

ABSTRACT
Among the numerous variants of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) that have been reported worldwide, the emergence of the Omicron variant has drastically changed the landscape of the coronavirus disease (COVID-19) pandemic. Here, we analyzed the genetic diversity of Moroccan SARS-CoV-2 genomes with a focus on Omicron variant after one year of its detection in Morocco in order to understand its genomic dynamics, features and its potential introduction sources. From 937 Omicron genomes, we identified a total of 999 non-unique mutations distributed across 92 Omicron lineages, of which 13 were specific to the country. Our findings suggest multiple introductory sources of the Omicron variant to Morocco. In addition, we found that four Omicron clades are more infectious in comparison to other Omicron clades. Remarkably, a clade of Omicron is particularly more transmissible and has become the dominant variant worldwide. Moreover, our assessment of Receptor-Binding Domain (RBD) mutations showed that the Spike K444T and N460K mutations enabled a clade higher ability of immune vaccine escape. In conclusion, our analysis highlights the unique genetic diversity of the Omicron variant in Moroccan SARS-CoV-2 genomes, with multiple introductory sources and the emergence of highly transmissible clades. The distinctiveness of the Moroccan strains compared to global ones underscores the importance of ongoing surveillance and understanding of local genomic dynamics for effective response strategies in the evolving COVID-19 pandemic.
KEYWORDS: SARS-CoV-2 genomic surveillance, variants of concern, omicron, phylogenetic analysis, spike protein, next-generation sequencing
1. Introduction
After SARS-CoV-2 emergence in December 2019, many genome variants have been discovered and documented, and they have been linked to a considerable rise in the mortality rate in numerous countries [1]. The RNA of SARS-CoV-2 exhibits a high mutation rate, resulting in more genetic diversity. Mutations, including those in the S glycoprotein, ORF1ab, ORF3a, nucleocapsid (N) gene, membrane (M) gene, envelope (E) protein, ORF6, ORF7a, ORF7b, ORF8, and ORF10, can influence all segments of the SARS-CoV-2 genome [2]. Mutations occurring in the S glycoprotein region can increase transmissibility and disease severity. Furthermore, these mutations might facilitate evasion from the immune system, treatments, and vaccines, and could potentially alter diagnostic procedures [3].
To date, the World Health Organization (WHO) has stated five Variants of Concern (VoCs), i.e., Alpha, Beta, Gamma, Delta, and Omicron (https://www.who.int/en/activities/tracking-SARS-CoV-2-variants). After its emergence on 26 November 2021 in Botswana, South Africa, Omicron (B.1.1.529) variant has been spreading rapidly across the world [4]. From the VoCs mentioned earlier, the Omicron variant, while inducing milder symptoms, exhibits heightened transmissibility and a swifter rate of spread compared to previous variants. It is worth highlighting that the Omicron variant, along with its subvariants, maintains its dominant position, retaining a notable presence up to the current day [5]. This is ascribed to the high number of mutations (more than 60 mutations) that are widely distributed on multiple proteins of the genome. More than 32 mutations are located at the RBD of the Spike protein. Omicron is the variant with the highest number of mutations among SARS-CoV-2 variants characterized to date [6,7].
Genomic surveillance is one of the strategies/tools that furnish real-time information on circulating variants of SARS-CoV-2. This information provides insights into its genomic diversity, dispersal and transmission mechanisms [8]. A Moroccan genomic monitoring consortium composed of several public laboratories was constructed in response to the COVID-19 pandemic to track the changes in the SARS-CoV-2 mutation profile in order to identify keystone events in the evolution of the virus and assess the outcomes of COVID-19 prevention measures taken by the Moroccan government [9].
Morocco is considered as a good example not only to understand the dynamics of the virus at the national level but also to get a global overview of the virus’ genome [10]. In fact, the strategic geographic localization of Morocco – being close to European, Middle-eastern and other African countries – and its tourism-based economy brings millions of visitors every year. All this has promoted higher heterogeneity of the viral population circulating in the kingdom despite the strict lockdown and restrictions implemented during the first 2 years of the pandemic to limit the spread of the virus. Indeed, previous studies described the Moroccan SARS-CoV-2 as heterogeneous and demonstrated multiple independent introductions of the virus from abroad [11–13]. The aim of this work is to study the genomic dynamics of SARS-CoV-2 genomes with a focus on Omicron variant in Morocco and its sublineages.
2. Materials and methods
2.1. RNA collection and next-generation sequencing
Encoded RNA samples were obtained from diagnostic laboratories as part of the consortium in Rabat, Morocco. Between November 2020 and November 2022, we sequenced 415 SARS-CoV-2 genomes within the framework of the national genomic surveillance strategy.
cDNA was prepared using a SuperScript VILO cDNA synthesis kit (Invitrogen, Thermo Fisher Scientific, U.S.A.). The libraries were adjusted to 30 pM and loaded onto the Ion Chef instrument (Thermo Fisher Scientific, U.S.A.) for emulsion PCR, enrichment, and subsequent loading onto the Ion S5 530 chip. Whole-Genome Sequencing (WGS) was performed using the Ion Ampliseq SARS-CoV-2 research panel (Invitrogen, Thermo Fisher Scientific) for complete viral genome sequencing according to the instructions for use on an Ion GeneStudio S5 Prime series system. With an amplicon length range of 125–275 bp, the panel provides >99% coverage at 20X of the SARS-CoV-2 genome (~30 kb), with a mean depth >1000. The software Torrent Suite (v5.12.0) was used to analyze the raw data. Short and poor-quality reads were eliminated using the NGS QC Toolkit (v2.3.3) [14]. The consensus sequence was produced using IRMAreport (v1.3.0.2) [15] and mapped to the reference genome (Wuhan-Hu-1, GenBank accession number MN908947.3). Variations were found using Variant Caller (v5.10.1.19) [16].
2.2. Data collection
A total of 1822 SARS-CoV-2 genomes were sequenced in Morocco and deposited in GISAID repository, including the aforementioned 415 genomes sequenced in Laboratory of Biotechnology. The genomic sequences were submitted to GISAID between March 2020 and November 2022. After filtering, 1407 genomes with high-coverage and complete sequences, along with complete collection date were kept for further analysis. Metadata of SARS-CoV-2 genomes sequenced in Morocco is provided in Supplementary Table 1.
Furthermore, to study the genetic characteristics of Omicron variant comparatively to the other VoCs and within its own sublineages in Morocco, we analyzed a total of 937 Omicron genomes sequenced in Morocco between November 2021 and November 2022, including 80 Omicron genomes sequenced for the purpose of this study (Supplementary Table 2).
