

Abstract
Increased sitting time, the most common form of sedentary behavior, is an independent risk factor for all-cause and cardiovascular disease mortality; however, the mechanisms linking sitting to cardiovascular risk remain largely elusive. Studies over the last decade have led to the concept that excessive time spent in the sitting position and the ensuing reduction in leg blood flow-induced shear stress cause endothelial dysfunction. This conclusion has been mainly supported by studies using flow-mediated dilation in the lower extremities as the measured outcome. In this review, we summarize evidence from classic studies and more recent ones that collectively support the notion that prolonged sitting-induced leg vascular dysfunction is likely also attributable to changes occurring in vascular smooth muscle cells (VSMCs). Indeed, we provide evidence that prolonged constriction of resistance arteries can lead to modifications in the structural characteristics of the vascular wall, including polymerization of actin filaments in VSMCs and inward remodeling, and that these changes manifest in a time frame that is consistent with the vascular changes observed with prolonged sitting. We expect this review will stimulate future studies with a focus on VSMC cytoskeletal remodeling as a potential target to prevent the detrimental vascular ramifications of too much sitting.
Keywords: cytoskeletal remodeling, endothelium-independent vasodilation, inward remodeling leg vascular dysfunction, prolonged sitting, vascular smooth muscle
INTRODUCTION
Sedentary behavior, defined as low energy expenditure (≤1.5 METs) while in a seated, reclined, or lying position during waking hours (1), is on the rise worldwide and particularly prevalent in modern societies (2–4). Sedentary behavior represents an independent risk factor for all-cause and cardiovascular disease mortality (2–10), and emerging epidemiological evidence suggests that this increased cardiovascular risk associated with sedentary behavior exists even independently of time spent in physical activity and structured exercise (11–18). However, the mechanisms linking sedentary behavior and cardiovascular disease risk remain largely unknown. Sitting is the most common form of sedentary behavior and, over the last decade, we and others have consistently demonstrated that prolonged, uninterrupted sitting (e.g., from 1 to 6 h) causes a transient reduction in leg vascular function (19–32), referred to as sitting leg vasculopathy (33). Recurring and extended episodes of leg vascular dysfunction associated with sitting may contribute to the genesis of leg peripheral artery disease (PAD) (33–35). Consistent with this idea, epidemiological data are available showing that sedentary time is associated with low levels of ankle-brachial index, a noninvasive clinical predictor of lower-extremity PAD (36).
Most studies documenting the detrimental vascular effects of prolonged sitting have used the Doppler ultrasound-based flow-mediated dilation (FMD) technique, a noninvasive approach to assess conduit artery function (19–25, 37–44). Because FMD is endothelial dependent (45–49), its assessment is commonly used as a barometer of endothelial function. As a result, the conviction is that prolonged sitting causes endothelial dysfunction and that this is the predominant feature of sitting leg vasculopathy (33). The purpose of this review is to challenge this current view by aggregating evidence from basic science and clinical studies that collectively support the possibility that prolonged sitting-induced leg vascular dysfunction is likely also attributable to adaptations beyond the endothelium, e.g., changes occurring in the underlying vascular smooth muscle cells (VSMCs).
EVIDENCE THAT PROLONGED SITTING BLUNTS REACTIVE HYPEREMIA, PASSIVE LIMB MOVEMENT-INDUCED HYPEREMIA, AND ENDOTHELIUM-INDEPENDENT VASODILATION IN THE LOWER LIMBS
The assessment of FMD involves a transient suprasystolic occlusion of the lower limb to generate a reactive hyperemic shear stress response that serves as a stimulus for vasodilation, which can be detected at the level of the conduit artery (e.g., femoral, popliteal, or tibial artery) via ultrasound. Notably, although FMD is reflective of conduit artery endothelial function (50–54), the magnitude of reactive hyperemia elicited by transient limb ischemia reflects the amount of dilation in downstream resistance arteries and, as such, it can serve as an indicator of resistance vessel function (55).
In the forearm, the significance of postocclusive reactive hyperemia was first documented by Mitchell et al. (56). They showed that cardiovascular risk factors are more closely related to reactive hyperemia than to FMD, a finding corroborated by a subsequent study (57). Evidence is also available suggesting that forearm reactive hyperemia is an independent predictor (i.e., beyond other risk factors) of adverse cardiovascular events and a measure that can discriminate subjects with increased cardiovascular disease risk (58–64). Despite its prognostic value, forearm reactive hyperemia is minimally dependent on nitric oxide (NO). Indeed, intra-arterial blockade of NO synthase using NG-monomethyl-l-arginine (l-NMMA) does not influence forearm reactive hyperemia or it only marginally blunts it (65–69). Work by Crecelius et al. (69) demonstrated that activation of inwardly rectifying potassium channels and Na+/K+-ATPase, largely expressed in VSMCs of resistance arteries (70), are the primary determinants of the reactive hyperemic response. Based on the above, it is prudent to deduce that, unlike FMD, postocclusive reactive hyperemia is not an endothelium-specific test (66, 69, 71–75) or at minimum not an endothelium NO-dependent phenomenon. Reactive hyperemia is rather likely mediated by a myriad of vasodilator substances (e.g., metabolites). More specifically, the magnitude of the hyperemic response is dependent on the amount of tissue hypoxia generated during occlusion and consequent release of vasodilators, as well as dependent on the capacity of the VSMCs to relax and effectively increase lumen diameter of resistance arteries (55, 76, 77).
Although most vascular sitting studies have primarily focused on the FMD response as the main outcome variable, a consistent observation by our group and others is that lower limb reactive hyperemia is also markedly blunted after prolonged sitting (21, 23, 40, 78–80) (Fig. 1). Because reduced leg FMD following prolonged sitting persists even after statistically controlling for the reduction in hyperemic shear stress stimulus (19–22, 43), most of such papers arrived at the conclusion that prolonged sitting impairs lower limb endothelial function. Accordingly, as mentioned earlier, the prevailing belief in the field is that impaired leg vascular function after prolonged sitting is attributable to endothelial defects. However, through the lenses of this review, we consider the consistent finding that prolonged sitting blunts reactive hyperemia (a largely endothelium-independent response) in the lower limbs as evidence to support the possibility that other factors beyond changes in endothelial function may also occur with sitting.
Figure 1.

Prolonged sitting is associated with pronounced reduction in leg blood flow (A), impaired reactive hyperemia (B), and blunted flow-mediated dilation (FMD; C) in the popliteal artery in healthy young adults. Values are presented as means ± SE. *P < 0.05 vs. presitting. Figure redrawn from Restaino et al. (21) with permission.
An important consideration here is that, as already stated, the extent of vasodilators released during occlusion is likely proportional to the magnitude of tissue hypoxia (i.e., oxygen deficit) that is achieved. This is particularly relevant because inactivity associated with sitting reduces the skeletal muscle metabolic rate, which in turn reduces the oxygen deficit during occlusion (based on measurements using near-infrared spectroscopy) and thus the stimulus for vasodilators to be released (81–84). Therefore, the blunted reactive hyperemia after prolonged sitting may be mediated, at least in part, by the reduced oxygen deficit (76). As recently and nicely put forth by Anderson and Park (76), future studies should examine whether dampened postocclusive reactive hyperemia after prolonged sitting remains observed when the amount of oxygen deficit is experimentally matched, e.g., by manipulating the duration of cuff occlusion (77).
Passive limb movement (PLM) is another approach to induce hyperemia, which can be captured using Doppler ultrasound, and used to assess resistance vessel function. Extensive reviews and guidelines describing this technique exist (55, 85, 86). In brief, PLM involves the manipulation of a limb (e.g., the leg) without voluntary muscle contraction; thus, the increase in skeletal muscle activation and metabolism is negligeable. The hyperemic response is elicited by mechanical deformation and stretch of the skeletal muscle, resulting in a cascade of vasodilatory events in the resistance arteries. Unlike reactive hyperemia, intra-arterial l-NMMA studies in the leg demonstrate that PLM-induced hyperemia is ∼80% NO dependent (87, 88). Given the NO dependency of this hyperemic response, this maneuver is increasingly used to assess endothelial function of the skeletal muscle resistance arteries. Notably, and relevant to the present review, a series of studies led by Garten et al. (27–29) demonstrate that PLM-induced hyperemia is blunted following 1.5 to 3 h of sitting (Fig. 2), again further emphasizing the detrimental effects of prolonged sitting on the vasculature of the lower limbs. No studies have yet been performed to determine whether the magnitude of l-NMMA-induced suppression (i.e., the NO contribution) of hyperemia during PLM is affected by prolonged sitting. Stated differently, it remains unknown whether the prolonged sitting-related reduction in vasodilation to PLM is indeed due to diminished NO bioavailability, for example, as it has been demonstrated with aging (89, 90). It is possible that NO production by endothelial cells during PLM remains intact but that the underlying VSMCs of resistance arteries become less responsive to NO and/or that resistance arteries have inwardly remodeled. Although more work is needed to examine these questions, information presented below reinforces the possibility of these scenarios.
Figure 2.

Evidence that sitting for 3-h blunts passive limb movement (PLM)-induced hyperemia in the leg of young healthy aerobically untrained adults. Values are presented as means ± SE. *P < 0.05 vs. presitting. Figure redrawn from Garten et al. (27) with permission.
With the impetus to better understand the vascular endothelium-independent effects of prolonged sitting, Liu et al. (30) recently conducted a much-needed study to examine whether prolonged sitting (3 h) influences the sensitivity of VSMCs to NO using the well-established nitroglycerin test. The authors reported that prolonged sitting not only impairs popliteal artery FMD, but it also blunts its vasodilation response to nitroglycerin (Fig. 3). Interestingly, data from a prior study by the same group provided evidence that popliteal artery FMD and nitroglycerin-induced vasodilation were positively correlated with the amount of moderate-to-vigorous physical activity (r = 0.85 and r = 0.59, respectively) assessed over a 5-day period in a small cohort of older men and women (91). When sedentary time was used instead as the outcome variable, these correlations (although inversely related as expected; r = −0.15 and −0.18) did not reach statistical significance (91), suggesting findings from laboratory-based sitting studies may not always align with observations from free-living conditions. Nevertheless, taken all together, it can be concluded that endothelium-independent mechanisms likely contribute to the diminished leg vasodilatory capacity following prolonged sitting.
