

Abstract
Advanced cancers often present with the cachexia syndrome that impacts peripheral tissues leading to involuntary weight loss and reduced prognosis. The central tissues undergoing depletion are skeletal muscle and adipose, but recent findings reveal an expanding tumor macroenvironment involving organ crosstalks that underlie the cachectic state.
The Origin of the Tumor Macroenvironment in Cancer Cachexia
Cancer patients often suffer from a syndrome called cachexia which is manifested by involuntary weight loss1. In cases when the weight loss is so severe the condition becomes invariably fatal. There are several principals of cancer cachexia that have been well documented in the literature and serve as a basis for this commentary2, especially for those less familiar with the topic. They include:
Weight loss is a predictor of survival and patients with cachexia generally respond more poorly to therapy leading to a lower quality of life.
Cachexia is commonly associated with end stage disease, but there are exceptions such as in pancreatic cancer where weight loss frequently occurs prior to time of diagnosis.
As little as 5% weight loss can negatively impact prognosis, and although measuring weight is not as accurate as recording lean body mass by radiographic imaging, body weight remains a standard diagnostic assessment for cachexia in cancer patients.
Anorexia contributes to cachexia, but hyper feeding does not restore body weight or improve survival for cancer patients. Thus, cancer cachexia should not be solely considered a nutritional disorder.
Weight loss predominantly derives from the catabolism of two tissues, adipose and skeletal muscle. The depletion of these tissues is thought to reflect the metabolic dysregulation of a selective group of tumor types.
The past three decades of cancer research has revealed great insights in the cellular and molecular interactions of the tumor microenvironment. In parallel studies along the same timeline, cancer cachexia research has focused on identifying circulating factors and mechanisms underlying tissue atrophy and weight loss within the tumor macroenvironment. Early work proposed that dysregulated metabolism in this macroenvironment is due to the high demand for glucose as a main nutrient source to sustain tumor growth and survival, which comes at the cost of depleting host tissues2. In this wasting scenario, glucose is generated from the breakdown of adipose and skeletal muscle that release glycerol and amino acids, respectively. These metabolites are then transported to the liver where they are utilized in gluconeogenesis to generate glucose via the Cori cycle2. Other breakdown products of adipose, in the form of fatty acids, and separate amino acids released from skeletal muscle can be taken up directly by the tumor and utilized to synthesize nucleic acids and proteins, required for cell division and cell migration. For the cancer patient suffering from cachexia, these catabolic processes generate a high degree of energy loss leading to weakness and fatigue.
Following this initial characterization of the tumor macroenvironment in cachexia, studies began to focus in on the underlying molecular mechanisms leading to the catabolism of adipose and skeletal muscle, the two tissues primarily accounting for weight loss in cancer patients suffering from cachexia1. Catabolism here refers to the loss of tissue mass resulting from atrophy rather than from apoptosis or necrosis, given that there is relevatively little cumulative evidence to show that cell death in either myonuclei of mature skeletal muscle or adipocytes in fat contributes to tumor-induced tissue wasting1.
