

Abstract
Therapy resistance and metastatic progression are primary causes of cancer-related mortality. Disseminated tumor cells possess adaptive traits that enable them to reprogram their metabolism, maintain stemness, and resist cell death, facilitating their persistence to drive recurrence. The survival of disseminated tumor cells also depends on their ability to modulate replication stress in response to therapy while colonizing inhospitable microenvironments. In this study, we discovered that the nuclear translocation of AXL, a TAM receptor tyrosine kinase, and its interaction with WRNIP1, a DNA replication stress response factor, promotes the survival of HER2+ breast cancer cells that are resistant to HER2-targeted therapy and metastasize to the brain. In preclinical models, knocking down or pharmacologically inhibiting AXL or WRNIP1 attenuated protection of stalled replication forks. Furthermore, deficiency or inhibition of AXL and WRNIP1 also prolonged metastatic latency and delayed relapse. Together, these findings suggest that targeting the replication stress response, which is a shared adaptive mechanism in therapy-resistant and metastasis-initiating cells, could reduce metachronous metastasis and enhance the response to standard-of-care therapies.
INTRODUCTION
Despite significant progress in cancer treatment, therapy resistance, and the emergence of metachronous metastases or metastatic relapse remain major clinical challenges, often resulting in patient mortality even after successful primary treatment (1–3). Metastatic relapses arise from adaptations to therapy and/or constraints imposed by the nutrient and microenvironmental conditions, leading to the emergence of resistant clones from disseminated tumor cells that persist in distal organs (4,5). These adaptations can cause changes in cellular transcriptomic and metabolic profiles, resulting in increased cellular reactive oxygen species, depletion of cellular nucleotide pools, and altered replication timing and progression, ultimately leading to replication stress (6,7). Thus, the survival of disseminated therapy-resistant tumor cells depends on their ability to activate DNA damage tolerance pathways and mitigate replication stress. Despite this critical role of replication stress response in the survival of metastasis-initiating cells, the precise mechanisms that trigger this response remain unclear. Identifying and understanding these survival dependencies could provide insights into the development of novel therapeutic strategies to prevent metachronous metastasis and improve patient outcomes.
Metachronous brain metastases frequently occur in patients with HER2-amplified breast cancer following primary tumor diagnosis and treatment. Although multiple agents with distinct mechanisms of action have been approved for the treatment of early-stage and metastatic HER2+ disease, their efficacy has been limited to the primary tumor and extracranial metastasis, with little effect on cranial metastasis (8,9). The lack of treatment response could be influenced by several factors that mediate survival of disseminated tumor cells in the brain microenvironment. Ongoing clinical trials exploring novel approaches to manage HER2+ central nervous system metastasis represent an important step toward this goal (10–15). To develop effective therapeutic strategies for HER2+ brain metastases, it is crucial to improve our understanding of their biology, not only in relation to the primary tumor but also considering divergence in genetic, epigenetic, metabolic and signaling dependencies in breast cancer brain metastatic cells and differences in the central nervous system tumor microenvironment (16,17).
Despite the benefits of HER2-targeted therapies, patients eventually develop resistance, and the survival benefits from mono or combination therapies are short-lived. Several mechanisms, such as incomplete inhibition of HER family receptors, the emergence of HER2 splice variants, activating mutations, and activation of compensatory pathways, have been reported to promote resistance to HER2-targeted therapies (18,19). In addition, receptor tyrosine kinases (RTKs) have been shown to facilitate therapy resistance in HER2+ breast cancer (20). Among them, AXL, a RTK that belongs to the TAM (Tyro3, Axl, Mer) family, has been shown to be enriched in lapatinib-resistant breast cancer cells (21). Furthermore, studies have shown that AXL inhibition in ovarian, melanoma and non-small cell lung cancer cells induces DNA damage and replication stress, suggesting that AXL is crucial in the cellular response to replication stress (22–24). Similarly, AXL-positive glioma-initiating progenitor cells that overcome replication stress are capable of initiating tumors (25). How AXL promotes replication stress response remains unknown.
In this study, we observed an enriched nuclear translocation of AXL in brain metastases compared to matched human primary tumors, and in therapy resistant HER2+ latent and metachronous brain metastatic mouse models. Further investigation revealed that nuclear AXL provides a stable protective effect on replication stress-induced DNA damage by interacting with a DNA replication stress response modulator, WRNIP1, thereby promoting therapy resistance in latent and metachronous brain metastases. Depleting AXL or WRNIP1 decreased the survival of Lat and M-BM cells in the brain microenvironment. Additionally, we demonstrated that inhibiting AXL with the pharmacologic inhibitor bemcentinib (BGB324) reduced the metastatic potential of tumor cells and enhanced NK cell-mediated immunosurveillance, resulting in decreased metastatic burden in preclinical models.
METHODS
Patient Samples
Female HER2+ breast cancer patient specimens (matched primary tumor with brain met) were obtained from the Breast Cancer Research Program at Vanderbilt Ingram Cancer Center. Patient consent was voluntary, the age was undisclosed, and we obtained a written informed consent from Vanderbilt Ingram Cancer Center institutional review board-approved protocol (Vanderbilt Ingram Cancer Center no. 160606) for the use of deidentified tumor tissue for research purposes (26). Tumor sections of 5μm embedded in paraffin were provided and subsequently stained for AXL immunohistochemistry analysis.
Cell Lines
HER2+ breast cancer cell lines HCC1954 and SKBR3 (Parental cells, PA) and their brain metastatic derivatives Lat, S-BM and M-BM cells were cultured in RPMI-1640 (Cat.R8758, Sigma). Media was supplemented with 2mM glutamine, 10% FBS, 1 mg/mL amphotericin B, 100 units/L penicillin and 100 mg/L streptomycin. Cells were maintained at 37°C with 5% CO2 and passaged every 2–3 days. For more information on isolation of isogenic Lat and M-BM cells, please refer to our previous publications on how we generate the cell lines to Malladi et al., Cell 2016 (27) and Parida et al., Cell Metabolism 2022 (26). Lenti-X-HEK 293T cells were obtained from Takara (cat# 632180) and grown in DMEM, high glucose media supplemented with 1mM sodium pyruvate, 100units/L penicillin, 100mg/L streptomycin and 10% FBS. Human NK cells were obtained from ATCC (NK-92 cat# CRL-2107) and cultured in αMEM media (without ribonucleosides and deoxyribonucleosides, 2mM L-glutamine, 1.5g/L sodium bicarbonate, 0.2mM inositol; 0.1mM β-mercaptoethanol; 0.02mM folic acid and human IL-2 (200U/mL). Cell line authentitication was confirmed by short tandem repeat (STR) profiling (Promega PowerPlex® Fusion System). Mycoplasma was routinely checked by PCR using the Universal Mycoplasma Detection Kit (ATCC cat # 30–1012K)
Animals
All animal experiments were performed in strict accordance with the Guide for Care and Use of Laboratory Animals approved by the UT Southwestern Institutional Animal Care and Use Committee (animal protocol number 2017–102099). 4–5 weeks old female athymic mice (Hsd: Athymic nude mice-Foxn1nu) were purchased from Envigo (Cat. 069). For acclimatization animals were housed in the UT Southwestern animal facility room, maintained in a 12/12-hour light/dark cycle at a temperature of 20–26°C with 30–50% humidity and fed with standard Teklad diet (Envigo, Cat. 2916). For inducible knockdown experiments mice were fed with doxycycline diet (Envigo, Cat. TD.08541). Ad libitum access to food and water was always provided to mice. On course of the experiment animal health was monitored every day. Vehicle control (0.2% Methylcellulose and 5% DMSO in water) or Tucatinib (Selleckchem cat# S8362) and BGB324 (MedChemExpress cat# HY-15150) were administrated through oral gavage.
Oncosphere Assay
For oncosphere assay, 200 tumor cells were seeded in ultra-low attachment plates (96 well format) with oncosphere media (HuMEC serum free media with 1X B27 supplement, 5mg/ml insulin, 20ng/ml EGF and 10ng/ml bFGF) for a week at 37°C with 5% CO2. Oncosphere images were taken with EVOS M5000 microscope and quantified with ImageJ software.
Analysis of AXL Expression by Flow Cytometry
1×106 cells were cultured for 24hrs in RPM1 with 3% FBS. Cells were detached, washed with FACS buffer and resuspended in 100uL FACS solution for 20min with AXL-APC (R and D Systems cat# FAB154A, RRID: AB_2934005) and IgG-APC (R and D Systems cat# IC002A, RRID: AB_357239) conjugated antibodies. Cells were washed twice with FACS buffer and then proceed for intracellular staining using eBioscience™ Foxp3 / Transcription Factor Staining Buffer Set (Invitrogen™ cat# 00–5523-00) to permeabilize cell and nuclear membrane after extracellular staining. In brief, fixation/permeabilization concentrate (00–5123-43) and fixation/permeabilization diluent (00–5223-56) were mixed to a 1:4 ratio and administered to cells for 20min. Next, 10x Permeabilization Buffer (00–8333-56) was diluted to 1X in Fixation/Permeabilization Diluent (00–5223-56) and used to wash cells and incubate AXL-APC and IgG-APC antibodies for 20min. Cells were then washed, suspended in FACS buffer and flow cytometric analysis was performed using BD LSR Fortessa machine. FlowJo software was used for data analysis.
