

Abstract
γ-methylene C(sp3)─H functionalization of linear free carboxylic acids remains a significant challenge. Here in we report a Pd(II)-catalyzed tandem γ-arylation and γ-lactonization of aliphatic acids enabled by a L,X-type CarboxPyridone ligand. A wide range of γ-arylated γ-lactones are synthesized in a single step from aliphatic acids in moderate to good yield. Arylated lactones can readily be converted into disubstituted tetrahydrofurans, a prominent scaffold amongst bioactive molecules.
Keywords: carboxylic acids, C─H activation, arylation, lactonization, palladium
Graphical Abstract
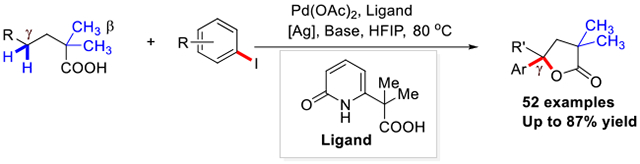
A Pd(II)-catalyzed tandem γ-methylene C-H arylation and lactonization of aliphatic acids was enabled by a L,X-type CarboxPyridone ligand. A wide range of γ-arylated -γ-lactones are synthesized in a single step from aliphatic acids in moderate to good yield. Arylated lactones can readily be converted into disubstituted tetrahydrofurans, a prominent scaffold amongst bioactive molecules.
Lactones are estimated to be present in nearly one-third of natural products and medicinally active molecules.1 In particular, γ-butyrolactones with a tertiary center adjacent to the oxygen atom are commonly encountered in natural products (Scheme 1.A) and can be used as valuable synthetic intermediates for bioactive targets. For instance, γ-ethyl-γ-phenyl-butyrolactone (EFBL) exhibits both anticonvulsant and hypnotic properties,2 various tulipaline A derivatives have shown anticancer effects,3 while Boivinianin A4 and 3-hydroxyboivinic acids were found in natural products. Generally, these γ-tertiary arylated γ-butyrolactone derivatives are synthesized from unsaturated carboxylic acids via electrophilic lactonization5 or hydro lactonization6. Therefore, the development of new catalytic methods for the synthesis of arylated γ-butyrolactones from simple starting materials is of high interest in medicinal chemistry. A transition metal catalyzed double γ-C(sp3)─H functionalization of commercially available aliphatic acids would be highly desirable for rapid diverse synthesis of γ-butyrolactones.
Scheme 1.

Pd(II)-Catalyzed γ-Methylene C─H functionalization of Aliphatic Acids
Over the past decades following the discovery of MPAA ligands in Pd-catalyzed C─H activation/functionalization, a series of bifunctional ligands have been developed to functionalize inert C(sp3)-H bonds of aliphatic substrates, broadening native substrate scope and enabling new transformations.7 Aliphatic acids have attracted much attention for the C(sp3)─H functionalization via Pd-catalysis due to their abundant occurrence in bioactive molecules and organic molecules.8 A series of catalytic systems have been developed for various ligand enabled transformations of terminal β- or γ-methyl C(sp3)─H bonds to access a diverse range of reactions. 9-16 Despite these advances, selective activation of methylene C─H bonds over methyl C-H bonds remains challenging, owing to the higher reactivity of the latter towards palladium catalyzed C─H activation reactions. Recently our group developed a series of pyridine-pyridone bidentate ligands17 that delivered α,β-unsaturated carboxylic acids and γ-alkylidene butenolides via β- methylene C(sp3)─H activation of aliphatic acids.18,19-22 These findings prompted us to design new catalysts/ligands and reaction conditions23,24 to achieve the previously inaccessible γ-methylene C(sp3)-H bond functionalization.25
In our initial report in 2022, a method for Pd-catalyzed γ-methylene C─H lactonization relied on the use of dicarboxylic acids and a pyridine-pyridone ligand (Scheme 1.B).26 Recently, we also demonstrated Pd-catalyzed transannular C(sp3)─H arylation of cyclic carboxylic acids using a newly developed quinuclidine-pyridone ligand (Scheme 1.B).27 However, γ-lactonization was limited to diacids, while γ-C(sp3)─H arylation was achieved for cyclic substrates only. The development of efficient catalysts and strategies to functionalize γ-methylene C(sp3)─H bonds in linear aliphatic acid remains an unanswered challenge.