In order to compare Moroccan samples with a global SARS-CoV-2 dataset we used the genomic epidemiology dataset of SARS-CoV-2 from Nextstrain (https://nextstrain.org/ncov/). The dataset included in addition to 1822 genomes sequenced in Morocco, 3839 genomes collected between December 2019 and December 2022 by 595 submitting laboratories from 186 countries around the world (Supplementary Table 3).
2.3. Phylogenetic analysis
The phylogenetic analysis was performed using the Nextstrain pipeline (version November 2022: https://github.com/nextstrain/ncov) [17]. The resulting narrative and associated datasets (JSONs) were visualized in Auspice (https://auspice.us/) in a Newick format [18]. Samples were classified phylogenetically and studied as clades according to Nextclade annotation (v2.9.1) as it provides a time-oriented and less-exclusive clustering. We also used the Pangolin annotation of lineages to describe more specific lineages (cov-lineages.org) [19]. The alignment of genomic sequences for the identification of related samples was performed using the Basic Local Alignment Search Tool (BLAST) from the GISAID platform that includes over 14 million SARS-CoV-2 genomes. To further enhance the robustness of our analysis, we performed another alignment using the AudacityInstant tool [20]. This tool was specifically built to search the entire EpiCoV database, providing access to closely related sequences and valuable metadata. Mutations and lineages’ first appearance and annotations were extracted from the IDbSV database [21].
2.4. Prediction of mutational escape
We estimated the potential mutational escape of SARS-CoV-2 Omicron variant using a mutational antigenic profiling of SARS-CoV-2 RBD against a panel of antibodies targeting diverse RBD epitopes [22]. This model predicts polyclonal antibody binding to SARS-CoV-2 lineages based on deep mutational scanning of RBD targeting antibodies (https://jbloomlab.github.io/SARS2_RBD_Ab_escape_maps/escape-calc/). Then, the complementary plots and figures were generated using ggplot2, dplyr, reshape2 and tidyverse packages in a custom R-script that can be retrieved from our GitHub repository (https://github.com/mouneem/SARS-CoV-2-Morocco) [23,24].
3. Results
3.1. Genetic characteristics of SARS-CoV-2 variants in Morocco
A total of 1407 SARS-CoV-2 genomes were included in this analysis. The phylogenetic clustering and geographic distribution of SARS-CoV-2 samples in Morocco are provided in Supplementary Figure 1. We began our analysis by characterizing the mutational landscape of these genomes, as illustrated in Figure 1a. This analysis revealed a significant number of mutations present in the genomic regions that code for structural proteins such as the Spike (S), Membrane (M), Envelope (E), and Nucleocapsid (N) proteins. The high rate of mutations in these regions is particularly noteworthy, as structural proteins play a crucial role in the replication and transmission of the virus. As demonstrated in Figure 1b, the Moroccan landscape was heavily influenced by VoCs over a prolonged period of 32 months. These emerging strains have caused peaks of infections in the country during different phases of the pandemic. Furthermore, Figure 1c offers a visual representation of the genetic diversity within the Omicron variant by using a Lollipop plot. The plot illustrates the distribution of mutations in the Spike gene, a key protein that enables the virus to enter human cells. By showing the position of mutations within the gene, this plot gives insight into the genetic makeup of the Omicron variant, and how it has evolved over time. Additionally, the distribution of mutations can also provide information on how the variant might respond to existing therapies and vaccines, and how it might behave in the future. The projection of mutations on the gene annotation reveals that the two regions with relatively higher rate of mutations correspond to the N-terminal domain (NTD) of the S1 subunit of the Spike and the RBD.
Figure 1.
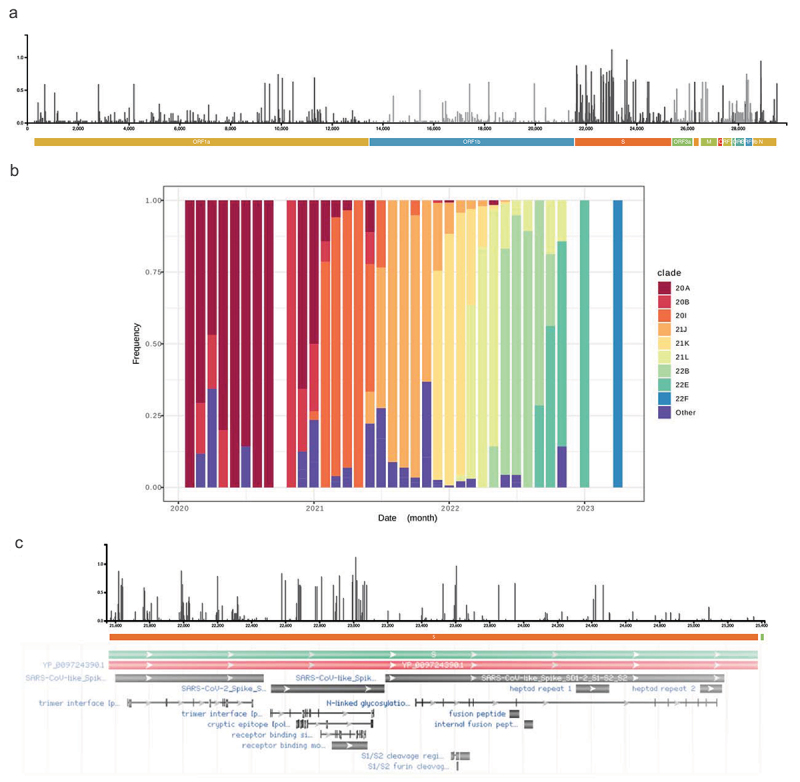
(a) Mutational landscape of the 1407 SARS-CoV-2 whole genomes. (b) Distribution of SARS-CoV-2 clades in Morocco three years into the pandemic. (c) Mutational landscape across the Spike gene of SARS-CoV-2 Omicron variant.
Next, we focus on the comparative genomic analysis of the main variants observed in Morocco (Alpha, Delta, and Omicron) to identify the key features that gave Omicron such characteristics. It is noteworthy to mention that variants such as Beta were identified in Morocco as well; however, we decided not to include them in this analysis due to their low frequency and short lifespan. The study of mutational rate of VoCs revealed outstanding differences. First of all, while the median number of substitutions in Alpha and Delta was estimated at 32 and 35 substitutions per sample respectively, Omicron samples expressed over the double of the previous number with 72 mutations per sample on average. These observations have been noted when analyzing Insertions/Deletions (INDELs). Omicron samples have over 5 INDELs per sample on average, while the other VoCs had less than 3 (Figure 2a).
Figure 2.
