

Abstract
Long-read sequencing technologies substantially overcome the limitations of short-reads but have not been considered a feasible replacement for population-scale projects, being a combination of too expensive, not scalable enough, or too error-prone. Here, we develop an efficient and scalable wet lab and computational protocol for Oxford Nanopore Technologies (ONT) long-read sequencing that seeks to address those limitations. We applied our protocol to cell lines and brain tissue samples as part of a pilot project for the NIH Center for Alzheimer’s and Related Dementias (CARD). Using a single PromethION flow cell, we can detect single nucleotide polymorphisms (SNPs) with F1-score comparable to Illumina short-read sequencing. Small indel calling remains difficult within homopolymers and tandem repeats, but achieves good concordance to Illumina indel calls elsewhere. Further, we can discover structural variants with F1-score on par with state-of-the-art de novo assembly methods. Our protocol phases small and structural variants at megabase scales and produces highly accurate, haplotype-specific methylation calls.
Introduction
Most current large-scale genomics projects rely on reference mapping of short-reads to detect and genotype variants, such as single nucleotide polymorphisms (SNPs), small insertions and deletions (indels), or structural variations (SVs) 1. For example, short-read whole genome sequencing is routinely used in population-scale studies to discover variation in human populations 2,3, or to perform disease associations studies 4, including in cancer 5,6.
However, a substantial part of the variation in the human genome is not accessible to short reads 7. This is because it is difficult to detect structural variants that are comparable to or longer than an individual read length 8,9, resulting in missingness and error rates much higher than for small variant detection 10,11. In addition, variation inside the repetitive regions of the genome is difficult to profile with short-reads 12,13 due to reference mapping ambiguity and bias 14. Read-based phasing of heterozygous variants into long haplotypes is also limited by read length 15. Previous studies mostly relied on reference haplotype panels to phase known variants 16, but this method is not applicable to rare and de novo mutations.
Long-read sequencing, such as those from Pacific Biosciences (PacBio) or Oxford Nanopore Technologies (ONT), can overcome the limitations of short-reads and has been shown to substantially improve structural variant calling performance 17,18 and small variant detection inside difficult-to-map parts of the genome 19. Long-read methods can also phase small and structural variants into megabase-scale phase blocks 19–21.
In addition to variant calling, several studies have used long-read sequencing to generate complete or nearly complete de novo genome assemblies 22,23. Notably, the Telomere-to-Telomere (T2T) Consortium produced the complete de novo assembly of a human genome, including centromeres and long segmental duplications 24. Recently, the Human Pangenome Reference Consortium (HPRC) released 47 nearly-complete haplotype-resolved human genomes with diverse genetic backgrounds 25.
Despite these advances, cost and scalability have remained prohibitive barriers to the use of long-read sequencing in population-scale studies. For example, recent projects that generated high-quality genome assemblies, such as those from the Vertebrate Genomes Project (VGP), T2T, and HPRC, all used an expensive combination of multiple sequencing technologies at high coverage, including PacBio HiFi, ultra-long ONT, Hi-C, and parental sequencing 26.
Here, we show that it is possible to achieve state-of-the-art small and structural calling performance using only ONT reads produced by a single flow cell at high throughput. First, we developed specialized ONT sequencing protocols that balance read length and yield. Second, we complement the sequencing protocols with a computational pipeline called Napu (Nanopore Analysis Pipeline) that produces haplotype-resolved de novo assemblies, along with phased small variants, structural variants, and methylation calls. We make our sequencing and informatics pipelines openly available, the latter as a complete, easily runnable open-source software package.
Results
Scalable ONT sequencing of cell lines and brain tissue.
Several recent studies utilized ultra-long (100 kb+) ONT sequencing to produce high-quality de novo assemblies of human genomes 27–31. However, multiple flow cells were used to achieve sufficient genomic coverage, as ultra-long DNA preparation protocols typically see lower sequencing yields. In this work, we optimized a DNA processing and library preparation protocol to yield high data output (>100 Gb, corresponding to >33X coverage) in a scalable manner from a single PromethION flow-cell, while still maintaining read lengths sufficient for de novo assembly and long-range phasing.
The DNA processing and library preparation protocol (Methods) are publicly available through the protocols.io for the frontal cortex 32 and cell-lines 33. Overall, per sample, the DNA processing step yields ~10 ug of sheared DNA. Together, the DNA processing and library preparation take approximately 20 hours over two days to process up to 16 samples in a single batch, and the PromethION whole genome sequencing takes 72 hours (Figure 1).
Figure 1. Single flow cell Oxford Nanopore Technologies (ONT) sequencing protocol.
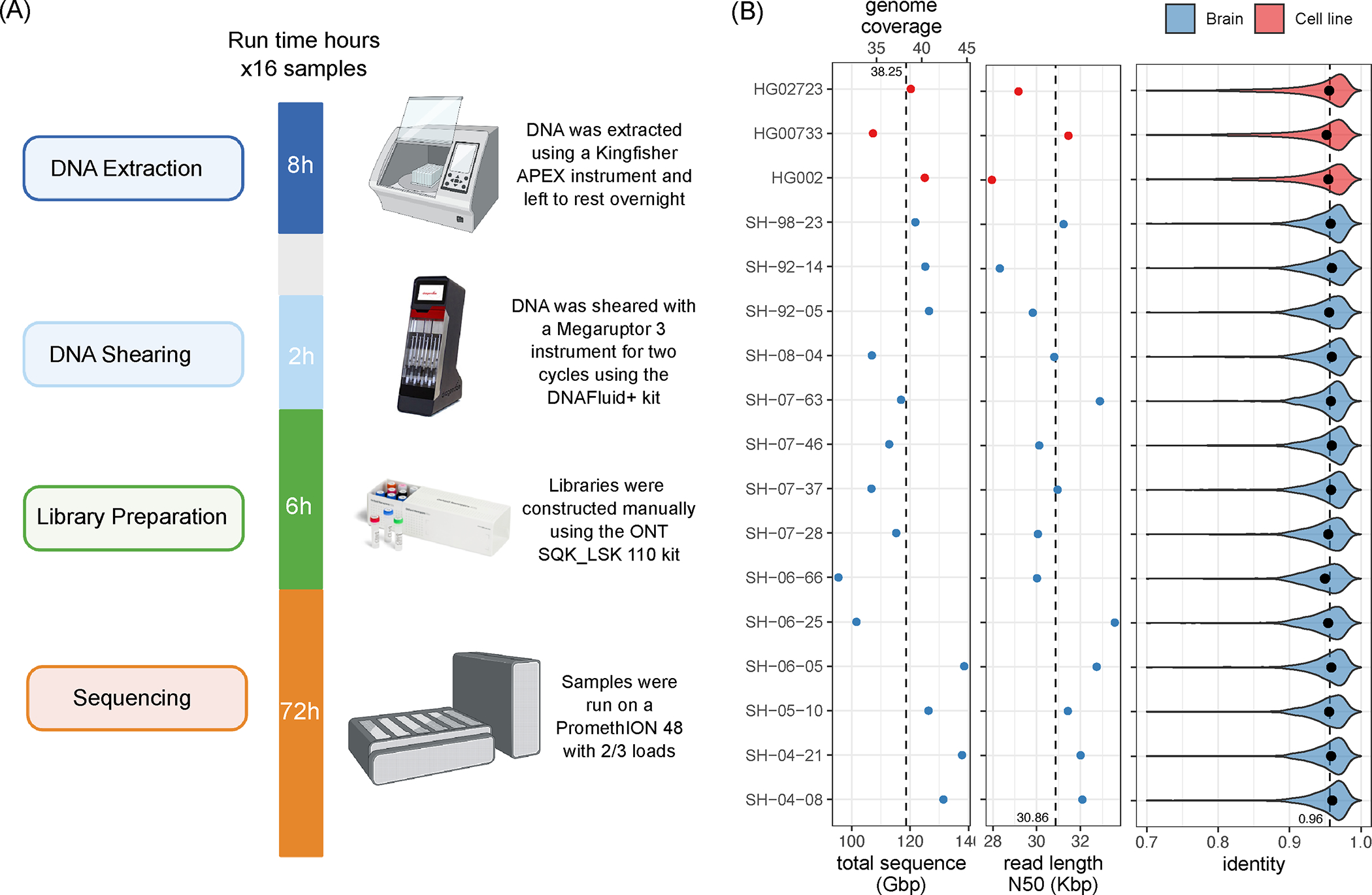
(Left) Overview of the sequencing protocol, indicating all processes from DNA extraction to sequencing. In brief, the DNA is extracted using a Kingfisher Apex instrument using the Nanobind Tissue Big DNA kit. The DNA is then sheared on a Megaruptor3 instrument, and libraries are constructed using an SQK-LSK 110 kit and sequenced on a PromethION for 72 hours. Panel was created using BioRender.com. (Right) from left-to-right: total sequenced bases / haploid human genome coverage (assuming a 3.1GB genome) from PASS reads (with estimated QV>=10) for each sample. The vertical dotted line marks the average yield across samples. Read length N50 of PASS reads, i.e., the read length (y-axis) such that reads of this length or longer represent 50% of the total sequence. The vertical dotted line marks the average N50 across samples. Distribution of PASS read identities when aligned to T2T-CHM13 v2.0. The dots mark the median identity in each sample, and the vertical dotted line is the average across samples. Source data is available at Supplementary Table 1.