Herein, we report a tandem γ-C(sp3)─H arylation/lactonization of linear aliphatic acid enabled by an L,X-type CarboxPyridone ligand. A wide range of free aliphatic acids could be converted to γ-arylated γ-lactone products with high yields and mono-selectivity using various aryl iodides as coupling partners. The γ-arylated γ-lactones can also be used as a key synthetic precursor to access medicinally significant disubstituted tetrahydrofuran derivatives. 28
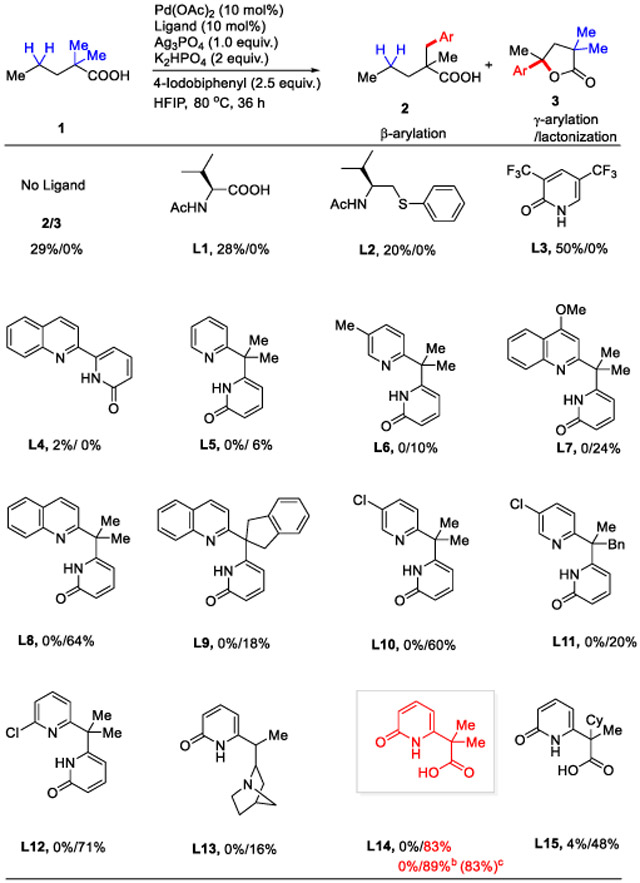
We started our investigation by using 2,2-dimethyl pentanoic acid (1a) as a model substrate and 4-iodobiphenyl as a coupling partner29 for the identification of suitable ligands and conditions (Table 1). In the absence of any external ligand, only β-methyl C(sp3)─H arylation was observed. Monoprotected amino acids (MPAA) (L1), mono-protected aminoethyl phenyl thioether (MPAThio) (L2) and monodentate pyridone (L3) ligands also resulted in only β-methyl C(sp3)─H arylation and failed to give any methylene functionalized product. We then turn to the recently discovered pyridine-pyridone ligand class. Employment of ligand L4 that forms 5-membered chelation was unsuccessful and resulted in β-methyl C(sp3)─H arylation only. Encouragingly, switching to the six-membered chelating bidentate pyridine-pyridone ligand L5 afforded 6% yield of the desired double γ-C(sp3)─H arylation/lactonization product. These observations indicate that ligands play a key role in controlling the regioselectivity between the β-methyl and γ-methylene C─H bonds. A slight improvement was seen with L6 and L7 ligands, producing the desired products in 10% and 24% yields respectively. To our delight, the quinoline-pyridone ligand (L8) further improved the yield to 64%. 5-chloro ligand (L10) showed comparable results with L8. While increasing the steric bulk at the methylene junction was not fruitful (L9 & L11) and resulted in a drop of yield. Introducing a chloro substituent at the 6-position of the pyridine ring in the ligand (L12) further improved the yield to 71%. Our recently developed quinuclidine-pyridone ligand27 (L13) for γ-C(sp3)─H arylation of cyclic acids was also tested, albeit affording poor yield.
Table 1.
