

Abstract
In addition to the primary cell surface receptor CD4, CCR5 or another coreceptor is necessary for infections by human immunodeficiency virus type 1 (HIV-1), yet the mechanisms of coreceptor function and their stoichiometries in the infection pathway remain substantially unknown. To address these issues, we studied the effects of CCR5 concentrations on HIV-1 infections using wild-type CCR5 and two attenuated mutant CCR5s, one with the mutation Y14N at a critical tyrosine sulfation site in the amino terminus and one with the mutation G163R in extracellular loop 2. The Y14N mutation converted a YYT sequence at positions 14 to 16 to an NYT consensus site for N-linked glycosylation, and the mutant protein was shown to be glycosylated at that position. The relationships between HIV-1 infectivity values and CCR5 concentrations took the form of sigmoidal (S-shaped) curves, which were dramatically altered in different ways by these mutations. Both mutations shifted the curves by factors of approximately 30- to 150-fold along the CCR5 concentration axis, consistent with evidence that they reduce affinities of virus for the coreceptor. In addition, the Y14N mutation specifically reduced the maximum efficiencies of infection that could be obtained at saturating CCR5 concentrations. The sigmoidal curves for all R5 HIV-1 isolates were quantitatively consistent with a simple mathematical model, implying that CCR5s reversibly associate with cell surface HIV-1 in a concentration-dependent manner, that approximately four to six CCR5s assemble around the virus to form a complex needed for infection, and that both mutations inhibit assembly of this complex but only the Y14N mutation also significantly reduces its ability to successfully mediate HIV-1 infections. Although several alternative models would be compatible with our data, a common feature of these alternatives is the cooperation of multiple CCR5s in the HIV-1 infection pathway. This cooperativity will need to be considered in future studies to address in detail the mechanism of CCR5-mediated HIV-1 membrane fusion.
Infections by human immunodeficiency virus type 1 (HIV-1) are initiated by virus binding followed by fusion of the viral membrane with the host cell plasma membrane. This process is dependent on “knobs” in the viral envelope that consist of gp41 integral membrane trimers and associated gp120 surface subunits (reviewed in reference 10). Binding of HIV-1 onto the primary receptor, CD4, induces a conformational change in gp120 that exposes a previously buried site for association with a coreceptor (32, 33, 50, 58, 61). Although several chemokine receptors can function at least weakly as HIV-1 coreceptors, CCR5 and CXCR4 are believed to be most important in vivo (1, 12, 15–17, 21, 38, 65). Viruses (termed R5) that use CCR5 are involved in transmission between individuals, whereas variants that use CXCR4 (termed X4) often form during disease progression (13, 39, 53, 54). Following HIV-1 interaction with a coreceptor, a trimeric coiled coil is extended from the gp41 subunits into the cellular membrane (10). It is believed that membrane fusion is subsequently initiated by formation of a small pore (10).
Several extracellular regions of CCR5 have been implicated in its coreceptor function (2, 7, 31, 46, 51, 52). An amino-terminal region containing sulfated tyrosines at Y3, Y10, Y14, and Y15 is critical for gp120 binding and infectivity (18–20, 31, 49). Previously, we identified a Y14N polymorphism in the CCR5 of African green monkeys that severely disrupts HIV-1 coreceptor activity (31). Another important region occurs in extracellular loop 2. Wild-type African green monkey CCR5 differs from human CCR5 by a G163R substitution that severely inhibits R5 gp120 binding and HIV-1 infections without affecting chemokine binding or signaling (56). Monoclonal antibody 2D7 recognizes a nearby epitope in extracellular loop 2 of CCR5 that includes K171 and E172, and it also inhibits R5 gp120 binding and HIV-1 infections (36, 43, 56, 62). The results of studies of CCR5 chimeras are also compatible with these conclusions (2, 7, 31, 52, 60).
Information concerning the effects of CCR5 cell surface concentrations on its coreceptor activity would potentially elucidate the dynamic cell surface interactions that control HIV-1 infections and disease. Although natural variation in CD4 and coreceptor concentrations and changes in their levels during activation and differentiation of T lymphocytes and macrophages correlate with cellular susceptibilities to HIV-1 infections (41, 45, 59, 63), the basic mechanisms that underlie these effects have not been analyzed. Indeed, standard assays for coreceptors have employed transiently transfected cell populations that contain unknown and heterogeneous quantities of CD4, CCR5, and/or CXCR4 (2, 7, 8, 18, 19, 31, 46, 49, 51, 52, 56, 60). In a previous quantitative study we found that CCR5 functions more efficiently when cell surface concentrations of CD4 are increased (48). Thus, in human HeLa-CD4 cells (clone HI-J) that contain a large quantity of CD4 (ca. 1.5 × 105 CD4 molecules/cell) (47) a low trace threshold of wild-type CCR5 (ca. 103 CCR5 molecules/cell) is sufficient for maximally efficient R5 HIV-1 infections (48). In contrast, at a lower CD4 concentration, larger amounts of CCR5 are necessary to achieve the same high efficiency of infection. These results were consistent with the idea that HIV-1 initially adsorbs onto HeLa-CD4 cells and reversibly binds CD4 to form virus-(CD4)n complexes, where n is a positive function of CD4 concentration. Since gp120-CD4 interactions induce exposure of coreceptor binding sites (32, 33, 50, 58, 61), the number of these sites also increases as a function of CD4 concentration. Consequently, at higher CD4 concentrations, a lower concentration of CCR5 is required to form the assemblages necessary for infection. These and other results strongly suggest that coreceptors generally function after HIV-1 adsorbs onto cell surfaces (e.g., see references 32, 33, 40, 50, 58, and 61) and that they reversibly associate with the virus in a concentration-dependent manner to mediate infections.
Unfortunately, because wild-type CCR5 functions as an efficient coreceptor at barely detectable trace concentrations, we were unable in our previous study (48) to accurately determine the relationship between its concentration and its coreceptor function. To overcome this limitation in the present study, we have quantitatively analyzed the efficiencies of R5 HIV-1 infections of HeLa-CD4 (clone HI-J) cells as functions of the cell surface concentrations of wild-type CCR5 and of the mutant proteins CCR5(Y14N) and CCR5(G163R). Because these mutant proteins were highly attenuated, much larger concentrations were required for maximally efficient infections. This enabled us to readily isolate cell clones that expressed these CCR5s in discrete amounts over a broad concentration range and to more accurately measure the relationships between CCR5 concentrations and R5 HIV-1 infectivities. Interestingly, the resulting curves had sigmoidal shapes that were all quantitatively compatible with the hypothesis that fusion of the viral and cellular membranes requires the cooperation of multiple CCR5 molecules.
MATERIALS AND METHODS
Cells and viruses.
293T cells were from the American Type Culture Collection (Manassas, Va.) and maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 4.5 g of glucose per liter. All other cells were maintained in DMEM with 10% FBS. HeLa-CD4 (clone HI-J), and HeLa-CD4-CCR5 (clones JC.53 and JC.10) cells were described previously (28, 48). HeLa-CD4-CCR5 cells expressing the Y14N and G163R variants of human CCR5 were obtained by ligating the BamHI-XhoI fragments from the pcDNA3 expression vectors for the mutant CCR5s (31, 56) into pSFF (6) cut with the same enzymes. The retroviral vector SFF-CCR5 was prepared and used to infect HI-J cells as previously described (48). Cell clones were isolated by limiting dilution. Clones were screened for expression and clonality by immunofluorescence microscopy, and expression levels were quantitated by radioimmunoassay and quantitative immunofluorescent flow cytometry as described below.
The R5 SF162, JRFL, ADA, and BaL isolates of HIV-1 were obtained from the AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, National Institutes of Health (NIAID, NIH), and were contributed by Jay Levy, by Irvin Chen, by Howard Gendelman, and by Suzanne Gartner, Mikulas Popovic, and Robert Gallo, respectively. The JRCSF isolate was obtained as an infectious molecular clone, pYK-JRCSF, from the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH, and was contributed by Irvin Chen and Yoshio Koyanagi. High-titer stocks of these HIV-1 isolates were prepared as previously described (56).
Transient expression of CCR5s in HeLa-CD4 cells.
Single amino acid substitutions in human CCR5 were introduced in the pKS(+)-CCR5 vector (31) using a Quickchange site-directed mutagenesis kit (Stratagene, La Jolla, Calif.) as directed by the manufacturer. The coding region was sequenced to ensure that only the desired mutation was introduced, excised with BamHI and XhoI, and ligated into pcDNA3 (Invitrogen Corp., San Diego, Calif.). HI-J cells were transiently transfected with the pcDNA3-CCR5 expression vectors using Superfect reagent (Qiagen Inc., Santa Clara, Calif.) according to the manufacturer's instructions. Expression of mutant CCR5 cells was confirmed after 48 h by immunofluorescent flow cytometry, and coreceptor function was determined by focal-infectivity assay as described below.
Focal-infectivity assays.
The focal-infectivity assay (11) was used to titer preparations of R5 HIV-1. Briefly, cells were plated at 5 × 103 cells/well in 48-well cluster plates. Twenty-four hours later the cells were pretreated with DEAE-dextran (8 μg/ml) at 37°C for 20 min and then incubated with 0.1 ml of virus diluted 10-, 100-, or 1,000-fold in DMEM–0.1% FBS at 37°C. After 2 h the cells were fed with 1 ml of DMEM–10% FBS and incubated at 37°C for 72 h. The cells were then fixed in ethanol, and infected foci were visualized by an immunoperoxidase assay (11), using as the primary antibody the filtered (pore size, 0.45 μm) supernatant from the anti-p24 hybridoma 183-H12-5C (AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH; contributed by Bruce Chesebro and Hardy Chen). Titers on specific cell lines were determined by counting the stained foci in the well with the lowest dilution that contained less than 200 foci/well and multiplying by the dilution factor. Titers were normalized to those on JC.53 cells expressing high levels of CD4 and CCR5, where the titers of the concentrated stocks were approximately 106 focus-forming units (FFU)/ml for JRCSF and 5 × 105 FFU/ml for BaL (or 100 and 50 foci/well at the 1,000-fold dilution). The lower limit of detection was 100 FFU/ml or approximately 0.01 to 0.05% of the JC.53 titer. Syncytia were scored by microscopic examination of the foci in a given well for the presence of multinucleated cells as described previously (30).