Using our optimized protocol, we sequenced 17 human genomes, including three cell lines (HG002, HG00733, HG02723) that have been extensively used in other benchmarking studies, and 14 brain tissue samples that were obtained as a part of NABEC cohort 34. Each dataset was sequenced using a single PromethION R9 flow cell, yielding on average 116 Gb of base-called reads (~37X coverage assuming 3.1Gb genome) with an estimated average across bases read quality above Q10 (Figure 1; Supplementary Table 1). The average read N50 was 31 kb (average maximum = 769 kb), which is overall lower than other long DNA preparation protocols. However, the libraries with longer DNA fragments require multiple flow cells to achieve sufficient read coverage (Shafin et al., 2020). Read N50 is a maximum number such that reads longer than the N50 cover at least half of the total read length. We used the R9.4.1 pore version and the Guppy version 6.1.2 in super accuracy mode for base and methylation calls, median read identity mapped to GRCh38 was 96%.
Napu variant and methylation calling pipeline overview.
We adapted existing tools and developed additional methods for high-quality variant calling, haplotype-specific methylation profiling, and diploid de novo assembly (Methods; Extended Data Figure 1). The pipeline begins by generating a diploid de novo assembly using a combination of Shasta, which produces a haploid assembly and Hapdup (https://github.com/KolmogorovLab/hapdup), which generates locally phased diploid contigs. We then use the generated assemblies to call structural variants (at least 50 bp in size) against a given reference genome using an assembly-to-reference tool called hapdiff (https://github.com/KolmogorovLab/hapdiff; Methods).
Ideally, small variants could also be recovered from diploid contigs, as has been successfully done for HiFi-based assemblies 12,25. Our Shasta-Hapdup assemblies had mean substitution error rates of ~8 per 100 kb, an improvement over the previous ONT-based assemblies 19,29,30, but higher than current contig assemblies produced with PacBio HiFi (<1 per 100kb) 25. Reference-based variant calling methods can abstain from making a call when the read alignment is ambiguous, which can reduce the false-positive error (Shafin et al., 2021). Thus, in Napu we used an updated version of the PEPPER-Margin-DeepVariant software 19 to call small variants against a reference.
Given a set of structural variants produced with de novo assemblies, and reference-based small variant calls, Napu phases them into a harmonized variant call set using newly introduced functionality in the Margin tool. In addition, given the phased reference alignment with methylation tags (produced by Guppy), we produce haplotype-specific calls of hypo- and hyper-methylated regions of the genome.
We publish the complete Napu pipeline written in WDL in the Dockstore repository (https://dockstore.org/organizations/NIHCARD; Methods) to encourage easy reuse. Our analysis was run on the cloud using the Terra compute environment 35. Analysis of a single ONT human sample at 30–40x coverage took 22–25 wall-clock hours if run on the Terra environment (2200–2500 CPU hours; estimated Google Cloud computing cost about 100$; Supplementary Table 2).
Small variant calling and benchmarking.
We benchmarked the performance of small variant (substitutions and indels shorter than 50 bp) calls produced by the updated PEPPER-Margin-DeepVariant and compared them to Illumina-based small variant calls produced by DeepVariant (Figure 2; Supplementary Table 3). Comparisons are reported using false positives (FP) and false negative (FN) and F1 score (harmonic mean of precision and recall) metrics. First, we used the Genome in a Bottle (GIAB) small variant benchmark (v4.2.1) that provides a curated set of small variant calls for the HG002 genome. Inside the Tier1-confident regions, both ONT and Illumina-based SNP calls were highly concordant with the GIAB benchmark (F1-scores 0.997 and 0.996, respectively). The residual false-negative error was substantially lower for ONT SNP calls, compared to Illumina calls (9,993 and 20,281, respectively). On the other hand, ONT SNP calls had substantially more false-positive errors, compared to Illumina (5,923 and 2,922, respectively).
Figure 2. Small variant calling performance evaluation.
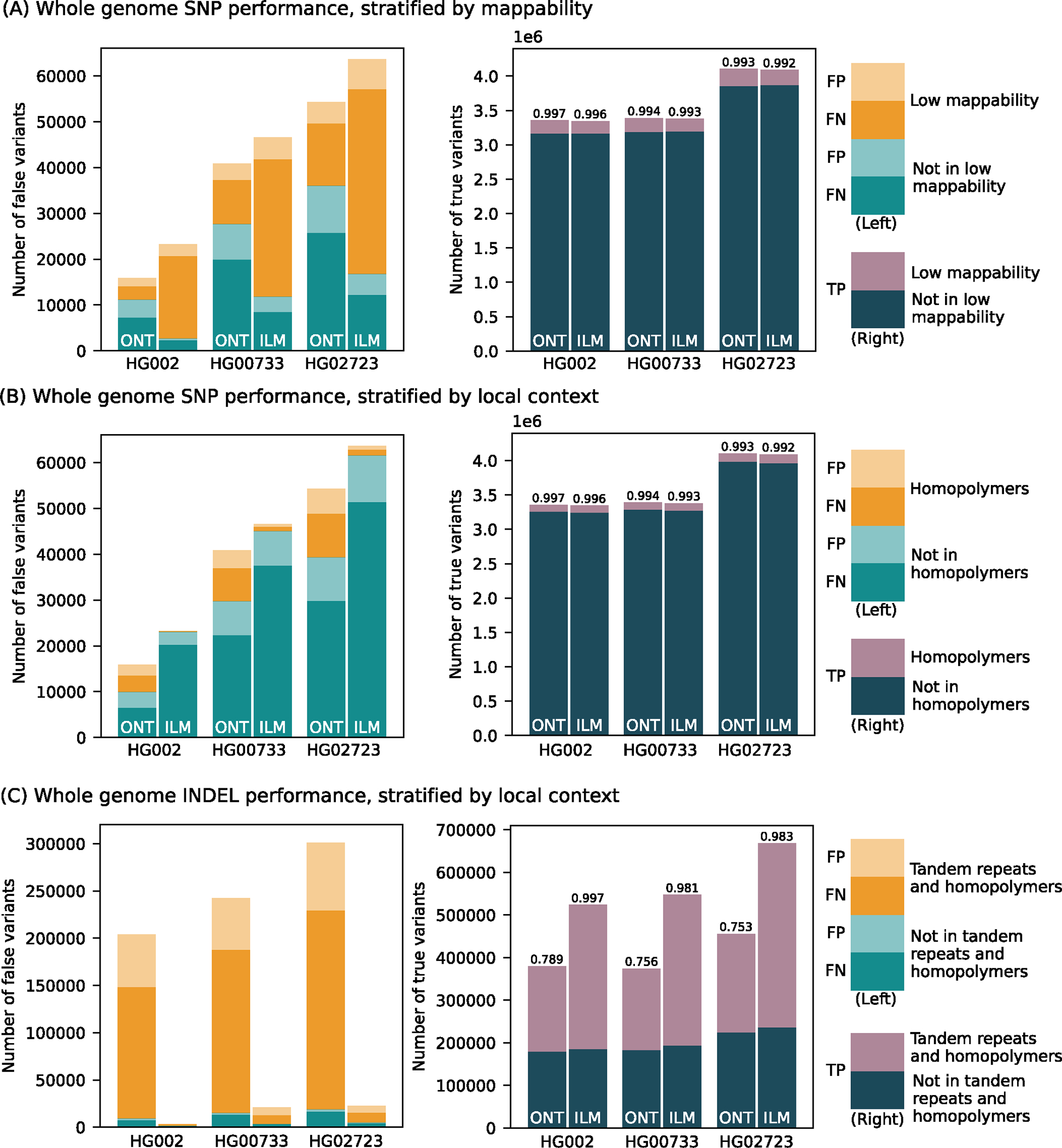
Number of false positive, false negative, and true positive small variant calls made by PEPPER-Margin-DeepVariant (PMDV) using either ONT reads or DeepVariant using Illumina reads. (A) SNP performance, stratified by genomic regions mappability. (B) SNP performance, stratified by homopolymer context. (C) INDEL performance stratified by homopolymers and tandem repeats context. F1-scores are reported on top of the true positive bars. Statistics computed against the Genome in a Bottle v4.2.1 benchmark for HG002; for other cell lines (HG00733, HG02723) small variant calls generated by DeepVariant with HiFi reads are used. Source data is available at Supplementary Table 3.
Since the GIAB benchmark is only available for the HG002 genome, we used small variant calls from HiFi reads (produced by DeepVariant) as ground truth for the HG00733 and HG02723 genomes. The results were consistent with the GIAB benchmark analysis, confirming the reduced SNP error rate of ONT-based methods, compared to Illumina within the Tier1 benchmark regions (Figure 2; Supplementary Table 3).
The difference in recall and precision between ONT and Illumina SNP calls was dependent on the genomic context (Figure 2). The major source of error in Illumina SNP calls is due to false negatives within regions of known low mappability. Inside those regions, ONT SNP calls had noticeably higher SNP F1-score (0.987) compared to Illumina (0.944). Conversely, Illumina performance was better in homopolymers of size at least 7bp (SNP F1-score was 0.999 for Illumina and 0.970 for ONT) and, to a lesser extent, within tandem repeats (SNP F1-score 0.997 for Illumina and 0.992 for ONT).