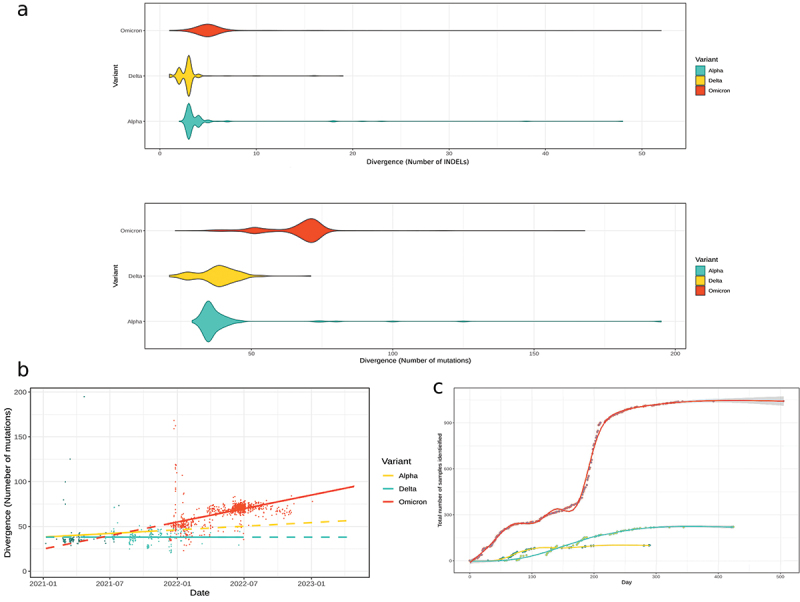
(a) Divergence of substitutions and INDELs within variants of concern in Morocco. (b) Linear evolution Model based on the mutation rate in SARS-CoV-2 variants. (c) Evolution of variants of concern 365 days after their introduction to Morocco.
Furthermore, we observed that Omicron sequences are less stable compared to other VoCs. We estimate that Omicron samples pile-up new mutations every 11 days (0.09 new mutations per day). This rate is higher than the rate estimated for Alpha and Delta (0.02 and 0.005 new mutations per day respectively) (Figure 2b). This not only highlights the important divergence of Omicron but also its high evolution pace compared to other variants. With such an unprecedented mutation rate, Omicron has become the most prevalent variant of the virus only one month after its introduction (Figure 2c). It is noteworthy to mention that the spread of Omicron in Morocco and in the rest of the world has not been dominated by only one lineage (Supplementary Figure 2). It is apparent from Figure 1c that multiple sub-lineages of Omicron have been dominating along the year 2022, amongst which we featured four notable Clades (21K, 21 L, 22B and 22E).
3.2. Potential introduction sources of Omicron variant to Morocco
Here, we focused only on Omicron lineages through the study of their lifespan and their possible introduction sources to Morocco. As observed in the previous results, the clades 21K, 21 L, 22B and 22E were in competition for dominance across 2022. Although two other clades (22A and 22C) have been also identified amongst the studied samples, they had very low abundance and very short lifespan in Morocco (Table 1).
Table 1.
Lifespan and spread of Omicron clades in Morocco and their possible introduction sources.
| Clade | Lifespan | No of samples | Potential source of the first introduction |
|---|---|---|---|
| 21K | Dec 2021 and Jun 2022 | 172 | EPI_ISL_10740491Europe/UK/England Europe/UK/England |
| 21 L | Feb 2022 and Aug 2022 | 178 | EPI_ISL_13588936South America/Brazil/Rio de Janeiro/Resende South America/Brazil/Rio de Janeiro/Resende |
| 22A | Jun 2022 and Jul 2022 | 5 | EPI_ISL_15701296Europe/Switzerland/Basel-Landschaft Europe/Switzerland/Basel-Landschaft |
| 22B | May 2022 and Sep 2022 | 496 | EPI_ISL_15701252Europe/Russia/Moscow Region/Solnechnogorsk Europe/Russia/Moscow Region/Solnechnogorsk |
| 22C | Jun 2022 and Jul 2022 | 9 | EPI_ISL_12413598North America/U.S.A./Ohio North America/U.S.A./Ohio |
| 22E | Oct 2022 and Nov 2022 | 20 | EPI_ISL_15501517Europe/Switzerland/Zurich Europe/Switzerland/Zurich |
Clade 21K was introduced to Morocco in December 2021, only a few weeks after its first appearance in South Africa. Our phylogenetic clustering suggests that the spread of this lineage was through multiple independent introductions from abroad. The BLAST alignment of the first Moroccan Omicron samples revealed high similarity with several samples from England, United Kingdom. Yet, the alignment of other samples revealed different potential sources such as Quebec, Canada (EPI_ISL_15387263), South Dakota, U.S.A. (EPI_ISL_10070711) or Okinawa, Japan (EPI_ISL_13421945). These results were confirmed with the AudacityInstant that showed the high similarity between Moroccan Omicron sequence and samples from the United Kingdom (UK) and United States of America (U.S.A.).
Clade 21L was the second Omicron variant identified in Morocco. The first introduction of this clade dates back to February 2022. The alignment of the first 21L sample in Morocco revealed high similarity with several samples from Rio de Janeiro, Brazil, although this variant is suspected to have originated in South Africa. One interesting fact about 21L is that it has evolved after a few weeks to most of the known Omicron variants circulating to this day i.e., 22A, 22B, 22C, 22D, 22E and 22F. While 22C has barely appeared among the studied samples (1.01%), 22B has widely spread in the Moroccan kingdom, representing over half (56.2%) of Moroccan Omicron samples. According to the samples deposited in the GISAID database, 22B has likely arisen in early 2022 in South Africa. Initially, the sequences were mainly from South Africa, although its detection became more widespread, particularly in Europe, by April 2022. Interestingly, the first known 22B sample in Morocco (EPI_ISL_13408047) shares similarities with samples from the Moscow Region, Russia.
The last interesting Omicron clade in Morocco is 22E, which appeared in the last quarter of 2022 and became dominant by October 2022. This variant is also a 21L descendant. Omicron variant 22E has likely appeared in mid-2022 in Central or Western Africa. Phylogenetic trees of all Omicron clades are available in Supplementary Figure 3.
3.3. Comparative mutation analysis within Omicron lineages
Omicron variants were divided into three generations; i. The first Omicron generation includes 21K and 21L clades that appeared by the end of 2021. ii. The second Omicron generation includes 22A, 22B and 22C clades which appeared in the first half of 2022 as a result of the genetic evolution of 21L that harbors 65 to 85 mutations. iii. The third Omicron generation includes lineages hosting more than 75 mutations and evolving from 22B and 22C that spread starting from the second half of 2022.