Measurement of cell surface CCR5.
To estimate the CCR5 antigen density on HeLa-CD4-CCR5 cells, a Dako Qifikit (Dako Corporation, Carpenteria, Calif.) for quantitative analysis of indirect immunofluorescence staining in flow cytometry was used according to the manufacturer's instructions. Cells were lifted in 8 mM EDTA–0.9% (wt/vol) NaCl. Primary-antibody staining was with 25 μg of mouse anti-human CCR5 monoclonal antibody 2D7 (PharMingen, San Diego, Calif.) per ml. The FACScalibur flow cytometry system (Becton Dickinson Immunocytometry Systems, San Jose, Calif.) was used to record all flow cytometry data. The results were processed according to the manufacturer's instructions. In addition to flow cytometry, a quantitative radioimmunoassay was used to quantitate cell surface CCR5. Anti-CCR5 monoclonal antibody 2D7 (5 μg/ml) was incubated with cells seeded at 2 × 105 cells/well in 24-well cluster plates for 1.5 h at 37°C. The cells were then sequentially incubated with a 1:250 dilution of goat anti-mouse immunoglobulin G serum (Organon Teknika, Durham, N.C.) and a 1:250 dilution of 125I-protein A (0.4 μCi/ml, 2 to 10 μCi/μg; NEN Life Science Products, Boston, Mass.) at 37°C for 1 h each. Cells were washed, lysed in 0.1 N NaOH, and counted in a gamma counter. Counts were normalized to the protein concentration determined by the Coomassie blue method (Bio-Rad Laboratories, Hercules, Calif.). In each experiment, cell lines were assayed in duplicate wells and multiple cell lines from the HeLa-CD4-CCR5 panel previously described (48) were assayed to ensure the colinearity of this assay with that previously described using a saturating concentration of a polyclonal rabbit anti-CCR5 serum (48).
[35S]sulfate labeling, glycosidase treatment, and electrophoretic analysis of CCR5.
293T cells (2 × 106) in 100-mm-diameter dishes were transfected with the appropriate pcDNA3-CCR5 construct by using Superfect reagent as instructed by the manufacturer, and labeling was performed 24 h later. Metabolic labeling with [35S]sulfate was performed as described previously (20) with the following modifications. Approximately-75%-confluent cultures of transfected 293T cells or HeLa-CD4-CCR5 (clone JC.53) cells were incubated with 1 mCi of [35S]sulfate (NEN Life Science Products) for 16 h in 8 ml of sulfate-free minimum essential medium with 5% FBS dialyzed against phosphate-buffered saline (PBS). The minimum essential medium was prepared from a Selectamine kit (Life Technologies, Grand Island, N.Y.) with a modified 10× Eagle's balanced salt solution with MgCl2 substituted for MgSO4. After being labeled, the crude cellular membrane fraction was prepared as described previously (31). Membrane proteins were solubilized in lysis buffer (0.5% NP-40 in PBS with protease inhibitors) and immunoprecipitated with 5C7 mouse-anti human CCR5 monoclonal antibody (AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH; contributed by LeukoSite, Inc.) and then with protein A-Sepharose (Sigma, St. Louis, Mo.) as described previously (20). Alternatively, labeled cells were solubilized in lysis buffer, the nuclei were removed by centrifugation at 500 × g for 20 min, and CCR5 was immunoprecipitated with 5C7. In both cases, the immunoprecipitated proteins were separated by 0.1% sodium dodecyl sulfate (SDS)–8% polyacrylamide gel electrophoresis (PAGE) and separated proteins were transferred to NitroPure nitrocellulose membranes (Micron Separations, Inc., Westboro, Mass.). The 35S-labeled proteins were detected by autoradiography of the nitrocellulose membranes. In other experiments, membrane fractions from unlabeled cells were electrophoresed and transferred to nitrocellulose and CCR5 was detected by Western immunoblotting with rabbit anti-human CCR5 serum and chemiluminescence reagents as described previously (31). For experiments in which O-glycosidase treatment was required, CCR5 immunoprecipitates were resuspended in 30 μl of buffer (PBS, 1% Triton X-100, 0.5% SDS) and treated at 37°C for 2 h with a cocktail of glycosidases containing 50 mU of neuraminidase, 3 mU of O-glycosidase, 13 mU of β-galactosidase, and 100 mU of N-acetyl-β-d-glucosaminidase. N-Glycosidase treatment was performed by resuspending membrane preparations in 4 μl of 1% SDS in PBS and denaturing by heating to 65°C for 10 min. One unit of the peptide N-glycosidase F was added in 40 μl of 1% NP-40 in PBS and incubated at 37°C for 4 h. All enzymes were from Roche Molecular Biochemicals, Indianapolis, Ind.
gp120 and MIP1β binding.
The competition of [125I]MIP1β binding by BaL gp120 or by unlabeled MIP1β was performed essentially as previously described for 293T cells transiently transfected with CD4 and CCR5 (56). HeLa-CD4-CCR5 and HeLa-CD4-mutant CCR5 clones were seeded at 5 × 104 cells per well of a 24-well cluster plate and incubated 24 h later with 3 nM [125I]MIP1β (2,200 μCi/mmol; NEN Life Science Products); purified BaL gp120 (56) or unlabeled MIP1β (Peprotech, Rocky Hill, N.J.) was added, as required, in DMEM with 10% FBS for 1 h at 37°C, and the clones were washed three times and lysed in 0.1 M NaOH. The lysates were analyzed in a gamma counter.
Mathematical analysis of the infectivity results.
Our basic model is closely correspondent with our infectivity protocol, in which viruses are adsorbed for 2 h onto cell clones that contain different amounts of CCR5 but a constant amount of CD4, and the foci of infection are counted after 72 h. Thus, we measured the integrated total quantity of infections that occurred in the cultures rather than the rates of infection. This requires the integration of rate equations over the period of our assays. Nevertheless, as demonstrated below, our equations and results are also compatible with a quasikinetic interpretation (also, see Discussion). When preparations of HIV-1 are incubated with HeLa-CD4 (clone HI-J) cultures for 2 h at 37°C, only a small fraction of the infectious virions adsorb onto the cells. Thus, removing the virus-containing medium and placing it onto a fresh culture results in the same titer on both culture dishes (28). These titers are directly proportional to the concentration of virus, to the numbers of cells in the cultures, and to the adsorption times (28). As described in the introduction, our previous data (48) and other evidence (32, 33, 50, 58, 61) strongly imply that HIV-1 binds to coreceptors secondarily after the virus has initially adsorbed onto the cells to form virus-(CD4)n complexes. Although n is unknown, it is believed that immature HIV-1 virions contain 72 gp120-gp41 trimers and that gp120 shedding reduces the quantity of this glycoprotein in the mature infectious virions (23, 34, 44). In addition, the cell surface binding of CCR5 onto adsorbed R5 HIV-1 occurs in a concentration-dependent manner (48). Accordingly, we assume that R5 HIV-1 initially adsorbs onto all HI-J-derived clones of HeLa-CD4 cells in equal numbers and that infection requires a reversible cell surface interaction of the adsorbed virus with CCR5 that can be approximated as follows:
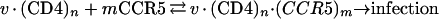 |
1 |
where v · (CD4)n is the adsorbed virus complexed with CD4, and m is the number of CCR5 molecules that associate with these virions to form fusion-competent complexes. Therefore, the rate of infection at any instant is
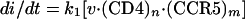 |
2 |
where k1 is the rate constant for the final reaction and di/dt is the rate of infection. Because of the low multiplicities of infection in our assays (less than 0.02 virus/cell), the attachment of virus should not significantly reduce the cell surface concentrations of CD4 or CCR5 which are not complexed to cell-associated virions. Thus, it is reasonable to assume that the concentration of free CCR5 is approximately equal to the total concentration of CCR5 expressed by a given cell clone. Based on this assumption, it follows from the equilibrium in equation 1 that
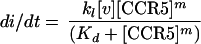 |
3 |
where Kd is the dissociation constant for the equilibrium in equation 1, [CCR5] is the total concentration of CCR5 expressed by a given cell clone, and [v] is the concentration of total infectious virions on the cell surfaces (i.e., [v] = [v · (CD4)n] + [v · (CD4)n · (CCR5)m]).
At any time (t) after initial adsorption, [v] declines due to infection and potentially also to inactivation (and/or dissociation). Hence,
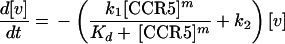 |
4 |
where k2 is the rate constant for inactivation. Integration of equation 4 gives
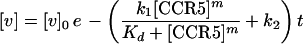 |
5 |
where [v]0 is the concentration of infectious virus that is initially adsorbed onto the cell surfaces.
