

Abstract
Disassembly is a key event of virus entry into cells. Here, we have investigated cellular requirements for the first step of adenovirus type 2 (Ad2) disassembly, the release of the fibers. Although fiber release coincides temporally with virus uptake, fiber release is not required for Ad2 endocytosis. It is, however, inhibited by actin-disrupting agents or soluble RGD peptides, which interfere with integrin-dependent endocytosis of Ad2. Fiber release occurs at the cell surface. Actin stabilization with jasplakinolide blocks Ad2 entry at extended cell surface invaginations and efficiently promotes fiber release, indicating that fiber release and virus endocytosis are independent events. Fiber release is not sufficient for Ad2 escape from endosomes, since inhibition of protein kinase C (PKC) prevents Ad2 escape from endosomes but does not affect virus internalization or fiber release. PKC-inhibited cells accumulate Ad2 in small vesicles near the cell periphery, indicating that PKC is also required for membrane trafficking of virus. Taken together, our data show that fiber release from incoming Ad2 requires integrins and filamentous actin. Together with correct subcellular transport of Ad2-containing endosomes, fiber release is essential for efficient delivery of virus to the cytosol. We speculate that fiber release at the surface might extend the host range of Ad2 since it is associated with the separation of a small fraction of incoming virus from the target cells.
Adenoviruses (Ads) are among the best-characterized viral systems (24, 49). Both past and ongoing Ad studies have significantly contributed to concepts in molecular and cellular biology and have facilitated the development of Ad vectors for preclinical and clinical trials (3). Ads are nonenveloped icosahedral particles of about 90 nm in diameter (8). The main capsid component is the facette-associated hexon protein, which is assembled and stabilized by various minor proteins. Hexon largely protects a double-stranded linear DNA genome, which is packed inside the capsid together with additional viral proteins including the cysteine protease L3/p23. The bases of the capsid vertices are built from the penton base protein from which fibers emanate. Of the six Ad subgroups, comprising almost 50 serotypes, the subgroup C viruses such as Ad2 and Ad5 have been studied the most extensively. It was realized relatively early that subgroup C Ad entry requires two receptors, a primary receptor, CAR (coxsackie-adenovirus receptor), for attachment (4, 56) and secondary receptors, αvβ5 or αvβ3 integrins, for internalization (62). While the primary receptor determines virus tropism and is a major aim in current retargeting studies of Ad (for a recent review, see reference 12), the secondary receptor plays more subtle but less well understood roles in the infection process. Besides facilitating virus internalization, most probably via clathrin-coated pits (41, 58), αvβ5 integrins contribute to Ad-dependent permeabilization of the plasma membrane (61), activation of phosphatidylinositol 3-OH kinase (29), and small G proteins of the Rho family (28) and the activation of the Raf/extracellular receptor kinase 1 and 2 (ERK1,2) pathway (6). While the mitogen-activated protein kinase pathway of ERK1,2 is not required for Ad entry, activated integrins play a role in virus uptake and intracellular transport.
Endocytosis of Ad requires integrins, assembly of clathrin-coated pits, and invagination and fission of the plasma membrane, followed by routing of the emerging vesicle toward early endosomes. The entire process is regulated by lipid kinases, actin-modulating small GTPases, and the large GTPase dynamin (28, 29). Like Ad endocytosis, integrin-mediated endocytosis is complex and can involve the recruitment of the clathrin internalization machinery, dynamin (38, 48), dynamic rearrangement of cortical actin filaments, and, finally, vesicular movement through the cell cortex controlled by lipid and protein kinases and small GTPases (27, 36, 51). Unlike physiological extracellular integrin ligands, internalized Ad2 escapes from acidified endocytic vesicles (5, 41). It is then translocated as a naked particle along microtubules to the nucleus (54) and docks at nuclear pore complexes (9), where the capsid is disassembled (20) and the DNA genome is imported into the nucleoplasm (19).
In contrast to wild-type (wt) Ad2, an Ad2 mutant, ts1, fails to escape from the endocytic system (33, 60). ts1 has a point mutation in the L3/p23 protease, preventing protease packaging and proper processing of the viral capsid structure at the restrictive temperature (for a review, see reference 18). After passing the cell cortex, ts1 endosomes are transported to a perinuclear region by microtubule-dependent motors (54) and ts1 is eventually degraded in lysosomes (20). Part of the reason why ts1 fails to escape from endosomes could be the failure to release fibers, although it binds to CAR and internalizes via integrins much like wt Ad2 (20, 29). Here we have analyzed cellular requirements for the first step of wt Ad2 disassembly, the release of the fibers. We show that besides cortical actin filaments, integrins play a crucial role in detaching fibers at the cell surface. Following fiber detachment, Ad2 is endocytosed and transported through the actin cortex in a protein kinase C (PKC)-dependent fashion to finally escape from endosomes and reach the cytosol.
MATERIALS AND METHODS
Cells, viruses, and infections.
HeLa cells (American Type Culture Collection) were grown on glass coverslips or 30-mm-diameter petri dishes in Dulbecco's modified Eagle's medium (DMEM) (Gibco-BRL) containing 7% fetal calf serum (HyClone) and l-glutamine in a humidified 5% CO2–air atmosphere. In some experiments, cells were treated for 30 min at 37°C in growth medium with 2 μM cytochalasin D (CD) (Calbiochem, Juro Supply) 180 nM jasplakinolide (Jas) (a gift from Phil Crews, Santa Cruz, Calif.), 0.5 mM cyclic arginine-glycine-aspartate peptide (cRGD) (a gift from J. Glass, Telios Pharmaceuticals, San Diego, Calif.), 1 mM linear arginine-glycine-glutamate-serine peptide (RGES) (Sigma), the PKC inhibitor calphostin C (5 μM) (Calbiochem, Juro Supply) or bisindolylmaleimide (BIM) (5 μM) (Sigma, Fluka), the myristoylated pseudosubstrate of PKCα and PKCβ (10 μg/ml) (Promega, Catalys AG), which inhibits all classical PKCs, or a control myristoylated autocamptide-related inhibitory peptide (10 μg/ml) (Calbiochem, Juro Supply). The cold-synchronized infection with Ad in RPMI-bovine serum albumin (BSA) medium containing drugs was followed by a warming period in DMEM–0.2% BSA containing drugs as indicated.
Ad2 (wt and ts1) were grown, isolated, and labeled with Texas red (TR) or [35S]methionine as described previously (20, 21, 37, 60). A 0.16-μg sample of fluorescent virus in 0.25 ml of cold RPMI-BSA was bound to subconfluent HeLa cells grown on 12-mm glass coverslips (multiplicity of infection [MOI], 400). Alternatively, 160,000 cpm of [35S]methionine-labeled Ad2 (1.9 × 106 PFU) was incubated with 3 × 105 HeLa cells in cold RPMI (MOI 6). Cells were washed, incubated with warm DMEM-BSA for various times, chilled in cold PBS, and treated with trypsin (2 mg/ml; Gibco-BRL) for 1 h in the cold as described earlier (21). After inactivation of trypsin, cells were lysed in 0.5 ml of 0.5% Empigen BB (Calbiochem, Juro Supply) in MNT buffer (0.02 M 2-N-morpholinoethansulfonic acid, 0.1 M sodium chloride, 0.03 M Tris-HCl) (pH 7.5) containing 0.001 M EDTA and the protease inhibitors 0.5 mM phenylmethylsulfonyl fluoride (Böhringer, Mannheim, Germany), 0.5 mM benzamidine (Fluka), 0.001 mg of leupeptin (ICN Biomedicals GmbH) per ml, 0.001 mg of chymostatin (Böhringer) per ml, and 0.001 mg of pepstatin (Fluka) per ml (21). An aliquot of the lysate was precipitated with trichloroacetic acid, dissolved in sodium dodecyl sulfate (SDS) sample buffer, and analyzed for intact and trypsin-cleaved hexon by SDS-polyacrylamide gel electrophoresis (PAGE) and PhosphorImager analysis (Molecular Dynamics, Bucher AG) using National Institutes of Health Image software for quantification (http://rsb.info.nih.gov/nih-image/index.html). The ratio of cleaved to uncleaved hexon is an indication of the cell surface accessibility of Ad2 and usually correlates with virus internalization (21).
