolism-specific transcription factors are frequently deregulated during cancer progression and metastasis. As such, the aberrant alteration in the activity of transcription factors affects the signaling landscape which triggers the metabolic reprogramming of cancer cells leading to cancer progression [3], [4], [5]. Therefore, it is important to investigate the regulatory networks of how the metabolic pathways are regulated through dysregulated transcription factors in order to develop novel strategies and agents for cancer targeting.
In this review, we comprehensively summarized the current knowledge about the important transcription factors and their aberrant activity in driving the metabolic reprogramming of the cancer. The first section introduced metabolic reprogramming through altered transcriptional regulation to meet the energy demand for cancer cell growth. The following section described the dysregulated transcription factors in cancer cell metabolism and their crosstalk and feedback loops that contribute to the transcriptional changes in the dynamic tumor microenvironment. Finally, we discussed the therapeutic interventions against cancer by targeting the dysregulated transcription factors and offered perspectives for future research.
2. The metabolic reprogramming in cancer
The homeostasis between the catabolic pathways, anabolic pathways, and waste disposal is crucial for cell survival, proliferation, and death [6]. Cancer cells maintain the metabolic homeostasis to grow and survive in hostile microenvironments. The metabolic reprogramming alters the metabolism in cancer cells with regard to normal cells, which supports the increased demand for energy, maintains the redox balance, and generates the building blocks for the rapid proliferation, growth, and survival of cancer in dynamic tumor microenvironments. The alternation of the metabolic pathways, such as aerobic glycolysis, pentose phosphate pathway (PPP), glutaminolysis, mitochondrial oxidative phosphorylation and biogenesis, amino acid metabolism, and lipid metabolism have been observed in various cancer types [7]. Thus, deregulated metabolism represents one of the key hallmarks in the cancers [8].
The normal cells utilize glucose for mitochondrial oxidative phosphorylation under aerobic conditions. In this oxidation, glucose is converted to pyruvate through glycolysis, which is then catalyzed to acetyl-CoA for mitochondrial tricarboxylic acid (TCA) cycle entry leading to the energy production of 36 ATPs. The normal cells under hypoxic conditions metabolize glucose through anaerobic glycolysis. In anaerobic glycolysis, glucose is converted to pyruvate, followed by lactate production in the cytoplasm for energy production of 2 ATPs. Strikingly, cancer cells often utilize aerobic glycolysis for glucose metabolism followed by lactate production to generate energy even in the presence of abundant oxygen. This phenomenon of increased glucose uptake and lactate production under oxygen supply is known as the Warburg effect discovered by Warburg in the 1920 s (Fig. 1) [9], [10]. However, it remains not well understood why cancer cells rely on this less efficient respiration pathway for energy production and how cancer cells could achieve aerobic glycolysis even in the presence of abundant oxygen. The mitochondrial dysfunction in cancer cells was the earlier explanation for this phenomenon, which was accepted by many scientific scholars at that time [11], [12], [13]. However, recent studies revealed that cancer cells have intact mitochondrial oxidative phosphorylation capabilities, arguing against the concept for defective mitochondrial dysfunction in cancer cells. The recent explanation for this phenomenon is that aerobic glycolysis provides adaptive advantages for highly proliferating cells. The increase in aerobic glycolysis not only supplies the energy, but also offers the glycolytic intermediates for the building blocks critical for the biosynthetic pathways [14]. Cancer cells elevate aerobic glycolysis to speed up the production of ATP by enhancing the uptake of glucose due to the upregulation of glucose transporters and various metabolic enzymes involved in glycolysis. Therefore, tumors can generate the energy and biomolecules needed for cancer cell growth and survival more efficiently [10]. Furthermore, the PPP pathway derived from aerobic glycolysis generates the ribose 5-phosphate (R5P) for nucleotide synthesis and NADPH for redox balance control. It has been shown that the PPP is highly regulated in normal cells and its dysregulation results in uncontrolled biosynthesis. To meet the higher biosynthetic demands, the PPP is frequently deregulated in cancer cells. In addition, the NADPH produced from the PPP protects cancer cells against oxidative stress from radiation and chemotherapy and participates in DNA damage repair [15].
Fig. 1.
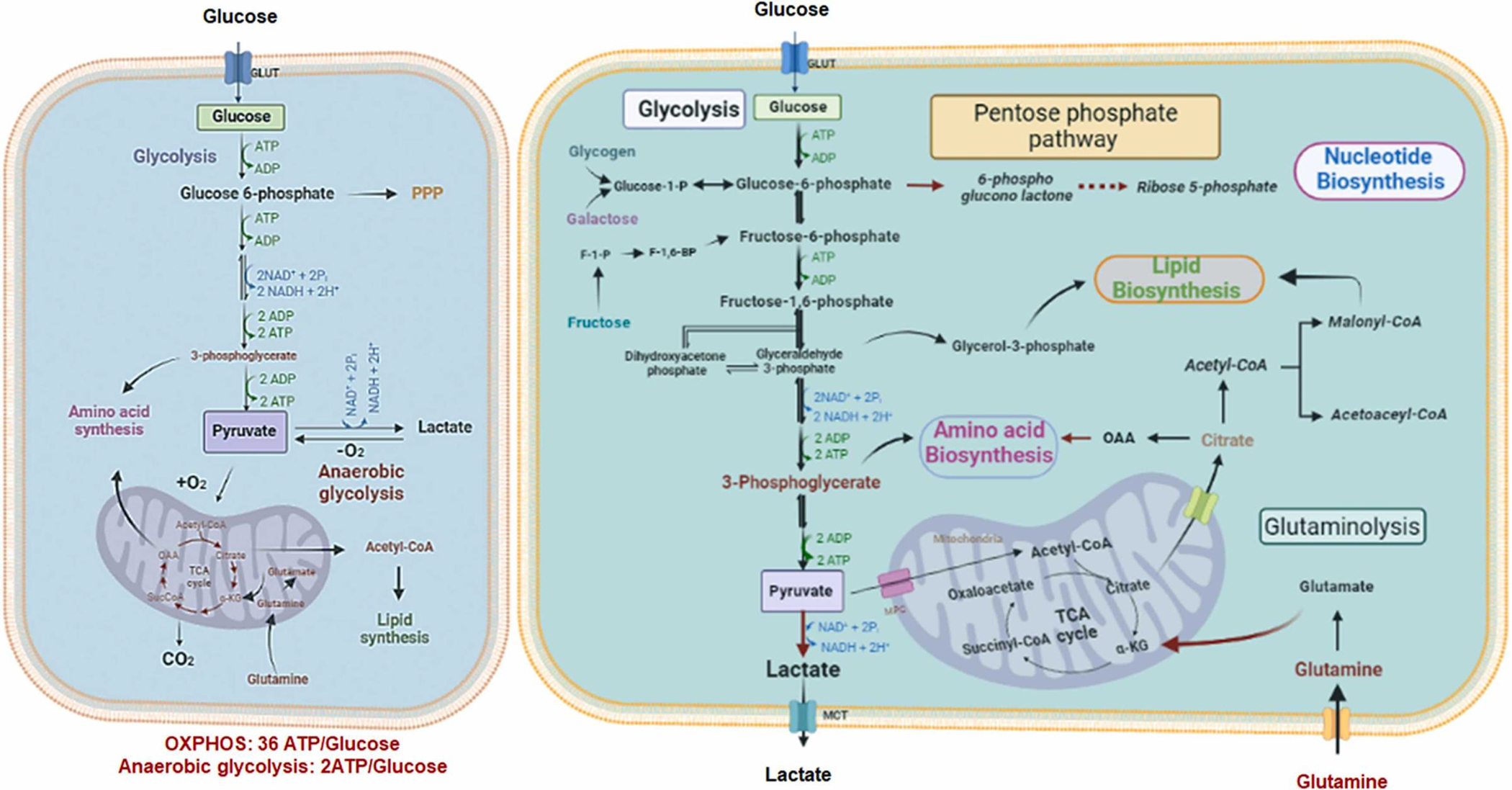
The metabolic reprogramming distinctly occurs in cancer cells compared to normal cells. Normal cells depend on mitochondrial oxidative phosphorylation for glucose metabolism (right panel). In contrast, cancer cells (left panel) convert glucose to lactate in cytoplasm for ATP synthesis. In addition, cancer cells increase their glucose and glutamine uptake to generate more metabolic intermediates to support anabolic pathways for amino acid, nucleotide and lipid biosynthesis.
Apart from glycolysis, glutaminolysis is also highly utilized for cancer cells to maintain their proliferation and survival [16], [17]. Glutamine, a nonessential amino acid that is abundantly present in the tumor microenvironment, provides nitrogen sources for protein and nucleotide synthesis. Glutamine is converted to glutamate through glutaminolysis, which is then converted to α-ketoglutarate (α-KG) to participate in mitochondrial TCA cycle for energy production and subsequent lipid synthesis [18]. Glutamine is also used by cells for glutathione synthesis to protect cells against oxidative damage and maintain redox balance. Glutamine is one of the most consumed amino acids in cancer and its metabolism is frequently altered in cancer. To enhance the uptake of glutamine, the glutamine transporter is upregulated in cancer [19]. In addition, the expression of the metabolic enzyme required for glutaminolysis is also increased in cancer cells to promote glutaminolysis. In some cancer cells, glutamine flux is converted toward de novo nucleotide synthesis (Fig. 1) [17].
The one-carbon (1 C) metabolism is also an important process for cancer cells to synthesize purine, amino acids, and phospholipids [20]. The 1 C metabolism also maintains redox homeostasis through ATP and NADH synthesis. The folate and methionine cycles are the important pathways of the 1 C metabolism [21]. 1 C metabolism generates S-adenosyl-methionine (SAM) from methionine, which serves as the global methyl donor for DNA and histone methylation in cells [22]. Serine and glycine, precursors for various molecules, are important carbon donors in the 1 C metabolism critical for purine synthesis. Cancer cells reprogram serine metabolism to increase serine biosynthesis through upregulation of various enzymes in serine biosynthetic pathways [23]. Similarly, glycine metabolism also reprogrammed in cancer cells for the synthesis of glutathione and purine to maintain cell proliferation and redox balance [24]. Cancer cells also require a higher level of SAM to maintain cell growth and proliferation [25]. Collectively, cancer cells utilize 1 C metabolism for their rapid proliferation.
Other amino acids are also the important precursors required for the synthesis of proteins, lipids and nucleic acids. Cancer cells require both non-essential amino acids and essential amino acids, such as branched-chain amino acids (BCAAs), to support their rapid growth [26], [27]. The enhancement of the uptake of amino acids by upregulating the amino acid transporters and the metabolic enzymes involved in amino acid synthesis and catabolism is also observed in cancer cells (Fig. 2) [28], [29].
Fig. 2.
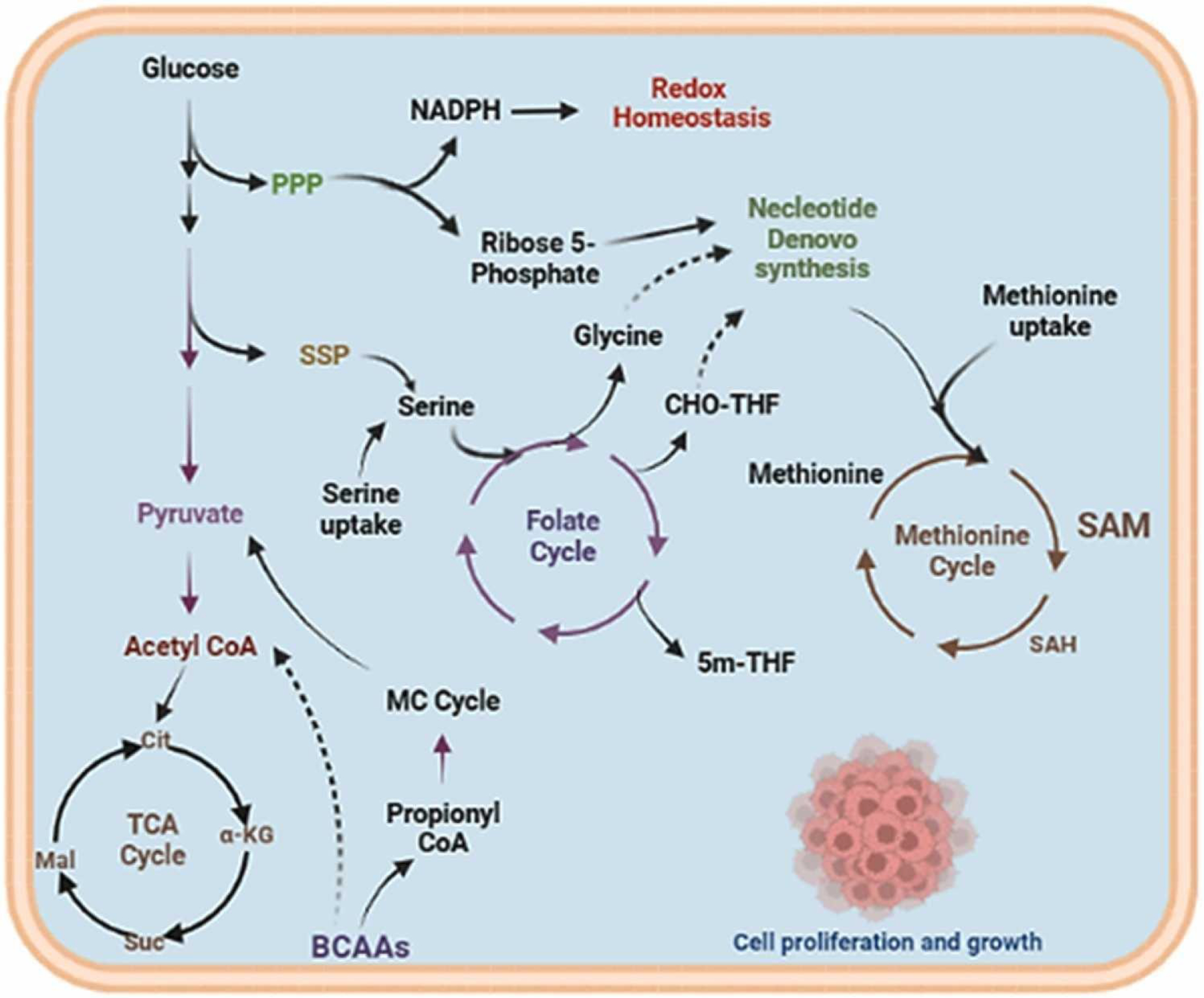
The one-carbon (1C) metabolism regulates the synthesis of purine, amino acids, and phospholipids to maintain the redox homeostasis and rapid cell growth. Cancer cells enhance the biosynthesis of amino acid serine and glycine. The amino acids are used as donors of one carbon unit, the 1C meta- bolism transfers this carbon unit for the biosynthesis of various metabolic outputs, such as nucleotides and redox homeostasis by folate and methio- nine cycles.
The lipids are required for the synthesis of biological membranes and also serve as a source of energy [30]. Cancer cells upregulate the fatty acid transporters to enhance the uptake of the exogenous fatty acids and cholesterol into the cells [31]. The cholesterol synthesis and de novo fatty acid synthesis pathways are also induced in cancer cells by activating various metabolic enzymes that participated in lipid biosynthesis pathways [32], [33], [34]. Excessive lipids and cholesterol are stored as lipid droplets in cancer cells. Notably, the accumulation of lipid droplets in various cancers provides the resistant mechanism to cancer therapy [35], [36]. Lipid droplets are breakdown under metabolic stress conditions through lipolysis and fatty acid oxidation to generate ATP and NADPH for cell survival maintenance [37], [38]. The lipolysis upregulated in tumors is used to generate free fatty acids to support the rapid growth of cancer cells [39].
The nucleotides are the building blocks for the synthesis of genetic materials required for the biosynthesis of DNAs and RNAs during cell proliferation. The nucleotide synthesis is achieved through numerous pathways, such as PPP, TCA cycle, 1 C metabolism and amino acid synthesis [40]. It is possible that the proper coordination of these pathways is highly regulated in normal cells. The R5P, an intermediate of the PPP and glycine, is required for the biosynthesis of inosine monophosphate (IMP) important for subsequent synthesis of the purine nucleotides. The methyl group derived from 1 C metabolism, non-essential amino acids, and NADPH is required for the synthesis of nucleotide bases [41]. The de novo biosynthetic pathways upregulated in cancers are the main pathways to synthesize nucleotides and related metabolites in cancer cells [42], [43].
While the exact mechanisms by which cancer cells leverage metabolic reprogramming remain to be explored, the alterations of the aforementioned metabolic pathways likely account in part for this process. It has been documented that the coordination of various signaling pathways for metabolic reprogramming involves the transcriptional programs in cancer cells upon the changes in the dynamic tumor microenvironments [44]. Cancer cells sense the availability of the various nutrients to rewire the metabolic program for their growth and survival in response to oxygen and nutrient-deprived microenvironments. To achieve this, the changes in the global gene expression and/or epigenetic landscape in cancer cells likely occur [45], [46]. As such, transcription factors act as a regulatory interface between the metabolic pathways and gene expression control, therefore playing a crucial role in the metabolic reprogramming [47], [48], [49]. Notably, the signaling pathways regulate the activity of the key transcription factors to orchestrate the gene expression of the metabolic enzymes for metabolism control. The well-known signaling pathway orchestrating metabolic reprogramming is the phosphatidylinositol-3-kinase (PI3K)/protein kinase B (Akt)/ mammalian target of rapamycin complex 1 (mTORC1) signaling pathway, which is activated by diverse mechanisms, such as mutations in oncogenes and tumor suppressor genes [7], [50], [51]. The transcription factor binds to the promoters of its target genes to stimulate or inhibit the gene expression. Transcription factors are altered in cancer cells by diverse mechanisms including gene amplifications and deletions, point mutations, chromosomal translocations, non-coding DNA mutations, and epigenetic mechanisms involving DNA methylation and histone modifications [52].
In the next section, we highlighted some of the key transcription factors that are dysregulated in cancer cells to participate in metabolic reprogramming, and such alterations can be harnessed for therapeutic targets to fight against cancer. These critical transcription factors including Myc, p53, nuclear factor erythroid 2-related factor 2 (NRF2), hypoxia-inducible factor 1 (HIF1), and forkhead box protein O (FOXO) are discussed in terms of the molecular mechanisms by which they reprogram the metabolic pathways in cancer and their conserved DNA recognizing motifs have been shown in Table 1.
Table 1.
Conserved DNA recognition motif of transcription factors.
| Transcription factor | Conserved DNA recognizing Motif |
|---|---|
| Myc | 5’-CACGTG-3’ |
| p53 | 5’-PuPuPuC(A/T)(T/A)GPyPyPy-3’ |
| NRF2 | 5’-TGACXXXGC-3’ |
| HIF-1α | 5’-RCGTG-3’ |
| FOXO | 5’-TT[G/A]TTTTG-3’ |
| 5’-TT[G/A]TTTAC-3’. |
2.1. Dysregulated transactional factors facilitate metabolic reprogramming in cancer cells
Cancer cells reprogram metabolic pathways to meet the biosynthetic, bioenergetics, and redox demands for their proliferation and survival. It is well documented that numerous transcription factors are deregulated in cancer cells to drive metabolic reprogramming. The key transcription factors that orchestrate the metabolic pathways are discussed below (Fig. 3).