The clades 21L and 21K share 38 nucleotide mutations coding for amino-acid mutations, these mutations are shared as well with their ancestor clade 21M. In addition to those, 21L and 21K have 27 and 20 exclusive mutations, respectively. At the Spike-level, both have 21 amino-acid mutations, with 21K having 12 exclusive amino-acid mutations and 21L having 6. The six additional Spike mutations in 21L are S:T19I, S:V213G, S:S371F, S:T376A, S:D405N, and S:R408S. On top of these, there is a deletion of nine nucleotides (from position 21,633 to 21,641) which lead to the mutations S:L24-, S:P25-, S:P26-, and S:A27S. Meanwhile, amongst the 20 specific mutations for 21K we observed a particular cluster of mutations at the S1-S2 furin cleavage site (S:H655Y, S:N679K, S:P681H). Moreover, we found that S:Q498R and S:N501Y mutations are shared between 21L, 22B and 22E.
Like the previously discussed Omicron clades, 22A and 22B harbor suspicious high-rate of mutations. Remarkably, although we characterized very similar Spike protein changes in 22A and 22B, and most Spike-focused studies often group them together. The clade 22A was very rare in comparison to 22B that represented 52% of Omicron samples in this study. In fact, the same pattern was observed in Morocco’s neighboring countries (Europe, North Africa and Middle-East) where 22B was more prevalent than 22A that has spread mainly in North America and Southern Africa. The high similarity between 22A and 22B can be explained by their shared ancestor 21L. Both 22A and 22B differ from 21L by S:L452R and S:F486V mutations in addition to the S:69–70 deletion that they share with 21K. Apart from the Spike mutations, 22B is characterized by M:D3N and ORF6:D61L. Every mutation found in 22B is also present in 22E, along with other mutations in 22E, namely S:K444T and S:N460K. It is noteworthy to mention that several 22E lineages, such as PANGO lineage BQ.1.1, have acquired the additional mutation S:R346T. Besides, 22E harbored extra mutations in regions aside the Spike like, ORF1a:Q556K, ORF1a:L3829F, ORF1b:Y264H, ORF1b:M1156I, ORF9b:P10F, and N:E136D (Figure 3a-b). A lollipop plot showing the distribution of mutations across the SARS-CoV-2 genome of each Omicron clade is provided in the Supplementary Figure 4.
Figure 3.
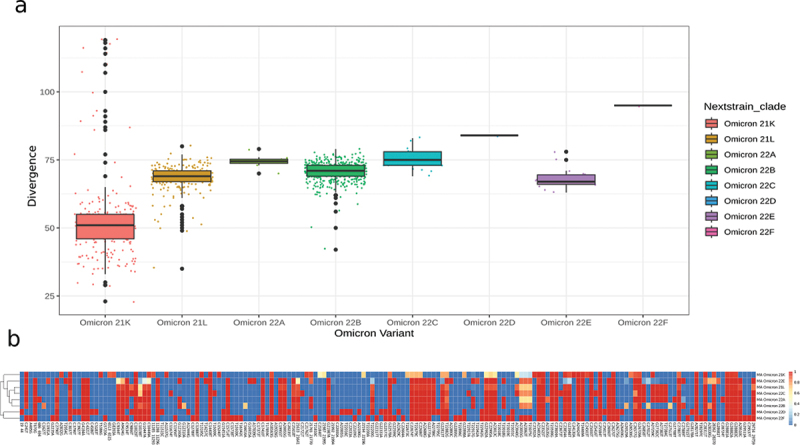
(a) Divergence of Omicron clades. (b) Mutational Heatmap of Omicron clades.
3.4. Genetic profiling of the locally spread Omicron clades
To characterize the genetic features of Omicron clades spread at the national level and how they differ from the rest of the world, we compared the mutations from clades identified in Morocco to samples collected from different regions worldwide (Supplementary Table 4 and 5). At a national level, our observations indicate that the majority of SARS-CoV-2 samples in Morocco are classified as either 22B or 21K, which is in line with global trends. Figure 4 shows that in cities like Casablanca, Tanger, and Kenitra, the 22B clade was found to be predominant, whereas in cities such as Rabat, Mohammedia, and Marrakech, a higher proportion of samples belonged to clades 21K and 21L. Such regional variation in clade distribution among different cities suggests the potential for distinct emergence patterns. However, the close proximity of cities like Casablanca and Mohammedia, as well as Rabat and Kenitra, indicates the absence of localized clusters. This proximity implies a broader and interconnected transmission of the Omicron variant that extends beyond individual cities. Although both local and international datasets contained the same number of clades (Nextstrain annotation), we noted that only 61 pangolin lineages descendant from Omicron have been identified in Morocco, instead of 250 in the rest of the world including 13 lineages exclusive to Morocco (Figure 5a). It is important to note that the pangolin classification is more sensitive to genetic changes compared to Nextstrain classification, which indicates the existence of specific mutations characterizing the Moroccan Omicron samples. Actually, we identified a total of 318 mutations (Occurrence > 1) exclusive to Moroccan Omicron samples (Figure 5b). Amongst the 318 mutations, 72 were located in the Spike region and 18 were INDELS (Figure 5b). Therefore, we devote this last part of the manuscript to study and describe the important characteristics of these mutations.
Figure 4.
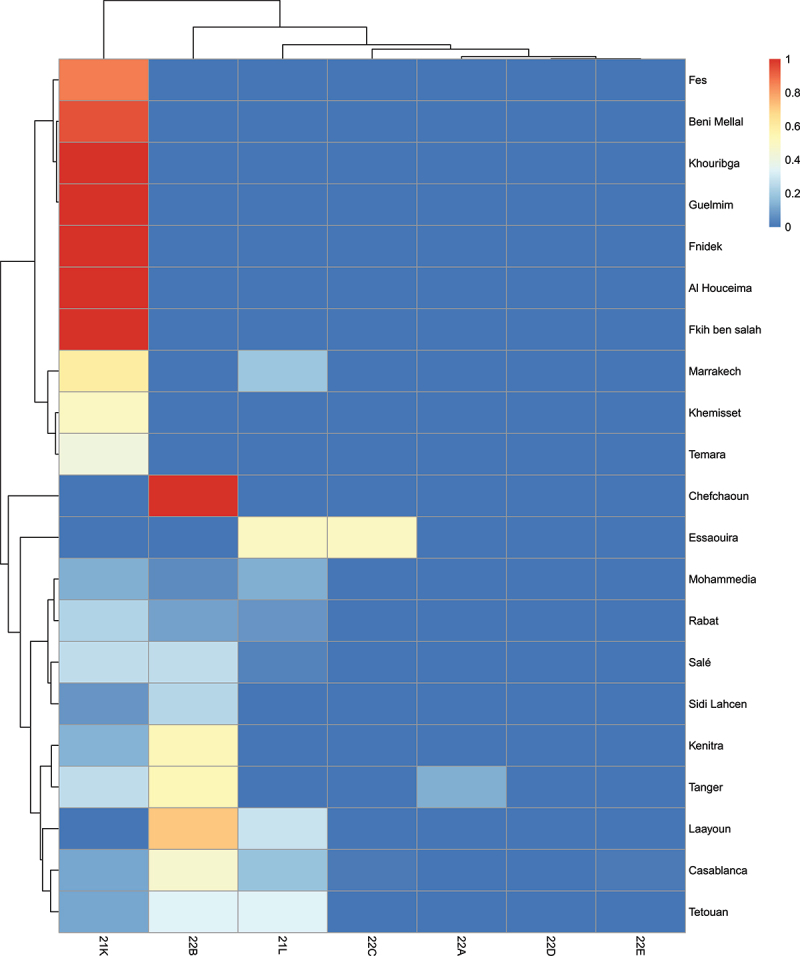
(a) Heatmap showing the distribution of Omicron clades in Moroccan cities.