Figure 3.
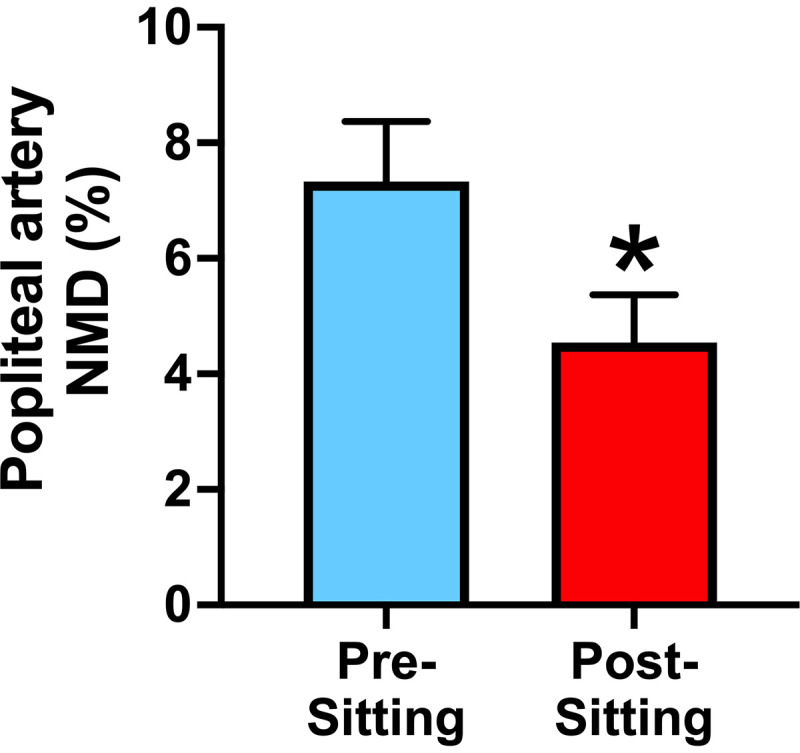
Nitroglycerin-mediated dilation (NMD), an assessment of endothelium-independent vasodilation, is impaired in the popliteal artery after 3 h of sitting in healthy young adults. Values are presented as means ± SE. *P < 0.05 vs. presitting. Figure redrawn from Liu et al. (30) with permission.
EVIDENCE THAT SITTING IS ASSOCIATED WITH VASOCONSTRICTION OF RESISTANCE ARTERIES AND REDUCED LEG BLOOD FLOW
Another consistent finding by us and others is that leg blood flow is notably reduced during sitting (19–23, 25). In fact, given that blood flow-induced shear stress is an important signal for maintaining optimal endothelial health, in prior studies we have ascribed the impairment in popliteal artery endothelial function after prolonged sitting to the sustained reduction in shear stress to which the vasculature is exposed during sitting (20, 21, 23, 33, 37, 43).
Several factors may contribute to the vasoconstriction of resistance arteries and suppressed leg blood flow during sitting. An evident mechanism is the low metabolic rate of inactive skeletal muscle (40, 77), as metabolic demand is matched by perfusion. However, other mechanisms specific to the sitting posture contribute. For example, it is plausible that, during sitting, increased hydrostatic pressure within the leg vasculature causes blood pooling within the venous circulation. Indices of augmented lower limb venous congestion, including increased calf (21, 27, 29, 39, 42, 79) and ankle (22) circumference, have been documented. This is likely aggravated by reduced skeletal muscle activity during sitting and, consequently, the loss of muscle pump effect that facilitates venous return to the heart (92). It is also possible that during sitting, venous return is further limited by the pressure exerted on the back of the thighs. Therefore, physical compression, venous distension-induced arterial constriction, and increased hydrostatic pressure-induced myogenic constriction (93) are likely key mechanisms underlying increased leg vascular resistance during sitting.
Furthermore, given that muscle sympathetic nerve activity (MSNA) is increased in the upright position (94), α-adrenergic vasoconstriction may also contribute to leg vascular resistance during sitting. In support of this, most studies provide evidence that blood pressure is increased during sitting, relative to the supine position (20, 22, 27, 29, 39, 95, 96); however, this is not a universal finding (97). The increase in blood pressure does not appear to be associated with an increase in cardiac output (95). Finally, we have also provided evidence that flexion of the hips and knees with sitting, and associated arterial bending, hinders blood flow to the lower limbs (20, 24, 33, 98). Decreased blood flow owing to arterial angulations or to other factors described earlier results in a reduction of shear stress stimulus in the leg vasculature, likely leading to reduced endothelium-derived NO production and consequently increased vascular tone. The next section discusses the vascular ramifications of prolonged vasoconstriction.
EVIDENCE THAT PROLONGED VASOCONSTRICTION CAUSES INWARD EUTROPHIC REMODELING
The purpose of this section is to present evidence that prolonged constriction of resistance arteries can lead to modifications in the structural characteristics of the vascular wall in a time frame that is consistent with vascular changes observed after prolonged sitting. The notion that structural changes in the vascular wall can occur within hours is largely underappreciated and this is what, in part, motivated the writing of this review. Arteries that exhibit inward eutrophic remodeling are distinguished by reduced passive luminal diameter with no change in wall cross-sectional area (99). A rapid reduction in vascular diameter is achieved by contraction of VSMCs via calcium-calmodulin-dependent phosphorylation of the regulatory myosin light chains and subsequent actomyosin cross-bridge cycling (100). Evidence indicates that calcium-independent processes are also in place in the acute control of vascular diameter, including calcium sensitization (101) and actin filament remodeling (102). These processes highlight the malleable nature of the VSMC machinery that regulates the structural and active diameters of resistance arteries.
Although the mechanisms implicated in the transition from acute to longer-term (minutes to hours) control of vascular diameter are less understood than the mechanisms controlling rapid changes in diameter, we provide compelling evidence that when arteries are exposed to neurohumoral vasoconstrictors, e.g., norepinephrine (NE) and angiotensin II (ANG II), for an extended period, they maintain a reduced diameter despite removal of the vasoconstrictor agonists and exposure to endothelium-independent vasodilators (103–105) (Fig. 4). This narrowing of the lumen is indicative that longer lasting changes in some cellular and/or extracellular determinants of vascular wall structure have occurred. Of significance, inward eutrophic remodeling of resistance arteries (e.g., mesenteric and cremasteric) is the most common structural change in hypertension (106–112) and its presence is predictive of life-threatening cardiovascular events (113–116).
Figure 4.

Prolonged exposure to norepinephrine and angiotensin II (NE + ANG II) causes inward eutrophic remodeling in rat (male Sprague–Dawley; weight range, 250–350 g)-isolated cremaster arterioles. Luminal diameter of arterioles exposed to NE (10−5.5 M) + ANG II (10−7 M) and their maximal relaxation responses under Ca2+-free conditions before and after exposure to NE + ANG II for 4 h. Mean trace is illustrated. *P < 0.05 vs. before agonist exposure. Figure redrawn from Martinez-Lemus et al. (104) with permission.
The precise mechanisms that control the initial stages of inward eutrophic remodeling are not fully elucidated; however, evidence indicates changes occur mainly at the level of the actin cytoskeleton and are associated with actin polymerization pathways (111, 117) in conjunction with the repositioning and stiffening of VSMCs in resistance vessels (103–105). It has been reported that during prolonged agonist-induced vasoconstriction, some but not all VSMCs reelongate from their constricted length and increase their degree of overlap by sliding over one another (105, 118). Why some cells reposition while others do not, remains to be determined. A potential explanation is that not all cells in the medial layer of arterioles are fully differentiated VSMCs. It has been well documented, particularly in the cerebral circulation, that different phenotypes of the cells known as pericytes populate the wall of blood vessels from the terminal arterioles to the capillaries and venules (119, 120). The presence of diverse morphologies of mural cells in arterioles suggest that pericyte-like mural cells may be present in the arteriolar wall and as such may respond differently to short- and long-term vasoconstrictor stimuli. Nonetheless, repositioning and stiffening processes of VSMCs likely involve the formation of more permanent actin cytoskeletal structures and new and stronger focal adhesion sites as the cell becomes repositioned in the arteriolar wall of resistance vessels (121–123). Indeed, evidence indicates that VSMCs exposed to vasoconstrictor agonists stiffen while their adhesion to the substrate is also strengthened (124, 125). We posit that these cellular changes prevent the resistance vessel from dilating to its original maximal passive diameter as alluded earlier (Fig. 4). In that regard, we have reported that during the initial stages of inward eutrophic remodeling, most of the reduction in passive diameter observed in isolated resistance arteries following prolonged (i.e., 4 h) vasoconstriction is reverted with actin cytoskeletal disruption (111) (Fig. 5). This finding contributes to the evidence that actin polymerization in VSMCs mediates vascular inward eutrophic remodeling caused by prolonged vasoconstriction in resistance vessels. The next section highlights some of the molecular mechanisms that participate in the regulation of VSMC actin polymerization and stiffening, as well as vascular inward remodeling.
Figure 5.

Disruption of the actin cytoskeleton reverts the inward remodeling caused by prolonged exposure to norepinephrine (NE) and angiotensin-II (ANG II) in rat (male Sprague–Dawley; weight range, 250–350 g)-isolated cremaster arterioles. Before and after the 4-h incubation with NE (10−5.5 M) + ANG II (10−7 M), arterioles were allowed to develop spontaneous myogenic tone and were exposed to Ca2+-free conditions to assess maximal relaxation. After 5 min under the second exposure to Ca+2-free conditions, arterioles were treated (1 h) with vehicle control or mycalolide-B (2 µM) to depolymerize actin fibers. Mean traces are illustrated. *P < 0.05 vs. control. Figure redrawn from Staiculescu et al. (111) with permission.