Small indels continue to be the major source of error for ONT small variant calls (indel F1 score of 0.789 for ONT vs. 0.997 for Illumina inside the Tier1 GIAB regions) (Figure 2). The large majority of errors in ONT indel calls occur within homopolymers and tandem duplications (F1 score of 0.676 for ONT vs. 0.997 for Illumina). Outside of homopolymer runs and tandem repeats (approximately 35% of all indels), F1-scores for ONT improved substantially (F1-score 0.975 for ONT), although still lower than Illumina (0.996 F1). In exons (defined by the GIAB benchmark and representing <0.1% of all indels) F1-scores were 0.9230 for ONT and 0.9923 for Illumina (Supplementary Table 3).
De novo haplotype-resolved assembly using only ONT reads.
Recent studies utilized HiFi-based de novo assemblies to produce highly accurate and complete structural variant calls 12,25. To profile the full spectrum of heterozygous variants, diploid assembly is required; HiFi-based studies used either trio or Hi-C information to produce phased assemblies. Although Shasta has a somewhat experimental phased assembly mode designed to work with ultra-long (>= 100 kb) reads, the default modes of current ONT assembly methods (such as Shasta or Flye) only generate haploid assemblies, representing a random mosaic of the paternal and maternal haplotypes. We therefore developed a method called Hapdup that takes a haploid assembly as input and produces a locally phased diploid assembly, sometimes also referred to as a dual assembly (Methods).
Using the Shasta + Hapdup combination, we generated de novo assemblies for 14 brain samples and three cell lines (Extended Data Figure 2; Supplementary Table 4). All assembly statistics were highly consistent among the samples, with the mean haploid assembly size of 2.88 Gb; mean NG50 = 22.0 Mb; mean NGA50 = 14.6 Mb (measured against the T2T-CHM13 2.0 reference). NG50 is the maximum number such that contigs longer than NG50 cover at least half of the genome length. NGA50 is defined similarly, but for reference alignment lengths. Mean QV (34.22) was estimated from k-mer frequencies using yak36. Mean NG50 of the phase blocks was ~2 Mb; switch error rate of cell line assemblies was 0.07–0.19% (Supplementary Table 5).
Compared to the trio-binned ONT HG002 assemblies produced from a recent long-read benchmarking study 26, our assemblies had slightly reduced contiguity (due to lower read depth and length), but better QV and phasing accuracy (Extended Data Figure 3; Supplementary Table 5). Compared to trio-binned HiFi HG002 assemblies, our assemblies had lower QV due to the residual error in the ONT-based consensus.
Structural variant calling and benchmarking.
To produce assembly-based SV calls, we developed a package called hapdiff, which is a combination of minimap2 and a modified version of SVIM-asm (Methods). In particular, we added functionality to group multiple indels inside a single VNTR element.
We benchmarked Hapdup and HPRC assembly-based SV calls, as well as reference mapping-based SV calls produced with Sniffles2 37 and CuteSV 18. We also included a comparison with short-read-based SV calls using Manta, one of the top performers in the recent short-read SV studies 10,11. We benchmarked these sets of SV calls using Truvari 38 against the recent manually curated structural variant benchmark produced by the Genome in a Bottle initiative for the HG002 genome 11.
Hapdup-based SV calls (F1-score 0.967) improved over Sniffles2 (0.953) and CuteSV (0.938) that were also generated using the ONT data. HPRC-based assembly concordance was only slightly higher (0.970), compared to Hapdup (Figure 3; Supplementary Table 6). Illumina-based SV calls had substantially lower performance (F1-score 0.402), in particular missing many insertions. In addition, long insertions are often misclassified as translocations by short-read methods 17.
Figure 3. Structural variant evaluations.
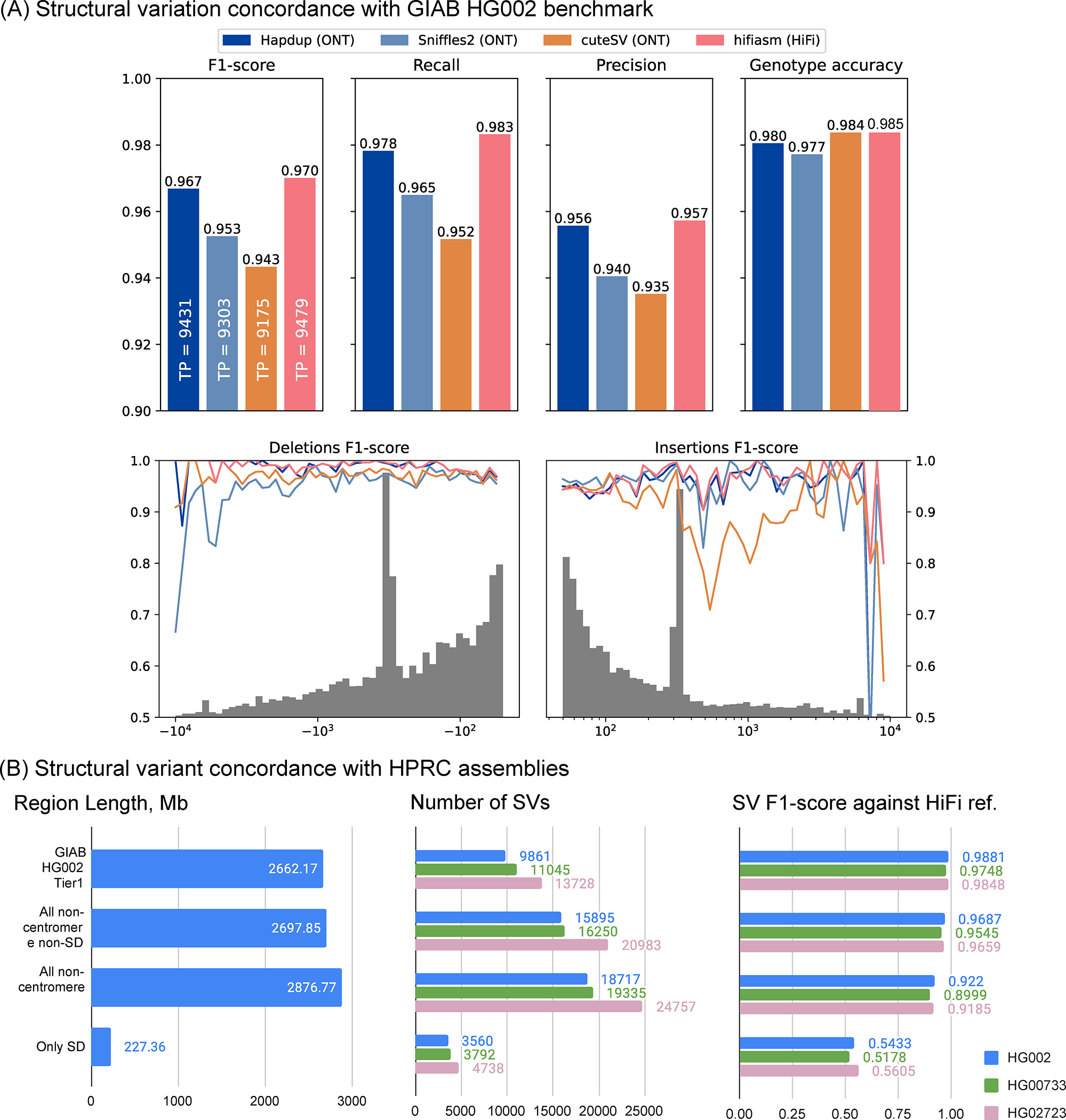
(A) Recall, precision and F1-scores computed for various tools and sequencing technologies with Genome in a Bottle Tier1 v0.6 benchmark as reference (defined on HG002). The gray histogram shows the distribution of SV sizes in the reference set. F1-scores computed for various SV size bins. (B) Structural variation call concordance with HiFi-based assemblies for various regions of the genome. Source data is available at Supplementary Tables 6–7.
The GIAB call set is limited to a set of high-confidence regions and lacks variant calls in many complex loci. To investigate the quality of our SV calls further, we used the assemblies generated by the HPRC as a benchmark on three cell lines and evaluated the concordance of SV calls inside various regions of the human genome (Supplementary Table 7). While the HPRC assemblies are not perfect, they do span nearly the entire genome. Using the T2T-CHM13 assembly as a reference, we filtered out annotated centromere satellite repeats and segmental duplications. The remaining regions contain 15,588 SVs in HG002 HPRC-based calls, a ~50% increase compared to the GIAB SV (v0.6) Tier1 regions. Hapdup retained a high agreement with HiFi assemblies on these regions (F1-score 0.95–0.97). In the regions with only satellite repeats filtered out, therefore keeping segmental duplications, Hapdup had a reduced F1-score of 0.93, and these regions contained 16,219 SVs total. Analysis with two other cell lines showed similar trends (Figure 3).
Analysis using the TT-Mars 39 tool that performs SV calls validation on the sequence level also confirmed the high quality of Hapdup-based SV calls relative to other tools (Extended Data Figure 4). Similarly, analysis using Flagger 25 confirmed high assembly accuracy (Supplementary Methods; Extended Data Figures 5–6). The assemblies also showed high concordance with the variants in challenging medically-relevant loci13 (Methods; Supplementary Tables 8 and 9).
Harmonization and phasing of small and structural variants.