For skeletal muscle atrophy, studies have primarily used rodent models of cancer cachexia to reveal the ubiquitin ligase proteasome pathway as a major regulator of skeletal muscle protein catabolism3. The first identified, and to this day the two most highly cited biomarkers of skeletal muscle atrophy in cancer as well as other muscle wasting conditions, are the E3 ubiquitin ligases, MuRF1/TRIM63 and Atrogin-1/MAFBx/FBX32. These enzymes promote the ubiquitination and subsequent degradation of selective sarcomeric proteins to mediate the reduction of muscle size and strength. Whether these enzymes can serve as cachexia biomarkers for human samples remains under investigation. Other more recently identified ubiquitin ligases that share comparable proteolysis-promoting activities in skeletal muscle are TRIM32, MUSA/FBXO30, SMART/FBXO21, and FBXO31. A separate regulatory pathway of muscle atrophy in cancer cachexia that perhaps exhibits a tighter correlation between rodent models and cancer patients is the autophagy system3. This system is characterized by the breakdown of damaged organelles, such as mitochondria, or macromolecules including proteins or RNA, which are selectively removed from cells. In cancer, autophagy can become hyperactivated in peripheral tissues, leading to loss of organelles and proteins that results in muscle atrophy, and like the E3 ubiquitin proteasome pathway, autophagy is also increased in other chronic illnesses associated with muscle loss. A third, more recently described underlying mechanism of skeletal muscle atrophy is mitochondrial dysfunction, which is evident in both animal models and patients with cancer cachexia3,4. This dysfunction can result from multiple aberations, including a reduction in the biogenesis of mitochondria, often linked to a decrease in the levels of the peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1α) and defects in mitochondrial dynamics, such as a hyperactivation of fission, which reduces oxadive capacity and ATP production and in turn causes muscle weakness and fatigue. Although each of these pathways are distinct in their ability to control skeletal muscle mass and function, they all have been shown in some form to be regulated by similar circulating tumor and host inflammatory factors including tumor necrosis factor alpha (TNFα), interleukin-6 (IL-6), myostatin, bone morphogenetic protein inhibitors, and transforming growth factor-beta (TGF-β). The same can be said for how these inflammatory mediators individually signal in muscle cells through a range of transcription factors such as FOXO, NF-κB, STAT3, SMAD2/3, or C/EBP to directly activate one or more of the proteasome, autophagy, or mitochondrial pathways leading to muscle atrophy.
In cancer cachexia, loss of adipose tissue occurs by an atrophy state referred as lipolysis, which in mouse models has been consistently reported in the literature to preceed muscle loss, and from genetic evidence has also been suggested to be a requisite for muscle loss1,5. Adipose tissue undergoes lipolysis through the hydrolysis of triglycerides that result in the formation of glycerol and fatty acids. This catabolic process is regulated by three enzymatic lipases: the adipocyte triglyceride lipase (ATGL), the hormone sensitive lipase (HSL), and the monoaceylglycerol lipase (MAGL)6. In cancer patients with cachexia, adipose loss is strongly correlated with elevated levels of HSL, which is activated in response to a β-adrenergic hormone signal that stimulates adenylate cyclase and produces cAMP. Another major regulatory pathway of lipolysis was found to occur through the inactivation of the AMP-activated protein kinase (AMPK)6. This well conserved energy sensor is typically activated in low states of ATP. However, even though intracellular ATP levels are low in cachectic adipocytes, AMPK was shown to be paradoxically inactivated in adipocytes from tumor bearing mice and cachectic cancer patients. Inactivation of AMPK promotes its own degradation and subsequent stimulation of lipolysis. Analogous to the drivers of skeletal muscle catabolism, a defined set of host hormones and tumor factors have been found to promote lipolysis, including zinc-alpha 2 glycoprotein (ZAG) and IL-6. Tumor-induced secreted factors also promote adipose remodeling and browning7, though the relevance of these to cachexia in patients with cancer is uncertain.
Expansion of the Macroenvironment of Cancer Cachexia
Beyond the original studies centered on the macroenvironment of tumor, skeletal muscle, adipose, and liver, as part of the Cori cycle2, more recent studies implicate multiple pathways of organ crosstalk with muscle and adipose leading to whole body wasting. This newly appreciated expanded macroenvironment includes the brain, bone, and gut microbiota (Figure 1).
Figure 1. The involvement of an expanded tumor macroenvironment in cancer cachexia.