MOLECULAR MECHANISMS IMPLICATED IN ACTIN POLYMERIZATION AND STIFFENING OF VSMCs AND VASCULAR INWARD REMODELING
Work by our group demonstrated that inward eutrophic remodeling in resistance arteries exposed to vasoconstrictor agonists (i.e., NE and ANG II) is accompanied by the formation of reactive oxygen species (ROS) and activation of metalloproteinases (MMPs) (110) (Fig. 6). It should be noted that the process of inward remodeling can start early, even within 1 h of exposure to vasoconstrictors (108), and it remains sustained with longer exposures (i.e., from hours to days) (109). Of note, broad MMP inhibition did not influence the production of ROS but prevented the remodeling induced by prolonged vasoconstriction. Conversely, inhibition of ROS (using apocynin or tempol) prevented both the activation of MMP and the inward remodeling, indicating that activation of MMP induced by ROS contributes to the remodeling process associated with vasoconstriction (110).
Figure 6.

Prolonged exposure to norepinephrine and angiotensin-II [NE (10−5.5 M) + ANG II (10−7 M), 4 h] increases reactive oxygen species (ROS) formation and matrix metalloproteinases (MMPs) activation in rat (male Sprague–Dawley; weight range, 250–350 g)-isolated cremaster arterioles. A: arterioles with or without NE + ANG II were coincubated with 5- (and 6-)carboxy-2′,7′-dichlorodihydrofluorescein diacetate (DCFH; 30 µM) and dihydroethidium (DHE; 5 µM) and imaged with a multiphoton microscope. Bar graphs represent percent changes in DCFH and DHE fluorescence intensities from 5 min to 4 h of incubation. B: arterioles with or without NE + ANG II were subjected to gel zymography. Bands representing gelatinolytic activity were analyzed by densitometry and expressed as fold changes from control for the activity of latent (72 kDa) and active (64 kDa) forms of MMP2. Values are presented as means ± SE. *P < 0.05 vs. control. Figure redrawn from Martinez-Lemus et al. (110) with permission.
Although MMP activity is typically associated with extracellular matrix (ECM) degradation, there are several mechanisms by which MMP may modulate cytoskeletal structures. For example, degradation of ECM structures by MMP creates protein fragments with exposed cryptic sites that activate integrins (126). Activation of integrins in turn activates intracellular signals that lead to cytoskeletal remodeling. In this regard, evidence exists indicating that activation of specific integrins also contributes to inward eutrophic remodeling (107). MMP can also induce cytoskeletal modifications by transactivation of the epidermal growth factor (EGF), which occurs upon stimulation with vasoconstrictor agonists such as NE and ANG II. After the shedding of EGF by MMP, EGF binds to the epidermal growth factor receptor (EGFR) and phosphorylates tyrosine kinase. This process leads to the downstream activation of several kinases (i.e., p38 MAPK and ERK1/2) that will be translocated to the nucleus to bind and phosphorylate nuclear transcription factors, stimulating gene transcription, protein synthesis, and cell growth. Among these kinases, p38 MAPK is known to participate in the signaling events, causing vasoconstriction-mediated EGFR activation (127, 128).
Another important enzyme in VSMC cytoskeletal remodeling is tissue transglutaminase (TG2) (129). Although TG2 is most known for its function as a crosslinker of the ECM, TG2 also has the capacity to function as G protein and cellular receptor scaffold (129, 130). TG2 activation stimulates downstream intracellular pathways in VSMCs (131), including the small GTPase Ras homolog family member A (RhoA), and its downstream effector Rho-associated protein kinase (ROCK) (130, 132–134). ROCK activates LIM kinase (LIMK), which is a critical regulator of actin dynamics and VSMC stiffness (134–139). Indeed, LIMK phosphorylates and inactivates cofilin, and as cofilin’s primary role is to sever F-actin stress fibers, cofilin inactivation enhances actin polymerization (140, 141). In this regard, we have shown that TG2 activation causes actin polymerization and stiffening in VSMCs and inward remodeling in isolated resistance arteries, which can be prevented by actin depolymerization (142) (Fig. 7). LIMK inhibition prevents VSMC stiffening and vascular inward remodeling induced by vasoconstrictor agonists, and these effects are accompanied by a reduction in phosphorylated cofilin (134, 139) and F-actin content (134). In vivo, ANG II infusion-induced inward remodeling is also prevented by LIMK inhibition (134). It should be noted that actin dynamics as modulated by the activity of cofilin have diverse effects on different cell types (134, 143, 144). As such, cofilin phosphorylation can promote both cell stiffening and cell migration in addition to modulating cellular phenotype switching (143), which is highly compatible with the reports indicating arteriolar inward remodeling involves both VSMC repositioning and stiffening.
Figure 7.

Actin depolymerization reverts tissue transglutaminase (TG2) activation-induced inward remodeling in rat-isolated cremaster arterioles. A: confocal images of isolated arterioles incubated with Alexa Fluor 488-cadaverine (green) and exposed for 4 h to vehicle control (left), to 200 µM of TG2 activator [dithiothreitol (DTT), middle], or TG2 activator in the presence of 1 mM of TG2 inhibitor cystamine (right). B: passive pressure diameter curves of arterioles before and after exposure to 200 µM of TG2 activator and cystamine (1 mM) for 4 h. *P < 0.05 vs. before TG2 activator; #P < 0.05 vs. TG2 activator. C: confocal images of isolated arterioles exposed to vehicle control (left) or 2 µM mycalolide-B (right) and subsequently stained with phalloidin-Alexa 546 to visualize the actin cytoskeleton. D: pressure-diameter curves of TG2 activator inwardly remodeled arterioles before and after exposure to 2 µM of mycalolide-B or its vehicle control for 1 h. *P < 0.05 vs. remodeled + mycalolide-B or remodeled + vehicle. Figure redrawn from Castorena-Gonzalez et al. (112) with permission.
Prior work indicates that NO is a key downregulator of TG2 activity (145–147). The endothelium is the primary source of NO in the vasculature and its production is largely stimulated by flow-induced shear stress (148–151). In accordance, previous preclinical studies show that TG2 contributes to inward remodeling induced by prolonged low blood flow (108, 142, 151). Along these lines, in a scenario of reduced blood flow as it occurs in the leg vasculature during sitting, it is plausible that diminished endothelium-derived NO production and associated increases in VSMC TG2 activity also contribute to actin polymerization and stiffening of VSMCs, as well as vascular inward remodeling. However, this hypothesis remains to be tested. Other common risk factors for PAD, including aging, obesity, and hypertension, are also associated with increased actin polymerization and stiffening in VSMCs, and TG2 and LIMK activation are key molecular events implicated in this process (134, 152, 153). As such, it is possible that an overabundance of sitting coupled with other cardiovascular risk factors accelerates VSMC cytoskeletal remodeling in the leg circulation of conduit and resistance vessels, thus increasing the susceptibility for PAD development in the lower extremities.
CONCLUSIONS AND PERSPECTIVES
The prevailing dogma is that impaired leg vascular function with prolonged sitting is attributable to endothelial defects. Work by our group has significantly contributed to this view. Without discrediting this prior work, herein we summarize evidence from classic studies and more recent ones that collectively support the notion that prolonged sitting-induced leg vascular dysfunction is likely also attributable to changes occurring in VSMCs (Fig. 8). In that context, evidence is provided that prolonged constriction of resistance arteries can lead to modifications in the structural characteristics of the vascular wall, including polymerization of actin filaments in VSMCs and inward remodeling, and that these changes occur in a time frame that is consistent with the micro- and macrovascular changes observed with prolonged sitting. In fact, it is conceivable that impaired postocclusion reactive hyperemia and PLM-induced hyperemia in the lower limb after uninterrupted sitting are reflective of inward remodeling of resistance arteries induced by prolonged leg vasoconstriction. Because actin polymerization stiffens the cell, it is also likely that VSMC stiffening contributes to the impaired vasorelaxation responses associated with sitting. Studies are now needed to mechanistically determine whether structural modifications in VSMCs (on both conduit and resistance arteries) contribute to the impaired vasodilatory capacity of the leg vasculature caused by prolonged sitting. Ultimately, the goal is to identify therapeutic targets for intervention that can alleviate the vascular burden of excessive sitting and thus prevent risk of PAD development in the lower limbs. Some dietary strategies (e.g., vitamin C and fish oil supplementation) have already been attempted with mixed results (29, 31, 154). We expect pharmacological studies targeting sitting leg vasculopathy are also forthcoming. However, interventions designed to reduce sitting behavior (i.e., removal of the “insult”) are likely to always be the most effective and sustainable.
Figure 8.

Summary of proposed vascular smooth muscle cell (VSMC)-related mechanisms contributing to leg vascular dysfunction with prolonged sitting and likely amplified with superimposition of cardiovascular disease risk factors such as advanced age, obesity, and hypertension. EC, endothelial cell; ECM, extracellular matrix; Kir, inwardly rectifying potassium channel; NE, norepinephrine; ANG II, angiotensin II; ROS, reactive oxygen species; MMP, matrix metalloproteinase; NO, nitric oxide; TG2, tissue transglutaminase; RhoA, Ras homolog family member A; LIMK, LIM kinase; ATR, angiotensin receptor; ADR-α, α-adrenergic receptor.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants R01HL137769 (to J.P.), R01HL151384 (to L.A.M.-L. and J.P.), and R01HL153264 (to L.A.M.-L. and J.P.) and University of Missouri-Columbia Research Excellence Program (to L.F.-S.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
L.F.-S., L.A.M.-L., and J.P. conceived and designed research; L.F.-S. and J.P. prepared figures; L.F.-S. and J.P. drafted manuscript; L.F.-S., L.A.M.-L., and J.P. edited and revised manuscript; L.F.-S., L.A.M.-L., and J.P. approved final version of manuscript.
