

Abstract
The orphan G protein-coupled receptor (GPCR) GPR161 plays a central role in development by suppressing Hedgehog signaling. The fundamental basis of how GPR161 is activated remains unclear. Here, we determined a cryogenic-electron microscopy structure of active human GPR161 bound to heterotrimeric Gs. This structure revealed an extracellular loop 2 that occupies the canonical GPCR orthosteric ligand pocket. Furthermore, a sterol that binds adjacent to transmembrane helices 6 and 7 stabilizes a GPR161 conformation required for Gs coupling. Mutations that prevent sterol binding to GPR161 suppress Gs-mediated signaling. Surprisingly, these mutants retain the ability to suppress GLI2 transcription factor accumulation in primary cilia, a key function of ciliary GPR161. By contrast, a protein kinase A-binding site in the GPR161 C-terminus is critical in suppressing GLI2 ciliary accumulation. Our work highlights how structural features of GPR161 interface with the Hedgehog pathway and sets a foundation to understand the role of GPR161 function in other signaling pathways.
Introduction
Orphan G protein-coupled receptors (GPCRs) coordinate diverse signaling pathways to control many aspects of human physiology1. The orphan GPCR GPR161 has been characterized as a unique example of a constitutively active receptor that is located within the primary cilium of cells, an organelle that protrudes from the cell surface and locally organizes signaling components2. In its best understood signaling role, GPR161 is a critical negative regulator of the Hedgehog pathway2. Knockout of Gpr161 in mice is embryonically lethal, and the embryos display severe limb, facial, and early nervous system defects indicative of hyperactive Hedgehog signaling3–7. GPR161 mutations in humans lead to developmental defects such as spina bifida8–10, pituitary stalk interruption syndrome11, and cancers such as medulloblastoma4,12. Overexpression of GPR161 has been linked to triple-negative breast cancer13. Like many other orphan GPCRs, however, fundamental mechanisms of GPR161 function remain unknown14, including what stimulus gives rise to GPR161 constitutive activity and how signaling activity downstream of GPR161 impinges on its biological function.
The primary function of GPR161 has been framed by its discovery as a Hedgehog pathway regulator2. Hedgehog signaling during vertebrate embryogenesis mediates multicellular development, including the proper formation of limbs, the face, and the nervous system15. In the presence of the Hedgehog signal, GLI2/3 transcriptional factors accumulate in the primary cilia and form activators (GLI-A)16. In the absence of the Hedgehog signal, GLI2/3 are constitutively phosphorylated by protein kinase A (PKA), which leads to proteolytic conversion of these proteins into Hedgehog pathway repressors (GLI-R). Because PKA is canonically activated by the GPCR second messenger cyclic adenosine monophosphate (cAMP), current models propose that elevated ciliary cAMP levels activate PKA to suppress the Hedgehog pathway17. Although many GPCRs localize to the primary cilium18,19, several observations have placed GPR161 as a unique Hedgehog pathway regulator. Loss of GPR161 function in mice and in fish causes phenotypes consistent with inappropriate Hedgehog pathway activation3–7,20. Furthermore, GPR161 is constitutively active in model cell lines and drives elevated cAMP via activation of Gs2,3,20–22,23,24. Upon Hedgehog pathway activation, GPR161 exits cilia by internalizing to the recycling endocytic compartment2,21.GPR161 is therefore thought to repress Hedgehog signaling by constitutive coupling to Gs, which elevates cAMP levels to drive PKA activity (Fig. 1a).
Figure 1: Structure-inspired deorphanization of GPR161.
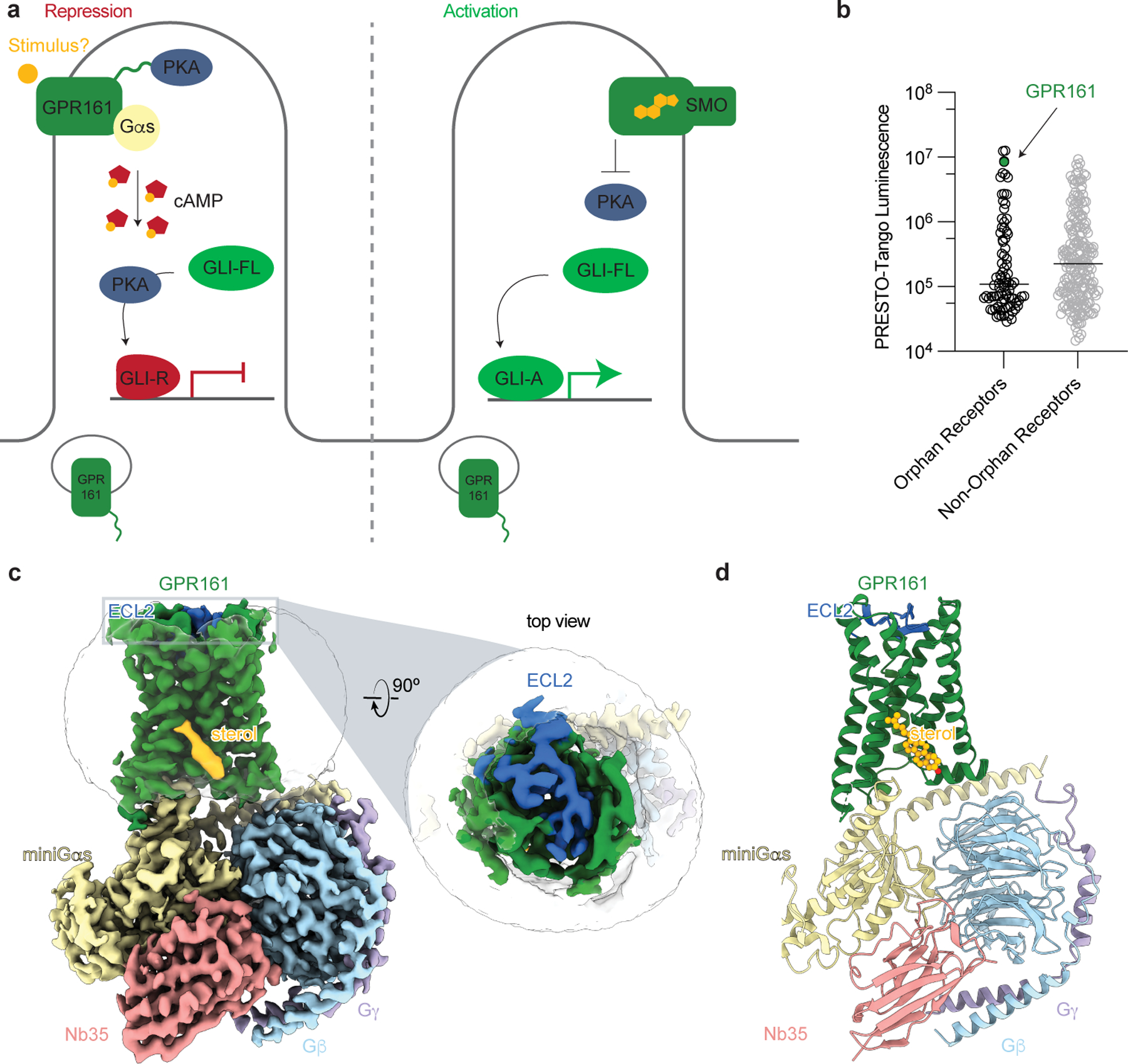
a) The Hedgehog pathway is regulated by two key GPCRs, GPR161 and Smoothened (SMO). In the absence of Hedgehog, GPR161 represses the pathway through constitutive cAMP signaling from an unknown stimulus. In the presence of Hedgehog, SMO activates the pathway by entering the cilia, binding cholesterol, and inhibiting PKA, while GPR161 exits cilia. b) GPR161 yields an exceptionally strong signal for β-arrestin recruitment in the PRESTO-Tango assay when compared across 314 GPCRs (data replotted from Kroeze WM et al 201522, n = 4 for each target, shown as mean ± s.e.m. of technical replicates). This assay is performed in a modified HEK293 cell line, suggesting that GPR161 is constitutively active under heterologous expression conditions. c) Cryo-EM density map of GPR161 in complex with Gs heterotrimer (miniGαs, Gβ, and Gγ) and stabilizing nanobody 35 (Nb35). The map reveals a density consistent with the shape of a sterol (yellow) and an extracellular loop 2 (ECL2, blue) that is packed within the seven transmembrane core of GPR161. d) Ribbon diagram of activated GPR161 heterotrimer complex. Cholesterol is modeled into the sterol density (yellow).
Several fundamental aspects of GPR161 function remain unclear - in particular, the potential stimulus that drives GPR161 activity remains unknown. The C-terminus of GPR161 binds specifically PKA type I regulatory subunits through an A-kinase anchoring protein domain (AKAP), a unique feature of GPR161 among hundreds of GPCRs25. The interdependent roles of Gs coupling and PKA binding, and their relative importance in Hedgehog signaling is also poorly defined. Here we use a combination of cryogenic-electron microscopy (cryo-EM), signaling studies, molecular dynamics simulations, and biochemical assays to determine the molecular mechanism of GPR161 activation and Hedgehog pathway repression. Our studies reveal that GPR161-induced Gs signaling is driven by a sterol-binding site. However, this signaling activity does not repress GLI2 ciliary accumulation, a key role of ciliary GPR161 in Hedgehog pathway repression. By contrast, the AKAP domain in GPR161 is necessary for repressing GLI2 accumulation in cilia. Together, these findings provide an activation mechanism for GPR161 and support PKA as a central downstream ciliary regulator of the Hedgehog pathway.
Results
Cryo-EM structure of active GPR161 bound to Gs heterotrimer
GPR161 is one of the most constitutively active GPCRs tested in the β-arrestin PRESTO-Tango assay22, which uses nonciliated HEK293 cells that normally do not support Hedgehog pathway signaling (Fig. 1b). We reasoned that purification of GPR161 from HEK293 cells may allow us to determine a structure of the active signaling state and may reveal potential stimuli for GPR161. Preparations of GPR161 in the absence of a signal transducer were of poor quality, suggesting that GPR161 alone may be structurally dynamic or otherwise unstable. To stabilize GPR161 in the active state and simultaneously increase the likelihood that the receptor would co-purify with an activating stimulus26, we C-terminally fused the receptor to a minimal version of the Gαs protein. This minimized “miniGs” construct retains the receptor-interacting GTPase domain of the Gαs subunit but is engineered to interact with a GPCR in the absence of a guanine nucleotide27. We purified GPR161-miniGs fusion protein to homogeneity, further complexed it with other heterotrimeric G protein subunits Gβ1 and Gγ2 as well as nanobody 35 (Nb35) to stabilize the interaction between Gαs and Gβ1γ2 (Extended Data Fig. 1)28. The resulting complex was imaged by cryo-EM to yield a reconstruction of the GPR161-miniGs complex at 2.7 Å resolution (Table 1 and Extended Data Fig. 2) and enabled model building for the seven transmembrane domain of GPR161, the Gs subunits, Nb35, and, most notably, a sterol-like molecule (Fig. 1c,d and Extended Data Fig. 3).
Table 1:
Cryo-EM data collection and model statistics.
| GPR161-Gs (EMDB-40603) (PDB 8SMV) | |
|---|---|
| Data collection and processing | |
| Magnification | 105,000 |
| Voltage (kV) | 300 |
| Electron exposure (e–/Å2) | 50.7 |
| Defocus range (μm) | −0.8 to −2.2 |
| Pixel size (Å) | 0.86 |
| Symmetry imposed | C1 |
| Initial particle images (no.) | 9,760,777 |
| Final particle images (no.) | 335,928 |
| Map resolution (Å) FSC threshold |
2.7 0.143 |
| Map resolution range (Å) | 2.7–5.4 |
| Refinement | |
| Initial model used | Alphafold2 model |
| Model resolution (Å) FSC threshold |
3.1 0.5 |
| Model resolution range (Å) | 3.1–50 |
| Map sharpening B factor (Å2) | −119 |
| Model composition Non-hydrogen atoms Protein residues Ligands |
8,169 1,034 1 |
|
B factors (Å2) Protein (GPR161) Protein (G protein/Nb35) Ligand (cholesterol) |
45.0 21.6 53.5 |
| R.m.s. deviations Bond lengths (Å) Bond angles (°) |
0.004 1.033 |
| Validation MolProbity score Clashscore Poor rotamers (%) |
1.12 1.86 0 |
| Ramachandran plot Favored (%) Allowed (%) Disallowed (%) |
96.96 3.04 0 |
Our structure of the GPR161-miniGs complex is similar to many other activated Class A GPCRs bound to heterotrimeric G proteins, like the prototypical β2-adrenergic receptor (β2AR) (Extended Data Fig. 4a). A key hallmark of Class A GPCR activation is outward displacement of transmembrane helix 6 (TM6) to accommodate the C-terminal α-helix of Gα subunit29. While we do not have an inactive structure of GPR161 for comparison, the conformation of TM6 in GPR161 bound to Gs is similar to the outward displaced conformation observed for β2AR (Extended Data Fig. 4a). We conclude that our structure of GPR161-miniGs captures the Gs coupled, active conformation of the receptor.
GPR161 extracellular loop 2 is self-activating
The structure of active GPR161-miniGs revealed a unique conformation of extracellular loop 2 (ECL2) compared to the majority of ligand-activated Class A GPCRs. Notably, the ECL2 of GPR161 forms a beta hairpin that folds over the extracellular face of the receptor to completely occlude the canonical orthosteric ligand binding pocket observed for many other Class A GPCRs (Fig. 1c,d and Fig. 2a). Two sets of interactions anchor ECL2 into the canonical Class A pocket of GPR161: 1) a distributed set of hydrophobic contacts between the deep portion of ECL2 and the GPR161 and 2) ionic interactions between D172/K175 in ECL2 and E293/K298 in ECL3 (Extended Data Figure 4b). Comparison to β2AR highlights that the GPR161 ECL2 occupies the same space that adrenaline occupies in β2AR (Fig. 2b). The conformation of ECL2 in GPR161 is reminiscent of the orphan GPCR GPR52, which contains an ECL2 that also occludes the extracellular face of the receptor (Fig. 2b)30. In GPR52, ECL2 serves as a key determinant of constitutive activity - in effect, GPR52 is “self-activated” by ECL2. Indeed structures of several other orphan GPCRs, including GPR21 and GPR17 have recently revealed similar conformations of ECL2 associated with self-activation (Extended Data Fig. 4c)31,32.
Figure 2: Extracellular loop 2 of GPR161 occupies classic GPCR orthosteric site.
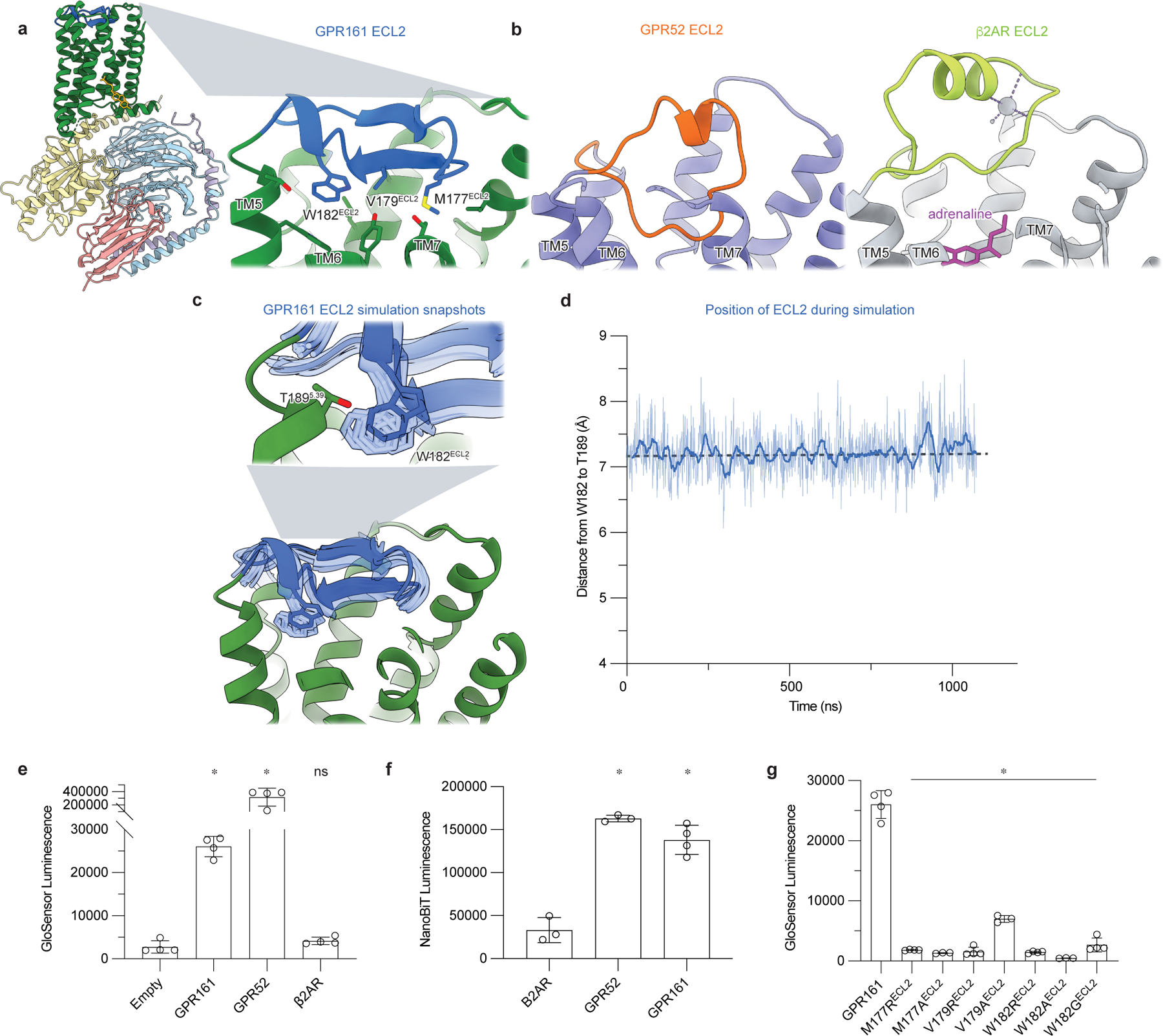
a) The ECL2 of GPR161 makes hydrophobic contacts with the core of the receptor. b) Comparison of ECL2 of the self-activating orphan GPCR GPR52 (PDB ID: 6LI330) and the prototypical agonist-activated GPCR β2-adrenergic receptor (β2AR) bound to agonist adrenaline (PDB ID: 4LDO78). c) In molecular dynamics simulations of GPR161, ECL2 remains in the canonical ligand binding pocket even when mini Gs is removed. Simulation snapshots of ECL2 are shown in light blue, and the cryo-EM structure in dark blue and green. d) Time trace of the distance between residues W182 in ECL2 and T189 in TM4 during a representative molecular dynamics simulation of GPR161 without mini Gs. Thick trace represents a 20 -ns moving average; thin trace represents unsmoothed values. Dashed horizontal line indicates the corresponding distance in the cryo-EM structure. See Methods. Data from remaining simulations is shown in Extended Data Figure 5. e) cAMP production assay for GPR161 (unfused, wildtype), GPR52, and β2AR. GPR161 is constitutively active for cAMP production. Data are mean ± sd, n=3 (for GPR52) or n=4 (for empty, GPR161 and β2AR) biologically independent samples (*P < 0.05; ns, not significant; one-way ANOVA followed by Dunnett’s multiple comparison tests; adjusted P values: GPR161 and GPR52 vs. empty=<0.0001, β2AR vs. empty= 0.9239). f) Nanoluc complementation assay for receptor recruitment of miniGs. Both GPR52 and GPR161 constitutively recruit miniGs. Data are mean ± sd, n=3 (for β2AR, P = <0.0001 and GPR52) or n=4 (for GPR161) biologically independent samples (*P < 0.05; ns, not significant; one-way ANOVA followed by Dunnett’s multiple comparison tests; adjusted P-values: GPR52 and GPR161 vs. β2AR=<0.0001). g) cAMP production assay assessing mutations in ECL2 of GPR161 for residues that make hydrophobic contacts with the transmembrane bundle. Data are mean ± sd, n=4 biologically independent samples (*P < 0.05; ns, not significant; one-way ANOVA followed by Dunnett’s multiple comparison tests; adjusted P values: <0.0001 for all mutants vs. WT GPR161).