Ligand Identificationa:
|
Conditions: 1a (0.1 mmol), 2a (2.5 equiv.), Pd(OAc)2 (10 mol %), ligand (10 mol%), K2HPO4 (2.0 equiv.), Ag3PO4 (1.0 equiv.), HFIP (1.0 mL), 80 °C, under air, 36 h. 20 mol% ligand used for L1 and L3. Here, Ar indicates 4-biphenyl group. 1H NMR yields, determined using CH2Br2 as an internal standard.
15 mol% ligand loading.
isolated yield.
Surprisingly, we found that CarboxPyridone ligand (L14) (which enabled ortho C─H activation and hydroxylation of a broad range of benzoic and phenylacetic acids30) was uniquely effective in improving both the yields and the scope of the coupling partners. Increasing to 15 mol% loading of the ligand resulted in further improvement and gave the optimum reaction conditions (89% yield). Further modification of the CarboxPyridone ligand backbone (L15) resulted in lower yield of desired product.
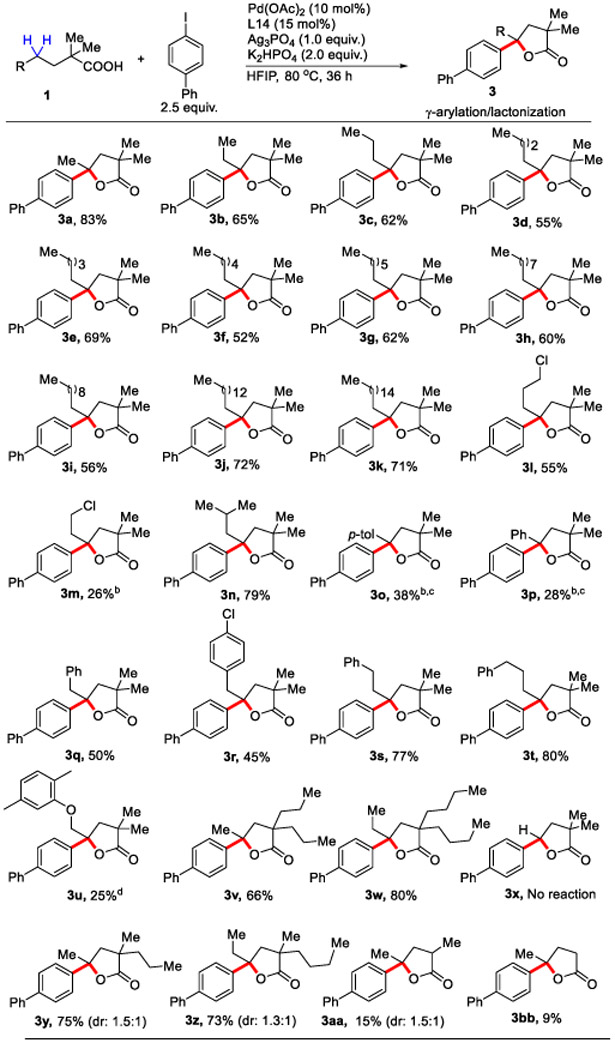
With the optimal ligand and reaction conditions in hand, we then explored the generality of this tandem γ-C(sp3)─H arylation/ lactonization by varying the structure of the linear carboxylic acid with 4-iodobiphenyl as the coupling partner (Table 2). A wide range of α-quaternary aliphatic carboxylic acids were compatible with the current reaction conditions. Linear aliphatic acids bearing various chain lengths ranging from 5, 6, 7 up to 20 (1a to 1k) were well tolerated with excellent yield and selectivity. The length of the aliphatic chain in the carboxylic acid does not affect the selectivity and yield of the reaction. Chloro substituents at the aliphatic chain (1l & 1m) and branched chain substrates (1n) were also well tolerated. Whilst substrates containing arene ring at the γ-position of the aliphatic chain (1o & 1p) worked with modest yields, substrates with more distant arene substituents (1q to 1t) afforded desired product in good to excellent yield. In case of 1a & 1p minor amounts of γ-lactone products observed without arylation. To show case the utility a marketed drug Gemfibrozil (1u) was tried under the reaction condition giving the desired product with modest yield. Variation of the alkyl substituent at the α-position (1v & 1w) also afforded the γ-arylated γ-lactone products with excellent yield and selectivity.