Western Blotting
Cells were grown in RPMI-1640 with 3% FBS for 24hrs and harvested with NP40 lysis buffer along with protease and phosphatase inhibitors. Protein was quantified using Thermofisher Pierce™ BCA protein assay kit (Thermo Scientific cat# 23225) and 20ug of protein was loaded in 7.5% or 10% SDS-PAGE gels, transferred on to nitrocellulose membrane using semi-dry transfer machine and blocked for 1hr in 5% TBST-milk. Primary antibodies were incubated overnight in 1% BSA-TBST solution. Membranes were washed with TBST, and secondary antibodies were incubated for 1hr in 5% TBST-milk. After TBST wash, membranes were developed using Thermofisher West Femto super signal and Bio-Rad imager. For primary antibodies we used AXL (Cell Signaling Technology cat# 8661, RRID:AB_11217435, 1:1000), EGFR (Cell Signaling Technology cat# 4267, RRID:AB_2864406, 1:1000), HER2 (Cell Signaling Technology cat# 2165, RRID:AB_10692490, 1:10,000), WRNIP1 (Santa Cruz Biotechnology cat# sc-377402, RRID:AB_2924675, 1:500) and Beta Actin (Abcam Cat# ab49900, RID:AB_867494,1:20,000). For secondary antibodies we used Goat anti-mouse IgG HRP conjugate (Millipore Cat# AP308P, RRID: AB_92635, 1:5000), and Goat anti-rabbit IgG HRP conjugate (Millipore Cat# AP307P, RRID: AB_11212848, 1:5000).
Immunoprecipitation
For immunoprecipitation experiments, 3.5×106 cells were cultured for 24hrs in RPM1 with 3% FBS, washed with cold 1XPBS and lysed with NP40 along with protease and phosphatase inhibitors for 30min. Lysate was spun down and the supernatant was collected for protein quantification using Thermofisher Pierce™ BCA protein assay kit (Thermo Scientific cat# 23225). 20ug protein was used as input and 500ug protein for immunoprecipitation with IgG control (Cell Signaling Technology cat # 2729, RRID: AB_1031062) and AXL (Cell Signaling Technology cat# 8661, RRID: AB_11217435, 1:50) antibodies overnight at 4°C with constant rotation. On the next day, 20uL of A/G agarose beads (Santa Cruz Biotechnology cat# sc-2003) were added and incubated for 5hrs at 4°C with constant rotation. Then, beads were spun down, washed 3X with NP40 buffer and heated to 95°C for 5min in 2X Laemmli Sample Buffer. Pull downs were subjected to Western blotting or processed for mass spectrometry for protein identification. To determine WRNIP1 interaction changes with AXL, cells were incubated for 5hrs with 4mM Hydroxyurea and 1uM BGB324 prior to collection for immunoprecipitation assay.
Mass Spectrometry
Gel band samples were digested overnight with trypsin (Pierce) following reduction and alkylation with DTT and iodoacetamide. The samples then underwent solid-phase extraction cleanup with an Oasis HLB plate and the resulting samples were injected into an Orbitrap Fusion Lumos mass spectrometer coupled to an Ultimate 3000 RSLC-Nano liquid chromatography system. Samples were then injected onto a 75 um i.d., 75-cm long EasySpray column (Thermo Scientific) and eluted with a gradient from 0–28% buffer B over 90 min. Buffer A contained 2% (v/v) ACN and 0.1% formic acid in water, and buffer B contained 80% (v/v) ACN, 10% (v/v) trifluoroethanol, and 0.1% formic acid in water. The mass spectrometer operated in positive ion mode with a source voltage of 1.5 kV and an ion transfer tube temperature of 275°C. MS scans were acquired at 120,000 resolution in the Orbitrap and up to 10 MS/MS spectra were obtained in the ion trap for each full spectrum acquired using higher-energy collisional dissociation (HCD) for ions with charges 2–7. Dynamic exclusion was set for 25s after an ion was selected for fragmentation. Raw MS data files were analyzed using Proteome Discoverer v2.4 (Thermo Scientific), with peptide identification performed using Sequest HT searching against the human protein database from UniProt. Fragment and precursor tolerances of 10 ppm and 0.6 Da were specified, and three missed cleavages were allowed. Carbamidomethylation of Cys was set as a fixed modification, with oxidation of Met set as a variable modification. The false-discovery rate (FDR) cutoff was 1% for all peptides.
Immunohistochemistry
Paraffin-embedded tumor sections of 5um thickness were sent to the Tissue Management Shared Resource core at UTSW for immunohistochemistry staining. First, the Paraffin-embedded tissue slides were baked for 20 minutes at 60°C, then deparaffinized and hydrated. Heat-induced antigen retrieval was performed at pH 9.0 for 20 minutes in a Dako PT Link. Post antigen retrieval peroxidase block was performed and then tissue was incubated with anti-AXL antibody (Cell Signaling Technology cat# 8661, RRID: AB_11217435) to a 1:300 dilution 35 minutes. The staining was visualized using the EVOS M5000 microscope to determine histoscore. AXL nuclear to non-nuclear ratio was analyzed using ImageJ software.
Immunocytochemistry
2.5×105 cells were cultured on cover slips for 24hrs in RPM1 with 3% FBS. Cells were treated with 4mM Hydroxyurea for 5hrs and then fixed for 20min using 4% paraformaldehyde. This was followed by permeabilization on ice with 0.1% Triton-X for 8 minutes. Coverslips were blocked with 3% BSA for 1hr and incubated with primary antibody overnight, γH2AX (Millipore cat# 05–636, RRID: AB_309864, 1:1000). Next day, corresponding secondary antibodies (1:500) were incubated for 1hr and Hoechst for 8 minutes (Thermo Scientific™ cat# 62249, 1:20,000) and mounted on slides. Slides were subjected to 0.5um thickness Z-stack serial microscopy imaging with confocal (Zeiss LSM 700) at 63X oil objective. γH2AX intensity analysis was done using maximal projection images and measuring γH2AX intensity per nuclei in Cell Profiler software.
Immunofluorescence
Collected mouse brains were fixed with 4% formaldehyde overnight, washed 3 times with 1X-PBS. Brains samples were then submitted to the Tissue Management core to put in paraffin blocks. Sections of 10μm thickness were made and incubated for 30min at 60°C, then de-paraffinized and rehydrated by serial incubations in xylene (2X for 3 min), 100% ethanol (2X for 3 min), 95% ethanol (2X for 3 min), 70% ethanol (2X for 3 min) and water (2X for 3 min). Slides were fixed for 15min in 50% Acetone solution and washed with 1X-PBS. Antigen retrieval was performed by placing slides on antigen retrieval buffer (10mM Tris HCl, 1mM EDTA, 10% glycerol, pH9.0) at 95°C for 25 min. The slides were then cooled to RT and rinsed with 1X-PBS (2X for 2 min) before being blocked in 10% horse and 10% goat serum in 0.1% PBST for 1hr at RT. The slides were incubated with primary antibody anti-GFP (Thermofisher Scientific cat# PA5–22688; RRID: AB_2540616, 1:300) in 2% serum PBST overnight at 4°C. The next day, the slides were washed with 1X-PBS for 10 min and incubated with secondary antibody for 1hr at RT. In addition, the nucleus was stained with Hoechst (Thermo Scientific™ cat# 62249, 1:20,000) for 10min. Evos M5000 microscope was used to determine GFP-positive cells for tumor quantification of mice brains bearing Lat cells. These brains were serially sectioned and 25% of the brain was quantified and adjusted to 100% of the whole brain.
Quantitative RT-PCR and RNA- Sequencing
RNA was isolated using the RNeasy Kit (Qiagen cat# 74004). 1 μg of extracted RNA was subjected to reverse transcription with iScript (Bio-Rad) reagents. Relative mRNA expression was determined by doing quantitative RT-PCR using SYBR green Supermix (Bio-Rad). mRNA data was normalized to β-actin. Primers used: AXL; Forward 5’- CCAGGACACCCCAGAGGTGCTAAT −3’ Reverse 5’- TGGTGGACTGGCTGTGCTTGC −3’ and β-actin; Forward 5’- AAAGACCTGTACGCCAACAC −3’ Reverse 5’- GTCATACTCCTGCTTGCTGAT-3’. For RNA-sequencing as previously reported in Parida et al., Cell Metabolism 2022 (26), total RNA was isolated from HCC1954 parental and metastatic derivatives. RNA quality was assessed using Agilent BioAnalyzer 2000 and samples with RNA integrity greater than 9.5 was used for subsequent analyses. RNA libraries were prepared using TruSeq RNA Sample Prep Kit v2 (Illumina) and sequencing was prepared following standards Illumina protocols. Fastqc (v0.11.2) and Fastq screen (v0.4.4) were used to check quality of Single-end 76 bp read length Fastq files. STAR (v2.5.3a) was used to mapped Fastq files to human reference genome (hg19). GENCODE(v19) and iGenomes were used for generation of read counts. Differentially expressed genes in Lat and M-BM cells were identified and shown as a volcano plot using normalized gene counts.
Peripheral Mouse NK Cell Analysis
100uL of blood was collected from the submandibular vein of athymic nude mice in EDTA tubes. RBCs were lysed with 8min of ACK lysis buffer. Samples were diluted with 1X-PBS and then centrifuged 600g. Cells were then washed with FACS buffer, blocked with TrueStain FcX (BioLegend cat#101319; RRID: AB_1574973) to avoid non-specific Fc receptor binding and dead cells were satin for 15min in Zombie Aqua (BioLegend cat# 423101). Live cells were subject to a 25min incubation with anti-mouse antibody-conjugated as follow: PerCP-CD45 (BioLegend cat#103129; RRID:AB_893343), FITC-CD3 (BioLegend cat#100203; RRID:AB_312660), FITC-CD19 (BioLegend cat# 152403; RRID:AB_2629812), APC-NKp46 (BioLegend cat# 137607; RRID:AB_10612749) and BV421-GZMB (BioLegend cat#396413; RRID:AB_2810602) for intracellular staining as described previously in the analysis of AXL expression by flow cytometry section method. Flow cytometry gating was performed using FlowJo software as described in supplementary figure 2C.