We next turned to examine whether the GPR161 ECL2 is stably anchored within the core of GPR161, even in the absence of miniGs. To explore this in atomic detail, we performed molecular dynamics simulations of GPR161 in which we removed miniGs. In these simulations, GPR161 drifts away from the miniGs-bound conformation of the cryo-EM structure, with the binding site for Gs α5 helix frequently occluded due to inward motion of TM6 and TM7 (Extended Data Fig. 5d). Yet, in all six simulations, we observed that ECL2 remains stably in a conformation similar to that observed in the cryo-EM structure (Fig. 2c,d and Extended Data Fig. 5a). The GPR161 ECL2 therefore persistently engages the canonical orthosteric pocket of GPR161, at least within the timescale of our simulations.
How does GPR161 ECL2 influence constitutive activity? To first verify the constitutive activity of GPR161, we used two different cellular assays: GloSensor assay to assess cAMP production and a miniGs recruitment assay33 using an optimized NanoLuciferase fragment complementation termed “NanoBiT”34. Expression of wild-type, unfused GPR161 in suspension adapted HEK293 cells gave consistently high levels of cAMP under basal conditions relative to empty vector and β2AR (13 and 8 fold, respectively) (Fig. 2e and Extended Data Fig. 6). However, GPR161 produced markedly less cAMP than GPR52 (Fig. 2e), perhaps highlighting that self-interaction of ECL2 is not sufficient to drive high basal activity. In a miniGs protein fragment complementation assay, GPR161 basally recruited more miniGs than β2AR, with levels that are more similar to GPR52 (Fig. 2f). The results from these two orthogonal assays demonstrate that GPR161 is constitutively active, albeit to a lesser extent than the self-activating orphan receptor GPR52.
Inspired by the example of GPR52, we examined the possible role of ECL2 in GPR161 activation. Several hydrophobic residues of the GPR161 ECL2 protrude into a region that overlaps with the canonical orthosteric pocket in other Class A GPCRs (Fig. 2a). We targeted several of these (M177ECL2, V179ECL2, W182ECL2) for mutagenesis experiments to understand whether the conformation of ECL2 in GPR161 is important for constitutive activity. We substituted each of these positions with either alanine (to test for simple loss of the side chain) or arginine (to introduce a large perturbation in local hydrophobic contacts). We also examined a W182GECL2 mutant, which has previously been associated with rare cases of spina bifida, a neural tube developmental defect35. Mutation of these hydrophobic residues in ECL2 to either alanine or arginine caused a near complete loss of cAMP generation by GPR161, suggesting that the cis interaction with ECL2 is essential for GPR161 activation (Fig. 2g). We therefore surmise that ECL2 contributes to self-activation of GPR161.
A sterol facilitates GPR161 coupling to Gs
A surprising finding in the cryo-EM map of GPR161-miniGs is the presence of a sterol-like density located at an extrahelical site near the cytoplasmic ends of transmembrane helices 6 and 7 (TM6 and TM7). Although the exact identity of this sterol is unclear, we tentatively modeled a cholesterol molecule in this density. We next sought to understand whether sterols engage this site and if sterol binding at this site leads to signaling output for GPR161. Given the importance of sterols in metazoan Hedgehog pathway signaling36–38, we first examined whether residues surrounding the putative sterol are conserved in evolution. Several key interacting residues (I3237.52, W3277.56 and R3328.51) are conserved from humans to the echinoderm Strongylocentrotus purpuratus (Fig. 3b and Extended Data Fig. 7b).
Figure 3: GPR161-miniGs stably and specifically binds cholesterol.
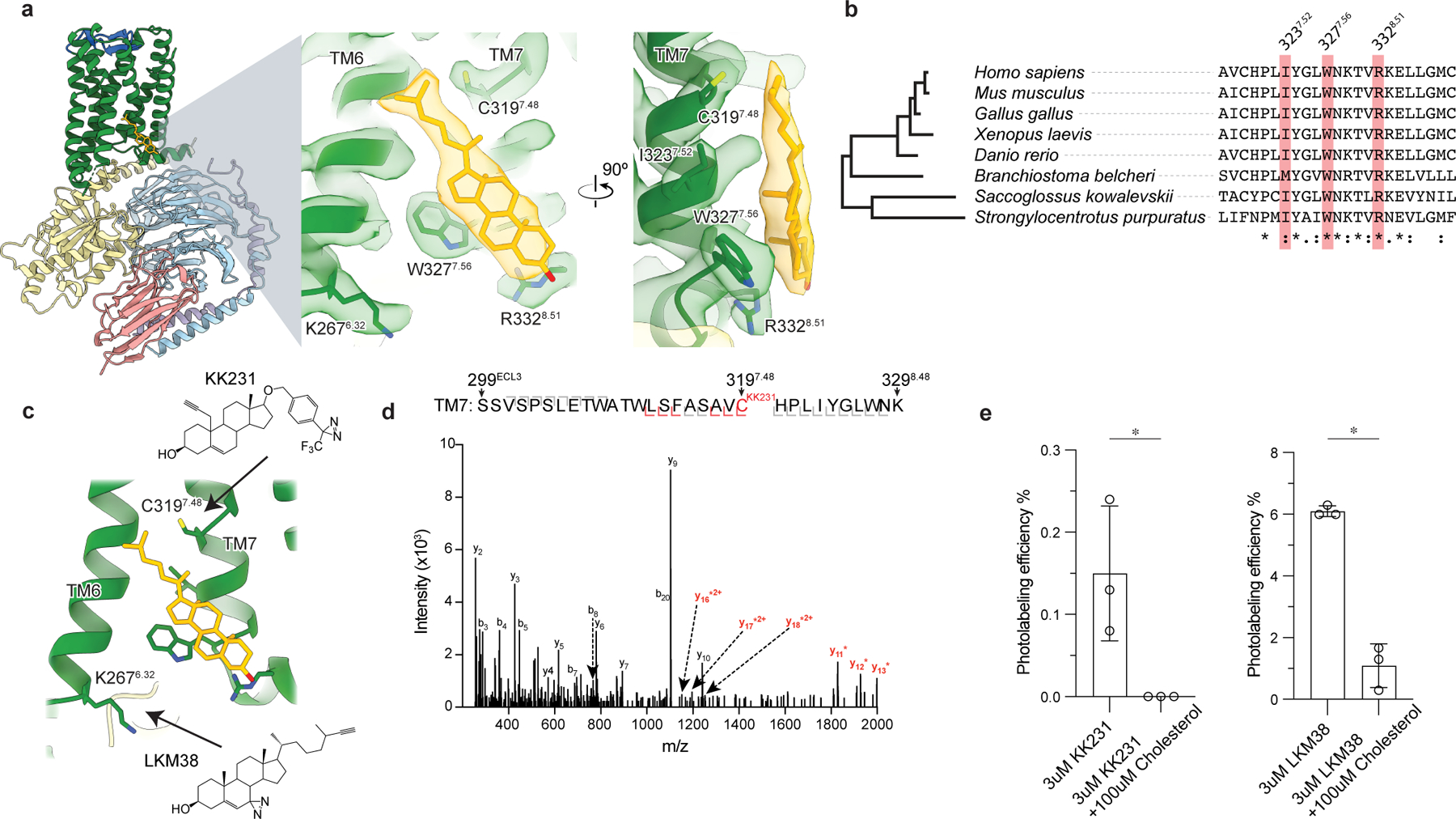
a) Close up view of cholesterol bound to the intracellular side of transmembrane helix 6 (TM6) and TM7. Three key interacting residues (I3237.52, W3277.56, R3328.51) are highlighted as sticks. b) GPR161 cholesterol binding site residues are conserved from humans to sea urchins (Strongylocentrotus purpuratus). A full alignment from these organisms used to define the dendrogram on left is shown in Extended Data Fig. 7. c) Detergent solubilized GPR161-miniGs purified without cholesterol hemisuccinate was incubated with either one of two distinct photoaffinity cholesterol analogs (KK231 or LKM38) and then crosslinked with >320 nm UV light. The resulting photo-labeled preparation was digested with trypsin and analyzed by collision-induced dissociation mass spectrometry, which revealed that KK231 labels position C3197.48 while LKM38 labels K2676.32. d) Product ion spectrum of KK231-labeled GPR161-miniGs sample with peptides mapped to TM7 and Helix 8. This peptide is modified with a mass consistent with KK231 at position C3197.48. Red brackets and peaks indicate product ions that contain the KK231 adduct. e) Photolabeling efficiency of GPR161-miniGs by KK231 and LKM238 in the absence or presence of excess unlabeled cholesterol. Data are mean ± s.d of n=3 technically independent replicates from two independently prepared protein samples (*P < 0.05, Student’s unpaired two-tailed t-test; P values: KK231 vs. KK231+cholesterol=0.0337, LKM38 vs. LKM38+cholesterol= 0.0003).
To determine whether the observed density is indeed a sterol, we performed photoaffinity labeling experiments with purified GPR161-miniGs and two cholesterol analogs: LKM38 and KK231. These sterol analogs contain an ultraviolet light activated diazirine group, either on the B-ring of the steroid (LKM38) or the aliphatic tail (KK231), that rapidly forms covalent adducts with proximal residues. Previous studies have demonstrated that these sterol photoaffinity analogs enable identification of functionally-relevant sterol binding sites in diverse membrane proteins39–42. After photoaffinity labeling GPR161-miniGs, adducted residues were identified by tryptic digestion followed by LC-MS/MS sequencing of the resultant peptides. We obtained 76% sequence coverage of GPR161, with full residue-level sequencing of six of the seven transmembrane helices (Extended Data Fig. 8b). Consistent with our binding pose for cholesterol, the diazirine group in the B-ring of LKM38 labeled K2676.32 in TM6 while the similar functional group in the tail of KK231 labeled residue C3197.48 in TM7 (Fig. 3c,d and Extended Data Fig. 8a). To determine whether the observed photolabeling is specific, we repeated this experiment in the presence of a 33-fold molar excess of unlabeled cholesterol. For both LKM38 and KK231 the presence of unlabeled cholesterol completely suppressed photoaffinity labeling, suggesting that cholesterol itself can bind at this site (Fig. 3e).
We next sought to understand whether cholesterol binding promotes interactions between GPR161 and Gs. In the absence of an inactive-state structure of GPR161, we turned to molecular dynamics simulations to assess if the presence of Gs is required for stable cholesterol binding (Fig. 4a,b and Extended Data Fig. 5b,c). We simulated GPR161 either restrained to remain in the miniGs conformation on the intracellular side or without any restraints. Each condition was simulated with six replicate simulations, each 1 µs in length. When GPR161 is restrained in the miniGs bound conformation, 5 of 6 simulations showed stable cholesterol association with W3277.56 (Extended Data Fig. 5c). By contrast, in 5 of 6 simulations of unrestrained GPR161, we observed that cholesterol rapidly disengages the extrahelical binding site and remains unbound for the remainder of the simulation (Extended Data Fig. 5b). Indeed, in the absence of any restraints, W3277.56 flipped inward into the seven transmembrane core of GPR161, thereby removing a key contact for cholesterol at the extrahelical binding site. Additionally, this rotamer of W3277.56 would occlude binding of the C-terminal α-helix of Gs (Extended Data Fig. 5d). These simulations therefore suggested that cholesterol binding to GPR161 at the TM6/TM7 extrahelical site is cooperative with Gs binding.
Figure 4: Cholesterol binding to GPR161 facilitates Gs coupling.
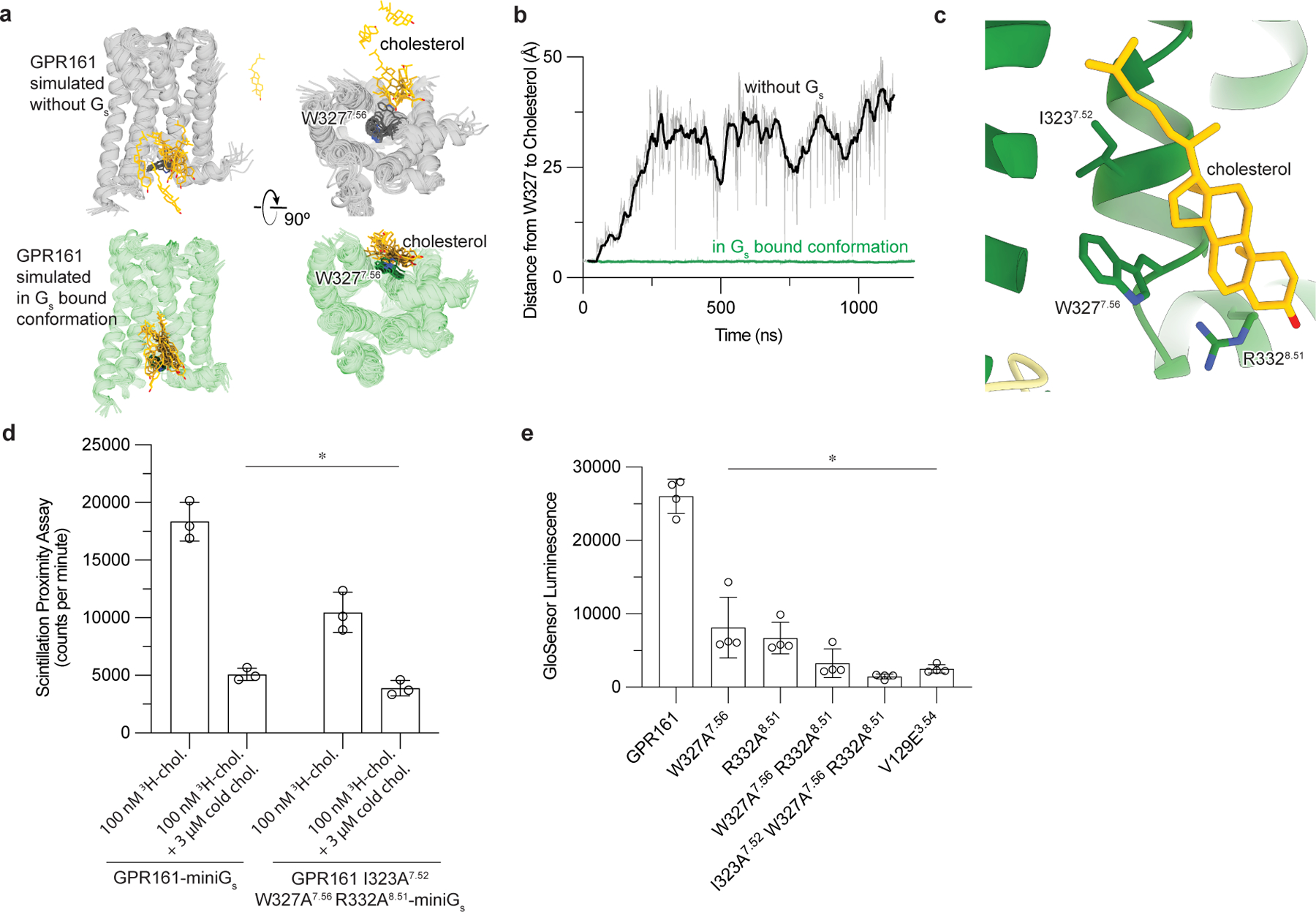
a)In unrestrained simulations of GP161 with miniGs removed, the initially bound cholesterol is highly dynamic and often dissociates from the receptor (top row; simulation snapshots shown in gray and yellow). By contrast, in simulations where GPR161 residues that contact miniGs are restrained to their miniGs bound conformation, cholesterol remains stably bound (bottom row; simulation snapshots shown in green and yellow). b) Time traces from representative simulations under each condition show that cholesterol dissociates in the former case but maintains its contact with residue W3277.56 in the latter case. See Methods. Data from remaining simulations is shown in Extended Data Figure 5. c) Close up view of cholesterol bound to the intracellular side of transmembrane helix 6 (TM6) and TM7. Three key interacting residues (I3237.52, W3277.56, R3328.51) are highlighted as sticks. d) 3H-cholesterol (3H-chol) binding assay for purified GPR161-miniGs and GPR161-AAA7.52, 7.56, 8.51-miniGs. Data are mean ± s.d. from n=3 biologically independent replicates (*P < 0.05; two-way ANOVA followed by Dunnett’s multiple comparison tests; adjusted P values: GPR161-miniGs vs.GPR161-AAA7.52, 7.56, 8.51-miniGs -miniGs=0.0002, GPR161-miniGs vs. GPR161-miniGs + cold chol. and GPR161-AAA7.52, 7.56, 8.51-miniGs + cold chol.=<0.0001). e) GloSensor cAMP production assay assessing mutations in cholesterol binding site of GPR161. Data are mean ± s.d. from n=4 biologically independent replicates (*P < 0.05; one-way ANOVA followed by Dunnett’s multiple comparison tests; adjusted P values: <0.0001 for all mutants vs. GPR161 WT).