Table 2.
Scope for the γ-Arylation/Lactonization Reactiona:
|
Conditions: 1a (0.1 mmol), 2a (2.5 equiv.), Pd(OAc)2 (10 mol %), ligand 14 (15 mol%), K2HPO4 (2.0 equiv.), Ag3PO4 (1.0 equiv.), HFIP (1.0 mL), 80 °C, under air, 36 h.
20 mol% Pd(OAc)2 & 20 mol% ligand used.
in case of 3o and 3p 6% and 4% respectively lactonized products obtained without arylation observed.
30 mol% Pd(OAc)2 & 30 mol% ligand was used. The diastereomeric ratio was determined from crude NMR analysis.
Interestingly, 2,2-dimethylbutanoic acid bearing primary γ-C─H bonds showed no reaction under the developed condition leaving only unreacted starting material in the reaction medium, suggesting high selectivity of the developed system towards γ-methylene C─H bond over γ-methyl C─H bond. Substrate with two different substituents at the alpha position (1y & 1z) was also found viable to give γ-arylated γ-lactone products with good yield. We then performed the reaction on carboxylic acids containing an α-hydrogen. Only γ-arylated γ-lactone products were observed for 2-methylpentanoic acid (1aa) and valeric acid (1bb) in 15% and 9% yields respectively. The low yields of these substrates were probably due to the absence of Thorpe-Ingold effect.
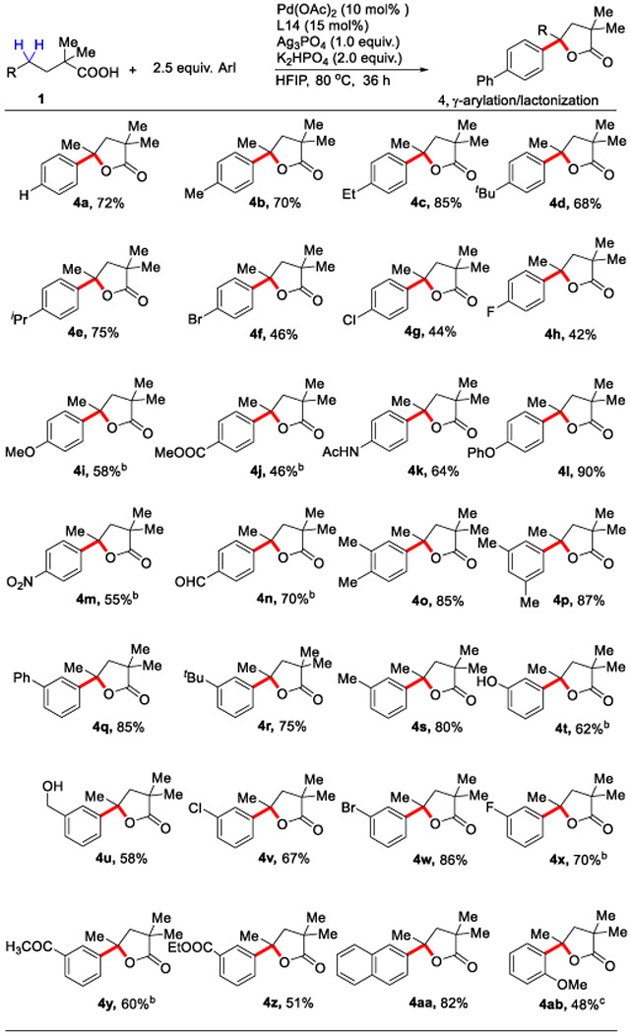
Next, a wide range of aryl iodide coupling partners were tested in our arylation/lactonization reaction using 2,2-dimethyl pentatonic acid as a model substrate (Table 3). Employment of unsubstituted iodobenzene afforded 72% yield of the desired product (4a). Various aliphatic substituents at the 3- and 4-positions of the aryl iodide provided excellent selectivity and good yield of the γ-arylated γ-lactone products. Halogen substituted aryl iodides including fluoro, chloro and bromo substituents showed good compatibility, with latter particularly useful for subsequent diversification via cross coupling reaction. A range of electron-donating (OMe & OPh) and electron-withdrawing (NHAc, CHO, CO2Me, NO2) substituents were well tolerated. Free hydroxy groups on the aryl iodides are also well tolerated under the reaction conditions to deliver the product (4t & 4u) without affecting the hydroxy group. 2-naphthyl iodide also works perfectly under the developed condition. 2-methoxy substituted aryl iodide gave the desired γ-arylated γ-lactone product (4ab), however, it required a higher catalyst loading.