Incorporation of equation 5 into equation 3 gives
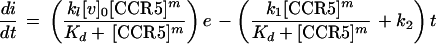 |
6 |
Integrating equation 6 between the initial uninfected state at time zero and the final state at time infinity (we can assume that all of the viable virus has been removed from the cell surfaces long before the cultures are fixed at 72 h) gives the result
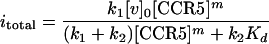 |
7 |
It is useful to define the term E = k1/(k1 + k2). From equation 7 it follows that itotal equals [v]0E at high levels of CCR5. Thus, E is the fraction of initially adsorbed virus that successfully infects the culture when the specific CCR5 is not limiting. We normalized the infectivity data for each assay relative to the infectivity obtained with the control culture (clone JC.53) that expresses a large amount of wild-type CCR5 and thus where itotal is [v]0Ewt (wt indicates the wild-type value). This gives
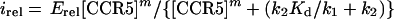 |
8 |
where irel is the normalized infectivity (relative to the virus titer in the control cells) for the assay and Erel is E/Ewt. Thus, Erel is the asymptote of the sigmoidal plot of irel versus [CCR5] for the specific panel of cell clones being assayed. Taking the logarithm of equation 8 gives
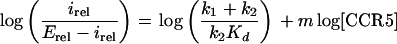 |
9 |
Consequently, the model predicts that a plot of log[irel/(Erel − irel)] versus log[CCR5] should give a straight line with a slope m and an intercept of log[(k1 + k2)/(k2Kd)]. It is important to realize that use of equations 8 and 9 is valid regardless of the time period used for virus adsorption. The 2-h adsorption time employed in our protocol could be considered to be the sum of multiple brief periods, each of which would of necessity yield the same irel versus [CCR5] plot. This occurs because the infectivity measurements are normalized relative to values obtained using highly susceptible JC.53 cells that express wild-type CCR5 and because all of the cell clones, including JC.53, adsorb the same quantities of HIV-1 (see above).
Although we have assumed in equation 4 that inactivation of cell surface-adsorbed virus might affect all species of virus, it could alternatively be postulated that inactivation would be accelerated in the fusion-competent assemblages that contain at least m CCR5s. Indeed, such an assumption has been considered previously for influenza A virus (42). According to this idea, assembly of a fusion-competent complex destabilizes the virus and sensitizes it to either infection or inactivation. This hypothesis predicts that irel equals Erel at all CCR5 concentrations. However, our results clearly show that irel values depend strongly on CCR5 concentrations (see Results). Hence, our results support the assumption inherent in equation 4.
It is important to realize that a kinetic analysis of the initial rates of infection could be done after a brief period of virus adsorption using equation 3 as an approximation. Normalizing such initial rates relative to the values observed with JC.53 cells that contain a large concentration of wild-type CCR5 would give the equation (di/dt)rel = k1,rel[CCR5]m/(Kd + [CCR5]m), which has a form similar to that of equation 8. It would be predicted that the asymptotes of the sigmoid plots of (di/dt)rel versus [CCR5] would equal k1,rel. Hence, a plot of log{(di/dt)rel/[(di/dt)rel,max − (di/dt)rel]} versus log[CCR5], where max indicates the asymptote of the sigmoidal data plot, would give a straight line with a slope m and an intercept of log(1/Kd). This result is strikingly similar to that of the analysis of the cumulative infection data as shown above in equation 9. This implies that the graphical methods used to analyze the sigmoid curves obtained from kinetic measurements or from cumulative infectivity data would be identical and would be expected to give the same estimates of m. The only difference in the graphical analysis would be the meaning of the intercepts. Although kinetic analyses would also be useful, they are more difficult because the infections would have to be synchronized and because estimates of di/dt require several infectivity assays. For these reasons, we used irel measurements and equation 9 for this initial investigation. These considerations clearly show that our analytical methods as currently applied could not distinguish between alternative models that yield mathematical solutions with the general forms of equations 3 and 8. Hence, although our data appears to be consistent with equation 8 (see below), it is also compatible with a quasikinetic interpretation (see Discussion).
RESULTS
Mutagenesis of CCR5 at positions Y14 and G163.
To evaluate the importance of CCR5 amino acids Y14 and G163 for R5 HIV-1 infections, we constructed several mutant CCR5s with substitution mutations at these positions and we tested their coreceptor activities by a standard transient-transfection–focal-infectivity assay with HeLa-CD4 (clone HI-J) cells (28, 31). These mutant CCR5s were all expressed at similar levels on cell surfaces (see the legend to Fig. 1). Figure 1 shows the normalized titers of infection mediated by these CCR5s relative to that of wild-type CCR5 for five different R5 isolates of HIV-1. In contrast to the relatively large functional differences revealed by the studies described below, most of these substitutions caused only slight two- to threefold reductions in these assays. Similar assays of alanine-scanning mutant constructs have previously also found only small effects, except with mutant constructs in which several sites in the critical amino-terminal region were mutated (8, 18, 24, 49). Despite the limitations of this methodology, the data in Fig. 1A confirm the importance of CCR5 residues Y14 and G163 in R5 HIV-1 infections. Y14F eliminates the anionic sulfated hydroxyl group and causes significant attenuation, and further inhibitions are caused by A, Q, or N substitutions, whereas replacement with the alternative anionic amino acid E has the least effect. At position 163, the greatest inhibitions occur when G is replaced with the two largest amino acids assayed, R and W.
FIG. 1.

Infections of HeLa-CD4 (clone HI-J) cells transiently transfected with site-directed mutant constructs of CCR5. Site-directed mutant constructs of CCR5 with alterations at Y14 and G163 in a mammalian expression vector were transiently transfected into HI-J cells. Titers of the five different R5 HIV-1 isolates indicated were determined after 48 h by a focal-infectivity assay which uses p24gag immunoperoxidase staining to detect foci. The titers are normalized to those determined on cells transfected with the wild-type CCR5 expression vector in the same assay. The infection assay was repeated three or four times, with error bars representing the standard errors of the means (SEM). Expression was measured by immunofluorescent flow cytometry, using 2D7 as the primary antibody. The percentage of transfected cells ranged from approximately 5 to 30% between assays; however, values were similar for different constructs within a given assay. The transfection efficiencies for one representative assay were as follows: 8.9% for the wild type, 10.8% for the Y14A construct, 9.0% for the Y14E construct, 10.3% for the Y14F construct, 10.1% for the Y14N construct, 10.0% for the Y14Q construct, 7.0% for the G163A construct, 10.1% for the G163E construct, 8.3% for the G163K construct, 7.0% for the G163P construct, 7.2% for the G163R construct, and 7.3% for the G163W construct. The mean expression level in transfected cells was approximately 6 × 104 CCR5 molecules/cell, with values being distributed over the range of 3 × 103 to 4 × 105 CCR5 molecules/cell, and the mutations did not alter the expression levels (data not shown).
Properties of the CCR5(Y14N) and CCR5(G163R) proteins.
As shown in Fig. 2A and B, we have confirmed the sulfation of CCR5 by labeling cells with [35S]sulfate, followed by immunoprecipitation of CCR5 and electrophoresis in the presence of 0.1% SDS. Consistent with previous evidence for tyrosine sulfation and O-linked glycosylation of CCR5 (20), the sulfate labeling of CCR5 was not reduced by enzymatic removal of O-linked oligosaccharides (e.g., see Fig. 2B). Interestingly, CCR5(Y14N) molecules are relatively heterogeneous in size, with approximately 60 to 70% of the molecules having larger apparent Mrs than that of wild-type CCR5 or CCR5(G163R). Accordingly, this mutation converts the YYT sequence at positions 14 to 16 into an NYT consensus site for N-linked glycosylation, and endoglycosidase F converts the larger CCR5(Y14N) components into the smaller-sized species (Fig. 2C). As discussed below, N-linked glycosylation of CCR5(Y14N) may have a small effect on our assays using this mutant protein.
FIG. 2.

Analysis of the sulfation of wild-type CCR5, CCR5(G163R), and CCR5(Y14N) and the N glycosylation of CCR5(Y14N). (A) 293T cells transfected with the indicated CCR5 plasmids were metabolically labeled with [35S]sulfate. CCR5 was immunoprecipitated from the crude membrane fraction with mouse anti-human CCR5 monoclonal antibody 5C7, separated by 0.1% SDS–8% PAGE, and transfered to a nitrocellulose membrane. 35S-labeled proteins were detected by autoradiography. Mock indicates cells transfected with the vector alone. wt, wild type; IP, immunoprecipitate. (B) [35S]sulfate-labeled CCR5 from HeLa-CD4-CCR5 cells (clone JC.53) or transfected 293T cells were immunoprecipitated from whole-cell lysates and treated with an O-glycosidase cocktail or treated under the same conditions in the absence of enzyme as indicated (see Materials and Methods). The treated immunoprecipitates were separated by SDS-PAGE, and labeled CCR5 was detected by autoradiography. (C) Membrane fractions from transfected 293T cells were separated by SDS-PAGE and analyzed by Western immunoblotting with polyclonal rabbit anti-human CCR5 serum. Samples were treated prior to SDS-PAGE with the peptide N-glycosidase F (see Materials and Methods) or mock treated as indicated. The polyclonal serum was raised against an amino-terminal peptide including the Y14 region (31) and detects the N-glycosylated form of CCR5(Y14N) (N-glyco CCR5) inefficiently. In contrast, the enzymatically deglycosylated form is more readily detected (compare the two lanes on the right side). In all panels the mobilities of protein standards with the indicated Mrs (in thousands) are shown on the left of the panels.
We also analyzed the effects of the G163R and Y14N mutations on interactions of CCR5 with MIP1β and with a previously characterized gp120 derived from the BaL isolate of R5 HIV-1 (29, 56). Interestingly, MIP1β bound strongly and specifically onto cells that expressed these mutant CCR5s. Moreover, the apparent affinities of MIP1β for the wild-type and mutant CCR5s were identical, as indicated by the displacement of [125I]MIP1β caused by increasing concentrations of unlabeled MIP1β (see Fig. 3B). However, the maximum level of MIP1β binding relative to the total quantity of cell surface CCR5 was lower for CCR5(Y14N) than for wild-type CCR5 or CCR5(G163R). Thus, in a representative experiment described in the legend of Fig. 3, the maximum binding of MIP1β normalized to the binding of 2D7 monoclonal antibody was 0.7 for wild-type CCR5, 0.7 for CCR5(G163R), and 0.3 for CCR5(Y14N). These data imply that, relative to wild-type CCR5 and CCR5(G163R), approximately 60% fewer of the CCR5(Y14N) molecules on the cell surfaces are able to interact with MIP1β, and we presume that these inactive molecules are the N-glycosylated components.