REFERENCES
- 1. Tremblay MS, Aubert S, Barnes JD, Saunders TJ, Carson V, Latimer-Cheung AE, Chastin SFM, Altenburg TM, Chinapaw MJM; SBRN Terminology Consensus Project Participants. Sedentary Behavior Research Network (SBRN) - terminology consensus project process and outcome. Int J Behav Nutr Phys Act 14: 75, 2017. doi: 10.1186/s12966-017-0525-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dempsey PC, Biddle SJH, Buman MP, Chastin S, Ekelund U, Friedenreich CM, Katzmarzyk PT, Leitzmann MF, Stamatakis E, van der Ploeg HP, Willumsen J, Bull F. New global guidelines on sedentary behaviour and health for adults: broadening the behavioural targets. Int J Behav Nutr Phys Act 17: 151, 2020. doi: 10.1186/s12966-020-01044-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dempsey PC, Friedenreich CM, Leitzmann MF, Buman MP, Lambert E, Willumsen J, Bull F. Global public health guidelines on physical activity and sedentary behavior for people living with chronic conditions: a call to action. J Phys Act Health 18: 76–85, 2021. doi: 10.1123/jpah.2020-0525. [DOI] [PubMed] [Google Scholar]
- 4. Bull FC, Al-Ansari SS, Biddle S, Borodulin K, Buman MP, Cardon G, Carty C, Chaput JP, Chastin S, Chou R, Dempsey PC, DiPietro L, Ekelund U, Firth J, Friedenreich CM, Garcia L, Gichu M, Jago R, Katzmarzyk PT, Lambert E, Leitzmann M, Milton K, Ortega FB, Ranasinghe C, Stamatakis E, Tiedemann A, Troiano RP, van der Ploeg HP, Wari V, Willumsen JF. World Health Organization 2020 guidelines on physical activity and sedentary behaviour. Br J Sports Med 54: 1451–1462, 2020. doi: 10.1136/bjsports-2020-102955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ekelund U, Tarp J, Steene-Johannessen J, Hansen BH, Jefferis B, Fagerland MW, Whincup P, Diaz KM, Hooker SP, Chernofsky A, Larson MG, Spartano N, Vasan RS, Dohrn I-M, Hagströmer M, Edwardson C, Yates T, Shiroma E, Anderssen SA, Lee I-M. Dose-response associations between accelerometry measured physical activity and sedentary time and all cause mortality: systematic review and harmonised meta-analysis. BMJ 366: l4570, 2019. doi: 10.1136/bmj.l4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Grøntved A, Hu FB. Television viewing and risk of type 2 diabetes, cardiovascular disease, and all-cause mortality: a meta-analysis. JAMA 305: 2448–2455, 2011. doi: 10.1001/jama.2011.812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Patterson R, McNamara E, Tainio M, de Sá TH, Smith AD, Sharp SJ, Edwards P, Woodcock J, Brage S, Wijndaele K. Sedentary behaviour and risk of all-cause, cardiovascular and cancer mortality, and incident type 2 diabetes: a systematic review and dose response meta-analysis. Eur J Epidemiol 33: 811–829, 2018. doi: 10.1007/s10654-018-0380-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bailey DP, Hewson DJ, Champion RB, Sayegh SM. Sitting time and risk of cardiovascular disease and diabetes: a systematic review and meta-analysis. Am J Prev Med 57: 408–416, 2019. doi: 10.1016/j.amepre.2019.04.015. [DOI] [PubMed] [Google Scholar]
- 9. Wilmot EG, Edwardson CL, Achana FA, Davies MJ, Gorely T, Gray LJ, Khunti K, Yates T, Biddle SJH. Sedentary time in adults and the association with diabetes, cardiovascular disease and death: systematic review and meta-analysis. Diabetologia 55: 2895–2905, 2012. doi: 10.1007/s00125-012-2677-z. [DOI] [PubMed] [Google Scholar]
- 10. Dunstan DW, Dogra S, Carter SE, Owen N. Sit less and move more for cardiovascular health: emerging insights and opportunities. Nat Rev Cardiol 18: 637–648, 2021. doi: 10.1038/s41569-021-00547-y. [DOI] [PubMed] [Google Scholar]
- 11. Motuma A, Gobena T, Roba KT, Berhane Y, Worku A. Long sedentary time is associated with worsened cardiometabolic risk factors among university employees in Eastern Ethiopia. Sci Rep 12: 22431, 2022. doi: 10.1038/s41598-022-26762-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Thorp AA, Healy GN, Owen N, Salmon J, Ball K, Shaw JE, Zimmet PZ, Dunstan DW. Deleterious associations of sitting time and television viewing time with cardiometabolic risk biomarkers. Diabetes Care 33: 327–334, 2010. doi: 10.2337/dc09-0493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chomistek AK, Manson JE, Stefanick ML, Lu B, Sands-Lincoln M, Going SB, Garcia L, Allison MA, Sims ST, LaMonte MJ, Johnson KC, Eaton CB. Relationship of sedentary behavior and physical activity to incident cardiovascular disease. J Am Coll Cardiol 61: 2346–2354, 2013. doi: 10.1016/j.jacc.2013.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Krishnan S, Rosenberg L, Palmer JR. Physical activity and television watching in relation to risk of type 2 diabetes: the Black Women’s Health Study. Am J Epidemiol 169: 428–434, 2008. doi: 10.1093/aje/kwn344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Healy GN, Matthews CE, Dunstan DW, Winkler EAH, Owen N. Sedentary time and cardio-metabolic biomarkers in US adults: NHANES 200306. Eur Heart J 32: 590–597, 2011. doi: 10.1093/eurheartj/ehq451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dunstan DW, Barr ELM, Healy GN, Salmon J, Shaw JE, Balkau B, Magliano DJ, Cameron AJ, Zimmet PZ, Owen N. Television viewing time and mortality: the Australian diabetes, obesity and lifestyle study (AusDiab). Circulation 121: 384–391, 2010. doi: 10.1161/CIRCULATIONAHA.109.894824. [DOI] [PubMed] [Google Scholar]
- 17. Wijndaele K, Healy GN, Dunstan DW, Barnett AG, Salmon J, Shaw JE, Zimmet PZ, Owen N. Increased cardiometabolic risk is associated with increased TV viewing time. Med Sci Sports Exerc 42: 1511–1518, 2010. doi: 10.1249/MSS.0b013e3181d322ac. [DOI] [PubMed] [Google Scholar]
- 18. Healy GN, Dunstan DW, Salmon J, Shaw JE, Zimmet PZ, Owen N. Television time and continuous metabolic risk in physically active adults. Med Sci Sports Exerc 40: 639–645, 2008. doi: 10.1249/MSS.0b013e3181607421. [DOI] [PubMed] [Google Scholar]
- 19. Thosar SS, Bielko SL, Mather KJ, Johnston JD, Wallace JP. Effect of prolonged sitting and breaks in sitting time on endothelial function. Med Sci Sports Exerc 47: 843–849, 2015. doi: 10.1249/MSS.0000000000000479. [DOI] [PubMed] [Google Scholar]
- 20. Morishima T, Restaino RM, Walsh LK, Kanaley JA, Padilla J. Prior exercise and standing as strategies to circumvent sitting-induced leg endothelial dysfunction. Clin Sci (Lond) 131: 1045–1053, 2017. doi: 10.1042/CS20170031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Restaino RM, Holwerda SW, Credeur DP, Fadel PJ, Padilla J. Impact of prolonged sitting on lower and upper limb micro- and macrovascular dilator function. Exp Physiol 100: 829–838, 2015. doi: 10.1113/EP085238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Morishima T, Restaino RM, Walsh LK, Kanaley JA, Fadel PJ, Padilla J. Prolonged sitting-induced leg endothelial dysfunction is prevented by fidgeting. Am J Physiol Heart Circ Physiol 311: H177–H182, 2016. doi: 10.1152/ajpheart.00297.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Restaino RM, Walsh LK, Morishima T, Vranish JR, Martinez-Lemus LA, Fadel PJ, Padilla J. Endothelial dysfunction following prolonged sitting is mediated by a reduction in shear stress. Am J Physiol Heart Circ Physiol 310: H648–H653, 2016. doi: 10.1152/ajpheart.00943.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Walsh LK, Restaino RM, Martinez-Lemus LA, Padilla J. Prolonged leg bending impairs endothelial function in the popliteal artery. Physiol Rep 5: e13478, 2017. doi: 10.14814/phy2.13478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mcmanus AM, Ainslie PN, Green DJ, Simair RG, Smith K, Lewis N. Impact of prolonged sitting on vascular function in young girls. Exp Physiol 100: 1379–1387, 2015. doi: 10.1113/EP085355. [DOI] [PubMed] [Google Scholar]
- 26. Thosar SS, Johnson BD, Johnston JD, Wallace JP. Sitting and endothelial dysfunction: the role of shear stress. Med Sci Monit 18: RA173–RA180, 2012. doi: 10.12659/msm.883589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Garten RS, Hogwood AC, Weggen JB, Fralin RC, LaRosa K, Lee D, Michael A, Scott M. Aerobic training status does not attenuate prolonged sitting-induced lower limb vascular dysfunction. Appl Physiol Nutr Metab 44: 425–433, 2019. doi: 10.1139/apnm-2018-0420. [DOI] [PubMed] [Google Scholar]
- 28. Garten RS, Scott MC, Zúñiga TM, Hogwood AC, Fralin RC, Weggen J. A prior high-intensity exercise bout attenuates the vascular dysfunction resulting from a prolonged sedentary bout. J Phys Act Health 16: 916–924, 2019. doi: 10.1123/jpah.2018-0568. [DOI] [PubMed] [Google Scholar]
- 29. Decker KP, Feliciano PG, Kimmel MT, Hogwood AC, Weggen JB, Darling AM, Richardson JW, Garten RS. Examining sex differences in sitting-induced microvascular dysfunction: insight from acute vitamin C supplementation. Microvasc Res 135: 104147, 2021. doi: 10.1016/j.mvr.2021.104147. [DOI] [PubMed] [Google Scholar]