Table 3.
Aryl Iodide Scope for the γ-Arylation/Lactonization Reactiona:
|
Conditions: 1a (0.1 mmol), 2a (2.5 equiv.), Pd(OAc)2 (10 mol %), ligand L14 (15 mol%), K2HPO4 (2.0 equiv.), Ag3PO4 (1.0 equiv.), HFIP (1.0 mL), 80 °C, under air, 36 h.
20 mol% Pd(OAc)2 & 20 mol% ligand used.
30 mol% Pd(OAc)2 & 30 mol% ligand was used.
To showcase the utility of these reactions, a larger scale reaction (1 .0 mmol scale) was carried out with 80% isolated yield of the desired product (Scheme 2). This γ-arylated γ-lactone can be readily converted to disubstituted tetrahydrofuran in a single step using Lithium Aluminum hydride (LAH) as a reducing agent. This substituted tetrahydrofurans are very important scaffolds in various natural products and medicinally significant molecules. 28
Scheme 2.

1.0 mmol Scale Synthesis and Further Transformation to Tetrahydrofuran. Step-1: Conditions: 1a (1.0 mmol), 2a (2.5 equiv.), Pd(OAc)2 (10 mol %), ligand L14 (15 mol%), K2HPO4 (2.0 equiv.), Ag3PO4 (1.0 equiv.), HFIP (1.0 mL), 80 °C, under air, 36 h. Step-2: 3a (0.5 mmol), LAH (1.5 equiv.), THF, 0 °C - rt, 6h.
To investigate the mechanism of this Pd-catalyzed tandem γ-C(sp3)─H arylation/lactonization reaction, several control experiments were conducted (Scheme 3). First, no reaction was observed in absence of aryl iodide, which indicates the crucial role of the aryl iodide. Then several possible intermediates as substrates were subjected to the reaction conditions. 4% yield of the desired γ-arylated γ-lactone was obtained from γ-arylated acid 6a, and no reaction occurred with γ-butyrolactone 6b, suggesting that neither of those intermediates were likely to be involved in the catalytic cycle. On the other hand, β,γ-dehydrogenated intermediate 6c & γ-arylated β,γ-dehydrogenated intermediate 6d gave 23% & 28% yield of the desired product under the developed reaction conditions indicating their potential involvement as intermediate in the reaction. γ,δ-dehydrogenated intermediate 6e resulted in only 5% yield of the desired product with 30% yield of the corresponding di-isomer, implying that γ,δ-dehydrogenation pathway is unlikely. We have performed another control experiment in presence of 20 mol% 6c as additive (Scheme 3C) and observed only 4% yield of the desired product, which might have resulted from catalyst inhibition by the excess olefine.18
Scheme 3.

Control Experiments with Possible Intermediate.
[a] Standard conditions: 1a (0.1 mmol), 4-Iodobiphenyl (2.5 equiv.), Pd(OAc)2 (10 mol %), L14 (15 mol%), K2HPO4 (2.0 equiv.), Ag3PO4 (1.0 equiv.), 4-Iodobiphenyl (2.5 equiv.), HFIP (1.0 mL), 80 °C, under air, 36 h. Here, Ar means 4-biphenyl group.
To get more insight into the mechanistic details of the reaction, we have conducted H/D exchange experiments under standard conditions (Scheme 4). In the absence of aryl iodide, we observed 40% deuteration at the methyl C─H bonds of 1a but no deuterium incorporation on methylene C─H bonds, which means that a reversible β-methyl C─H activation is taking place under these conditions (Scheme 4.A).
Scheme 4.

H/D Exchange Experiments.