FIG. 3.

Binding of gp120 and MIP1β to wild-type (wt), G163R, and Y14N CCR5s expressing HeLa-CD4 clones. (A) Cells were incubated with 3 nM [125I]MIP1β in the presence of increasing concentrations of purified BaL gp120. The [125I]MIP1β bound is plotted as a percentage of the [125I]MIP1β bound in the absence of gp120 ± the standard deviation. The cell lines used were JC.53 (wild-type CCR5), YB8 [CCR5(Y14N)], and JGR.H11 [CCR5(G163R)]. For expression levels of CCR5 on these clones, see Table 1. (B) The same clones used to obtain the results shown in panel A were incubated with 3 nM [125I]MIP1β in the presence of increasing concentrations of unlabeled MIP1β, and bound [125I]MIP1β is plotted as a percentage of binding in the absence of unlabeled MIP1β ± the standard deviation. These data were analyzed by the Scatchard method as previously described (48) (analysis not shown). The estimates of the Km and Bmax values generated by this analysis for each cell line are, respectively, 18 ± 4 nM and 1.4 × 105 ± 0.3 × 105 MIP1β molecules/cell for JC.53 cells, 22 ± 8 nM and 0.5 × 105 ± 0.2 × 105 MIP1β molecules/cell for YB8 cells, and 8 ± 2 nM and 0.7 × 105 ± 0.2 × 105 MIP1β molecules/cell for JGR.H11 cells. These values are the dissociation constants and the numbers of binding sites per cell, respectively.
In agreement with previous reports (56, 58, 61), we also found that BaL gp120 was able to competitively displace [125I]MIP1β from the surfaces of cells that contained CD4 and wild-type CCR5 (Fig. 3A). In contrast, the displacement of [125I]MIP1β from CCR5(G163R) was highly attenuated, in agreement with previous evidence (56), and we were unable to detect any significant gp120-induced displacement of this labeled chemokine from cells that expressed CCR5(Y14N). These results suggest that both the Y14N and G163R mutations reduce CCR5 affinity for R5 gp120.
Dependencies of R5 HIV-1 infections on cell surface concentrations of wild-type CCR5, CCR5(Y14N), and CCR5(G163R).
Previously, we used the HI-J clone of HeLa-CD4 cells (ca. 1.5 × 105 CD4 molecules/cell) (47) to construct derivative clones that stably express discrete amounts of wild-type CCR5 over a broad range of concentrations (48); we have now employed the same methods to construct panels of HeLa-CD4-CCR5(Y14N) and HeLa-CD4-CCR5(G163R) cell clones. Table 1 shows the CCR5 expression levels on these cell clones as well as data for two previously characterized clones (JC.10 and JC.53) that express widely different amounts of wild-type CCR5 (48).
TABLE 1.
CCR5-expressing clones derived from HI-J cells
| Cell line | CCR5 expressiona (104 molecules/cell ± SEM) |
|---|---|
| CCR5(Y14N) clones | |
| JYN.8 | 4.2 ± 0.5 |
| JYN.5 | 5.6 ± 0.4 |
| JYN.4c | 6.0 ± 0.4 |
| JYN.15 | 8.3 ± 0.1 |
| YD2 | 9.4 ± 0.6 |
| JYN.2 | 10 ± 0.7 |
| JYN.3 | 10 ± 0.2 |
| JYN.14 | 13 ± 0.6 |
| JYN.2-19b | 16 ± 1.1 |
| YB8bc | 17 ± 1.4 |
| JYN.2-15b | 17 ± 1.6 |
| JYN.2-27b | 25 ± 3.9 |
| CCR5(G163R) clones | |
| JGR.H5 | 1.8 ± 0.3 |
| JGR.H4c | 1.9 ± 0.2 |
| JGR.L2 | 2.3 ± 0.3 |
| JGR.H3 | 2.7 ± 0.2 |
| JGR.L10 | 3.2 ± 0.2 |
| JGR.L6 | 4.1 ± 0.2 |
| JGR.H7 | 5.7 ± 0.2 |
| JGR.H11bc | 9.8 ± 1.7 |
| Wild-type CCR5 clones | |
| JC.10b | 0.6 ± 0.1 |
| JC.53b | 19 ± 1.3 |
Expression was determined by radioimmunoassay as described in Materials and Methods except where noted. The background binding was determined on HI-J cells, which do not express CCR5, and was subtracted from the values shown. The background was equivalent to values obtained when the anti-CCR5 monoclonal antibody 2D7 was omitted and was approximately 15% of the level of binding measured on JC.10 cells. Cell lines on which the number of 2D7 antibody binding sites had been determined by quantitative flow cytometry were included in the same radioimmunoassay and used to generate a standard curve to convert the binding values to the equivalent number of antibody binding sites. Values are the means of results of four or five independent assays ± SEM.
Expression of CCR5 in these clones was determined by quantitative immunofluorescent flow cytometry as described in Materials and Methods. The background fluorescence was determined on HI-J cells, which do not express CCR5, and was subtracted from the values shown. The background was equivalent to values obtained when the anti-CCR5 monoclonal antibody 2D7 was omitted and was approximately 30% of the level of fluorescence measured on JC.10 cells. The values obtained were converted to the number of 2D7 antibody binding sites by using a calibration curve generated by beads conjugated with known quantities of mouse immunoglobulin G (see Materials and Methods). Values are the means of results of three to seven independent assays ± SEM.
The expression of CD4 was monitored in these cell clones by quantitative immunofluorescent flow cytometry. The CD4 levels did not differ from those of the parental HeLa-CD4 (clone HI-J) cells (results not shown).
We measured infectivities of the R5 HIV-1 isolates BaL and JRCSF using the cell clones listed in Table 1. Each cell clone was analyzed three to eight times with each virus in parallel with identical assays of the JC.10 and JC.53 cells that express wild-type CCR5, and the infectivities were normalized relative to the titers of the same viruses in the JC.53 cells. As illustrated by the representative results in Fig. 4A, the titers in the JC.53 and JC.10 control cells were not significantly different despite the 32-fold difference in their concentrations of CCR5, confirming our previous conclusion that a trace of wild-type CCR5 suffices for maximally efficient R5 HIV-1 infections (48). This is consistent with other evidence that all HI-J-derived clones initially adsorb equal amounts of HIV-1 and that wild-type CCR5 then functions efficiently even at low concentrations to facilitate the entry process (48). In contrast, efficient infections of these viruses in the CCR5(G163R) and CCR5(Y14N) panels required much higher concentrations of the coreceptor (Fig. 4), consistent with evidence in Fig. 3 that these mutations reduce coreceptor affinities for R5 gp120. The infectivity curves were half-maximal when the cells expressed approximately 3 × 104 CCR5(G163R) molecules/cell, which is at least 30 times higher than the threshold concentration of wild-type CCR5 required for the same efficiency of infection (48). In the Y14N clonal panel, the midpoints of the infectivity curves (ca. 1.3 × 105 to 1.7 × 105 CCR5(Y14N) molecules/cell) were shifted even further toward high concentrations. These functional differences between the wild-type and mutant CCR5s are extremely large compared with the differences observed in Fig. 1. In contrast to these results with R5 isolates of HIV-1, the presence or quantities of these CCR5s on the surfaces of the HI-J-derived cell clones had no effect on titers of the X4 control virus NL4-3 (48; also data not shown). Two other features of these results are notable. First, the plots appear to have sigmoidal shapes (Fig. 4). This was confirmed by experiments using other R5 HIV-1 viruses (see below). It implies that multiple CCR5 molecules may be required for R5 HIV-1 infections. Second, the sigmoidal curves plateau at different efficiencies of infection.
FIG. 4.

(A) Infections mediated by CCR5(G163R)-expressing clonal cell lines. The curves with open symbols show the levels of HIV-1 infections of HeLa-CD4-CCR5(G163R) cells as determined by a focal-infectivity assay which uses p24gag immunoperoxidase staining to detect foci. The filled symbols indicate infections of HeLa-CD4-CCR5 cells expressing wild-type (wt) CCR5 (clones JC.10 and JC.53, with ca. 6 × 103 and 2 × 105 CCR5 molecules/cell, respectively). The isolates used were JRCSF (diamonds) and BaL (squares). The titer on each cell clone was determined by dilution of virus stocks and multiplying the number of foci observed by the dilution factor as described in Materials and Methods. The relative infectivity was determined by dividing the titer determined on a given cell clone by the titer determined on HeLa-CD4-CCR5 (clone JC.53) cells with the same virus stocks in the same assay. Thus, the relative infectivity of the JC.53 cell clone shown in this graph was 1 by definition. Each point is the average of results of five to seven experiments, and the error bars represent the SEM. (B) Infections mediated by CCR5(Y14N)-expressing cell lines. The isolates used were JRCSF (diamonds) and BaL (squares). The titers were normalized as described for panel A. Each point is the average of results of three to eight experiments, and the error bars represent SEM.
Results of microscopic analyses of the immunoperoxidase-stained foci of infection in these cultures were consistent with the hypothesis that the limiting step in utilization of the Y14N mutant CCR5 occurred at or before the membrane fusion step of infection (Fig. 5). Specifically, the foci of infection in the control JC.10 and JC.53 cultures that have wild-type CCR5 consisted mostly of large syncytia. In contrast, the infected foci in cultures with CCR5(Y14N) consisted of small syncytia or single cells. This was especially striking in the clones, such as those with YD2, that contained low amounts of CCR5(Y14N), in which the foci were principally single infected cells. Although less striking, the sizes and frequencies of syncytia were also significantly correlated with cell surface concentrations of CCR5(G163R) (P ≤ 0.02, n = 4).