- 30. Liu H, O'Brien MW, Wu Y, Bustamante CM, Kimmerly DS. An acute bout of prolonged sitting blunts popliteal endothelium-independent dilation in young, healthy adults. J Appl Physiol (1985) 134: 521–528, 2023. doi: 10.1152/japplphysiol.00712.2022. [DOI] [PubMed] [Google Scholar]
- 31. Morishima T, Tsuchiya Y, Padilla J, Ochi E. Eight weeks of fish oil supplementation does not prevent sitting-induced leg endothelial dysfunction. Appl Physiol Nutr Metab 45: 55–60, 2020. Jan doi: 10.1139/apnm-2019-0138. [DOI] [PubMed] [Google Scholar]
- 32. Paterson C, Fryer S, Zieff G, Stone K, Credeur DP, Barone Gibbs B, Padilla J, Parker JK, Stoner L. The effects of acute exposure to prolonged sitting, with and without interruption, on vascular function among adults: a meta-analysis. Sports Med 50: 1929–1942, 2020. doi: 10.1007/s40279-020-01325-5. [DOI] [PubMed] [Google Scholar]
- 33. Padilla J, Fadel PJ. Prolonged sitting leg vasculopathy: contributing factors and clinical implications. Am J Physiol Heart Circ Physiol 313: H722–H728, 2017. doi: 10.1152/ajpheart.00326.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McLenachan JM, Williams JK, Fish RD, Ganz P, Selwyn AP. Loss of flow-mediated endothelium-dependent dilation occurs early in the development of atherosclerosis. Circulation 84: 1273–1278, 1991. doi: 10.1161/01.cir.84.3.1273. [DOI] [PubMed] [Google Scholar]
- 35. Widlansky ME, Gokce N, Keaney JF, Vita JA. The clinical implications of endothelial dysfunction. J Am Coll Cardiol 42: 1149–1160, 2003. doi: 10.1016/s0735-1097(03)00994-x. [DOI] [PubMed] [Google Scholar]
- 36. Kulinski JP, Sanghavi M, Ayers CR, Das SR, Banerjee S, Berry JD, Addo T, De Lemos JA, Kumbhani DJ. Association between low ankle-brachial index and accelerometer-derived sedentary and exercise time in the asymptomatic general population. Vasc Med 20: 332–338, 2015. doi: 10.1177/1358863X15573837. [DOI] [PubMed] [Google Scholar]
- 37. Boyle LJ, Credeur DP, Jenkins NT, Padilla J, Leidy HJ, Thyfault JP, Fadel PJ. Impact of reduced daily physical activity on conduit artery flow-mediated dilation and circulating endothelial microparticles. J Appl Physiol (1985) 115: 1519–1525, 2013. doi: 10.1152/japplphysiol.00837.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Padilla J, Sheldon RD, Sitar DM, Newcomer SC. Impact of acute exposure to increased hydrostatic pressure and reduced shear rate on conduit artery endothelial function: a limb-specific response. Am J Physiol Heart Circ Physiol 297: H1103–H1108, 2009. doi: 10.1152/ajpheart.00167.2009. [DOI] [PubMed] [Google Scholar]
- 39. Vranish JR, Young BE, Kaur J, Patik JC, Padilla J, Fadel PJ. Influence of sex on microvascular and macrovascular responses to prolonged sitting. Am J Physiol Heart Circ Physiol 312: H800–H805, 2017. doi: 10.1152/ajpheart.00823.2016. [DOI] [PubMed] [Google Scholar]
- 40. Vranish JR, Young BE, Stephens BY, Kaur J, Padilla J, Fadel PJ. Brief periods of inactivity reduce leg microvascular, but not macrovascular, function in healthy young men. Exp Physiol 103: 1425–1434, 2018. doi: 10.1113/EP086918. [DOI] [PubMed] [Google Scholar]
- 41. Ballard KD, Duguid RM, Berry CW, Dey P, Bruno RS, Ward RM, Timmerman KL. Effects of prior aerobic exercise on sitting-induced vascular dysfunction in healthy men. Eur J Appl Physiol 117: 2509–2518, 2017. doi: 10.1007/s00421-017-3738-2. [DOI] [PubMed] [Google Scholar]
- 42. Kruse NT, Hughes WE, Benzo RM, Carr LJ, Casey DP. Workplace strategies to prevent sitting-induced endothelial dysfunction. Med Sci Sports Exerc 50: 801–808, 2018. doi: 10.1249/MSS.0000000000001484. [DOI] [PubMed] [Google Scholar]
- 43. Teixeira AL, Padilla J, Vianna LC. Impaired popliteal artery flow-mediated dilation caused by reduced daily physical activity is prevented by increased shear stress. J Appl Physiol (1985) 123: 49–54, 2017. doi: 10.1152/japplphysiol.00001.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Carter SE, Draijer R, Holder SM, Brown L, Thijssen DHJ, Hopkins ND. Effect of different walking break strategies on superficial femoral artery endothelial function. Physiol Rep 7: e14190, 2019. doi: 10.14814/phy2.14190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tryfonos A, Cocks M, Mills J, Green DJ, Dawson EA. Exercise-induced vasodilation is not impaired following radial artery catheterization in coronary artery disease patients. J Appl Physiol (1985) 128: 422–428, 2020. doi: 10.1152/japplphysiol.00695.2019. [DOI] [PubMed] [Google Scholar]
- 46. Tryfonos A, Cocks M, Rasoul D, Mills J, Green DJ, Dawson EA. Impact of catheterization on shear-mediated arterial dilation in healthy young men. Eur J Appl Physiol 120: 2525–2532, 2020. doi: 10.1007/s00421-020-04473-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pohl U, Holtz J, Busse R, Bassenge E. Crucial role of endothelium in the vasodilator response to increased flow in vivo. Hypertension 8: 37–44, 1986. doi: 10.1161/01.hyp.8.1.37. [DOI] [PubMed] [Google Scholar]
- 48. Smiesko V, Kozík J, Dolezel S. Role of endothelium in the control of arterial diameter by blood flow. Blood Vessels 22: 247–251, 1985. [PubMed] [Google Scholar]
- 49. Rubanyi GM, Romero JC, Vanhoutte PM. Flow-induced release of endothelium-derived relaxing factor. Am J Physiol Heart Circ Physiol 250: H1145–H1149, 1986. doi: 10.1152/ajpheart.1986.250.6.H1145. [DOI] [PubMed] [Google Scholar]
- 50. Thijssen DHJ, Black MA, Pyke KE, Padilla J, Atkinson G, Harris RA, Parker B, Widlansky ME, Tschakovsky ME, Green DJ. Assessment of flow-mediated dilation in humans: a methodological and physiological guideline. Am J Physiol Heart Circ Physiol 300: H2–H12, 2011. doi: 10.1152/ajpheart.00471.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Thijssen DHJ, Bruno RM, van Mil ACCM, Holder SM, Faita F, Greyling A, Zock PL, Taddei S, Deanfield JE, Luscher T, Green DJ, Ghiadoni L. Expert consensus and evidence-based recommendations for the assessment of flow-mediated dilation in humans. Eur Heart J 40: 2534–2547, 2019. doi: 10.1093/eurheartj/ehz350. [DOI] [PubMed] [Google Scholar]
- 52. Harris RA, Nishiyama SK, Wray DW, Richardson RS. Ultrasound assessment of flow-mediated dilation. Hypertension 55: 1075–1085, 2010. doi: 10.1161/HYPERTENSIONAHA.110.150821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Corretti MC, Anderson TJ, Benjamin EJ, Celermajer D, Charbonneau F, Creager MA, Deanfield J, Drexler H, Gerhard-Herman M, Herrington D, Vallance P, Vita J, Vogel R; International Brachial Artery Reactivity Task Force. Guidelines for the ultrasound assessment of endothelial-dependent flow-mediated vasodilation of the brachial artery. J Am Coll Cardiol. 39: 257–265, 2002. doi: 10.1016/s0735-1097(01)01746-6. [DOI] [PubMed] [Google Scholar]
- 54. Kooijman M, Thijssen DHJ, de Groot PCE, Bleeker MWP, van Kuppevelt HJM, Green DJ, Rongen GA, Smits P, Hopman MTE. Flow-mediated dilatation in the superficial femoral artery is nitric oxide mediated in humans. J Physiol 586: 1137–1145, 2008. doi: 10.1113/jphysiol.2007.145722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Limberg JK, Casey DP, Trinity JD, Nicholson WT, Wray DW, Tschakovsky ME, Green DJ, Hellsten Y, Fadel PJ, Joyner MJ, Padilla J. Assessment of resistance vessel function in human skeletal muscle: guidelines for experimental design, Doppler ultrasound, and pharmacology. Am J Physiol Heart Circ Physiol 318: H301–H325, 2020. doi: 10.1152/ajpheart.00649.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mitchell GF, Parise H, Vita JA, Larson MG, Warner E, Keaney JF, Keyes MJ, Levy D, Vasan RS, Benjamin EJ. Local shear stress and brachial artery flow-mediated dilation. Hypertension 44: 134–139, 2004. doi: 10.1161/01.HYP.0000137305.77635.68. [DOI] [PubMed] [Google Scholar]
- 57. Philpott AC, Lonn E, Title LM, Verma S, Buithieu J, Charbonneau F, Anderson TJ. Comparison of new measures of vascular function to flow mediated dilatation as a measure of cardiovascular risk factors. Am J Cardiol 103: 1610–1615, 2009. doi: 10.1016/j.amjcard.2009.01.376. [DOI] [PubMed] [Google Scholar]
- 58. Anderson TJ, Charbonneau F, Title LM, Buithieu J, Rose MS, Conradson H, Hildebrand K, Fung M, Verma S, Lonn EM. Microvascular function predicts cardiovascular events in primary prevention. Circulation 123: 163–169, 2011. doi: 10.1161/CIRCULATIONAHA.110.953653. [DOI] [PubMed] [Google Scholar]
- 59. Heitzer T, Schlinzig T, Krohn K, Meinertz T, Münzel T. Endothelial dysfunction, oxidative stress, and risk of cardiovascular events in patients with coronary artery disease. Circulation 104: 2673–2678, 2001. doi: 10.1161/hc4601.099485. [DOI] [PubMed] [Google Scholar]