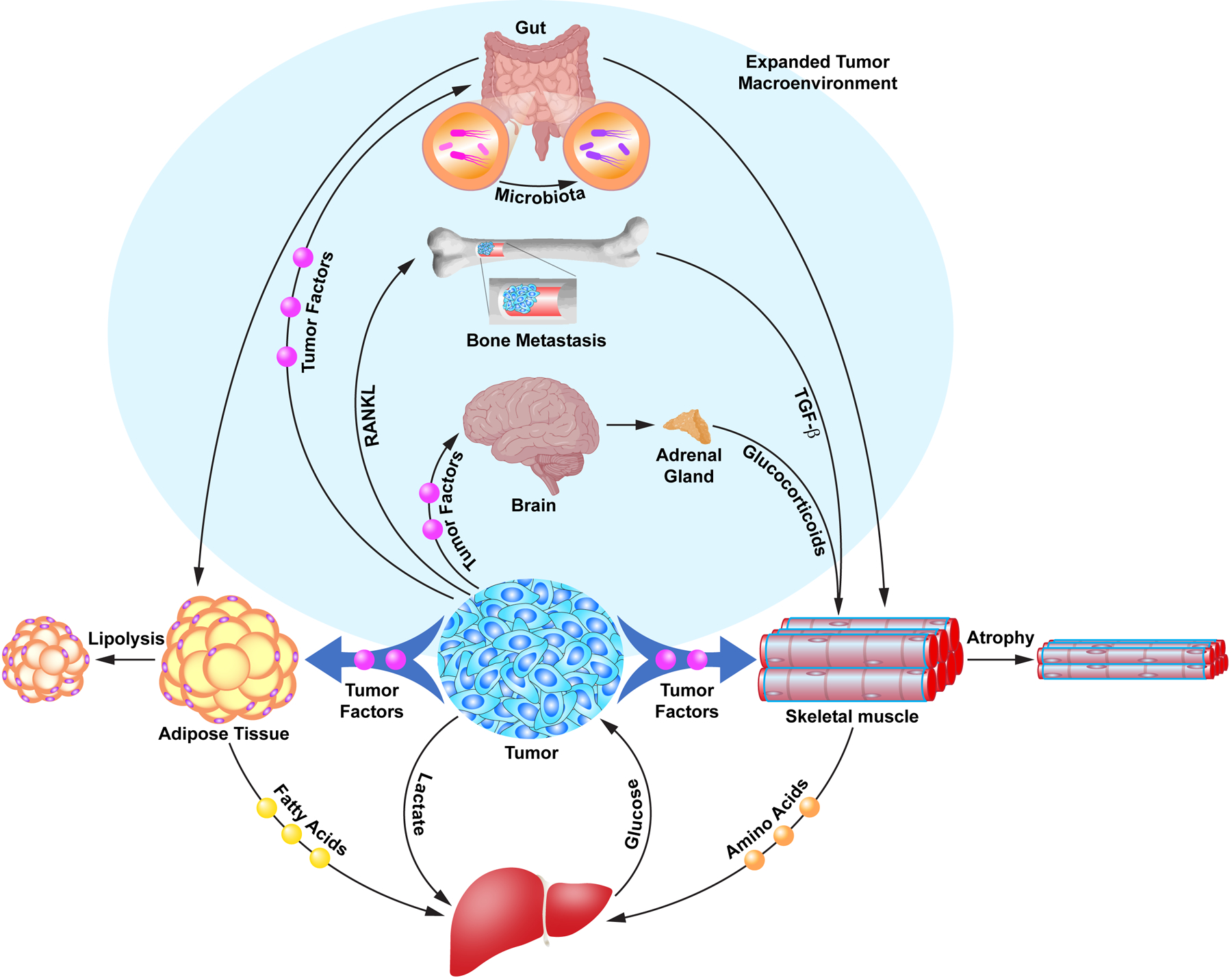
The illustration depicts the original tumor macroenvironment in cancer cachexia described as the Cori cycle involving metabolite and signaling crosstalks between tumor, adipose, skeletal muscle, and liver tissues leading to catabolism (atrophy and lipolysis) and weight loss (unshaded region). More recently, signaling axes have been identified originating from different tumor types that reveal an expanded tumor macroenvironment in cancer cachexia involving crosstalk between brain to muscle; bone to muscle; and gut microbiota to adipose and muscle that contributes to tissue catabolism (shaded region).