Fig. 3.
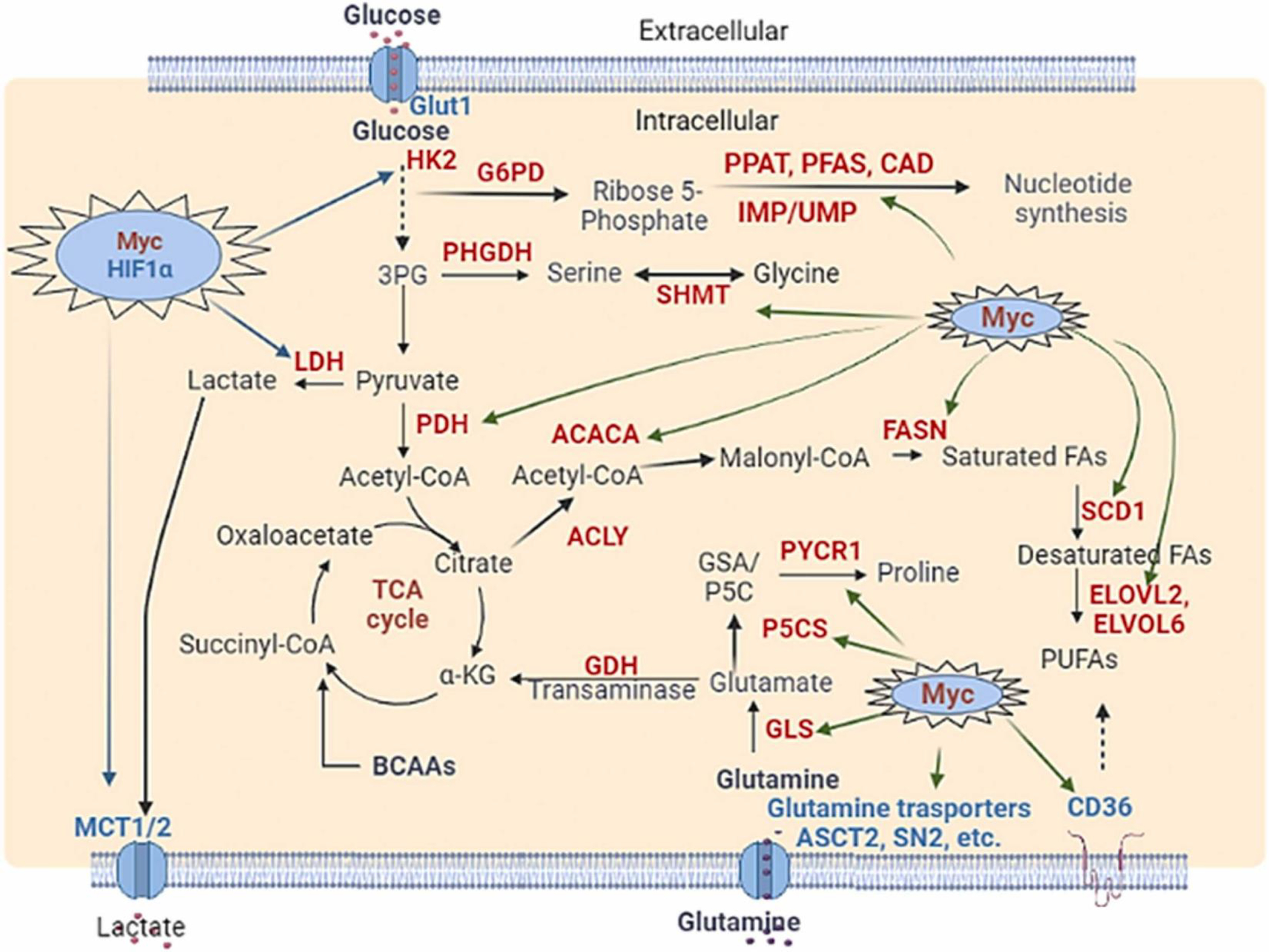
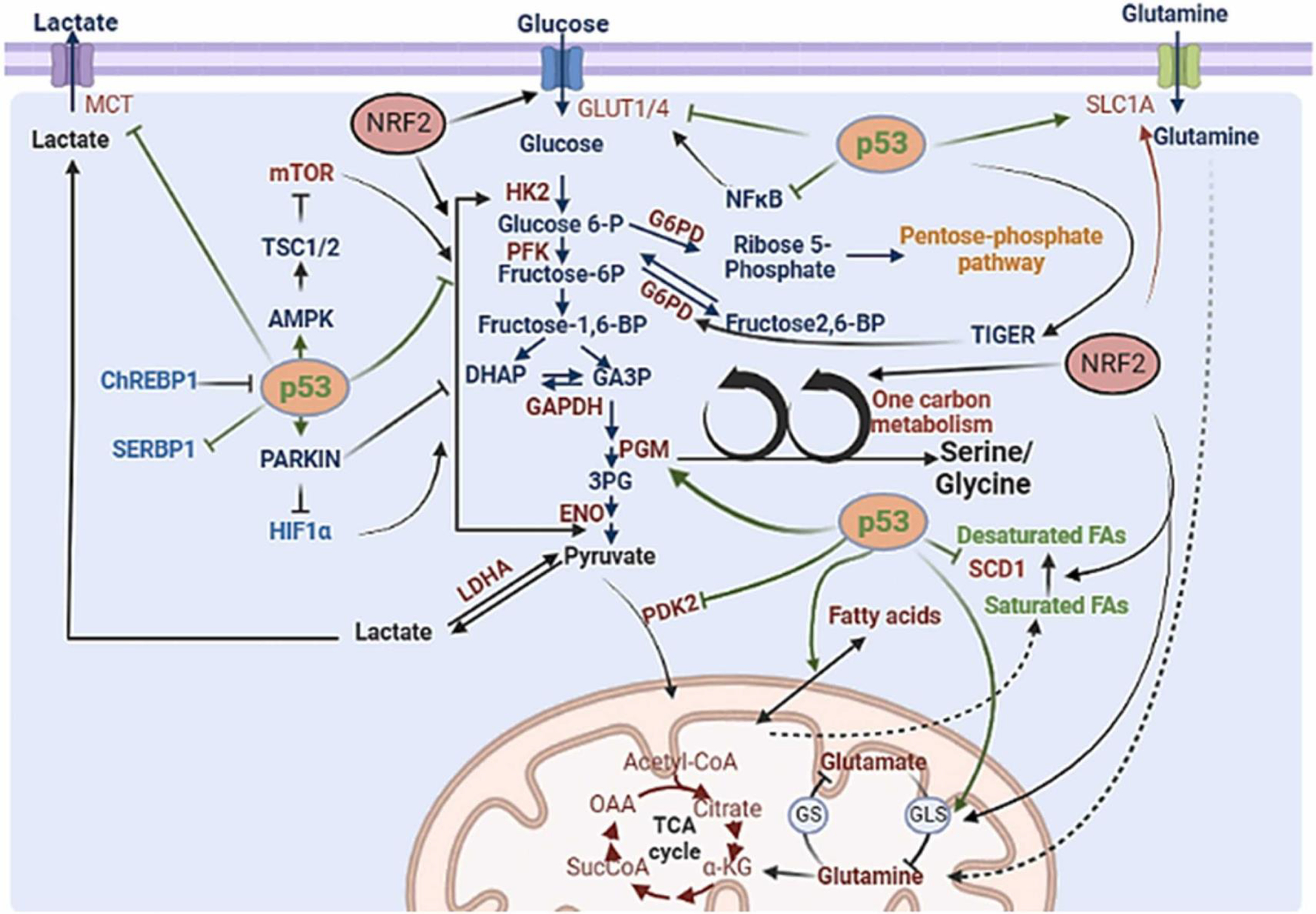
Dysregulated transcription factors impact cancer cell metabolism and reprogramming. (A) Myc and HIF1α regulate the expression of diverse metabolic enzymes. The green arrow indicated the Myc-upregulated enzymes (red) and transporters (blue). The enzymes elevated by Myc and HIF-1 were indicated by the blue arrow. (B) p53 and NRF2 regulate the expression of diverse metabolic enzymes. The green arrow indicates the p53 affected enzymes and transporters. The enzymes elevated by NRF2 were indicated by the black arrow. Inhibition is shown by Blunt arrow (┴), whereas activation is indicated by sharp arrow (→). Other transcription factors, such as chREBP1, SERBP1, and HIF1, are shown in blue.
2.2. Myc
Myc is one of the well-known transcription factors involved in cancer metabolism. Myc controls the transcription of various genes, which function in cell metabolism. Myc can either activates or represses the gene expression [53]. Importantly, the Myc is highly deregulated in a variety of human cancers. The increased Myc expression involves numerous mechanisms including gene amplifications, copy number gain, germline enhancer polymorphism, and alterations in its upstream signals. It has been shown that Myc plays a key role to reprogram virtually all aspects of metabolic pathways, such as glycolysis, PPP, glutaminolysis, TCA cycle, and lipid and protein metabolism in cancer cells (Fig. 3A) [54].
Cancer cells enhance glucose uptake and often rewire oxidative phosphorylation to aerobic glycolysis known as the Warburg effect. Myc increases the uptake of glucose by upregulating glucose transporter 1 (GLUT1). Furthermore, increased Myc expression orchestrates nearly all enzymes participated in glycolysis, such as hexokinase 2 (HK2), glucose-6-phosphate isomerase (GPI), phosphofructokinase (PFK), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), phosphoglycerate kinase (PGK), pyruvate dehydrogenase kinase 1 (PDK1), phosphofructokinase, muscle (PFKM), and enolase 1 (ENO1) [55], [56]. Moreover, increased Myc expression in cancer cells upregulates the expression of the lactate dehydrogenase A (LDH-A), which converts pyruvate to lactate [57]. In addition, Myc induces the expression of monocarboxylate transporters (MCT1/2) to modulate lactate transport in cancer cells [58], [59]. Myc also upregulates the PPP pathway by utilizing glucose for nucleotide synthesis by enhancing the expression of glucose-6-phosphate dehydrogenase (G6PD) and transketolase in cancer cells [60], [61]. Hence, Myc is one of the key transcription factors driving the Warburg effect.
For amino acid metabolism, glutamine is one of the most important amino acids that contributes to tumor growth [54]. Myc induces the expression of glutamine transporter alanine serine cysteine transporter 2 (ASCT2 /SLC1A5) and sodium coupled neutral amino acid transporter 5 (SNAT5/ SCL38A5) to promote the glutamine uptake [62], [63], [64]. Furthermore, Myc increases the expression of the glutaminase through suppression of miR-23, leading to the conversion of glutamine to glutamate for mitochondrial TCA cycle entry and ATP production [64]. Myc also regulates glutamate dehydrogenase (GDH) and glutamate-dependent transaminases [glutamic pyruvic transaminase (GPT2), glutamic oxaloacetic transaminases (GOT1/2), and phosphoserine aminotransferase (PSAT1)] to enhance the biosynthesis of amino acids [65], [66], [67]. In addition, Myc directs the glutamine Flux towards de novo Nucleotide Synthesis by upregulating the expression of phosphoribosyl pyrophosphate amidotransferase (PPAT), phosphoribosylformylglycinamidine synthase (PFAS), and carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase (CAD) genes that utilize glutamine towards IMP and uridine monophosphate (UMP) synthesis [42]. Depending on the availability of nutrients, Myc could change the mitochondrial metabolism from being dependent on glucose to glutamine. Myc also promotes glutathione synthesis under oxidative stress to maintain the survival of cancer cells under oxidative stress. Myc enhances serine and glycine metabolism by upregulating various enzymes such as serine hydroxymethyltransferases (SHMT1/2), and phosphoglycerate dehydrogenase (PHGDH) and phosphoserine phosphatase (PSPH) [68].
Amino acid serve as the building blocks critical for the synthesis of proteins, lipids and nucleic acids. Myc regulates the metabolism of essential and non-essential amino acids. To promote the transport of essential amino acids, Myc activates the amino acid transporters like solute carrier family member proteins (SLC7A5, SLC43A1, and SLC1A5) [69]. To break down the branched-chain amino acids (leucine, isoleucine, and valine), Myc upregulates the branched chain amino acid transaminase 1 (BCAT1) [70]. Myc also promotes proline synthesis by upregulating pyrroline-5-carboxylate synthase (P5CS) and pyrroline-5-carboxylate reductase 1 (PYCR), but downregulating proline oxidase/proline dehydrogenase (POX/PRODH) [71]. Myc enhances serine and glycine biosynthesis by activation or upregulation of PHGDHPSAT1, phosphoserine phosphatase (PSPH), and SHMT2 [54]. Myc also inhibits glycine N-methyltransferase (GNMT) expression to prevent glycine catabolism in cancer cells [72].
For lipid metabolism, Myc upregulates the expression of CD36 and carnitine palmitoyltransferase IA/2 (CPT1A/CPT2) for fatty acid uptake in mitochondria [73]. Myc also activates the acetyl-CoA carboxylase (ACC), fatty acid synthase (FASN), stearoyl-CoA desaturase (SCD), and ATP citrate lyase (ACYL) to stimulate the fatty acid synthesis [73], [74], [75]. To regulate cholesterol metabolism, Myc upregulates the 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase (HMGCR) for cholesterol synthesis in tumors. Myc also promotes fatty acid oxidation by activating the AMP-activated kinase (AMPK) [73]. In addition, Myc cooperates with the transcription factors SREBPs to regulate lipid synthesis and tumor growth in multiple cancer types [75].
Myc alters nucleotide synthesis by inducing various genes involved in purine and pyrimidine synthesis. First, Myc induces phosphoribosyl pyrophosphate synthetase 2 (PRPS2) to generate the nucleic acid intermediate, nuclephosphoribosyl pyrophosphate. Second, Myc activates numerous enzymes involved in purine synthesis, such as phosphoribosyl pyrophosphate amidotransferase (PPAT) and phosphoribosyl aminoimidazole succinocarboxamide synthetase (PAICS), and inosine monophosphate dehydrogenase 1, and 2 (IMPDH1 and IMPDH2). Third, Myc activates carbamoyl-phosphate synthetase aspartate transcarbamylase, and dihydroorotase (CAD) to elicit pyrimidine synthesis. Fourth, Myc also promotes nucleotide synthesis by regulating other metabolic pathways, such as PPP, folate cycle, and 1 C metabolism [61], [76], [77], [78]. Fifth, Myc upregulates glucose-6-phosphate dehydrogenase (G6PD) and transketolase in the PPP pathway to synthesize the ribose 5-phosphate the building block of nucleotide biosynthesis as described above. Finally, Myc enhances the biosynthesis of serine and glycine biosynthesis, which act as the precursors for purine and pyrimidine synthesis.
Several studies have shown that Myc-driven metabolic reprogramming may facilitate tumorigenesis. Hu et al. using in vivo transgenic tumor model of Myc-driven liver cancer showed that alterations in glycolysis is involved in Myc-driven tumor formation [79]. Another study revealed that glutaminase is responsible for tumorigenesis in Myc-driven hepatic cellular carcinoma (HCC) mouse model, which was suppressed by glutaminase inhibitor treatment [80]. Similar to the HCC model, Myc overexpression initiated renal cell carcinoma through glutamine metabolism using the Myc transgenic tumor model in kidney tissue [65]. In addition, Pacelli et al. demonstrated that the inhibitor of carnitine-palmitoyl transferase 1 A (CPT1A) involved in fatty acid uptake restricted Myc-driven lymphomagenesis in xenograft models and Eμ-Myc transgenic mice[81], suggesting the role of lipid metabolism in Myc-driven cancer development. Barna et al. showed that increased protein synthesis upon Myc overexpression accelerated cell growth and tumorigenesis, which was compromised by haploinsufficiency of the ribosomal protein in Eμ-Myc/+ transgenic mice with reduced protein synthesis [82]. Consistently, Myc-driven lymphomagenesis relied on phosphoribosyl-pyrophosphate synthetase 2 (PRPS2)-mediated increase in protein and nucleotide biogenesis in transgenic mice [83]. Notably, Myc-driven metabolic reprogramming was also demonstrated in human cancer samples. In colorectal cancer samples, Myc expression was highly correlated with the expression level of 231 metabolism-related genes in tumor tissue compared with adjacent normal tissue, and the correlation was reversed in HCT116 cells by Myc inhibition [84]. Collectively, these studies underscore the important role of metabolic reprogramming induced by Myc in tumorigenesis, placing Myc as a potential target for cancer therapy.
The abovementioned studies revealed that the transcription factor Myc reprogrammed different metabolic pathways in diverse cancer models. For example, Myc-mediated alteration in glycolysis was shown in the liver and small cell lung cancer models, while alteration in glutamine was observed in the liver and renal carcinomas and alteration in fatty acid and nucleotide metabolism was seen in lymphoma and colorectal cancer [65], [79], [80], [81], [83], [84], [85]. It is important to note that Myc overexpression displayed an opposite effect on metabolic pathways in different tumors, as Myc drives glutamine synthesis pathways in liver cancer but promotes glutamine degradation pathways in lung cancer [80], [86]. Therefore, comprehensive research on tumor-specific metabolic reprogramming by Myc transcription factor is warranted to better understanding the complexity of Myc-driven metabolic addictions in distinct cancer types. This effort will help to develop specific metabolic inhibitors for the treatment of Myc-driven tumors in different organs.
Recently, Tsakaneli et al. demonstrated that neuroblastoma cells expressing Myc-N, a Myc isoform, produce extracellular vesicles enriched with oncogenic glycolytic enzymes, hexokinase 2 (HK2) and pyruvate kinase M2 (PKM2), which induce glycolysis and c-Myc expression in recipient cells. Moreover, Myc-N-regulated extracellular vesicles also caused activation of c-Myc in recipient stromal cells surrounding Myc-N-positive neuroblastomas in the syngeneic mouse model [87]. This study supports the notion that Myc-mediated metabolism plays an important role to reshape the tumor microenvironment. However, it remains to be addressed 1) Whether c-Myc overexpressing tumors also secret extracellular vesicles enriched in glycolytic enzymes to modulate c-Myc expression in cells found in the tumor microenvironment (2) Whether c-Myc also regulates different metabolic enzymes related to fatty acid, nucleotide and amino acid metabolism. (3) Whether tumor models other than neuroblastomas also exhibit similar extracellular vesicle-mediated metabolic Myc activation in recipient cells. We speculate that tumor cells with Myc overexpression may release diverse extracellular vesicles enriched with various metabolic enzymes, which may be altered by distinct tumor tissues and tumor microenvironments.