- 60. Huang AL, Silver AE, Shvenke E, Schopfer DW, Jahangir E, Titas MA, Shpilman A, Menzoian JO, Watkins MT, Raffetto JD, Gibbons G, Woodson J, Shaw PM, Dhadly M, Eberhardt RT, Keaney JF, Gokce N, Vita JA. Predictive value of reactive hyperemia for cardiovascular events in patients with peripheral arterial disease undergoing vascular surgery. Arterioscler Thromb Vasc Biol 27: 2113–2119, 2007. doi: 10.1161/ATVBAHA.107.147322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Title LM, Lonn E, Charbonneau F, Fung M, Mather KJ, Verma S, Anderson TJ. Relationship between brachial artery flow-mediated dilatation, hyperemic shear stress, and the metabolic syndrome. Vasc Med 13: 263–270, 2008. doi: 10.1177/1358863X08095154. [DOI] [PubMed] [Google Scholar]
- 62. Ishibashi Y, Takahashi N, Shimada T, Sugamori T, Sakane T, Umeno T, Hirano Y, Oyake N, Murakami Y. Short duration of reactive hyperemia in the forearm of subjects with multiple cardiovascular risk factors. Circ J 70: 115–123, 2006. doi: 10.1253/circj.70.115. [DOI] [PubMed] [Google Scholar]
- 63. London GM, Pannier B, Agharazii M, Guerin AP, Verbeke FHM, Marchais SJ. Forearm reactive hyperemia and mortality in end-stage renal disease. Kidney Int 65: 700–704, 2004. doi: 10.1111/j.1523-1755.2004.00434.x. [DOI] [PubMed] [Google Scholar]
- 64. Paine NJ, Hinderliter AL, Blumenthal JA, Adams KF, Sueta CA, Chang PP, O'Connor CM, Sherwood A. Reactive hyperemia is associated with adverse clinical outcomes in heart failure. Am Heart J 178: 108–114, 2016. doi: 10.1016/j.ahj.2016.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tagawa T, Imaizumi T, Endo T, Shiramoto M, Harasawa Y, Takeshita A. Role of nitric oxide in reactive hyperemia in human forearm vessels. Circulation 90: 2285–2290, 1994. doi: 10.1161/01.cir.90.5.2285. [DOI] [PubMed] [Google Scholar]
- 66. Bank AJ, Sih R, Mullen K, Osayamwen M, Lee PC. Vascular ATP-dependent potassium channels, nitric oxide, and human forearm reactive hyperemia. Cardiovasc Drugs Ther 14: 23–29, 2000. doi: 10.1023/a:1007835003493. [DOI] [PubMed] [Google Scholar]
- 67. Engelke KA, Halliwill JR, Proctor DN, Dietz NM, Joyner MJ. Contribution of nitric oxide and prostaglandins to reactive hyperemia in the human forearm. J Appl Physiol (1985) 81: 1807–1814, 1996. doi: 10.1152/jappl.1996.81.4.1807. [DOI] [PubMed] [Google Scholar]
- 68. Nugent AG, McGurk C, McAuley D, Maguire S, Silke B, Johnston GD. Forearm reactive hyperaemia is not mediated by nitric oxide in healthy volunteers. Br J Clin Pharmacol 48: 457–459, 1999. doi: 10.1046/j.1365-2125.1999.00032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Crecelius AR, Richards JC, Luckasen GJ, Larson DG, Dinenno FA. Reactive hyperemia occurs via activation of inwardly rectifying potassium channels and Na+/K+-ATPase in humans. Circ Res 113: 1023–1032, 2013. doi: 10.1161/CIRCRESAHA.113.301675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ko EA, Han J, Jung ID, Park WS. Physiological roles of K+ channels in vascular smooth muscle cells. J Smooth Muscle Res 44: 65–81, 2008. doi: 10.1540/jsmr.44.65. [DOI] [PubMed] [Google Scholar]
- 71. Rosenberry R, Nelson MD. Reactive hyperemia: a review of methods, mechanisms, and considerations. Am J Physiol Regul Integr Comp Physiol 318: R605–R618, 2020. doi: 10.1152/ajpregu.00339.2019. [DOI] [PubMed] [Google Scholar]
- 72. Banitt PF, Smits P, Williams SB, Ganz P, Creager MA. Activation of ATP-sensitive potassium channels contributes to reactive hyperemia in humans. Am J Physiol Heart Circ Physiol 271: H1594–H1598, 1996. doi: 10.1152/ajpheart.1996.271.4.H1594. [DOI] [PubMed] [Google Scholar]
- 73. Jantzi MC, Brett SE, Jackson WF, Corteling R, Vigmond EJ, Welsh DG. Inward rectifying potassium channels facilitate cell-to-cell communication in hamster retractor muscle feed arteries. Am J Physiol Heart Circ Physiol 291: H1319–H1328, 2006. doi: 10.1152/ajpheart.00217.2006. [DOI] [PubMed] [Google Scholar]
- 74. Edwards G, Félétou M, Weston AH. Endothelium-derived hyperpolarising factors and associated pathways: a synopsis. Pflugers Arch 459: 863–879, 2010. doi: 10.1007/s00424-010-0817-1. [DOI] [PubMed] [Google Scholar]
- 75. Edwards G, Dora KA, Gardener MJ, Garland CJ, Weston AH. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature 396: 269–272, 1998. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- 76. Anderson CP, Park SY. Attenuated reactive hyperemia after prolonged sitting is associated with reduced local skeletal muscle metabolism: insight from artificial intelligence. Am J Physiol Regul Integr Comp Physiol 325: R380–R388, 2023. doi: 10.1152/ajpregu.00067.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Padilla J, Johnson BD, Newcomer SC, Wilhite DP, Mickleborough TD, Fly AD, Mather KJ, Wallace JP. Normalization of flow-mediated dilation to shear stress area under the curve eliminates the impact of variable hyperemic stimulus. Cardiovasc Ultrasound 6: 44, 2008. doi: 10.1186/1476-7120-6-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Horiuchi M, Stoner L. Macrovascular and microvascular responses to prolonged sitting with and without bodyweight exercise interruptions: a randomized cross-over trial. Vasc Med 27: 127–135, 2022. doi: 10.1177/1358863X211053381. [DOI] [PubMed] [Google Scholar]
- 79. Credeur DP, Miller SM, Jones R, Stoner L, Dolbow DR, Fryer SM, Stone K, McCoy SM. Impact of prolonged sitting on peripheral and central vascular health. Am J Cardiol 123: 260–266, 2019. doi: 10.1016/j.amjcard.2018.10.014. [DOI] [PubMed] [Google Scholar]
- 80. Shivgulam ME, O'Brien MW, Johns JA, Petterson JL, Wu Y, Frayne RJ, Kimmerly DS. Impact of habitual sedentary patterns on popliteal artery endothelial-dependent vasodilation in healthy adults. Vasc Med 27: 120–126, 2022. doi: 10.1177/1358863X211065494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Rosenberry R, Munson M, Chung S, Samuel TJ, Patik J, Tucker WJ, Haykowsky MJ, Nelson MD. Age-related microvascular dysfunction: novel insight from near-infrared spectroscopy. Exp Physiol 103: 190–200, 2018. doi: 10.1113/EP086639. [DOI] [PubMed] [Google Scholar]
- 82. Headid RJ, Pekas EJ, Wooden TK, Son W-M, Layec G, Shin J, Park S-Y. Impacts of prolonged sitting with mild hypercapnia on vascular and autonomic function in healthy recreationally active adults. Am J Physiol Heart Circ Physiol 319: H468–H480, 2020. doi: 10.1152/ajpheart.00354.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ross JM, Fairchild HM, Weldy J, Guyton AC. Autoregulation of blood flow by oxygen lack. Am J Physiol 202: 21–24, 1962. doi: 10.1152/ajplegacy.1962.202.1.21. [DOI] [PubMed] [Google Scholar]
- 84. Rosenberry R, Trojacek D, Chung S, Cipher DJ, Nelson MD. Interindividual differences in the ischemic stimulus and other technical considerations when assessing reactive hyperemia. Am J Physiol Regul Integr Comp Physiol 317: R530–R538, 2019. doi: 10.1152/ajpregu.00157.2019. [DOI] [PubMed] [Google Scholar]
- 85. Trinity JD, Richardson RS. Physiological impact and clinical relevance of passive exercise/movement. Sports Med 49: 1365–1381, 2019. doi: 10.1007/s40279-019-01146-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Gifford JR, Richardson RS. CORP: ultrasound assessment of vascular function with the passive leg movement technique. J Appl Physiol (1985) 123: 1708–1720, 2017. doi: 10.1152/japplphysiol.00557.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Mortensen SP, Askew CD, Walker M, Nyberg M, Hellsten Y. The hyperaemic response to passive leg movement is dependent on nitric oxide: a new tool to evaluate endothelial nitric oxide function. J Physiol 590: 4391–4400, 2012. doi: 10.1113/jphysiol.2012.235952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Trinity JD, Groot HJ, Layec G, Rossman MJ, Ives SJ, Runnels S, Gmelch B, Bledsoe A, Richardson RS. Nitric oxide and passive limb movement: a new approach to assess vascular function. J Physiol 590: 1413–1425, 2012. doi: 10.1113/jphysiol.2011.224741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Groot HJ, Trinity JD, Layec G, Rossman MJ, Ives SJ, Morgan DE, Bledsoe A, Richardson RS. The role of nitric oxide in passive leg movement‐induced vasodilatation with age: insight from alterations in femoral perfusion pressure. J Physiol 593: 3917–3928, 2015. doi: 10.1113/JP270195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Trinity JD, Groot HJ, Layec G, Rossman MJ, Ives SJ, Morgan DE, Gmelch BS, Bledsoe A, Richardson RS. Passive leg movement and nitric oxide-mediated vascular function: the impact of age. Am J Physiol Heart Circ Physiol 308: H672–H679, 2015. doi: 10.1152/ajpheart.00806.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. O'Brien MW, Robinson SA, Frayne R, Mekary S, Fowles JR, Kimmerly DS. Achieving Canadian physical activity guidelines is associated with better vascular function independent of aerobic fitness and sedentary time in older adults. Appl Physiol Nutr Metab 43: 1003–1009, 2018. doi: 10.1139/apnm-2018-0033. [DOI] [PubMed] [Google Scholar]
- 92. Delp MD, Laughlin MH. Regulation of skeletal muscle perfusion during exercise. Acta Physiol Scand 162: 411–419, 1998. doi: 10.1046/j.1365-201X.1998.0324e.x. [DOI] [PubMed] [Google Scholar]