NK Cytotoxicity Assay
Mouse NK cells were isolated from spleens of the vehicle and BGB324 treated mice at termination point. Mechanical spleen dissociation was done in sterile conditions and splenocytes were suspended in isolation buffer (1X-PBS, 0.5% BSA,2mM EDTA, pH 7.2). Mouse NK cells were isolated using NK Cell Isolation Kit, an LS Column, and a MidiMACS™ Separator (cat# 130–115-818). Isolated NK cells were maintained in RPMI 1640 containing 15% FBS, 50uM β−2ME, 2 mM L-glutamine and 500 U/mL recombinant mouse-IL-2 overnight. For co-culture cytotoxicity experiments, cancer cells were seeded overnight and stained with eBioscience™ Calcein Violet 450 AM Viability Dye (cat# 65–0854-39). Mouse NK cells were then added to culture plate with cancer cells in a ratio of 1:10 (Cancer cell: NK cell) for 5hrs at 37°C. Once co-culture was terminated, cells were spun down and supernatant was collected to measure Calcein-AM violet released by using microplate-reader. Cytotoxicity percentage (% of lysis) was calculated by using the formula: [(sample release - min release) / (max release - min release)] x100 = % Lysis, where max release is lysed target cells with 0.1% Triton-X and min release are target cells alone.
For cytotoxicity assay with human NK92 cells, cancer cells were seeded, stained with Calcein-AM violet and co-culture with human NK cells to a 1:5 target to effector ration with 1uM BGB324 for 5hr at 37°C. Cytotoxicity percentage was calculated same as described above.
shRNA Knockdown and Rescue
3×106 HEK293T cells were transfected for 5hrs with 8ug of target lentiviral transgenes (pTripz and SMARTvector) with packaging plasmid psPAX2 (7μg) and envelop plasmid MD2.G (2.7μg) using lipofectamine 3000 (Invitrogen cat# L3000015). Media was replaced the following day and virus was collected 48hrs later. Cancer cells were transduced with 1:1 ratio of harvested lentivirus to Opti-MEM Pro media along with 5ug/ml of polybrene overnight. Then, 1ug/ml of doxycycline was added to induce expression of turboRFP target shRNA and gene knockdown. The following lentiviral inducible shRNAs were used: for pTRIPz; control (Dharmacon cat# RHS4743) and AXL (Dharmacon cat# RHS4696–200673419), and for SMARTvector; control (Dharmacon cat# VSC11655) and WRNIP1 (Dharmacon cat# V3SH11252–225041462).
For AXL rescue experiment, wildtype and kinase dead full length (Supplementary Table 1 and 2) synthetic gBlock oligos (codon optimized without changing the amino acids) were procured from Integrated DNA Technology and cloned into pINDUCER21 (Addgene plasmid #46948) where AXL was in frame with HA tag. Virus was produced and infected as described above.
DNA fiber assay
2.5×105 cells were seeded for 24hr in RPM1 with 3% FBS and pulse-labelled with 20uM 5-iodo-2’-deoxyuridine (IdU) and 200uM 5-chloro-2’-deoxyuridine (CldU) for 20 minutes each. For fork degradation experiments, cells were incubated with 4mM of hydroxyurea (HU) for 4hrs immediately following CldU treatment. For DNA fiber staining, primary antibodies (Abcam anti-BrdU/CldU cat# ab6326, RRID: AB_305426, 1:100 and BD Bioscience anti-BrdU/IdU cat# 347580, RRID: AB_10015219, 1:10) were incubated overnight and corresponding secondary antibodies (1:1000) were incubated for 1hr. Mounted slides were imaged with EVOS M5000 microscope. Length of labelled tracks was measured using ImageJ, 200 individual fibers were analyzed per biological replicate. For experiment using AXL inhibitor, cells were pretreated for 2hrs with either 1uM BGB324 or vehicle DMSO prior labeling and maintained during CldU, IdU and HU treatments.
Subcellular Fractionation
8×106 cells were seeded for 24hr in RPM1 with 3% FBS. Cells were then subjected to subcellular protein fractionation according to manufacturer’s instructions (Thermo Scientific cat# 78840). Fractions were then processed for western blotting as described before. To identify the purity of each fractionation compartment we used GAPDH (Cell Signaling Technology cat# 97166, RRID:AB_2756824, 1:10,000) for cytoplasmic extract, TFRC (Santa Cruz Biotechnology cat# sc-32272, RRID:AB_627167, 1:1000). for membrane extract, Lamin-B1 (Cell Signaling Technology cat# 12586, RRID:AB_2650517, 1:2000) for soluble nuclear extract and Histone-3 (Cell Signaling Technology cat# 4499, RRID:AB_10544537 1:2000) for the chromatin-bound extract.
Statistical Analysis
Statistical analysis between two comparative groups was determined using the Student t test or Mann–Whitney U test. For analysis of statistical comparisons between multiple study groups, one-way ANOVA was used followed by Dunnett test. GraphPad Prism 9 software was used for all statistical analysis. Values are expressed as mean ± SEM. The values were considered statistically significant based on p values (*, p < 0.05, **, p < 0.01, ***, p < 0.001, ****, p < 0.0001 and ns: not significant).
Data Availability Statement
RNA-sequencing data generated in this study are deposited in the Gene Expression Omnibus (GEO) under the accession number GSE180098. All other raw data are available upon request from the corresponding author.
RESULTS
HER2+ latent and metachronous brain metastatic cells are resistant to targeted therapy
Improved HER2 targeted therapies are effective in extending survival of breast cancer patients with brain metastasis but are ineffective and not curative in a significant number of patients (28). To evaluate the efficacy of lapatinib and tucatinib, small molecule HER2 inhibitors that can cross the blood-brain barrier, on brain metastasis we used isogenic HCC1954 HER2+ breast cancer brain-tropic latent (Lat), synchronous metastatic (S-BM) cells and metachronous metastatic cells (M-BM) and SKBR3 Lat and M-BM cells (26). While no significant changes were observed in the expression of EGFR or HER2 in these cells (Supplementary Fig. S1A), oncosphere assay revealed that Parental (PA) and S-BM were susceptible to lapatinib and tucatinib, whereas Lat and M-BM cells were resistant to these inhibitors (Fig. 1A; Supplementary Fig. S1B).
Figure 1. Nuclear AXL is enriched in HER2+ therapy resistance latent and metachronous HER2+ breast cancer brain metastatic cells.
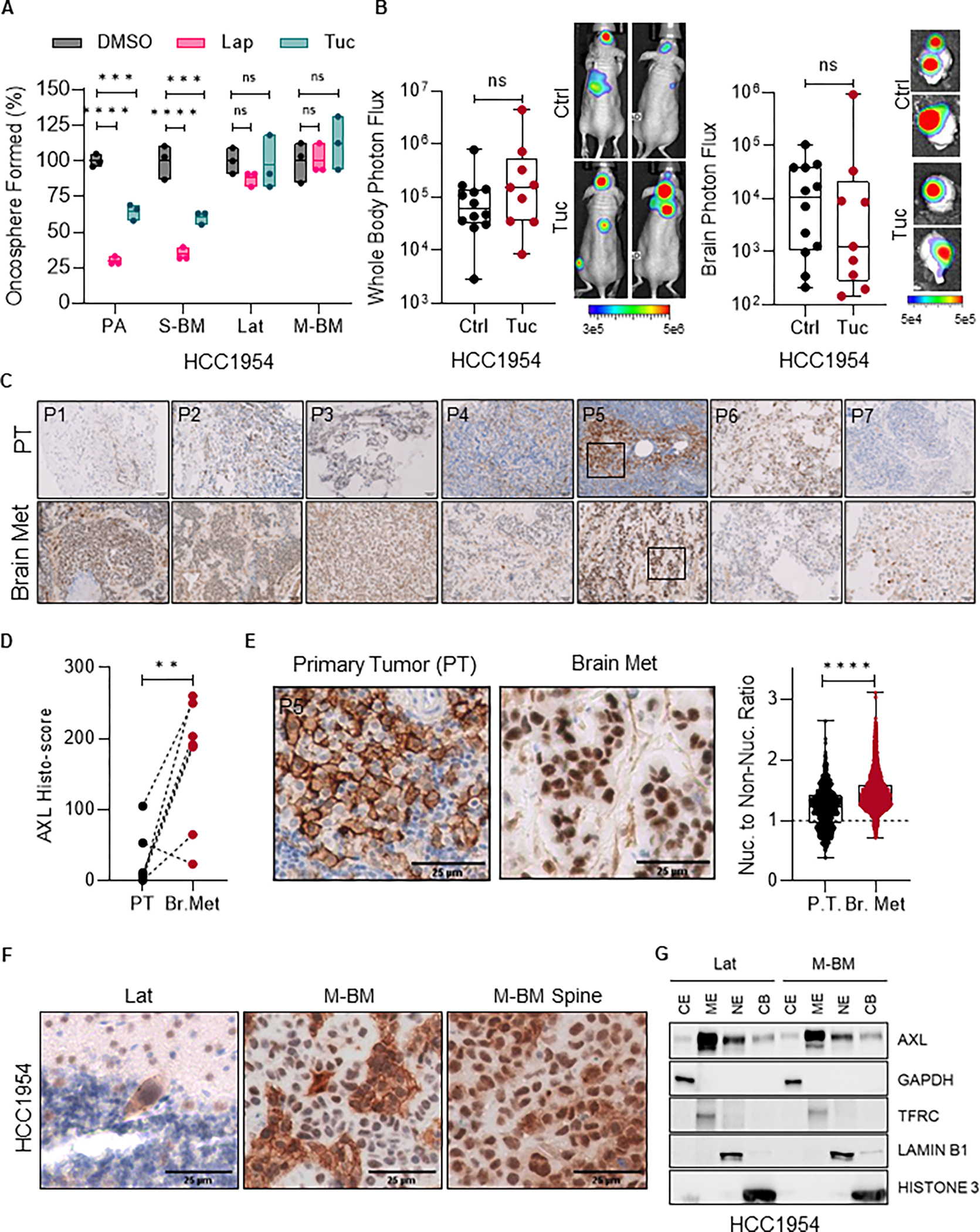
A, Relative quantification of oncospheres formed by HCC1954 PA, S-BM, Lat and M-BM cells seven days post-treatment with Tucatinib (3μM) and Lapatinib (1μM) compared to DMSO control. Data are presented as mean ± SEM. One-way ANOVA was used, followed by the Dunnett test. ****, P < 0.0001. ns: not significant. B, Whole body and ex-vivo brain metastasis incidence as measured by bioluminescence imaging (BLI) signal from athymic nude mice bearing HCC1954 M-BM cells that were intracardially injected. Vehicle (Ctrl) and Tucatinib (Tuc, 50mg/kg, once a day) was administrated orally 3 days post-injection daily up to 5 weeks. Control (n = 12) and Tucatinib (n = 9). Data are presented as mean ± SEM. Mann-Whitney U test, ns: not significant. Two representative BLI images of whole-body mice and ex-vivo brains of their respective conditions. C-E, AXL immunohistochemistry (IHC) in matched primary tumors and brain metastatic lesions (n = 7) from HER2+ breast cancer patients. (C). Representative AXL IHC images of matched primary and brain metastatic lesions from HER2+ breast cancer patients. (D). Histology-score analysis of AXL IHC. Data are presented as mean ± SEM. Paired Student t test. **, P < 0.01 (E). Representative images from patient-5 (P5) of Figure 1C highlighting nuclear AXL expression and quantification of AXL nuclear to non-nuclear ratio in matched primary and brain metastatic lesions. Data are presented as mean ± SEM. Unpaired Student t test. ****, P < 0.001. PT: primary tumor. Nuc.: nuclear. F, Representative images of AXL IHC on brain and spinal metastatic lesions in mice bearing HCC1954 Lat and M-BM cells. G, Immunoblot analysis of subcellular fractions from HCC1954 Lat and M-BM cells showing AXL localization and purity of subcellular fractions. GAPDH (CE, cytoplasmic extract), TFRC (ME, membrane extract), Lamin-B1 (NE, soluble nuclear extract) and Histone-3 (CB, chromatic bound extract).