To analyze the extent of fiber association with viral capsid, cells were lysed in Empigen-MNT buffer and fiber antigens were collected by incubating the lysate with an immunoglobulin G fraction of the polyclonal antifiber antibody R72 (5 μg/ml) (2) and Zysorbin (Zymed, Mächler AG). Immunocomplexes were washed, fractionated by SDS-PAGE, and analyzed by PhosphorImager analysis. The ratio of [35S]methionine in hexon to that in fiber was taken as a measure of the number of fiber-containing Ad capsids (21).
Quantitative fluorescence microscopy.
Cell-associated Ad2-TR was quantitated in the cell periphery, in the cytoplasm, and at the nucleus as described previously (37). In brief, HeLa cells were infected with Ad2-TR, fixed with 3% paraformaldehyde in phosphate-buffered saline, permeabilized for 2 min with 0.5% Triton X-100 in phosphate-buffered saline including 20 mM ammonium chloride to reduce autofluorescence, and stained for the nuclear DNA with 4,6-diamidino-2-phenylindole (DAPI) (0.5 μg/ml) (Sigma), and for the plasma membrane Ca-ATPase 1 with an affinity-purified antibody kindly provided by D. Guerini (ETH Zurich) (53) and goat anti-rabbit immunoglobulin G coupled to Alexa 488 (Molecular Probes, Leiden, The Netherlands). Specimens were embedded in 0.2% p-phenylendiamine–0.02 M Tris-HCl–85% glycerol (pH 8.8) and analyzed by fluorescence microscopy using an inverted motorized Leica DMIRBE microscope equipped with a 100× objective (N.A. 1.3 PL Fluotar) and a back-illuminated MicroMax-controlled charge-coupled device camera (800 by 1,000 pixels of 15 by 15 μm [Princeton Instruments, Visitron GmbH]), which was operated in 16-bit mode at −25°C. Excitation occurred through band-pass excitation filters for DAPI (330/80; Omega XF03), Alexa 488 (475/40; Omega XF100), and TR (560/55; Omega XF101), and emitted light was collected either through a single-pass (DAPI, 425LP, dichroic 400; Omega XF03) or a double-pass (green and red, 528/30 and 633/60; dichroic 490/575; Omega XF53) emission filter. The TR images were recorded sequentially throughout the entire cell sample (z-stack) at 1-μm steps. Alexa 488, DAPI, and differential interference contrast images were obtained from sections toward the cell bottom or the middle to indicate cell and nuclear dimensions, respectively. Each TR image was processed by fast Fourier transformation (FFT) to remove out-of-focus information and to correct for uneven illumination as described previously (37). FFT-processed images were merged using the summation function, and background was subtracted by setting the lowest pixel value to 0. Pixel values of TR were determined in regions of interest, which were defined by a thresholding function using the plasma membrane and the nuclear strains, respectively.
CLSM.
Confocal laser-scanning microscopy (CLSM) was performed with a confocal unit (TCS SP; Leica), on an inverted microscope (Leica DMIRBE) using the 63× lens (N.A. 1.32). Optical sections were collected at step sizes of 0.5 μm. For DAPI imaging, a UV laser with excitation lines at 351 and 364 nm was used. For detection of the emission, the spectrophotometer was set to 410 to 530 nm. The plasma membrane marker Ca ATPase 1 was visualized by indirect immunofluorescence using an Alexa 488-tagged secondary antibody. Alexa 488- and TR-coupled Ad2 were recorded with the 488- and 568-nm laser lines of an argon/krypton laser, respectively. The settings of the spectrophotometer were 495 to 560 nm for Alexa 488 and 580 to 640 nm for TR. The pinhole for all recordings was set to 1.0. Images of the three fluorochromes were taken sequentially to exclude cross talk between the channels.
EM.
Electron microscopy (EM) was carried out as described previously (54). For quantification, micrographs were recorded in sections across the middle of the cells by a slow-scan charge-coupled device camera (Gatan, Gloor AG) at ×30,000 magnification using the DigitalMicrograph software package (version 3.3.1; Gatan, Pleasanton, Calif.). Morphometry of single sections from 11 to 14 randomly selected entire cells was carried out for each of the drug conditions as specified. All the cell-associated virus particles were quantitated. This required 90 to 179 micrographs depending on the number of virus particles present in the analyzed cells.
Statistics.
Statistical analyses were conducted using a one-sided t test with indicated confidence intervals, standard error of the mean (SEM), and sample size (n).
RESULTS
Dissociation of the fibers from the capsid is the first event in the stepwise uncoating program of Ad2 (21). Although it coincides with virus endocytosis, the cellular requirements or functional implications are unknown.
Cellular requirements for fiber release.
Since Ad uptake into cells requires an intact actin cytoskeleton and is facilitated by αv integrins (28, 42), we first analyzed whether filamentous actin (F-actin) and integrins were implicated in fiber release from incoming Ad2. To disrupt actin filaments, HeLa cells were treated with CD, which caps F-actin, stimulates ATP hydrolysis on globular actin, and also affects the small G protein rho A (44, 47). Alternatively, cells were incubated in low concentrations of Jas, which binds to and stabilizes F-actin in a similar fashion to the cell-impermeable phalloidin (7, 11). The efficiency of CD in disrupting F-actin and the ability of Jas to bind to F-actin were examined by staining cells with phalloidin-Alexa 488 following fixation with paraformaldehyde and permeabilization with Triton X-100. Jas-treated cells showed no phalloidin staining at all, although stress fibers and cortical actin were readily visualized using anti-actin antibodies (data not shown). CD-treated cells contained large amounts of phalloidin-positive actin aggregates. To measure fiber release, [35S]methionine-labeled Ad2 was bound to drug-treated cells in the cold and internalized at 37°C for 0, 15, or 30 min. Immunocomplexes containing fiber epitopes were collected from Empigen-lysed cells using a specific antifiber antibody (R72,2), which recognizes intact Ad2 particles (21). The amounts of immunoadsorbed [35S]methionine-labeled fiber and coprecipitated hexon were quantitated by SDS-PAGE and PhosphorImager analysis as described earlier (21). The hexon-to-fiber ratio derived from intact [35S]methionine-labeled virus (not incubated with cells) was set to 100% (Fig. 1, lane 1). As expected, control cells efficiently promoted the dissociation of fiber from hexon within 30 min (more than sixfold) and with a half-maximal efficiency at 12 min postinfection (p.i.) (lanes 2 to 4). In contrast, fiber dissociation from hexon was slowed in CD-treated cells, reaching half-maximal efficiencies after 30 min of warming (lanes 5 to 7). However, no delay of fiber release was detected in infections in Jas-treated cells (lanes 8 to 10), suggesting that fiber release is promoted by an intact rather than dynamic F-actin cytoskeleton. We then asked if integrins were directly or indirectly involved in triggering fiber release. Cells were incubated with cRGD peptides or control RGE peptides to inhibit Ad2 endocytosis (1, 62), and fiber dissociation was determined as described above. The results demonstrated that cRGD peptides were efficient inhibitors, since control RGE peptides had no effect on fiber release (lanes 11 to 16). The data showed that fiber release correlated with virus uptake and required an intact actin cytoskeleton.
FIG. 1.

Fiber release from incoming Ad2 depends on intact F-actin and is inhibited by RGD peptides but not PKC inhibitors. HeLa cells were treated with the indicated inhibitors for 30 min at 37°C and then incubated with [35S]methionine-labeled Ad2 in the cold for 1 h and warmed for different times as indicated. Cell lysates were immunoprecipitated with antifiber antibodies under nondissociating conditions and fractionated by SDS-PAGE. [35S]methionine in hexon (Hex) and fiber (Fib) was quantitated and expressed as a ratio of hexon to fiber by using purified [35S]methionine-labeled Ad2 as a standard (100%). The data are representative of at least two independent experiments.