More interestingly, Liu et al. revealed that inspiratory hyperoxia therapy decreases lung cancer metastasis in the lung cancer model through inhibiting Myc/SLC1A5-dependent metabolic pathway without impact on primary tumor in mice [88]. The authors anticipated that the lung-specific tumor microenvironment may play a role in inspiratory hyperoxia therapy for lung tumors. However, they did not investigate whether xenograft tumors had any alteration of Myc expression [88]. This work raises the question whether Myc suppression by inspiratory hyperoxia therapy depends on the tumor microenvironment. Hence, more studies are required to fully understand Myc-mediated metabolic reprogramming in the tumor microenvironment.
2.3. p53
p53 is a well-known tumor suppressor whose inactivation is the most frequent event in cancer. p53 is a transcription factor that regulates a number of the genes associated with metabolism to orchestrate its tumor suppressive activity [89]. p53 gene mutations are mostly missense and the resulting p53 mutants display the gain-of-function activities. Interestingly, many of these p53 mutants reprogram cancer metabolism towards cell growth and proliferation in the tumor microenvironments [90], [91]. Similar to Myc, p53 also acts through its transcriptional regulation to control various metabolic pathways including glucose, fatty acid, protein, and nucleotide metabolism [92]. In a normal cell, p53 supports oxidative phosphorylation while inhibiting glycolysis. However, p53 loss in tumors causes metabolic reprogramming by shifting oxidative phosphorylation to glycolysis [91], [93], [94]. To achieve this, p53 represses the transcription of glucose transporters (GLUT1 and GLUT4) to reduce the glucose uptake for glycolysis. p53 can also repress the expression of GLUT3 and GLUT1 indirectly to reduce the glucose uptake [95]. p53 inhibits lactate export and import from the cells by suppressing the expression of malic enzymes (ME1 and ME2) [96]. p53 activates AMPK, which negatively regulates mTOR and mTOR-mediated glycolysis and anabolic biosynthetic processes [92]. p53 also regulates glycolysis by directly targeting the transcription of the metabolic enzymes or indirectly modulating signaling pathways. For instance, p53 decreases the expression of phosphoglycerate mutase 1 (PGM1) and HK2, but increases the expression of parkin RBR E3 ubiquitin protein ligase (PARK2), which ubiquitinates and degrade HIF-1α, to inhibit glycolysis [97], [98], [99], [100], [101]. Furthermore, p53 transcriptionally represses 6-phosphofructo-2-kinase/fructose-2,6-biphosphatases (PFKFB3 and PFKFB4), which reduces intracellular levels of fructose-2,6-bisphosphate [102]. Of note, p53 increases the expression of TP53-induced glycolysis regulatory phosphatase (TIGAR), which functions as fructose-2,6-bisphosphatase to reduce intracellular levels of fructose-2,6-bisphosphate and inhibits the glycolysis enzyme, phosphofructokinase (PFK-1), to redirect glucose to the PPP pathway [103]. p53 binds and inactivates glucose-6-phosphate dehydrogenase (G6PD), which is the rate-limiting enzyme of PPP, to inhibit the PPP shunt [104]. For glycolysis suppression, p53 also induces the expression of various genes, such as Sestrin 1/2 and TSC complex subunit 1/2 (TSC1/2), which negatively regulate Akt/mTOR activation involved in the glycolysis [105], [106], [107]. Hence, p53 loss results in the reprogramming of glucose metabolism towards glycolysis.
p53 maintains mitochondrial oxidative phosphorylation by regulating the expression of other targets. p53 induces synthesis of cytochrome c oxidase 2 (SCO2) and expression of AIF (apoptosis-inducing factor) and Parkin [108]. Parkin upregulation increases pyruvate dehydrogenase E1α1 (PDHA1), which then induces the cytochrome C oxidase complex leading to the increased levels of oxidative phosphorylation [99]. In contrast, p53 represses the expression of pyruvate dehydrogenase kinase 2 (PDK2), which serves as a negative regulator of pyruvate dehydrogenase (PDH), thus promoting TCA cycle entry [109]. p53 also promotes mitochondrial oxidation by inducing the expression of mitochondrial glutaminase 2 (GLS2), which enhances glutathione (GSH) synthesis and α-ketoglutarate [110]. Under glutamine starvation conditions, p53 activates aspartate/glutamate transporter, Solute Carrier Family 1 Member 3 (SLC1A3), to enhance glutamine transport [111]. p53 inhibits de novo serine biosynthesis by transcriptionally repressing first and rate-limiting enzyme, PHGDH, for serine synthesis [112]. p53 can upregulate the expression of POX, which is a catabolic enzyme of proline synthesis [113].
In addition to regulating glucose and amino acid metabolism, p53 also regulates lipid metabolism [89], [114]. In this respect, p53 activates fatty acid oxidation but represses fatty acid synthesis by regulating the expression of diverse enzymes involved in lipid metabolism. For instance, p53 represses the expression of Stearoyl-CoA-desaturase 1 (SCD1), protein kinase AMP-activated catalytic subunit alpha 2 (PRKAA2), SERPINE1 mRNA Binding Protein 1 (SERBP1), but induces the expression of malonyl CoA decarboxylase (MCD), lipin 1 (LPIN1), carnitine palmitoyltransferase 1 C (CPT1C), pantothenate kinase 1 (PANK1), and Guanidinoacetate methyltransferase (GAMT) [91]. However, p53 could also inhibit lipid synthesis indirectly by inhibiting the PPP for NADPH production as mentioned above. p53 also plays an important role in nucleotide metabolism. p53 inhibits purine synthesis by suppressing ribose 5 phosphate formation in the PPP pathway and repressing GMP synthetase (GMPS)[89], [115]. However, p53 enhances nucleotide synthesis by induction of Ribonucleotide Reductase Regulatory TP53 Inducible Subunit M2B (p53R2/RRM2B) to support DNA damage repair in cells during DNA damage [91], [116].
Although p53 has been shown to regulate diverse metabolic pathways as mentioned above, whether these metabolic changes are linked to p53-mediated tumor suppression has not been well established. Interestingly, Li et al. showed that while the mice expressing acetylation-defective p53 mutant display impaired p53-mediated cell cycle arrest, apoptosis, and senescence, these mice do not show early onset of tumorigenesis as p53−/− mice, correlated with defective in glycolysis and reactive oxygen species (ROS), implying that the metabolic regulation and antioxidant function of p53 may be involved in p53 tumor-suppressive role in cancer [117]. A recent study revealing that p53 suppresses SREBP-2-mediated mevalonate pathway to restrict tumorigenesis has begun to shine light on the role of metabolism regulation by p53 in mediating p53’s tumor suppression [118]. In addition to impacting on cancer cell phenotypes, p53 also plays an important role in regulating inflammation and immunity. Recent study revealed that p53 affects the immune response by enhancing cytokines secretion and that loss of p53 promotes the recruitment of T cells and myeloid cells [119]. Other study further demonstrated that loss of p53 in T cells enhances glycolysis and PPP pathway [120]. It will be interesting to dissect whether metabolic regulation such as glycolysis regulated by p53 is indeed involved in p53-mediated cancer immunity.
Cancer-associated p53 mutant has been shown to activate RhoA/ROCK/GLUT1 signaling leading to the increases of glucose uptake and Warburg effect in cells and mouse models [121]. Furthermore, activated RhoA/ROCK signaling promotes tumorigenesis in mouse models [121], but the direct evidence that elevated Warburg effect by activated RhoA/ROCK/GLUT1 axis contributes to oncogenic activity of mutant p53 is lacking. Intriguingly, Siolas et al. showed intratumoral p53 mutation drives neutrophil accumulation and confers resistance to immunotherapy in pancreatic cancer [122]. However, whether mutant p53 acts through metabolism regulation to orchestrate neutrophil accumulation and immunotherapy resistance remains to be determined. Like p53 loss, cancer-associated p53 mutant has been shown to drive SREBP-mediated mevalonate pathway, which is required for the effect of mutant p53 on breast tissue architecture [123].
Collectively, p53 loss and p53 mutations found in diverse human cancers likely reprogram cancer metabolism towards the aerobic glycolysis and lipid biogenesis pathways to support cancer progression (Fig. 3B).
2.4. NRF2
Nuclear factor erythroid 2-related factor 2 (NRF2) is a key regulator of antioxidants that protect against oxidative stress and electrophilic injury to maintain intrinsic redox homeostasis [124]. NRF2 is a transcription factor that plays a dual role in cancer [125], [126], [127]. On one hand, activation of NRF2 protects against chemical and radiation-induced carcinogenesis by activating antioxidant detoxifying and cytoprotective genes in response to environmental stress. On the other hand, constitutively active NRF2 provides survival benefits in already established cancers to promote cancer progression and metastasis [128]. Similar to Myc and p53, NRF2 also participates in reprogramming various metabolic pathways, as discussed below (Fig. 3B). Hence, NRF2 activation may drive cellular reprograming to promote cancer growth and survival for tumorigenesis.
NRF2 increases the expression of glucose transporter GLUT1 to enhance glucose uptake. NRF2 also diverts respiration towards aerobic glycolysis by modulating the expression of various enzymes in glycolysis. NRF2 induces the expression of various glycolysis enzymes, such as glucose phosphate isomerase 1 (GPI1), hexokinase (HK1, HK2), 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatase 2 (PFK2), 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 4 (PFK4), ENO1, ENO4, fructose-bisphosphate aldolase A (ALDA), and pyruvate kinase M (PKM) [129], [130]. Interestingly, NRF2 can shift glucose metabolism into the PPP by upregulation of key enzymes involved in PPP including G6PD, 6-phosphogluconate dehydrogenase (PGD), PPAT, methylenetetrahydrofolate dehydrogenase 2 (MTHFD2), phosphogluconate dehydrogenase (PGD), transaldolase 1 (TALDO1) and transketolase (TKT) [129], [131].
NRF2 also regulates amino acid metabolism by upregulating the expression of glutamine transporter SLC1A5 and SLC1A4 for glutamine uptake and upregulating activating GLS2 and GPT2 for glutaminolysis, which provides the intermediates for the synthesis of nucleotide and non-essential amino acids [132], [133]. NRF2 regulates the serine/glycine biosynthesis by inducing the expression of PHGDH, PSAT1, and SHMT2 through ATF4 [134]. Alternatively, NRF2 can indirectly promotes the de novo synthesis of serine. Interestingly, SUMOylation of NRF2 stimulates PHGDH through scavenging the ROS accumulation from the cell [135]. In addition, NRF2 upregulates glutamate-cystine antiporter solute carrier family 7, member 11 (SLC7A11) to enhance cysteine transport [136], [137].
NRF2 also regulates lipid metabolism by promoting fatty acid oxidation while inhibiting lipid biosynthesis. To promote fatty acid oxidation, NRF2 modulates the expression of CPT1, CPT2 and acyl-CoA oxidase 1 and 2 (ACOX1, ACOX2), the key enzymes involved in fatty acid oxidation [138]. For the inhibition of fatty acid synthesis, NRF2 downregulates the expression of the key enzymes in fatty acid synthesis, such as fatty acid elongases (Elovl2,3,5,6 and Cyb5r3), acetyl-CoA carboxylase 1 (ACC1), FASN, Scd1 and ATP-citrate lyase (ACYL)[139], [140], [141]. Finally, NRF2 enhances nucleotide synthesis by activating PPP pathway, glutaminolysis and serine-glycine synthesis, as described above. In this respect, NRF2 upregulates the enzymes involved in de novo nucleotide biosynthesis, such as PHGDH, MTHFD2, and inorganic pyrophosphatase (PPA).
It is important to note that NRF2-mediated metabolic reprogramming is correlated with tumorigenesis. Singh et al. showed that NRF2 suppresses the expression of miR-1 and miR-206 to reprogram metabolism towards the PPP and TCA cycle in lung cancer cells. Furthermore, the authors demonstrated that miR-1 and miR206 expression significantly downregulated, whereas the expression of NRF2 target genes and PPP genes is significantly upregulated in adenocarcinoma tumors compared to matched adjacent normal lung tissues and associated with poor survival outcome [142]. In breast cancer cells, Liu et al. showed that B7-H3 promotes aerobic glycolysis by suppressing NRF2, and NRF2 suppression causes stabilizing HIF-1 and HIF-1-mediated activation of enzymes in glycolytic pathways leading to increased glucose uptake in tumor xenografts [143]. Mitsuishi et al. showed that sustained activation of the PI3K/Akt pathway enhances the nuclear accumulation of NRF2 and enables NRF2 to drive metabolic reprogramming by redirecting glucose and glutamine to biosynthetic pathways for maintaining cell proliferation [131]. Saito et al. showed that phosphorylation of p62/SQSTM1 activates NRF2 to direct glucose towards the gluconate pathway to provide tolerance of HCC cells against anticancer drugs [144]. Thus, NRF2-mediated metabolic reprogramming may contribute to drug resistance in cancer.
Cancer cells are largely dependent on non-essential amino acids including glutamine during cancer progression and metastasis. To meet the requirement of glutamine, serine and cysteine, cancer cells upregulate the expression of glutamine, serine and cysteine transporters through NRF2. NRF2 then directs the glutamine and serine in the formation of glutathione, which helps with scavenging the accumulation of ROS [145]. Collectively, NRF2-mediated metabolic reprogramming through uptake of the non-essential amino acids from extracellular environments may contribute to tumor progression, thus offering a potential strategy for cancer targeting.
2.5. HIF1
Cancer cells grow in hypoxic conditions due to inadequate vascularization in solid tumor mass. The tumor cells adapt to the low oxidative stress conditions by inducing and activating HIF1α. The activated HIF1α plays a pivotal role in protecting tumor cells under hypoxic conditions by activating numerous downstream pathways essential for cell survival [146], [147]. HIF1α helps tumor cells to switch from highly efficient oxidative phosphorylation to less efficient glycolytic pathways[146]. HIF1α is a transcription factor that forms a heterodimeric form with HIF1β. Interestingly, HIF1α is induced under oxygen deprivation due to the reduction of its ubiquitination and degradation, while HIF1β is constitutively expressed irrespectively of oxygen availability [148], [149]. HIF1α consists of two major domains, an N terminal domain that essentially stabilizes the HIF1α protein and a C terminal domain through which HIF1α interacts with several co-activators such as CREB binding protein (CBP)/ E1A binding protein (P300) to regulate the expression of HIF1α under hypoxic conditions [150]. The expression of HIF1α protein is tightly regulated by numerous proteins under steady-state conditions. The HIF1α protein is degraded through oxygen-dependent and oxygen independent manners. In an oxygen-dependent manner, the HIF1α protein undergoes hydroxylation by Prolyl 4- hydroxylases (PHD), which requires 2-oxoglutarate, ascorbate, oxygen, and Fe2+ ion [151], [152]. Moreover, the HIF1α also undergoes acetylation at K532 by arrest defective-1 (ARD-1) enzyme [153]. These post-translational modifications recruit von Hippel–Lindau (VHL) E3 ligase to drive ubiquitination and degradation of HIF1α. In an oxygen-independent manner, HIF1α interacts with p53, which recruits mouse double minute 2 homolog (MDM2) E3 ligase to degrade HIF1α protein [154].
HIF1α protein regulates a myriad of the genes involved in glucose metabolism. HIF1α promotes the glycolysis by inducing the expression of glucose transporters (GLUT1 and GLUT3) and the glycolytic enzymes, such as HK1, HK2, PKM2, and lactate dehydrogenase (LDH) [149], [155], [156], [157], [158], [159], [160]. Similar to HIF1α, HIF2α and HIF3α are also induced under hypoxic conditions. HIF2α, which shares 48% sequence homology with HIF1α, also induces the expression of GLUT1 (Fig. 3A). However, the functional role of HIF3α has not been well understood thus far [161], [162]. In addition, HIF1 transactivates the expression of PDK1 and ADM, which may help with chemoresistance of the cancer cells. Numerous studies showed that metabolic reprograming may be responsible for HIF1-mediated tumorigenesis and metastasis. For example, Becker et al. showed that human breast cancer-associated fibroblasts display metabolic reprogramming towards glycolysis to support the biosynthetic pathways of cancer cells through epigenetic HIF1 activation. They further demonstrated that this metabolic shift in cancer-associated fibroblasts fuels cancer cells and promotes tumor growth [163]. Zheng et al. showed that transient overexpression of isocitrate dehydrogenase 3α (IDH3α) increases metabolic reprogramming and angiogenesis induced by HIF1 to enhance xenograft tumor growth in nude mice [164]. In addition, Zhao et al. using a lung metastases tumor model showed that HIF1 activation induced the expression of PDK1 and lactate dehydrogenase A (LDHA) to divert oxidative phosphorylation towards anaerobic glycolysis, and the HIF1 inhibitor reversed the metabolic reprogramming and suppressed lung metastasis [165]. By inducing the expression of PDK1 that suppresses PDH activity and its conversion of pyruvate to acetyl-CoA, HIF1α ensures the cells to utilize glycolysis instead of oxidative phosphorylation for energy production during hypoxia [166].
Highly glycolytic cancer cells release lactate outside the cells, which causes acidity in the tumor microenvironment. The acidification in the tumor microenvironment impairs the anti-tumor activity of T cells and NK cells partly by blocking the glycolytic enzymes including GAPDH and PHGDH for the production of 3-phosphoglycerate derivative serine essential for the proliferation of the T cells. Furthermore, it has been recently observed that lactate could potentially suppress the production of interleukin 2 (IL-2) and interferon γ (IFN-γ) from T cells [167]. In pancreatic cancer, lactic acid could induce the infiltration of myeloid derived suppresser cells (MDSCs) to suppress the anti-tumor activity of T cells. Moreover, the acidic environment of tumor stabilizes the HIF1α, which promotes the haphazardly vascularization through inducing vascular endothelial growth factor (VEGF) and recruits MDSCs to suppress the anti-tumor activity of T cells, resulting in tumor progression [168].
2.6. FOXO
FOXO family proteins are the transcription factors, which are categorized as evolutionarily conserved winged-helix DNA binding proteins (FOX proteins). These proteins act as tumor suppressors by orchestrating several biological processes, such as cell cycle arrest, apoptosis, and senescence. Moreover, these proteins help with scavenging the reactive oxygen species to maintain cellular homeostasis. To date, around 50 FOX proteins and 19 subfamilies have been reported and categorized based on their sequence homology [169], [170], [171], [172]. Despite the sequence homology, these proteins play a specific role due to their differential expression in distinct tissues. Conversely, these proteins also show redundancy in terms of their functions. Additionally, FOXO proteins consist of four members including FOXO1, FOXO3, FOXO4 and FOXO6 in mammalian cells. They can act as pioneer factors by binding to a consensus sequence and ensuring the active chromatin for transcription (Table 1) [173], [174]. Although the role of these proteins in regulating cancer progression and metastasis is well studied, there are limited studies showing how FOXO proteins directly regulate the metabolic pathway. FOXO1 has been shown to suppress the expression of enolase and pyruvate kinase as well as blocks the transcription of glucose 6-phosphatase and phosphoenolpyruvate kinase in hepatocyte cells [175], [176]. However, a rigorous study is needed to elucidate the exact mechanism by which FOXO1 regulates glycolysis.