Brain to Skeletal Muscle Crosstalk in Cancer Cachexia
The brain has widely been recognized to play a significant role in cancer cachexia, primarily as a regulator of the appetite/anorexia response contributing to weight loss. Historically, feeding was known to be regulated by the hypothalamus, influenced by such inflammatory ctyokines as interleukin-1beta (IL-1β) and TNFα8. However, IL-1β activity in the central nervous system (CNS) also causes peripheral increases of circulating glucocorticoids, which correlates with skeletal muscle atrophy in association with induction of the ubiquitin proteasome system. Use of a corticosteroid antagonist, mifepristone, or removal of the adrenal gland following direct IL-1β administration into the brain inhibits proteolysis and restores skeletal muscle mass. Furthermore, muscle mass is also spared in mice challenged with glucocoriticoid treatment, when the glucocorticoid receptor is genetically ablated from skeletal muscles. Thus, IL-1β activation regulates a CNS-adrenal-skeletal muscle signaling axis that leads to muscle loss8 (Figure 1). More recent findings in an orthotopic mouse model of pancreatic cancer cachexia extend this local brain inflammation to an infiltrating population of myeloid innate immune cells derived predominantly of neutrophils8. These cells were found to accumulate in the velum interpositum, a structure adjacent to the hippocampus and the habenula, the latter being a pair of small nuclei located above the thalamus. Ablation of neutrophils using either pharmacological and genetic means improves appetite and attenuates skeletal muscle loss in mice with pancreas tumors8. It is possible that in a cachexia setting, accumulation of neutrophils to the CNS mediates a similar peripheral signal as was observed upon direct administration of IL-1β into the hypothalamus, but whether a CNS-adrenal-skeletal muscle signaling axis is also involved remains to be investigated.
Signaling Axes from Bone Loss to Muscle Loss in Cancer Cachexia
In tumor types that metastasize to bone and where osteolysis occurs, TGF-β is released from the mineralized bone matrix and increases in the circulation, initiating crosstalk with skeletal muscle9. TGF-β signaling in skeletal muscle activates SMAD3, leading to alterations in the ryanodine receptor, which in turn triggers an imbalance in Ca++ homeostasis. This imbalance alters normal Ca++ binding to the sarcomeric protein, troponin, causing impaired muscle contractions and eventual muscle weakness9. Although breast cancers often metastasize to bone, these patients are not commonly diagnosed with weight loss, with perhaps the exception of end stage disease1. Thus, these findings indicate that this type of bone to muscle crosstalk might be meaningful in understanding muscle weakness and fatigue in cancer patients regardless of changes to muscle mass or body weight. Separate findings reported with ovarian cancer patients found a strong correlation with circulating levels of the receptor activator of nuclear factor kappa B ligand (RANKL) and cachexia10. Although bone metastases do not commonly present in these patients, bone loss still occurs, which associates with overall whole body wasting. Experimental studies suggest that tumor-derived RANKL is sufficient to promote bone resorption through its well described activation of osteoclasts. Studies in a mouse model of ovarian-induced cancer cachexia and cultured myotubes indicate that RANKL directly regulates skeletal muscle atrophy, signaling through an NF-κB pathway and the E3 ubiquitin ligase proteasome system. In addition, administration of zoledronic acid, a bone resorptive agent, in tumor-bearing mice, preserves trabecular bone while also partially preserving skeletal muscle function9 and mass11. Since dosing with zoledronic acid also reduces circulating levels of RANKL, it suggests that bone loss induced by ovarian tumors contributes to elevated RANKL levels and catabolism of skeletal muscle. Thus, TGF-β and RANKL add to the list of circulatory factors in the macroenvironment of cancer cachexia regulating bone to muscle crosstalk (Figure 1). Moreover, platinum-based chemotherapy, including carboplatin for the treatment of breast cancer12 or cisplatin for the treatment of ovarian cancer10, promotes both bone loss and reduction of skeletal muscle mass in mice. At least in the setting of cisplatin, these phenotypes can be mitigated by the treatment of zoledronic acid. Such findings imply that platinum-based chemotherapies also participate in bone to muscle crosstalk, which could involve the circulatory activities of such factors as TGF-β and RANKL.