FIG. 5.

Syncytium formation in infected cultures is affected by Y14N and G163R substitutions. The number of foci that contained syncytia were counted from four independent experiments for each virus (JRCSF and BaL) on two cell lines from each CCR5 panel. The wild-type cell lines were JC.10 and JC.53, the Y14N cell lines were YD2 and YB8, and the G163R cell lines were JGR.H4 and JGR.H11. For expression levels of wild-type or mutant CCR5 in each cell line, see Table 1. The percentages of foci that contained syncytia from each assay were averaged and are shown ± SEM. ND, not done (no value is reported for the infections of YD2 cells by the JRCSF isolate because insufficient foci were available to accurately determine the percentage of syncytia in any given experiment). The percentage of foci that contained syncytia determined on a given cell clone was independent of the absolute number of foci or the virus dilution within a given experiment (results not shown). The number of foci observed for each cell line in a single experiment ranged from approximately 50 to 200. Qualitatively similar results were obtained for the number of nuclei per syncytium (results not shown).
Quantitative evaluation of the infectivity data.
The mathematical modeling that is described in Materials and Methods provides a simple quantitative approach for interpreting the infectivity results shown in Fig. 4. This model predicts that a plot of log[irel/(Erel − irel)] versus log[CCR5] should give a straight line with a slope m, where irel is the titer of the virus in a cell clone divided by the titer in the control JC.53 cells that express wild-type CCR5, Erel is the asymptote for each sigmoid curve in Fig. 4, and m is the number of CCR5s required to mediate R5 HIV-1 infection.
The data for several infections of our clonal panels are plotted in this manner in Fig. 6, and a compilation of the midpoints of the sigmoidal curves (50% effective concentrations [EC50s]) and of the estimated m values is shown in Table 2. Because the efficiencies of JRCSF virus infection in the majority of the CCR5(Y14N) cell clones were near or below the limit of detection (Fig. 4), these results were not analyzed for Fig. 6. However, we isolated three adapted variants of the JRCSF virus that were better able to infect the CCR5(Y14N) clonal panel (unpublished results), and the data obtained using these adapted variants are included in Fig. 6 and Table 2. Consistent with our model, the data points for different viruses in different clonal panels appear to fall on straight lines. Moreover, the slopes of these straight lines fall within a narrow range of values (ca. 4 to 6). This is a prediction of the model because the number of CCR5s (m) required to form a fusion-competent complex should be independent of the specific R5 isolates of HIV-1 or of the CCR5 that is analyzed.
FIG. 6.

Mathematical analysis of infections mediated by CCR5(G163R) and CCR5(Y14N). The data in Fig. 4 were analyzed according to the mathematical model (equation 9) derived in Materials and Methods. The relative infectivity at the highest concentration of mutant CCR5 that was assayed was used as the value of Erel. These Erel values for viruses assayed on the G163R panel were 0.87 and 0.74 for the JRCSF and BaL viruses, respectively. The Erel values for viruses assayed on the Y14N panel were 0.10, 0.37, 0.46, and 0.97 for the BaL, JRCSF (no. 1 YB8-adapted [YB8#1]), JRCSF (no. 2 YB8-adapted [YB#2]), and JRCSF (JYN.4-adapted) viruses, respectively. All irel values which were defined were plotted as log[irel(Erel − irel)] versus log[CCR5]. Only data points obtained when [CCR5] was at subsaturating concentrations can be plotted in this analysis, because where Erel equals irel, equation 9 is undefined. In addition, not all of the cell lines shown in Table 1 were assayed with the CCR5(Y14N)-adapted variants of JRCSF described below. Therefore, the number of data points in Fig. 6 is not the same for each virus isolate but represents all of the informative data available. The values derived from the CCR5(G163R) panel are shown with filled symbols; circles indicate BaL values, and squares indicate JRCSF values. From the CCR5(Y14N) panel (open symbols), the isolates used were BaL (circles) and three independent isolates of JRCSF which have increased efficiencies of fusion (Erel) on the CCR5(Y14N) panel (YB8 and JYN.4 adapted [squares and triangles, respectively]) (unpublished results). The JRCSF-JYN.4-adapted isolate also exhibits an increased apparent affinity for CCR5(Y14N) (unpublished results), as demonstrated by the upward shift of the curve.
TABLE 2.
Mathematical analysis of infectivity dataa
| Clonal panel | Virus | R | m | EC50 (no. of CCR5 molecules/cell) |
|---|---|---|---|---|
| CCR5(G163R) | JRCSF | 0.91 | 5.4 ± 1.1 | 3.2 × 104 ± 0.3 × 104 |
| BaL | 0.92 | 3.0 ± 0.6 | 3.0 × 104 ± 0.2 × 104 | |
| CCR5(Y14N) | JRCSF(YB8-adapted virus 1) | 0.96 | 6.1 ± 0.7 | 1.4 × 105 ± 0.1 × 105 |
| JRCSF(YB8-adapted virus 2) | 0.96 | 6.5 ± 0.7 | 1.3 × 105 ± 0.1 × 105 | |
| JRCSF(JYN.4-adapted) | 0.83 | 4.3 ± 1.7 | 6.4 × 104 ± 0.7 × 104 | |
| BaL | 0.96 | 6.6 ± 0.7 | 1.2 × 105 ± 0.1 × 105 |
Data plotted in Fig. 6 were fit to equation 9 as described in Materials and Methods using Kaleidagraph software (Synergy Software, Reading, Pa.). The calculated linear correlation coefficients are reported in the R column. The EC50 value (the CCR5 concentration where irel equals Erel/2) is calculated based on EC50m of k2Kd/(k1 + k2), which allows the comparison of the midpoints of curves with different estimates of m. The EC50 and m values are reported ± SEM as calculated with Kaleidagraph.
DISCUSSION
Effects of coreceptor concentrations on HIV-1 infections.
We have attempted to quantitatively analyze the effects of CCR5 cell surface concentrations on its function as a coreceptor for R5 HIV-1 isolates. As described previously (48) and illustrated in Fig. 4, this was very difficult to do using wild-type CCR5, which efficiently mediates infections even at trace concentrations that were difficult to accurately measure. To overcome this limitation and to learn more about the functional roles of different CCR5 domains, we used the Y14N and G163R mutant CCR5s, which bind to R5 gp120s weakly (Fig. 3) and function as coreceptors only at relatively high and easily measured cell surface concentrations (Fig. 4). Interestingly, the relationships between viral infectivities and CCR5 concentrations were nonlinear functions with sigmoidal shapes that were altered in distinct ways by the mutations. Both mutations dramatically shifted the infectivity curves toward higher CCR5 concentrations, suggesting that infections require the concentration-dependent reversible association of CCR5s with the adsorbed virions. In addition, the Y14N mutation substantially reduced the maximum infectivities that were obtained at saturating coreceptor concentrations (i.e., the asymptotes in Fig. 4). These mutations also reduced the sizes and numbers of syncytia in the infectivity assays (Fig. 5), consistent with other evidence that they inhibit the membrane fusion process. The last-named results were less striking than the infectivity data (Fig. 4), in agreement with other indications that syncytial and infectivity assays are limited by different factors (see below).
Possible influence of HIV-1 inactivation (or dissociation) on infectivity assays.
Our results also strongly suggest that inactivation or dissociation of cell surface-adsorbed virions is a significant process that can influence HIV-1 titers, especially in cells that contain low concentrations of weak or mutant coreceptors. Because natural R5 HIV-1 isolates bind first to CD4 and only secondarily to CCR5 (32, 33, 50, 58, 61), it is very likely that all of the cell clones we used would initially adsorb the same quantity of virus. Indeed, the fact that R5 HIV-1 titers were identical in the JC.10 and JC.53 cells despite the enormous difference in their concentrations of wild-type CCR5 (Table 1 and Fig. 4) supports this idea. This conclusion is also strongly supported by a previous study of HIV-1 using HeLa cells (40) and by theoretical considerations (5, 25, 55). In essence, the HeLa-CD4 cells (clone HI-J) that we used function as polyvalent adsorbers to efficiently bind HIV-1 that diffuses into contact with the cell surfaces, and CCR5 then influences a postadsorption step of the infection pathway. Consequently, the relatively inefficient infections of many cell clones (Fig. 4) implies that inactivation or dissociation of cell surface-adsorbed virus must occur at a rate that significantly competes with infection. This inference is further discussed below.
Modeling the role of CCR5 in R5 HIV-1 infections.
Although our data are quantitatively compatible with the specific model derived in Materials and Methods, it is also clear from that derivation that our results would also be consistent with a related quasikinetic interpretation. Consequently, in this section we will describe these alternative models.
The model implicit in the derivation of equation 9 is based on the ideas mentioned above that R5 HIV-1 virions which adsorb onto the surfaces of CD4-positive cells begin to reversibly associate with CCR5 to form oligomeric complexes and that a complex containing m CCR5s is necessary for infection. If the CCR5 concentration is very low or if the coreceptor has a mutation that reduces its affinity for the virus, only a small proportion of the virions would occur in infection-competent complexes at any instant and the infection would proceed slowly. However, because the interactions with CCR5 are reversible, all of the adsorbed virions would spend a fraction of their time in competent complexes that contain at least m CCR5s. Consequently, if there were no competing process of virus inactivation (or dissociation), all of the adsorbed virions would eventually infect the cells and the titers measured in all our cell clones would be equivalent. Clearly, however, our data show that some cell clones are inefficiently infected. According to this model, therefore, the efficiency of infecting any cell clone is determined by competition between infection and inactivation or dissociation. If infection occurs slowly for any reason, inactivation would become more predominant. This model predicts that cell clones that are inefficiently infected would be more slowly infected than more efficiently infected clones. Although there is no previous evidence for inactivation of HIV-1 on cell surfaces, such inactivation occurs spontaneously in culture media (see, e.g., reference 28) and dissociation of HIV-1 from cell surfaces has also been described (26). According to this model, the efficiency of infection mediated by high concentrations of CCR5(Y14N) is low because this mutation decreases k1 (the rate constant for infection) or, less likely, increases k2 (the rate constant for inactivation).