Furthermore, FOXO1 interacts with PPARγ coactivator 1α (PGC-1α) and activates gluconeogenic genes. FOXO1 also regulates the expression of PGC-1α at the transcriptional level, indicating the feedback mechanism for oxidative stress regulation [177], [178], [179], [180]. Similarly, FOXO1 can interact with C/EBPα and stimulate the transcription of gluconeogenic genes like phosphoenolpyruvate carboxykinase, which promotes the growth of cancer cells [181]. It is important to note that FOXO3a inhibits tumor growth and metastasis by regulating LINC00926, a long non-coding RNA that suppresses the phosphoglycerate kinase 1 (PGK1), an enzyme essential for glycolysis. It will be of great interest to explore whether FOXO3a impacts on cancer phenotypes through PGK1 regulation.
In cancer cells, the expression of FOXO3a is upregulated, leading to a decrease in oxygen consumption to support cancer cell growth in the anaerobic condition. Intriguingly, the upregulation of FOXO3a is correlated with the activation of PDK, which inhibits the pyruvate dehydrogenase complex (PDC) for oxidative phosphorylation in cells. Moreover, the overexpression of FOXO3a is also correlated with the downregulation of several enzymes in TCA cycle and oxidative phosphorylation, such as fumarate hydratase (FH), NADH dehydrogenase (ND1) and cytochrome c oxidase (COX). Under normoxia, FOXO3a induces mitophagy that reduces the mitochondrial pool by promoting the expression of BCL2/adenovirus E1B 19 kd-interacting protein 3 (BNIP3) [182], [183], [184]. FOXO3A arrests the cell cycle progression in response to DNA damage and stimulates the DNA repair by modulating the growth arrest and DNA damage response gene (Gadd45a) (157). Meanwhile, cancer cells stabilize several transcription factors, such as c-Myc, PGC-1α and NRF1, leading to promoting mitochondrial biogenesis.
As the crucial role of FOXO in metabolic reprogramming, there are a few studies highlighting the role of FOXO-mediated metabolic reprogramming in tumorigenesis. Yan et al. demonstrated that FOXO1-mediated metabolic reprogramming regulates macrophage function, and that FOXO deletion in macrophages inhibited glycolysis leading to impaired phagocytic function of macrophages for inhibiting tumor cell growth and survival [185]. Nagarajan et al. showed that p53 transcriptionally represses paraoxonase 2 (PON2) and regulates GLUT1-mediated glucose transport for pancreatic cancer growth and metastasis. They further showed that PON2 loss activates AMPK→FOXO3A→PUMA signaling pathway to suppress pancreatic tumor growth [186].
Additionally, FOXO proteins show antagonism of Myc function by directly inhibiting the transcription and translational level of Myc. FOXO proteins regulate the function of Myc transcription factors through several ways. One way is that FOXO proteins recognize the promoters of Myc target proteins to inhibit their expression or simultaneously induce expression of MAX interacting proteins (MAXI 1 and MXD) [187]. Furthermore, activation of FOXO3a can promote Myc phosphorylation at phosphodegron motif and subsequent ubiquitination and degradation through FBW7 ubiquitin ligase [188]. Interestingly, the stabilization of FOXO acts a tumor suppressive during early stage of cancer development likely by suppressing Myc and HIF1α and their downstream gene expression, whereas overexpression of FOXO in the later stage of cancer could exacerbate tumor growth likely by activating the metabolic genes for ROS [188]. Hence, understanding the upstream factors that control the expression of FOXO proteins and interplay between FOXO proteins and Myc determining the fate and survival of cancer cells may open up the new avenue for cancer intervention.
2.7. Other transcription factors
In addition to the above-mentioned transcription factors, other important transcription factors that participate in metabolic reprogramming in cancer cells including Carbohydrate-response element-binding protein (ChREBP), Sterol regulatory element-binding protein 1 (SREBP-1), The peroxisome proliferator-activated receptors (PPARs), signal transducer and activator of transcription 3 (STAT3), estrogen receptors (ERs), E2 factor (E2F), and androgen receptor (AR) were briefly described below:
ChREBP, a key regulator of enzymes involved in fatty acid synthesis, is a glucose-regulated transcription factor [189]. ChREBP translocates from the cytoplasm to the nucleus in response to glucose and binds to carbohydrate response element of lipogenic genes in conjunction with MLX1. However, the activity of ChREBP is suppressed by adenosine monophosphate (AMP), ketone bodies, and cAMP [190]. The ChREBP/MLX heterodimer orchestrates glucose and lipid metabolism through regulating the expression of glycolytic enzymes (PKLR, FK, GLUT2, GLUT4), gluconeogenic enzyme (G6PC), and lipogenic enzymes (FASN, ACC1, SCD1, Elovl6) [190], [191], [192], [193]. ChREBP increases the aerobic glycolysis by modulating the expression of PDK, glycolytic enzyme (phospho-fructokinase, Pklr) and PPP pathway enzymes (G6PDH, TKT) [194], [195]. Interestingly, ChREBP inhibition leads to p53 activation, which is involved in metabolic reprogramming as mentioned above (Figs. 3B and 4) [193], [196]. Whether ChREBP acts through p53 regulation to control metabolic reprogramming and tumorigenesis remains to be explored.
Fig. 4.
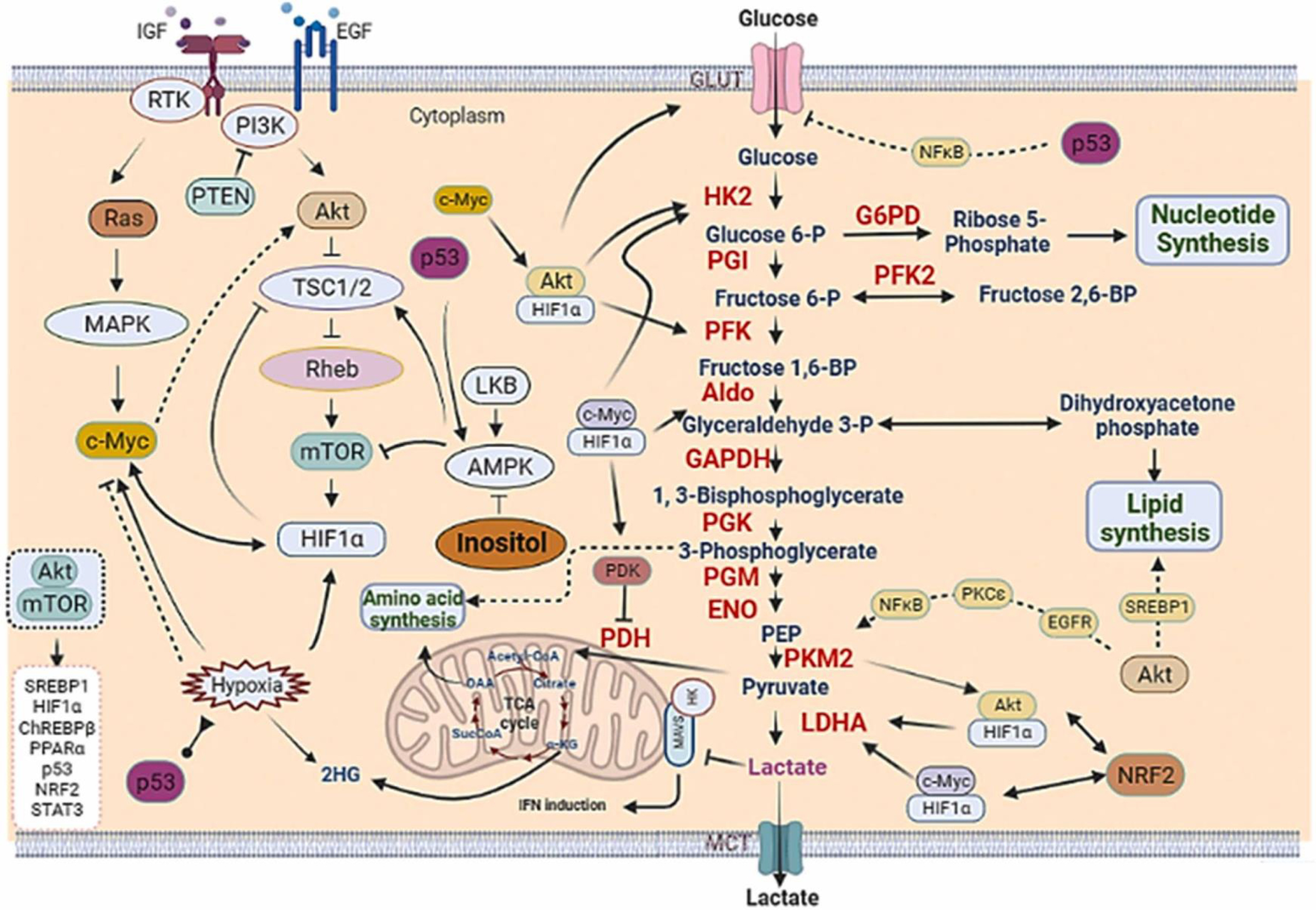
The crosstalk between transcriptional factors, signaling pathways and signaling metabolites regulates metabolic reprogramming. Tumor cells displayed dysregulated numerous transcription factors, which crosstalk with multiple signaling pathways to orchestrate distinct metabolic processes, thus facilitating cancer cell survival and cancer progression. The crosstalk between transcription factors indicated by the double headed arrows and enzymes are shown in red.
Lipid metabolism has emerged to play a key role in cancer progression and metastasis. SREBP-1 serves as a transcription factor, which reprograms tumor metabolism by regulating lipid metabolism [197]. SREBP regulates the gene involved in cholesterol synthesis and lipid metabolism [198], [199]. In normal tissues, SREBPs levels and activity are tightly controlled by endogenous sterol levels via a negative feedback regulation. However, cancer cells leverage high glucose uptake to synthesize fatty acids and cholesterol for the generation of new cell membranes and lipid rafts [200]. SREBP-1 has been identified as a key player for integrating the metabolic flux from glycolysis driven by PI3K/Akt signaling and fatty acid synthesis [201]. SREBP-1 could also regulate glutamine metabolism to drive the synthesis of the lipids (Fig. 3B and Fig. 4) [197], [202]. Furthermore, it has been shown that SREBP-1 is up-regulated in numerous cancers and contributes to tumor growth [203]. However, in order to reveal a direct connection between SREBP-1-mediated metabolic reprograming and tumorigenesis, more investigations are necessary. Collectively, SREBP-1 plays a critical role in linking lipid metabolism to oncogenic signaling. In addition, PPARs, a group of transcription factors, belonging to a nuclear receptor family of proteins also play important role in lipid metabolism [204]. These PPARs consist of three major isoforms (PPAR-α, PPAR-β and PPAR-γ). These proteins are differentially expressed in different tissues, which confer distinct noncanonical functions in diverse cells. PPAR-α expressed in adipose tissues, liver and heart regulates the fatty acid β-oxidation of fats. PPAR-β is also ubiquitously expressed in different types of tissues and involved in β-oxidation of fat [205], [206]. However, PPAR-γ expressed in adipose tissues induces the differentiation of adipose tissue and controls the lipogenesis (Fig. 4) [207], [208]. These proteins have been shown to regulate the activity of various mitochondrial proteins, such as carnitine palmitoyltransferase, citrate synthase and cytochrome oxidases [209], [210], [211], [212]. However, the role of PPAR proteins in cancer is still questionable [213]. As more evidence linking metabolic reprograming to cancer progression, PPAR-mediated lipid metabolism in cancer might provide valuable insight for cancer therapeutics.
Signal transducers and activators of transcription (STATs) are a group of transcription factors that play a crucial role in various biological processes, including innate immunity and cell proliferation and survival [214]. STAT3 have shown to induce glycolysis by upregulating HIF1α [215]. Interestingly, STAT3 can cooperate with HIF1 to regulate gene expression [216]. STAT3 upregulated in several cancers and promote tumorigenesis. The STAT3 transcription factor regulates diverse target genes to orchestrate cancer phenotypes [217]. It has been shown that STAT3 protects the NADH dehydrogenase (ND1) and succinate dehydrogenase (SDH) protein from ischemic shock under hypoxia and maintains the oxidative phosphorylation flux in cancer cells [218]. However, it is unclear whether these metabolic gene changes indeed contribute to cancer phenotypes induced by STAT3 activation.
Estrogen receptors consist of two isoforms, ERα and ERβ, which belong to the steroid/nuclear receptor superfamily of transcription factors. In essence, these transcription factors form a homodimer and/or heterodimer to regulate the expression of diverse genes associated with female sexual phenotypes and breast cancer progression [219]. It has been shown that upon its ligand 17-β-estradiol binding, ERs bind to the promoter region of the COX gene in mitochondrial DNA and induces the expression of COX, leading to promoting oxidative phosphorylation [220], [221]. In addition to the canonical transcriptional regulation, ERs could regulate mitochondrial metabolism by promoting overexpression and activating the NRF1[222]. Moreover, the ER also binds to β-hydroxyacyl coA- dehydrogenase (HACoADH) and modulates the β-oxidation of fat [223]. However, it remains unclear whether these regulations indeed contribute to cancer phenotypes regulated by ERs.
Additionally, other transcription factors, such as E2F and androgen receptor (AR), have been shown to participate in metabolic regulation. E2F overexpression regulates numerous genes such as NADH dehydrogenase subunit 1 (ND1), aconitase, and fumarate hydratase (FH) involved in oxidative phosphorylation and TCA cycle [224]. Like ER, AR is a transcription factor that also belongs to the steroid/nuclear receptor superfamily. AR upregulated in advanced prostate cancer plays a key role in prostate cancer cell proliferation and progression by regulating expression of its target genes. AR has been shown to repress numerous metabolic enzymes including FH and ND1 involved in oxidative phosphorylation and TCA cycle [225], although the functional role of these regulation in cancer remains to be established.
We speculate that metabolic reprogramming regulated by ChREBP, SREBP-1, PPARs, and STAT3 may be partly involved in their role in regulating tumorigenesis. For example, Tong at el. showed that ChREBP induction is required for the proliferation of colorectal cancer cells, and siRNA-mediated inhibition of ChREBP leads to a metabolic shift from aerobic glycolysis to mitochondrial oxidative phosphorylation, followed by tumor growth inhibition in vivo [196]. Wen et al. showed that downregulation of SREBP-1-mediated metabolic reprogramming restricts colon cancer growth both in vitro and in vivo [226]. Phan et al. showed that PPARγ sumoylation induced lipid accumulation and tumor growth in vivo [227]. STAT3 has been shown to increase fatty acid oxidation in CD8+ T cells, which plays a critical role in obesity-associated breast cancer progression [228]. Finally, Patel et al. revealed that STAT3-driven metabolic switch promotes tyrosine kinase inhibitor persistence in chronic myeloid leukemia [229]. However, further studies are needed to firmly understand the role of metabolic reprogramming regulated by these transcription factors in cancer progression and metastasis.
2.8. The crosstalk between transcriptional factors, signaling pathways and signaling metabolites in regulating metabolic reprogramming
Metabolic reprogramming has emerged to play key roles in regulating cancer cell growth, progression and metastasis and therapy resistance. Accumulating evidence indicates that distinct tumor microenvironments composed of diverse cell types, such as stromal cells, fibroblasts, immune cells, macrophages, cancer cells, and endothelial cells, can impact on metabolic features of cancer cells [230]. Tumor tissues are generally heterogeneous, which are composed of distinct cancer cell populations with different genetic and/or epigenetic backgrounds [231]. Cancer cells stay in a microenvironment different from normal cells, which may contain low nutrients and oxygen availability and high immunosurveillance. In order for cancer cells to survive under these hostile tumor microenvironments during cancer progression, cancer cells have evolved to reshape their metabolic states towards metabolic reprogramming [232], [233]. Interestingly, the crosstalk between transcription factors, multiple signaling pathways and signaling metabolites is an important mechanism by which tumor cells can adjust their metabolic states to achieve rapid growth and survival in the dynamic and stressed cancer microenvironments [234], [235].
One of the most important signaling pathways is the PI3K/Akt/mTOR signaling network. Growth factors, such as IGF-1 and epidermal growth factor. (EGF), activate PI3K/Akt/mTOR signaling, which is antagonized by phosphatase and tensin homolog (PTEN) tumor suppressor, to increase the anabolic program for maintaining cell proliferation and survival [236]. Notably, activation of PI3K/Akt/mTOR signaling is frequently observed in cancers partly because of gain of function mutations of PI3K and/or loss of function mutations of PTEN [237], [238], [239]. Activation of PI3K/Akt/mTOR signaling shifts oxidative phosphorylation to aerobic glycolysis through elevated GULT1 expression and activation of glycolysis enzymes, hexokinase and PFK1 [50], [240]. Notably, it has been shown that Akt and mTOR activation enhances robust anabolic biosynthetic program through regulating the activity of various aforementioned transcription factors, such as SREBP-1, HIF1α, ChREBPß, and PPARα, p53, NRF2, and STAT3 [241], [242], [243], [244], [245], [246], [247]. In addition to PI3K/Akt/mTOR signaling, KRAS, frequently activated in many cancers, promotes Myc-dependent metabolic reprograming in a manner dependent on PI3K and MAPK activation (Fig. 4) [248].
HIF1α is stabilized, translocated to the nucleus and turns on its target genes for regulating cell growth, survival, angiogenesis, and metabolic adaption in response to hypoxia conditions [249]. The crosstalk between HIF1α and signaling pathways is well documented and leads to metabolic reprogramming and cancer growth. The PI3K/Akt signaling and RAS drive the HIF1α synthesis through mTOR activation [250]. The HIF1α is also activated by the growth factor IGF-1 and cytokine TNF-α [251], [252]. Interestingly, HIF1α could induce gene expression of IGF-1 and TNF-α in cancer cells, thereby offering the positive feedback loop to further enhance HIF1α induction and activation. Apart from the crosstalk between transcription factors and signaling pathways, there is the interplay between HIF1α and Myc [253], [254]. Overexpression of Myc induces the expression and activation of HIF1α in cancer [255]. Myc and HIF1α modulate many common enzymes in glycolysis and amino acid and lipid metabolism [256], [257]. The crosstalk between Myc and HIF1α shapes the metabolic reprogramming leading to tumor growth and proliferation. However, it is important to note that HIF1α inhibits Myc activity in hypoxia, which can be overridden by Myc overexpression, although the underlying mechanism remains to be explored [253], [254]. HIF1α also interacts or cross-talks with other transcription factors, such as p53, NRF2, and NF-κb, although their functional link needs to be fully explored [258], [259]. Of note is that p50 and p65 subunits of NF-κB interact with HIF1α and enhance HIF1α expression under hypoxia or stimulation of reactive oxidation species [260]. The crosstalk between NRF2 and HIF1α, both of which orchestrate some similar signaling pathways in response to hypoxia, was also documented [261]. It has been shown that the NRF2 knockdown resulted in the decreased levels of HIF1α, whereas HIF1α activation resulted in elevated NRF2 activity [262], [263]. While high expression of p53 inhibits HIF1α expression and activity, gain of function mutation of p53 in cancer activates HIF1α [258].