Figure 5.
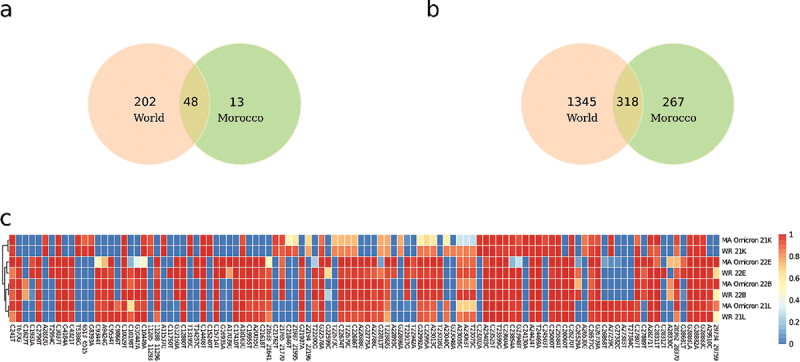
(a) Venn Diagram of Omicron clades in Morocco and worldwide. (b) Venn Diagram of Omicron mutations in Morocco and worldwide (frequency <1 excluded). (c) Heatmap of mutations within Omicron lineages.
Next, we compared samples at a clade-specific level (Figure 5c). While no significant difference was observed in samples classified as clade 21K in Morocco and worldwide, the clade 21L included slight changes at both ends of the genome. The most notable difference is that more than 35% of Moroccan samples included a deletion of 25 nucleotides at the 29,734–29759 positions. Meanwhile the C241T mutation that was observed in 98% of Moroccan samples is one of the most frequent 5’ UTR mutations in SARS-CoV-2 with a frequency higher than 95% in 21L, 22B and 22E.
Our findings showed that 86.1% and 85% of Moroccan samples harbored two different synonymous mutations ORF1ab:L454L and N:G16G respectively. These mutations were observed in relatively less frequencies worldwide data (55.9% and 54.4%). The PANGO lineage analysis of these mutations showed that they are associated with some sublineages of 22B mainly BA.5.2 that emerged in the beginning of July 2022 and spread mainly in the U.S.A., Japan and Europe and was introduced to Morocco in May 2022.
Moroccan 22E samples were characterized by the mutations ORF1ab:N5592S, S:del 69–70 and S:T19I. Although these mutations were already very frequent worldwide, they were identified more frequently in Moroccan samples by 14%, 13% and 11%, respectively. In contrast, ORF1ab:L454L, which was also observed in higher frequency in 22B, was identified exclusively in 18% of 22E Moroccan samples. This lineage analysis showed the association of this mutation to BQ.1.1 sublineage more specifically.
3.5. Predicted mutational escape from antibodies
In this section, we estimated the potential mutational escape of each Omicron clade using a mutational antigenic profiling of SARS-CoV-2 RBD against a panel of antibodies targeting diverse RBD epitopes [16]. This estimation allowed us to visualize the cumulative effect of the mutation in each variant in regard to its bind-affinity to three monoclonal antibodies: LY-CoV016 (etesevimab), LY-CoV555 (bamlanivimab), and REGN10987 (imdevimab). The clades 21K, 21L, 22B and 22E harbored 10, 16, 17 and 20 mutations respectively in the RBD region of the Moroccan strains. Amongst the studied samples, we identified specific mutations in the Moroccan samples. The C1419T substitution was present in 7 sequences (Clade 21K). Notably, these samples exhibited a high degree of relatedness, as they were identified in the same region and during the same period (November–December 2021, Rabat). Similarly, we observed mutations in Casablanca region, where six samples from clade 21K harbored the G2246A mutation, five samples from clade 22B had the A13859G mutation, and five samples from clade 21K contained the G1747A mutation. This clustering of mutations in specific geographic regions further supports the notion of relatedness among the samples introduced to Morocco. While both mutations S:S375F and S:T376A have unknown impact on the studied structure, the other 22 mutations yielded an average escape score of 0.04 (with a median of 0.03), which is in line with the findings of Bloom et al., [25]. The mutations S:E484A, S:F486V and S:Q493R had the greatest impacts on the structure stability with 0.15, 0.08 and 0.05 escape scores, respectively (Figure 6a-b). Notably, these mutations were all identified in the 22E clade. In fact, amongst the four Omicron clades studied, we observed that 22E accumulated the highest escape score (Supplementary Table 4).
Figure 6.
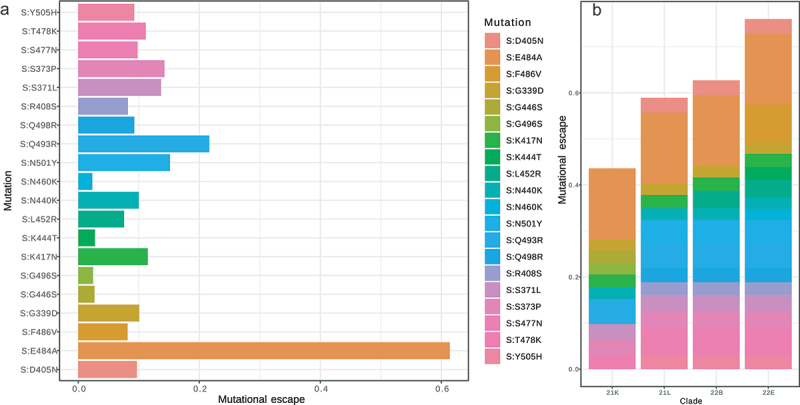
(a) Individual escape score of each mutation in Omicron variant Spike protein. (b) Cumulative mutational impact on immune escape from antibodies for each Omicron clade.