If cholesterol potentiates Gs binding, we predicted that disrupting the cholesterol binding site would both decrease cholesterol binding and cAMP production downstream of GPR161-induced Gs activation. Closer examination of the conserved residues in the sterol site highlighted that I3237.52 interacts with the iso-octyl tail of cholesterol, W3277.56 binds cholesterol central rings, and R3328.51 engages the hydroxyl group (Fig. 4c). We generated a GPR161-miniGs construct substituting alanine at these conserved positions (GPR161-I323A7.52-W327A7.56-R332A8.51 and hereafter referred to as GPR161-AAA7.52, 7.56, 8.51) and tested the ability of purified receptor preparations to bind 3H-cholesterol using a scintillation proximity assay (Fig. 4d). In this assay, GPR161-AAA7.52, 7.56, 8.51-miniGs bound cholesterol less than GPR161-miniGs. We observe some residual binding of 3H-cholesterol to GPR161-AAA7.52, 7.56, 8.51-miniGs, suggesting that other residues in GPR161-AAA7.52, 7.56, 8.51-miniGs may interact with cholesterol as well. Supporting the importance of the sterol binding site, alanine mutation of W3277.56 and R3328.51 showed decreased cAMP production in a GloSensor assay compared to wild-type, while the GPR161-AAA7.52, 7.56, 8.51 mutant ablates cAMP production (Fig. 4e). Importantly the GPR161-AAA7.52, 7.56, 8.51 mutant showed even lower levels of cAMP production compared to V129E3.54, a mutant previously designed to directly disrupt the predicted GPR161-Gs interaction2. Similarly, GPR161-AAA7.52, 7.56, 8.51 mutant did not recruit miniGs in a NanoLuciferase complementation assay33 (Fig. 5d) while GPR161-V129E showed a more moderate decrease in miniGs recruitment (Extended Data Fig. 9e). Our combined biochemical, simulation, and signaling studies show that cholesterol, and potentially other sterols, can bind GPR161 to support interactions with Gs, thereby promoting cAMP production.
Figure 5: GPR161 PKA-RI binding, but not cAMP generation, is necessary to repress ciliary trafficking of GLI2.
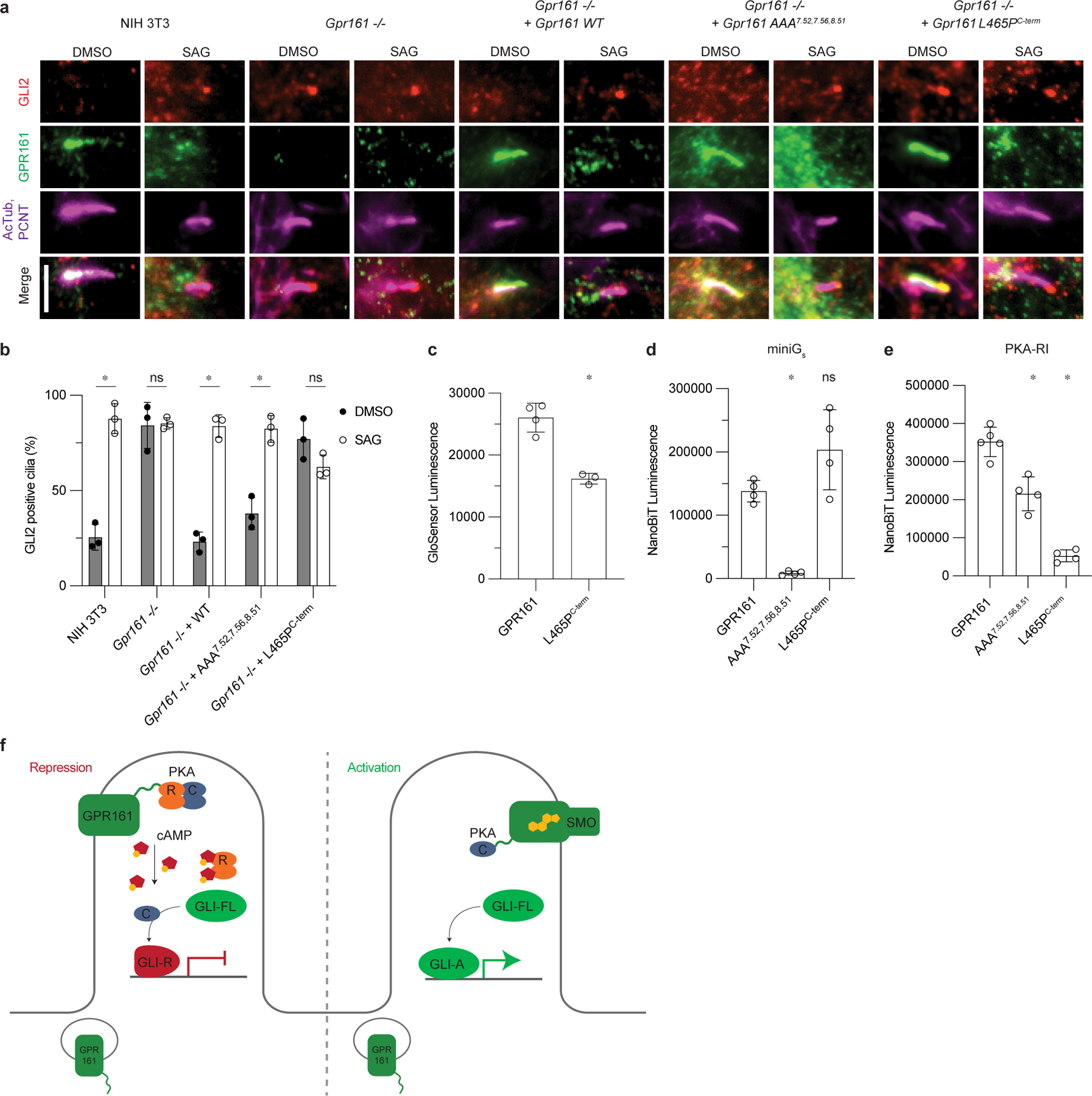
a) Representative images of GPR161 and mutants on ciliary localization and GLI2 repression in ciliary tips in NIH 3T3 cells. NIH 3T3 Flp-In CRISPR based Gpr161−/− cells stably expressing untagged mouse wild-type or Gpr161 mutants were starved for 24 hr upon confluence and were treated for further 24 hr ± SAG. Cells were immunostained with anti-GLI2 (red), anti-GPR161 (green), anti-acetylated, and centrosome (AcTub; PCNT purple) antibodies. Scale bar, 5 µm. b) Quantification GLI2 positive cilia indicating Hedgehog pathway activation. AAA7.52, 7.56, 8.51 rescues function similar to WT, and L465PC-term does not, similar to Gpr161−/−. Data are shown from n=3 biologically independent experiments from images taken from 2–3 different regions/experiment and counting 15–30 cells/region. Data are mean±s.d. (*P < 0.05; ns, not significant; two-way ANOVA followed by Šidák’s multiple comparison tests; adjusted P values for DMSO vs. SAG: NIH3T3=<0.0001, Gpr161−/−=>0.9999, Gpr161−/− + WT or AAA7.52, 7.56, 8.51=<0.0001, Gpr161−/− + L465PC-term=0.1459). c) cAMP production assay assessing L465PC-term mutation. Data are mean±sd, n=3 (for L465PC-term) or n=4 (for GPR161) biologically independent samples (*P < 0.05; one-way ANOVA followed by Dunnett’s multiple comparison tests; adjusted P value for GPR161 vs. L465PC-term=<0.0001). d) Nanoluc complementation assay for receptor recruitment of miniGs. Both GPR161 and L465PC-term constitutively recruit miniGs while AAA7.52,7.56,8.51 does not. Data are mean±sd, n=4 biologically independent samples (*P < 0.05; ns, not significant; one-way ANOVA followed by Dunnett’s multiple comparison tests; adjusted P values: GPR161 vs. AAA7.52,7.56,8.51=0.0018, GPR161 vs. L465PC-term=0.0656). e) Nanoluc complementation assay for receptor recruitment of PKA-RI. GPR161, AAA7.52, 7.56, 8.51, and L465PC-term each recruit less PKA-RI, respectively. Data are mean±sd, n=4 (for AAA7.52,7.56,8.51 and L465PC-term) or n=5 (for GPR161) biologically independent samples (*P < 0.05; ns, not significant; one-way ANOVA followed by Dunnett’s multiple comparison tests; adjusted P values; GPR161 vs. AAA7.52,7.56,8.51= 0.0004, GPR161 vs. L465PC-term=<0.0001). f) PKA-centric model of Hedgehog pathway repression in cilia. In the absence of Hedgehog, GPR161 represses the pathway in cilia through coupling PKA. GPR161 also functions in periciliary endosomal compartments in regulating GLI-R formation3. In the presence of Hedgehog, SMO activates the pathway by entering the cilia, binding cholesterol, and sequestering PKA-C, while GPR161 exits cilia.
cAMP from GPR161 not required for repression of ciliary GLI2
We next aimed to understand how activation of GPR161 leads to Hedgehog pathway repression in cilia. Upon Hedgehog pathway activation, GLI2 proteins accumulate at the tips of cilia43–45. We previously showed removal of GPR161 from cilia, either as a Gpr161 gene knockout or from disruption of GPR161 trafficking to cilia results in accumulation of GLI2 in ciliary tips of resting cells3. We therefore used GLI2 ciliary accumulation as a sensitive test for GPR161 function in this cellular compartment.
GPR161 localizes to primary cilia in Hedgehog pathway responsive NIH 3T3 cells in the basal condition, assessed by co-localization with the ciliary markers acetylated tubulin (AcTub) and pericentrin (PCNT). Concordant with Hedgehog pathway inhibition, GLI2 does not accumulate in the primary cilium in the basal condition (DMSO treatment). Addition of the small molecule Hedgehog pathway agonist SAG leads to exit of GPR161 from cilia and accumulation of GLI2 in the ciliary tip (Fig. 5a). In Gpr161−/− NIH 3T3 cells, GLI2 accumulates in the ciliary tip in the basal condition and addition of SAG does not further increase ciliary GLI2 levels; this phenotype is consistent with loss of ciliary Hedgehog pathway repression by GPR161. Expression of wild-type GPR161 in Gpr161−/− NIH 3T3 cells rescues repression of Hedgehog pathway in cilia, as indicated by a low level of GLI2 positive cilia in the basal condition.
We first examined whether GPR161 ECL2 mutants, which are defective for cAMP production, can rescue GLI2 ciliary suppression. Here, we found that ECL2 mutants of GPR161 failed to accumulate in the primary cilium, suggesting that these mutants are either defective in biogenesis or are impaired in normal trafficking to the primary cilium (Extended Data Fig. 9b). Consistent with a lack of GPR161 localization in cilia, GPR161 ECL2 mutants also failed to suppress GLI2 in ciliary tips.
We next turned to the GPR161-AAA7.52, 7.56, 8.51 and GPR161-V129E3.54 mutants to understand whether cholesterol-dependent Gs activation is important for GPR161 repression of the Hedgehog pathway in cilia. Unlike the ECL2 mutants, Gpr161-AAA7.52, 7.56, 8.51 and Gpr161-V129E3.54 expressed in Gpr161−/− NIH 3T3 cells showed robust ciliary localization similar to wild-type GPR161 (Fig. 5a. and Extended Data Fig. 9a,b). Like wild-type GPR161, activation of the Hedgehog pathway by SAG led to exit of GPR161-AAA7.52, 7.56, 8.51 from the primary cilium. By contrast, GPR161-V129E3.54 did not exit cilia upon Hedgehog pathway activation (Extended Data Fig. 9a,b). Our prior studies with GPR161-V129E3.54 suggested that this mutant reduced cAMP production2; we found here that Gpr161-V129E3.54 did not suppress ciliary GLI2 when expressed in Gpr161−/− NIH 3T3 cells (Extended Data Fig. 9a,c). However, the inability of GPR161-V129E3.54 to exit cilia, combined with residual interaction with miniGs, suggests that this mutation may have consequences beyond decreased cAMP production. Indeed, the V129E3.54 mutation maps to regions of the intracellular GPCR cavity that recent structures have demonstrated are important for engaging GPCR kinases46,47 and β-arrestin48,49. Surprisingly, expression of the Gpr161-AAA7.52, 7.56, 8.51 mutant suppressed GLI2 accumulation, consistent with repression of the Hedgehog pathway in the basal state (Fig. 5a,b). This unexpected result suggests that a Gs signaling-independent function of GPR161 is the predominant mediator for GLI2 repression in cilia in NIH 3T3 cells.
GPR161 is unique among many GPCRs in that it contains a PKA-binding AKAP domain. Previous studies have identified an amphipathic helix in the C-terminus of GPR161 that directly binds PKA regulatory subunits type I (RI)25; introduction of a single point mutant, L465PC-term, breaks this amphipathic helix and prevents PKA type I holoenzyme binding. Compared to the sterol-binding site, this PKA-binding site in GPR161 is less well conserved (Extended Data Fig. 7b). We assessed whether disruption of PKA binding by GPR161 influences Hedgehog pathway repression. In HEK293 cells, expression of GPR161-L465PC-term led to constitutive cAMP production, albeit to a lesser extent than wild-type GPR161 (Fig. 5c). Indeed, GPR161-L465PC-term robustly recruited miniGs in a NanoLuciferase fragment complementation assay33 (Fig. 5d), indicating that this mutation does not influence GPR161 interactions with Gs. Consistent with previous reports, GPR161-L465PC-term recruited less PKA-RI than wild-type GPR161 (Fig. 5e). By contrast, the cAMP deficient mutants GPR161-AAA7.52, 7.56, 8.51 and GPR161-V129E3.54 recruited PKA-RI, albeit to slightly lower levels than the wild-type GPR161 (Fig. 5e and Extended Data Fig. 9f).
Having validated that GPR161-L465PC-term attenuates interaction with PKA-RI, we tested whether this mutant represses the ciliary Hedgehog pathway in Gpr161−/− NIH 3T3 cells. We validated that GPR161-L465PC-term was located in primary cilia in the basal condition (Fig. 5a). Like wild-type GPR161, GPR161-L465PC-term exited cilia upon activation of the Hedgehog pathway by SAG (Fig. 5a and Extended Data Fig. 9b). However, GPR161-L465PC-term was unable to repress GLI2 localization to the ciliary tip in the basal condition, indicating that PKA anchoring by GPR161 is critical for ciliary Hedgehog pathway control (Fig. 5a,b). Indeed, a double mutant combining disruption of Gs coupling and PKA binding (GPR161-AAA7.52, 7.56, 8.51-L465PC-term) was also unable to repress ciliary GLI2 in the basal condition (Extended Data Fig. 9c). We conclude that GPR161 binding to PKA-RI is essential for Hedgehog repression in the primary cilia, while GPR161-induced Gs signaling is dispensable.
Discussion
Our studies illuminate mechanisms of GPR161 activation, and how these mechanisms relate to GPR161-mediated regulation of Hedgehog signal transduction. Our cryo-EM structure of GPR161 revealed two stimuli contributing to GPR161 constitutive activity: first, a self-activating ECL2; and second, a sterol-like density at an extrahelical site. We demonstrate that ECL2 occludes the canonical Class A GPCR orthosteric site and is required for GPR161 trafficking to the primary cilia and cAMP signaling. Indeed, a structural analysis of constitutively active orphan GPCRs published while this work was under review also found a similar conformation of the GPR161 ECL250. We show that cholesterol can bind at the sterol site, that sterol-binding site availability is dependent upon the G protein-bound conformation of GPR161, and that the sterol-binding site regulates cAMP signaling. These two features of GPR161 activation illuminate the basis for Gs-induced cAMP constitutive activity observed in previous studies2,3,21. Neither of these two regions likely accommodates binding of spexin, a peptide recently suggested to activate GPR16124,51. Because ECL2 occludes the classic Class A GPCR orthosteric ligand binding pocket, it is unlikely that this site can accommodate small molecules or peptides. By contrast, the identification of many extrahelical binding sites for small molecule modulators of GPCRs52–56 suggests that this cholesterol binding site may be a potentially druggable site for GPR161 and related orphan GPCRs.
With these fundamental activation properties of GPR161, we provide context into how GPR161 regulates the Hedgehog signaling pathway. A central model for GPR161 function in Hedgehog pathway repression is the importance of constitutive cAMP generation2. Optogenetic and chemogenetic triggers that elevate ciliary cAMP levels repress Hedgehog signaling17. We directly tested the importance of GPR161-induced cAMP production in one aspect of Hedgehog pathway repression, namely suppression of GLI2 transcription factor accumulation in the primary cilium in NIH 3T3 cells. We previously showed exclusion of GPR161 from cilia alone causes an aberrant increase in GLI2 levels in the primary cilium3. Thus, this assay is a precise read out of GPR161 activity in cilia. Surprisingly, a GPR161 mutant that is unable to couple to Gs and support cAMP production (GPR161-AAA) retains the ability to suppress ciliary GLI2 accumulation in NIH 3T3 cells, suggesting that cAMP production of GPR161 is less crucial for Hedgehog pathway repression in cilia than current models suggest. Rather, we find that anchoring of the PKA type I complexes to GPR161 plays a more important role in suppressing GLI2 levels in cilia.