Recent studies indicate that the metabolites derived from metabolism also contribute the metabolic reprogramming by regulating signaling pathways and/or transcriptional control. As a result, the metabolites secreted by cancer cells or other cell types in tumor microenvironments could impact cancer metabolism leading to the changes in cancer phenotypes [264]. The intermediates metabolites derived from mitochondrial TCA cycle, such as succinate, fumarate, and α-KG are known to act as signaling metabolites to regulate cancer cells and stem cells[265], [266]. α-KG serves as a cofactor for activation of the Jumonji histone demethylases and ten-eleven translocation (TET) DNA demethylases to orchestrate epigenetic reprogramming and transcriptional controls. Accumulating evidence reveals that α-ketoglutarate regulates cancer progression and metastasis through controlling epigenetic reprogramming and transcription. D-2-hydroxyglutarate (D-2HG), a reduced form of the TCA cycle intermediate α-KG, serves as an oncometabolite for cancer progression. Notably, accumulation of D-2HG in tumor tissues due to gain of function mutation of Isocitrate dehydrogenases (IDH) that converts isocitrate to α-KG, promotes cancer phenotypes by antagonizing α-KG-mediated epigenetic reprogramming and transcriptional controls. Similar to D-2HG, succinate and fumarate also interfere with the α-KG-dependent epigenetic modifications. The loss of function mutation of the succinate dehydrogenase mutation (SDH) and fumarate hydratase (FH) causes the accumulation of succinate and fumarate, respectively [267]. The accumulation of succinate or fumarate in the cytoplasm causes pseudohypoxia by competing with α-KG in binding to the PHD enzyme leading to stabilization of HIF1α [268]. Succinate also inhibits α-KG-dependent TET regulation, leading to promoter hypermethylation of several genes and transcriptional dysregulation [269].
Glucose metabolism through glycolysis not only produces energy for cancer cell proliferation and survival, but also generate metabolites and/or intermediate metabolites for signaling and translational regulation. Lactate, the end product of glycolysis, is ubiquitously present in the cancer microenvironments and previously thought to be a waste. Numerous outstanding studies published recently highlight the important role of lactate in cancer and immune regulation by multiple mechanisms [270], [271]. Lactate can serve as a fuel to promote cancer cell growth by shunting into TCA cycle to generate energy [272]. Lactate enriched and secreted from tumor microenvironments acts as an immune suppressive signal by limiting T cell activation, although the underlying mechanisms are not well understood [273]. Lactate has been shown to promote M2 macrophage polarization by activating HIF1α [274]. We demonstrated that lactate acts as a signaling metabolite to restrict innate immunity through direct MAVS binding [275]. Importantly, lactate can aslo modulate cellular metabolism through histone lactylation-mediated gene expression [276]. It is important to note that myo-inositol generated from glucose metabolism can also serve as a signaling metabolite that directly binds to and suppress AMPK activation leading to restricting AMPK-dependent mitochondrial fission and mitochondrial dysfunctions [277].
Collectively, these studies demonstrate numerous metabolites and/or intermediate metabolites derived from various metabolic processes serve as signaling metabolites to orchestrate the crosstalk between metabolism, signaling activation and transcriptional control.
2.9. Targeting metabolic vulnerability driven by deregulated transcription factors for cancer therapy
A plethora of studies highlight the crucial role of metabolic reprogramming in facilitating cancer progression and metastasis, immune escape and drug resistance. Metabolic reprogramming serves as a key adaptation mechanism for disseminated cancer cells to survive and prorogate in hostile tumor microenvironments, thus representing an actionable vulnerability of cancer cells that can be harnessed for developing strategies and/or agents for cancer targeting [278], [279]. While it has been well documented that directly targeting various metabolic enzymes such that involved in glycolysis and lipid metabolism critical for oncogenic processes is quite robust and effective in preclinical tumor models, the fact that many of the metabolic enzymes are also essential for normal cell functions may greatly limit the efficacy of this targeting strategy in clinical practice due to the intolerable side effects [280], [281]. Since numerous transcriptional factors, such as Myc, HIF1a, p53, STAT3, and NRF2 as described above, are deregulated in human cancers and play a key role in cancer progression and metastasis partly through their metabolic regulation, targeting these deregulated transcription factors involved in metabolic control may provide the alternative strategy for cancer targeting with the better therapeutic window.
Although directly targeting transcription factors by developing specific small molecular inhibitors against them is still technically challenging, numerous small molecule inhibitors blocking the transcription factor networks have been developed and shown to be effective in preclinical models [52], [282]. The examples are that BET bromodomain inhibitor I-BET 762 or JQ1 has been shown to suppress Myc activity partly by inducing Myc downregulation and displays anticancer effect against various cancers in in vivo tumor models [283], [284], [285]. Notably, Zhang et al. demonstrated that treatment of JQ1 suppresses cancer cell proliferation through inhibiting Myc-mediated glycolysis in Leukemia cells [286]. MDM2 inhibitor RG7112, which activates the p53 pathway by inhibiting MDM2-mediated p53 degradation, induces tumor regression of various tumor panels of pediatric cancer [287], although the role of p53-mediated metabolic reprogramming in the tumor-suppressive effect of RG7112 has not been examined [287]. Another example is that STAT3 inhibitor ODZ10117 suppresses tumor growth in glioblastoma tumor models [288]. An important breakthrough from a recent study indicated that a potent and selective small-molecule STAT3 degrader (SD-36) developed from the proteolysis targeting chimera (PROTAC) technology has been shown to achieve the robust tumor repression in multiple xenograft models at well-tolerable dose schedules [289]. NRF2 inhibitor, ML385, has been shown to target non-small cell lung cancer (NSCLC) with Keap1 mutations and sensitizes NSCLC to the carboplatin chemotherapy agent both in vitro and in vivo [290]. Collectively, these studies highlight the potential efficacy of targeting transcription factors for cancer suppression. However, further studies are needed to explore whether metabolic reprogramming in cancer is indeed altered and involved in tumor suppression by pharmacologically targeting these deregulated transcriptional factors.
3. Conclusions and future directions
Recent studies in last two decades have made the tremendous progress in cancer metabolism. Dysregulation of the transcription factors has been shown to regulate various metabolic pathways to facilitate cancer cell growth and survival in the dynamic tumor microenvironments. The transcription factors, such as Myc and HIF1α, could work in concert to regulate cancer metabolism through orchestrating the expression of the distinct metabolic genes. Notably, the activity of transcription factors is regulated by the key signaling pathways (e.g. PI3K/Akt/mTOR) and signaling metabolites (e.g. lactate). Some small molecules targeting transcription factors have been shown to be quite effective in preclinical models. However, since some transcription factors can regulate similar metabolic targets and/or metabolic pathways, targeting a particular transcription factor may be compensated by another transcription factor, which affects the same set of target genes in metabolic pathways. Thus, the combinatory inhibition of these transcriptions may need to be considered. Despite the important advances in better understanding of cancer metabolism, several outstanding questions need to be further addressed.
First, while deregulated transcription factors have been shown to play important roles in cancer regulation, whether this regulation acts through metabolic control remains unclear. Second, while numerous small molecule inhibitors targeting transcription factors are effective in preclinical models, their efficacy and safety in clinical trials are unknown. Third, how signaling metabolites, such as lactate and D-2HG, regulate cancer and immune cell properties is not well understood. Fourth, most studies described in this review are mainly based on xenograft models in nude mice without proper tumor microenvironments and intact immunity. It will be important to use syngenetic models and/or genetic models with intact immunity to further validate the conclusions. Fifth, since the tumor microenvironment plays a key role in cancer progression and therapy resistance, it is important to understand how transcription factor-mediated metabolic reprogramming may impact on tumor microenvironment to facilitate cancer progression. Finally, the concept of oncometabolite is just in its early stage, and only a few oncometabolites are identified. More systematic studies are required to globally identify additional oncometabolites for their roles and underling mechanisms in cancer regulation. Addressing these questions will not only significantly advances the metabolism field, but also offers the potential strategies for effective cancer targeting.
Acknowledgments
We thank the members of the Lin’s lab for their critical reading and comments on our manuscript. We apologize to many investigators whose important works were not cited in this review due to the space limitations. This work is supported in part by NIH grants (R01CA248037 and R01CA256158) and Anderson Discovery Endowed Professorship Funds to H.K.L.
Abbreviations
- ACC1
Acetyl-CoA carboxylase 1
- ACOX1/2
acyl-CoA oxidase 1 and 2
- ACYLATP
citrate lyase
- Akt
protein kinase B
- AMPKAMP
activated protein kinase
- AR
androgen receptor
- ASCT2/SLC1A5
alanine serine cysteine transporter 2/solutecarrier family 1 member 5
- BCA
Asbranched-chain amino acids
- BCAT1
branched chain amino acid transaminase 1
- BNIP3BCL2/
adenovirus E1B 19 kd-interacting protein 3
- CAD
carbamoyl-phosphate synthetase 2, aspartatetranscarbamylase, and dihydroorotase
- CBPCREB
binding protein
- ChREBP
carbohydrate-response element-binding protein
- COX
cytochrome c oxidase
- CPT1A/CPT2
carnitine palmitoyltransferase IA/2
- E2FE2
factor
- ELOVL6ELOVL
Fatty Acid Elongase 6
- ENO1
enolase 1
- ER
sestrogen receptors
- FASN
fatty acid synthase
- FOXO
forkhead box protein O
- G6PD
glucose-6-phosphate dehydrogenase
- GAMT
guanidinoacetate N-methyltransferase
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GDH
glutamate dehydrogenase
- GLS2
glutaminase 2
- GLUT1
glucose transporter 1
- GMPS
guanine monophoshate synthetase
- GNMT
glycine N-Methyltransferase
- GPI
glucose-6-phosphate isomerase
- GPT2
glutamic-pyruvic transaminase 2
- HIF1
hypoxia-inducible factor 1
- HK2
hexokinase 2
- HMGCR3
hydroxy-3-methylglutaryl-CoA reductase
- IDH3
αisocitrate dehydrogenase 3α
- IFN
γInterferon γ
- IL2
interleukin 2
- IMP
inosine monophosphate
- LDH
Alactate dehydrogenase A
- MCD
malonyl-CoA decarboxylase
- MCT1/2
monocarboxylate transporter 1/2
- MDM2
mouse double minute 2 homolog
- MDSCs
myeloid-derived suppresser cells
- ME1/2
malic enzyme 1/2
- MTHFD2
methylenetetrahydrofolate dehydrogenase 2
- mTORC1
mammalian target of rapamycin complex 1
- ND1NADH
dehydrogenase subunit 1
- NRF2
nuclear factor erythroid 2-related factor 2
- NSCLC
non-small cell lung cancer
- p300E1A
binding protein p300
- p53R2/RRM2
Bribonucleotide reductase regulatory TP53inducible subunit M2B
- P5CS
pyrroline-5-carboxylate synthase
- PANK1
pantothenate kinase 1
- PARK2
Parkin RBR E3 Ubiquitin Protein Ligase
- PDC
pyruvate dehydrogenase complex
- PDK
pyruvate dehydrogenase kinase 1
- PFAS
phosphoribosylformylglycinamidine synthase
- PFK
phosphofructokinase
- PFKFB6
phosphofructo-2-kinase/fructose-2,6-biphosphatase
- PFKM
phosphofructokinase, muscle
- PGK
phosphoglycerate kinase
- PGM1
phosphoglucomutase 1
- PHGDH
phosphoglycerate dehydrogenase
- PI3K
phosphatidylinositol-3-kinase
- PKM1/2
Pyruvate kinase M1/2
- PON2
paraoxonase 2
- PPA
inorganic pyrophosphatase
- PPARs
peroxisome proliferator-activated receptors
- PPAT
phosphoribosyl pyrophosphate amidotransferase
- PPP
pentose phosphate pathway
- PRKAA2
protein kinase AMP-activated catalytic subunitalpha 2
- PRODH/POX
proline dehydrogenase/proline oxidase
- PROTAC
Proteolysis-targeting chimera
- PRPS2
phosphoribosyl pyrophosphate synthetase 2
- PSAT1
phosphoserine aminotransferase 1
- PSPH
phosphoserine phosphatase
- PTEN
phosphatase and tensin homolog
- PYCR
pyrroline-5-carboxylate reductase 1
- SAMS
adenosyl-methionine
- SCD
stearoyl-CoA desaturase
- SCO2
synthesis of cytochrome c oxidase 2
- SHMT1/2
serine hydroxymethyltransferases 1/2
- SLC43A1
solute carrier family 43 member 1
- SLC7A5
solute carrier family 7 member 5
- SNAT5/SCL38A5
sodium-coupled neutral amino acid transporter5/solute carrier family 38 member 5
- SREBP-1
sterol regulatory element-binding protein 1
- STAT3
signal transducer and activator of transcription3
- TALDO1
transaldolase 1
- TIGARTP53
induced glycolysis regulatory phosphatase
- TKT
transketolase
- TP53
mutant tumor protein 53
- TSC1/2
tuberous sclerosis complex subunit 1
- UMP
uridine monophosphate
- VEGF
vascular endothelial growth factor
- VHL
von Hippel–Lindau tumor suppressor
Footnotes
Declaration of Competing Interest
H.K.L. is a consultant for Stablix, Inc. All other authors declare no competing interests.
Data availability
No data was used for the research described in the article.