- 93. Kitano A, Shoemaker JK, Ichinose M, Wada H, Nishiyasu T. Comparison of cardiovascular responses between lower body negative pressure and head-up tilt. J Appl Physiol (1985) 98: 2081–2086, 2005. Jun doi: 10.1152/japplphysiol.00563.2004. [DOI] [PubMed] [Google Scholar]
- 94. Ray CA, Rea RF, Clary MP, Mark AL. Muscle sympathetic nerve responses to dynamic one-legged exercise: effect of body posture. Am J Physiol Heart Circ Physiol 264: H1–H7, 1993. doi: 10.1152/ajpheart.1993.264.1.H1. [DOI] [PubMed] [Google Scholar]
- 95. Shvartz E, Gaume JG, White RT, Reibold RC. Hemodynamic responses during prolonged sitting. J Appl Physiol Respir Environ Exerc Physiol 54: 1673–1680, 1983. doi: 10.1152/jappl.1983.54.6.1673. [DOI] [PubMed] [Google Scholar]
- 96. Paterson C, Fryer S, Stone K, Zieff G, Turner L, Stoner L. The effects of acute exposure to prolonged sitting, with and without interruption, on peripheral blood pressure among adults: a systematic review and meta-analysis. Sports Med 52: 1369–1383, 2022. doi: 10.1007/s40279-021-01614-7. [DOI] [PubMed] [Google Scholar]
- 97. Cicolini G, Pizzi C, Palma E, Bucci M, Schioppa F, Mezzetti A, Manzoli L. Differences in blood pressure by body position (supine, Fowler’s, and sitting) in hypertensive subjects. Am J Hypertens 24: 1073–1079, 2011. doi: 10.1038/ajh.2011.106. [DOI] [PubMed] [Google Scholar]
- 98. McDaniel J, Ives SJ, Richardson RS. Human muscle length-dependent changes in blood flow. J Appl Physiol (1985) 112: 560–565, 2012. doi: 10.1152/japplphysiol.01223.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Mulvany MJ, Baumbach GL, Aalkjaer C, Heagerty AM, Korsgaard N, Schiffrin EL. Vascular remodeling. Hypertension 28: 505–506, 1996. [PubMed] [Google Scholar]
- 100. Hirano K, Hirano M, Kanaide H. Regulation of myosin phosphorylation and myofilament Ca2+ sensitivity in vascular smooth muscle. J Smooth Muscle Res 40: 219–236, 2004. doi: 10.1540/jsmr.40.219. [DOI] [PubMed] [Google Scholar]
- 101. Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev 83: 1325–1358, 2003. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]
- 102. Gunst SJ, Fredberg JJ. The first three minutes: smooth muscle contraction, cytoskeletal events, and soft glasses. J Appl Physiol (1985) 95: 413–425, 2003. doi: 10.1152/japplphysiol.00277.2003. [DOI] [PubMed] [Google Scholar]
- 103. Hill MA, Potocnik SJ, Martinez-Lemus LA, Meininger GA. Delayed arteriolar relaxation after prolonged agonist exposure: functional remodeling involving tyrosine phosphorylation. Am J Physiol Heart Circ Physiol 285: H849–H856, 2003. doi: 10.1152/ajpheart.00986.2002. [DOI] [PubMed] [Google Scholar]
- 104. Martinez-Lemus LA. Persistent agonist-induced vasoconstriction is not required for angiotensin II to mediate inward remodeling of isolated arterioles with myogenic tone. J Vasc Res 45: 211–221, 2008. doi: 10.1159/000112513. [DOI] [PubMed] [Google Scholar]
- 105. Martinez‐Lemus LA, Hill MA, Bolz SS, Pohl U, Meininger GA. Acute mechanoadaptation of vascular smooth muscle cells in response to continuous arteriolar vasoconstriction: implications for functional remodeling. FASEB J 18: 708–710, 2004. doi: 10.1096/fj.03-0634fje. [DOI] [PubMed] [Google Scholar]
- 106. Li JS, Schiffrin EL. Effect of calcium channel blockade or angiotensin-converting enzyme inhibition on structure of coronary, renal, and other small arteries in spontaneously hypertensive rats. J Cardiovasc Pharmacol 28: 68–74, 1996. doi: 10.1097/00005344-199607000-00011. [DOI] [PubMed] [Google Scholar]
- 107. Heerkens EHJ, Shaw L, Ryding A, Brooker G, Mullins JJ, Austin C, Ohanian V, Heagerty AM. alphaV integrins are necessary for eutrophic inward remodeling of small arteries in hypertension. Hypertension 47: 281–287, 2006. doi: 10.1161/01.HYP.0000198428.45132.02. [DOI] [PubMed] [Google Scholar]
- 108. Bakker ENTP, Buus CL, Spaan JAE, Perree J, Ganga A, Rolf TM, Sorop O, Bramsen LH, Mulvany MJ, Vanbavel E. Small artery remodeling depends on tissue-type transglutaminase. Circ Res 96: 119–126, 2005. doi: 10.1161/01.RES.0000151333.56089.66. [DOI] [PubMed] [Google Scholar]
- 109. Bakker ENTP, Buus CL, VanBavel E, Mulvany MJ. Activation of resistance arteries with endothelin-1: from vasoconstriction to functional adaptation and remodeling. J Vasc Res 41: 174–182, 2004. doi: 10.1159/000077288. [DOI] [PubMed] [Google Scholar]
- 110. Martinez-Lemus LA, Zhao G, Galiñanes EL, Boone M. Inward remodeling of resistance arteries requires reactive oxygen species-dependent activation of matrix metalloproteinases. Am J Physiol Heart Circ Physiol 300: H2005–H2015, 2011. doi: 10.1152/ajpheart.01066.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Staiculescu MC, Galiñanes EL, Zhao G, Ulloa U, Jin M, Beig MI, Meininger GA, Martinez-Lemus LA. Prolonged vasoconstriction of resistance arteries involves vascular smooth muscle actin polymerization leading to inward remodelling. Cardiovasc Res 98: 428–436, 2013. doi: 10.1093/cvr/cvt034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Castorena-Gonzalez JA, Staiculescu MC, Foote C, Martinez-Lemus LA. Mechanisms of the inward remodeling process in resistance vessels: is the actin cytoskeleton involved? Microcirculation 21: 219–229, 2014. doi: 10.1111/micc.12105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Rizzoni D, Porteri E, Boari GEM, De Ciuceis C, Sleiman I, Muiesan ML, Castellano M, Miclini M, Agabiti-Rosei E. Prognostic significance of small-artery structure in hypertension. Circulation 108: 2230–2235, 2003. doi: 10.1161/01.CIR.0000095031.51492.C5. [DOI] [PubMed] [Google Scholar]
- 114. Rizzoni D, Porteri E, Giustina A, De Ciuceis C, Sleiman I, Boari GEM, Castellano M, Muiesan ML, Bonadonna S, Burattin A, Cerudelli B, Agabiti-Rosei E. Acromegalic patients show the presence of hypertrophic remodeling of subcutaneous small resistance arteries. Hypertension 43: 561–565, 2004. doi: 10.1161/01.HYP.0000114604.52270.95. [DOI] [PubMed] [Google Scholar]
- 115. Porteri E, Rizzoni D, Mulvany MJ, De Ciuceis C, Sleiman I, Boari GE, Castellano M, Muiesan ML, Zani F, Rosei EA. Adrenergic mechanisms and remodeling of subcutaneous small resistance arteries in humans. J Hypertens 21: 2345–2352, 2003. doi: 10.1097/00004872-200312000-00024. [DOI] [PubMed] [Google Scholar]
- 116. Mathiassen ON, Buus NH, Sihm I, Thybo NK, Mørn B, Schroeder AP, Thygesen K, Aalkjaer C, Lederballe O, Mulvany MJ, Christensen KL. Small artery structure is an independent predictor of cardiovascular events in essential hypertension. J Hypertens 25: 1021–1026, 2007. doi: 10.1097/HJH.0b013e32805bf8ed. [DOI] [PubMed] [Google Scholar]
- 117. Rembold CM, Tejani AD, Ripley ML, Han S. Paxillin phosphorylation, actin polymerization, noise temperature, and the sustained phase of swine carotid artery contraction. Am J Physiol Cell Physiol 293: C993–C1002, 2007. doi: 10.1152/ajpcell.00090.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Martinez-Lemus LA, Hill MA, Meininger GA. The plastic nature of the vascular wall: a continuum of remodeling events contributing to control of arteriolar diameter and structure. Physiology (Bethesda) 24: 45–57, 2009. doi: 10.1152/physiol.00029.2008. [DOI] [PubMed] [Google Scholar]
- 119. Hartmann DA, Coelho-Santos V, Shih AY. Pericyte control of blood flow across microvascular zones in the central nervous system. Annu Rev Physiol 84: 331–354, 2022. doi: 10.1146/annurev-physiol-061121-040127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Klug NR, Sancho M, Gonzales AL, Heppner TJ, O'Brien RIC, Hill-Eubanks D, Nelson MT. Intraluminal pressure elevates intracellular calcium and contracts CNS pericytes: role of voltage-dependent calcium channels. Proc Natl Acad Sci USA 120: e2216421120, 2023. doi: 10.1073/pnas.2216421120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Fernstrom K, Farmer P, Ali MS. Cytoskeletal remodeling in vascular smooth muscle cells in response to angiotensin II-induced activation of the SHP-2 tyrosine phosphatase. J Cell Physiol 205: 402–413, 2005. doi: 10.1002/jcp.20436. [DOI] [PubMed] [Google Scholar]
- 122. Hu H, Bliss JM, Wang Y, Colicelli J. RIN1 is an ABL tyrosine kinase activator and a regulator of epithelial-cell adhesion and migration. Curr Biol 15: 815–823, 2005. doi: 10.1016/j.cub.2005.03.049. [DOI] [PubMed] [Google Scholar]