Structural and small variant calls were harmonized and phased with Margin 2.3.1 to produce a complete representation of the sample variants (Methods). Mean phase block NG50 was 1.08 Mb among the brain samples (Figure 4; Supplementary Table 10). HG002 and HG0073 cell lines had similar statistics, but HG02723 had noticeably higher phase block length (NG50 2.83 Mb). This individual has a higher proportion of African ancestry, and as a result, fewer apparent blocks of autozygosity that prevent phasing.
Figure 4. Combined, phased small and structural variants improve the profiling of complex genomic regions.
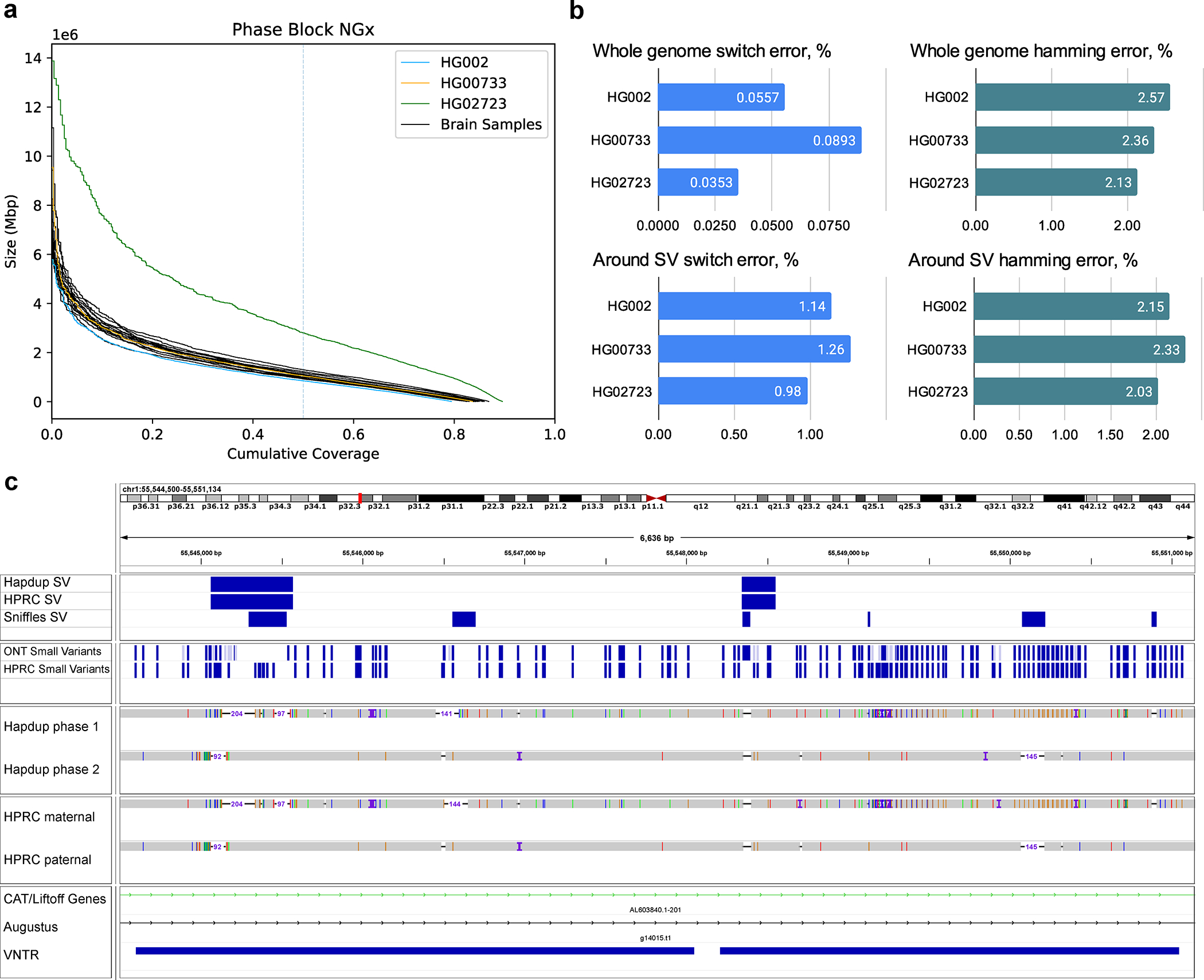
(A) Variant phasing evaluation. Left plot shows the phased block NGx, reported by Margin. HG02723 has an increased phase block length due to higher heterozygosity. (B) SNP hamming and switch error computed against the small variants in HiFi-based assemblies. Evaluations are also shown for a subset of SNPs that are within 100 bp of structural variants (C) An example of a Hapdup and hifiasm representations of complex clusters with small and structural variants at chr1:55,544,500–55,551,000 (in CHM13 reference), visualized using IGV. Top tracks show phased SNPs and SVs produced by Napu and derived from HPRC assemblies (using dipcall). A few inconsistencies between SNP positions are explained by ambiguities between read and contig alignments around SV sites. Source data is available at Supplementary Table 10.
A harmonized, phased view of all variants can facilitate the characterization of complex regions that contain multiple small and structural variants on both haplotypes. Figure 4 presents an example of such a region in the HG002 genome; our variant representation of this region provides an integrated view of variation that is consistent with the HPRC assemblies.
Analysis of rare structural variants in brain genomes.
We next analyzed the distribution of structural variants in the 14 brain samples, which have not been previously sequenced with long-reads. The analyzed samples represent control cases from the NABEC cohort (age range, 68 – 95 years), which at the time of death did not show signs of neurodegenerative disorders. On average, 19,255 SVs were identified in the most confident regions of the genome (Figure 5A; Extended Data Figure 7; Supplementary Table 11). The SVs were matched to three SV catalogs to annotate their frequency in the population. In each sample, about 304 (1.6%) SVs were absent from the public SV catalogs or matched with rare variants. Among those rare variants, about 14 per sample are located around genes, including ~4 on average overlapping coding regions (Figure 5A, right).
Figure 5. Structural variant landscape summary.
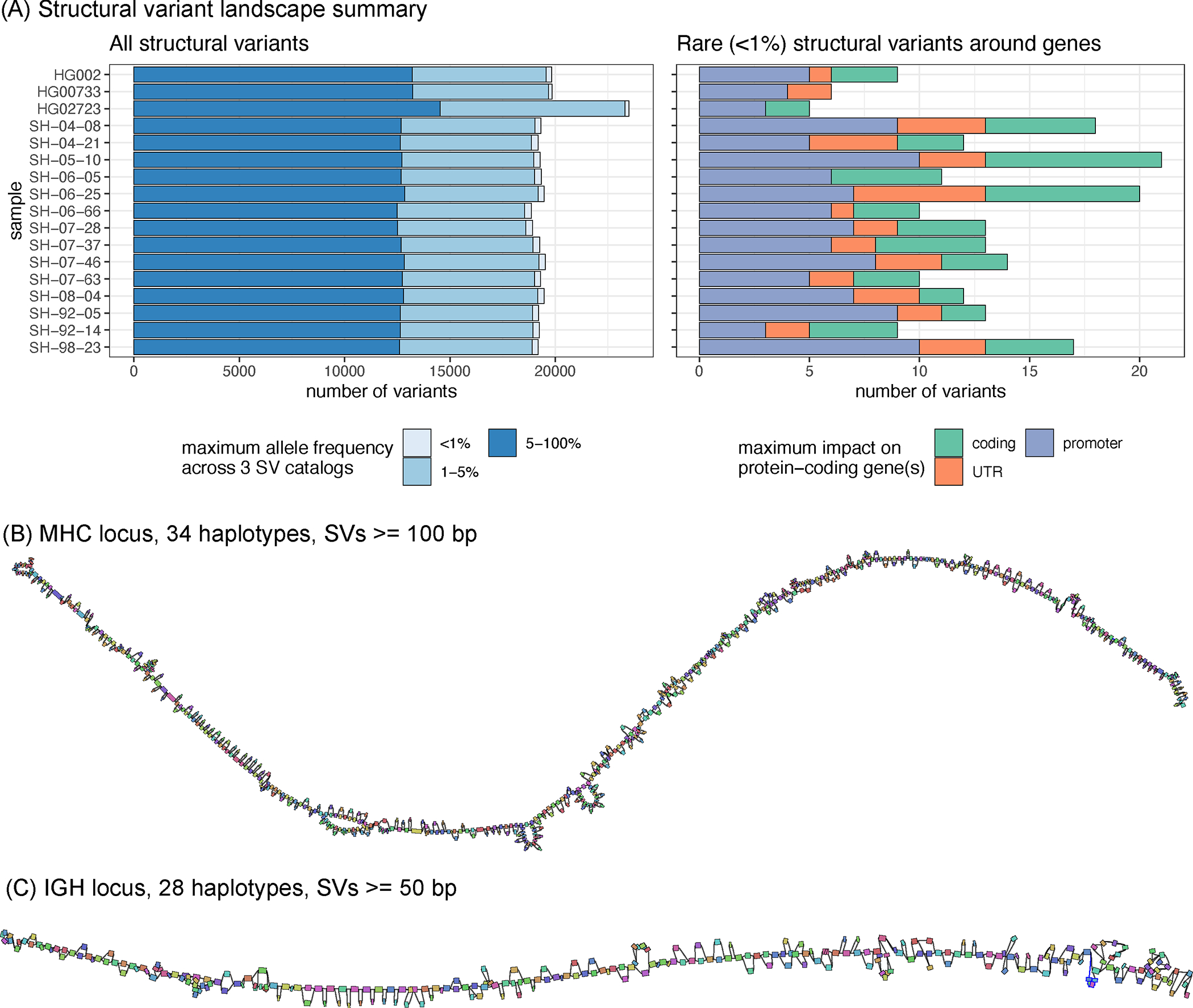
(A) The number of structural variants across samples. In the left panel, structural variants were annotated with three SV catalogs (the gnomAD-SV database, a long-read-based SV catalog, and the HPRC v1.0 SV catalog). SVs are matched if they have at least 10% genomic overlap. SVs close to centromeres, telomeres, or within segmental duplications were removed. The colors highlight the maximum frequency across these catalogs, the lighter blue showing “rare” SVs (with an allele frequency below 1%) in the catalogs, or unmatched. SVs may be unmatched, either because they are novel or due to the difficulties in the database comparison. The right panel shows the number of rare structural variants in protein-coding genes, grouped by their impact on the gene structure. (B) MHC pangenome built from 28 brain and 6 cell line haplotypes, containing 640 nodes, SVs over 100bp are shown. (C) IGH pangenome built from 28 brain haplotypes containing 268 nodes. Pangenome graphs were visualized using Bandage. Each graph node represents an allele, and two nodes are connected if the corresponding alleles are linked in at least one of the haplotypes. Colors are assigned at random. Nodes are randomly laid out along the reference coordinates. Source data is available at Supplementary Table 11.