Similarly, in the presence of aryl iodide, we also observed 62% deuteration at the methyl C─H bonds, while no deuteration was observed at the β-methylene C─H bonds. These results suggest that the (1) the methyl C─H activation was reversible, but it fails to generate any product under the standard reaction conditions; (2) whereas methylene C─H activations seems irreversible and as soon as C-H activation occurs it converted to product. In another interesting experiment, we performed a reaction with a substrate containing 52% deuteriation (52% deuterium for each β-methylene C─H bond) at the β-methylene C─H and observed that two β-methylene C─H bonds contain 45% and 7% D respectively (Scheme 4.C). These results suggest that the β,γ-dehydrogenation may be acting as a transient intermediate as we didn’t observe any dehydrogenated product in our reaction at any time interval (for details see SI).
In summary, we have developed a Pd(II)-catalyzed tandem γ-C(sp3)─H arylation and γ-lactonization of linear aliphatic acids. The key to the success of this reaction is the use of the bidentate CarboxPyridone ligand. This new reaction enables diverse synthesis of γ-arylated γ-butyrolactones from the simple carboxylic acids in a single step. The scalability and compatibility of this transformation with many functional groups make it suitable for synthetic applications. These γ-arylated γ-butyrolactone products can be readily converted to a wide range of disubstituted tetrahydrofuran derivatives.
Supplementary Material
Acknowledgements
We gratefully acknowledge the NIH (NIGMS R01GM084019) and The Scripps Research Institute for financial support. We thank Dr. Daniel Strassfeld & Dr. Sam Chan for valuable discussion. We thank Nik Chekshin for proof reading. We thank Dr. Laura Pasternack for spectroscopic services. Dr. Jason Chen, Brittany Sanchez, Quynh Nguyen Wong from the Scripps Automated Synthesis Center are acknowledged for their assistance in the high-resolution mass spectrometry analysis.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References:
- (1).(a) Kang EJ, Lee E, Chem. Rev 2005, 105, 4348–4378. [DOI] [PubMed] [Google Scholar]; (b) Janecka A, Wyrębska A, Gach K, Fichna J, Janecki T, T. Drug Discovery Today 2012, 17, 561–572. [DOI] [PubMed] [Google Scholar]; (c) Geske GD, O’Neill JC, Blackwell HE, Chem. Soc. Rev 2008, 37, 1432–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kitson RRA, Millemaggi A, Taylor RJK, Angew. Chem., Int. Ed 2009, 48, 9426–9451. [DOI] [PubMed] [Google Scholar]; (e) Corkery JM, Loi B, Claridge H, Goodair C, Corazza O, Elliott S, Schifano F, Neurosci. Biobehav. Rev 2015, 53, 52–78. [DOI] [PubMed] [Google Scholar]
- (2).(a) Chamorro G, Vega F, Madrigal E, Salazar M, Arzneim.-Forsch 2002, 52, 524–528; [DOI] [PubMed] [Google Scholar]; (b) Chamorro G, Vega F, Madrigal E, Mercado EN, Salazar MA, Toxicol. Lett 2003, 142, 37–43; [DOI] [PubMed] [Google Scholar]; (c) Rasgado LAV, Villanueva I, Diaz FV, Acta Pharmaceutica 2017, 67, 215–226. [DOI] [PubMed] [Google Scholar]
- (3).(a) Wang T-C, Lee K-H, Chen Y-L, Liou S-S, Tzeng C-C, Bioorg. Med. Chem. Lett 1998, 8, 2773–2776; [DOI] [PubMed] [Google Scholar]; (b) Tzeng C-C, Lee K-H, Wang T-C, Han C-H, Chen Y-L, Pharm. Res 2000, 17, 715–719; [DOI] [PubMed] [Google Scholar]; (c) Lee K-H, Huang B-R, Eur. J. Med. Chem 2002, 37, 333–338; [DOI] [PubMed] [Google Scholar]; (d) Huang P-J, Lee K-H, Med. Chem. Res 2011, 20, 1081–1090. [Google Scholar]
- (4).Mulholland DA, McFarland K, Randrianarivelojosia M, Biochem. Syst. Ecol 2006, 34, 365–369. [Google Scholar]