Fiber release occurs at the cell surface.
To test if Ad uptake was required for fiber release from incoming capsids, we examined Ad2 endocytosis by three different assays, a biochemical cell surface trypsinization assay, a quantitative combined CLSM-fluorescence microscopy assay, and quantitative EM. In the first assay, [35S]methionine-labeled Ad2 was bound to CD- or Jas-treated cells and the amount of trypsin-resistant hexon was determined at different times of infection (21). Since this assay measures the trypsin accessibility of the major capsid protein hexon at 4°C, it estimates the amount of protease-sensitive virus on the cell surface. Note, however, that the trypsinizations did not remove virus particles from control cells (Fig. 2A, and D), although the hexons were quantitatively cleaved at the potential trypsin cleavage sites in loop 1 near the N terminus (26). Using this assay, typical times for half-maximal Ad2 internalization (t1/2) into control cells were around 10 to 12 min and efficiencies were near 80% at 60 min p.i. (Fig. 2A and G). Disruption of the actin cytoskeleton by CD, however, completely blocked the appearance of trypsin-resistant hexon up to 60 min p.i. (Fig. 2B and G), consistent with earlier results (28).
FIG. 2.

Cell surface trypsinization assay of incoming [35S]methionine-labeled Ad2 in cells treated with actin and PKC inhibitors. HeLa cells were treated with various inhibitors for 30 min, and then [35S]methionine-labeled Ad2 was bound for 60 min in the cold. The cells were warmed for different times as indicated and treated with cold trypsin to probe for surface accessibility of hexon. The ratio of intact (Hex) to cleaved (Hex′) hexon was determined after SDS-PAGE (A to F) and plotted for each condition, taking the sum of the cleaved and uncleaved hexons from cells not treated with trypsin (−T) as 100% (G and H). Controls not treated with trypsin are shown in panels A, B, D, and F. Dashed lines in panels G and H indicate the total hexon radioactivity, and solid lines show the trypsin-resistant hexon determined by PhosphorImager analysis. Symbols for different drug treatments are used as indicated in panels G and H. Representative data from at least two independent experiments are shown.
These results were in agreement with CLSM data demonstrating that fluorescent Ad2-TR did not effectively reach the nuclei of CD-treated cells up to 60 min p.i. (Fig. 3C, whole en face projections). In control cells, Ad2-TR was efficiently translocated to the nucleus at 60 min but not at 0 min p.i. (Fig. 3A and B). We have quantitated fluorescent Ad2 by using an FFT image-processing routine described earlier (37). Ad2-TR near the cell periphery, the cytoplasm, and the nuclei of control or CD-treated cells was determined at 0 and 70 min p.i. (Fig. 4A). In control cells, the fraction of Ad2-TR in the periphery decreased about fivefold (P < 0.01), but in CD-treated cells it was only slightly reduced (P = 0.025). Conversely, the CD-treated cells contained more Ad2-TR in both the peripheral and the cytoplasmic areas (P < 0.01) but much less virus in the nuclear area (P < 0.01) compared to the control cells at 70 min p.i.
FIG. 3.
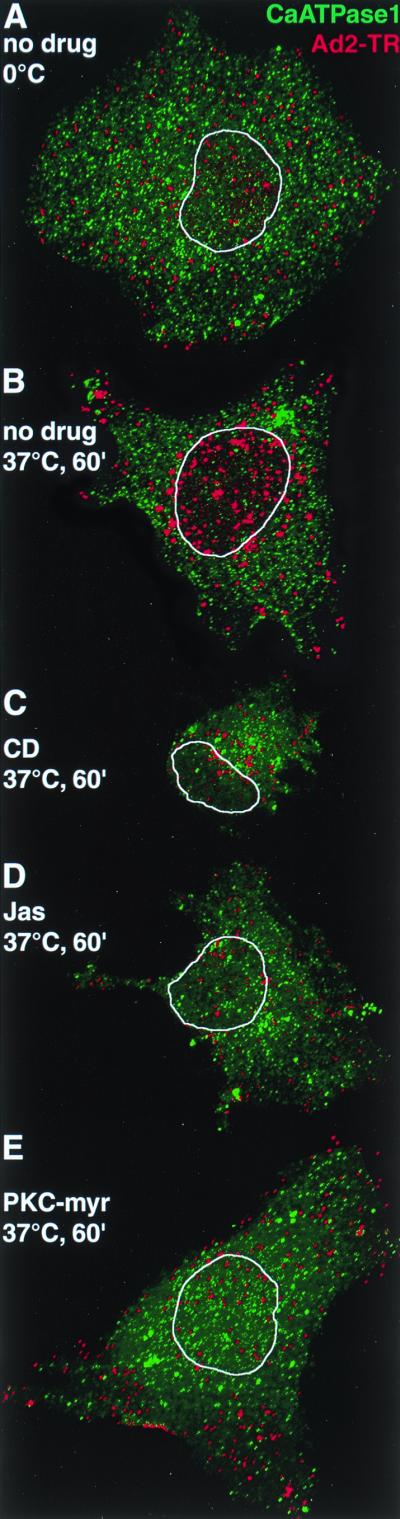
CLSM analysis of incoming Ad2-TR in cells treated with actin and PKC inhibitors reveals the actin and PKC requirements for nuclear transport of Ad2. TR-labeled Ad2 (red) was bound to HeLa cells pretreated with drugs or not pretreated and internalized for 0 min (A) or 60 min (B to E) as indicated in Materials and Methods. CLSM sections sampling the entire cell were generated for the TR channel and the Alexa 488 channel (plasma membrane Ca ATPase 1 [green]) and projected en face. The limits of the nucleus as determined by DAPI staining (results not shown) are indicated by a white trace. A single representative cell for each of the conditions is shown.
FIG. 4.

Quantitative subcellular analysis of incoming fluorescently labeled wt Ad2 and ts1 Ad2 in cells treated with actin- or PKC-directed inhibitors. Ad2-TR was cold bound to inhibitor-treated or control HeLa cells and internalized for 0 min (open bars) or 60 or 70 min (solid bars) as described in Materials and Methods. The cells were fixed and analyzed for virus fluorescence in the cell periphery, the cytoplasm, and the nucleus by using an image deconvolution routine described previously (37). Results are shown as mean fluorescence values, with the corresponding SEM derived from the indicated number of analyzed cells (n). (A) Results for wt Ad2-TR in actin-inhibited cells. (B and C) Results for wt and ts1 Ad2-TR in cells not treated with drugs (solid bars) or treated with the PKC inhibitor PKC-myr (stippled bars) or a control myristoylated autocamptide (autocamp) directed against Cam kinase II (striped bars).
EM confirmed that Ad2 was largely excluded from the cytoplasm and enriched at the plasma membrane of CD-treated cells at 30 min p.i. and was typically associated with plasma membrane extensions (Fig. 5). Quantifications of electron micrographs indicated that only 4% of the cell-associated Ad2 particles were found in the cytoplasm of these cells and 6% were in intracellular vesicles, compared to 31% cytoplasmic (P < 0.01) and 11% vesicular (P = 0.1) in control cells (Fig. 6A and B). Note that in CD-treated cells all the vesicular virus particles were found in small vesicles with diameters less than 200 nm and 90% of the particles were associated with the plasma membrane compared to 58% in control cells (P < 0.01). The levels of extracellular Ad2 in the control cells at 30 min p.i. were about twice as high as in the low-MOI infections using [35S]methionine-labeled Ad2 (Fig. 2) suggesting that the Ad2 uptake system is saturated in the high-MOI EM experiments. Interestingly, 7% of the viruses at the surface of CD-treated cells were associated with coated pits compared to 13% in control cells (P = 0.025), indicating that CD did not block the formation of clathrin-coated pits containing Ad2, in agreement with earlier studies on Semliki Forest virus entry (30).