We propose the following model for repression of the Hedgehog pathway by GPR161 in cilia (Fig. 5f). In the absence of Hedgehog, GPR161 bound to PKA is localized to primary cilia. PKA within the cilia phosphorylates GLI resulting in processing into its repressor form. The general presence of ciliary cAMP is important for this process but could be generated by other ciliary GPCRs23, by receptor independent activation57 of adenylyl cyclases by Gs in cilia, or by G-protein independent activity of adenylyl cyclase58. A complete disruption of transport of adenylyl cyclases into cilia from upstream maturation defects during ER-Golgi transit in the secretory pathway, as seen in the Ankmy2 knockout, prevents GLI-R formation in embryos and results in GLI2 accumulation in cilia59. In addition, although basal levels of cAMP in cilia are controversial, with reports ranging from levels comparable to that in the cytoplasm60 to supraphysiological levels (~4.5 µM)58, PKA-C can be activated with sub-optimal cAMP levels61. In the presence of Hedgehog, GPR161 traffics out of the cilia, removing PKA-RI with it62.
Our model does not exclude the role of extraciliary GPR161, particularly of the receptor in the periciliary endosomal compartment3, in GLI-R processing and thereby regulating tissue-specific morpho-phenotypes. We have recently demonstrated that GPR161 is not only localized to primary cilia but is also located in periciliary endosomes3. Both ciliary and extraciliary pools of GPR161 contribute to GLI-R formation and regulate tissue-specific repression of Hedgehog pathway in mice3. Although we show that GPR161 AKAP activity is critical for suppression of GLI2 trafficking in cilia, the AKAP function of GPR161 is not fully necessary for its suppression of Hedgehog pathway phenotypes in zebrafish20. The most parsimonious model explaining these paradoxical results would be that while ciliary AKAP function of GPR161 is critical for suppression of GLI2 trafficking to cilia, GPR161 functions outside cilia through sterol-mediated cAMP signaling. Production of cAMP has been reported for other GPCRs in the endosomal compartment63–66. In resting cells with Hedgehog pathway off, levels of accessible cholesterol are low in the ciliary membrane, but higher in the plasma membrane67. Sterols are also enriched in the endocytic recycling compartment68. Therefore, both the plasma membrane and periciliary endosomal pools could support cAMP production by GPR161. Further experiments in organismal models will be needed to test the role of cAMP, sterols, and AKAP signaling by GPR161 in the extraciliary endosomal compartment in tissue-specific Hedgehog signaling.
Our revised model for GPR161 provides a compelling parallel to recent reports that highlight direct interactions between Smoothened and PKA-C in Hedgehog signaling. As a Gi-coupled GPCR, Smoothened suppression of cAMP generation was initially described as critical for Hedgehog signaling in the fly69. Subsequent studies in vertebrates have called into question the importance of Smoothened-induced Gi signaling in cilia in vertebrates19,70. More recently, the identification of a PKA-inhibitory motif in the Smoothened C-terminus suggests that activated Smoothened directly sequesters the catalytic subunits of PKA (PKA-C) to suppress enzymatic activity71. Instead of acting via cAMP on PKA, we propose that two GPCRs important to Hedgehog signaling, GPR161 and Smoothened, predominantly depend on binary interactions with PKA-C or RI subunits to regulate the Hedgehog pathway in cilia (Fig. 5f).
Our identification of a conserved sterol binding site in GPR161 raises fundamental questions about the role of sterols in control of GPR161 signaling. We compared relative conservation of the GPR161 sterol binding site to the PKA-binding helix in the C-terminus (Extended Data Fig. 7b). Although the sterol binding site is conserved in deuterostome genomes, the PKA-binding motif is not clearly identified in echinoderms (e.g., S. purpuratus), early branching chordates (e.g., B. belcheri), and hemichordates (e.g., S. kowalevskii). The conservation of the cholesterol binding site, and the importance of this site for GPR161 to couple to Gs and generate cAMP, points to sterol driven cAMP generation of GPR161 having an important biological function.
Our finding that the conserved sterol binding site is not critical for controlling GLI2 levels in the primary cilium suggests several possibilities. First, as noted above, it is possible that extraciliary GPR161 uses a mechanism distinct from its ciliary AKAP function to control the Hedgehog pathway. For example, extraciliary GPR161 may depend on cholesterol or other sterols to promote cAMP formation to control GLI-R formation. Second, it is possible that cAMP production by GPR161 has roles in adult tissues outside of the Hedgehog pathway. Indeed, GPR161 is expressed in many different cell types in adult tissues that are not hedgehog regulated, such as the adult hippocampus CA1 region72. GPR161 has also been reported to localize to cilia of hippocampal neurons2,72,73. Ciliary peptidergic GPCR signaling in the CA1 pyramidal neurons has been recently shown to regulate chromatin accessibility74, but the role of cAMP signaling mediated by cilia remains unknown. Third, GPR161 could also have roles in cancers beyond its role in Hedgehog pathway repression in medulloblastoma4,12. For example, GPR161 is overexpressed in triple-negative breast cancer and has been proposed to promote cell proliferation and invasiveness in tumor cells by forming a signaling complex with β-arrestin2 and IQGAP113,75. Our identification of a GPR161 mutant that specifically attenuates cAMP production will enable a careful dissection of these potential roles of GPR161, irrespective of its function in cilia and beyond the role proposed in the Hedgehog pathway.
Our study highlights how GPCR cAMP-PKA signaling establishes precise signaling microdomains in primary cilia. Such restrictive signaling in nanodomains has been an emerging feature of subcellular signaling by cAMP76,77. Most broadly, our work highlights that orphan GPCRs may have functions beyond the biological pathway where they are first encountered. Directly observing the stimuli that activate orphan GPCRs will enable precise approaches to dissect the functional relevance of a specific signaling pathway in a biological outcome. The advent of structure-based methods to interrogate orphan GPCRs will therefore broaden views on the biology coordinated by this fascinating family of understudied proteins.
Methods
Expression and purification of GPR161 active-state complex
The human GPR161 gene with an N-terminal influenza hemagglutinin signal sequence and FLAG epitope was cloned into a pcDNA3.1 vector with zeocin resistance and a tetracycline inducible cassette79. The construct contains an N-terminal influenza hemagglutinin signal sequence and a FLAG epitope The miniGs399 protein26 was fused to the C terminus of GPR161 preceded by a glycine/serine linker and rhinovirus 3C protease recognition site. The resulting fusion construct was transfected into inducible Expi293F-TetR cells (unauthenticated and untested for mycoplasma contamination, Thermo Fisher) using the ExpiFectamine transfection reagent per manufacturer instructions. After 18 h, protein expression was induced with 1 µg/mL doxycycline hyclate for 28 h before collection by centrifugation. Pelleted cells were washed with 50 mL phosphate buffered saline, pH 7.5 before storage at −80 °C.
For complex purification for cryo-EM, frozen cells were hypotonically lysed in a buffer comprised of 20 mM HEPES pH 7.5, 1 mM EDTA, 160 µg/mL benzamidine, 160 µg/mL leupeptin, and 100 µM TCEP for 10 min at 25˚C. The membrane fraction was collected by centrifugation, and the fusion protein was extracted with a buffer comprised of 20 mM HEPES, pH 7.5, 300 mM NaCl, 1% (w/v) lauryl maltose neopentyl glycol (L-MNG, Anatrace), 0.1% (w/v) cholesteryl hemisuccinate (CHS, Steraloids), 2 mM MgCl2, 2 mM CaCl2, 160 µg/mL benzamidine, 2 µg/mL leupeptin, and 100 µM TCEP with dounce homogenization and incubation with stirring for one hour at 4 °C. The soluble fraction was separated from the insoluble fraction by centrifugation and was applied to a column of homemade M1–FLAG antibody-conjugated Sepharose beads at a rate of 1 mL/min. Sepharose resin was then washed with ten column volumes of 20 mM HEPES pH 7.5, 300 mM NaCl, 0.1% (w/v) L-MNG, 0.01% (w/v) CHS, 2 mM MgCl2, 2 mM CaCl2, 100 µM TCEP and then washed with 10 column volumes of 20 mM HEPES pH 7.5, 150 mM NaCl, 0.0075% (w/v) L-MNG, 0.00075% (w/v) CHS, 2 mM MgCl2, 2 mM CaCl2, 100 µM TCEP prior to elution with 20 mM HEPES pH 7.5, 150 mM NaCl, 0.0075% (w/v) L-MNG, 0.00075% (w/v) CHS, 100 µM TCEP, 5 mM EDTA, and 0.2 mg/mL FLAG peptide. The eluted GPR161-miniGs fusion protein was concentrated in a 100 kDa MWCO Amicon spin concentrator, and injected onto a Superdex 200 Increase 10/300GL (Cytiva) gel filtration column equilibrated in 20 mM HEPES pH 7.5, 150 mM NaCl, 100 µM TCEP, 0.0075% (w/v) L-MNG, 0.0025% glyco-diosgenin (GDN, Anatrace), and 0.0005% CHS. Monodisperse fractions of GPR161-miniGs were complexed with Gβ1γ2 heterodimer and Nb35 (purified as described previously80: briefly, Nb35 was expressed in BL21 Rosetta Escherichia coli cells and purified using anti-Protein C antibody-coupled Sepharose, then purified further by size exclusion chromatography using a Superdex S75 Increase 10/300 GL column) at 2-fold molar excess overnight at 4˚C. The next day, the complex was concentrated with a 100 kDa MWCO spin concentrator and excess Gβ1γ2 and Nb35 was removed via size-exclusion chromatography, using a Superdex200 Increase 10/300 GL column (GE Healthcare) equilibrated in 20 mM HEPES pH 7.5, 150 mM NaCl, 100 µM TCEP, 0.0075% (w/v) L-MNG, 0.0025% (w/v) GDN, and 0.00075% CHS. The resulting complex was concentrated to 2.9 mg/mL with a 100 kDa MWCO spin concentrator for preparation of cryo-EM grids.
Two separate preparations of GPR161-miniGs were made for biochemical experiments that deviated slightly from the purification protocol for cryoEM. For cholesterol photolabeling experiments, GPR161-miniGs was expressed and purified using the above protocol except CHS and GDN was excluded from all buffers. For 3H-cholesterol binding experiments, n-Dodecyl-𝜷-D-Maltopyranoside (DDM, Anatrace) was used in lieu of LMNG at a final concentration of 0.02% and CHS and GDN were excluded from all buffers. The resulting size exclusion chromatography-purified protein samples were flash frozen in liquid nitrogen for downstream assay use.
Cryo-EM vitrification, data collection, and processing
The GPR161-miniGs-Gβγ-Nb35 complex was concentrated to 3 mg/mL and 3 µl was applied onto a glow-discharged 300 mesh 1.2/1.3 gold grid covered in a holey carbon film (Quantifoil). Excess sample was removed with a blotting time of 3 s and a blotting force of 1 at 4 °C prior to plunge freezing into liquid ethane using a Vitrobot Mark IV (Thermo Fisher). A total of 8,294 movies were recorded with a K3 detector (Gatan) on a Titan Krios (Thermo Fisher) microscope operated at 300 keV with a BioQuantum post-column energy filter set to a zero-loss energy selection slit width set of 20 eV. The 60-frame movies were recorded for 2.6 s at a physical pixel size of 0.86 Å per pixel and a defocus range of −0.8 to − 2.2 µm for a total dose of 50.7 e−/Å2. Exposure areas were acquired with an automated image shift collection strategy using EPU (Thermo Fisher).
Movies were motion-corrected and dose-fractionated on-the-fly during data collection using UCSF MotionCor281. Corrected micrographs were imported into cryoSPARC v3.1 (Structura Biotechnology)82 for CTF estimation via the Patch CTF Estimation job type. Micrographs with a CTF fit resolution of > 5 Å were excluded from further processing. Templates for particle picking were generated using a 20 Å low-pass filtered model generated from an ab initio model made from blob-picked and 2D classified particles. Template picking yielded 9,760,777 particles, which were extracted in a 288-pixel box and Fourier cropped to 72 pixels. Particles were classified in 3D with alignment using the 20 Å low-pass filtered ab initio model and three “random” reconstructions generated from a prematurely terminated ab initio reconstruction job, called “garbage collectors,” with the Heterogeneous Refinement job type. Two rounds of Heterogeneous Refinement yielded 3,033,326 particles that were re-extracted in the same box sized cropped to 144 pixels. Additional Heterogeneous refinement and extraction without binning yielded 1,143,566 particles that were refined using the Non-Uniform Refinement job type. Particles were exported using csparc2star.py from the pyem script package83, and a mask covering the 7TM domain of GPR161 was generated using the Segger tool in UCSF ChimeraX84 and the mask.py script in pyem. The particles and mask were imported into Relion v3.085 and classified in 3D without alignment. Particles comprising the three highest resolution classes were reimported into cryoSPARC for Non-Uniform Refinement. Finally, particles were exported into cisTEM86 for two local refinements using the Manual Refinement job type and low-pass filtering outside of masks. In the first local refinement, the previous 7TM mask was used, and the second local refinement used a full-particle mask.
Model building and refinement
Model building and refinement began with the Alphafold287 predicted structure as the starting model, which was fitted into the experimental cryoEM map using UCSF ChimeraX. The model was iteratively refined with real space refinement in Phenix88 and manually in Coot89. The cholesterol model and rotamer library were generated with the PRODRG server90, docked using Coot, and refined in Phenix. Final map-model validations were carried out using Molprobity and EMRinger in Phenix.
cAMP signaling assays
We measured cAMP production to determine activation of Gs signaling by GPR161. For each GPR161 construct (wild-type (WT), M177R, V179R, W182G, W182R, W327A, W327R, R332A, AA, AAA, L465P), a 2 mL suspension culture of Expi293F-TetR cells was co-transfected with a 3:1 ratio of a pcDNA3.1 plasmid expressing GPR161 and a luciferase-based cAMP biosensor, pGlosensor-22F (Promega). Surface expression levels of constructs were titrated to similar levels with doxycycline and measured by flow cytometry using an Alexa-647 conjugated anti-M1 Flag antibody. Cells were collected 24 h post-induction, resuspended in Expi293 expression media (Gibco) supplemented with 10% DMSO, and gradually frozen to −80 ˚C in a Mr. Frosty Freezing container for future use. To perform the assay, frozen cells were rapidly thawed in a 37˚C water bath and resuspended in fresh Expi293 expression medium. Cells were diluted to a final concentration of 1e6 cells per mL in Expi293 expression medium plus 2% (v/v) Glosensor assay reagent (Promega) and intubated for 75 min at room temperature with gentle rotation. Cells were then plated in a white 384-well plate (Greiner) to a final density of 15,000 cells per well. Immediately after cell addition, luminescence was measured using a CLARIOstar instrument. Statistical analyses were performed using one-way ANOVA followed by Dunnett’s multiple comparison tests between all possible pairs using GraphPad Prism 9 (Dotmatics).
3H-Cholesterol binding assay
To measure cholesterol binding to GPR161, we developed a scintillation proximity assay (SPA) using purified receptor and 3H-cholesterol (PerkinElmer). To capture M1-FLAG tagged receptor, we used Protein A coated beads and purified M1-FLAG antibody. Each binding reaction was performed in a final volume of 100 µL in a binding buffer comprised of 0.01% dodecylmaltoside, 20 mM HEPES pH 7.5, 150 mM NaCl, 100 nM purified M1-FLAG antibody, and 2 mM CaCl2 (to enable M1-FLAG tag binding to antibody). SPA beads were added to a final concentration of 0.675 mg/mL, 3H-cholesterol was added to 100 nM, and 100 nM of purified GPR161-miniGs or GPR161-AAA7.52, 7.56, 8.51-miniGs was added to start the reaction. For competition with cold cholesterol a 3 µM final concentration was used. The reactions were incubated with shaking at room temperature for 24 hrs, and bound 3H-cholesterol was measured in a scintillation counter (Perkin Elmer). Statistical analyses were performed using two-way ANOVA followed by Dunnett’s multiple comparison tests between all possible pairs using GraphPad Prism 9 (Dotmatics).
Photolabeling and MS analysis
Photolabeling reagents were synthesized as previously described39–41 and stored in the dark at −20˚C as 10 mM stocks in ethanol. Aliquots of the photolabeling reagents (KK231 or LKM38) were air dried in the dark at room temperature and resolubilized in 20 mM HEPES pH 7.5, 150 mM NaCl, 0.0075% LMNG (no CHS or GDN) containing 20 µg of purified GPR161-miniGs in a total volume of 50 µL. The protein was incubated with the photolabeling regent for one hour in the dark at 4 ˚C and then irradiated in a quartz cuvette with >320 nm UV light as previously described91. Briefly, a photoreactor using a 450-watt Hanovia medium pressure mercury lamp was used as the light source and a 1.5 cm thick saturated copper sulfate solution was used as a filter to absorb light of wavelength <315 nm. For site identification experiments, the photolabeling reagent concentration was 100 µM. For cholesterol competition experiments, aliquots of ethanolic stocks of the photolabeling reagent and cholesterol (10 mM stock) were added into the same tube and air dried prior to solubilization with GPR161-miniGs. The final concentration of the photolabeling reagents was 3 µM and of cholesterol 100 µM.
For mass spectrometric analysis, the samples were desalted using Biospin 6 columns (BioRad, CA), equilibrated with 50 mM triethylammonium bicarbonate and 0.02%(w/v) n-Dodecyl-β-D-Maltoside. The proteins were sequentially reduced with 5 mM tris(2-carboxyethyl)phosphine (TCEP) for 30 mins, alkylated with 5 mM N-ethylmaleimide (NEM) in the dark for 45 mins, and quenched with 5 mM dithiothreitol (DTT) for 15 mins. These three steps were done at room temperature. The proteins were digested with 8 µg trypsin at 4 ˚C for one week at which time the digestions were terminated by addition of formic acid (FA) to a final concentration of 1%.
The resultant peptides were analyzed with an OrbiTrap ELITE mass spectrometer (Thermo Fisher Scientific). Briefly, 15 μL samples were injected by an autosampler (UltiMate 3000 UHPLC system; ThermoFisher) onto a home-packed polymeric reverse phase PLRP-S (Agilent, Santa Clara, CA) column (10 cm × 75 μm, 300 Å) at a flow rate of 800 nL/min. A 10%-95% acetonitrile (ACN) gradient was applied for 150 minutes to separate peptides. Solvent A was 0.1% FA/water, and solvent B was 0.1% FA/ACN. The following gradient was applied: isocratic elution at 10% solvent B, 1–60 minutes; 10%–95% solvent B, 60–110 minutes; 95% solvent B, 110–140 minutes; 95%–10% solvent B, 140–145 minutes; 10% solvent B, 145–150 minutes. For the first 60 minutes, a built-in divert valve on the mass spectrometer was used to remove the hydrophilic contaminants from the mass spectrometer. Mass spectra (MS1) were acquired at high resolution (resolution of 60,000) in the range of m/z = 100–2,000. Top 20 ion precursors in MS1 were selected for MS2 using data-dependent acquisition with exclusion of singly charged precursors. Fragmentation was performed with high-energy dissociation (HCD) using a normalized collision energy of 35%. Product ion spectra (MS2) were acquired at a resolution of 15,000.