To explore the effects of tucatinib on non-responsive metachronous brain metastasis in vivo, we intracardially injected HCC1954 M-BM cells and treated mice with tucatinib (50 mg/kg once a day for 5 weeks). Consistent with the results of the oncosphere assay, we did not observe any significant differences in metastatic incidence between the control and tucatinib treated groups, further demonstrating that these brain-tropic cells are resistant to HER2 kinase inhibition (Fig. 1B).
Nuclear AXL is enriched in HER2+ latent and metachronous brain metastatic cells
Next, we proceeded to examine the potential resistance mechanisms by analyzing transcriptomic profiles of the brain-tropic HCC1954 Lat and M-BM cells. Transcriptomic analysis revealed a significant enrichment of the receptor tyrosine kinase, AXL in both HCC1954 Lat and M-BM cells compared to the PA cells (Supplementary Fig. S1C–D). We also observed highly elevated AXL expression at both RNA and protein levels in Lat and M-BM cells, but not in synchronous brain metastatic cells and PA cells (Supplementary Fig. S1E–H). AXL is known to interact with other RTKs such as EGFR, HER2, HER3, MET, PDGFR and VEGFR-2, thereby promoting downstream signaling and resistance to therapies targeted such as EGFR, HER2, and PDGF (29,30). In addition, we did not detect any interaction between AXL and HER2 or EGFR in HCC1954 Lat and M-BM cells (Supplementary Fig. S1I).
To validate these findings in patient samples, we performed immunohistochemical analyses using a validated AXL antibody on matched primary tumor and brain metastatic lesions from HER2+ breast cancer patients. AXL was enriched in brain metastatic lesions from 6 out of 7 patients compared to its expression in matched primary tumors (Fig. 1C–D). Notably, nuclear AXL expression was predominantly observed in brain metastatic lesions (Fig. 1E). Consistent with these observations, we also detected an increase in nuclear AXL expression in brain and spinal metastatic lesions in mice injected with Lat and M-BM cells (Fig. 1F; Supplementary Fig. S1J).
Moreover, the subcellular fractionation and flow cytometry analysis further confirmed increased of full-length nuclear AXL in Lat and M-BM cells (Fig. 1G; Supplementary Fig. S1K–L). Collectively, these results indicate the enrichment of nuclear AXL in HER2+ latent and metachronous brain metastatic cells.
AXL inhibition limits metastatic latency and relapse
Clinical trials for various types of cancer, including melanoma, TNBC, NSCLC, GBM, and pancreatic cancer, are currently assessing therapeutic potential of several AXL inhibitors (31). To investigate the efficacy of the potent and selective small molecule inhibitor, BGB324, on Lat and M-BM cells, we performed oncosphere assays. BGB324 alone or in combination with HER2 kinase inhibitors lapatinib or tucatinib significantly reduced the number of oncospheres formed compared to the control (Fig. 2A; Supplementary Fig. S2A). As expected, BGB324 was ineffective in AXL low HCC1954 PA and S-BM (Supplementary Fig. S2B). We evaluated the effect of BGB324 on disseminated tumors in the brain by injecting HCC1954 Lat and M-BM cells intracardially and administering BGB324 (50 mg/kg; oral gavage; BID for 5 weeks) to mice three days post intra-cardiac injection. AXL inhibition significantly decreased the survival of latent brain metastatic cells as measured by GFP positive events in mice injected with Lat cells compared to the control (Fig. 2B). Additionally, whole-body photon flux and ex-vivo brain bioluminescence imaging (BLI) revealed a significant reduction in brain metastatic burden in mice bearing M-BM cells treated with BGB324 (Fig. 2C–D).
Figure 2. AXL inhibition attenuates brain metastasis and enhances NK cytotoxicity.
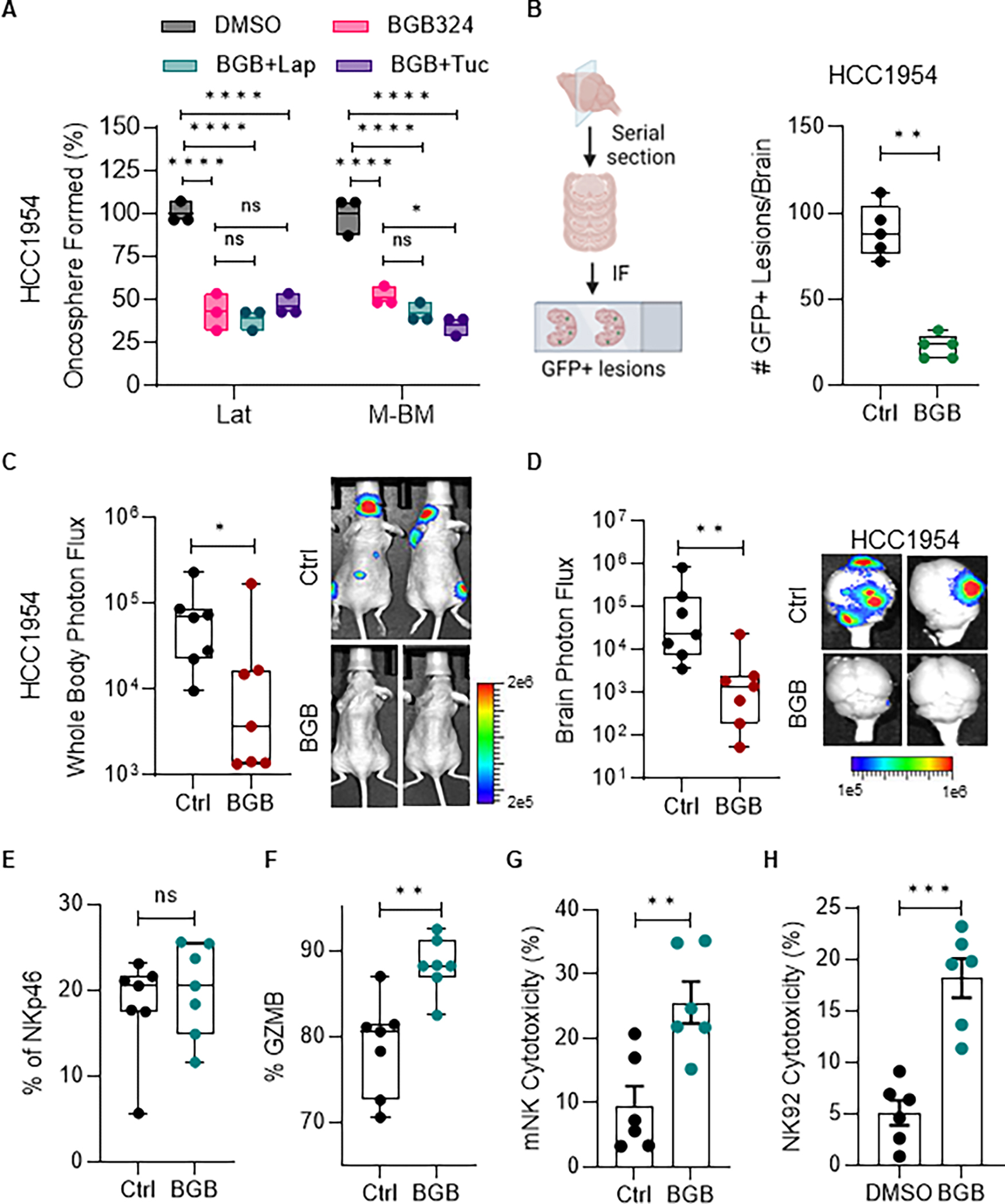
A, Relative quantification of oncospheres formed by HCC1954 Lat and M-BM cells treated with BGB324 (BGB, 1μM) alone or in combination with Lapatinib (Lap, 1μM) and Tucatinib (Tuc, 3μM) for seven days. Data are presented as mean ± SEM. One-way ANOVA was used, followed by the Dunnett test. *, P < 0.05, ****, P < 0.0001, ns: not significant. B, Illustration showing brain tissue processing to quantify GFP+ lesions. Quantification of GFP+ cells/lesions from brains of mice bearing HCC1954 Lat cells (n = 5 in both groups). C-D, Whole body and ex-vivo brain metastasis incidence as measured by BLI signal from athymic nude mice bearing HCC1954 M-BM cells. BGB324 (BGB, 50mg/kg, BID) or vehicle (Ctrl) was administrated orally 3 days post-injection for 5 weeks (n = 7 in both groups). Data are presented as mean ± SEM. Mann–Whitney U test, *, P < 0.05, **, P < 0.01. Two representative BLI images of whole-body mice and ex-vivo brains of their respective conditions. E-F, Quantification of NK cells (NKp46) and cytotoxic NK cells (GZMB) in peripheral blood collected from mice experiment in Figure 2C–D. Data are presented as mean ± SEM. Unpaired Student t test. ns: not significant. G, Quantification of mouse NK cell cytotoxicity against HCC1954 PA cells. Mouse NK cells were isolated from spleens of mice treated with vehicle or BGB324 in figure 2C–D. Data are presented as mean ± SEM. Unpaired Student t test. **, P < 0.01. H, Quantification of human NK cell (NK92) cytotoxicity after BGB324 (1μM) treatment on HCC1954 PA cells. Data are presented compared to DMSO (Ctrl) as mean ± SEM. Unpaired Student t test. ***, P < 0.001. (B, Created with BioRender.com).