FIG. 5.

Thin-section electron microscopy of wt Ad2-infected HeLa cells treated with actin- or PKC-directed inhibitors. Cells were treated with drugs as described in Materials and Methods and incubated with 50 μg of purified Ad2 per ml for 60 min in the cold. Unbound virus (approximately 98 to 99% of input virus) was washed off, and the cells were incubated with or without inhibitors for 30 min at 37°C, fixed, and processed for thin-section EM. (A) control cell infected without drugs. (B) CD-treated cell. (C) Jas-treated cell. (D) PKC-myr-treated cell. The inset in panel D shows an enlargement of a region proximal to the plasma membrane. Thin arrows indicate extracellular Ad2 particles, large arrows indicate cytoplasmic Ad2, small arrowheads show coated vesicles containing Ad2 (A) and also Ad2 particles within small vesicles (D), and the large arrowhead depicts Ad2 within a medium-sized vesicle (D), while ∗ indicates a large vesicle (D). Note the enrichment of Ad2 particles in plasma membrane invaginations of cells treated with the actin stabilizer Jas (C) and the localization of Ad2 within small vesicles of PKC-myr-treated cells (D). Bar = 500 nm.
FIG. 6.

Quantification of Ad2 particles at the plasma membrane, in intracellular vesicles, and in the cytoplasm of HeLa cells treated with actin- and PKC-directed drugs or not treated. HeLa cells were infected and analyzed by thin-section EM as in Fig. 5. (A) Control cells. (B) CD-treated cells. (C) Jas-treated cells. (D) PKC-myr-treated cells. Viruses were counted in smooth (sh), invaginated (iv), and coated-pit (cp) regions of the plasma membrane, within small (s), medium (m), or large (l) vesicles, and also in the cytosol (solid bars). The number of Ad particles on the entire plasma membrane (open bars) and within the entire vesicle population (striped bars) is also indicated (tot). Mean values are expressed as the percentage of total virus particles, and the corresponding SEM values are indicated based on the analyzed number of cells (ranging from 11 to 14) and virus particles (ranging from 339 to 607). The corresponding number of electron micrographs (ranging from 90 to 179) is also indicated for each of the conditions.
A different picture emerged for Jas-treated cells, where [35S]methionine labeled Ad2 became rapidly resistant to surface trypsinization, with half-maximal efficiencies at 10 min p.i. and overall efficiencies of nearly 80% at 60 min p.i., similar to control cells (Fig. 2C and G). However, unlike control cells, the Jas-treated cells did not dissociate Ad2 from their surface, as indicated by the total amount of cell-associated hexon protein (Fig. 2G). It is possible that either enhanced endocytosis precluded virus shedding to the medium or Ad2 became trapped in plasma membrane structures prior to endocytosis. To distinguish between these two possibilities, we analyzed the entry of Ad2-TR into Jas-treated and control cells by CLSM. The data suggested that Ad2-TR remained near the periphery of Jas-treated cells at 60 min p.i. (Fig. 3D). Little nuclear Ad2-TR was detected in these cells at 60 min p.i. (Fig. 3). Quantifications of Ad2-TR fluorescence in different subcellular regions indicated that the differences in Ad2-TR levels in both the periphery and the cytoplasm of Jas-treated cells compared to control cells were highly significant at 70 min p.i. (P < 0.01) (Fig. 4A). Likewise, the differences in nuclear fluorescence were highly significant at 70 min p.i. (P < 0.01). Interestingly, peripheral Ad2-TR in the Jas-treated cells was slightly lower at 70 min p.i. than at 0 min p.i. (P < 0.01) and was somewhat increased in the nuclear region at 70 min p.i. compared to 0 min (P < 0.01).
To test if the inhibition of nuclear targeting by Jas was leaky or, alternatively, if the cell surface structure was altered by Jas treatment, we used EM to analyze the subcellular localization of Ad. As indicated in Fig. 5, many cell surface invaginations were detected in Jas-treated cells (Fig. 5B, arrows). These invaginations often (but not always) contained Ad particles and were also found in noninfected Jas-treated HeLa cells (data not shown). A quantitative analysis of Ad2 particles in randomly selected Jas-treated cells indicated that 85% of the virus particles were at the extracellular face of the plasma membrane, 10% were within intracellular vesicles, and 5% were in the cytosol, even though these cells displayed trypsin resistance of the viral hexon protein (Fig. 6C). In contrast, 58 and 31% of particles were found extracellularly and in the cytoplasm of control cells (Fig. 6A). Interestingly, in Jas-treated cells, 33% of the particles were within extended surface invaginations, 49% were on smooth plasma membrane regions, and 3% were in coated-pit-containing plasma membrane domains. Compared to the values for control cells, these numbers were highly significant (P < 0.01) and were in full agreement with a reduced infection of Jas-treated cells (data not shown). Collectively, the data indicated that Jas blocked Ad2 uptake but still allowed efficient fiber release from the particles, implying that fiber release occurs at the cell surface independently of endocytosis.
Fiber release is not sufficient for virus escape from endosomes.
Vitronectin is a physiological ligand of the Ad coreceptor αvβ5 integrin (39). Endocytosis of a conformationally altered vitronectin has been reported to be αvβ5 integrin dependent, and subsequent vitronectin degradation in lysosomes requires PKC (40). We therefore asked if HeLa cells treated with PKC inhibitors were able to induce fiber release from Ad2 and deliver Ad2 to the cytosol. As indicated in Fig. 1 (lanes 17 to 22), the PKC effector site inhibitor calphostin C or a competitive inhibitor for the ATP binding site BIM had no drastic effect on the efficiency of fiber release from [35S]methionine-labeled Ad2, although both drugs slightly lowered the rate of fiber release. Likewise, the PKC inhibitors did not significantly affect the efficiency of Ad2 uptake at 30 min p.i., although they slightly reduced the internalization rate measured by the trypsin cleavage assay (Fig. 2D to F). Interestingly, the total amount of Ad2 associated with PKC-inhibited cells decreased to about 60% at 60 min p.i. whereas control cells contained 85% of the originally cell-bound virus (Fig. 2H). This loss of cell-associated virus was most probably not due to intracellular degradation, since we did not detect any of the typical hexon proteolysis products due to lysosomal degradation (20). It is possible that Ad2 was endocytosed and then delivered back to the surface, where it was removed by trypsin as originally described for ts1 Ad2 (20).
To test if PKC was required for Ad escape from endosomes, we treated HeLa cells with a third PKC inhibitor, an N-terminally myristoylated pseudosubstrate of PKCα (PKC-myr) (14), infected cells with Ad2 at high MOI for 30 min, and subjected them to EM analysis (Fig. 5D). The quantitated data indicated that 34% the virus particles found in PKC-myr-treated cells were located within vesicles compared to 11% in control cells (P < 0.01) (Fig. 6D). Correspondingly, the levels of cytosolic Ad2 were significantly reduced (14%) compared to those in the control cells (31%) (P < 0.01). As expected from the biochemical data (Fig. 2), the amounts of extracellular virus particles were not significantly different in control and PKC-inhibited cells (58 and 52%, respectively) and the numbers of Ad2 in plasma membrane invaginations and coated pits were comparable to those in control cells (P = 0.1). Similar results were obtained with other PKC inhibitors, such as calphostin C and BIM (data not shown). Our results are consistent with the notion that PKC inhibition had no severe effect on Ad2 endocytosis but inhibited Ad escape from endosomes. A close inspection of electron micrographs of PKC-inhibited cells revealed that half of the intracellular particles (vesicular plus cytosolic Ad2), namely, 24% of all the cell-associated Ad2, were located within vesicles smaller than 200 nm (Fig. 6D), reminiscent of endocytic transport vesicles (22).