The data were searched against a database containing the sequence of GPR161-miniGs using PEAKS Studio X pro (Bioinformatics Solutions Inc, Waterloo, ON, Canada) with the following settings: precursor ions mass accuracy of 20 ppm, fragmentation ion accuracy of 0.1 Da, up to three missed cleavages on either side of peptide with trypsin digestion; methionine oxidation, cysteine alkylation with NEM and DTT, any amino acids with adduct of LKM38 (mass = 396.34) and KK231 (mass = 484.26) were included as variable modifications. The searched results were filtered with a 1% false discovery rate and the detected peptides were confirmed by manual analysis for monoisotopic mass accuracy and retention time with Xcalibur 2.2 (ThermoFisher). Fragment ions were also manually confirmed and were accepted based on the presence of a monoisotopic mass within 20 ppm mass accuracy. Photolabeling efficiency was estimated by generating selected ion chromatograms (SIC) of both unlabeled and photolabeled peptides, determining the area under the curve and calculating efficiency as: labeled peptide / (unlabeled peptide + labeled peptide). Statistical significance was analyzed with Student’s unpaired t-test using GraphPad Prism 9 (Dotmatics).
NanoBiT recruitment assays
We measured protein recruitment to determine functionality of GPR161 mutants. For each GPR161 construct (wild-type (WT), AAA, L465P, V129E), as well as for β2AR and GPR52, a 2-mL suspension culture of Expi293F-TetR cells was co-transfected with a 500 ng of a pcDNA3.1 plasmid expressing receptor fused C-terminally to smBiT and 100 ng of transducer fused to lgBiT (miniGs: N-terminally, PKA-RI: C-terminally). Surface expression levels of constructs were titrated to similar levels with doxycycline and measured by flow cytometry using an Alexa-647 conjugated anti-M1 FLAG antibody. After 24 h of induction, cells were centrifuged at 300xg, and resuspended in DPBS at a concentration of ~55,000 cells per 200 µL. 40 µL of 30 µM coelenterazine-h diluted in PBS was added to cells for a final concentration of 5 µM. Cells were incubated for ~30 min at room temperature with gentle shaking. Luminescence was measured using a CLARIOstar instrument. Statistical analyses were performed using one-way ANOVA followed by Dunnett’s multiple comparison tests between all possible pairs using GraphPad Prism 9 (Dotmatics).
Molecular dynamics simulations
Simulation setup
We performed simulations of GPR161 with cholesterol under two conditions: A) GPR161 unrestrained (6 independent simulations, roughly 1 μs each) B) GPR161 restrained to its G protein—bound conformation (6 independent simulations, roughly 1 μs each). For all conditions, the initial structures were based on the cryoEM structure reported [MOU1] in this paper and were prepared using Maestro (Schrödinger, LLC). In both conditions, the nanobody and G protein were removed from the structure.
Missing amino acid side chains were modeled using Prime (Schrödinger, LLC). Neutral acetyl and methylamide groups were added to cap the N- and C-termini, respectively, of the protein chains. Titratable residues were kept in their dominant protonation state at pH 7.4. Histidine residues were modeled as neutral, with a hydrogen atom bound to either the delta or epsilon nitrogen depending on which tautomeric state optimized the local hydrogen-bonding network. Dowser 92 was used to add water molecules to protein cavities. GPR161 was aligned on the receptor in the crystal structure of Prostaglandin E2 receptor EP2 subtype (PDB ID: 7CX4) 93 in the Orientation of Proteins in Membranes (OPM) database 94. The aligned structures were inserted into a pre-equilibrated palmitoyloleoyl-phosphatidylcholine (POPC) membrane bilayer using Dabble 95. Sodium and chloride ions were added to neutralize each system at a concentration of 150 mM. To simulate the G protein–bound conformation in condition B, 0.5 kcal∙mol-1 ∙Å-2 restraints were applied throughout the production simulation on non-hydrogen atoms of GPR161 residues that are within 4 Å of the G protein in the experimentally determined structure. These residues were: 125, 128, 129, 130, 131, 132, 133, 135, 136, 137, 211, 214, 215, 218, 219, 267, 268, 271, 272, and 327. Both final systems consist of 52716 atoms, including 140 lipid molecules and 9810 water molecules (initial system dimensions: 85 Å x 80 Å x 82 Å).
Simulation protocols
For each simulation, initial atom velocities were assigned randomly and independently. We employed the CHARMM36m force field for protein molecules, the CHARMM36 parameter set for lipid molecules and salt ions, and the associated CHARMM TIP3P model for water 96,97. Simulations were run using the AMBER20 software 98 under periodic boundary conditions with the Compute Unified Device Architecture (CUDA) version of Particle-Mesh Ewald Molecular Dynamics (PMEMD) 99 on one GPU.
The systems were first heated over 12.5 ps from 0 K to 100 K in the NVT ensemble using a Langevin thermostat with harmonic restraints of 10.0 kcal∙mol-1 ∙Å-2 on the non-hydrogen atoms of the lipids, protein, and cholesterol. Initial velocities were sampled from a Boltzmann distribution. The systems were then heated to 310 K over 125 ps in the NPT ensemble. Equilibration was performed at 310 K and 1 bar in the NPT ensemble, with harmonic restraints on the protein and cholesterol non-hydrogen atoms tapered off by 1.0 kcal∙mol-1 ∙Å-2 starting at 5.0 kcal∙mol-1 ∙Å-2 in a stepwise manner every 2 ns for 10 ns, and finally by 0.1 kcal∙mol-1 ∙Å-2 every 2 ns for an additional 18 ns. Except for the restrained residues listed above in condition B, all restraints were completely removed during production simulation. Production simulations were performed at 310 K and 1 bar in the NPT ensemble using the Langevin thermostat and Berendsen barostat.
Lengths of bonds to hydrogen atoms were constrained using SHAKE, and the simulations were performed using a timestep of 4.0 fs while using hydrogen mass repartitioning 100. Non-bonded interactions were cut off at 9.0 Å, and long-range electrostatic interactions were calculated using the particle-mesh Ewald (PME) method with an Ewald coefficient (β) of approximately 0.31 Å and B-spline interpolation of order 4. The PME grid size was chosen such that the width of a grid cell was approximately 1 Å. Snapshots of the trajectory were saved every 200 ps.
Simulation analysis protocols
The AmberTools17 CPPTRAJ package 101 was used to reimage trajectories at 1 ns per frame, Visual Molecular Dynamics (VMD) 102 was used for visualization and analysis, and PyMOL (The PyMOL Molecular Graphics System, Schrödinger, LLC) was used for renderings.
Plots of time traces from simulations were generated with Matplotlib 103 and show both original, unsmoothed traces (thin, transparent lines) and traces smoothed with a moving average (thick lines), using an averaging window of 20 ns. All traces include the initial equilibration with harmonic restraints on the protein and cholesterol non-hydrogen atoms.
To monitor ECL2 movement, we measure the minimal distance between all atoms of W182 and T189 in each simulation frame(Fig. 2d and Extended Data Fig. 5a). To capture cholesterol motion, we measure the minimal distance between all non-hydrogen atoms of cholesterol and W327 (Fig. 4b and Extended Data Fig. 5b,c).
Ciliary localization and Hedgehog pathway activation
Cell lines
NIH 3T3-FlpIn cells were authenticated by and purchased from Thermo Fisher Scientific. They have tested negative for Mycoplasma. The Gpr161−/− NIH 3T3 Flp-In cell line was a gift from Rajat Rohatgi23. The cells were cultured in DMEM-high glucose media (D5796; Sigma) with 10% BCS (Sigma-Aldrich), 0.05 mg/ml penicillin, 0.05 mg/ml streptomycin, and 4.5 mM glutamine. Stable expression in knockout cells were generated by retroviral infection with pBABE constructs having untagged wild type or mutant mouse Gpr161 inserts followed by antibiotic selection. Single or multiple amino acid mutations in full-length Gpr161 were generated using Q5 site-directed mutagenesis kit (NEB).
Reverse transcription, quantitative PCR
RNA was extracted using the GenElute mammalian total RNA purification kit (RTN350; Sigma) from the cultured cells. Genomic DNA was eliminated by DNase I (D5307; Sigma). qRT-PCR was performed with the SYBR Green Quantitative RT-qPCR Kit (QR0100; Sigma). Primer sequences were: Hprt-F 5’- AGG GAT TTG AAT CAC GTT TG-3’, Hprt-R 5’- TTT CAT GGC AAC ATC AAC AG-3’, Gpr161-F 5’- TGC CAT CGA TCG CTA CTA CG-3’, Gpr161-R 5’- CAC TTG AAT TCG TCG AAT TCG ACA G-3’. The Gpr161-F/R oligos were specifically designed to detect a wobbled siRNA-resistant version of the mouse Gpr161 open reading frame that we use for rescue experiments. Reactions were run in CFX96 Real time System (Bio Rad).
Immunostaining and microscopy
For immunofluorescence experiments in cell lines, cells were cultured on coverslips until confluent and starved for 48 h. To quantify ciliary GLI2 and GPR161 levels, cells were treated with 500 nM SAG or DMSO for 24 h after 24 h of serum starvation. Cells were fixed with 4% PFA for 10 min at room temperature. After blocking with 5% normal donkey serum, the cells were incubated with primary antibody solutions for 1 h at room temperature followed by treatment with secondary antibodies for 30 min along with DAPI. Primary antibodies used were against GPR161 (1:200, custom-made)21, acetylated α-tubulin (mAb 6-11B-1, Sigma; 1:2000), GLI2 (1:500, gift from Jonathan Eggenschwiler)104, pericentrin (611814, BD Biosciences; 1:500). Coverslips were mounted with Fluoromount-G and images were acquired with a Zeiss AxioImager.Z1 microscope using a 40x oil immersion objective lens.
Quantification and statistical analysis
Cilia positive for GLI2 or GPR161 in Gpr161−/− cells expressing untagged wild-type or mutant GPR161 were counted. Statistical analyses were performed using two-way ANOVA followed by Šidák’s multiple comparison tests between all possible pairs using GraphPad Prism 9 (Dotmatics).
Extended Data
Extended Data Fig. 1. Biochemical preparation of GPR161-miniGs complex.
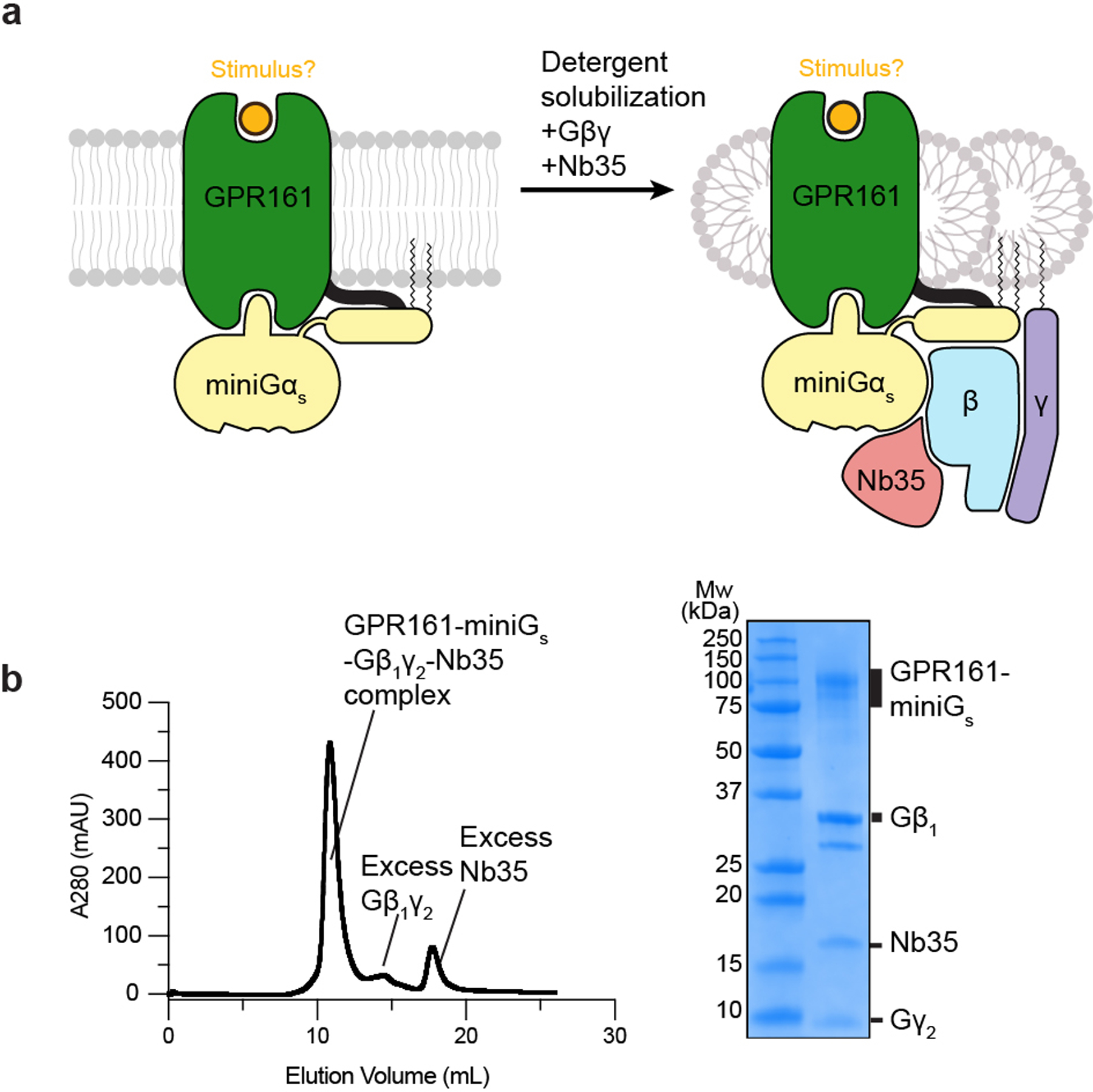
a) Cartoon depiction of GPR161 stabilization, solubilization, and purification. b) Size-exclusion chromatogram (left) and SDS-PAGE gel (right) of purified GPR161-Gs complex with Nb35. Purification and SDS-PAGE gel were done once and not repeated. Chromatogram and SDS-PAGE gel are shown for preparation used for cryo-EM analysis.
Extended Data Fig. 2. Cryogenic electron microscopy processing of GPR161.
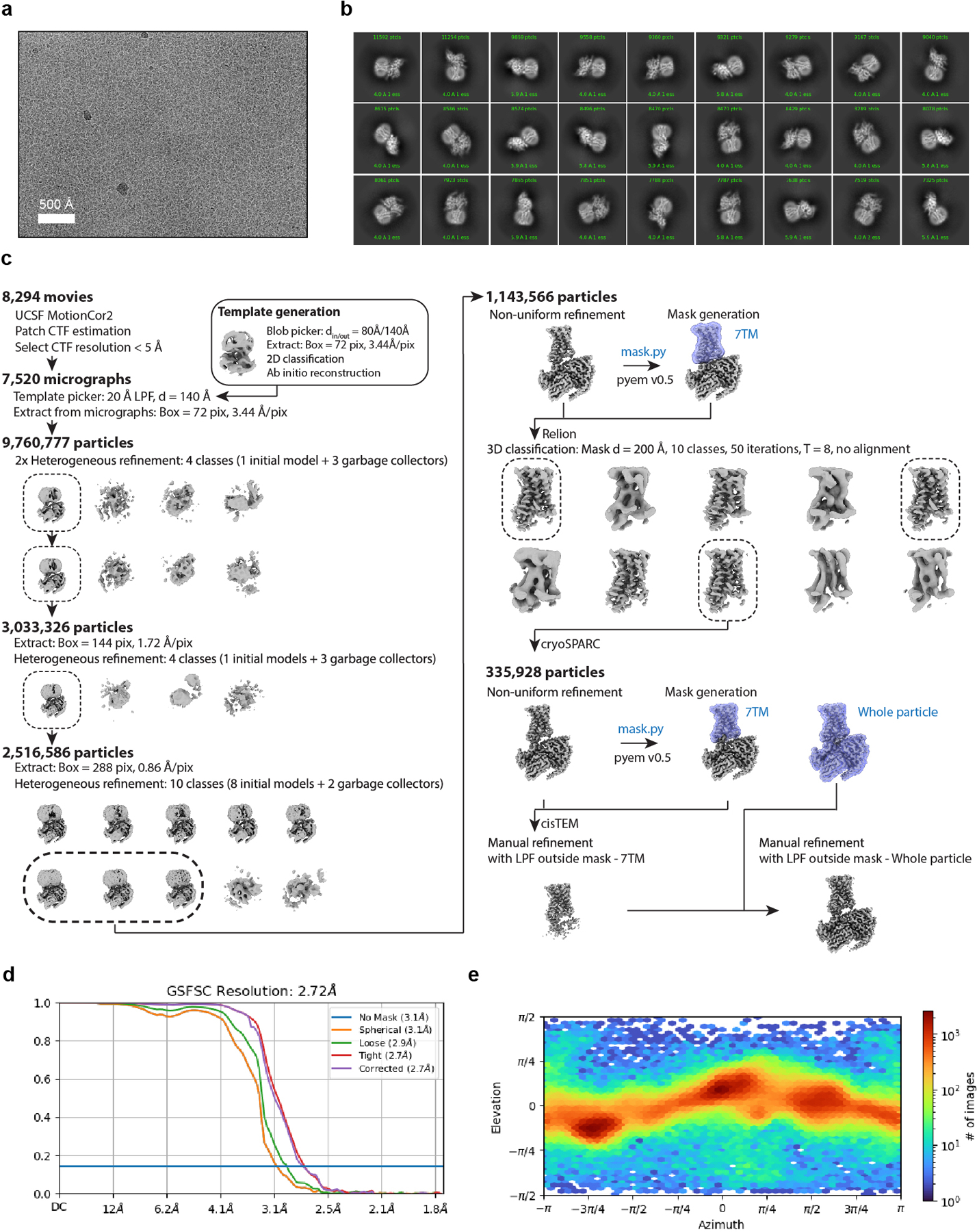
a) A representative motion-corrected cryogenic electron microscopy (cryo-EM) micrograph obtained from a Titan Krios microscope (n = 8,294). b) A subset of highly populated, reference-free 2D-class averages. c) Schematic showing the cryo-EM data processing workflow. Initial processing was performed using UCSF MotionCor2 and cryoSPARC. Particles were transferred using the pyem script package to RELION for alignment-free 3D classification. Finally, particles were processed in cisTEM using the manual refinement job type with a 7TM mask followed by a full particle mask. Dashed boxes indicated selected classes. d) Gold-standard Fourier Shell Correlation (GSFSC) curve for final refined and sharpened map computed in cryoSPARC. e) Euler angle distribution of final refined map computed in cryoSPARC.