Despite being control samples, we found 3 SVs in total amongst the SVs coinciding with coding regions of genes that are predicted to be intolerant to loss-of-function variants or to be haplo-insufficient. One such example is a 4.2 Kbp heterozygous deletion of a transcription start site and exon of RBFOX1, a gene that may be involved in spinocerebellar ataxia type 2, a rare neurological condition (Extended Data Figure 8).
Our brain genome assemblies contained contiguous sequences of highly polymorphic loci. To investigate such loci, we looked at the major histocompatibility complex (MHC) and immunoglobulin heavy chain (IGH) loci, which were both assembled in single haplotype-specific contigs in all our samples. Reference-based characterization of these regions is difficult because of high heterozygosity and repetitiveness. We instead constructed pangenome graphs (using minigraph) that represent all structural variants 25. The MHC pangenome graph was built from 28 brain and 6 cell line haplotypes and consists of 1,294 nodes reflecting SVs larger than 50 bp; we also built a “lower resolution” graph with 640 nodes containing SVs over 100 bp (Figure 5B). An IGH pangenome was built from 28 brain haplotypes and contained 268 nodes with SVs larger than 50 bp (Figure 5C). Cell lines are typically derived from B-cell lymphocytes and contain extensive somatic rearrangements in this locus and are therefore not suitable for the germline variant comparison.
Haplotype-resolved methylation calls.
ONT sequencing also allows the identification of base modifications. Here, we produced phased 5-methylcytosine calls aligned to the GRCh38 and T2T-CHM13 references. Initial methylation calls were produced de novo using Remora (https://github.com/nanoporetech/remora).
For the HG002 genome, our methylation calls covered 28.83 million CpG sites (98.8% of total GRCh38 CpG sites) and had a high correlation to calls made using the standard whole genome bisulfite sequencing (WGBS) in regions covered by both technologies (R=0.949, RMSE=11.314; Figure 6a). We calculated correlations between all other samples and HG002 WGBS data to understand the level of ‘background’ methylation between all samples; these correlations ranged between 0.84–0.86 (Supplementary Table 12). HG002 WGBS calls were collected from the ONT open data repository (https://labs.epi2me.io/gm24385-5mc).
Figure 6. Haplotype-specific methylation profiling.
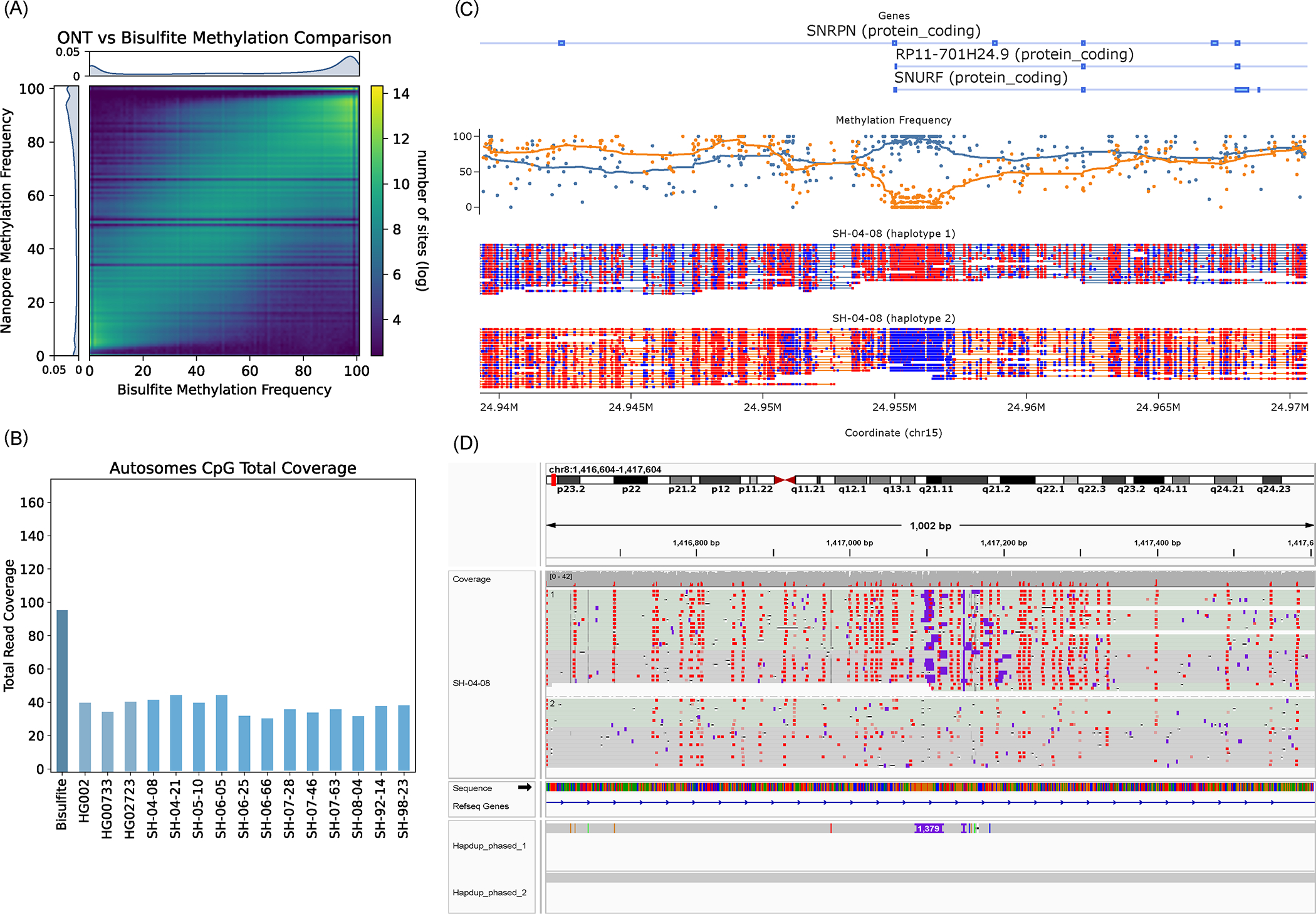
(A) Heatmap of concordance between Bisulfite whole genome sequencing and ONT Remora Methylation calls in HG002 at sites shared by both technologies covered by at least 5 reads. The lower coverage of ONT data causes striping in the heatmap at specific frequencies. (B) Read depth of Bisulfite and ONT samples. CpG sites are one position apart in the sense and antisense DNA strands due to C-G base pairing. Since this read coverage is counted per CpG location the actual coverage was doubled to account for the neighboring strand locations and estimate actual genome wide coverage. (C) An example of differential methylation pattern in the SNRPN locus in the brain sample SH-04–08. Red CpG sites are methylated and blue sites are unmethylated. Above the reads is a plot of methylation frequency and gene locations, visualized using modbamtools (D) IGV visualization of phased methylated ONT reads and the phased assemblies of brain sample SH-04–08 at the DLGAP2 gene locus that shows a 1,379 base pair insertion that is differentially methylated across haplotypes. Source data is available at Supplementary Table 12.
A unique feature of ONT-based methylation calls, compared to WGBS is haplotype-level resolution (Figure 6c). To explore this, we identified haplotype-specific differentially methylated regions in gene promoter regions and regions flanking structural variants. Here we consider gene promoters that have a difference in average methylation between haplotypes that are more than three deviations away from the absolute median difference.
Differential haplotype methylation patterns were found in 4.73% (690) of autosomal protein-coding gene promoters in HG002. Similarly, HG00733 and HG02723 had 4.43% (662) and 3.57% (519) of gene promoters differentially methylated, respectively. The brain samples had 2.8% - 3.8% of promoters differentially methylated. Differential haplotype methylation for the UCSC GRCh38 CpG islands track ranged from 6% - 7.6% (1651 – 2109) across brains and cell lines. Structural variants also showed differential haplotype methylation patterns in 5.7% - 6.2% (532 – 731) of deletions in cell lines and 4.6%-5.8% (454 −554) of deletions in brain samples. Figure 6D shows an example of gene DLG associated protein 2 (DLGAP2) in the SH-04–08 sample with a 1,379 bp heterozygous insertion that coincides with haplotype-specific methylation (Supplementary Table 12).