To address the possibility that PKC inhibition affected the intracellular transport of Ad2-containing vesicles, we determined the subcellular location of fluorescently labeled Ad2-TR. CLSM analysis at 60 min p.i. suggested an inhibition of nuclear transport of wt Ad2 (Fig. 3E). Quantitative subcellular analysis of Ad2-TR indicated that PKC-myr decreased Ad transport from the periphery to the cytoplasm and also to the nucleus at 60 min p.i. (Fig. 4B) (P = 0.05). The amounts of Ad2 in the cytoplasmic area were not affected by PKC inhibition and remained in the same range as those observed at 0 min p.i. The effects of PKC-myr were specific since a myristoylated pseudosubstrate for calmodulin (Cam) kinase II (autocamptide [25]) had no effects on the transport of wt Ad2 (Fig. 4B).
To further test if PKC was specifically required for transport of endosomal Ad2, we analyzed the subcellular localization of mutant Ad2 ts1, which is known to remain in the endocytic system (33). ts1-containing vesicles are transported to a perinuclear region by microtubule-dependent motors, but ts1 does not escape or bind to the nuclear envelope (37, 54). Results in Fig. 4C indicate that PKC inhibition decreased the clearance of ts1 from the cell periphery (P = 0.025) but had no effect on ts1 transport to or from the cytoplasmic and nuclear regions. Cytoplasmic and nuclear levels of ts1 in PKC-inhibited cells were not significantly different from the levels at 0 min p.i. (Fig. 4C). Surprisingly, autocamptide inhibition of Cam kinase II enhanced the clearance of ts1 but not wt Ad2 from the cell periphery (P < 0.01). Since autocamptide did not affect the cytoplasmic and perinuclear levels of ts1 and since ts1 is known to cycle from an endocytic compartment back to the surface even in control cells (20), the data might suggest that Cam kinase II is involved in the transport of ts1 vesicles in the periphery. Additional studies are, however, needed to clarify the precise role of Cam kinase II in Ad entry. Taken together, our data showed that PKC is not involved in fiber release and virus endocytosis but is specifically required for transport of endosomal Ad2-containing vesicles in the periphery. Possibly, the transport deficiency of PKC-inhibited cells is the underlying reason for the failure of Ad2 to escape from endosomes. Alternatively, PKC might activate a cell-based membrane lysis machinery.
Fiber release correlates with virus shedding from the target cell.
In cold-synchronized infections, Ad2 internalization coincides with the shedding of 15 to 20% of the cell-bound virus particles into the medium (21). To ask if virus shedding correlated with fiber release, we overexpressed dominant negative dynamin I K44A, which has a reduced rate of GTP hydrolysis affecting both clathrin- and non-clathrin-dependent endocytosis (13, 48) including Ad uptake and gene expression (58). K44A or wt dynamin I expression was induced by derepressing a transfected HeLa cell line for 4 days in tetracycline-free medium (13). The cells were then incubated with TR-labeled wt Ad2 for 1 h in the cold and warmed for 5 or 60 min. Cells overexpressing dominant negative dynamin were identified by the lack of internalized transferrin-Alexa 488, which was present during the warming period at 100 μg/ml. With this assay, we found that about 40% of the K44A dynamin I-transfected cells actually overexpressed the K44A mutant protein (data not shown). Total cell-associated Ad2-TR was then quantitated in single cells by fluorescence microscopy (37). At 5 and 60 min p.i., control cells expressing wt dynamin contained an average of 23 and 17 TR fluorescence units, respectively (Fig. 7A). The difference was significant (P = 0.05) and was in good agreement with earlier results measuring the dissociation of radiolabeled Ad2 particles from cells (21). In contrast, K44A dynamin-expressing cells contained similar amounts of Ad2-TR at 5 and 60 min p.i., namely, 12.4 and 10.7 fluorescence units per area (Fig. 7A). Likewise, there was no difference in the amount of cell-associated Ad2-TR on RGD peptide-incubated cells at 5 or 60 min p.i., suggesting that integrins are involved in the shedding of Ad2 (data not shown). Importantly, the mutant ts1 was not dissociated from wt or K44A dynamin-expressing cells (Fig. 7B), consistent with its failure to release fibers upon entry (20). Together, the data suggested that Ad2 shedding from the target cells required functional endocytosis and correlated with fiber release.
FIG. 7.

Dissociation of Ad2 from target cells requires functional dynamin. wt or ts1 Ad2-TR was bound in the cold to HeLa cells expressing either wt dynamin I (wt) or the GTP hydrolysis-defective K44A dynamin mutant. Following warming for 5 min (open bars) or 60 min (solid bars), cells were fixed and the cell-associated TR fluorescence was determined by FFT as described earlier (37). Results are expressed as mean values normalized to the corresponding cell areas including the SEM and the number of cells analyzed (n).
DISCUSSION
Despite ongoing progress in elucidating virus cell interactions, there are still major deficits in our understanding of Ad biology, including entry and disassembly of the capsid. In this study we have analyzed cellular factors involved in the initial step of Ad2 disassembly, the release of the fibers. Fiber release is a hallmark of the stepwise Ad disassembly program culminating in the dissociation of the capsid and the release of the viral DNA into the nucleoplasm (21, 55).
Surface events for fiber release.
We show here that fiber release requires an intact actin cytoskeleton and proper contacts of the incoming capsid to cell surface integrins but is independent of virus endocytosis. Fiber release from incoming Ad2 capsids occurs at the plasma membrane following attachment of the distal fiber knob domains of virus to the surface receptor CAR (4, 15). The proximal fiber region is anchored in the capsid via the vertex protein penton base. While Ad2 binding to CAR occurs independently of physiological temperatures, fiber release requires warming of the cells and proper contacts of the penton base protein with cell surface integrins. These contacts can be inhibited by soluble RGD peptides or the calcium chelator EGTA inhibiting Ad2 endocytosis (19, 20, 62). These treatments also inhibit fiber release, suggesting that integrin contacts with the penton base in the context of CAR-bound virus can trigger the dissociation of fibers. It is possible but unlikely that the penton base is released together with fibers, since the penton base but not the fiber was detected by indirect immunofluorescence and cryoimmuno-EM on Ad particles near the nuclear envelope (data not shown). That the underlying actin cytoskeleton is also instrumental for fiber release is indicated by the strong inhibition of fiber release in cells treated with actin-destabilizing agents, including CD and latrunculin B (data not shown).
Earlier studies indicated that fiber release coincides with virus internalization (21). These results and our present data showing that fiber release occurs in the absence of virus internalization, e.g., in cells treated with the actin-stabilizing drug Jas, clearly establish that the trigger for fiber release occurs at the cell surface. Virus binding to the primary receptor CAR alone is unlikely to trigger fiber release, since fiber release can be effectively prevented by conditions which inhibit proper contacts of virus with αv integrins without affecting virus binding to CAR. CAR may, however, be required to hold the virus particle in a proper position, allowing αv integrins to contact the penton base. CAR and integrins may in fact synergize the efficiency of infection, as suggested by fiber swap experiments where the fiber of a slowly infecting subgroup B virus (Ad7) was transferred to a quickly infecting subgroup C virus particle (Ad5). The result was somewhat surprising in that the chimeric particle now had the slow nuclear transport characteristics of the subgroup B particles (34). Analysis of whether fiber was released from the chimeric particles and particles had escaped to the cytosol will be interesting. In any case, fiber release is expected to generate particles which are infectious provided that they attach to a suitable receptor, as suggested by experiments with fiberless Ad2 particles infecting integrin-positive monocytic cells (57). Soluble αvβ5 integrin fragments have been visualized as diffuse densities in cryoelectron micrographs of isolated Ad2 (32), but it is not clear if integrins play a direct or an accessory role in triggering fiber release and internalization.