Extended Data Fig. 3. Cryo-EM local density.
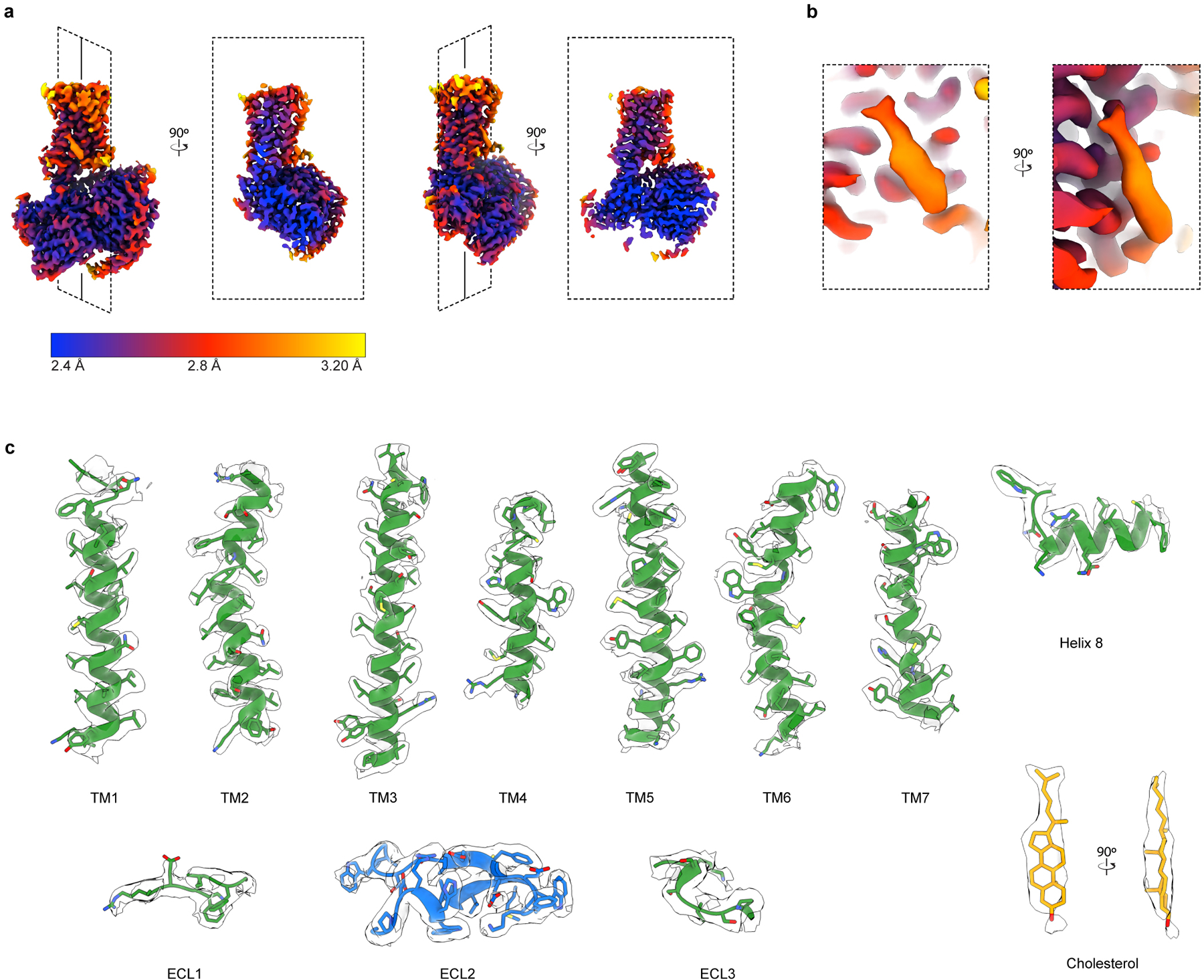
a) Orthogonal views of local resolution for the sharpened, final map of GPR161-Gs complex computed with local resolution in cryoSPARC. b) Close up of local resolution for sterol density. c) Isolated cryo-EM densities from the unsharpened, final map of GPR161 complex. Shown are the transmembrane (TM) helices, extracellular loops, and cholesterol-like density.
Extended Data Fig. 4. Comparison to additional GPCR structures.
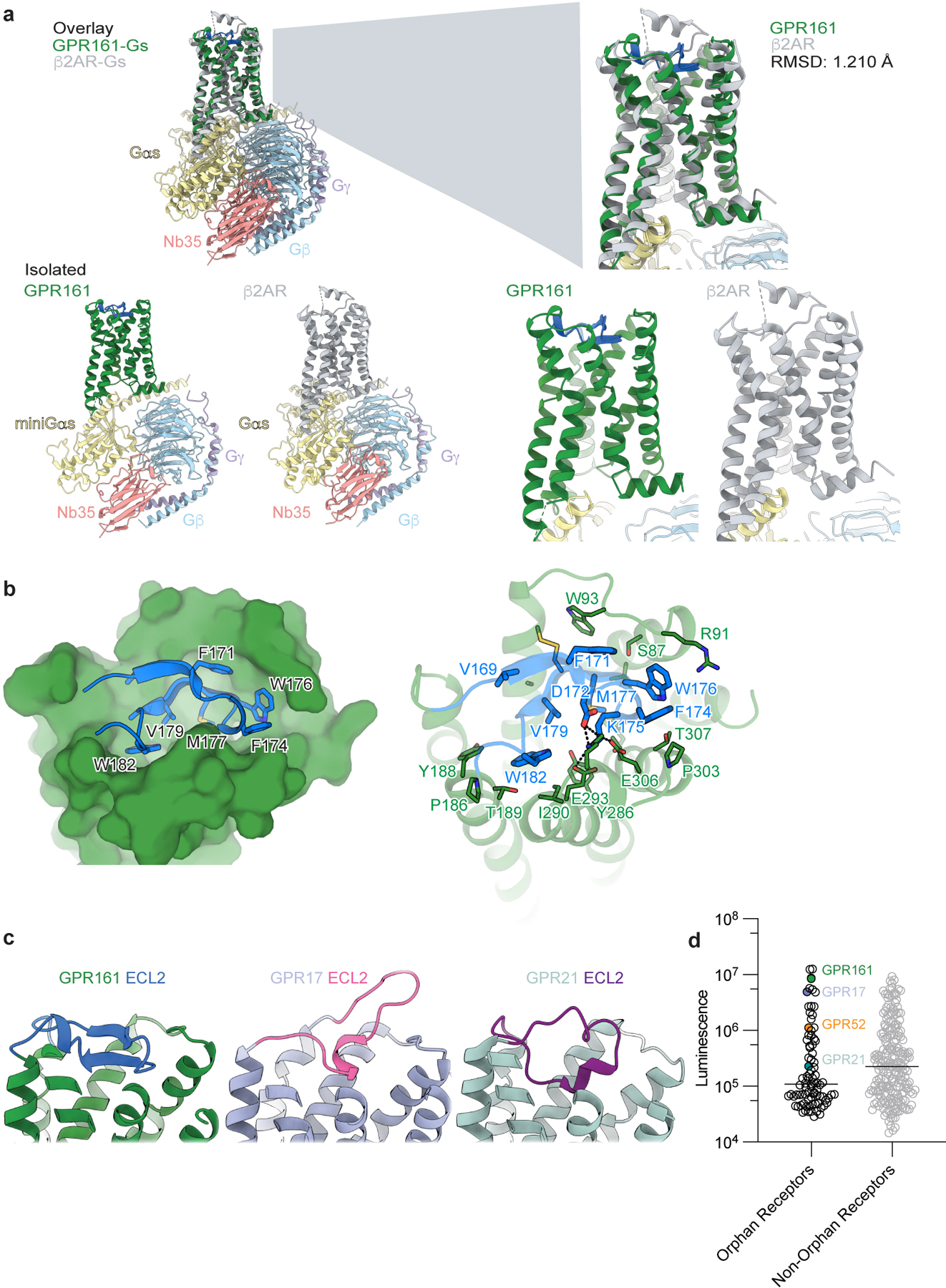
a) Structural comparison of GPR161 heterotrimer complex and β2AR heterotrimer complex (PDB ID: 3SN628). GPR161 has the same hallmarks of GPCR activation as the prototypical receptor, β2AR. b) View of the GPR161 ECL2 inside the canonical Class A GPCR binding site. ECL2 makes multiple hydrophobic interactions deep within the pocket. The superficial part of the pocket harbors ionic interactions between ECL2 and the binding pocket. c) Structural comparison of GPR161 to other orphan GPCRs with self-activating ECL2, including GPR17 (PDB ID: 7Y89) and GPR21 (PDB ID: 8HMV)31,32. The cis-interaction of ECL2 with the canonical ligand-binding site is seen across self-activating orphan GPCRs but the precise loop conformation changes between receptors. c) Luminescence for β-arrestin recruitment in the PRESTO-Tango assay when compared across 314 GPCRs (data replotted from Kroeze WM et al 201522, n = 4 for each target, shown as mean ± s.e.m. of technical replicates). GPR21 yields a signal slightly above the median. GPR52 yields a signal one order of magnitude above the median. GPR161 and GPR17 yield a signal about two orders of magnitude above the median.
Extended Data Fig. 5. GPR161 molecular dynamics simulation trajectories.
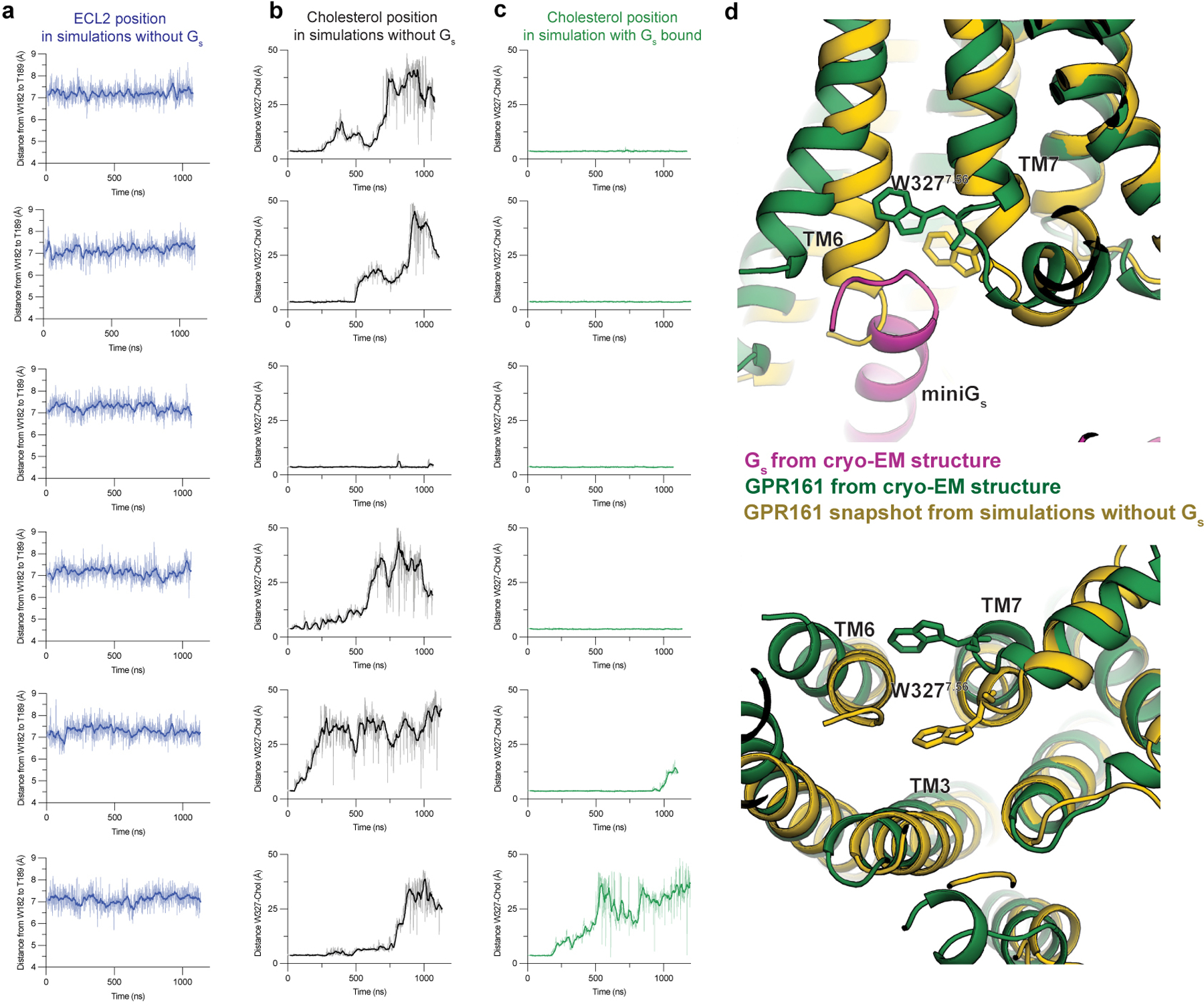
a) Time traces of ECL2 position in all six unrestrained simulations of GPR161 with miniGs removed. ECL2 position is represented by distance between W182 and T189. b) Time traces of distance between cholesterol and GPR161 residue W3277.56 in all six unrestrained simulations of GPR161 with miniGs removed. c) Time traces of distance between cholesterol and GPR161 residue W3277.56 in all six simulations where GPR161 residues that contact miniGs are restrained to their miniGs bound conformation. d) A comparison of the cryo-EM structure (green and magenta) to a representative snapshot from an unrestrained simulation of GPR161 with miniGs removed shows that, in the absence of miniGs, the intracellular ends of TM6 and TM7 move inwards, obstructing the Gs binding site.
Extended Data Fig. 6. Surface expression of GPR161 mutants.
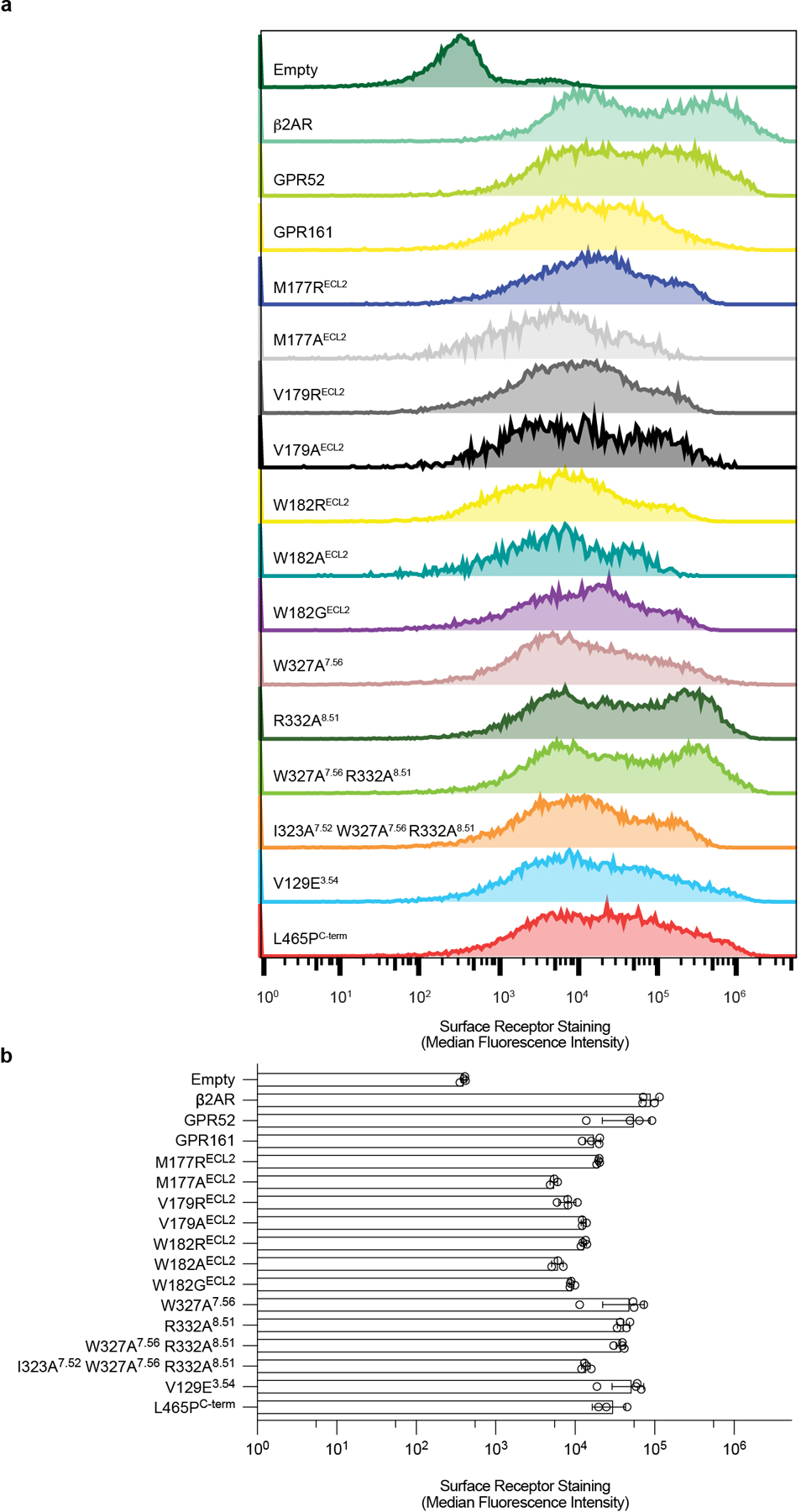
a) Representative flow cytometry surface expression histograms for receptors and mutants used in cell-based assays. b) Surface expression of receptors and mutants quantified by anti-FLAG-A647 median fluorescence intensity ± sd from n=3 (for L465PC-term) or n=4 (for rest) biologically independent samples.