4. Discussion
In the current study, we identified a total of 21 clades (92 lineages) from 1407 samples analyzed, amongst four VoCs (Alpha, Beta, Delta and Omicron). Our findings show that the genomic dynamics of SARS-CoV-2 in Morocco is characterized by the dominance of VoCs circulating across 32 months. In fact, the first introduced lineage to Morocco in March 2020 was 20A (Lineage B.1.1) characterized by the D614G mutation at the RBD, which was widely spread in Europe, Middle-East and Africa and became the origin of most circulating lineages worldwide by the end of 2020. By the beginning of 2021, the Alpha variant (a mutant version of 20A) was introduced to Morocco in January 2021 [26] and rapidly spread to become dominant after 2 months. This dominance was interrupted by Delta and Omicron variants by August 2021 and March 2022 respectively [8]. Moreover, our findings suggest that the Spike protein harbors a higher rate of mutations as compared to other genomic regions of the virus, more particularly in the RBD and the NTD. This is the result of the relatively higher affinity of some Spike-mutant strains to the human Angiotensin-Converting Enzyme 2 (ACE2), and several studies have demonstrated this computationally and experimentally [5,27,28]. Interestingly, these regions have been extensively analyzed in previous studies, and several molecular assays linked changes in these regions specifically to increased affinity and transmission rate [29]. Furthermore, we mainly identified four dominant Omicron clades in Morocco (21K, 21L, 22B and 22E) originating from multiple potential sources around the world. We also noted that 22E was particularly more transmissible and has become the dominant variant in Morocco by October 2022.
While analyzing the mutational profile of Omicron lineages, we found that clades 21L and 21K share several mutations in addition to some exclusive mutations at the Spike level and other regions of the viral genome. Omicron clade 21K is characterized by a cluster of mutations in the S1-S2 furin cleavage. These mutations seem to be associated with increased transmissibility [27]. We also found that clades 22B and 22E harbor the same mutations except for three mutations only present in 22E (S:K444T; S:N460K; S:R346T). S:R346T is known to increase viral neutralizing antibodies escape [30]. In fact, a significant decrease in neutralization titers was observed in a study that examined the ability of the virus to neutralize antibodies in vaccinated and unvaccinated individuals with Omicron-22E [31]. Breakthrough infections have also highlighted that 22E can escape the monoclonal antibodies (mAbs) Evusheld and Bebtelovimab [30]. Meanwhile, S:N460K was reported to promote neutralizing antibodies escape [32]. In other studies, the same mutation was linked to neutralization resistance and enhanced fusogenicity in 22E as compared to its ancestral 22B [31,33]. We also found that the mutations S:E484A shared by the four clades of interest have been linked to immune escape in other variants i.e., 20H (Beta), 20J (Gamma), and 21B (Kappa) [34,35]. Additionally, S:Q498R and S:N501Y mutations shared by 21L, 22B and 22E have been linked to the increase in the binding affinity to ACE2 according to several in vitro studies [36].
Next, we compared the genetic profile of Omicron clades in Morocco and worldwide. We found that there is no significant difference in sequences classified as clade 21K. However, in clade 21L, we noticed that 35% of Moroccan sequences included a deletion of 25 nucleotides at the 29,734–29759 positions. This region corresponds to the 3’UTR s2m. Recent studies have explained how it forms homodimeric kissing complexes that the viral N protein converts into a stable structure [37]. Whereas, other studies showed that it contains two binding sites for the host miR1307p, which has been proposed to control a variety of cytokines and their receptors, including IL18 and IL6R [38]. Remarkably, the projection of this deletion over time shows that most of the sequences harboring this deletion have appeared in the latter stage of the clade 21L, which suggests that the sequences identified in Morocco are the mutant form of 21L that acquired this deletion before mutating into one of the second-generation Omicron lineages (22A, 22B or 22C). C241T is another mutation that was found in 98% of Moroccan sequences and reported to reduce replication efficiency which is correlated with an increase in recovery rates and a decrease in mortality rates of SARS-CoV-2 cases worldwide [39]. Two synonymous mutations namely ORF1ab:L454L and N:G16G were identified in more than 85% of 22B Moroccan sequences and reported in a study to contribute in enhancing the transmissibility of SARS-CoV-2 [40]. This high prevalence can be explained by the rapid viral spread in the country, especially as 87.1% of sequences harboring these mutations were collected in the Casablanca region which is the most populated area in Morocco. Meanwhile, the mutations located in 22E are slightly more frequent in Moroccan sequences as compared to sequences in the rest of world. This indicates the relative homogeneity of the analyzed sequences as well as the introduction of this lineage to Morocco after acquiring these mutations (Supplementary Figure 5). In contrast, we identified the ORF1ab:L454L mutation exclusively in 22E Moroccan sequences, more particularly in BQ.1.1 sublineage, therefore we suggest the accumulation of this mutation during the expansion and evolution of this sublineage in Morocco.
In the last part of this study, we estimated the potential mutational escape from antibodies of the four Omicron clades. We noticed that 22E accumulated the highest escape score. This result could explain the rapid expansion of this particular clade by the end of 2022 despite the high vaccination rate in the country. The projection of the antibody-escape scores on the spread timeline of different lineages in Figure 1c demonstrates that Omicron clades with high antibody-escape scores were able to replace their predecessors due to the specific RBD mutations in each Omicron clade (21L: T376A, D405N, and R408S; 22B: L452R and F486V; 22E: L452R and F486V). This finding suggests that genetic selection influenced by the introduction of vaccines has led to positive selection of lineages with high rate of mutations located at structural genes, particularly the Spike protein with a potential to increase the ability of the virus to bind tohuman ACE2 and reduce its ability to be cleaved by antibodies. However, despite the outstanding traits of Omicron, a drastic decline in the number of fatal cases and deaths was observed during the spread of Omicron variants in Morocco where most of the vulnerable populations are vaccinated. This observation was also true worldwide, given the fact that the overall genetic landscape is not very different from Morocco (Supplementary Figure 6). In fact, Omicron causes less severe symptoms, even though it is more transmissible than other variants [41]. These results were confirmed by a proteomic analysis, which demonstrated how immune responses to Omicron infection in vaccinated individuals were less severe in comparison to individuals infected with other variants [42].