Consistent with earlier studies with SW480 cells (28), our inhibition experiments with HeLa cells using the actin stabilizer Jas confirmed that filamentous or growing actin alone did not support Ad2 endocytosis. In contrast to CD, which depolymerizes F-actin by decreasing the rate of actin growth at the barbed end of the filament and also severs filaments, Jas potently induces de novo actin nucleation and polymerization, decreasing monomeric actin and lowering the dynamics of actin filaments (7). Both inhibitors blocked Ad2 endocytosis in several different cell types, indicating that filamentous actin plays an obligatory role in Ad2 endocytosis. Interestingly, Ad2 was trapped in extensive plasma membrane invaginations of Jas-treated cells (Fig. 5). Similar cell surface changes were also observed in noninfected polarized cells treated with Jas (50). Viruses in these invaginations were not accessible to the extracellular trypsin used in standard internalization assays (21). Only direct analysis of the infected cells by quantitative EM and measurements of infectivity revealed the effect of Jas on Ad infection. Although CD and Jas were almost equally effective at restricting Ad2 entry at the surface, Jas-treated cells showed some apparent nuclear transport of fluorescent Ad2 (Fig. 4). This was most probably not due to intracellular Ad2 transport but, rather, was caused by cell contraction in response to Jas treatment. Notably, CD caused cell contraction as well, but this contraction occurred much faster than with Jas treatment and was essentially complete after the preincubation period before Ad2 was added to the cells.
While a disrupted actin cytoskeleton did not allow fiber release, filamentous or growing actin (Jas treatment) supported fiber release at the cell surface. This event was associated with the shedding of 15 to 25% of the initially bound Ad2 particles to the medium. It was unlikely that the shed particles had incorrectly attached to the cell surface, since virus binding to HeLa cells was inhibited by more than 95% on preincubation with soluble fiber knob. About half of the released capsids were able to bind to and infect new target cells, but the rest did not bind back to CAR-positive cells, suggesting that these particles had lost most of their fibers. Virus shedding to the medium was completely blocked by overexpression of the dominant negative dynamin I mutant (K44A), which inhibits both clathrin- and caveola-dependent endocytosis and might also affect intracellular transport of vesicles (48). Possibly, K44A dynamin expression interfered with integrin transport to the cell surface or alternatively altered the affinity of cell surface receptors for Ad2, by analogy to observations with epidermal growth factor receptor and epidermal growth factor (45).
Relevance of fiber release.
Fiber shedding at the cell surface may have several functions. The first function could be to promote capsid internalization independently of the primary receptor CAR or perhaps of major histocompatibility complex class I (4, 23, 56). It is not known whether the CAR receptor has the capacity to internalize, but the intracellular CAR domain is not required for infection (59). However, this does not exclude the notion that CAR remains permanently at the plasma membrane. Ad2 ts1 mutants bound to CAR-positive HeLa cells can be internalized without the apparent loss of fibers, indicating that fiber release is not required for interaction with an internalization receptor (20). Another reason why Ad2 would want to release its fibers at the surface could be to shed particles from a target cell and thus have the potential to infect neighboring cells expressing a different set of cell surface receptors. Fiber release is also associated with the ability of incoming Ad to escape from endosomes, as indicated by the results with the ts1 mutant, which fails to release fibers but enters by an integrin-dependent pathway (20, 29, 33). The reason for the failure of ts1 to release its fibers is not clear. Possibly, there are multiple contact sites between integrins and the penton base, one for uptake and another perhaps for fiber release. Alternatively, ts1 might fail to release its fibers due to its nonprocessed capsid structure or because it lacks contacts with unknown surface molecules.
PKC is required for vesicular trafficking and Ad escape from endosomes but not fiber release.
Following fiber release, Ad is internalized by an integrin dependent pathway. Although fiber release per se is not required for internalization it may be required for capsid escape from endosomes. Carboxylic ionophores or primary amines neutralizing the acidic pH of intracellular compartments have no effects on the efficiency and the kinetics of fiber release (21), but they inhibit Ad escape from endosomes if the virus is present at low MOI (17, 46), suggesting that fiber release is not sufficient for Ad2 escape from endosomes. In contrast to pH-neutralizing procedures, inhibition of cellular PKCs was an effective treatment to limit Ad2 escape from endosomes even when Ad2 was present at high MOI. Given the observations that αv integrins contribute to Ad-mediated release of cytosolic contents (61) and PKC can be a downstream effector of αv integrins (38), it is possible that combined signaling of integrins and PKC activates a membrane lytic function effecting Ad release to the cytosol. Alternatively, but not exclusively, Ad2-mediated endosome lysis may require PKC-dependent trafficking of virus-containing endosomes and may be related to proper endosome acidification, which can be increased by PKC stimulation (63). This scenario would be in agreement with earlier studies suggesting that subgroup C Ads escape from a mildly acidic compartment, possibly the tubulovesicular early endosomal compartment (5, 22, 41), and that membrane trafficking in the cell periphery can be limited by the cortical actin cytoskeleton (16, 31, 35). Consistently, the early endosomal marker rab5 has been suggested to interact with actin filaments (36) and affect Ad5-mediated gene delivery (43). More recently, increasing evidence indicates that actin-dependent endocytic events in the cell cortex can also be regulated by calcium and PKC (38, 52). For example, movement of intracellular vesicles containing αvβ3 integrins seems to be affected by calcium (27), and calcium has been implicated in endosomal fusion (10). Given our observations that three different inhibitors of classical PKCs, namely, an ATP analogue (BIM), an effector site inhibitor (calphostin C), and a PKCα pseudosubstrate mimicking the catalytic site (PKC-myr peptide), all inhibited Ad escape from endosomes without affecting virus endocytosis or fiber release, we speculate that a calcium-dependent classical PKC plays a role in peripheral trafficking of endocytic vesicles containing Ad2. In addition, our EM analysis indicated that the disruption of the actin cytoskeleton gave rise to a modest number of small peripheral endocytic vesicles containing Ad2, which were indistinguishable from those present in the PKC-inhibited cells. Together, these results may suggest that cytosolic calcium can be involved in Ad infection, despite the report that global calcium influx does not occur during the first 10 to 15 min of subgroup C Ad infections (43). It is, however, possible that local increases in the concentration of cytosolic calcium, perhaps in the vicinity of endocytic vesicles, are sufficient, together with additional unknown factors, to execute the proper endocytic trafficking and stimulate virus escape to the cytosol.
ACKNOWLEDGMENTS
The first two authors contributed equally to this work.
We thank Stephan Keller, Danilo Guerini, Marshall Horwitz, Christophe Lamaze, Sandy Schmid, and Joe Weber for the gifts of viruses, cell lines, and antibodies; Oliver Meier for discussions and for sharing unpublished data; and Urs Ziegler and Peter Groscurth for generous access to the CLSM instrument. Jasplakinolide was kindly provided by Phil Crews (UCSC).
The work was supported by a grant from the Swiss National Science Foundation and the Kanton of Zürich (to U.F.G.).