Inhibition of AXL has been reported to activate innate and adaptive immune responses (32–34). Previous studies have shown that latent cells are able to survive in equilibrium with NK cells, and depletion of NK cells in mice bearing latent cells results in increased metastatic outbreaks (27). On the other hand, M-BM cells can evade NK cell-mediated immune surveillance and initiate metastasis (26). Therefore, we investigated whether AXL inhibition could enhance NK cell function and limit metastatic burden. Although there was no difference in the number of NK cells observed in peripheral blood, treatment with BGB324 significantly increased the percentage of Granzyme B positive cytotoxic NK cells in peripheral blood, indicating enhanced NK cell function (Fig. 2E–F; Supplementary Fig. S2C). Moreover, NK cells isolated from spleens of mice treated with BGB324 were more cytotoxic than those from control mice (Fig. 2G). Similarly, human NK cells treated with BGB324 showed enhanced cytotoxicity in vitro compared to controls (Fig. 2H). Taken together, these results suggest AXL inhibition can not only attenuate the metastasis-initiating capacity of tumor cells but also augment NK cell-mediated innate immune surveillance.
AXL depletion in Lat and M-BM cells attenuates metastatic fitness
To assess the impact of reducing AXL function in tumor cells, we used doxycycline-inducible shRNAs (Supplementary Fig. S3A). Depleting AXL in Lat and M-BM cells decreased oncosphere formation, similar to AXL inhibition. Oncosphere formation was further reduced in the presence of lapatinib and tucatinib (Fig. 3A; Supplementary Fig. S3B). To evaluate the impact of AXL depletion on brain-tropic cells, we administered doxycycline diet to mice 3 days after injecting Lat and M-BM cells intracardially (Fig. 3B). AXL depletion resulted in a significant decrease in surviving latent brain metastatic cells, as measured by GFP positive events in mice injected with Lat cells compared to controls (Fig. 3C; Supplementary Fig. S3C). Likewise, AXL depletion led to a significant decrease in brain metastatic incidence, as measured by whole body and ex-vivo brain BLI in mice injected with M-BM cells compared to controls (Fig. 3D–E; Supplementary Fig. S3D–E). Therefore, we conclude that AXL promotes survival and progression of brain-tropic HER2+ metastatic cells and that its depletion or inhibition attenuates metastatic fitness.
Figure 3. AXL depletion attenuates latent and metachronous brain metastasis.
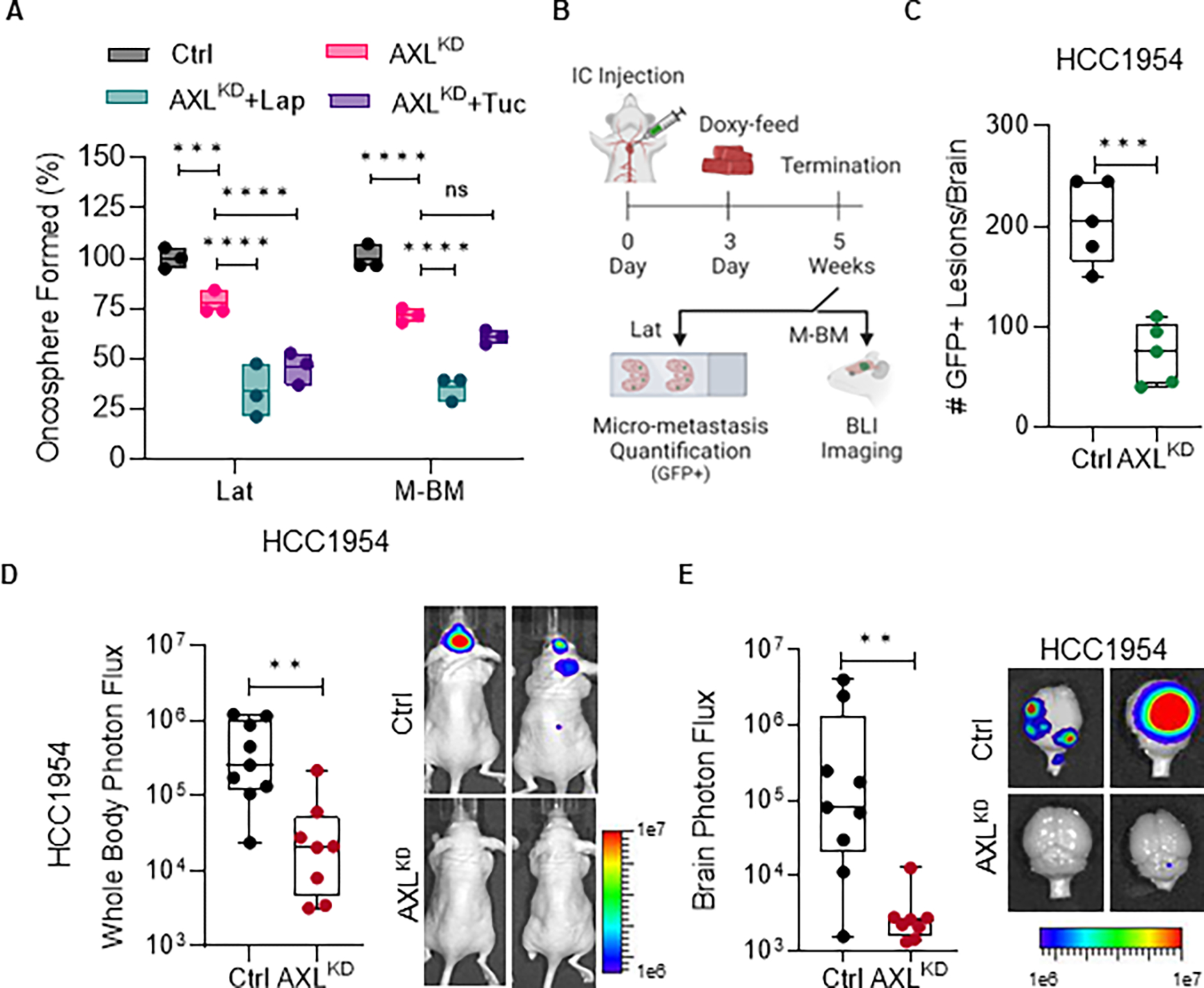
A, Relative quantification of oncospheres formed by HCC1954 Lat and M-BM cells upon AXL knockdown (AXLKD) and Lapatinib (1μM) or Tucatinib (3μM) treatment for seven days. Data are presented as mean ± SEM. One-way ANOVA was used, followed by the Dunnett test. *, P < 0.05, ***, P < 0.001, ****, P < 0.0001, ns: not significant. B, Experimental scheme to assess the impact of AXL depletion on metastatic latency and relapse. C, Quantification of GFP+ brain metastatic lesions in mice bearing HCC1954 AXLKD Lat cells (n = 5 per group). Mice were on doxycycline feed 3 days post injection until study termination. Data are presented as mean ± SEM. Mann–Whitney U test, ***, P < 0.001. D-E, Whole body and ex-vivo brain metastasis incidence as measured by BLI signal from athymic nude mice bearing HCC1954 AXLKD M-BM cells. (Ctrl group n = 9 and AXLKD group n = 8). Mice were on doxycycline feed 3 days post injection until study termination. Data are presented as mean ± SEM. Mann–Whitney U test, **, P < 0.01. Two representative BLI images of whole-body mice and ex-vivo brains of their respective conditions. (B, Created with BioRender.com).
AXL-WRNIP1 interaction promotes replication fork stabilization
To investigate the role of nuclear AXL in promoting metastatic latency and recurrence, we conducted AXL immuno-precipitation and mass spectrometry analysis on HCC1954 and SKBR3 Lat and M-BM cells. We identified Werner Helicase Interacting Protein 1 (WRNIP1), a replication stress response protein, as one of the proteins that interacted with AXL in these cells (Supplementary Fig. S4A). We confirmed this interaction by performing endogenous co-immunoprecipitation in brain-tropic cells (Fig. 4A). Moreover, consistent with the function of WRNIP1 in DNA replication stress, we found that AXL-WRNIP interaction was enhanced when cells were treated with hydroxyurea (HU), a ribonucleotide reductase inhibitor that induces DNA replication stress through depletion of deoxyribonucleotides (Supplementary Fig. S4B). WRNIP1 is known to protect stalled replication forks from degradation and promote replication fork restart (35). Therefore, we assessed replication fork stability in HCC1954 and SKBR3 parental and brain-tropic cells in the presence of HU. DNA fiber assays demonstrated increased replication fork stability in AXL high Lat and M-BM cells compared to PA cells (Fig. 4B; Supplementary Fig. S4C). Under similar assay conditions, depletion of WRNIP1 in Lat and M-BM cells resulted in nascent DNA degradation and no change in the replication fork speed was observed when compared to controls, consistent with previous reports (35) (Fig. 4C; Supplementary Fig. S4D–E). Analogous to WRNIP1 depletion, loss of AXL results in nascent DNA degradation, however an increase in replication fork speed and DNA damage was observed in AXL depleted cells compared to their respective controls under stress conditions (Fig. 4D–E; Supplementary Fig. S4F–I).