We underline the crucial synergy between phylogenetic analysis and epidemiological data in revealing viral transmission patterns. Phylogenetic research enables the reconstruction of evolutionary links, identification of transmission clusters, and likely sources of infection. Meanwhile, epidemiological data provide insights into disease spread among populations and the demographic factors influencing transmission dynamics [43,44]. However, we acknowledge that the imbalanced sampling size across countries is a significant challenge that necessitates cautious interpretation of our findings. These discrepancies in sampling attempts can generate biases, resulting in inadequate representations of transmission patterns across areas. To overcome this constraint, future research should concentrate on enhancing data collection efforts in underrepresented regions, increasing collaboration between researchers and public health organizations and conducting sensitivity analysis to test the robustness of our findings. By recognizing these limitations, we aim to emphasize the necessity of taking a comprehensive and global perspective when interpreting our findings to guide successful public health initiatives and treatments.
5. Conclusions
The SARS-CoV-2 Omicron variant that was first identified in South Africa in November 2021 and has become the dominant strain globally. This variant has a higher number of mutations in the Spike protein compared to other SARS-CoV-2 variants. Our study focused on analyzing four specific clades of Omicron, 21K, 21L, 22B and 22E, which were found to be more infectious and able to evade the immune system more effectively. These clades possess mutations in the Spike RBD, such as S477N, Q498R, N440K, Y505H, N679K, N764K, D796Y, and Q954H that contributed to their increased infectivity. Notably, the 22E clade was particularly more transmissible and has become the dominant variant in Morocco and many other countries. Furthermore, our analysis indicated that the 22E clade possesses specific Spike RBD mutations K444T and N460K that enable it to evade immunity from vaccines. However, it is worth noting that the spread of Omicron variants has been associated with a decrease in the fatality rate in Morocco and worldwide, consistent with studies indicating that Omicron causes less severe illness, especially in vaccinated individuals.
Supplementary Material
Acknowledgements
This work was carried out with the assistance of national funding from the Moroccan Ministry of Higher Education and Scientific Research (COVID-19 program) to A.I. and scholarship of excellence from the National Center for Scientific and Technical Research in Morocco. We would like to acknowledge the GISAID database for providing publicly available data, which greatly contributed to our research.
Funding Statement
This research was funded by Moroccan Ministry of Higher Education and Scientific Research (COVID-19 Program).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data generated during the current study are available from the
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/20477724.2023.2250942
References
- [1].Dhawan M, Saied ARA, Mitra S, et al. Omicron variant (B.1.1.529) and its sublineages: What do we know so far amid the emergence of recombinant variants of SARS-CoV-2? Biomed Pharmacother. 2022. Oct 1;154:113522. Elsevier Masson s.r.l. doi: 10.1016/j.biopha.2022.113522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Alquraan L, Alzoubi KH, Rababa’h SY.. Mutations of SARS-CoV-2 and their impact on disease diagnosis and severity. Inf Med Unlocked. 2023;39:101256. doi: 10.1016/j.imu.2023.101256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Galani A, Markou A, Dimitrakopoulos L, et al. Delta SARS-CoV-2 variant is entirely substituted by the omicron variant during the fifth COVID-19 wave in Attica region. Sci Total Environ. 2023. Jan;856:159062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].El Mazouri S, Bendani H, Boumajdi N, et al. Report of SARS-CoV-2 BA.1 lineage in Morocco. Microbiol Resour Announc. 2022. May;11(5). doi: 10.1128/mra.00169-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ren W, Zhu Y, Lan J, et al. Susceptibilities of human ACE2 genetic variants in Coronavirus infection. J Virol. 2022;96(1):e0149221. doi: 10.1128/JVI.01492-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].He X, Hong W, Pan X, et al. SARS-CoV-2 Omicron variant: characteristics and prevention. MedComm. 2(4): Blackwell Publishing Inc.:838–845. 2021. Dec 1. doi: 10.1002/mco2.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Weng S, Shang J, Cheng Y, et al. Genetic differentiation and diversity of SARS-CoV-2 Omicron variant in its early outbreak. Biosaf Heal. 2022. Jun;4(3):171–178. doi: 10.1016/J.BSHEAL.2022.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Oude Munnink BB, Worp N, Nieuwenhuijse DF, et al. The next phase of SARS-CoV-2 surveillance: real-time molecular epidemiology. Nature Med. 2021. Sep 1;27(9): 1518–1524. Nature Research. doi: 10.1038/s41591-021-01472-w. [DOI] [PubMed] [Google Scholar]
- [9].El Mazouri S, Bendani H, Boumajdi N, et al. Phylogeography and genomic analysis of sars-cov-2 delta variant in morocco. J Infect Dev Ctries. 2022;16(8):1258–1268. doi: 10.3855/jidc.16513 [DOI] [PubMed] [Google Scholar]
- [10].Wilkinson E, Giovanetti M, Tegally H, et al. A year of genomic surveillance reveals how the SARS-CoV-2 pandemic unfolded in Africa. Science. 2021;374(6566):423–431. doi: 10.1126/science.abj4336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Meriem L, Essabbar A, Alouane T, et al. Do the Moroccan SARS-CoV-2 genetic diversity hamper the use of the developed universal vaccines in Morocco? bioRxiv [Preprint]. 2020. doi: 10.1101/2020.06.30.181123 [DOI] [Google Scholar]
- [12].Chaouni B, Allali I, Sehli S, et al. Genomic evidence of multiple SARS-CoV-2 introductions into Morocco. J Obstet Gynaecol. 2020. Jul;9:679. [Google Scholar]
- [13].Badaoui B, Sadki K, Talbi C, et al. Genetic diversity and genomic epidemiology of SARS-CoV-2 in Morocco. Biosaf Heal. 2021. Apr;3(2):124–127. doi: 10.1016/J.BSHEAL.2021.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Patel RK, Jain M, Liu Z. NGS QC toolkit: a toolkit for quality control of next generation sequencing data. PLoS One. 2012;7(2):e30619. doi: 10.1371/journal.pone.0030619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Shepard SS, Meno S, Bahl J, et al. Viral deep sequencing needs an adaptive approach: IRMA, the iterative refinement meta-assembler. BMC Genomics. 2016;17(1). doi: 10.1186/s12864-016-3030-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Aanniz T, Ouadghiri M, Bendahou MA, et al. First Report of a SARS-CoV-2 genome sequence with a Spike His69-Val70 deletion and an Asn439Lys mutation in Morocco; 2021. doi: 10.3390/pathogens9100829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hadfield J, Megill C, Bell SM, et al. NextStrain: real-time tracking of pathogen evolution. Bioinformatics. 2018. Dec;34(23):4121–4123. doi: 10.1093/bioinformatics/bty407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Cardona G, Rosselló F, Valiente G. Extended Newick: it is time for a standard representation of phylogenetic networks. BMC Bioinf. 2008. Dec;9: doi: 10.1186/1471-2105-9-532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Rambaut A, Holmes EC, O’Toole Á, et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat Microbiol. 2020. Nov;5(11):1403–1407. doi: 10.1038/s41564-020-0770-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].“GISAID - Audacity instant app.” (Cited 2023. Jul 4). https://gisaid.org/database-features/audacity-instant-app/.