As described in Materials and Methods, equation 8 has the same general form as the kinetic equation 3. Moreover, we have shown that the graphical method (i.e., that illustrated in Fig. 6) that we used to analyze our results would also be applicable to analysis of sigmoidal curves derived from kinetic data. Consequently, we believe that our results would also be compatible with the following quasikinetic interpretation. This model also assumes that CCR5 binds reversibly to the adsorbed virion to form a requisite complex that contains at least m CCR5s, in accordance with equations 1 to 3. However, the quasikinetic model then assumes that the attachment of virus to CCR5 triggers a fairly rapid irreversible conformational change in the virus glycoproteins that essentially determines whether the infection succeeds or is aborted. Specifically, if the irreversible changes occur when the virion is complexed with at least m CCR5s, then infection proceeds with an efficiency (k1) that is dependent on the virus and the particular coreceptor. In contrast, if the adsorbed virion is associated with fewer than m CCR5s at the time it undergoes irreversible changes, then the infection would be aborted. We refer to this model as quasikinetic because the efficiency of infection is determined according to equation 3 by the equilibrium distribution of complexes that occur on the cell surfaces at any instant and by the efficiency constant, k1. In this model, the efficiencies of infections mediated by high saturating concentrations of CCR5(Y14N) are low (Fig. 4) because this mutation reduces k1. In contrast to the previous model, this model predicts that low efficiency infections would be completed as rapidly as infections mediated by wild-type CCR5. The quasikinetic model is consistent with evidence that coreceptor binding can cause rapid irreversible conformational changes in gp120-gp41 complexes (27). However, the principle of microscopic reversibility implies that these irreversible changes are preceded by a period of reversible interactions of virions with CCR5.
It is notable that both models predict a requirement for the same number (m) of CCR5s for infections by R5 isolates of HIV-1 and that both invoke a process of viral inactivation or dissociation. Furthermore, in both models the CCR5(Y14N) mutation has a major inhibitory effect not only on the assembly of complexes necessary for infection but also on the efficiency with which these complexes mediate infections. Consequently, we believe that our results strongly support these conclusions and that the amino-terminal region of CCR5 may be especially important for the membrane fusion reaction.
Stoichiometry of CCR5 in mediation of HIV-1 infections.
Consistent with the mathematical models described above, the data points in Fig. 6 for different viruses in different clonal panels appear to fall on straight lines that have the same slopes, within experimental error. These data imply that R5 HIV-1 infections require the concerted actions of multiple CCR5s and indicate that the required number (m) of CCR5s is probably in the range of 4 to 6 (Table 2). Interestingly, this conclusion is consonant with results of studies of influenza A virus which have suggested that the membrane fusion step of infection may require a collar of approximately three to six viral hemagglutinin-receptor complexes surrounding a membrane pore (4, 9, 14) and with evidence that multiple env trimers are necessary for HIV-1 infection (22, 34, 35). In addition, receptor clustering may be required for infections by other retroviruses (57).
Despite this consistency with our models, we emphasize that the m values in Table 2 should be considered to be approximations. Furthermore, only a portion of the cell surface CCR5 may be available for HIV-1 infections (36), perhaps because of sequestration in different microenvironments. This is especially likely with CCR5(Y14N) because a fraction (ca. 60 to 70%) of these molecules contain N-linked oligosaccharides (e.g., Fig. 2). This glycosylation appears to prevent interactions with MIP1β (Fig. 3), and it seems likely that such a large modification would also prevent interactions of R5 HIV-1 with this region of CCR5, which is known to be critical for coreceptor activity (18–20, 31, 49). Consequently, we believe that the CCR5(Y14N) molecules that lack N-linked oligosaccharides are responsible for the weak infectivity mediated by this mutant coreceptor. If the functionally relevant fraction of CCR5 was reduced by any mechanism, our estimated EC50s would be correspondingly lowered whereas our estimates of m would be unaltered (equation 9). Similarly, this factor would not affect our estimates of the asymptotes in Fig. 4. Consequently, this type of error would not affect any of our conclusions about infection mechanisms. However, there are indications that CCR5 may dimerize on cell surfaces (3), and we have observed CCR5 complexes in some of our electrophoresis experiments (e.g., Fig. 2). If this is correct, our estimates of m would imply that this number of dimers rather than monomers must associate with R5 HIV-1 to mediate infections. Recent evidence has also implied that a small proportion of CCR5 may be associated with CD4 in uninfected cells (64). Presumably, the cell surface densities of such complexes would be dependent on the concentrations of CD4 and CCR5 on the cell surfaces, but their roles in infection remain uncertain. By confocal immunofluorescence microscopy we have been unable to confirm any significant colocalization of CD4 and CCR5 in the JC.53 clone of HeLa-CD4-CCR5 cells (unpublished results). These uncertainties will require additional investigations.
We emphasize that the sigmoidal plots of HIV-1 infectivities versus CCR5 concentrations were obtained using mutant CCR5s. Because wild-type CCR5 functions efficiently even at barely detectable trace concentrations, we were unable to resolve the low concentration region for this coreceptor (e.g., see Fig. 4 and reference 48). Nevertheless, we believe that the cooperativities and stoichiometries of coreceptors in the infection pathway should be identical for all CCR5s and HIV-1 isolates, and our results are consistent with this hypothesis (Fig. 6 and Table 2).
General conclusions.
Our results suggest that CCR5 reversibly associates in a concentration-dependent manner with R5 HIV-1 on the surfaces of CD4-positive cells to form a complex that is essential for infections, that this complex contains at least four to six CCR5 molecules, and that virus dissociation or inactivation occurs at a competitively significant rate that can substantially influence HIV-1 titers in cell culture assays. Since lymphocytes and macrophages also express widely different amounts of CD4 and coreceptors at distinct stages of activation and differentiation (37, 41, 59), it seems possible that similar HIV-1 inactivation processes may occur in vivo. This possibility deserves careful analysis because it may influence the selection of viral tropism variants in vivo. Our results also reveal that mutations in CCR5 can strongly inhibit assembly of the essential complexes and/or the functional activities of the complexes. Similarly, distinct viruses can interact with CCR5 with different affinities and with different fusion efficiencies. However, our results do not establish whether the number of CCR5 molecules necessary for infection must all coordinately interact with a single gp120-gp41 trimer or with distinct trimers. We favor the latter hypothesis because similar estimates have been made for the number of influenza A virus hemagglutinin trimers required to form a fusion pore (4, 9, 14) and because evidence suggests that the mechanisms for membrane fusion are highly concordant for these viruses (10). Consistent with these conclusions, it appears that multiple gp120-gp41 trimers are required for HIV-1 infections (22, 34, 35).
This analysis suggests that careful control of CD4 and coreceptor expression levels will be important for understanding the functions of these molecules and for evaluating drugs that may interfere with these early steps of infection. As illustrated by comparison of Fig. 1 and 4, inhibitory effects of CCR5 mutations and their mechanistic implications can be obscured at high expression levels or by using conventional coreceptor assays. In addition, because infectivity assays appear to be more sensitively affected by virus inactivation than syncytial assays (see above), these methods should not be assumed to be correspondent. Syncytial assays are also dependent on cellular contacts, which are relatively complex and difficult to control. Finally, our data imply that the effectiveness of drugs targeted at CCR5 likely depend on the cell surface concentrations of CD4 (48) and CCR5 in the specific cells. Drugs that inhibit affinities of coreceptors for HIV-1 would probably be less beneficial than drugs that also block the functional activities of the assembled coreceptor complexes.
ACKNOWLEDGMENTS
This research was supported by NIH grant 2 RO1 CA67358. E.J.P. was supported in part by NRSA postdoctoral fellowship I F32 AI09735 from the NIH.
We are grateful to our coworkers and colleagues Navid Madani, Chetankumar Tailor, Mariana Marin, and Jean-Michel Heard for encouragement and critical advice. We are additionally grateful to Antony Bakke and Randy Smith for assistance with flow cytometry and to Adriana Weissman for conducting preliminary experiments. We also gratefully acknowledge the helpful discussions and criticisms of Joseph Bentz, who initially suggested that a variant of the quasikinetic model would be compatible with our data.