Comparing ONT sequencing using R9 and R10 protocols.
During the preparation of this manuscript, an updated version of the nanopore sequencing protocol (R10.4.1 kit V14) became commercially available. To evaluate the benefits of the updated protocol, we re-sequenced HG002, HG00733 and HG02723 cell lines and compared the results with the R9 protocol. We generated 110–121 Gb of reads with N50 ~29 kb using single flow cells (Extended Data Figure 9; Supplementary Table 13). The median read identity was 99%, compared to 96% using R9 sequencing.
As expected, the R10 protocol resulted in substantial improvements in reference-based indel calls. Inside GIAB Tier1 regions, F1-score improved from 0.79 to 0.87 (R9 and R10, respectively). Notably, outside of homoploymers and tandem repeats, R10 indel calls also improved substantially and were similar to Illumina calls (F1-scores 0.997 and 0.996, respectively). Similarly, indel F1-scores in exons improved from 0.923 to 0.985 (ONT R9 and R10, respectively). Reference-based SNP calls were similar between R9 and R10.
Assemblies and variant calls using R10 showed better or similar performance in almost all benchmark categories (Extended Data Figure 9). Notably, Shasta+Hapdup assembly QV improved from 34.3 to 42.8. This opens the possibility for calling SNP variants directly from diploid assemblies (Methods).
Discussion
In this work, we designed an ONT sequencing protocol that produces over 100Gb of ONT reads using a single PromethION flow cell. We developed methods and adapted existing tools, combined into an end-to-end Napu pipeline implemented in WDL, which is freely available to use and adapt without restriction. Using the sequencing data from 3 human cell lines and 14 post-mortem brain tissue samples, we showed that Napu produces state-of-the-art SNP, structural variant and methylation calls. This makes large-scale long-read sequencing projects feasible; the protocol is currently being used to sequence thousands of brain genomes as a part of the NIH CARD initiative.
Our SNP calls produced with PEPPER-Margin-DeepVariant were comparable to state-of-the-art short-read-based methods. As expected, the most noticeable improvement was associated with the regions of low short-read mappability. We also added a new functionality to Margin that can phase small and structural variant calls into megabase-scale haplotypes and reduces phasing switch error. Our methylation calls were highly concordant with the standard bisulfite sequencing, but in addition had haplotype-specific resolution, highlighted by our analysis of differentially methylated promoters.
Although an improvement over the previous ONT benchmarks, small indels inside homopolymers and low-complexity repeats remain the major source of the residual errors. This constitutes approximately two-thirds of all small indels in a human genome, but a minor fraction of overall variation in protein-coding sequence. Our evaluation of the R10 sequencing protocol showed substantial improvements in indel accuracy compared to the R9 protocol, in particular inside protein-coding regions. However, residual errors in long homopolymer and tandem repeat regions remain a challenge.
We developed a Hapdup method that generates de novo diploid assemblies from ONT sequencing only. Our assemblies had high structural quality through most of the human genome, but did not reconstruct many long segmental duplications. This is primarily due to the reduced read length, compared to standard ONT protocols (most unassembled segmental duplications are longer than our read’s N50). However, it may be possible to reconstruct some of the missing duplications using a haplotype clustering approach 40.
Our analysis of structural variation calls highlighted that different methods may represent the same genomic variation differently, for example, by splitting or merging multiple indels in close proximity, or shifting alignment coordinates inside VNTRs. This results in difficulties in variant comparison across multiple samples or methods 38. De novo assemblies implicitly encode the genomic variation, and new pangenome graph methods 25 aim to provide an alternative representation of small and structural variations. However, reference mapping is still required for matching the variant calls against the existing databases 41,42. Further improvements in structural variant representation and comparison models will be critical for the next large-scale, long-read genomic studies.
Overall, R9 and R10 benchmarks were highly concordant. Although an improvement, the R10 protocol has not been extensively evaluated on a wide range of human tissues. We expect that within the next few years other research groups will use both R9 and R10 chemistries, as it may be difficult to switch chemistry versions for an ongoing large sequencing project.
Methods
Ethics oversight.
The NABEC study has been originally approved by the Joint Addiction, Aging, and Mental Health Data Access Committee and more information can be found at the dbGaP website under the study accession id: phs001300.v4.p1. The National Institutes of Health (NIH) considers the research using post-mortem material as non-human subject research and therefore no additional Institutional Review Board (IRB) approval was required.
Sample collection.
For the NABEC brain samples, frozen tissue was sampled from the frontal cortex for 14 neurologically normal individuals. All samples were obtained from the Banner Sun Health Research Institute (https://www.bannerhealth.com/services/research/ locations/sun-health-institute/programs/body-donation/tissue). All individuals were of European ancestry and had no clinical history of neurological or cerebrovascular disease, or a diagnosis of cognitive impairment during life. Demographics, tissue source and cause of death for each subject are shown in Supplementary Table 14. Average age at time of death was 85.2 years of age (range, 68 – 95 years) and 8 were male and 6 were female.
For the cell lines, high molecular weight (HMW) DNA was extracted from the following cultured cell lines purchased from Coriell (https://www.coriell.org/): HG002 (Ashkenazi Jewish ancestry, cat. no. GM24385), HG02723 (African ancestry, cat. no. HG02723) and HG00733 (American ancestry, cat. no. HG00733). For these three cell-lines, cell culture was performed using Epstein–Barr virus (EBV)-transformed B lymphocyte culture from the cell-lines in RPMI-1640 media with 2 mM L-glutamine and 15 % fetal bovine serum at 37 °C.
DNA processing.
The frontal cortex 32 and cell-lines 33 protocols are explained in detail and are both publicly available on protocols.io. In brief, for the frontal cortex samples ~40 mg of frozen tissue was homogenized with a Tissueruptor instrument (Qiagen). HMW DNA was then extracted using a Kingfisher Apex instrument (Thermofisher) with a custom script and Nanobind Tissue Big DNA kit, which uses 3 mm Nanobind disks (Circulomics/ PacBio, US). The HMW DNA was sheared to a target size of 30kb with the DNA Fluid + needles at speed 45 for two cycles on a Megaruptor3 instrument (Diagenode).
The cell-line HMW DNA was extracted manually from 4×106 of cells using a Nanobind Tissue Big DNA kit (Circulomics/PacBio, US). An enrichment of short DNA fragments was observed for the Coriell cell-lines post DNA extraction. Therefore, to yield data comparable to the brain sequencing, using the Short Read Eliminator kit (SS-100–101-01) from Circulomics/Pacbio, a size selection step was included to deplete DNA fragments up to 25kb. Finally, the HMW DNA was then sheared to a target size of 30kb with the DNA Fluid + needles at speed 45 for two cycles (Diagenode). For all samples, DNA length was assessed by running 1ul on a genomic screentape on the TapeStation 4200 (Agilent). DNA concentration was assessed using the dsDNA BR assay on a Qubit fluorometer (Thermo Fisher).
Library preparation.
Libraries were constructed using an SQK-LSK 110 kit (ONT). To ensure minimal DNA loss during library preparation, and to retain long DNA fragments, the following modifications were made to the standard SQK-LSK 110 (ONT) protocol; 1) 4.5 ug of DNA was used as starting input. This is higher than the recommended amount due to the fact that around 60–75% of the starting material is lost during library preparation. Therefore, starting with 4.5ug DNA input ensured we could do three loads of 400 ng with the final library, 2) to reach the 4.5ug DNA input the starting DNA volume was usually higher than the recommended 47ul. In this case, the volume of AMPure XP beads was modified to match the input DNA volume, i.e. if 58ul of DNA was added then 70ul was added of AMPure XP beads, 3) during DNA repair and end-prep, 75% ethanol was used for washing rather than 70% (per ONT, anything between 70–80% is acceptable), 4) at step 16 of DNA repair and end-prep, the original elution time was 2 minutes at room temperature. This was modified to 3 minutes at 37°C with light shaking on a Thermomixer instrument (Eppendorf) at 450 rpm, 5) during Adapter ligation and clean-up, 45uL of AMPure XP beads were used 6) SFB was used, and finally 7) in step 16 of the Adapter ligation and clean-up, the final elution conditions were changed to 20 minutes at 37°C.
For comparison, the cell lines (HG002, HG02723 and HG00733) were resequenced with R10. For this libraries were constructed using an SQK-LSK 114 kit (ONT), however the following modifications were made to the standard protocol; 1) 2.5ug of DNA was used as starting input and 2) in the Adapter ligation and clean-up step, the final elution conditions were changed to 20 minutes at 37°C.
PromethION sequencing.
For R.9, PromethION sequencing was performed as per manufacturer’s guidelines (ONT, FLO-PRO002) with minor adjustments, such as 400 ng of the library was loaded onto each primed R9.4.1 flow cell to maximize data output. For R.10, PromethION sequencing was performed loading 180 ng of the library onto a primed R.10.4.1 flow cell. Over the three-day run, most samples only required one additional load (usually around 48 hours). However, some runs hit a pore occupancy of ~2000 earlier, which was usually due to the variability across PromethION flow cells. In these cases, two reloads were required, which usually meant one reload every 24 hours.