- [21].Essabbar A, Kartti S, Alouane T, et al. IDbSV: an open-access repository for monitoring SARS-CoV-2 variations and evolution. Front Med. 2021. Dec;8. doi: 10.3389/fmed.2021.765249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Greaney AJ, Loes AN, Crawford KHD, et al. Comprehensive mapping of mutations in the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human plasma antibodies. Cell Host Microbe. 2021. Mar;29(3):463–476.e6. doi: 10.1016/J.CHOM.2021.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wickham H, Averick M, Bryan J, et al. Welcome to the Tidyverse. J Open Source Softw. 2019. Nov;4(43):1686. doi: 10.21105/JOSS.01686 [DOI] [Google Scholar]
- [24].Wickham H. ggplot2. Wiley Interdiscip Rev Comput Stat. 2011. Mar;3(2):180–185. doi: 10.1002/WICS.147 [DOI] [Google Scholar]
- [25].Bloom JD, Beichman AC, Neher RA, et al. Evolution of the SARS-CoV-2 mutational spectrum. bioRxiv [Preprint]. 2022. doi: 10.1101/2022.11.19.517207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Mouna O, Aanniz T, Essabbar A, et al. Report of SARS-CoV-2 B1.1.7 lineage in Morocco. Microbiol Resour Announc. 2021. Apr;10(16):e00240–21. doi: 10.1128/MRA.00240-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Rodriguez JA, Gonzalez J, Arboleda-Bustos CE, et al. Computational modeling of the effect of five mutations on the structure of the ACE2 receptor and their correlation with infectivity and virulence of some emerged variants of SARS-CoV-2 suggests mechanisms of binding affinity dysregulation. Chem Biol Interact. 2022. Dec;368:110244. doi: 10.1016/J.CBI.2022.110244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Chan MC, Chan KK, Procko E, et al. Machine learning guided design of high affinity ACE2 decoys for SARS-CoV-2 neutralization. bioRxiv Prepr Serv Biol. 2021. Dec. doi: 10.1101/2021.12.22.473902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Haslwanter D, Dieterle ME, Wec AZ, et al. A combination of receptor-binding domain and N-Terminal domain neutralizing antibodies limits the generation of SARS-CoV-2 Spike neutralization-escape mutants. MBio. 2021;12(5):e0247321. doi: 10.1128/mBio [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Cao Y, Jian F, Wang J, et al. Imprinted SARS-CoV-2 humoral immunity induces convergent Omicron RBD evolution. Nature. 2022. doi: 10.1101/2022.09.15.507787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Qu P, Evans JP, Faraone JN, et al. Distinct neutralizing antibody escape of SARS-CoV-2 Omicron subvariants BQ.1, BQ.1.1, BA.4.6, BF.7 and BA.2.75.2. bioRxiv Prepr Serv Biol. 2022. doi: 10.1101/2022.10.19.512891 [DOI] [Google Scholar]
- [32].Kaku CI, Starr TN, Zhou P, et al. Evolution of antibody immunity following Omicron BA.1 breakthrough infection. bioRxiv [Preprint]. 2022. doi: 10.1101/2022.09.21.508922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Qu P, Evans JP, Faraone JN, et al. Enhanced neutralization resistance of SARS-CoV-2 Omicron subvariants BQ.1, BQ.1.1, BA.4.6, BF.7, and BA.2.75.2. Cell Host Microbe. 2022. Nov;31(1):9–17.e3. doi: 10.1016/J.CHOM.2022.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Greaney AJ, Starr TN, Bloom JD. An antibody-escape estimator for mutations to the SARS-CoV-2 receptor-binding domain. Virus Evol. 2022;8(1): doi: 10.1093/ve/veac021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wang Z, Schmidt F, Weisblum Y, et al. mRNA vaccine-elicited antibodies to SARS-CoV-2 and circulating variants. Nature. 2021. Apr;592(7855):616–622. doi: 10.1038/s41586-021-03324-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zahradník J, Marciano S, Shemesh M, et al. SARS-CoV-2 variant prediction and antiviral drug design are enabled by RBD in vitro evolution. Nat Microbiol. 2021. Sep;6(9):1188–1198. doi: 10.1038/s41564-021-00954-4 [DOI] [PubMed] [Google Scholar]
- [37].Frye CJ, Shine M, Makowski JA, et al. Bioinformatics analysis of the s2m mutations within the SARS-CoV-2 Omicron lineages. J med virol. 2022;95(1):John Wiley and Sons Inc. doi: 10.1002/jmv.28141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Imperatore JA, Cunningham CL, Pellegrene KA, et al. Highly conserved s2m element of SARS-CoV-2 dimerizes via a kissing complex and interacts with host miRNA-1307-3p. Nucleic Acids Res. 2022. Jan;50(2):1017–1032. doi: 10.1093/nar/gkab1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Chaudhari A, Chaudhari M, Mahera S, et al. In-silico analysis reveals lower transcription efficiency of C241T variant of SARS-CoV-2 with host replication factors MADP1 and hnRNP-1. IMU. 2021. Jan;25:100670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Jian F, Yu Y, Song W, et al. Further humoral immunity evasion of emerging SARS-CoV-2 BA.4 and BA.5 subvariants. Lancet Infect Dis. 2022. Nov 1;22(11): 1535–1537. NLM (Medline). doi: 10.1016/S1473-3099(22)00642-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wolter N, Jassat W, Walaza S, et al. Early assessment of the clinical severity of the SARS-CoV-2 omicron variant in South Africa: a data linkage study. Lancet. 2022. Jan;399(10323):437–446. doi: 10.1016/S0140-6736(22)00017-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Bao J, Sun R, Ai J, et al. Proteomic characterization of Omicron SARS-CoV-2 host response. Cell Discov. 2022;8(1):46. doi: 10.1038/s41421-022-00418-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Attwood SW, Hill SC, Aanensen DM, et al. Phylogenetic and phylodynamic approaches to understanding and combating the early SARS-CoV-2 pandemic. Nat Rev Genet. 2022;23(9):547–562. doi: 10.1038/s41576-022-00483-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Duault H, Durand B, Canini L. Methods combining genomic and epidemiological data in the reconstruction of transmission trees: a systematic review. Pathogens. 2022;11(2):252. doi: 10.3390/pathogens11020252 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated during the current study are available from the