- (5.Dowle MD, Davies DI, Chem. Soc. Rev 1979, 8, 171–197. [Google Scholar]
- (6).Maji R, Ghosh S, Grossmann O, Zhang P, Leutzsch M, Tsuji N, List B, J. Am. Chem. Soc 2023, 145, 8788–8793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) For selected reviews on C─H bond functionalization, see: Chen X, Engle KM, Wang D-H, Yu J-Q, Angew.Chem. Int. Ed 2009, 48, 5094–5115. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Daugulis O, Roane J, Tran LD, Acc. Chem. Res 2015, 48, 1053–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lyons W, Sanford MS, Chem. Rev 2010, 110, 1147–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Lam NYS, Wu K, Yu J-Q, Angew. Chem. Int. Ed 2021, 60, 15767–15790. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Lucas EL, Lam NYS, Zhuang Z, Chan HSS, Strassfeld DA, Yu J-Q Acc. Chem. Res 2022, 55, 537–550. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) He G, Wang B, Nack WA, Chen G, Acc. Chem. Res 2016, 49, 635–645. [DOI] [PubMed] [Google Scholar]; (g) Baudoin O, Chem. Soc. Rev 2011, 40, 4902–4911. [DOI] [PubMed] [Google Scholar]; (h) Zhang Q, Shi B-F, Chem. Sci, 2021, 12, 841–852. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Das J, Mal DK, Maji S, Maiti D, ACS Catal. 2021, 11, 4205–4229. [Google Scholar]; (j) Font M, Gulías M, Mascarenas JL, Angew.Chem.Int.Ed 2022,61, e202112848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) Falbe J, Bauer W, Büchel KH, Houben J, Weyl T, Methods of Organic Chemistry Vol. 5; Thieme, 1985. [Google Scholar]; (b) Goossen LJ, Rodríguez N, K. Angew. Chem. Int. Ed 2008, 47, 3100–3120. [DOI] [PubMed] [Google Scholar]
- (9).(a) Giri R, Maugel N, Li J-J, Wang D-H, Breazzano SP, Saunders LB, Yu J-Q, J. Am. Chem. Soc 2007, 129, 3510–3511. [DOI] [PubMed] [Google Scholar]; (b) Chen G, Zhuang Z, Li G-C, Saint-Denis TG, Hsiao Y, Joe CL, Yu J-Q, Angew. Chem., Int. Ed 2017, 56, 1506–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhu Y, Chen X, Yuan C, Li G, Zhang J, Zhao Y, Nat. Commun 2017, 8, 14904–14101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ghosh KK, van Gemmeren M, Chem. - Eur. J 2017, 23, 17697–17700. [DOI] [PubMed] [Google Scholar]; (e) Shen P-X, Hu L, Shao Q, Hong K, Yu J-Q, J. Am. Chem. Soc 2018, 140, 6545–6549. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Hu L, Shen P-X, Shao Q, Hong K, Qiao JX, Yu J-Q, Angew. Chem., Int. Ed 2019, 58, 2134–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Meng G, Hu L, Tomanik M, Yu J-Q Angew. Chem. Int. Ed 2022, e202214459. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Das J, Pal T, Ali W, Sahoo SR, Maiti D, ACS Catal. 2022,12, 11169–11176. [Google Scholar]
- (10).(a) Dolui P, Das J, Chandrashekar HB, Anjana SS, Maiti D, Angew. Chem. Int. Ed 2019, 58, 13773–13777. [DOI] [PubMed] [Google Scholar]; (b) Liu L, Liu YH, Shi BF, Chem. Sci 2020, 11, 290–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Zhuang Z, Yu C-B, Chen G, Wu Q-F, Hsiao Y, Joe CL, Qiao JX, Poss MA, Yu J-Q, J. Am. Chem. Soc 2018, 140, 10363–10367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).(a) Das J, Dolui P, Ali W, Biswas JP, Chandrashekar HB, Prakash G, Maiti D, Chem. Sci 2020, 11, 9697–9702. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ghosh KK, Uttry A, Mondal A, Ghiringhelli F, Wedi P, Gemmeren M, Angew. Chem. Int. Ed 2020, 59, 12848–12852; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Park HS, Fan Z, Zhu R, Yu J, Angew. Chem. Int. Ed 2020, 59, 12853–12859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).