Extended Data Fig. 7. Phylogenetic analysis of GPR161.
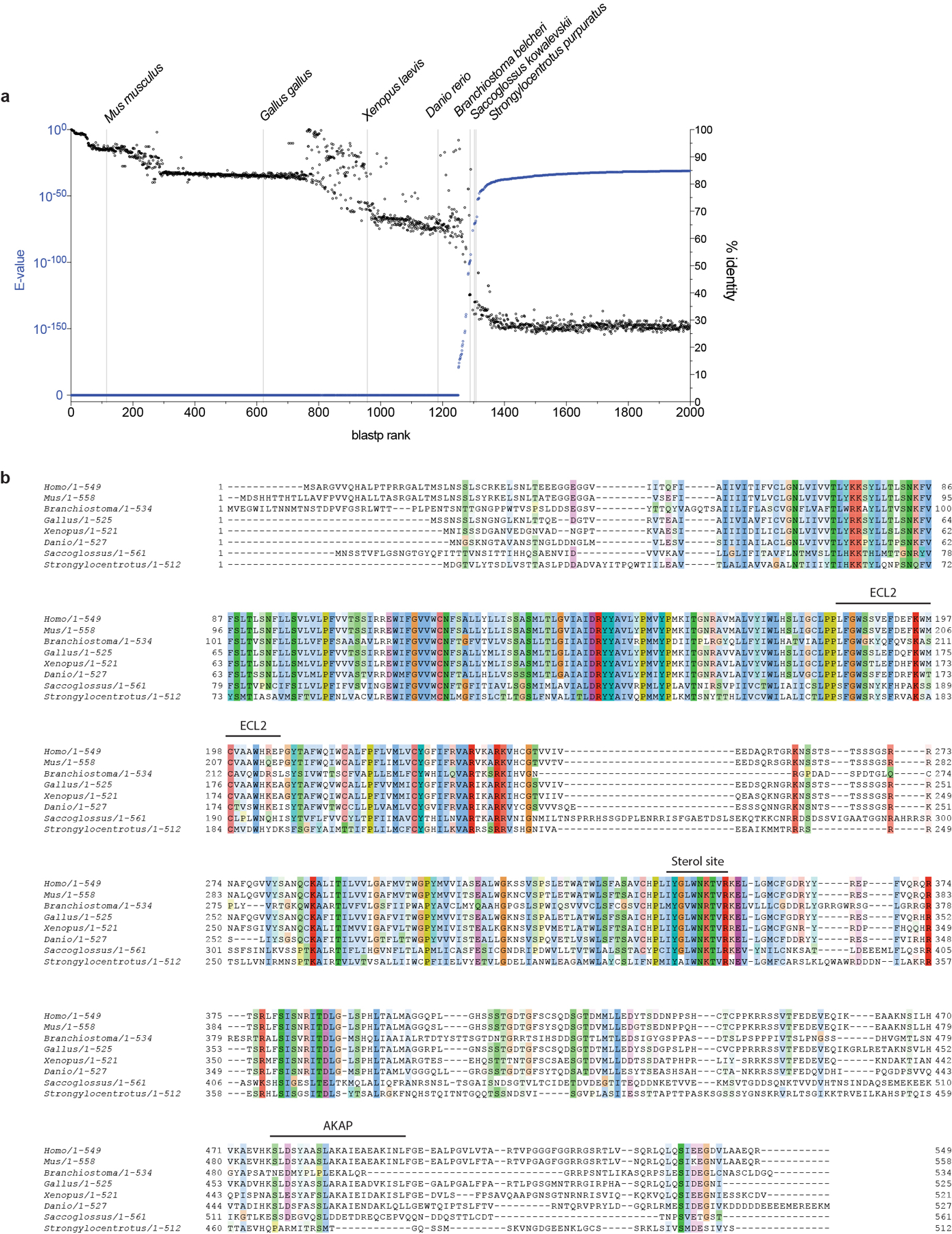
a) BLAST search results for Human GPR161 (Uniprot: Q8N6U8). Sequences are plotted from highest confidence (E-Value) and highest sequence identity (% identity) to lowest. Representative organisms spanning the full range of homologous GPR161 sequences are listed. b) Full sequence alignment of eight GPR161 model organism sequences identified in BLAST search.
Extended Data Fig. 8. Photolabeling with LKM238 and mass spectrometry sequence coverage.
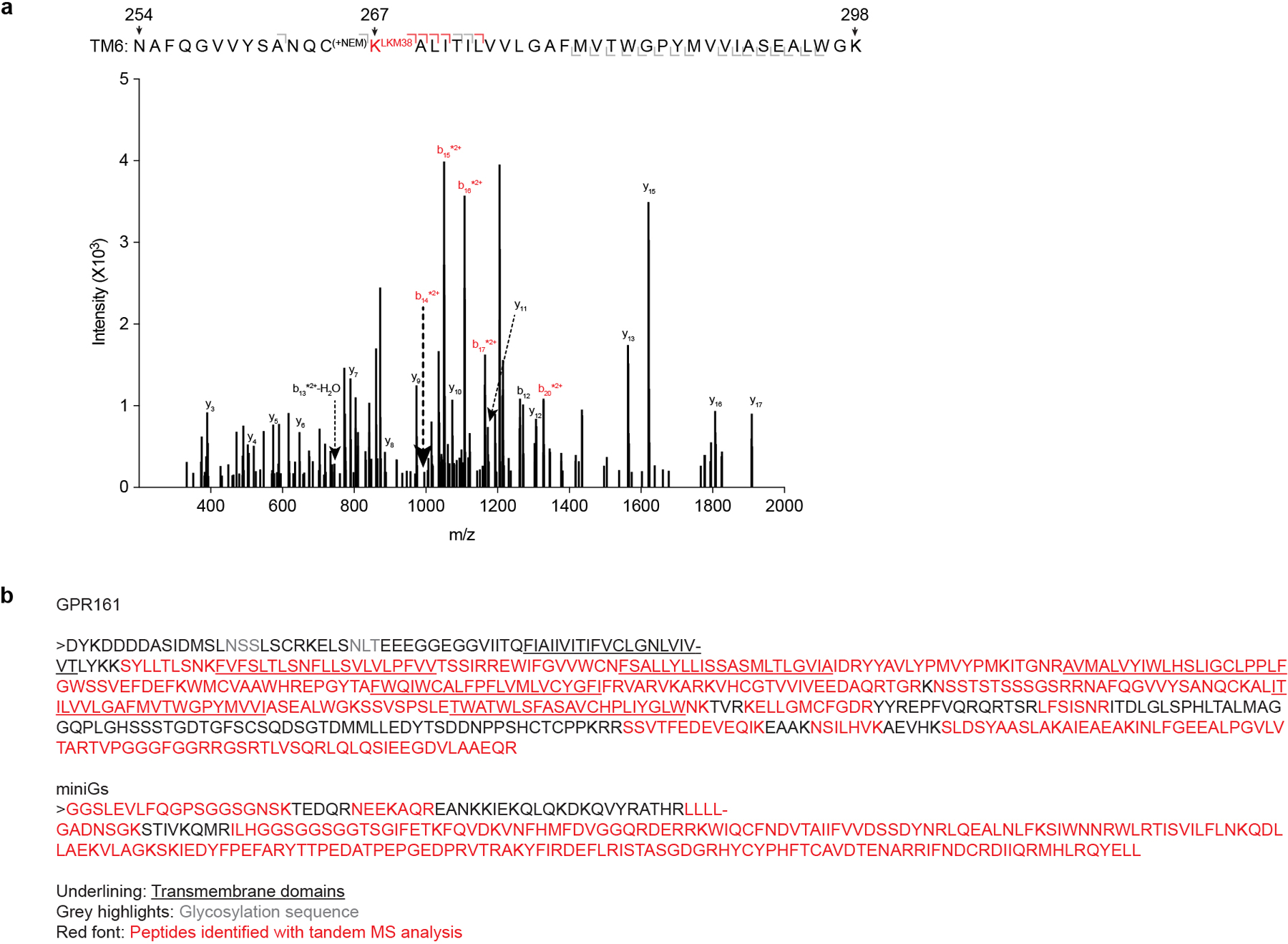
a) Product ion spectrum of LKM238-labeled GPR161-miniGs with peptides mapped to TM6. This peptide is modified with a mass consistent with LKM238 at position K2676.32. Red brackets and peaks indicate product ions that contain the LKM238 adduct. b) Mass spectrometric sequence coverage of GPR161-miniGs. Underlined segments indicate transmembrane spanning helices, red font indicates peptides identified by tandem MS analysis and gray font indicates glycosylation sites.
Extended Data Fig. 9. GPR161 localization and repression of GLI2 ciliary trafficking.
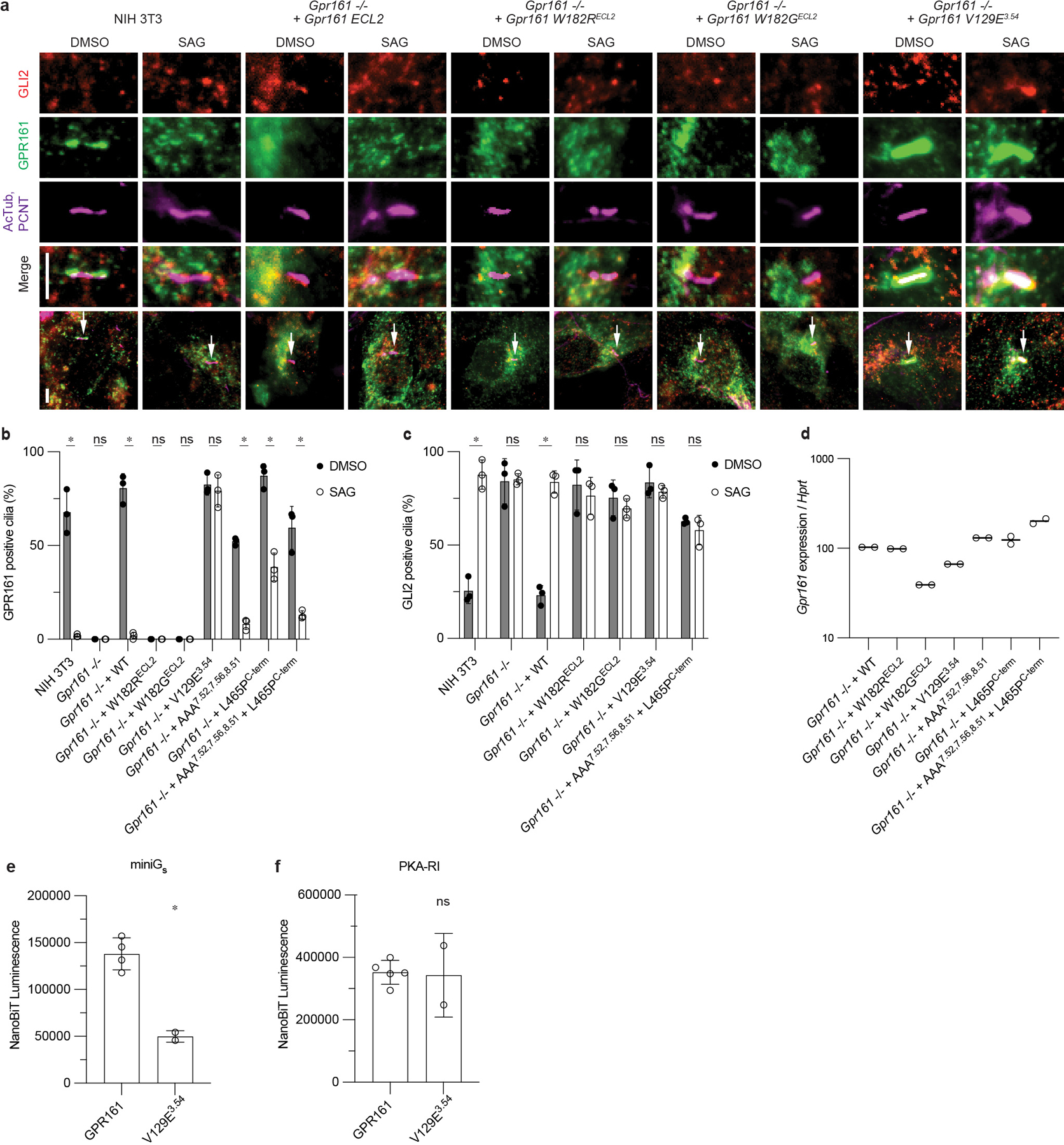
a) Representative images of GPR161 mutants on ciliary localization and GLI2 repression in ciliary tips in NIH 3T3 cells. NIH 3T3 Flp-In CRISPR based Gpr161−/− cells stably expressing untagged mouse wild-type or Gpr161 mutants were starved for 24 hr upon confluence and were treated for further 24 hr ± SAG (500 nM). After fixation, cells were immunostained with anti-GLI2 (red), anti-GPR161 (green), anti-acetylated, and centrosome (AcTub; PCNT purple) antibodies. Whole cell images with an arrow indicating imaged cilia. Scale bar, 5 µm. b) Quantification of GPR161 positive cilia indicating trafficking and egress of GPR161 from cilia in the pathway off and on state, respectively. ECL2 mutants do not traffic to cilia suggesting impaired biogenesis. GPR161-V129E3.54 does not egress from cilia following pathway activation and GPR161-L465PC-term has reduced egress compared to GPR161. (*P < 0.05; ns, not significant; two-way ANOVA followed by Šidák’s multiple comparison tests; Adjusted P values for DMSO vs. SAG: NIH3T3, Gpr161−/− +WT, +AAA7.52, 7.56, 8.51, +L465PC-term, + AAA7.52, 7.56, 8.51 L465PC-term=<0.0001, Gpr161−/− , Gpr161−/− + W182RECL2, +W182GECL2=>0.9999, Gpr161−/− + V129E3.54=0.997) c) Quantification GLI2 positive cilia indicating Hedgehog pathway activation. ECL2 mutants and GPR161-V129E3.54 do not rescue, similar to Gpr161−/−. For b,c, data are shown from n=3 independent experiments from images taken from 2–3 different regions/experiment and counting 15–30 cells/region. Data are mean ± s.d. (*P < 0.05; ns, not significant; two-way ANOVA followed by Šidák’s multiple comparison tests; Adjusted P values for DMSO vs. SAG: NIH3T3, Gpr161−/− + WT = <0.0001, Gpr161−/− = >0.9999, Gpr161−/− + W182RECL2= 0.9705, Gpr161−/− + W182GECL2= 0.9724, Gpr161−/− + V129E3.54= 0.9882, Gpr161−/− + AAA7.52, 7.56, 8.51 + L465PC-term= 0.9917). d) Transcript abundance of wild-type and mutant Gpr161 constructs stably expressed in Gpr161−/− NIH3T3 cells quantified by quantitative RT-PCR. e) GPR161-V129E3.54 has reduced recruitment of miniGs compared to WT. Nanoluc complementation assay for receptor recruitment of miniGs. Data are mean ± s.d., n=2 (for V129E3.54) or n =4 (for GPR161) biologically independent samples (*P < 0.05; ns, not significant; Unpaired two-tailed t test with Welch's correction; Adjusted P value: GPR161 vs. V129E3.54=0.0008). f) GPR161-V129E3.54 has similar recruitment of PKA-RI compared to GPR161. Nanoluc complementation assay for receptor recruitment of PKA-RI. Data are mean ± s.d., n=2 (for V129E3.54) or n=5 (for GPR161) biologically independent samples (*P < 0.05; ns, not significant; Unpaired two-tailed t test with Welch's correction; Adjusted P value: GPR161 vs. V129E3.54=0.9406).
Supplementary Material
{kind=link}
Acknowledgments
This work was supported by the National Institutes of Health (NIH) grants R01GM108799 (A.S.E.), 1R35GM149287 (A.S.E.), Grant 1 P50 MH122379 (D.C), R01AR054396 and R01HD089918 (J.F.R.), R35GM144136 (S.M.) and R01GM138992 (R.O.D. and A.M.). Additional support was from NSF Graduate Research Fellowship (M.K.) and Human Frontier Science Program Long-Term Fellowship LT000916/2018-L (C.-M.S.). Cryo-EM equipment at UCSF is partially supported by NIH grants S10OD020054 and S10OD021741. Some of this work was performed at the Stanford-SLAC Cryo-EM Center (S2C2), which is supported by the National Institutes of Health Common Fund Transformative High-Resolution Cryo-Electron Microscopy program (U24 GM129541). The authors would also like to thank the following S2C2 personnel for their invaluable support and assistance: Corey Hecksel. We are indebted to Jonathan Eggenschwiler for his generous gift of anti-Gli2 antibody. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. A.M. acknowledges support from the Edward Mallinckrodt, Jr. Foundation and the Vallee Foundation. A.M. is a Chan Zuckerberg Biohub Investigator.
Footnotes
Competing Interests
A.M. is a founder of Epiodyne and Stipple Bio, consults for Abalone, and serves on the scientific advisory board of Septerna. R.O.D. serves on the scientific advisory board of Septerna. The remaining authors declare no competing interests.
Data Availability
Coordinates for the GPR161-miniGs-Gβγ-Nb35 protein complex have been deposited in the RCSB PDB under accession code 8SMV. EM density maps for GPR161-miniGs-Gβγ-Nb35 complex have been deposited in the Electron Microscopy Data Bank under accession code EMD-40603. The MD simulation trajectories have been deposited in the Zenodo database under doi https://doi.org/10.5281/zenodo.7887650. Publicly available PDB entries used in this study are 6LI3, 4LDO, 3SN6, 7Y89, 8HMV. Protein sequence data for sequence alignments are available from NCBI RefSeq. Sequences used in the alignment in Extended Data Fig. 7 are NP_001254539, NP_001074595, XP_004938289, XP_041427552, NP_001007200, XP_019638841, XM_002731669 and XP_782439. Mass spectrometry data and source data are available with the manuscript online.