REFERENCES
- 1.Bai M, Harfe B, Freimuth P. Mutations that alter an Arg-Gly-Asp (RGD) sequence in the adenovirus type 2 penton base protein abolish its cell-rounding activity and delay virus reproduction in flat cells. J Virol. 1993;67:5198–5205. doi: 10.1128/jvi.67.9.5198-5205.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baum S G, Horwitz M S, Maizel J V. Studies of the mechanism of enhancement of human adenovirus infection in monkey cells by simian virus 40. J Virol. 1972;10:211–219. doi: 10.1128/jvi.10.2.211-219.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benihoud K, Yeh P, Perricaudet M. Adenovirus vectors for gene delivery. Curr Opin Biotechnol. 1999;10:440–447. doi: 10.1016/s0958-1669(99)00007-5. [DOI] [PubMed] [Google Scholar]
- 4.Bergelson J M, Cunningham J A, Droguett G, Kurt-Jones E A, Krithivas A, Hong J S, Horwitz M S, Crowell R L, Finberg R W. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science. 1997;275:1320–1323. doi: 10.1126/science.275.5304.1320. [DOI] [PubMed] [Google Scholar]
- 5.Blumenthal R, Seth P, Willingham M C, Pastan I. pH-dependent lysis of liposomes by adenovirus. Biochemistry. 1986;25:2231–2237. doi: 10.1021/bi00356a057. [DOI] [PubMed] [Google Scholar]
- 6.Bruder J T, Kovesdi I. Adenovirus infection stimulates the Raf/MAPK signaling pathway and induces interleukin-8 expression. J Virol. 1997;71:398–404. doi: 10.1128/jvi.71.1.398-404.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bubb M R, Spector I, Beyer B B, Fosen K M. Effects of jasplakinolide on the kinetics of actin polymerization. An explanation for certain in vivo observations. J Biol Chem. 2000;275:5163–5170. doi: 10.1074/jbc.275.7.5163. [DOI] [PubMed] [Google Scholar]
- 8.Burnett R M. The structure of adenovirus. In: Chiu W, Burnett R M, Garcea R L, editors. Structural biology of viruses. Oxford, United Kingdom: Oxford University Press; 1997. pp. 209–238. [Google Scholar]
- 9.Chardonnet Y, Dales S. Early events in the interaction of adenoviruses with HeLa cells. I. Penetration of type 5 and intracellular release of the DNA genome. Virology. 1970;40:462–477. doi: 10.1016/0042-6822(70)90189-3. [DOI] [PubMed] [Google Scholar]
- 10.Colombo M I, Beron W, Stahl P D. Calmodulin regulates endosome fusion. J Biol Chem. 1997;272:7707–7712. doi: 10.1074/jbc.272.12.7707. [DOI] [PubMed] [Google Scholar]
- 11.Crews P, Manes L V, Boehler M. Jasplakinolide, a cyclodepsipeptide from the marine sponge Jaspis SP. Tetrahedron Lett. 1986;27:2797–2800. [Google Scholar]
- 12.Curiel D T. Strategies to adapt adenoviral vectors for targeted delivery. Ann N Y Acad Sci. 1999;886:158–171. doi: 10.1111/j.1749-6632.1999.tb09409.x. [DOI] [PubMed] [Google Scholar]
- 13.Damke H, Baba T, Warnock D E, Schmid S L. Induction of mutant dynamin specifically blocks endocytic coated vesicle formation. J Cell Biol. 1994;127:915–934. doi: 10.1083/jcb.127.4.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eichholtz T, de Bont D B, de Widt J, Liskamp R M, Ploegh H L. A myristoylated pseudosubstrate peptide, a novel protein kinase C inhibitor. J Biol Chem. 1993;268:1982–1986. [PubMed] [Google Scholar]
- 15.Freimuth P, Springer K, Berard C, Hainfeld J, Bewley M, Flanagan J. Coxsackievirus and adenovirus receptor amino-terminal immunoglobulin V-related domain binds adenovirus type 2 and fiber knob from adenovirus type 12. J Virol. 1999;73:1392–1398. doi: 10.1128/jvi.73.2.1392-1398.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geli M I, Riezman H. Endocytic internalization in yeast and animal cells: similar and different. J Cell Sci. 1998;111:1031–1037. doi: 10.1242/jcs.111.8.1031. [DOI] [PubMed] [Google Scholar]
- 17.Greber U F. Delivery of animal virus DNA into the nucleus. In: Seymour L, Kabanov A, Felgner P, editors. Self-assembling complexes for gene delivery: from chemistry to clinical trial. Chichester, United Kingdom: John Wiley & Sons, Ltd.; 1998. pp. 89–114. [Google Scholar]
- 18.Greber U F. Virus assembly and disassembly: the adenovirus cysteine protease as a trigger factor. Rev Med Virol. 1998;8:213–222. doi: 10.1002/(sici)1099-1654(1998100)8:4<213::aid-rmv225>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 19.Greber U F, Suomalainen M, Stidwill R P, Boucke K, Ebersold M, Helenius A. The role of the nuclear pore complex in adenovirus DNA entry. EMBO J. 1997;16:5998–6007. doi: 10.1093/emboj/16.19.5998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greber U F, Webster P, Weber J, Helenius A. The role of the adenovirus protease in virus entry into cells. EMBO J. 1996;15:1766–1777. [PMC free article] [PubMed] [Google Scholar]
- 21.Greber U F, Willetts M, Webster P, Helenius A. Stepwise dismantling of adenovirus 2 during entry into cells. Cell. 1993;75:477–486. doi: 10.1016/0092-8674(93)90382-z. [DOI] [PubMed] [Google Scholar]
- 22.Gruenberg J, Maxfield F R. Membrane transport in the endocytic pathway. Curr Opin Cell Biol. 1995;7:552–563. doi: 10.1016/0955-0674(95)80013-1. [DOI] [PubMed] [Google Scholar]
- 23.Hong S S, Karayan L, Tournier J, Curiel D T, Boulanger P A. Adenovirus type 5 fiber knob binds to MHC class I alpha2 domain at the surface of human epithelial and B lymphoblastoid cells. EMBO J. 1997;16:2294–2306. doi: 10.1093/emboj/16.9.2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Horwitz M S. Adenoviruses. In: Fields B N, Knipe D M, editors. Virology. 2nd ed. Vol. 1. New York, N.Y: Raven Press; 1990. pp. 1723–1740. [Google Scholar]
- 25.Ishida A, Kameshita I, Okuno S, Kitani T, Fujisawa H. A novel highly specific and potent inhibitor of calmodulin-dependent protein kinase II. Biochem Biophys Res Commun. 1995;212:806–812. doi: 10.1006/bbrc.1995.2040. [DOI] [PubMed] [Google Scholar]
- 26.Jörnvall H, Philipson L. Limited proteolysis and a reactive cysteine residue define accessible regions in the native conformation of the adenovirus hexon protein. Eur J Biochem. 1980;104:237–247. doi: 10.1111/j.1432-1033.1980.tb04421.x. [DOI] [PubMed] [Google Scholar]
- 27.Lawson M A, Maxfield F R. Ca(2+)- and calcineurin-dependent recycling of an integrin to the front of migrating neutrophils. Nature. 1995;377:75–79. doi: 10.1038/377075a0. [DOI] [PubMed] [Google Scholar]
- 28.Li E, Stupack D, Bokoch G M, Nemerow G R. Adenovirus endocytosis requires actin cytoskeleton reorganization mediated by Rho family GTPases. J Virol. 1998;72:8806–8812. doi: 10.1128/jvi.72.11.8806-8812.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li E, Stupack D, Klemke R, Cheresh D A, Nemerow G R. Adenovirus endocytosis via α(V) integrins requires phosphoinositide-3-OH kinase. J Virol. 1998;72:2055–2061. doi: 10.1128/jvi.72.3.2055-2061.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marsh M, Bolzau E, Helenius A. Penetration of Semliki Forest virus from acidic prelysosomal vacuoles. Cell. 1983;32:931–940. doi: 10.1016/0092-8674(83)90078-8. [DOI] [PubMed] [Google Scholar]
- 31.Marsh M, Bron R. SFV infection in CHO cells: cell-type specific restrictions to productive virus entry at the cell surface. J Cell Sci. 1997;110:95–103. doi: 10.1242/jcs.110.1.95. [DOI] [PubMed] [Google Scholar]
- 32.Mathias P, Galleno M, Nemerow G R. Interactions of soluble recombinant integrin αv β5 with human adenoviruses. J Virol. 1998;72:8669–8675. doi: 10.1128/jvi.72.11.8669-8675.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miles B D, Luftig R B, Weatherbee J A, Weihing R R, Weber J. Quantitation of the interaction between adenovirus types 2 and 5 and microtubules inside infected cells. Virology. 1980;105:265–269. doi: 10.1016/0042-6822(80)90177-4. [DOI] [PubMed] [Google Scholar]