Figure 4. AXL-WRNIP1 interaction mediates replication stress response in Lat and M-BM cells.
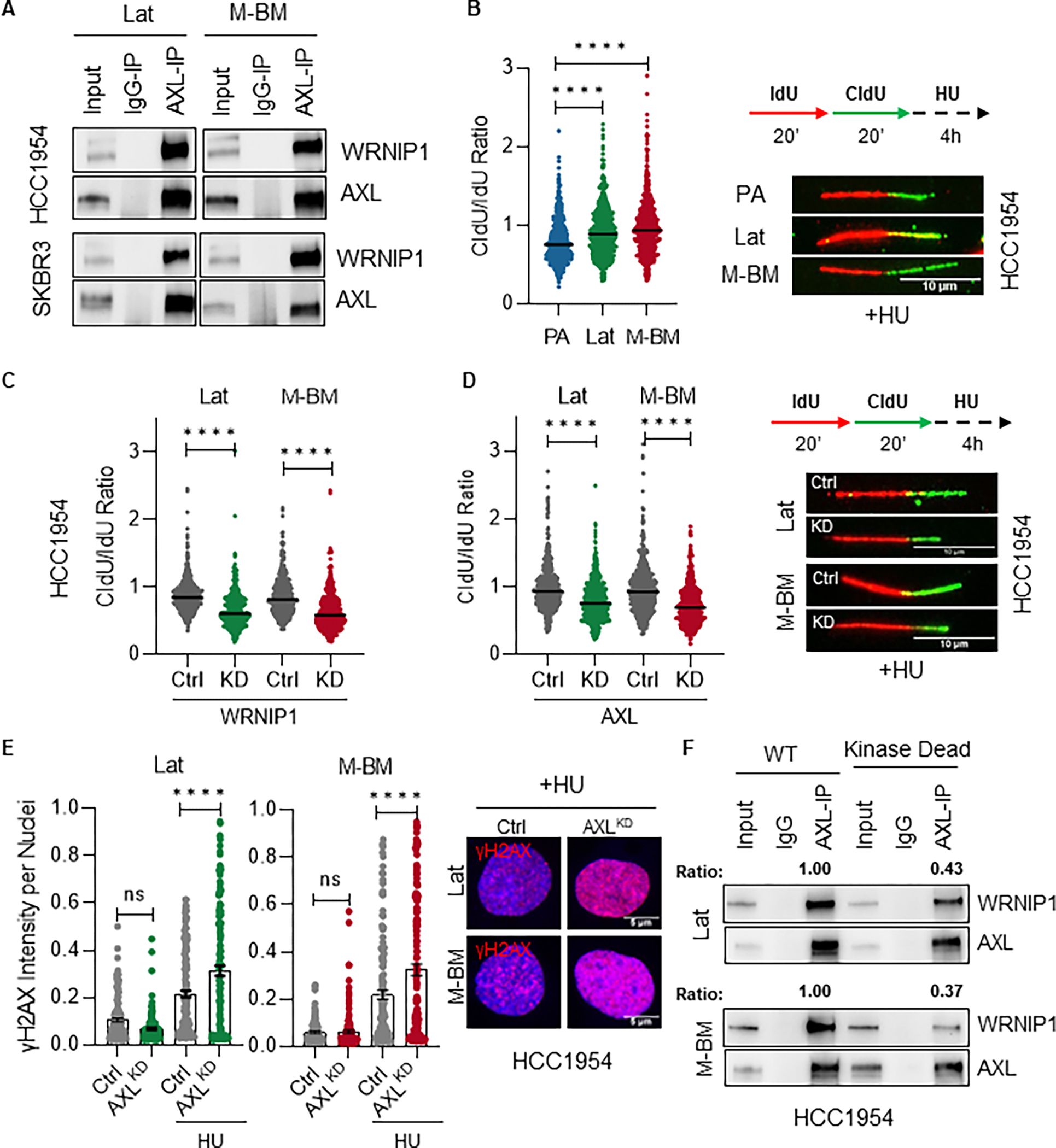
A, Immunoblot showing AXL-WRNIP1 interaction in HCC1954 Lat and M-BM cells under normal (Ctrl) and replication stress induced condition (in presence of 4mM HU for 5hrs). B, Relative fork degradation measured by the CldU/IdU ratio in presence of 4mM HU. Representative images of DNA fibers from HCC1954 PA, Lat and M-BM cells are also shown. IdU (red color) and CldU (green color). N = 3 for all experiments. Data are presented as mean ± SEM. Mann–Whitney U test, ****, P < 0.0001. C-D, Relative fork degradation measured by the CldU/IdU ratio in presence of 4mM HU upon WRNIP1 knockdown (WRNIP1KD) and AXL knockdown (AXLKD) in HCC1954 Lat and M-BM cells. Representative images of DNA fibers are also shown. IdU (red color) and CldU (green color). N = 3 for all experiments. Data are presented as mean ± SEM. Mann–Whitney U test, ****, P < 0.0001. E, γH2AX intensity quantification of HCC1954 Lat and M-BM cells treated with 4mM HU for 5hrs. Representative image of γH2AX intensity under HU. Data are presented as mean ± SEM. One-way ANOVA was used, followed by the Dunnett test. ****, P < 0.001, ns: not significant. F, Immunoblot showing reduced interaction between kinase dead AXL and WRNIP1. This assay was performed by reintroducing AXL (WT) and kinase dead mutant in HCC1954 AXLKD Lat and M-BM cells. AXL immunoprecipitation was performed and ratio was determined by normalizing WRNIP1 expression to immune-precipitated AXL.
To further investigate the role of AXL kinase in promoting AXL-WRNIP1 interaction, we generated full length wildtype and kinase dead AXL constructs and reintroduced them in AXL depleted HCC1954 Lat and M-BM cells (Supplementary Fig. S4J). We observed a significantly reduced WRNIP1 interaction with AXL in kinase dead mutant compared to wildtype AXL (Fig. 4F). These data demonstrate that the AXL-WRNIP1 interaction, which depends on the kinase activity of AXL, plays a critical role in facilitating the replication fork stabilization in Lat and M-BM cells.
WRNIP1 depletion in Lat and M-BM cells attenuates metastatic fitness
Despite the known role of WRNIP1 in the replication stress response, its contribution to tumorigenesis is not fully understood. To investigate the function of WRNIP1 in promoting brain metastasis, we depleted it in Lat and M-BM cells (Supplementary Fig. S5A). Depletion of WRNIP1 resulted in reduced oncosphere formation or metastasis initiating capacity (Supplementary Fig. S5B). Additionally, the survival of latent brain metastatic cells was significantly decreased in mice injected with HCC1954 Lat cells that lacked WRNIP1 (Fig. 5A). Similarly, in mice bearing HCC1954 M-BM cells, the incidence of brain metastasis was also reduced after the depletion of WRNIP1 compared to the control group (Fig. 5B–C). Taken together, depletion of AXL or WRNIP1 or limiting their interaction leads to reduced brain metastatic incidence.
Figure 5. Augmenting replication stress attenuates metastatic latency and recurrence.
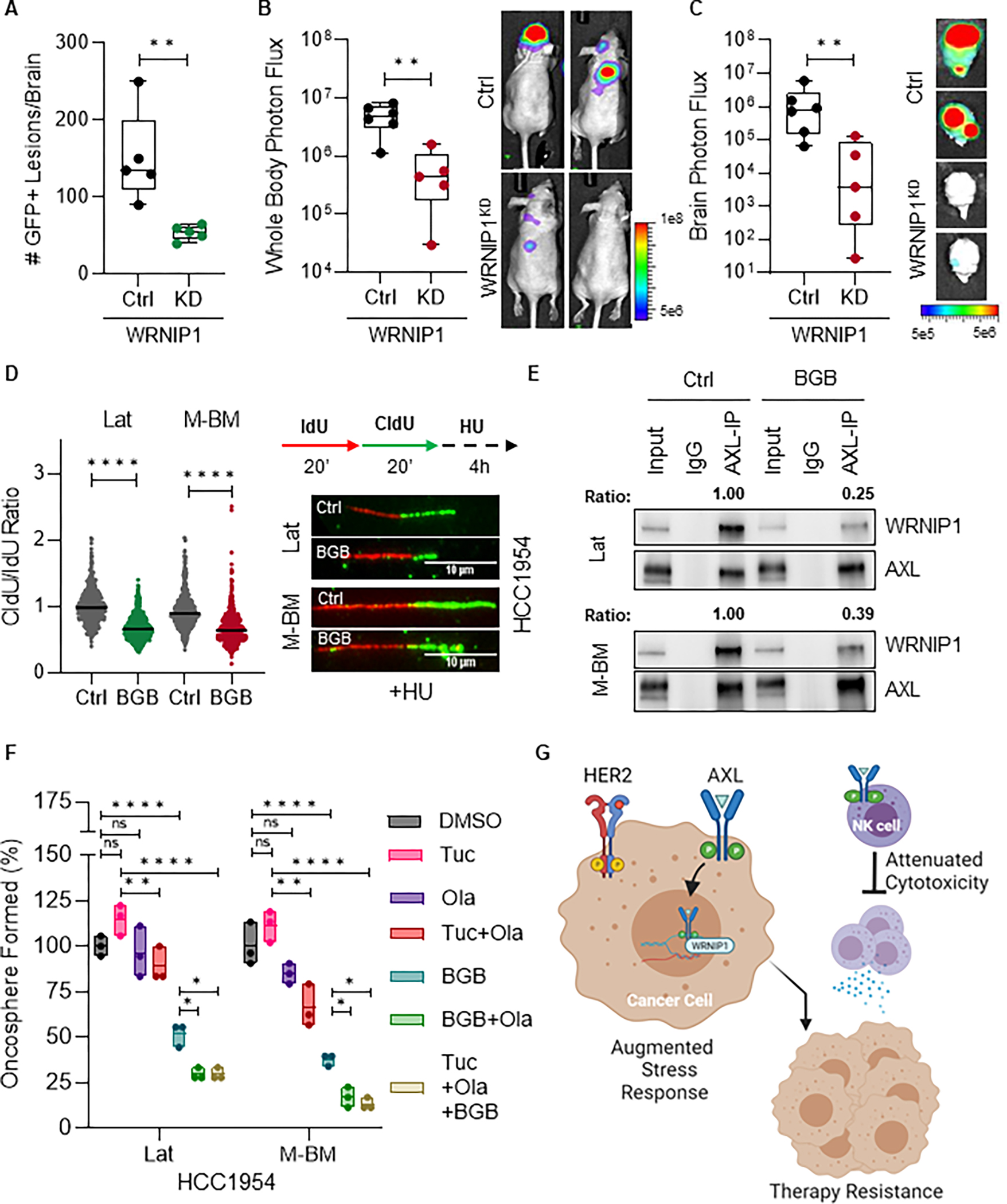
A, Quantification of GFP+ metastatic lesions in mice bearing HCC1954 WRNIP1 knockdown (WRNIP1KD) Lat cells (n = 5 per group). Doxycycline feed was given 3 days post injection and continued till study termination. Data are presented as mean ± SEM. Mann–Whitney U test, ***, P < 0.001. B-C, Whole body and ex-vivo brain metastasis incidence as measured by BLI signal from athymic nude mice bearing HCC1954 WRNIP1KD M-BM cells. (Ctrl group n = 6 and WRNIP1KD group n = 5). Doxycycline feed was given 3 days post injection and continued till study termination. Data are presented as mean ± SEM. Mann–Whitney U test, **, P < 0.01. Two representative BLI images of whole-body mice and ex-vivo brains of their respective conditions. D, Relative fork degradation measured by the CldU/IdU ratio in presence of 4mM HU upon BGB324 treatment in HCC1954 Lat and M-BM cells. Representative images of DNA fibers are also shown. IdU (red color) and CldU (green color). N = 3 for all experiments. Data are presented as mean ± SEM. Mann–Whitney U test, ****, P < 0.0001. E, Immunoblot showing reduced AXL-WRNIP1 interaction in the presence of BGB324 (1μM for 5 hours). Ratio was determined by normalizing WRNIP1 expression to immunoprecipitated AXL. F, Relative quantification of oncospheres formed by HCC1954 Lat and M-BM cells treated with DMSO, BGB324 (BGB: 1μM), Olaparib (Ola: 5μM), Tucatinib (Tuc: 3μM) and combinations. Data are presented as mean ± SEM. One-way ANOVA was used, followed by the Dunnett test. *, P < 0.05, **, P < 0.01, ****, P < 0.0001, ns: not significant. G, Graphical abstract summarizing key findings of AXL-WRNIP1 mediated replication stress response promoting latent and metachronous metastasis. (G, Created with BioRender.com).