Small variant benchmarking.
We used the Genome in a Bottle (GIAB) small variant benchmark to evaluate the performance on the HG002 genome, using benchmark v4.2.1 inside the confident intervals defined in “HG002_GRCh38_1_22_v4.2.1_benchmark_noinconsistent.bed”. SNP recall and precision of ONT and Illumina variant calls were computed using hap.py v0.3.14 (https://github.com/Illumina/hap.py). Since the GIAB benchmark is only available for HG002, we also used variant calls produced with DeepVariant using HiFi reads as ground truth for HG002, HG00733, and HG02723. On HG002, both GIAB and DeepVariant call sets resulted in very similar statistics (Supplementary Table 3). We used the confident regions defined for HG002 for the other cell lines, which explains the slight decreases in recall and precision for both ONT and Illumina for HG00733 and HG02723.
To benchmark phase switch and hamming error rates, we used small variant calls produced from HPRC assemblies using dipcall 43 v0.3 as ground truth. Error rates were then computed using “whatshap compare” v1.5 15 module.
Illumina data was obtained from NYGC 1000 genomes sequencing project, and was sequenced with Novaseq 6000 with a median insert size per sample of 433 bp, and 30x coverage, using the TruSeq DNA PCR-Free High Throughput Library Prep Kit 44.
Hapdup method and structural variant calling.
We developed a tool called Hapdup to generate diploid assemblies using only ONT reads. Hapdup takes as input (i) a set of haploid contigs produced by any long-read assembler, such as Shasta or Flye and (ii) alignment of the original long-reads against the assembly produced by minimap2 45. Such assembly only contains ~50% of the heterozygous variants, and our goal is to convert it into a locally phased diploid assembly that contains the complete set of structural variants in the genome.
Because de novo assemblies may leave some repetitive parts of the genome unassembled, reads originating from these unassembled repeat copies may misalign to their paralogs (if they happen to be assembled). Because the copies of long repeats often are not exact, misaligned reads may create artificial “haplotypes”, in addition to the (correctly mapped) paternal and maternal alleles. It is important to filter out misaligned reads to ensure that the subsequent diploid phasing is correct. Hapdup filters out reads with either (i) large unaligned parts or (ii) high alignment error. Afterwards, PEPPER is used to call SNPs, SNPs are phased using Margin, and reads are haplotagged according to their phases.
Next, Hapdup runs two instances of the Flye polisher, using only the aligned reads from either first or second phase. The polisher algorithm separates the input alignment into small chunks, and each chunk is polished reference-free using the maximum likelihood approach 46. To recover a large heterozygous variant, it is important that reads containing the variant are consistently aligned, which may be difficult in VNTR regions. To ensure the correct splitting of the original alignment, we added new functionality to the polishing algorithm that identifies regions with indels with inconsistent coordinates between reads, and ensures that the problematic regions are contained inside a single mini-alignment.
The polishing procedure can recover indels and substitution variants, but was not designed to detect structural variants that are created by genomic rearrangements (for example, inversions). Instead, Hapdup detects genomic rearrangement signatures from the read alignments, infers their phases, and applies the rearrangement to the corresponding set of contigs.
Hapdup outputs phased haplotypes in two different formats. First, it generates a “dual” assembly, that has the same contiguity as the original haploid set of contigs, but may contain occasional phase switches if phase blocks are shorter than contigs. Alternatively, Hapdup can split the contigs at the regions that lack proper phasing (indicated by Margin), so that every contig represents a contiguous paternal or maternal haplotype.
We used Shasta v0.10.0 with config “Nanopore-CARD-Jan2022.conf” and Hapdup v0.11 assemblies. Assemblies were also evaluated using QUAST v5.2.0 47, yak (https://github.com/lh3/yak), asmgene (a part of paftools v2.24-r1152-dirty; 48,49. We called SNPs from assemblies using dipcall v0.3 and confirmed a low switch error rate of 0.07–0.18% using the “whatshap compare” v1.5 15 module. Pangenomes were constructed using minigraph 50 with default parameters and visualized using Bandage 51.
To evaluate Hapdup as a standalone tool rather than a part of Napu, we performed additional benchmarks against the alternative assembly approaches, using the R9 data from 3 cell lines. Since Hapdup is currently the only method for diploid de novo assembly using only ONT data, we compared it to the ONT + trio method combined with Shasta or Flye (the current state-of-the-art for haploid ONT assembly). Hapup produced assemblies with the best NG50 and QV, likely because using parental data to separate the reads effectively halves the read depth for a haploid assembler (Supplementary Table 15). SV F1-scores were similar for Hapdup and trio-Flye assemblies (0.9687 and 0.9755 for HG002, respectively). Hapdup was faster than trio-Flye and slower than trio-Shasta (but produced substantially better assemblies).
Hapdiff, structural variant calling and benchmarking.
To produce structural variant calls from diploid assemblies, we developed a tool called hapdiff, which is based on a modified version of SVIM-asm 52. The package incorporates minimap2 with predefined alignment parameters, which were optimized for alignment of regions containing long structural variants (“-ax asm20 -B 2 -E 3,1 -O 6,100”). Having a fixed set of alignment parameters is also critical for reproducibility. Second, we added the functionality to group the variants inside the same VNTR together, if the annotation is provided. As illustrated in Extended Data Figure 10, VNTR grouping substantially improves the agreement between Hapdup and HPRC assemblies. This highlights the challenge of ensuring alignment consistency inside VNTR regions.
Sniffles v2.0.7 was run using default parameters, with a VNTR annotation file provided. CuteSV 2.0.2 was run with parameters recommended for ONT datasets by the developers. To benchmark HPRC SV calls against the GIAB callset, we changed genotypes of the HPRC chrX and chrY calls from heterozygous to homozygous to be consistent with the GIAB genotypes. We used hapdiff v.0.7. Multiple sets of structural variants were compared using Truvari v3.3.0 with added “-r 2000” parameter that controls the maximum linear distance between two variants. We also tested Truvari with default parameters and found that it has only a small effect on the resulting F1 scores and affected different tools similarly (Supplementary Table 16). In addition, we tested the effect of “--multimatch” option that allows to match multiple SVs in one set to one SV in another set. On the GIAB benchmark, it resulted in very minor increase in F1-scores for all tools (Supplementary Table 16). The difference was more noticeable in the comparison against HPRC SV calls, with Hapdup F1-scores increasing by approximately 2%, and Sniffles2 by approximately 6%. This highlights the issue of inconsistent SV representation.
To illustrate the improvement, we compared hapdiff to dipcall and SVIM-asm methods using the GIAB HG002 benchmark (Supplementary Table 17). Hapdif had better F1-score (0.9668), compared to dipcall (0.9174) and SVIM-asm (0.9427). All three tools had comparable running time. The hapdiff improvements are likely explained by VNTR grouping functionality and more conservative approach to alignment filtering in dipcall.
Extended Data Figure 10 Illustrates that using VNTR SV grouping improves the concordance between SV sets produced by Hapdup and HPRC assemblies. Sniffles2 had similar F1-score against the HPRC-based SV calls inside GIAB Tier1 regions, but the concordance was substantially reduced for the extended genomic intervals relative to the Shasta+Hapdup calls (Supplementary Table 7). This may partly be explained by the ambiguities in SV representation rather than truly erroneous calls, in particular representation of indels inside VNTRs. These ambiguities may also result in genotyping errors.
Benchmarking variants in challenging medically-relevant loci.
We further assessed the ability of our assemblies to represent and cover medically challenging genes 12,13. Here we assessed 389 genes that we previously postulated as highly complex and repetitive (e.g., LPA, SMN1, SMN2). We first measured the number of contigs observed per region to identify the coverage, and we observed that 359 and 356 genes are covered by at least one contig from the first haplotype and second haplotype, respectively (see Supplementary Table 8). Given that we cover these medically challenging genes, we next assessed the variant calling ability in these genes. For this, we used the recent benchmark from GIAB (CMRG v1.0) for SNPs and SVs. We were particularly interested in the ability of our method to recover indels and SVs. For substitutions, our reference-based pipeline obtained a high accuracy (F1-score: 0.97). For smaller insertions and deletions (<50 bp), we obtained an accuracy of 0.68 in these repetitive challenging genes. Next, we assessed the SV performance. Given the limited set of genes in this benchmark, we only could assess the performance based on 252 SVs. Our assemblies achieved a high accuracy (F1-score: 0.91); notably, SV calling using hapdiff achieved higher F1-score compared to SV calls made using dipcall (F1-score: 0.88). Sniffles2 had a similar F1-score on this benchmark (0.91; Supplementary Table 9).
Harmonizing and phasing of small and structural variants.
Margin was originally described in (Shafin et al., 2021). Here we describe the modifications to Margin that were made to support joint phasing of small and structural variants. The underlying phasing algorithm remained the same: allele support from reads is modeled by (i) selecting a configurable number of basepairs up- and downstream from the variant site from the reference, modifying this sequence to reflect each proposed allele, (ii) selecting a read subsequence based on the pairwise alignment to the extracted reference subsequence, and (iii) aligning each read subsequence to all alleles at each locus to generate emission probabilities for the HMM.