REFERENCES
- 1.Alkhatib G, Combadiere C, Broder C C, Feng Y, Kennedy P E, Murphy P M, Berger E A. CC CKR5: a RANTES, MIP-1α, MIP-1β receptor as a fusion cofactor for macrophage-tropic HIV-1. Science. 1996;272:1955–1958. doi: 10.1126/science.272.5270.1955. [DOI] [PubMed] [Google Scholar]
- 2.Atchison R E, Gosling J, Monteclaro F S, Franci C, Digilio L, Charo I F, Goldsmith M A. Multiple extracellular elements of CCR5 and HIV-1 entry: dissociation from response to chemokines. Science. 1996;274:1924–1926. doi: 10.1126/science.274.5294.1924. [DOI] [PubMed] [Google Scholar]
- 3.Benkirane M, Jin D Y, Chun R F, Koup R A, Jeang K T. Mechanism of transdominant inhibition of CCR5-mediated HIV-1 infection by ccr5Δ32. J Biol Chem. 1997;272:30603–30606. doi: 10.1074/jbc.272.49.30603. [DOI] [PubMed] [Google Scholar]
- 4.Bentz J. Minimal aggregate size and minimal fusion unit for the first fusion pore of influenza hemagglutinin mediated membrane fusion. Biophys J. 2000;78:227–245. doi: 10.1016/S0006-3495(00)76587-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berg H C, Purcell E M. Physics of chemoreception. Biophys J. 1977;20:193–219. doi: 10.1016/S0006-3495(77)85544-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bestwick R K, Kozak S L, Kabat D. Overcoming interference to retroviral superinfection results in amplified expression and transmission of cloned genes. Proc Natl Acad Sci USA. 1988;85:5404–5408. doi: 10.1073/pnas.85.15.5404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bieniasz P D, Fridell R A, Aramori I, Ferguson S S, Caron M G, Cullen B R. HIV-1-induced cell fusion is mediated by multiple regions within both the viral envelope and the CCR-5 co-receptor. EMBO J. 1997;16:2599–2609. doi: 10.1093/emboj/16.10.2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blanpain C, Lee B, Vakili J, Doranz B J, Govaerts C, Migeotte I, Sharron M, Dupriez V, Vassart G, Doms R W, Parmentier M. Extracellular cysteines of CCR5 are required for chemokine binding, but dispensable for HIV-1 coreceptor activity. J Biol Chem. 1999;274:18902–18908. doi: 10.1074/jbc.274.27.18902. [DOI] [PubMed] [Google Scholar]
- 9.Blumenthal R, Sarkar D P, Durell S, Howard D E, Morris S J. Dilation of the influenza hemagglutinin fusion pore revealed by the kinetics of individual cell-cell fusion events. J Cell Biol. 1996;135:63–71. doi: 10.1083/jcb.135.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan D C, Kim P S. HIV entry and its inhibition. Cell. 1998;93:681–684. doi: 10.1016/s0092-8674(00)81430-0. [DOI] [PubMed] [Google Scholar]
- 11.Chesebro B, Wehrly K. Development of a sensitive quantitative focal assay for human immunodeficiency virus infectivity. J Virol. 1988;62:3779–3788. doi: 10.1128/jvi.62.10.3779-3788.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choe H, Farzan M, Sun Y, Sullivan N, Rollins B, Ponath P D, Wu L, Mackay C R, LaRosa G, Newman W, Gerard N, Gerard C, Sodroski J. The β-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell. 1996;85:1135–1148. doi: 10.1016/s0092-8674(00)81313-6. [DOI] [PubMed] [Google Scholar]
- 13.Connor R I, Sheridan K E, Ceradini D, Choe S, Landau N R. Change in coreceptor use correlates with disease progression in HIV-1 infected individuals. J Exp Med. 1997;185:621–628. doi: 10.1084/jem.185.4.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Danieli T, Pelletier S L, Henis Y I, White J M. Membrane fusion mediated by the influenza virus hemagglutinin requires the concerted action of at least three hemagglutinin trimers. J Cell Biol. 1996;133:559–569. doi: 10.1083/jcb.133.3.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton R E, Hill C M, Davis C B, Peiper S C, Schall T J, Littman D R, Landau N R. Identification of a major co-receptor for primary isolates of HIV-1. Nature. 1996;381:661–666. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- 16.Doranz B J, Rucker J, Yi Y, Smyth R J, Samson M, Peiper S C, Parmentier M, Collman R G, Doms R W. A dual-tropic primary HIV-1 isolate that uses fusion and the β-chemokine receptors CKR-5, CKR-3, and CKR-2b as fusion cofactors. Cell. 1996;85:1149–1158. doi: 10.1016/s0092-8674(00)81314-8. [DOI] [PubMed] [Google Scholar]
- 17.Dragic T, Litwin V, Allaway G P, Martin S R, Huang Y, Nagashima K A, Cayanan C, Maddon P J, Koup R A, Moore J P, Paxton W A. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature. 1996;381:667–673. doi: 10.1038/381667a0. [DOI] [PubMed] [Google Scholar]
- 18.Dragic T, Trkola A, Lin S W, Nagashima K A, Kajumo F, Zhao L, Olson W C, Wu L, Mackay C R, Allaway G P, Sakmar T P, Moore J P, Maddon P J. Amino-terminal substitutions in the CCR5 coreceptor impair gp120 binding and human immunodeficiency virus type 1 entry. J Virol. 1998;72:279–285. doi: 10.1128/jvi.72.1.279-285.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Farzan M, Choe H, Vaca L, Martin K, Sun Y, Desjardins E, Ruffing N, Wu L, Wyatt R, Gerard N, Gerard C, Sodroski J. A tyrosine-rich region in the N terminus of CCR5 is important for human immunodeficiency virus type 1 entry and mediates an association between gp120 and CCR5. J Virol. 1998;72:1160–1164. doi: 10.1128/jvi.72.2.1160-1164.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Farzan M, Mirzabekov T, Kolchinsky P, Wyatt R, Cayabyab M, Gerard N P, Gerard C, Sodroski J, Choe H. Tyrosine sulfation of the amino terminus of CCR5 facilitates HIV-1 entry. Cell. 1999;96:667–676. doi: 10.1016/s0092-8674(00)80577-2. [DOI] [PubMed] [Google Scholar]
- 21.Feng Y, Broder C C, Kennedy P E, Berger E A. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science. 1996;272:872–877. doi: 10.1126/science.272.5263.872. [DOI] [PubMed] [Google Scholar]
- 22.Frey S, Marsh M, Gunther S, Pelchen-Matthews A, Stephens P, Ortlepp S, Stegmann T. Temperature dependence of cell-cell fusion induced by the envelope glycoprotein of human immunodeficiency virus type 1. J Virol. 1995;69:1462–1472. doi: 10.1128/jvi.69.3.1462-1472.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gelderblom H R, Ozel M, Pauli G. Morphogenesis and morphology of HIV. Structure-function relations. Arch Virol. 1989;106:1–13. doi: 10.1007/BF01311033. [DOI] [PubMed] [Google Scholar]
- 24.Genoud S, Kajumo F, Guo Y, Thompson D, Dragic T. CCR5-mediated human immunodeficiency virus entry depends on an amino-terminal gp120-binding site and on the conformational integrity of all four extracellular domains. J Virol. 1999;73:1645–1648. doi: 10.1128/jvi.73.2.1645-1648.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goldstein B. Diffusion limited effects of receptor clustering. Comments Theor Biol. 1989;1:109–127. [Google Scholar]
- 26.Hlavacek W S, Wofsy C, Perelson A S. Dissociation of HIV-1 from follicular dendritic cells during HAART: mathematical analysis. Proc Natl Acad Sci USA. 1999;96:14681–14686. doi: 10.1073/pnas.96.26.14681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones P L, Korte T, Blumenthal R. Conformational changes in cell surface HIV-1 envelope glycoproteins are triggered by cooperation between cell surface CD4 and co-receptors. J Biol Chem. 1998;273:404–409. doi: 10.1074/jbc.273.1.404. [DOI] [PubMed] [Google Scholar]
- 28.Kabat D, Kozak S L, Wehrly K, Chesebro B. Differences in CD4 dependence for infectivity of laboratory-adapted and primary patient isolates of human immunodeficiency virus type 1. J Virol. 1994;68:2570–2577. doi: 10.1128/jvi.68.4.2570-2577.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kozak S L, Kuhmann S E, Platt E J, Kabat D. Roles of CD4 and coreceptors in binding, endocytosis, and proteolysis of gp120 envelope glycoproteins derived from human immunodeficiency virus type 1. J Biol Chem. 1999;274:23499–23507. doi: 10.1074/jbc.274.33.23499. [DOI] [PubMed] [Google Scholar]
- 30.Kozak S L, Platt E J, Madani N, Ferro F E, Jr, Peden K, Kabat D. CD4, CXCR-4, and CCR-5 dependencies for infections by primary patient and laboratory-adapted isolates of human immunodeficiency virus type 1. J Virol. 1997;71:873–882. doi: 10.1128/jvi.71.2.873-882.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuhmann S E, Platt E J, Kozak S L, Kabat D. Polymorphisms in the CCR5 genes of African green monkeys and mice implicate specific amino acids in infections by simian and human immunodeficiency viruses. J Virol. 1997;71:8642–8656. doi: 10.1128/jvi.71.11.8642-8656.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kwong P D, Wyatt R, Robinson J, Sweet R W, Sodroski J, Hendrickson W A. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature. 1998;393:648–659. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lapham C K, Ouyang J, Chandrasekhar B, Nguyen N Y, Dimitrov D S, Golding H. Evidence for cell-surface association between fusin and the CD4-gp120 complex in human cell lines. Science. 1996;274:602–605. doi: 10.1126/science.274.5287.602. [DOI] [PubMed] [Google Scholar]
- 34.Layne S P, Merges M J, Dembo M, Spouge J L, Conley S R, Moore J P, Raina J L, Renz H, Gelderblom H R, Nara P L. Factors underlying spontaneous inactivation and susceptibility to neutralization of human immunodeficiency virus. Virology. 1992;189:695–714. doi: 10.1016/0042-6822(92)90593-e. [DOI] [PubMed] [Google Scholar]