(a) Ghosh KK, Uttry A, Koldemir A, Ong M, van Gemmeren M, Org. Lett 2019, 21, 7154–7157. [DOI] [PubMed] [Google Scholar]; (b) Zhuang Z, Herron AN, Fan Z, Yu J-Q, J.-Q. J. Am. Chem. Soc 2020, 142, 6769–6776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).(a) Zhuang Z, Yu J-Q, Nature 2020, 577, 656–659. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tomanik M, Qian S, Yu J-Q, J. Am. Chem. Soc 2022, 144, 11955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Uttry A, Mal S, van Gemmeren M, J. Am. Chem. Soc 2021, 143, 10895–10901. [DOI] [PubMed] [Google Scholar]
- (16).Hu L, Meng G, Chen X, Yoon JS, Shan J-R, Chekshin N, Strassfeld DA, Sheng T, Zhuang Z, Jazzar R, Bertrand G, Houk KN, Yu J-Q, J. Am. Chem. Soc 2023, 145, 30, 16297–16304. [DOI] [PubMed] [Google Scholar]
- (17).(a) Li Z, Wang Z, Cheksin N, Qian SQ, Qiao JX, Cheng P, Yeung K-S, Ewing WR, Yu J-Q, Science 2021, 372, 1452–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Wang Z, Hu L, Chekshin N, Zhuang Z, Qian S, Qiao JX, Yu J-Q, Science 2021, 374, 1281–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Meng G, Hu L, Chan HSS, Qiao JX, Yu J-Q, J. Am. Chem. Soc 2023, 145, 13003–13007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Hu L, Meng G, Yu J-Q, J. Am. Chem. Soc 2022, 144, 45, 20550–20553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Yang J-M, Lin Y-K, Sheng T, Hu L, Cai X-P, Yu J-Q, Science 2023, 380, 639–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).(a) Sheng T, Zhuang Z, Wang Z, Hu L, Herron AN, Qiao JX, Yu J-Q, J. Am. Chem. Soc 2022, 144, 12924–12933. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sheng T, Kang G, Zhuang Z, Chekshin N, Wang Z, Hu L, Qiao JX, Yu J-Q, J. Am. Chem. Soc 2023, 145, 20951–20958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).(a) He J, Wasa M, Chan KSL, Shao Q, Yu J-Q, Chem. Rev 2017, 117, 8754–8786. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shao Q, Wu K, Zhuang Z, Qian S, Yu J-Q, Acc. Chem.Res 2020, 53, 833–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).(a) He J, Li S, Deng Y, Fu H, Laforteza BN, Spangler JE, Homs A, Yu J-Q, Science 2014, 343, 1216–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chen Y-Q, Wang Z, Wu Y, Wisniewski SR, Qiao JX, Ewing WR, Eastgate MD, Yu J-Q, J. Am. Chem.Soc 2018,140,17884–17894. [DOI] [PubMed] [Google Scholar]
- (25).Das J, Ali W, Ghosh A, Pal T, Mandal A, Teja C, Dutta S, Pothikumar R, Ge H, Zhang X, Maiti D, Nature Chemistry 2023, 10.1038/s41557-023-01295-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Chan HSS, Yang J-M, Yu J-Q, Science 2022, 376, 1481–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Kang G, Strassfeld DA, Sheng T, Chen C-Y, Yu J-Q, Nature 2023, 618, 519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).(a) Fernández-Peña L, Díez-Poza C, González-Andrés P, Barbero A, Drugs. Mar. Drugs 2022, 20, 120.35200649 [Google Scholar]; (b) González-Andrés P, Fernández-Peña L, Díez-Poza C, Barbero A Mar. Drugs 2022, 20, 642. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lin Y-S, Lin J-H, Chang C-C, Lee S-S, J. Nat. Prod 2015, 78, 181–187. [DOI] [PubMed] [Google Scholar]
- (29).In our reaction, we chose 4-iodo-1,1’-biphenyl as a model aryl iodide since it gives good yield to the desired product and all the characteristic 1H NMR peaks for arylated lactone products are well distinguished to monitor the reaction.
- (30).Li Z, Park HS, Qiao JX, Yeung K-S, Yu J-Q J. Am. Chem. Soc 2022, 144, 39, 18109–18116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.