Augmenting replication stress attenuates metastatic latency and recurrence
Next, we investigated the impact of enhancing replication stress through pharmacological inhibitors on metastatic latency and recurrence. Inhibition of AXL kinase activity using BGB324 not only attenuated fork protection (Fig. 5D) but also disrupted AXL and WRNIP1 interaction, further reinforcing the role of AXL kinase activity in promoting interaction with WRNIP1 and mediating the replication stress response (Fig. 5E). Likewise, AXL was predominantly non-nuclear in brain lesions that persisted in mice treated with BGB324 (Supplementary Fig. S5C). To expand on these observations, we explored the effects of inhibiting poly ADP ribose polymerase 1 (PARP1), a protein involved in DNA damage repair and replication stress. Given that PARP1 inhibitors are known to enhance replication stress (36), we evaluated the impact of the PARP1 inhibitor, Olaparib alone and in combination with BGB324 and tucatinib on the oncosphere forming capacity of HCC1954 Lat and M-BM cells. While Olaparib alone was not sufficient to limit oncosphere formation, combination of Olaparib with BGB324 or tucatinib led to a significant reduction in oncosphere formation (Fig. 5F). In summary, these findings collectively demonstrate that actively augmenting replication stress and/or targeting AXL-WRNIP1-driven replication stress response alone and in combination with HER2 targeted therapies attenuates metastatic latency and metachronous metastasis.
DISCUSSION
We have discovered a novel non-canonical function for AXL in the nucleus, where it enhances a protective replication stress response by interacting with WRNIP1 thereby promoting therapy resistance. Our study has revealed that AXL is predominantly localized in the nucleus of HER2+ breast cancer brain metastatic lesions, and its depletion or treatment with an AXL inhibitor limits effective replication stress response and weakens the metastatic fitness of disseminated tumor cells in the brain microenvironment. In addition, AXL inhibition boosts NK cell cytotoxicity and innate immune surveillance, exerting potent anti-metastatic activity in mice. By investigating the nuclear function of AXL, we have discovered its critical interaction with WRNIP1, an important regulator of replication stress response. We have shown that the kinase dead AXL and pharmacologic AXL inhibition disrupts the AXL-WRNIP1 interaction and leads to replication fork destabilization. Similarly, depleting WRNIP1 or augmenting replication stress with PARP inhibitors reduces metastatic fitness (Fig. 5G).
Studies in the past have demonstrated that AXL promotes therapy resistance and dormancy, which is dependent on cell surface signal transduction and varies with tumor type. For instance, in dormant myeloma and prostate cancer cells, AXL is enriched, and its inhibition results in increased proliferation (37,38). On the other hand, in melanoma cells, AXL and TAM receptors promote invasion and reactivation from dormancy (39,40). Moreover, AXL and its downstream signaling response are enriched in chemo and targeted therapy resistant such as AML, NSCLC, HER2+ breast cancer and prostate cancer (21,41–44). Additionally, AXL is enriched in tumor infiltrating immune populations including macrophages, NK cells and regulatory T cells resulting in suppressed immune surveillance (32–34). Our findings support these observations, and we offer new insights into the mechanisms underlying how AXL modulates replication fork stabilization in tumor cells and promotes therapy resistance and metastatic latency (Fig. 5G).
Targeted therapies often lead to acquired resistance, which is difficult to overcome. Our findings suggest that AXL inhibition has dual effects: it attenuates tumor cell survival and enhances innate immune surveillance, resulting in longer sustained disease control. Based on these findings, it is likely that clinical trials evaluating AXL inhibition in combination with immunotherapy for the treatment of AXL positive and advanced metastatic NSCLC patients (NCT03184571) will lead to improved clinical outcomes. We propose that patient stratification based on AXL, GAS6 and expression of AXL downstream effectors will improve the therapeutic outcomes of these therapies. Moreover, our studies suggest that inhibitors that augment replication stress can be combined with AXL and/or HER2 targeted therapies to limit brain metastasis and prolong durable responses.
Finally, our findings reveal that AXL promotes therapy resistance and brain metastasis through novel mechanisms. Further research is necessary to (45) elucidate how AXL translocates to the nucleus in tumor cells, (16) to determine mechanistically how AXL-WRNIP1 promote replication fork stabilization and alleviate stress (3) identify factors driving AXL expression in tumor-exposed NK cells, (4) understand better the mechanisms by which AXL promotes NK cell dysfunction, and (5) investigate the distinct AXL signaling dynamics in disseminated tumor cells and infiltrating immune cells. Our preclinical models provide a valuable platform to address these questions. In summary, our study suggests that AXL along with WRNIP1 aids replicating fork stability and promotes tolerance in response to stress, and promotes therapy resistance along with survival of latent and metachronous HER2+ breast cancer cells. Inhibiting AXL has the potential to eradicate metastasis-initiating cells and enhance innate immune surveillance.
Supplementary Material
STATEMENT OF SIGNIFICANCE.
Nuclear AXL and WRNIP1 interact and mediate replication stress response, promote therapy resistance, and support metastatic progression, indicating that targeting the AXL/WRNIP1 axis is a potentially viable therapeutic strategy for breast cancer.
ACKNOWLEDGMENTS
This work was supported by the following grants: National Science Foundation grant 2019281049 (M. Marquez-Palencia), The Welch Foundation grant I-2087-20210327 (A. W. Whitehurst), Cancer Center Support Grant (P30 CA142543), CPRIT (RR170003), ACS (RSG-20-47-01-CSM), METAvivor (GAA202106-0027), Susan G. Komen Career Catalyst Grant (CCR22902470), Breast Cancer- Bone initiative from Charles Y.C. Pak Foundation grants, Vanderbilt-Ingram Cancer Center SPORE in Breast Cancer (P50CA098131) to (S. Malladi). Illustrations were created with BioRender.com. We thank Huiming Lu for providing reagents to assess replication stress response. We gratefully acknowledge the Small Animal Imaging and Tissue Management Cores supported by Cancer Center Support Grant P30 CA142543 and the UTSW Proteomics Core for proteomics assistance.
Footnotes
Authors declare no potential conflicts of interest.
AUTHORS’ DISCLOSURES
A. B. Hanker receives or has received research grant support from Takeda and Lilly. C. L. Arteaga receives or has received research grant support from Pfizer, Lilly, and Takeda, holds stock options in Provista and Y-TRAP, and serves or has served in an advisory role to Novartis, Lilly, TAIHO Oncology, Daiichi Sankyo, Merck, AstraZeneca, OrigiMed, Immunomedics, and Susan G. Komen Foundation.