References
- [1].Phan LM, Yeung SC, Lee MH, Cancer metabolic reprogramming: importance, main features, and potentials for precise targeted anti-cancer therapies, Cancer Biol. Med. 11 (2014) 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Rodriguez-Enriquez S, Marin-Hernandez A, Gallardo-Perez JC, Pacheco- Velazquez SC, Belmont-Diaz JA, Robledo-Cadena DX, et al. , Transcriptional regulation of energy metabolism in cancer cells, Cells (2019) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Islam Z, Ali AM, Naik A, Eldaw M, Decock J, Kolatkar PR, Transcription factors: the fulcrum between cell development and carcinogenesis, Front. Oncol. 11 (2021), 681377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kim DW, Kim KC, Kim KB, Dunn CT, Park KS, Transcriptional deregulation underlying the pathogenesis of small cell lung cancer, Transl. Lung Cancer Res. 7 (2018) 4–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Weidemuller P, Kholmatov M, Petsalaki E, Zaugg JB, Transcription factors: bridge between cell signaling and gene regulation, Proteomics 21 (2021), e2000034. [DOI] [PubMed] [Google Scholar]
- [6].Gomes AP, Blenis J, A nexus for cellular homeostasis: the interplay between metabolic and signal transduction pathways, Curr. Opin. Biotechnol. 34 (2015) 110–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].DeBerardinis RJ, Chandel NS, Fundamentals of cancer metabolism, Sci. Adv. 2 (2016), e1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ward PS, Thompson CB, Metabolic reprogramming: a cancer hallmark even warburg did not anticipate, Cancer Cell 21 (2012) 297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Warburg O, Wind F, Negelein E, The metabolism of tumors in the body, J. Gen. Physiol. 8 (1927) 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Vander Heiden MG, Cantley LC, Thompson CB, Understanding the Warburg effect: the metabolic requirements of cell proliferation, Science 324 (2009) 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Crabtree HG, Observations on the carbohydrate metabolism of tumours, Biochem. J. 23 (1929) 536–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Warburg O, On the origin of cancer cells, Science 123 (1956) 309–314. [DOI] [PubMed] [Google Scholar]
- [13].Racker E, Bioenergetics and the problem of tumor growth, Am. Sci. 60 (1972) 56–63. [PubMed] [Google Scholar]
- [14].Hsu PP, Sabatini DM, Cancer cell metabolism: Warburg and beyond, Cell 134 (2008) 703–707. [DOI] [PubMed] [Google Scholar]
- [15].Jiang P, Du W, Wu M, Regulation of the pentose phosphate pathway in cancer, Protein Cell 5 (2014) 592–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Jin L, Alesi GN, Kang S, Glutaminolysis as a target for cancer therapy, Oncogene 35 (2016) 3619–3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Yang L, Venneti S, Nagrath D, Glutaminolysis: a hallmark of cancer metabolism, Annu. Rev. Biomed. Eng. 19 (2017) 163–194. [DOI] [PubMed] [Google Scholar]
- [18].Brose SA, Marquardt AL, Golovko MY, Fatty acid biosynthesis from glutamate and glutamine is specifically induced in neuronal cells under hypoxia, J. Neurochem. 129 (2014) 400–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bhutia YD, Ganapathy V, Glutamine transporters in mammalian cells and their functions in physiology and cancer, Biochim. Biophys. Acta 1863 (2016) 2531–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ducker GS, Rabinowitz JD, One-carbon metabolism in health and disease, Cell Metab. 25 (2017) 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Clare CE, Brassington AH, Kwong WY, Sinclair KD, One-carbon metabolism: linking nutritional biochemistry to epigenetic programming of long-term development, Annu. Rev. Anim. Biosci. 7 (2019) 263–287. [DOI] [PubMed] [Google Scholar]
- [22].Mentch SJ, Locasale JW, One-carbon metabolism and epigenetics: understanding the specificity, Ann. N. Y. Acad. Sci. 1363 (2016) 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Amelio I, Cutruzzola F, Antonov A, Agostini M, Melino G, Serine and glycine metabolism in cancer, Trends Biochem. Sci. 39 (2014) 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Newman AC, Maddocks ODK, One-carbon metabolism in cancer, Br. J. Cancer 116 (2017) 1499–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Villa E, Sahu U, O’Hara BP, Ali ES, Helmin KA, Asara JM, et al. , mTORC1 stimulates cell growth through SAM synthesis and m(6)A mRNA-dependent control of protein synthesis, Mol. Cell 81 (2021) 2076–2093, e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ananieva EA, Wilkinson AC, Branched-chain amino acid metabolism in cancer, Curr. Opin. Clin. Nutr. Metab. Care 21 (2018) 64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wei Z, Liu X, Cheng C, Yu W, Yi P, Metabolism of amino acids in cancer, Front. Cell Dev. Biol. 8 (2020), 603837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Broer S, Amino acid transporters as targets for cancer therapy: why, where, when, and how, Int. J. Mol. Sci. (2020) 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lieu EL, Nguyen T, Rhyne S, Kim J, Amino acids in cancer, Exp. Mol. Med. 52 (2020) 15–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Houten SM, Violante S, Ventura FV, Wanders RJ, The biochemistry and physiology of mitochondrial fatty acid beta-oxidation and its genetic disorders, Annu. Rev. Physiol. 78 (2016) 23–44. [DOI] [PubMed] [Google Scholar]
- [31].Jeong H, Oh HE, Kim H, Lee JH, Lee ES, Kim YS, et al. , Upregulation of fatty acid transporters is associated with tumor progression in non-muscle- invasive bladder cancer, Pathol. Oncol. Res. (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Giacomini I, Gianfanti F, Desbats MA, Orso G, Berretta M, Prayer-Galetti T, et al. , Cholesterol metabolic reprogramming in cancer and its pharmacological modulation as therapeutic strategy, Front. Oncol. 11 (2021), 682911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Huang B, Song BL, Xu C, Cholesterol metabolism in cancer: mechanisms and therapeutic opportunities, Nat. Metab. 2 (2020) 132–141. [DOI] [PubMed] [Google Scholar]
- [34].Mashima T, Seimiya H, Tsuruo T, De novo fatty-acid synthesis and related pathways as molecular targets for cancer therapy, Br. J. Cancer 100 (2009) 1369–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Germain N, Dhayer M, Boileau M, Fovez Q, Kluza J, Marchetti P, Lipid metabolism and resistance to anticancer treatment, Biology (2020) 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Rak S, De Zan T, Stefulj J, Kosovic M, Gamulin O, Osmak M, FTIR spectroscopy reveals lipid droplets in drug resistant laryngeal carcinoma cells through detection of increased ester vibrational bands intensity, Analyst 139 (2014) 3407–3415. [DOI] [PubMed] [Google Scholar]
- [37].Petan T, Jarc E, Jusovic M, Lipid droplets in cancer: guardians of fat in a stressful world, Molecules (2018) 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Koundouros N, Poulogiannis G, Reprogramming of fatty acid metabolism in cancer, Br. J. Cancer 122 (2020) 4–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zaidi N, Lupien L, Kuemmerle NB, Kinlaw WB, Swinnen JV, Smans K, Lipogenesis and lipolysis: the pathways exploited by the cancer cells to acquire fatty acids, Prog. Lipid Res. 52 (2013) 585–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Khedkar GD, Prakash B, Khedkar CD, Chopade BA, Nucleic acids, in: Caballero B, Finglas PM, Toldrá F (Eds.), Encyclopedia of Food and Health, Academic Press, Oxford, 2016, pp. 84–92. [Google Scholar]
- [41].Nygaard P, Saxild HH, Nucleotide metabolism, in: Schaechter M (Ed.), Encyclopedia of Microbiology, third ed., Academic Press, Oxford, 2009, pp. 296–307. [Google Scholar]
- [42].Villa E, Ali ES, Sahu U, Ben-Sahra I, Cancer cells tune the signaling pathways to empower de novo synthesis of nucleotides, Cancers (2019) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Robinson AD, Eich ML, Varambally S, Dysregulation of de novo nucleotide biosynthetic pathway enzymes in cancer and targeting opportunities, Cancer Lett. 470 (2020) 134–140. [DOI] [PubMed] [Google Scholar]
- [44].Morrison AJ, Cancer cell metabolism connects epigenetic modifications to transcriptional regulation, FEBS J. 289 (2022) 1302–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Miranda-Goncalves V, Lameirinhas A, Henrique R, Jeronimo C, Metabolism and epigenetic interplay in cancer: regulation and putative therapeutic targets, Front. Genet. 9 (2018) 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Sun L, Zhang H, Gao P, Metabolic reprogramming and epigenetic modifications on the path to cancer, Protein Cell (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].van der Knaap JA, Verrijzer CP, Undercover: gene control by metabolites and metabolic enzymes, Genes Dev. 30 (2016) 2345–2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Hough KP, Chisolm DA, Weinmann AS, Transcriptional regulation of T cell metabolism, Mol. Immunol. 68 (2015) 520–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Horhold F, Eisel D, Oswald M, Kolte A, Roll D, Osen W, et al. , Reprogramming of macrophages employing gene regulatory and metabolic network models, PLoS Comput. Biol. 16 (2020), e1007657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Lien EC, Lyssiotis CA, Cantley LC, Metabolic reprogramming by the PI3K-Akt- mTOR pathway in cancer, Recent Results Cancer Res. 207 (2016) 39–72. [DOI] [PubMed] [Google Scholar]
- [51].Vadlakonda L, Pasupuleti M, Pallu R, Role of PI3K-AKT-mTOR and Wnt signaling pathways in transition of G1-S phase of cell cycle in cancer cells, Front. Oncol. 3 (2013) 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Bushweller JH, Targeting transcription factors in cancer - from undruggable to reality, Nat. Rev. Cancer 19 (2019) 611–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Walz S, Lorenzin F, Morton J, Wiese KE, von Eyss B, Herold S, et al. , Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles, Nature 511 (2014) 483–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Dong Y, Tu R, Liu H, Qing G, Regulation of cancer cell metabolism: oncogenic MYC in the driver’s seat, Signal Transduct. Target. Ther. 5 (2020) 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, et al. , Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc, J. Biol. Chem. 275 (2000) 21797–21800. [DOI] [PubMed] [Google Scholar]
- [56].Kim JW, Zeller KI, Wang Y, Jegga AG, Aronow BJ, O’Donnell KA, et al. , Evaluation of myc E-box phylogenetic footprints in glycolytic genes by chromatin immunoprecipitation assays, Mol. Cell Biol. 24 (2004) 5923–5936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Shim H, Dolde C, Lewis BC, Wu CS, Dang G, Jungmann RA, et al. , c-Myc transactivation of LDH-A: implications for tumor metabolism and growth, Proc. Natl. Acad. Sci. USA 94 (1997) 6658–6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Doherty JR, Yang C, Scott KE, Cameron MD, Fallahi M, Li W, et al. , Blocking lactate export by inhibiting the Myc target MCT1 Disables glycolysis and glutathione synthesis, Cancer Res. 74 (2014) 908–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Gan L, Xiu R, Ren P, Yue M, Su H, Guo G, et al. , Metabolic targeting of oncogene MYC by selective activation of the proton-coupled monocarboxylate family of transporters, Oncogene 35 (2016) 3037–3048. [DOI] [PubMed] [Google Scholar]
- [60].Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. , The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation, Immunity 35 (2011) 871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Morrish F, Isern N, Sadilek M, Jeffrey M, Hockenbery DM, c-Myc activates multiple metabolic networks to generate substrates for cell-cycle entry, Oncogene 28 (2009) 2485–2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, et al. , Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction, Proc. Natl. Acad. Sci. USA 105 (2008) 18782–18787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Penugurti V, Khumukcham SS, Padala C, Dwivedi A, Kamireddy KR, Mukta S, et al. , HPIP protooncogene differentially regulates metabolic adaptation and cell fate in breast cancer cells under glucose stress via AMPK and RNF2 dependent pathways, Cancer Lett. 518 (2021) 243–255. [DOI] [PubMed] [Google Scholar]
- [64].Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, et al. , c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism, Nature 458 (2009) 762–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Shroff EH, Eberlin LS, Dang VM, Gouw AM, Gabay M, Adam SJ, et al. , MYC oncogene overexpression drives renal cell carcinoma in a mouse model through glutamine metabolism, Proc. Natl. Acad. Sci. USA 112 (2015) 6539–6544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Korangath P, Teo WW, Sadik H, Han L, Mori N, Huijts CM, et al. , Targeting glutamine metabolism in breast cancer with aminooxyacetate, Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 21 (2015) 3263–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Tambay V, Raymond VA, Bilodeau M, MYC rules: leading glutamine metabolism toward a distinct cancer cell phenotype, Cancers (2021) 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Sun L, Song L, Wan Q, Wu G, Li X, Wang Y, et al. , cMyc-mediated activation of serine biosynthesis pathway is critical for cancer progression under nutrient deprivation conditions, Cell Res. 25 (2015) 429–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Yue M, Jiang J, Gao P, Liu H, Qing G, Oncogenic MYC activates a feedforward regulatory loop promoting essential amino acid metabolism and tumorigenesis, Cell Rep. 21 (2017) 3819–3832. [DOI] [PubMed] [Google Scholar]
- [70].Zhou W, Feng X, Ren C, Jiang X, Liu W, Huang W, et al. , Over-expression of BCAT1, a c-Myc target gene, induces cell proliferation, migration and invasion in nasopharyngeal carcinoma, Mol. Cancer 12 (2013) 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Liu W, Hancock CN, Fischer JW, Harman M, Phang JM, Proline biosynthesis augments tumor cell growth and aerobic glycolysis: involvement of pyridine nucleotides, Sci. Rep. 5 (2015) 17206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Kant R, Yen C-H, Hung J-H, Lu C-K, Tung C-Y, Chang P-C, et al. , Induction of GNMT by 1,2,3,4,6-penta-O-galloyl-beta-D-glucopyranoside through proteasome-independent MYC downregulation in hepatocellular carcinoma, Sci. Rep. 9 (2019) 1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Casciano JC, Perry C, Cohen-Nowak AJ, Miller KD, Vande Voorde J, Zhang Q, et al. , MYC regulates fatty acid metabolism through a multigenic program in claudin-low triple negative breast cancer, Br. J. Cancer 122 (2020) 868–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Edmunds LR, Sharma L, Kang A, Lu J, Vockley J, Basu S, et al. , c-Myc programs fatty acid metabolism and dictates acetyl-CoA abundance and fate, J. Biol. Chem. 289 (2014) 25382–25392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Gouw AM, Margulis K, Liu NS, Raman SJ, Mancuso A, Toal GG, et al. , The MYC oncogene cooperates with sterol-regulated element-binding protein to regulate lipogenesis essential for neoplastic growth, Cell Metab. 30 (2019) 556–572, e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Mannava S, Grachtchouk V, Wheeler LJ, Im M, Zhuang D, Slavina EG, et al. , Direct role of nucleotide metabolism in C-MYC-dependent proliferation of melanoma cells, Cell Cycle 7 (2008) 2392–2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Barfeld SJ, Fazli L, Persson M, Marjavaara L, Urbanucci A, Kaukoniemi KM, et al. , Myc-dependent purine biosynthesis affects nucleolar stress and therapy response in prostate cancer, Oncotarget 6 (2015) 12587–12602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Agarwal S, Chakravarthi B, Behring M, Kim HG, Chandrashekar DS, Gupta N, et al. , PAICS, a purine nucleotide metabolic enzyme, is involved in tumor growth and the metastasis of colorectal cancer, Cancers (2020) 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Hu S, Balakrishnan A, Bok RA, Anderton B, Larson PE, Nelson SJ, et al. , 13C-pyruvate imaging reveals alterations in glycolysis that precede c-Myc- induced tumor formation and regression, Cell Metab. 14 (2011) 131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Xiang Y, Stine ZE, Xia J, Lu Y, O’Connor RS, Altman BJ, et al. , Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis, J. Clin. Investig. 125 (2015) 2293–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Pacilli A, Calienni M, Margarucci S, D’Apolito M, Petillo O, Rocchi L, et al. , Carnitine-acyltransferase system inhibition, cancer cell death, and prevention of myc-induced lymphomagenesis, J. Natl. Cancer Inst. 105 (2013) 489–498. [DOI] [PubMed] [Google Scholar]
- [82].Barna M, Pusic A, Zollo O, Costa M, Kondrashov N, Rego E, et al. , Suppression of Myc oncogenic activity by ribosomal protein haploinsufficiency, Nature 456 (2008) 971–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Cunningham JT, Moreno MV, Lodi A, Ronen SM, Ruggero D, Protein and nucleotide biosynthesis are coupled by a single rate-limiting enzyme, PRPS2, to drive cancer, Cell 157 (2014) 1088–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Satoh K, Yachida S, Sugimoto M, Oshima M, Nakagawa T, Akamoto S, et al. , Global metabolic reprogramming of colorectal cancer occurs at adenoma stage and is induced by MYC, Proc. Natl. Acad. Sci. USA 114 (2017) E7697–E7706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Cargill KR, Stewart CA, Park EM, Ramkumar K, Gay CM, Cardnell RJ, et al. , Targeting MYC-enhanced glycolysis for the treatment of small cell lung cancer, Cancer Metab. 9 (2021) 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Yuneva MO, Fan TW, Allen TD, Higashi RM, Ferraris DV, Tsukamoto T, et al. , The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type, Cell Metab. 15 (2012) 157–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Tsakaneli A, Carregari VC, Morini M, Eva A, Cangemi G, Chayka O, et al. , MYC regulates metabolism through vesicular transfer of glycolytic kinases, Open Biol. 11 (2021), 210276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Liu X, Qin H, Li Z, Lv Y, Feng S, Zhuang W, et al. , Inspiratory hyperoxia suppresses lung cancer metastasis through a MYC/SLC1A5-dependent metabolic pathway, Eur. Respir. J. (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Liu J, Zhang C, Hu W, Feng Z, Tumor suppressor p53 and metabolism, J. Mol. Cell Biol. 11 (2019) 284–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Mantovani F, Collavin L, Del Sal G, Mutant p53 as a guardian of the cancer cell, Cell Death Differ. 26 (2019) 199–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Lacroix M, Riscal R, Arena G, Linares LK, Le L Cam, Metabolic functions of the tumor suppressor p53: Implications in normal physiology, metabolic disorders, and cancer, Mol. Metab. 33 (2020) 2–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Puzio-Kuter AM, The role of p53 in metabolic regulation, Genes Cancer 2 (2011) 385–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Ma W, Sung HJ, Park JY, Matoba S, Hwang PM, A pivotal role for p53: balancing aerobic respiration and glycolysis, J. Bioenerg. Biomembr. 39 (2007) 243–246. [DOI] [PubMed] [Google Scholar]
- [94].Blandino G, Valenti F, Sacconi A, Di Agostino S, Wild type- and mutant p53 proteins in mitochondrial dysfunction: emerging insights in cancer disease, Semin. Cell Dev. Biol. 98 (2020) 105–117. [DOI] [PubMed] [Google Scholar]
- [95].Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E, The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression, Cancer Res. 64 (2004) 2627–2633. [DOI] [PubMed] [Google Scholar]
- [96].Jiang P, Du W, Mancuso A, Wellen KE, Yang X, Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence, Nature 493 (2013) 689–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Kondoh H, Lleonart ME, Gil J, Wang J, Degan P, Peters G, et al. , Glycolytic enzymes can modulate cellular life span, Cancer Res. 65 (2005) 177–185. [PubMed] [Google Scholar]
- [98].Wang L, Xiong H, Wu F, Zhang Y, Wang J, Zhao L, et al. , Hexokinase 2- mediated Warburg effect is required for PTEN- and p53-deficiency-driven prostate cancer growth, Cell Rep. 8 (2014) 1461–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Zhang C, Lin M, Wu R, Wang X, Yang B, Levine AJ, et al. , Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect, Proc. Natl. Acad. Sci. USA 108 (2011) 16259–16264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Viotti J, Duplan E, Caillava C, Condat J, Goiran T, Giordano C, et al. , Glioma tumor grade correlates with parkin depletion in mutant p53-linked tumors and results from loss of function of p53 transcriptional activity, Oncogene 33 (2014) 1764–1775. [DOI] [PubMed] [Google Scholar]