- 123. Rembold CM. Force suppression and the crossbridge cycle in swine carotid artery. Am J Physiol Cell Physiol 293: C1003–C1009, 2007. doi: 10.1152/ajpcell.00091.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Hong Z, Reeves KJ, Sun Z, Li Z, Brown NJ, Meininger GA. Vascular smooth muscle cell stiffness and adhesion to collagen I modified by vasoactive agonists. PLoS One 10: e0119533, 2015. doi: 10.1371/journal.pone.0119533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Hong Z, Sun Z, Li M, Li Z, Bunyak F, Ersoy I, Trzeciakowski JP, Staiculescu MC, Jin M, Martinez-Lemus L, Hill MA, Palaniappan K, Meininger GA. Vasoactive agonists exert dynamic and coordinated effects on vascular smooth muscle cell elasticity, cytoskeletal remodelling and adhesion. J Physiol 592: 1249–1266, 2014. doi: 10.1113/jphysiol.2013.264929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Martinez-Lemus LA, Wu X, Wilson E, Hill MA, Davis GE, Davis MJ, Meininger GA. Integrins as unique receptors for vascular control. J Vasc Res 40: 211–233, 2003. doi: 10.1159/000071886. [DOI] [PubMed] [Google Scholar]
- 127. Wang X, Chow FL, Oka T, Hao L, Lopez-Campistrous A, Kelly S, Cooper S, Odenbach J, Finegan BA, Schulz R, Kassiri Z, Lopaschuk GD, Fernandez-Patron C. Matrix metalloproteinase-7 and ADAM-12 (a disintegrin and metalloproteinase-12) define a signaling axis in agonist-induced hypertension and cardiac hypertrophy. Circulation 119: 2480–2489, 2009. doi: 10.1161/CIRCULATIONAHA.108.835488. [DOI] [PubMed] [Google Scholar]
- 128. Nagareddy PR, Chow FL, Hao L, Wang X, Nishimura T, MacLeod KM, McNeill JH, Fernandez-Patron C. Maintenance of adrenergic vascular tone by MMP transactivation of the EGFR requires PI3K and mitochondrial ATP synthesis. Cardiovasc Res 84: 368–377, 2009. doi: 10.1093/cvr/cvp230. [DOI] [PubMed] [Google Scholar]
- 129. Nurminskaya MV, Belkin AM. Cellular functions of tissue transglutaminase. Int Rev Cell Mol Biol 294: 1–97, 2012. doi: 10.1016/B978-0-12-394305-7.00001-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Gundemir S, Colak G, Tucholski J, Johnson GVW. Transglutaminase 2: a molecular Swiss army knife. Biochim Biophys Acta 1823: 406–419, 2012. doi: 10.1016/j.bbamcr.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Guilluy C, Rolli-Derkinderen M, Tharaux PL, Melino G, Pacaud P, Loirand G. Transglutaminase-dependent RhoA activation and depletion by serotonin in vascular smooth muscle cells. J Biol Chem 282: 2918–2928, 2007. doi: 10.1074/jbc.M604195200. [DOI] [PubMed] [Google Scholar]
- 132. Calò LA, Lenzini L, Rossi GP. Aldosterone-induced oxidative stress. J Hypertens 32: 2280–2281, 2014. doi: 10.1097/HJH.0000000000000375. [DOI] [PubMed] [Google Scholar]
- 133. Nuno DW, England SK, Lamping KG. RhoA localization with caveolin-1 regulates vascular contractions to serotonin. Am J Physiol Regul Integr Comp Physiol 303: R959–R967, 2012. doi: 10.1152/ajpregu.00667.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Morales-Quinones M, Ramirez-Perez FI, Foote CA, Ghiarone T, Ferreira-Santos L, Bloksgaard M, Spencer N, Kimchi ET, Manrique-Acevedo C, Padilla J, Martinez-Lemus LA. LIMK (LIM Kinase) inhibition prevents vasoconstriction- and hypertension-induced arterial stiffening and remodeling. Hypertension 76: 393–403, 2020. doi: 10.1161/HYPERTENSIONAHA.120.15203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Yi R, Xiao-Ping G, Hui L. Atorvastatin prevents angiotensin II-induced high permeability of human arterial endothelial cell monolayers via ROCK signaling pathway. Biochem Biophys Res Commun 459: 94–99, 2015. doi: 10.1016/j.bbrc.2015.02.076. [DOI] [PubMed] [Google Scholar]
- 136. Dai YP, Bongalon S, Mutafova-Yambolieva VN, Yamboliev IA. Distinct effects of contraction agonists on the phosphorylation state of cofilin in pulmonary artery smooth muscle. Adv Pharmacol Sci 2008: 362741–362749, 2008. doi: 10.1155/2008/362741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Bagnato A, Rosanò L. Endothelin-1 receptor drives invadopodia: exploiting how β-arrestin-1 guides the way. Small GTPases 9: 394–398, 2018. doi: 10.1080/21541248.2016.1235526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Chen YL, Ren Y, Xu W, Rosa RH, Kuo L, Hein TW. Constriction of retinal venules to endothelin-1: obligatory roles of ET A receptors, extracellular calcium entry, and rho kinase. Invest Ophthalmol Vis Sci 59: 5167–5175, 2018. doi: 10.1167/iovs.18-25369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Foote CA, Castorena-Gonzalez JA, Staiculescu MC, Clifford PS, Hill MA, Meininger GA, Martinez-Lemus LA. Brief serotonin exposure initiates arteriolar inward remodeling processes in vivo that involve transglutaminase activation and actin cytoskeleton reorganization. Am J Physiol Heart Circ Physiol 310: H188–H198, 2016. doi: 10.1152/ajpheart.00666.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Bernard O. Lim kinases, regulators of actin dynamics. Int J Biochem Cell Biol 39: 1071–1076, 2007. doi: 10.1016/j.biocel.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 141. Ostrowska Z, Moraczewska J. Cofilin – a protein controlling dynamics of actin filaments. Postepy Hig Med Dosw 71: 0–0, 2017. doi: 10.5604/01.3001.0010.3818. [DOI] [PubMed] [Google Scholar]
- 142. Castorena-Gonzalez JA, Staiculescu MC, Foote CA, Polo-Parada L, Martinez-Lemus LA. The obligatory role of the actin cytoskeleton on inward remodeling induced by dithiothreitol activation of endogenous transglutaminase in isolated arterioles. Am J Physiol Heart Circ Physiol 306: H485–H495, 2014. doi: 10.1152/ajpheart.00557.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Swiatlowska P, Sit B, Feng Z, Marhuenda E, Xanthis I, Zingaro S, Ward M, Zhou X, Xiao Q, Shanahan C, Jones GE, Yu C-H, Iskratsch T. Pressure and stiffness sensing together regulate vascular smooth muscle cell phenotype switching. Sci Adv 8: eabm3471, 2022. doi: 10.1126/sciadv.abm3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Hollósi A, Pászty K, Bunta BL, Bozó T, Kellermayer M, Debreczeni ML, Cervenak L, Baccarini M, Varga A. BRAF increases endothelial cell stiffness through reorganization of the actin cytoskeleton. FASEB J 36: e22478, 2022. doi: 10.1096/fj.202200344R. [DOI] [PubMed] [Google Scholar]
- 145. Santhanam L, Tuday EC, Webb AK, Dowzicky P, Kim JH, Oh YJ, Sikka G, Kuo M, Halushka MK, Macgregor AM, Dunn J, Gutbrod S, Yin D, Shoukas A, Nyhan D, Flavahan NA, Belkin AM, Berkowitz DE. Decreased S-nitrosylation of tissue transglutaminase contributes to age-related increases in vascular stiffness. Circ Res 107: 117–125, 2010. Jul 9 doi: 10.1161/CIRCRESAHA.109.215228. [DOI] [PubMed] [Google Scholar]
- 146. Jandu SK, Webb AK, Pak A, Sevinc B, Nyhan D, Belkin AM, Flavahan NA, Berkowitz DE, Santhanam L. Nitric oxide regulates tissue transglutaminase localization and function in the vasculature. Amino Acids 44: 261–269, 2013. doi: 10.1007/s00726-011-1090-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Kurt-Celep İ, Nihan Kilinc A, Griffin M, Telci D. Nitrosylation of tissue transglutaminase enhances fibroblast migration and regulates MMP activation. Matrix Biol 105: 1–16, 2022. doi: 10.1016/j.matbio.2021.10.005. [DOI] [PubMed] [Google Scholar]
- 148. Cunningham KS, Gotlieb AI. The role of shear stress in the pathogenesis of atherosclerosis. Lab Invest 85: 9–23, 2005. [Erratum in Lab Invest 85: 942, 2005]. doi: 10.1038/labinvest.3700215. [DOI] [PubMed] [Google Scholar]
- 149. Langille BL, O'Donnell F. Reductions in arterial diameter produced by chronic decreases in blood flow are endothelium-dependent. Science 231: 405–407, 1986. doi: 10.1126/science.3941904. [DOI] [PubMed] [Google Scholar]
- 150. Rudic RD, Shesely EG, Maeda N, Smithies O, Segal SS, Sessa WC. Direct evidence for the importance of endothelium-derived nitric oxide in vascular remodeling. J Clin Invest 101: 731–736, 1998. doi: 10.1172/JCI1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Bakker ENTP, Pistea A, Spaan JAE, Rolf T, de Vries CJ, van Rooijen N, Candi E, VanBavel E. Flow-dependent remodeling of small arteries in mice deficient for tissue-type transglutaminase: possible compensation by macrophage-derived factor XIII. Circ Res 99: 86–92, 2006. doi: 10.1161/01.RES.0000229657.83816.a7. [DOI] [PubMed] [Google Scholar]
- 152. Ramirez-Perez FI, Cabral-Amador FJ, Whaley-Connell AT, Aroor AR, Morales-Quinones M, Woodford ML. Cystamine reduces vascular stiffness in Western diet-fed female mice. Am J Physiol Heart Circ Physiol 322: H167–H180, 2022. doi: 10.1152/ajpheart.00431.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Soares RN, Ramirez-Perez FI, Cabral-Amador FJ, Morales-Quinones M, Foote CA, Ghiarone T, Sharma N, Power G, Smith JA, Rector RS, Martinez-Lemus LA, Padilla J, Manrique-Acevedo C. SGLT2 inhibition attenuates arterial dysfunction and decreases vascular F-actin content and expression of proteins associated with oxidative stress in aged mice. Geroscience 44: 1657–1675, 2022. doi: 10.1007/s11357-022-00563-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Thosar SS, Bielko SL, Wiggins CS, Klaunig JE, Mather KJ, Wallace JP. Antioxidant vitamin C prevents decline in endothelial function during sitting. Med Sci Monit 21: 1015–1021, 2015. doi: 10.12659/MSM.893192. [DOI] [PMC free article] [PubMed] [Google Scholar]