REFERENCES
- 1.Kim K, Marquez-Palencia M, Malladi S. Metastatic Latency, a Veiled Threat. Front Immunol 2019;10:1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lambert AW, Pattabiraman DR, Weinberg RA. Emerging Biological Principles of Metastasis. Cell 2017;168:670–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Massague J, Ganesh K. Metastasis-Initiating Cells and Ecosystems. Cancer Discov 2021;11:971–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pantel K, Hayes DF. Disseminated breast tumour cells: biological and clinical meaning. Nat Rev Clin Oncol 2018;15:129–31 [DOI] [PubMed] [Google Scholar]
- 5.Peitzsch C, Tyutyunnykova A, Pantel K, Dubrovska A. Cancer stem cells: The root of tumor recurrence and metastases. Semin Cancer Biol 2017;44:10–24 [DOI] [PubMed] [Google Scholar]
- 6.Gaillard H, Garcia-Muse T, Aguilera A. Replication stress and cancer. Nat Rev Cancer 2015;15:276–89 [DOI] [PubMed] [Google Scholar]
- 7.da Costa A, Chowdhury D, Shapiro GI, D’Andrea AD, Konstantinopoulos PA. Targeting replication stress in cancer therapy. Nat Rev Drug Discov 2023;22:38–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001;344:783–92 [DOI] [PubMed] [Google Scholar]
- 9.Stavrou E, Winer EP, Lin NU. How we treat HER2-positive brain metastases. Esmo Open 2021;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kulukian A, Lee P, Taylor J, Rosler R, de Vries P, Watson D, et al. Preclinical Activity of HER2-Selective Tyrosine Kinase Inhibitor Tucatinib as a Single Agent or in Combination with Trastuzumab or Docetaxel in Solid Tumor Models. Mol Cancer Ther 2020;19:976–87 [DOI] [PubMed] [Google Scholar]
- 11.Lee VDDASWJWKKPA. ARRY-380, a potent, small molecule inhibitor of ErbB2, increases survival in intracranial ErbB2+ xenograft models in mice. Cancer Res 2012;72 [Google Scholar]
- 12.Swain SM, Shastry M, Hamilton E. Targeting HER2-positive breast cancer: advances and future directions. Nat Rev Drug Discov 2023;22:101–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin NU, Borges V, Anders C, Murthy RK, Paplomata E, Hamilton E, et al. Intracranial Efficacy and Survival With Tucatinib Plus Trastuzumab and Capecitabine for Previously Treated HER2-Positive Breast Cancer With Brain Metastases in the HER2CLIMB Trial. J Clin Oncol 2020;38:2610–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murthy R, Borges VF, Conlin A, Chaves J, Chamberlain M, Gray T, et al. Tucatinib with capecitabine and trastuzumab in advanced HER2-positive metastatic breast cancer with and without brain metastases: a non-randomised, open-label, phase 1b study. Lancet Oncol 2018;19:880–8 [DOI] [PubMed] [Google Scholar]
- 15.Murthy RK, Loi S, Okines A, Paplomata E, Hamilton E, Hurvitz SA, et al. Tucatinib, Trastuzumab, and Capecitabine for HER2-Positive Metastatic Breast Cancer. N Engl J Med 2020;382:597–609 [DOI] [PubMed] [Google Scholar]
- 16.Brastianos PK, Carter SL, Santagata S, Cahill DP, Taylor-Weiner A, Jones RT, et al. Genomic Characterization of Brain Metastases Reveals Branched Evolution and Potential Therapeutic Targets. Cancer Discov 2015;5:1164–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fecci PE, Rao G, Brastianos PK, Dunn GP, Anders CK. Editorial: It takes a village: The expanding multi-disciplinary approach to brain metastasis. Front Oncol 2022;12:1054490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vernieri C, Milano M, Brambilla M, Mennitto A, Maggi C, Cona MS, et al. Resistance mechanisms to anti-HER2 therapies in HER2-positive breast cancer: Current knowledge, new research directions and therapeutic perspectives. Crit Rev Oncol Hematol 2019;139:53–66 [DOI] [PubMed] [Google Scholar]
- 19.Schlam I, Tarantino P, Tolaney SM. Overcoming Resistance to HER2-Directed Therapies in Breast Cancer. Cancers (Basel) 2022;14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alexander PB, Chen R, Gong C, Yuan L, Jasper JS, Ding Y, et al. Distinct Receptor Tyrosine Kinase Subsets Mediate Anti-HER2 Drug Resistance in Breast Cancer. J Biol Chem 2017;292:748–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu L, Greger J, Shi H, Liu Y, Greshock J, Annan R, et al. Novel mechanism of lapatinib resistance in HER2-positive breast tumor cells: activation of AXL. Cancer Res 2009;69:6871–8 [DOI] [PubMed] [Google Scholar]
- 22.Flem-Karlsen K, McFadden E, Omar N, Haugen MH, Oy GF, Ryder T, et al. Targeting AXL and the DNA Damage Response Pathway as a Novel Therapeutic Strategy in Melanoma. Mol Cancer Ther 2020;19:895–905 [DOI] [PubMed] [Google Scholar]
- 23.Mullen MM, Lomonosova E, Toboni MD, Oplt A, Cybulla E, Blachut B, et al. GAS6/AXL Inhibition Enhances Ovarian Cancer Sensitivity to Chemotherapy and PARP Inhibition through Increased DNA Damage and Enhanced Replication Stress. Mol Cancer Res 2022;20:265–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ramkumar K, Stewart CA, Cargill KR, Della Corte CM, Wang Q, Shen L, et al. AXL Inhibition Induces DNA Damage and Replication Stress in Non-Small Cell Lung Cancer Cells and Promotes Sensitivity to ATR Inhibitors. Mol Cancer Res 2021;19:485–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sutcliffe MD, Galvao RP, Wang L, Kim J, Rosenfeld LK, Singh S, et al. Premalignant Oligodendrocyte Precursor Cells Stall in a Heterogeneous State of Replication Stress Prior to Gliomagenesis. Cancer Res 2021;81:1868–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parida PK, Marquez-Palencia M, Nair V, Kaushik AK, Kim K, Sudderth J, et al. Metabolic diversity within breast cancer brain-tropic cells determines metastatic fitness. Cell Metab 2022;34:90–105 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Malladi S, Macalinao DG, Jin X, He L, Basnet H, Zou Y, et al. Metastatic Latency and Immune Evasion through Autocrine Inhibition of WNT. Cell 2016;165:45–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Curigliano G, Mueller V, Borges V, Hamilton E, Hurvitz S, Loi S, et al. Tucatinib versus placebo added to trastuzumab and capecitabine for patients with pretreated HER2+ metastatic breast cancer with and without brain metastases (HER2CLIMB): final overall survival analysis. Ann Oncol 2022;33:321–9 [DOI] [PubMed] [Google Scholar]
- 29.Colavito SA. AXL as a Target in Breast Cancer Therapy. J Oncol 2020;2020:5291952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goyette MA, Duhamel S, Aubert L, Pelletier A, Savage P, Thibault MP, et al. The Receptor Tyrosine Kinase AXL Is Required at Multiple Steps of the Metastatic Cascade during HER2-Positive Breast Cancer Progression. Cell Rep 2018;23:1476–90 [DOI] [PubMed] [Google Scholar]
- 31.Zhu C, Wei Y, Wei X. AXL receptor tyrosine kinase as a promising anti-cancer approach: functions, molecular mechanisms and clinical applications. Mol Cancer 2019;18:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paolino M, Choidas A, Wallner S, Pranjic B, Uribesalgo I, Loeser S, et al. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature 2014;507:508–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Terry S, Abdou A, Engelsen AST, Buart S, Dessen P, Corgnac S, et al. AXL Targeting Overcomes Human Lung Cancer Cell Resistance to NK- and CTL-Mediated Cytotoxicity. Cancer Immunol Res 2019;7:1789–802 [DOI] [PubMed] [Google Scholar]
- 34.Tirado-Gonzalez I, Descot A, Soetopo D, Nevmerzhitskaya A, Schaffer A, Kur IM, et al. AXL Inhibition in Macrophages Stimulates Host-versus-Leukemia Immunity and Eradicates Naive and Treatment-Resistant Leukemia. Cancer Discov 2021;11:2924–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leuzzi G, Marabitti V, Pichierri P, Franchitto A. WRNIP1 protects stalled forks from degradation and promotes fork restart after replication stress. EMBO J 2016;35:1437–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rose M, Burgess JT, O’Byrne K, Richard DJ, Bolderson E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front Cell Dev Biol 2020;8:564601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khoo WH, Ledergor G, Weiner A, Roden DL, Terry RL, McDonald MM, et al. A niche-dependent myeloid transcriptome signature defines dormant myeloma cells. Blood 2019;134:30–43 [DOI] [PubMed] [Google Scholar]
- 38.Axelrod HD, Valkenburg KC, Amend SR, Hicks JL, Parsana P, Torga G, et al. AXL Is a Putative Tumor Suppressor and Dormancy Regulator in Prostate Cancer. Mol Cancer Res 2019;17:356–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Revach OY, Sandler O, Samuels Y, Geiger B. Cross-Talk between Receptor Tyrosine Kinases AXL and ERBB3 Regulates Invadopodia Formation in Melanoma Cells. Cancer Res 2019;79:2634–48 [DOI] [PubMed] [Google Scholar]
- 40.Fane ME, Chhabra Y, Alicea GM, Maranto DA, Douglass SM, Webster MR, et al. Stromal changes in the aged lung induce an emergence from melanoma dormancy. Nature 2022;606:396–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hong CC, Lay JD, Huang JS, Cheng AL, Tang JL, Lin MT, et al. Receptor tyrosine kinase AXL is induced by chemotherapy drugs and overexpression of AXL confers drug resistance in acute myeloid leukemia. Cancer Lett 2008;268:314–24 [DOI] [PubMed] [Google Scholar]
- 42.Lin JZ, Wang ZJ, De W, Zheng M, Xu WZ, Wu HF, et al. Targeting AXL overcomes resistance to docetaxel therapy in advanced prostate cancer. Oncotarget 2017;8:41064–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet 2012;44:852–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adam-Artigues A, Arenas EJ, Martinez-Sabadell A, Braso-Maristany F, Cervera R, Tormo E, et al. Targeting HER2-AXL heterodimerization to overcome resistance to HER2 blockade in breast cancer. Sci Adv 2022;8:eabk2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li BT, Smit EF, Goto Y, Nakagawa K, Udagawa H, Mazieres J, et al. Trastuzumab Deruxtecan in HER2-Mutant Non-Small-Cell Lung Cancer. N Engl J Med 2022;386:241–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-sequencing data generated in this study are deposited in the Gene Expression Omnibus (GEO) under the accession number GSE180098. All other raw data are available upon request from the corresponding author.