- [101].Liu J, Zhang C, Zhao Y, Yue X, Wu H, Huang S, et al. , Parkin targets HIF- 1alpha for ubiquitination and degradation to inhibit breast tumor progression, Nat. Commun. 8 (2017) 1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Franklin DA, He Y, Leslie PL, Tikunov AP, Fenger N, Macdonald JM, et al. , p53 coordinates DNA repair with nucleotide synthesis by suppressing PFKFB3 expression and promoting the pentose phosphate pathway, Sci. Rep. 6 (2016) 38067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, et al. , TIGAR, a p53-inducible regulator of glycolysis and apoptosis, Cell 126 (2006) 107–120. [DOI] [PubMed] [Google Scholar]
- [104].Jiang P, Du W, Wang X, Mancuso A, Gao X, Wu M, et al. , p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase, Nat. Cell Biol. 13 (2011) 310–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Budanov AV, Karin M, p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling, Cell 134 (2008) 451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Matthew EM, Hart LS, Astrinidis A, Navaraj A, Dolloff NG, Dicker DT, et al. , The p53 target Plk2 interacts with TSC proteins impacting mTOR signaling, tumor growth and chemosensitivity under hypoxic conditions, Cell Cycle 8 (2009) 4168–4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Liu Y, Gu W, The complexity of p53-mediated metabolic regulation in tumor suppression, Semin. Cancer Biol. (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Won KY, Lim SJ, Kim GY, Kim YW, Han SA, Song JY, et al. , Regulatory role of p53 in cancer metabolism via SCO2 and TIGAR in human breast cancer, Hum. Pathol. 43 (2012) 221–228. [DOI] [PubMed] [Google Scholar]
- [109].Contractor T, Harris CR, p53 negatively regulates transcription of the pyruvate dehydrogenase kinase Pdk2, Cancer Res. 72 (2012) 560–567. [DOI] [PubMed] [Google Scholar]
- [110].Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z, Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function, Proc. Natl. Acad. Sci. USA 107 (2010) 7455–7460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Tajan M, Hock AK, Blagih J, Robertson NA, Labuschagne CF, Kruiswijk F, et al. , A role for p53 in the adaptation to glutamine starvation through the expression of SLC1A3, Cell Metab. 28 (2018) 721–736, e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Ou Y, Wang SJ, Jiang L, Zheng B, Gu W, p53 Protein-mediated regulation of phosphoglycerate dehydrogenase (PHGDH) is crucial for the apoptotic response upon serine starvation, J. Biol. Chem. 290 (2015) 457–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Donald SP, Sun XY, Hu CA, Yu J, Mei JM, Valle D, et al. , Proline oxidase, encoded by p53-induced gene-6, catalyzes the generation of proline-dependent reactive oxygen species, Cancer Res. 61 (2001) 1810–1815. [PubMed] [Google Scholar]
- [114].Parrales A, Iwakuma T, p53 as a regulator of lipid metabolism in cancer, Int. J. Mol. Sci. (2016) 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Holzer K, Drucker E, Roessler S, Dauch D, Heinzmann F, Waldburger N, et al. , Proteomic analysis reveals GMP synthetase as p53 repression target in liver cancer, Am. J. Pathol. 187 (2017) 228–235. [DOI] [PubMed] [Google Scholar]
- [116].Tanaka H, Arakawa H, Yamaguchi T, Shiraishi K, Fukuda S, Matsui K, et al. , A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage, Nature 404 (2000) 42–49. [DOI] [PubMed] [Google Scholar]
- [117].Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, et al. , Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence, Cell 149 (2012) 1269–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Moon SH, Huang CH, Houlihan SL, Regunath K, Freed-Pastor WA, Morris J.P. t., et al. , p53 Represses the mevalonate pathway to mediate tumor suppression, Cell 176 (2019) 564–580, e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Blagih J, Zani F, Chakravarty P, Hennequart M, Pilley S, Hobor S, et al. , Cancer-specific loss of p53 leads to a modulation of myeloid and T cell responses, Cell Rep. 30 (2020) 481–496, e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Banerjee A, Thyagarajan K, Chatterjee S, Chakraborty P, Kesarwani P, Soloshchenko M, et al. , Lack of p53 augments antitumor functions in cytolytic T cells, Cancer Res. 76 (2016) 5229–5240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Zhang C, Liu J, Liang Y, Wu R, Zhao Y, Hong X, et al. , Tumour-associated mutant p53 drives the Warburg effect, Nat. Commun. 4 (2013) 2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Siolas D, Vucic E, Kurz E, Hajdu C, Bar-Sagi D, Gain-of-function p53(R172H) mutation drives accumulation of neutrophils in pancreatic tumors, promoting resistance to immunotherapy, Cell Rep. 36 (2021), 109578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Freed-Pastor WA, Mizuno H, Zhao X, Langerod A, Moon SH, Rodriguez- Barrueco R, et al. , Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway, Cell 148 (2012) 244–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Ma Q, Role of nrf2 in oxidative stress and toxicity, Annu. Rev. Pharmacol. Toxicol. 53 (2013) 401–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Ramos-Gomez M, Kwak MK, Dolan PM, Itoh K, Yamamoto M, Talalay P, et al. , Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice, Proc. Natl. Acad. Sci. USA 98 (2001) 3410–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Satoh H, Moriguchi T, Takai J, Ebina M, Yamamoto M, Nrf2 prevents initiation but accelerates progression through the Kras signaling pathway during lung carcinogenesis, Cancer Res. 73 (2013) 4158–4168. [DOI] [PubMed] [Google Scholar]
- [127].Lau A, Villeneuve NF, Sun Z, Wong PK, Zhang DD, Dual roles of Nrf2 in cancer, Pharmacol. Res 58 (2008) 262–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Wang H, Liu X, Long M, Huang Y, Zhang L, Zhang R, et al. , NRF2 activation by antioxidant antidiabetic agents accelerates tumor metastasis, Sci. Transl. Med. 8 (2016) 334ra51. [DOI] [PubMed] [Google Scholar]
- [129].Fu J, Xiong Z, Huang C, Li J, Yang W, Han Y, et al. , Hyperactivity of the transcription factor Nrf2 causes metabolic reprogramming in mouse esophagus, J. Biol. Chem. 294 (2019) 327–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].He F, Ru X, Wen T, NRF2, a transcription factor for stress response and beyond, Int. J. Mol. Sci. (2020) 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, et al. , Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming, Cancer Cell 22 (2012) 66–79. [DOI] [PubMed] [Google Scholar]
- [132].Romero R, Sayin VI, Davidson SM, Bauer MR, Singh SX, LeBoeuf SE, et al. , Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis, Nat. Med. 23 (2017) 1362–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].He F, Antonucci L, Karin M, NRF2 as a regulator of cell metabolism and inflammation in cancer, Carcinogenesis 41 (2020) 405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].DeNicola GM, Chen PH, Mullarky E, Sudderth JA, Hu Z, Wu D, et al. , NRF2 regulates serine biosynthesis in non-small cell lung cancer, Nat. Genet. 47 (2015) 1475–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Guo H, Xu J, Zheng Q, He J, Zhou W, Wang K, et al. , NRF2 SUMOylation promotes de novo serine synthesis and maintains HCC tumorigenesis, Cancer Lett. 466 (2019) 39–48. [DOI] [PubMed] [Google Scholar]
- [136].Feng L, Zhao K, Sun L, Yin X, Zhang J, Liu C, et al. , SLC7A11 regulated by NRF2 modulates esophageal squamous cell carcinoma radiosensitivity by inhibiting ferroptosis, J. Transl. Med. 19 (2021) 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Sayin VI, LeBoeuf SE, Singh SX, Davidson SM, Biancur D, Guzelhan BS, et al. , Activation of the NRF2 antioxidant program generates an imbalance in central carbon metabolism in cancer, eLife (2017) 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Pang S, Lynn DA, Lo JY, Paek J, Curran SP, SKN-1 and Nrf2 couples proline catabolism with lipid metabolism during nutrient deprivation, Nat. Commun. 5 (2014) 5048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Wu KC, Cui JY, Klaassen CD, Beneficial role of Nrf2 in regulating NADPH generation and consumption, Toxicol. Sci. 123 (2011) 590–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Ludtmann MH, Angelova PR, Zhang Y, Abramov AY, Dinkova-Kostova AT, Nrf2 affects the efficiency of mitochondrial fatty acid oxidation, Biochem. J. 457 (2014) 415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Panieri E, Buha A, Telkoparan-Akillilar P, Cevik D, Kouretas D, Veskoukis A, et al. , Potential applications of NRF2 modulators in cancer therapy, Antioxidants (2020) 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Singh A, Happel C, Manna SK, Acquaah-Mensah G, Carrerero J, Kumar S, et al. , Transcription factor NRF2 regulates miR-1 and miR-206 to drive tumorigenesis, J. Clin. Investig. 123 (2013) 2921–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Lim S, Liu H, Madeira da Silva L, Arora R, Liu Z, Phillips JB, et al. , Immunoregulatory protein B7-H3 reprograms glucose metabolism in cancer cells by ROS-mediated stabilization of HIF1alpha, Cancer Res. 76 (2016) 2231–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Saito T, Ichimura Y, Taguchi K, Suzuki T, Mizushima T, Takagi K, et al. , p62/ Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming, Nat. Commun. 7 (2016) 12030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Okazaki K, Papagiannakopoulos T, Motohashi H, Metabolic features of cancer cells in NRF2 addiction status, Biophys. Rev. 12 (2020) 435–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Masoud GN, Li W, HIF-1alpha pathway: role, regulation and intervention for cancer therapy, Acta Pharm. Sin. B 5 (2015) 378–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Sharma A, Sinha S, Shrivastava N, Therapeutic targeting hypoxia-inducible factor (HIF-1) in cancer: cutting gordian knot of cancer cell metabolism, Front. Genet. 13 (2022), 849040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Corrado C, Fontana S, Hypoxia and HIF signaling: one axis with divergent effects, Int. J. Mol. Sci. (2020) 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, et al. , Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha, Genes Dev. 12 (1998) 149–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Yamashita K, Discher DJ, Hu J, Bishopric NH, Webster KA, Molecular regulation of the endothelin-1 gene by hypoxia. Contributions of hypoxia- inducible factor-1, activator protein-1, GATA-2, AND p300/CBP, J. Biol. Chem. 276 (2001) 12645–12653. [DOI] [PubMed] [Google Scholar]
- [151].Infantino V, Santarsiero A, Convertini P, Todisco S, Iacobazzi V, Cancer cell metabolism in hypoxia: role of HIF-1 as key regulator and therapeutic target, Int. J. Mol. Sci. (2021) 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Strowitzki MJ, Cummins EP, Taylor CT, Protein hydroxylation by hypoxia- inducible factor (HIF) hydroxylases: unique or ubiquitous? Cells (2019) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Albanese A, Daly LA, Mennerich D, Kietzmann T, See V, The role of hypoxia- inducible factor post-translational modifications in regulating its localisation, stability, and activity, Int. J. Mol. Sci. (2020) 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Ravi R, Mookerjee B, Bhujwalla ZM, Sutter CH, Artemov D, Zeng Q, et al. , Regulation of tumor angiogenesis by p53-induced degradation of hypoxia- inducible factor 1alpha, Genes Dev. 14 (2000) 34–44. [PMC free article] [PubMed] [Google Scholar]
- [155].Kim JW, Gao P, Liu YC, Semenza GL, Dang CV, Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1, Mol. Cell Biol. 27 (2007) 7381–7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Semenza GL, Roth PH, Fang HM, Wang GL, Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1, J. Biol. Chem. 269 (1994) 23757–23763. [PubMed] [Google Scholar]
- [157].Kim JW, Tchernyshyov I, Semenza GL, Dang CV, HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia, Cell Metab. 3 (2006) 177–185. [DOI] [PubMed] [Google Scholar]
- [158].Fantin VR, St-Pierre J, Leder P, Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance, Cancer Cell 9 (2006) 425–434. [DOI] [PubMed] [Google Scholar]
- [159].Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P, et al. , Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia- inducible factor 1, J. Biol. Chem. 271 (1996) 32529–32537. [DOI] [PubMed] [Google Scholar]
- [160].Kierans SJ, Taylor CT, Regulation of glycolysis by the hypoxia-inducible factor (HIF): implications for cellular physiology, J. Physiol. 599 (2021) 23–37. [DOI] [PubMed] [Google Scholar]
- [161].Koh MY, Powis G, Passing the baton: the HIF switch, Trends Biochem. Sci. 37 (2012) 364–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Wu Q, You L, Nepovimova E, Heger Z, Wu W, Kuca K, et al. , Hypoxia- inducible factors: master regulators of hypoxic tumor immune escape, J. Hematol. Oncol. 15 (2022) 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Becker LM, O’Connell JT, Vo AP, Cain MP, Tampe D, Bizarro L, et al. , Epigenetic reprogramming of cancer-associated fibroblasts deregulates glucose metabolism and facilitates progression of breast cancer, Cell Rep. 31 (2020), 107701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Zeng L, Morinibu A, Kobayashi M, Zhu Y, Wang X, Goto Y, et al. , Aberrant IDH3alpha expression promotes malignant tumor growth by inducing HIF-1- mediated metabolic reprogramming and angiogenesis, Oncogene 34 (2015) 4758–4766. [DOI] [PubMed] [Google Scholar]
- [165].Dong Y, Yin S, Jiang C, Luo X, Guo X, Zhao C, et al. , Involvement of autophagy induction in penta-1,2,3,4,6-O-galloyl-beta-D-glucose-induced senescence-like growth arrest in human cancer cells, Autophagy 10 (2014) 296–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Hao X, Ren Y, Feng M, Wang Q, Wang Y, Metabolic reprogramming due to hypoxia in pancreatic cancer: Implications for tumor formation, immunity, and more, Biomed. Pharmacother. 141 (2021), 111798. [DOI] [PubMed] [Google Scholar]
- [167].Rostamian H, Khakpoor-Koosheh M, Jafarzadeh L, Masoumi E, Fallah- Mehrjardi K, Tavassolifar MJ, et al. , Restricting tumor lactic acid metabolism using dichloroacetate improves T cell functions, BMC Cancer 22 (2022) 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Husain Z, Huang Y, Seth P, Sukhatme VP, Tumor-derived lactate modifies antitumor immune response: effect on myeloid-derived suppressor cells and NK cells, J. Immunol. 191 (2013) 1486–1495. [DOI] [PubMed] [Google Scholar]
- [169].Myatt SS, Lam EW, The emerging roles of forkhead box (Fox) proteins in cancer, Nat. Rev. Cancer 7 (2007) 847–859. [DOI] [PubMed] [Google Scholar]
- [170].Kaestner KH, Knochel W, Martinez DE, Unified nomenclature for the winged helix/forkhead transcription factors, Genes Dev. 14 (2000) 142–146. [PubMed] [Google Scholar]
- [171].Jiramongkol Y, Lam EW, FOXO transcription factor family in cancer and metastasis, Cancer Metastasis Rev. 39 (2020) 681–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Herman L, Todeschini AL, Veitia RA, Forkhead transcription factors in health and disease, Trends Genet. 37 (2021) 460–475. [DOI] [PubMed] [Google Scholar]
- [173].Calnan DR, Brunet A, The FoxO code, Oncogene 27 (2008) 2276–2288. [DOI] [PubMed] [Google Scholar]
- [174].Li J, Dai S, Chen X, Liang X, Qu L, Jiang L, et al. , Mechanism of forkhead transcription factors binding to a novel palindromic DNA site, Nucleic Acids Res. 49 (2021) 3573–3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Gross DN, Wan M, Birnbaum MJ, The role of FOXO in the regulation of metabolism, Curr. Diabetes Rep. 9 (2009) 208–214. [DOI] [PubMed] [Google Scholar]
- [176].Link W, Fernandez-Marcos PJ, FOXO transcription factors at the interface of metabolism and cancer, Int. J. Cancer 141 (2017) 2379–2391. [DOI] [PubMed] [Google Scholar]
- [177].Puigserver P, Rhee J, Donovan J, Walkey CJ, Yoon JC, Oriente F, et al. , Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction, Nature 423 (2003) 550–555. [DOI] [PubMed] [Google Scholar]
- [178].Schilling MM, Oeser JK, Boustead JN, Flemming BP, O’Brien RM, Gluconeogenesis: re-evaluating the FOXO1-PGC-1alpha connection, Nature 443 (2006) E10–E11. [DOI] [PubMed] [Google Scholar]
- [179].Daitoku H, Yamagata K, Matsuzaki H, Hatta M, Fukamizu A, Regulation of PGC-1 promoter activity by protein kinase B and the forkhead transcription factor FKHR, Diabetes 52 (2003) 642–649. [DOI] [PubMed] [Google Scholar]
- [180].Khalil SM, Goswami S, Mo X, Muthusamy N, Protein phosphatase 2A activation dependent down regulation of PGC1-Alpha and FOXO1 phosphorylation with OSU-2S in human lymphoma cells, Blood 134 (2019) 3977. [Google Scholar]
- [181].Sekine K, Chen YR, Kojima N, Ogata K, Fukamizu A, Miyajima A, Foxo1 links insulin signaling to C/EBPalpha and regulates gluconeogenesis during liver development, EMBO J. 26 (2007) 3607–3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Tran H, Brunet A, Grenier JM, Datta SR, Fornace AJ Jr., DiStefano PS, et al. , DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45, Protein Sci. 296 (2002) 530–534. [DOI] [PubMed] [Google Scholar]
- [183].Ju Y, Xu T, Zhang H, Yu A, FOXO1-dependent DNA damage repair is regulated by JNK in lung cancer cells, Int. J. Oncol. 44 (2014) 1284–1292. [DOI] [PubMed] [Google Scholar]
- [184].Chaanine AH, Kohlbrenner E, Gamb SI, Guenzel AJ, Klaus K, Fayyaz AU, et al. , FOXO3a regulates BNIP3 and modulates mitochondrial calcium, dynamics, and function in cardiac stress, Am. J. Physiol. Heart Circ. Physiol. 311 (2016) H1540–H1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Yan K, Da TT, Bian ZH, He Y, Liu MC, Liu QZ, et al. , Multi-omics analysis identifies FoxO1 as a regulator of macrophage function through metabolic reprogramming, Cell Death Dis. 11 (2020) 800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Nagarajan A, Dogra SK, Sun L, Gandotra N, Ho T, Cai G, et al. , Paraoxonase 2 facilitates pancreatic cancer growth and metastasis by stimulating GLUT1- mediated glucose transport, Mol. Cell 67 (2017) 685–701, e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Delpuech O, Griffiths B, East P, Essafi A, Lam EW, Burgering B, et al. , Induction of Mxi1-SR alpha by FOXO3a contributes to repression of Myc- dependent gene expression, Mol. Cell Biol. 27 (2007) 4917–4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Peck B, Ferber EC, Schulze A, Antagonism between FOXO and MYC regulates cellular powerhouse, Front. Oncol. 3 (2013) 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Uyeda K, Yamashita H, Kawaguchi T, Carbohydrate responsive element-binding protein (ChREBP): a key regulator of glucose metabolism and fat storage, Biochem Pharmacol. 63 (2002) 2075–2080. [DOI] [PubMed] [Google Scholar]
- [190].Ortega-Prieto P, Postic C, Carbohydrate sensing through the transcription factor ChREBP, Front. Genet. 10 (2019) 472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Iizuka K, Bruick RK, Liang G, Horton JD, Uyeda K, Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis, Proc. Natl. Acad. Sci. USA 101 (2004) 7281–7286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Ishii S, Iizuka K, Miller BC, Uyeda K, Carbohydrate response element binding protein directly promotes lipogenic enzyme gene transcription, Proc. Natl. Acad. Sci. USA 101 (2004) 15597–15602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Lei Y, Zhou S, Hu Q, Chen X, Gu J, Carbohydrate response element binding protein (ChREBP) correlates with colon cancer progression and contributes to cell proliferation, Sci. Rep. 10 (2020) 4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Iizuka K, Horikawa Y, ChREBP: a glucose-activated transcription factor involved in the development of metabolic syndrome, Endocr. J. 55 (2008) 617–624. [DOI] [PubMed] [Google Scholar]
- [195].Iizuka K, The transcription factor carbohydrate-response element-binding protein (ChREBP): a possible link between metabolic disease and cancer, Biochim. Biophys. Acta Mol. Basis Dis. 1863 (2017) 474–485. [DOI] [PubMed] [Google Scholar]