REFERENCES:
- 1.Civelli O, Saito Y, Wang Z, Nothacker H-P & Reinscheid RK Orphan GPCRs and their ligands. Pharmacol. Ther. 110, 525–532 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Mukhopadhyay S et al. The Ciliary G-Protein-Coupled Receptor Gpr161 Negatively Regulates the Sonic Hedgehog Pathway via cAMP Signaling. Cell 152, 210–223 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Hwang S-H, Somatilaka BN, White K & Mukhopadhyay S Ciliary and extraciliary Gpr161 pools repress hedgehog signaling in a tissue-specific manner. Elife 10, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shimada IS et al. Basal Suppression of the Sonic Hedgehog Pathway by the G-Protein-Coupled Receptor Gpr161 Restricts Medulloblastoma Pathogenesis. Cell Rep. 22, 1169–1184 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hwang S-H et al. The G protein-coupled receptor Gpr161 regulates forelimb formation, limb patterning and skeletal morphogenesis in a primary cilium-dependent manner. Development 145, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim S-E et al. Wnt1 Lineage Specific Deletion of Gpr161 Results in Embryonic Midbrain Malformation and Failure of Craniofacial Skeletal Development. Front. Genet. 12, 761418 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shimada IS et al. Derepression of sonic hedgehog signaling upon Gpr161 deletion unravels forebrain and ventricular abnormalities. Dev. Biol. 450, 47–62 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim S-E et al. Dominant negative GPR161 rare variants are risk factors of human spina bifida. Hum. Mol. Genet. 28, 200–208 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li BI et al. The orphan GPCR, Gpr161, regulates the retinoic acid and canonical Wnt pathways during neurulation. Dev. Biol. 402, 17–31 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Matteson PG et al. The orphan G protein-coupled receptor, Gpr161, encodes the vacuolated lens locus and controls neurulation and lens development. Proceedings of the National Academy of Sciences 105, 2088–2093 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karaca E et al. Whole-exome sequencing identifies homozygous GPR161 mutation in a family with pituitary stalk interruption syndrome. J. Clin. Endocrinol. Metab. 100, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Begemann M et al. Germline GPR161 Mutations Predispose to Pediatric Medulloblastoma. J. Clin. Oncol. 38, 43–50 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feigin ME, Xue B, Hammell MC & Muthuswamy SK G-protein–coupled receptor GPR161 is overexpressed in breast cancer and is a promoter of cell proliferation and invasion. Proceedings of the National Academy of Sciences 111, 4191–4196 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Website. https://bpspubs.onlinelibrary.wiley.com/doi/10.1111/bph.16053.
- 15.McMahon AP, Ingham PW & Tabin CJ 1 Developmental roles and clinical significance of Hedgehog signaling. in Current Topics in Developmental Biology vol. 53 1–114 (Academic Press, 2003). [DOI] [PubMed] [Google Scholar]
- 16.Kopinke D, Norris AM & Mukhopadhyay S Developmental and regenerative paradigms of cilia regulated hedgehog signaling. Semin. Cell Dev. Biol. 110, 89–103 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Truong ME et al. Vertebrate cells differentially interpret ciliary and extraciliary cAMP. Cell 184, 2911–2926.e18 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hilgendorf KI, Johnson CT & Jackson PK The primary cilium as a cellular receiver: organizing ciliary GPCR signaling. Curr. Opin. Cell Biol. 39, 84–92 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mukhopadhyay S & Rohatgi R G-protein-coupled receptors, Hedgehog signaling and primary cilia. Semin. Cell Dev. Biol. 33, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tschaikner PM et al. Feedback control of the Gpr161-Gαs-PKA axis contributes to basal Hedgehog repression in zebrafish. Development 148, dev192443 (2021). [DOI] [PubMed] [Google Scholar]
- 21.Pal K et al. Smoothened determines β-arrestin–mediated removal of the G protein–coupled receptor Gpr161 from the primary cilium. J. Cell Biol. 212, 861–875 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kroeze WK et al. PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat. Struct. Mol. Biol. 22, 362–369 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pusapati GV et al. G protein–coupled receptors control the sensitivity of cells to the morphogen Sonic Hedgehog. Sci. Signal. 11, eaao5749 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Foster SR et al. Discovery of human signaling systems: Pairing peptides to G protein-coupled receptors. Cell 179, 895–908.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bachmann VA et al. Gpr161 anchoring of PKA consolidates GPCR and cAMP signaling. Proc. Natl. Acad. Sci. U. S. A. 113, 7786–7791 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nehmé R et al. Mini-G proteins: Novel tools for studying GPCRs in their active conformation. PLoS One 12, e0175642 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carpenter B & Tate CG Engineering a minimal G protein to facilitate crystallisation of G protein-coupled receptors in their active conformation. Protein Eng. Des. Sel. 29, 583–594 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rasmussen SGF et al. Crystal structure of the β2 adrenergic receptor–Gs protein complex. Nature 477, 549–555 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou Q et al. Common activation mechanism of class A GPCRs. Elife 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin X et al. Structural basis of ligand recognition and self-activation of orphan GPR52. Nature 579, 152–157 (2020). [DOI] [PubMed] [Google Scholar]
- 31.Ye F et al. Cryo-EM structure of G-protein-coupled receptor GPR17 in complex with inhibitory G protein. MedComm (2020) 3, e159 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wong T-S et al. Cryo-EM structure of orphan G protein-coupled receptor GPR21. MedComm (2020) 4, e205 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wan Q et al. Mini G protein probes for active G protein-coupled receptors (GPCRs) in live cells. J. Biol. Chem. 293, 7466–7473 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dixon AS et al. NanoLuc Complementation Reporter Optimized for Accurate Measurement of Protein Interactions in Cells. ACS Chem. Biol. 11, 400–408 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Copp AJ et al. Spina bifida. Nat Rev Dis Primers 1, 1–18 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eaton S Multiple roles for lipids in the Hedgehog signalling pathway. Nat. Rev. Mol. Cell Biol. 9, 437–445 (2008). [DOI] [PubMed] [Google Scholar]
- 37.Luchetti G et al. Cholesterol activates the G-protein coupled receptor Smoothened to promote Hedgehog signaling. Elife 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cooper MK et al. A defective response to Hedgehog signaling in disorders of cholesterol biosynthesis. Nat. Genet. 33, 508–513 (2003). [DOI] [PubMed] [Google Scholar]
- 39.Budelier MM et al. Photoaffinity labeling with cholesterol analogues precisely maps a cholesterol-binding site in voltage-dependent anion channel-1. J. Biol. Chem. 292, 9294–9304 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krishnan K et al. Validation of Trifluoromethylphenyl Diazirine Cholesterol Analogues As Cholesterol Mimetics and Photolabeling Reagents. ACS Chem. Biol. 16, 1493–1507 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Castellano BM et al. Lysosomal cholesterol activates mTORC1 via an SLC38A9-Niemann-Pick C1 signaling complex. Science 355, 1306–1311 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shin HR et al. Lysosomal GPCR-like protein LYCHOS signals cholesterol sufficiency to mTORC1. Science 377, 1290–1298 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen M-H et al. Cilium-independent regulation of Gli protein function by Sufu in Hedgehog signaling is evolutionarily conserved. Genes Dev. 23, 1910–1928 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Haycraft CJ et al. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 1, e53 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim J, Kato M & Beachy PA Gli2 trafficking links Hedgehog-dependent activation of Smoothened in the primary cilium to transcriptional activation in the nucleus. Proc. Natl. Acad. Sci. U. S. A. 106, 21666–21671 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen Q et al. Structures of rhodopsin in complex with G-protein-coupled receptor kinase 1. Nature 595, 600–605 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Duan J et al. GPCR activation and GRK2 assembly by a biased intracellular agonist. Nature 620, 676–681 (2023). [DOI] [PubMed] [Google Scholar]
- 48.Huang W et al. Structure of the neurotensin receptor 1 in complex with β-arrestin 1. Nature 579, 303–308 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Staus DP et al. Structure of the M2 muscarinic receptor-β-arrestin complex in a lipid nanodisc. Nature 579, 297–302 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nie Y et al. Specific binding of GPR174 by endogenous lysophosphatidylserine leads to high constitutive Gs signaling. Nat. Commun. 14, 5901 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Patel K & Smith NJ Primary cilia, A-kinase anchoring proteins and constitutive activity at the orphan G protein-coupled receptor GPR161: A tale about a tail. Br. J. Pharmacol. (2023) doi: 10.1111/bph.16053. [DOI] [PubMed] [Google Scholar]
- 52.Yang X et al. Molecular mechanism of allosteric modulation for the cannabinoid receptor CB1. Nat. Chem. Biol. 18, 831–840 (2022). [DOI] [PubMed] [Google Scholar]
- 53.Draper-Joyce CJ et al. Positive allosteric mechanisms of adenosine A1 receptor-mediated analgesia. Nature 597, 571–576 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Song G et al. Human GLP-1 receptor transmembrane domain structure in complex with allosteric modulators. Nature 546, 312–315 (2017). [DOI] [PubMed] [Google Scholar]
- 55.Liu X et al. An allosteric modulator binds to a conformational hub in the β2 adrenergic receptor. Nat. Chem. Biol. 16, 749–755 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Robertson N et al. Structure of the complement C5a receptor bound to the extra-helical antagonist NDT9513727. Nature 553, 111–114 (2018). [DOI] [PubMed] [Google Scholar]
- 57.Dessauer CW Adenylyl cyclase--A-kinase anchoring protein complexes: the next dimension in cAMP signaling. Mol. Pharmacol. 76, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moore BS et al. Cilia have high cAMP levels that are inhibited by Sonic Hedgehog-regulated calcium dynamics. Proc. Natl. Acad. Sci. U. S. A. 113, 13069–13074 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Somatilaka BN et al. Ankmy2 Prevents Smoothened-Independent Hyperactivation of the Hedgehog Pathway via Cilia-Regulated Adenylyl Cyclase Signaling. Dev. Cell 54, 710–726.e8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jiang JY, Falcone JL, Curci S & Hofer AM Direct visualization of cAMP signaling in primary cilia reveals up-regulation of ciliary GPCR activity following Hedgehog activation. Proceedings of the National Academy of Sciences 116, 12066–12071 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Smith FD et al. Local protein kinase A action proceeds through intact holoenzymes. Science 356, 1288–1293 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.May EA et al. Time-resolved proteomics profiling of the ciliary Hedgehog response. J. Cell Biol. 220, e202007207 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Calebiro D et al. Persistent cAMP-signals triggered by internalized G-protein-coupled receptors. PLoS Biol. 7, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Crilly SE & Puthenveedu MA Compartmentalized GPCR Signaling from Intracellular Membranes. J. Membr. Biol. 254, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Irannejad R et al. Conformational biosensors reveal GPCR signalling from endosomes. Nature 495, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vilardaga J-P, Jean-Alphonse FG & Gardella TJ Endosomal generation of cAMP in GPCR signaling. Nat. Chem. Biol. 10, 700–706 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kinnebrew M et al. Cholesterol accessibility at the ciliary membrane controls hedgehog signaling. (2019) doi: 10.7554/eLife.50051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mesmin B & Maxfield FR Intracellular sterol dynamics. Biochim. Biophys. Acta 1791, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ogden SK et al. G protein Gαi functions immediately downstream of Smoothened in Hedgehog signalling. Nature 456, 967–970 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ayers KL & Thérond PP Evaluating Smoothened as a G-protein-coupled receptor for Hedgehog signalling. Trends Cell Biol. 20, 287–298 (2010). [DOI] [PubMed] [Google Scholar]
- 71.Happ JT et al. A PKA inhibitor motif within SMOOTHENED controls Hedgehog signal transduction. Nat. Struct. Mol. Biol. 29, 990–999 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stubbs T, Bingman JI, Besse J & Mykytyn K Ciliary signaling proteins are mislocalized in the brains of Bardet-Biedl syndrome 1-null mice. Frontiers in cell and developmental biology 10, (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Badgandi HB, Hwang SH, Shimada IS, Loriot E & Mukhopadhyay S Tubby family proteins are adapters for ciliary trafficking of integral membrane proteins. J. Cell Biol. 216, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sheu SH et al. A serotonergic axon-cilium synapse drives nuclear signaling to alter chromatin accessibility. Cell 185, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chou C-H et al. Bisdemethoxycurcumin Promotes Apoptosis and Inhibits the Epithelial-Mesenchymal Transition through the Inhibition of the G-Protein-Coupled Receptor 161/Mammalian Target of Rapamycin Signaling Pathway in Triple Negative Breast Cancer Cells. J. Agric. Food Chem. 69, 14557–14567 (2021). [DOI] [PubMed] [Google Scholar]
- 76.Bock A et al. Optical Mapping of cAMP Signaling at the Nanometer Scale. Cell 182, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang JZ et al. Phase Separation of a PKA Regulatory Subunit Controls cAMP Compartmentation and Oncogenic Signaling. Cell 182, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ring AM et al. Adrenaline-activated structure of β2-adrenoceptor stabilized by an engineered nanobody. Nature 502, 575–579 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Staus DP et al. Sortase ligation enables homogeneous GPCR phosphorylation to reveal diversity in β-arrestin coupling. Proc. Natl. Acad. Sci. U. S. A. 115, 3834–3839 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Faust B et al. Autoantibody mimicry of hormone action at the thyrotropin receptor. Nature 609, 846–853 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zheng SQ et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Punjani A, Rubinstein JL, Fleet DJ & Brubaker MA cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017). [DOI] [PubMed] [Google Scholar]
- 83.Asarnow D, Palovcak E & Cheng Y asarnow/pyem: UCSF pyem v0.5. (2019). doi: 10.5281/zenodo.3576630. [DOI] [Google Scholar]
- 84.Pettersen EF et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zivanov J et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Grant T, Rohou A & Grigorieff N cisTEM, user-friendly software for single-particle image processing. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jumper J et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Emsley P & Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 90.Schüttelkopf AW & van Aalten DMF PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 60, 1355–1363 (2004). [DOI] [PubMed] [Google Scholar]
- 91.Darbandi-Tonkabon R et al. Photoaffinity labeling with a neuroactive steroid analogue. 6-azi-pregnanolone labels voltage-dependent anion channel-1 in rat brain. J. Biol. Chem. 278, 13196–13206 (2003). [DOI] [PubMed] [Google Scholar]
- 92.Zhang L & Hermans J Hydrophilicity of cavities in proteins. Proteins 24, 433–438 (1996). [DOI] [PubMed] [Google Scholar]
- 93.Qu C et al. Ligand recognition, unconventional activation, and G protein coupling of the prostaglandin E2 receptor EP2 subtype. Sci Adv 7, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lomize MA, Lomize AL, Pogozheva ID & Mosberg HI OPM: orientations of proteins in membranes database. Bioinformatics 22, 623–625 (2006). [DOI] [PubMed] [Google Scholar]
- 95.Betz R Dabble. (2017). doi: 10.5281/zenodo.836914. [DOI] [Google Scholar]
- 96.Huang J et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Klauda JB et al. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B 114, 7830–7843 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Case DA, Aktulga HM, Belfon K, Ben-Shalom IY, Berryman JT, Brozell SR, Cerutti DS, Cheatham TE III, Cisneros GA, Cruzeiro VWD, Darden TA, Duke RE, Giambasu G, Gilson MK, Gohlke H, Goetz AW, Harris R, Izadi S, Izmailov SA, Kasavajhala K, Kaymak MC, King E, Kovalenko A, Kurtzman T, Lee TS, LeGrand S, Li P, Lin C, Liu J, Luchko T, Luo R, Machado M, Man V, Manathunga M, Merz KM, Miao Y, Mikhailovskii O, Monard G, Nguyen H, O’Hearn KA, Onufriev A, Pan F, Pantano S, Qi R, Rahnamoun A, Roe DR, Roitberg A, Sagui C, Schott-Verdugo S, Shajan A, Shen J, Simmerling CL, Skrynnikov NR, Smith J, Swails J, Walker RC, Wang J, Wang J, Wei H, Wolf RM, Wu X, Xiong Y, Xue Y, York DM, Zhao S, and Kollman PA. Amber21. University of California, San Francisco: (2022). [Google Scholar]
- 99.Salomon-Ferrer R, Götz AW, Poole D, Le Grand S & Walker RC Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 9, 3878–3888 (2013). [DOI] [PubMed] [Google Scholar]
- 100.Hopkins CW, Le Grand S, Walker RC & Roitberg AE Long-Time-Step Molecular Dynamics through Hydrogen Mass Repartitioning. J. Chem. Theory Comput. 11, 1864–1874 (2015). [DOI] [PubMed] [Google Scholar]
- 101.Roe DR & Cheatham TE 3rd. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 9, 3084–3095 (2013). [DOI] [PubMed] [Google Scholar]
- 102.Humphrey W, Dalke A & Schulten K VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–8, 27–8 (1996). [DOI] [PubMed] [Google Scholar]
- 103.Hunter JD Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 9, 90–95 (2007). [Google Scholar]
- 104.Norman RX et al. Tubby-like protein 3 (TULP3) regulates patterning in the mouse embryo through inhibition of Hedgehog signaling. Hum. Mol. Genet. 18, 1740–1754 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Coordinates for the GPR161-miniGs-Gβγ-Nb35 protein complex have been deposited in the RCSB PDB under accession code 8SMV. EM density maps for GPR161-miniGs-Gβγ-Nb35 complex have been deposited in the Electron Microscopy Data Bank under accession code EMD-40603. The MD simulation trajectories have been deposited in the Zenodo database under doi https://doi.org/10.5281/zenodo.7887650. Publicly available PDB entries used in this study are 6LI3, 4LDO, 3SN6, 7Y89, 8HMV. Protein sequence data for sequence alignments are available from NCBI RefSeq. Sequences used in the alignment in Extended Data Fig. 7 are NP_001254539, NP_001074595, XP_004938289, XP_041427552, NP_001007200, XP_019638841, XM_002731669 and XP_782439. Mass spectrometry data and source data are available with the manuscript online.