- 34.Miyazawa N, Leopold P L, Hackett N R, Ferris B, Worgall S, Falck-Pedersen E, Crystal R G. Fiber swap between adenovirus subgroups B and C alters intracellular trafficking of adenovirus gene transfer vectors. J Virol. 1999;73:6056–6065. doi: 10.1128/jvi.73.7.6056-6065.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muallem S, Kwiatkowska K, Xu X, Yin H L. Actin filament disassembly is a sufficient final trigger for exocytosis in nonexcitable cells. J Cell Biol. 1995;128:589–598. doi: 10.1083/jcb.128.4.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murphy C, Saffrich R, Grummt M, Gournier H, Rybin V, Rubino M, Auvinen P, Lutcke A, Parton R G, Zerial M. Endosome dynamics regulated by a Rho protein. Nature. 1996;384:427–432. doi: 10.1038/384427a0. [DOI] [PubMed] [Google Scholar]
- 37.Nakano M Y, Greber U F. Quantitative microscopy of fluorescent adenovirus entry. J Struct Biol. 2000;129:57–68. doi: 10.1006/jsbi.1999.4201. [DOI] [PubMed] [Google Scholar]
- 38.Ng T, Shima D, Squire A, Bastiaens P I, Gschmeissner S, Humphries M J, Parker P J. PKCalpha regulates beta1 integrin-dependent cell motility through association and control of integrin traffic. EMBO J. 1999;18:3909–3923. doi: 10.1093/emboj/18.14.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Panetti T S, McKeown-Longo P J. The alpha v beta 5 integrin receptor regulates receptor-mediated endocytosis of vitronectin. J Biol Chem. 1993;268:11492–11495. [PubMed] [Google Scholar]
- 40.Panetti T S, Wilcox S A, Horzempa C, McKeown-Longo P J. Alpha v beta 5 integrin receptor-mediated endocytosis of vitronectin is protein kinase C-dependent. J Biol Chem. 1995;270:18593–18597. doi: 10.1074/jbc.270.31.18593. [DOI] [PubMed] [Google Scholar]
- 41.Pastan I, Seth P, FitzGerald D, Willingham M. Adenovirus entry into cells: some new observations on an old problem. New York, N.Y: Springer-Verlag; 1986. [Google Scholar]
- 42.Patterson S, Russell W C. Ultrastructural and immunofluorescence studies of early events in adenovirus-HeLa cell interactions. J Gen Virol. 1983;64:1091–1099. doi: 10.1099/0022-1317-64-5-1091. [DOI] [PubMed] [Google Scholar]
- 43.Rauma T, Tuukkanen J, Bergelson J M, Denning G, Hautala T. rab5 GTPase regulates adenovirus endocytosis. J Virol. 1999;73:9664–9668. doi: 10.1128/jvi.73.11.9664-9668.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ren X D, Kiosses W B, Schwartz M A. Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J. 1999;18:578–585. doi: 10.1093/emboj/18.3.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ringerike T, Stang E, Johannessen L E, Sandnes D, Levy F O, Madshus I H. High-affinity binding of epidermal growth factor (EGF) to EGF receptor is disrupted by overexpression of mutant dynamin (K44A) J Biol Chem. 1998;273:16639–16642. doi: 10.1074/jbc.273.27.16639. [DOI] [PubMed] [Google Scholar]
- 46.Rodriguez E, Everitt E. Adenovirus uncoating and nuclear establishment are not affected by weak base amines. J Virol. 1996;70:3470–3477. doi: 10.1128/jvi.70.6.3470-3477.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sampath P, Pollard T D. Effects of cytochalasin, phalloidin, and pH on the elongation of actin filaments. Biochemistry. 1991;30:1973–1980. doi: 10.1021/bi00221a034. [DOI] [PubMed] [Google Scholar]
- 48.Schmid S L, McNiven M A, De Camilli P. Dynamin and its partners: a progress report. Curr Opin Cell Biol. 1998;10:504–512. doi: 10.1016/s0955-0674(98)80066-5. [DOI] [PubMed] [Google Scholar]
- 49.Shenk T. Adenoviridae. In: Fields B N, Knipe D M, Howley P M, editors. Fundamental virology. 3rd ed. New York, N.Y: Lippincott-Raven; 1996. pp. 979–1016. [Google Scholar]
- 50.Shurety W, Stewart N L, Stow J L. Fluid-phase markers in the basolateral endocytic pathway accumulate in response to the actin assembly-promoting drug Jasplakinolide. Mol Biol Cell. 1998;9:957–975. doi: 10.1091/mbc.9.4.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Siddhanta U, Mcllroy J, Shah A, Zhang Y, Backer J M. Distinct roles for the p110alpha and hVPS34 phosphatidylinositol 3′-kinases in vesicular trafficking, regulation of the actin cytoskeleton, and mitogenesis. J Cell Biol. 1998;143:1647–1659. doi: 10.1083/jcb.143.6.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Song J C, Hrnjez B J, Farokhzad O C, Matthews J B. PKC-epsilon regulates basolateral endocytosis in human T84 intestinal epithelia: role of F-actin and MARCKS. Am J Physiol. 1999;277:C1239–C1249. doi: 10.1152/ajpcell.1999.277.6.C1239. [DOI] [PubMed] [Google Scholar]
- 53.Stauffer T P, Guerini D, Carafoli E. Tissue distribution of the four gene products of the plasma membrane Ca2+ pump. A study using specific antibodies. J Biol Chem. 1995;270:12184–12190. doi: 10.1074/jbc.270.20.12184. [DOI] [PubMed] [Google Scholar]
- 54.Suomalainen M, Nakano M Y, Boucke K, Keller S, Stidwill R P, Greber U F. Microtubule-dependent minus and plus end-directed motilities are competing processes for nuclear targeting of adenovirus. J Cell Biol. 1999;144:657–672. doi: 10.1083/jcb.144.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sussenbach J S. Early events in the infection of adenovirus type 5 in HeLa cells. Virology. 1967;33:567–574. doi: 10.1016/0042-6822(67)90056-6. [DOI] [PubMed] [Google Scholar]
- 56.Tomko R P, Xu R, Philipson L. HCAR and MCAR: the human and mouse cellular receptors for subgroup C adenoviruses and group B coxsackieviruses. Proc Natl Acad Sci USA. 1997;94:3352–3356. doi: 10.1073/pnas.94.7.3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Von Seggern D J, Chiu C Y, Fleck S K, Stewart P L, Nemerow G R. A helper-independent adenovirus vector with E1, E3, and fiber deleted: structure and infectivity of fiberless particles. J Virol. 1999;73:1601–1608. doi: 10.1128/jvi.73.2.1601-1608.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang K N, Huang S, Kapoormunshi A, Nemerow G. Adenovirus internalization and infection require dynamin. J Virol. 1998;72:3455–3458. doi: 10.1128/jvi.72.4.3455-3458.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang X H, Bergelson J M. Coxsackievirus and adenovirus receptor cytoplasmic and transmembrane domains are not essential for coxsackievirus and adenovirus infection. J Virol. 1999;73:2559–2562. doi: 10.1128/jvi.73.3.2559-2562.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weber J. Genetic analysis of adenovirus type 2. III. Temperature sensitivity of processing of viral proteins. J Virol. 1976;17:462–471. doi: 10.1128/jvi.17.2.462-471.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wickham T J, Filardo E J, Cheresh D A, Nemerow G R. Integrin αvβ5 selectively promotes adenovirus mediated cell membrane permeabilization. J Cell Biol. 1994;127:257–264. doi: 10.1083/jcb.127.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wickham T J, Mathias P, Cheresh D A, Nemerow G R. Integrin-alpha-v-beta-3 and integrin-alpha-v-beta-5 promote adenovirus internalization but not virus attachment. Cell. 1993;73:309–319. doi: 10.1016/0092-8674(93)90231-e. [DOI] [PubMed] [Google Scholar]
- 63.Zen K, Biwersi J, Periasamy N, Verkman A S. Second messengers regulate endosomal acidification in Swiss 3T3 fibroblasts. J Cell Biol. 1992;119:99–110. doi: 10.1083/jcb.119.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]