- [196].Tong X, Zhao F, Mancuso A, Gruber JJ, Thompson CB, The glucose- responsive transcription factor ChREBP contributes to glucose-dependent anabolic synthesis and cell proliferation, Proc. Natl. Acad. Sci. USA 106 (2009) 21660–21665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Guo D, Bell EH, Mischel P, Chakravarti A, Targeting SREBP-1-driven lipid metabolism to treat cancer, Curr. Pharm. Des. 20 (2014) 2619–2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Horton JD, Goldstein JL, Brown MS, SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver, J. Clin. Investig. 109 (2002) 1125–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Shimano H, Shimomura I, Hammer RE, Herz J, Goldstein JL, Brown MS, et al. , Elevated levels of SREBP-2 and cholesterol synthesis in livers of mice homozygous for a targeted disruption of the SREBP-1 gene, J. Clin. Investig. 100 (1997) 2115–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Vona R, Iessi E, Matarrese P, Role of cholesterol and lipid rafts in cancer signaling: a promising therapeutic opportunity? Front. Cell Dev. Biol. 9 (2021), 622908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Ricoult SJ, Yecies JL, Ben-Sahra I, Manning BD, Oncogenic PI3K and K-Ras stimulate de novo lipid synthesis through mTORC1 and SREBP, Oncogene 35 (2016) 1250–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Jhu JW, Yan JB, Lin ZH, Lin SC, Peng IC, SREBP1-induced glutamine synthetase triggers a feedforward loop to upregulate SREBP1 through Sp1 O- GlcNAcylation and augments lipid droplet formation in cancer cells, Int. J. Mol. Sci. (2021) 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Cheng C, Geng F, Cheng X, Guo D, Lipid metabolism reprogramming and its potential targets in cancer, Cancer Commun. 38 (2018) 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Gervois P, Torra IP, Fruchart JC, Staels B, Regulation of lipid and lipoprotein metabolism by PPAR activators, Clin. Chem. Lab. Med. 38 (2000) 3–11. [DOI] [PubMed] [Google Scholar]
- [205].Abbott BD, Wood CR, Watkins AM, Das KP, Lau CS, Peroxisome proliferator-activated receptors alpha, Beta, and gamma mRNA and protein expression in human fetal tissues, PPAR Res. 2010. (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Ricote M, Glass CK, PPARs and molecular mechanisms of transrepression, Biochim. Biophys. Acta 1771 (2007) 926–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Corrales P, Vidal-Puig A, Medina-Gomez G, PPARs and metabolic disorders associated with challenged adipose tissue plasticity, Int. J. Mol. Sci. (2018) 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Fan W, Evans R, PPARs and ERRs: molecular mediators of mitochondrial metabolism, Curr. Opin. Cell Biol. 33 (2015) 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Grabacka M, Pierzchalska M, Reiss K, Peroxisome proliferator activated receptor alpha ligands as anticancer drugs targeting mitochondrial metabolism, Curr. Pharm. Biotechnol. 14 (2013) 342–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Botta M, Audano M, Sahebkar A, Sirtori CR, Mitro N, Ruscica M, PPAR agonists and metabolic syndrome: an established role? Int. J. Mol. Sci. (2018) 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Hagland HR, Nilsson LI, Burri L, Nikolaisen J, Berge RK, Tronstad KJ, Induction of mitochondrial biogenesis and respiration is associated with mTOR regulation in hepatocytes of rats treated with the pan-PPAR activator tetradecylthioacetic acid (TTA), Biochem. Biophys. Res. Commun. 430 (2013) 573–578. [DOI] [PubMed] [Google Scholar]
- [212].Wang X, Moraes CT, Increases in mitochondrial biogenesis impair carcinogenesis at multiple levels, Mol. Oncol. 5 (2011) 399–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Peters JM, Shah YM, Gonzalez FJ, The role of peroxisome proliferator- activated receptors in carcinogenesis and chemoprevention, Nat. Rev. Cancer 12 (2012) 181–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Loh CY, Arya A, Naema AF, Wong WF, Sethi G, Looi CY, Signal transducer and activator of transcription (STATs) proteins in cancer and inflammation: functions and therapeutic implication, Front. Oncol. 9 (2019) 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Demaria M, Giorgi C, Lebiedzinska M, Esposito G, D’Angeli L, Bartoli A, et al. , A STAT3-mediated metabolic switch is involved in tumour transformation and STAT3 addiction, Aging 2 (2010) 823–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Pawlus MR, Wang L, Hu CJ, STAT3 and HIF1alpha cooperatively activate HIF1 target genes in MDA-MB-231 and RCC4 cells, Oncogene 33 (2014) 1670–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Poli V, Camporeale A, STAT3-mediated metabolic reprograming in cellular transformation and implications for drug resistance, Front. Oncol. 5 (2015) 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Szczepanek K, Chen Q, Derecka M, Salloum FN, Zhang Q, Szelag M, et al. , Mitochondrial-targeted Signal transducer and activator of transcription 3 (STAT3) protects against ischemia-induced changes in the electron transport chain and the generation of reactive oxygen species, J. Biol. Chem. 286 (2011) 29610–29620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Yasar P, Ayaz G, User SD, Gupur G, Muyan M, Molecular mechanism of estrogen-estrogen receptor signaling, Reprod. Med. Biol. 16 (2017) 4–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Stacey W, Bhave S, Uht RM, Mechanisms by which 17beta-estradiol (E2) suppress neuronal cox-2 gene expression, PloS One 11 (2016), e0161430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Chen JQ, Delannoy M, Cooke C, Yager JD, Mitochondrial localization of ERalpha and ERbeta in human MCF7 cells, Am. J. Physiol. Endocrinol. Metab. 286 (2004) E1011–E1022. [DOI] [PubMed] [Google Scholar]
- [222].Mattingly KA, Ivanova MM, Riggs KA, Wickramasinghe NS, Barch MJ, Klinge CM, Estradiol stimulates transcription of nuclear respiratory factor-1 and increases mitochondrial biogenesis, Mol. Endocrinol. 22 (2008) 609–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Zhou Z, Zhou J, Du Y, Estrogen receptor alpha interacts with mitochondrial protein HADHB and affects beta-oxidation activity, Mol. Cell Proteom. 11 (2012). M111 011056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Ambrus AM, Islam AB, Holmes KB, Moon NS, Lopez-Bigas N, Benevolenskaya EV, et al. , Loss of dE2F compromises mitochondrial function, Dev. Cell 27 (2013) 438–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Audet-Walsh E, Yee T, McGuirk S, Vernier M, Ouellet C, St-Pierre J, et al. , Androgen-dependent repression of ERRgamma reprograms metabolism in prostate cancer, Cancer Res. 77 (2017) 378–389. [DOI] [PubMed] [Google Scholar]
- [226].Wen YA, Xiong X, Zaytseva YY, Napier DL, Vallee E, Li AT, et al. , Downregulation of SREBP inhibits tumor growth and initiation by altering cellular metabolism in colon cancer, Cell Death Dis. 9 (2018) 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Phan ANH, Vo VTA, Hua TNM, Kim MK, Jo SY, Choi JW, et al. , PPARgamma sumoylation-mediated lipid accumulation in lung cancer, Oncotarget 8 (2017) 82491–82505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Zhang C, Yue C, Herrmann A, Song J, Egelston C, Wang T, et al. , STAT3 activation-induced fatty acid oxidation in CD8(+) T effector cells is critical for obesity-promoted breast tumor growth, Cell Metab. 31 (2020) 148–161, e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Patel SB, Nemkov T, Stefanoni D, Benavides GA, Bassal MA, Crown BL, et al. , Metabolic alterations mediated by STAT3 promotes drug persistence in CML, Leukemia 35 (2021) 3371–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Gouirand V, Guillaumond F, Vasseur S, Influence of the tumor microenvironment on cancer cells metabolic reprogramming, Front. Oncol. 8 (2018) 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Zellmer VR, Zhang S, Evolving concepts of tumor heterogeneity, Cell Biosci. 4 (2014) 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Reina-Campos M, Moscat J, Diaz-Meco M, Metabolism shapes the tumor microenvironment, Curr. Opin. Cell Biol. 48 (2017) 47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Quail DF, Joyce JA, Microenvironmental regulation of tumor progression and metastasis, Nat. Med. 19 (2013) 1423–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Nazemi M, Rainero E, Cross-talk between the tumor microenvironment, extracellular matrix, and cell metabolism in cancer, Front. Oncol. 10 (2020) 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Hanahan D, Weinberg RA, Hallmarks of cancer: the next generation, Cell 144 (2011) 646–674. [DOI] [PubMed] [Google Scholar]
- [236].Hoxhaj G, Manning BD, The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism, Nat. Rev. Cancer 20 (2020) 74–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT, The PI3K pathway in human disease, Cell 170 (2017) 605–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [238].Rodriguez-Escudero I, Oliver MD, Andres-Pons A, Molina M, Cid VJ, Pulido R, A comprehensive functional analysis of PTEN mutations: implications in tumor- and autism-related syndromes, Hum. Mol. Genet. 20 (2011) 4132–4142. [DOI] [PubMed] [Google Scholar]
- [239].Carracedo A, Pandolfi PP, The PTEN-PI3K pathway: of feedbacks and cross- talks, Oncogene 27 (2008) 5527–5541. [DOI] [PubMed] [Google Scholar]
- [240].Fan H, Wu Y, Yu S, Li X, Wang A, Wang S, et al. , Critical role of mTOR in regulating aerobic glycolysis in carcinogenesis (Review), Int. J. Oncol. 58 (2021) 9–19. [DOI] [PubMed] [Google Scholar]
- [241].Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, et al. , SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth, Cell Metab. 8 (2008) 224–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Harada H, Itasaka S, Kizaka-Kondoh S, Shibuya K, Morinibu A, Shinomiya K, et al. , The Akt/mTOR pathway assures the synthesis of HIF-1alpha protein in a glucose- and reoxygenation-dependent manner in irradiated tumors, J. Biol. Chem. 284 (2009) 5332–5342. [DOI] [PubMed] [Google Scholar]
- [243].Chau GC, Im DU, Kang TM, Bae JM, Kim W, Pyo S, et al. , mTOR controls ChREBP transcriptional activity and pancreatic beta cell survival under diabetic stress, J. Cell Biol. 216 (2017) 2091–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [244].Blanchard PG, Festuccia WT, Houde VP, St-Pierre P, Brule S, Turcotte V, et al. , Major involvement of mTOR in the PPARgamma-induced stimulation of adipose tissue lipid uptake and fat accretion, J. Lipid Res. 53 (2012) 1117–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [245].Zhou J, Wulfkuhle J, Zhang H, Gu P, Yang Y, Deng J, et al. , Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance, Proc. Natl. Acad. Sci. USA 104 (2007) 16158–16163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [246].Papaiahgari S, Zhang Q, Kleeberger SR, Cho HY, Reddy SP, Hyperoxia stimulates an Nrf2-ARE transcriptional response via ROS-EGFR-PI3K-Akt/ERK MAP kinase signaling in pulmonary epithelial cells, Antioxid. Redox Signal. 8 (2006) 43–52. [DOI] [PubMed] [Google Scholar]
- [247].Cui D, Qu R, Liu D, Xiong X, Liang T, Zhao Y, The cross talk between p53 and mTOR pathways in response to physiological and genotoxic stresses, Front. Cell Dev. Biol. 9 (2021), 775507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Pupo E, Avanzato D, Middonti E, Bussolino F, Lanzetti L, KRAS-driven metabolic rewiring reveals novel actionable targets in cancer, Front. Oncol. 9 (2019) 848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Caro J, Hypoxia regulation of gene transcription, High. Alt. Med. Biol. 2 (2001) 145–154. [DOI] [PubMed] [Google Scholar]
- [250].Agani F, Jiang BH, Oxygen-independent regulation of HIF-1: novel involvement of PI3K/AKT/mTOR pathway in cancer, Curr. Cancer Drug Targets 13 (2013) 245–251. [DOI] [PubMed] [Google Scholar]
- [251].Coimbra IB, Jimenez SA, Hawkins DF, Piera-Velazquez S, Stokes DG, Hypoxia inducible factor-1 alpha expression in human normal and osteoarthritic chondrocytes, Osteoarthr. Cartil. 12 (2004) 336–345. [DOI] [PubMed] [Google Scholar]
- [252].Fukuda R, Hirota K, Fan F, Jung YD, Ellis LM, Semenza GL, Insulin-like growth factor 1 induces hypoxia-inducible factor 1-mediated vascular endothelial growth factor expression, which is dependent on MAP kinase and phosphatidylinositol 3-kinase signaling in colon cancer cells, J. Biol. Chem. 277 (2002) 38205–38211. [DOI] [PubMed] [Google Scholar]
- [253].Li Y, Sun XX, Qian DZ, Dai MS, Molecular crosstalk between MYC and HIF in cancer, Front. Cell Dev. Biol. 8 (2020), 590576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [254].Gnanaprakasam JNR, Sherman JW, Wang R, MYC and HIF in shaping immune response and immune metabolism, Cytokine Growth Factor Rev. 35 (2017) 63–70. [DOI] [PubMed] [Google Scholar]
- [255].Chen C, Cai S, Wang G, Cao X, Yang X, Luo X, et al. , c-Myc enhances colon cancer cell-mediated angiogenesis through the regulation of HIF-1alpha, Biochem. Biophys. Res. Commun. 430 (2013) 505–511. [DOI] [PubMed] [Google Scholar]
- [256].Nagao A, Kobayashi M, Koyasu S, Chow CCT, Harada H, HIF-1-dependent reprogramming of glucose metabolic pathway of cancer cells and its therapeutic significance, Int. J. Mol. Sci. (2019) 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [257].Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV, MYC, metabolism, and cancer, Cancer Discov. 5 (2015) 1024–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [258].Zhang C, Liu J, Wang J, Zhang T, Xu D, Hu W, et al. , The interplay between tumor suppressor p53 and hypoxia signaling pathways in cancer, Front. Cell Dev. Biol. 9 (2021), 648808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [259].Johansson K, Cebula M, Rengby O, Dreij K, Carlstrom KE, Sigmundsson K, et al. , Cross talk in HEK293 cells between Nrf2, HIF, and NF-kappaB activities upon challenges with redox therapeutics characterized with single-cell resolution, Antioxid. Redox Signal. 26 (2017) 229–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [260].Jiang Y, Zhu Y, Wang X, Gong J, Hu C, Guo B, et al. , Temporal regulation of HIF-1 and NF-kappaB in hypoxic hepatocarcinoma cells, Oncotarget 6 (2015) 9409–9419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [261].Toth RK, Warfel NA, Strange bedfellows: nuclear factor, erythroid 2-like 2 (Nrf2) and hypoxia-inducible factor 1 (HIF-1) in tumor hypoxia, Antioxidants (2017) 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [262].Malec V, Gottschald OR, Li S, Rose F, Seeger W, Hanze J, HIF-1 alpha signaling is augmented during intermittent hypoxia by induction of the Nrf2 pathway in NOX1-expressing adenocarcinoma A549 cells, Free Radic. Biol. Med. 48 (2010) 1626–1635. [DOI] [PubMed] [Google Scholar]
- [263].Kim TH, Hur EG, Kang SJ, Kim JA, Thapa D, Lee YM, et al. , NRF2 blockade suppresses colon tumor angiogenesis by inhibiting hypoxia-induced activation of HIF-1alpha, Cancer Res. 71 (2011) 2260–2275. [DOI] [PubMed] [Google Scholar]
- [264].Wang YP, Li JT, Qu J, Yin M, Lei QY, Metabolite sensing and signaling in cancer, J. Biol. Chem. 295 (2020) 11938–11946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [265].Raimundo N, Baysal BE, Shadel GS, Revisiting the TCA cycle: signaling to tumor formation, Trends Mol. Med. 17 (2011) 641–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [266].Etchegaray JP, Mostoslavsky R, Interplay between metabolism and epigenetics: a nuclear adaptation to environmental changes, Mol. Cell 62 (2016) 695–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [267].Abla H, Sollazzo M, Gasparre G, Iommarini L, Porcelli AM, The multifaceted contribution of alpha-ketoglutarate to tumor progression: an opportunity to exploit? Semin. Cell Dev. Biol. 98 (2020) 26–33. [DOI] [PubMed] [Google Scholar]
- [268].Kluckova K, Tennant DA, Metabolic implications of hypoxia and pseudohypoxia in pheochromocytoma and paraganglioma, Cell Tissue Res. 372 (2018) 367–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [269].Yang M, Soga T, Pollard PJ, Oncometabolites: linking altered metabolism with cancer, J. Clin. Investig. 123 (2013) 3652–3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [270].de la Cruz-Lopez KG, Castro-Munoz LJ, Reyes-Hernandez DO, Garcia- Carranca A, Manzo-Merino J, Lactate in the regulation of tumor microenvironment and therapeutic approaches, Front. Oncol. 9 (2019) 1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [271].Rawat D, Chhonker SK, Naik RA, Mehrotra A, Trigun SK, Koiri RK, Lactate as a signaling molecule: Journey from dead end product of glycolysis to tumor survival, Front. Biosci. 24 (2019) 366–381. [DOI] [PubMed] [Google Scholar]
- [272].Rabinowitz JD, Enerback S, Lactate: the ugly duckling of energy metabolism, Nat. Metab. 2 (2020) 566–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [273].Romero-Garcia S, Moreno-Altamirano MM, Prado-Garcia H, Sanchez- Garcia FJ, Lactate contribution to the tumor microenvironment: mechanisms, effects on immune cells and therapeutic relevance, Front. Immunol. 7 (2016) 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [274].Zhang L, Li S, Lactic acid promotes macrophage polarization through MCT- HIF1alpha signaling in gastric cancer, Exp. Cell Res. 388 (2020), 111846. [DOI] [PubMed] [Google Scholar]
- [275].Zhang W, Wang G, Xu ZG, Tu H, Hu F, Dai J, et al. , Lactate is a natural suppressor of RLR signaling by targeting MAVS, Cell 178 (2019) 176–189, e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [276].Jiang J, Huang D, Jiang Y, Hou J, Tian M, Li J, et al. , Lactate modulates cellular metabolism through histone lactylation-mediated gene expression in non- small cell lung cancer, Front. Oncol. 11 (2021), 647559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [277].Hsu CC, Zhang X, Wang G, Zhang W, Cai Z, Pan BS, et al. , Inositol serves as a natural inhibitor of mitochondrial fission by directly targeting AMPK, Mol. Cell 81 (2021) 3803–3819, e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [278].Herst PM, Carson GM, Eccles DA, Berridge MV, Bioenergetic and metabolic adaptation in tumor progression and metastasis, Front. Oncol. 12 (2022), 857686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [279].Faubert B, Solmonson A, DeBerardinis RJ, Metabolic reprogramming and cancer progression, Science (2020) 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [280].Stine ZE, Schug ZT, Salvino JM, Dang CV, Targeting cancer metabolism in the era of precision oncology, Nat. Rev. Drug Discov. 21 (2022) 141–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [281].Luengo A, Gui DY, Vander Heiden MG, Targeting metabolism for cancer therapy, Cell Chem. Biol. 24 (2017) 1161–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [282].Chen A, Koehler AN, Transcription factor inhibition: lessons learned and emerging targets, Trends Mol. Med. 26 (2020) 508–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [283].Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. , BET bromodomain inhibition as a therapeutic strategy to target c-Myc, Cell 146 (2011) 904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [284].Shao Q, Kannan A, Lin Z, Stack BC Jr., J.Y. Suen, L. Gao, BET protein inhibitor JQ1 attenuates Myc-amplified MCC tumor growth in vivo, Cancer Res. 74 (2014) 7090–7102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [285].Xie F, Huang M, Lin X, Liu C, Liu Z, Meng F, et al. , The BET inhibitor I-BET762 inhibits pancreatic ductal adenocarcinoma cell proliferation and enhances the therapeutic effect of gemcitabine, Sci. Rep. 8 (2018) 8102. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [286].Zhang MY, Liu SL, Huang WL, Tang DB, Zheng WW, Zhou N, et al. , Bromodomains and extra-terminal (BET) inhibitor JQ1 suppresses proliferation of acute lymphocytic leukemia by inhibiting c-Myc-mediated glycolysis, Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 26 (2020), e923411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [287].Carol H, Reynolds CP, Kang MH, Keir ST, Maris JM, Gorlick R, et al. , Initial testing of the MDM2 inhibitor RG7112 by the pediatric preclinical testing program, Pediatr. Blood Cancer 60 (2013) 633–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [288].Kim BH, Lee H, Park CG, Jeong AJ, Lee SH, Noh KH, et al. , STAT3 inhibitor ODZ10117 suppresses glioblastoma malignancy and prolongs survival in a glioblastoma xenograft model, Cells (2020) 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [289].Zhou H, Bai L, Xu R, Zhao Y, Chen J, McEachern D, et al. , Structure-based discovery of SD-36 as a potent, selective, and efficacious PROTAC degrader of STAT3 protein, J. Med. Chem. 62 (2019) 11280–11300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [290].Singh A, Venkannagari S, Oh KH, Zhang YQ, Rohde JM, Liu L, et al. , Small molecule inhibitor of NRF2 selectively intervenes therapeutic resistance in KEAP1-deficient NSCLC tumors, ACS Chem. Biol. 11 (2016) 3214–3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data was used for the research described in the article.