Microbiotia Crosstalk to Skeletal Muscle and Adipose in Cancer Cachexia
As our understanding of the role of the microbiome in cancer cachexia continues to evolve, a growing number of studies have pinpointed mechanisms by which the gut microbiota modulates skeletal muscle physiology. These findings suggest that crosstalk between the gut microbiome and skeletal muscle exists, and that such crosstalk benefits skeletal muscle13. Signaling from the microbiota occurs from secreted metabolites in the form of short-chain and branched-chain fatty acids, bile acids, and amino acids. For example, the short-chain fatty acid, butyrate, when administered to aged mice protects against skeletal muscle atrophy and oxidative stress13. In a colon (C-26) and acute leukemia (BaF3) model of cancer cachexia, gut microbiomes are modified, eliciting a common anorexia-independent microbial signature, which correlates with cachectic phenotypes. In cancer cachexia patients, certain bacterial taxa exist, which is consistent with animal data. Interventional studies support that addition of prebiotics (substrates that are not digestable by the host but utilized by microorganisms and confer a health benefit to the host) and probiotics (live microorganisms that confer a health benefit to the host) reduces muscle and adipose loss and increases survival of mice with cachexia-inducing tumors. In mice with acute leukemia-induced cachexia, supplementation with the probiotic lactobacilli reduces systemic inflammation and preserves skeletal muscle mass, while in this same model, addition of a prebiotic, sugar beet pectin-derived oligosaccharides, stimulates microbiota content and production of the metabolite acetate, and preservation of adipose tissue14. This rescue in fat mass also correlates with lower expression of the lipolysis regulating enzyme, HSL, and overall blunts the effect on fatty acid catabolism. However, increases in metabolites are restricted to the gut, suggesting that the anti-cachexia benefits of oligosaccharides are unlikely to function through direct signaling on adipocytes14. In general, the microbiome clearly appears to play a role in the expanding tumor macroenvironment of cancer cachexia, but much remains to be learned in how tumor factors regulate the microbiota and how this regulation impacts on specific signaling axes between the gut and muscle and adipose to contribute to a cachexia phenotype.
Conclusion
The relatively recent discovery that organs such as the brain, bone and gut microbiota crosstalk with skeletal muscle and adipose tissues in cancer cachexia has significantly advanced our understanding of the underlying mechanisms of weight loss in this catabolic syndrome. An advantage of elucidating this expanding macroenvironment is the newly identified therapeutic targets that await to be tested. Key to successfully translating these efforts is identifying comparable targets in cachectic cancer patients and utilizing suitable animal models to confirm the efficacy of anti-cachexia agents. Lastly, although not discussed in this commentary, there is growing acceptance in the cancer cachexia field that numerous mechanisms driving skeletal muscle and adipose catabolism are sexually dimorphic15. Thus, attention will need to be given to factors involved in the signaling axes described within the tumor macroenvironment and whether activities within these axes differ between sexes, especially when considering future clinical trials.
ACKNOWLEDGMENTS
D.C.G is funded by an NIH/NIAMS grant R01 AR072714. T.A.Z. is supported by NIH/NCI grants R01 CA122596 and CA194593, VA awards I01BX004177 and I01CX002046, and an NIH/NCI Cancer Center Support Grant P30 CA082709. M.C.O and D.C.G. are funded by an NIH/NCI Cancer Center Support Grant P30 CA13813. T.A.Z., M.C.O. and D.C.G. are also supported by an NIH/NCI P01 CA236778 grant. We apologize to those colleagues we were unable to cite due to space limitations.
DECLARATION OF INTERESTS
D.C.G. has received research funding from Pfizer and has consulted for Novartis. T.A.Z is a scientific advisory board member of Emmyon, Inc and PeleOS, LL. Other authors have no conflicts of interest to declare.