- 35.Layne S P, Merges M J, Dembo M, Spouge J L, Nara P L. HIV requires multiple gp120 molecules for CD4-mediated infection. Nature. 1990;346:277–279. doi: 10.1038/346277a0. [DOI] [PubMed] [Google Scholar]
- 36.Lee B, Sharron M, Blanpain C, Doranz B J, Vakili J, Setoh P, Berg E, Liu G, Guy H R, Durell S R, Parmentier M, Chang C N, Price K, Tsang M, Doms R W. Epitope mapping of CCR5 reveals multiple conformational states and distinct but overlapping structures involved in chemokine and coreceptor function. J Biol Chem. 1999;274:9617–9626. doi: 10.1074/jbc.274.14.9617. [DOI] [PubMed] [Google Scholar]
- 37.Lee B, Sharron M, Montaner L J, Weissman D, Doms R W. Quantification of CD4, CCR5, and CXCR4 levels on lymphocyte subsets, dendritic cells, and differentially conditioned monocyte-derived macrophages. Proc Natl Acad Sci USA. 1999;96:5215–5220. doi: 10.1073/pnas.96.9.5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Littman D R. Chemokine receptors: keys to AIDS pathogenesis? Cell. 1998;93:677–680. doi: 10.1016/s0092-8674(00)81429-4. [DOI] [PubMed] [Google Scholar]
- 39.Liu R, Paxton W A, Choe S, Ceradini D, Martin S R, Horuk R, MacDonald M E, Stuhlmann H, Koup R A, Landau N R. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell. 1996;86:367–377. doi: 10.1016/s0092-8674(00)80110-5. [DOI] [PubMed] [Google Scholar]
- 40.Mondor I, Ugolini S, Sattentau Q J. Human immunodeficiency virus type 1 attachment to HeLa CD4 cells is CD4 independent and gp120 dependent and requires cell surface heparans. J Virol. 1998;72:3623–3634. doi: 10.1128/jvi.72.5.3623-3634.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Naif H M, Li S, Alali M, Sloane A, Wu L, Kelly M, Lynch G, Lloyd A, Cunningham A L. CCR5 expression correlates with susceptibility of maturing monocytes to human immunodeficiency virus type 1 infection. J Virol. 1998;72:830–836. doi: 10.1128/jvi.72.1.830-836.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nir S, de Lima M P, Larsen C, Wilschut J, Hoekstra D, Düzgünes N. Kinetics and extent of virus-cell aggregation and fusion. In: Bentz J, editor. Viral fusion mechanisms. Boca Raton, Fla: CRC Press, Inc.; 1993. pp. 437–452. [Google Scholar]
- 43.Olson W C, Rabut G E, Nagashima K A, Tran D N, Anselma D J, Monard S P, Segal J P, Thompson D A D, Kajumo F, Guo Y, Moore J P, Maddon P J, Dragic T. Differential inhibition of human immunodeficiency virus type 1 fusion, gp120 binding, and CC-chemokine activity by monoclonal antibodies to CCR5. J Virol. 1999;73:4145–4155. doi: 10.1128/jvi.73.5.4145-4155.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ozel M, Pauli G, Gelderblom H R. The organization of the envelope projections on the surface of HIV. Arch Virol. 1988;100:255–266. doi: 10.1007/BF01487688. [DOI] [PubMed] [Google Scholar]
- 45.Pesenti E, Pastore C, Lillo F, Siccardi A G, Vercelli D, Lopalco L. Role of CD4 and CCR5 levels in the susceptibility of primary macrophages to infection by CCR5-dependent HIV type 1 isolates. AIDS Res Hum Retrovir. 1999;15:983–987. doi: 10.1089/088922299310494. [DOI] [PubMed] [Google Scholar]
- 46.Picard L, Simmons G, Power C A, Meyer A, Weiss R A, Clapham P R. Multiple extracellular domains of CCR5 contribute to human immunodeficiency virus type 1 entry and fusion. J Virol. 1997;71:5003–5011. doi: 10.1128/jvi.71.7.5003-5011.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Platt, E. J., S. L. Kozak, and D. Kabat. Critical role of enhanced CD4 affinity in laboratory-adaptation of human immunodeficiency virus type-1. AIDS Res. Hum. Retrovir., in press. [DOI] [PubMed]
- 48.Platt E J, Wehrly K, Kuhmann S E, Chesebro B, Kabat D. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J Virol. 1998;72:2855–2864. doi: 10.1128/jvi.72.4.2855-2864.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rabut G E, Konner J A, Kajumo F, Moore J P, Dragic T. Alanine substitutions of polar and nonpolar residues in the amino-terminal domain of CCR5 differently impair entry of macrophage- and dualtropic isolates of human immunodeficiency virus type 1. J Virol. 1998;72:3464–3468. doi: 10.1128/jvi.72.4.3464-3468.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rizzuto C D, Wyatt R, Hernandez-Ramos N, Sun Y, Kwong P D, Hendrickson W A, Sodroski J. A conserved HIV gp120 glycoprotein structure involved in chemokine receptor binding. Science. 1998;280:1949–1953. doi: 10.1126/science.280.5371.1949. [DOI] [PubMed] [Google Scholar]
- 51.Ross T M, Bieniasz P D, Cullen B R. Multiple residues contribute to the inability of murine CCR-5 to function as a coreceptor for macrophage-tropic human immunodeficiency virus type 1 isolates. J Virol. 1998;72:1918–1924. doi: 10.1128/jvi.72.3.1918-1924.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rucker J, Samson M, Doranz B J, Libert F, Berson J F, Yi Y, Smyth R J, Collman R G, Broder C C, Vassart G, Doms R W, Parmentier M. Regions in β-chemokine receptors CCR5 and CCR2b that determine HIV-1 cofactor specificity. Cell. 1996;87:437–446. doi: 10.1016/s0092-8674(00)81364-1. [DOI] [PubMed] [Google Scholar]
- 53.Samson M, Libert F, Doranz B J, Rucker J, Liesnard C, Farber C M, Saragosti S, Lapoumeroulie C, Cognaux J, Forceille C, Muyldermans G, Verhofstede C, Burtonboy G, Georges M, Imai T, Rana S, Yi Y, Smyth R J, Collman R G, Doms R W, Vassart G, Parmentier M. Resistance to HIV-1 infection in Caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature. 1996;382:722–725. doi: 10.1038/382722a0. [DOI] [PubMed] [Google Scholar]
- 54.Scarlatti G, Tresoldi E, Björndal A, Fredricksson R, Colognesi C, Deng H K, Malnati M S, Plebani A, Siccardi A G, Littman D R, Fenyö E M, Lusso P. In vivo evolution of HIV-1 co-receptor usage and sensitivity to chemokine-mediated suppression. Nat Med. 1997;3:1259–1265. doi: 10.1038/nm1197-1259. [DOI] [PubMed] [Google Scholar]
- 55.Schwartz M. The adsorption of coliphage lambda to its host: effect of variations in the surface density of receptor and in phage-receptor affinity. J Mol Biol. 1976;103:521–536. doi: 10.1016/0022-2836(76)90215-1. [DOI] [PubMed] [Google Scholar]
- 56.Siciliano S J, Kuhmann S E, Weng Y, Madani N, Springer M S, Lineberger J E, Danzeisen R, Miller M D, Kavanaugh M P, DeMartino J A, Kabat D. A critical site in the core of the CCR5 chemokine receptor required for binding and infectivity of human immunodeficiency virus type 1. J Biol Chem. 1999;274:1905–1913. doi: 10.1074/jbc.274.4.1905. [DOI] [PubMed] [Google Scholar]
- 57.Siess D C, Kozak S L, Kabat D. Exceptional fusogenicity of Chinese hamster ovary cells with murine retroviruses suggests roles for cellular factor(s) and receptor clusters in the membrane fusion process. J Virol. 1996;70:3432–3439. doi: 10.1128/jvi.70.6.3432-3439.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Trkola A, Dragic T, Arthos J, Binley J M, Olson W C, Allaway G P, Cheng-Mayer C, Robinson J, Maddon P J, Moore J P. CD4-dependent, antibody-sensitive interactions between HIV-1 and its co-receptor CCR-5. Nature. 1996;384:184–187. doi: 10.1038/384184a0. [DOI] [PubMed] [Google Scholar]
- 59.Tuttle D L, Harrison J K, Anders C, Sleasman J W, Goodenow M M. Expression of CCR5 increases during monocyte differentiation and directly mediates macrophage susceptibility to infection by human immunodeficiency virus type 1. J Virol. 1998;72:4962–4969. doi: 10.1128/jvi.72.6.4962-4969.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang Z, Lee B, Murray J L, Bonneau F, Sun Y, Schweickart V, Zhang T, Peiper S C. CCR5 HIV-1 coreceptor activity. Role of cooperativity between residues in N-terminal extracellular and intracellular domains. J Biol Chem. 1999;274:28413–28419. doi: 10.1074/jbc.274.40.28413. [DOI] [PubMed] [Google Scholar]
- 61.Wu L, Gerard N P, Wyatt R, Choe H, Parolin C, Ruffing N, Borsetti A, Cardoso A A, Desjardin E, Newman W, Gerard C, Sodroski J. CD4-induced interaction of primary HIV-1 gp120 glycoproteins with the chemokine receptor CCR-5. Nature. 1996;384:179–183. doi: 10.1038/384179a0. [DOI] [PubMed] [Google Scholar]
- 62.Wu L, LaRosa G, Kassam N, Gordon C J, Heath H, Ruffing N, Chen H, Humblias J, Samson M, Parmentier M, Moore J P, Mackay C R. Interaction of chemokine receptor CCR5 with its ligands: multiple domains for HIV-1 gp120 binding and a single domain for chemokine binding. J Exp Med. 1997;186:1373–1381. doi: 10.1084/jem.186.8.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu L, Paxton W A, Kassam N, Ruffing N, Rottman J B, Sullivan N, Choe H, Sodroski J, Newman W, Koup R A, Mackay C R. CCR5 levels and expression pattern correlate with infectability by macrophage-tropic HIV-1, in vitro. J Exp Med. 1997;185:1681–1691. doi: 10.1084/jem.185.9.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xiao X, Wu L, Stantchev T S, Feng Y R, Ugolini S, Chen H, Shen Z, Riley J L, Broder C C, Sattentau Q J, Dimitrov D S. Constitutive cell surface association between CD4 and CCR5. Proc Natl Acad Sci USA. 1999;96:7496–7501. doi: 10.1073/pnas.96.13.7496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang Y J, Moore J P. Will multiple coreceptors need to be targeted by inhibitors of human immunodeficiency virus type 1 entry? J Virol. 1999;73:3443–3448. doi: 10.1128/jvi.73.4.3443-3448.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]