REFERENCES
- 1.Baracos VE, Martin L, Korc M, Guttridge DC, and Fearon KCH (2018). Cancer-associated cachexia. Nat Rev Dis Primers 4, 17105. 10.1038/nrdp.2017.105. [DOI] [PubMed] [Google Scholar]
- 2.Tisdale MJ (2002). Cachexia in cancer patients. Nat Rev Cancer 2, 862–871. 10.1038/nrc927. [DOI] [PubMed] [Google Scholar]
- 3.Sartori R, Romanello V, and Sandri M (2021). Mechanisms of muscle atrophy and hypertrophy: implications in health and disease. Nat Commun 12, 330. 10.1038/s41467-020-20123-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beltra M, Pin F, Ballaro R, Costelli P, and Penna F (2021). Mitochondrial Dysfunction in Cancer Cachexia: Impact on Muscle Health and Regeneration. Cells 10. 10.3390/cells10113150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Das SK, Eder S, Schauer S, Diwoky C, Temmel H, Guertl B, Gorkiewicz G, Tamilarasan KP, Kumari P, Trauner M, et al. (2011). Adipose triglyceride lipase contributes to cancer-associated cachexia. Science 333, 233–238. 10.1126/science.1198973. [DOI] [PubMed] [Google Scholar]
- 6.Rohm M, Zeigerer A, Machado J, and Herzig S (2019). Energy metabolism in cachexia. EMBO Rep 20. 10.15252/embr.201847258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kir S, White JP, Kleiner S, Kazak L, Cohen P, Baracos VE, and Spiegelman BM (2014). Tumour-derived PTH-related protein triggers adipose tissue browning and cancer cachexia. Nature 513, 100–104. 10.1038/nature13528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olson B, Diba P, Korzun T, and Marks DL (2021). Neural Mechanisms of Cancer Cachexia. Cancers (Basel) 13. 10.3390/cancers13163990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Waning DL, Mohammad KS, Reiken S, Xie W, Andersson DC, John S, Chiechi A, Wright LE, Umanskaya A, Niewolna M, et al. (2015). Excess TGF-beta mediates muscle weakness associated with bone metastases in mice. Nat Med 21, 1262–1271. 10.1038/nm.3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huot JR, Pin F, Chatterjee R, and Bonetto A (2022). PGC1alpha overexpression preserves muscle mass and function in cisplatin-induced cachexia. J Cachexia Sarcopenia Muscle 13, 2480–2491. 10.1002/jcsm.13035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pin F, Jones AJ, Huot JR, Narasimhan A, Zimmers TA, Bonewald LF, and Bonetto A (2022). RANKL Blockade Reduces Cachexia and Bone Loss Induced by Non-Metastatic Ovarian Cancer in Mice. J Bone Miner Res 37, 381–396. 10.1002/jbmr.4480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hain BA, Xu H, Wilcox JR, Mutua D, and Waning DL (2019). Chemotherapy-induced loss of bone and muscle mass in a mouse model of breast cancer bone metastases and cachexia. JCSM Rapid Commun 2. [PMC free article] [PubMed] [Google Scholar]
- 13.Lefevre C, and Bindels LB (2022). Role of the Gut Microbiome in Skeletal Muscle Physiology and Pathophysiology. Curr Osteoporos Rep 20, 422–432. 10.1007/s11914-022-00752-9. [DOI] [PubMed] [Google Scholar]
- 14.Bindels LB, Neyrinck AM, Salazar N, Taminiau B, Druart C, Muccioli GG, Francois E, Blecker C, Richel A, Daube G, et al. (2015). Non Digestible Oligosaccharides Modulate the Gut Microbiota to Control the Development of Leukemia and Associated Cachexia in Mice. PLoS One 10, e0131009. 10.1371/journal.pone.0131009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhong X, and Zimmers TA (2020). Sex Differences in Cancer Cachexia. Curr Osteoporos Rep 18, 646–654. 10.1007/s11914-020-00628-w. [DOI] [PMC free article] [PubMed] [Google Scholar]