Using our ONT sequencing data, we found that the proposed breakpoints for SVs would not always match the exact read alignment determined by the aligner. To account for this, we modified Margin to have two parameters for the distance up- and downstream from the variant for subsequence extraction: one for small variants (referenceExpansionForSmallVariants) and one for structural variants (referenceExpansionForStructuralVariants). A third parameter (indelSizeForSVHandling) was introduced to distinguish between small and structural variants with a configured value of 50bp. The small variant reference expansion value was left unchanged at 12bp. After experimentation on HG002’s chr20, we found that the reference expansion for structural variants had the greatest effect on accuracy at 64bp. Larger expansions did not yield notable phasing improvements but had increased runtime, while smaller expansions reduced phasing accuracy. We used Margin (commit bb1e16a) with the config file “allParams.phase_vcf.ont.sv.json” to generate the final phased vcf files.
We evaluated phasing accuracy of the cell line small variant calls against the calls produced from HPRC-based assemblies using the Whatshap 15. Switch error (ratio of adjacent SNPs in a wrong phase) was 0.04–0.09%, comparable to assemblies produced with a combination of HiFi and Trio / Hi-C 48,49. Hamming error (ratio of all SNPs inside a phased block in a wrong phase) was also low (2.1–2.6%), albeit slightly higher compared to Trio / Hi-C-based assemblies.
Notably, the original phasing approach that only considers small variants had substantially higher switch error rates (0.17–0.21%; Supplementary Table 10). This highlights that considering SVs improves phasing quality, consistent with other recent studies 20. The switch error around SVs (within 100 bp from boundaries) was 1–1.26%, which was an improvement over 2–2.4% rate of the SV-unaware method (Figure 4; Supplementary Table 10), but nevertheless elevated compared to the rest of the genome. More than half of SVs coincide with difficult-to-map VNTR regions, which may explain the reduced performance.
Methylation calling.
We produced phased 5-methylcytosine calls aligned to GRCh38 and T2T-CHM13 references using Remora (https://github.com/nanoporetech/remora) incorporated in Guppy 6.1.2; afterward reads were aligned and phased using PEPPER-Margin-DeepVariant 19 and annotated with modbamtools v0.4.8 53 and modbam2bed v0.6.3 (https://github.com/epi2me-labs/modbam2bed). Regional haplotype methylation in gene promoters and SVs were calculated using “modbamtools calcMeth”.
Evaluations using the T2T-CHM13 reference genome.
In our evaluations, we used both GRCh38 and T2T-CHM13 references to call small and structural variants. T2T-CHM13 contains approximately 200 Mb of sequence that is missing, misrepresented, or simulated in GRCh38, allowing the mapping and calling of more variants, particularly in sequences rich in repetitive content. Reflecting this, in three cell lines, PEPPER-Margin-DeepVariant called 1.19 – 1.29 million variants in T2T-CHM13 sequence non-syntenic to GRCh38, compared to 0.26–0.28 million variants in GRCh38 sequence non-syntenic to T2T-CHM13 (Supplementary Table 18).
To evaluate the small variant consistency, we lifted over GIAB confident regions from GRCh38 to T2T-CHM13. Our ONT-based SNP calls had high F1-score similarity (0.9951) with the calls produced using DeepVariant and HiFi data against the T2T-CHM13 reference. The F1 similarity was slightly below the concordance between ONT SNP calls and HiFi calls using the GRCh38 reference (F1-score 0.9976), which is likely explained by a few liftover artifacts (Supplementary Table 18).
Towards generating SNP calls from de novo assemblies.
We also explored if the improvements in the assembly QV allow for better SNP calling directly from the diploid contigs. The resulting SNP calls from R10 assemblies (produced by dipcall) had substantially better F1-scores (0.9853), compared to R9 assemblies (0.9735) inside the GIAB HG002 confident regions (Supplementary Table 19). Notably, most of the residual errors were overlapping with segmental duplication regions. Outside of those regions (“GRCh38_notinsegdups” stratification), the error was reduced by an order of magnitude, improving F1-score to 0.9943. In comparison, alignment-based SNP F1-score (using DeepVariant) within these regions was 0.9985. The same trend was observed in the comparison of SNP assembly-based calls against the variants produced from the HPRC assemblies (HG002, HG00733, HG02723), with F1-scores >0.99 (Supplementary Table 19).
The improved SNP concordance opens the possibility of high-quality SNP calls directly from Hapdup assemblies, and we implemented an option to perform it as a part of Napu (using dipcall). At the moment, mapping-based small variant calling remains the recommended option because of the small but noticeable advantage in accuracy. The remaining discrepancy between assembly- and mapping-based approaches is likely explained by the reduced number of segmental duplication copies in de novo assemblies, compared to the reference, resulting in more read mismappings. We expect that better segmental duplication assembly methods will substantially reduce the remaining base errors in ONT assemblies.
Extended Data
Extended Data Fig. 1. Variant calling and methylation analysis using Napu.
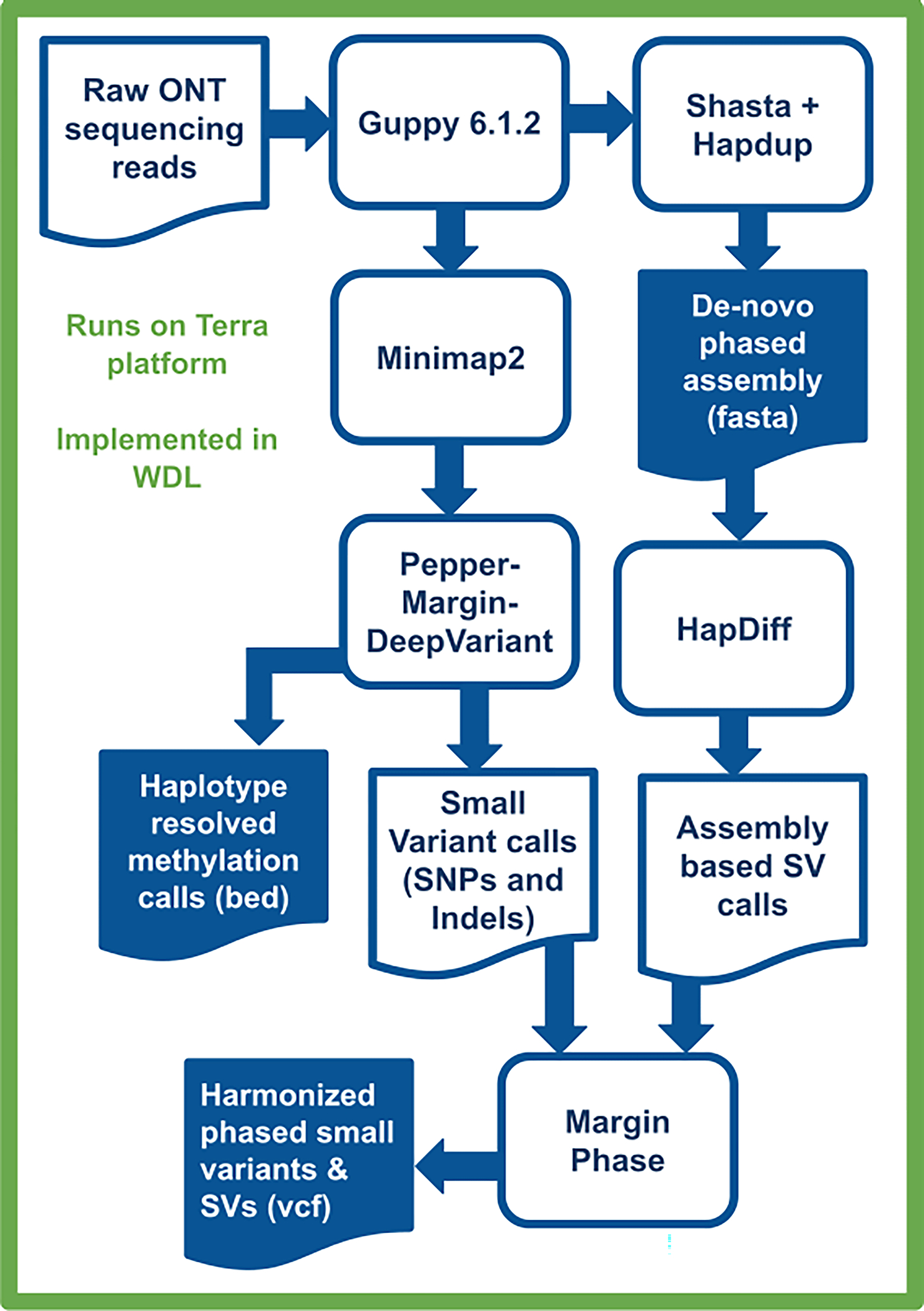
Raw ONT sequencing reads are basecalled by Guppy 6.1.2, which simultaneously produces methylation tags. A diploid, de-novo phased assembly is produced using a combination of Shasta and Hapdup. These assemblies are used to call SVs with Hapdiff. Small variants are called against a reference genome with Pepper-Margin-DeepVariant. The phased alignment file generated by Margin is used to produce haplotype-resolved methylation calls. Small variants and SVs are jointly phased by Margin, producing a single harmonized vcf.
Extended Data Fig. 2. Assemblies of 14 brain tissues and 3 cell lines generated by Shasta+Hapdup.
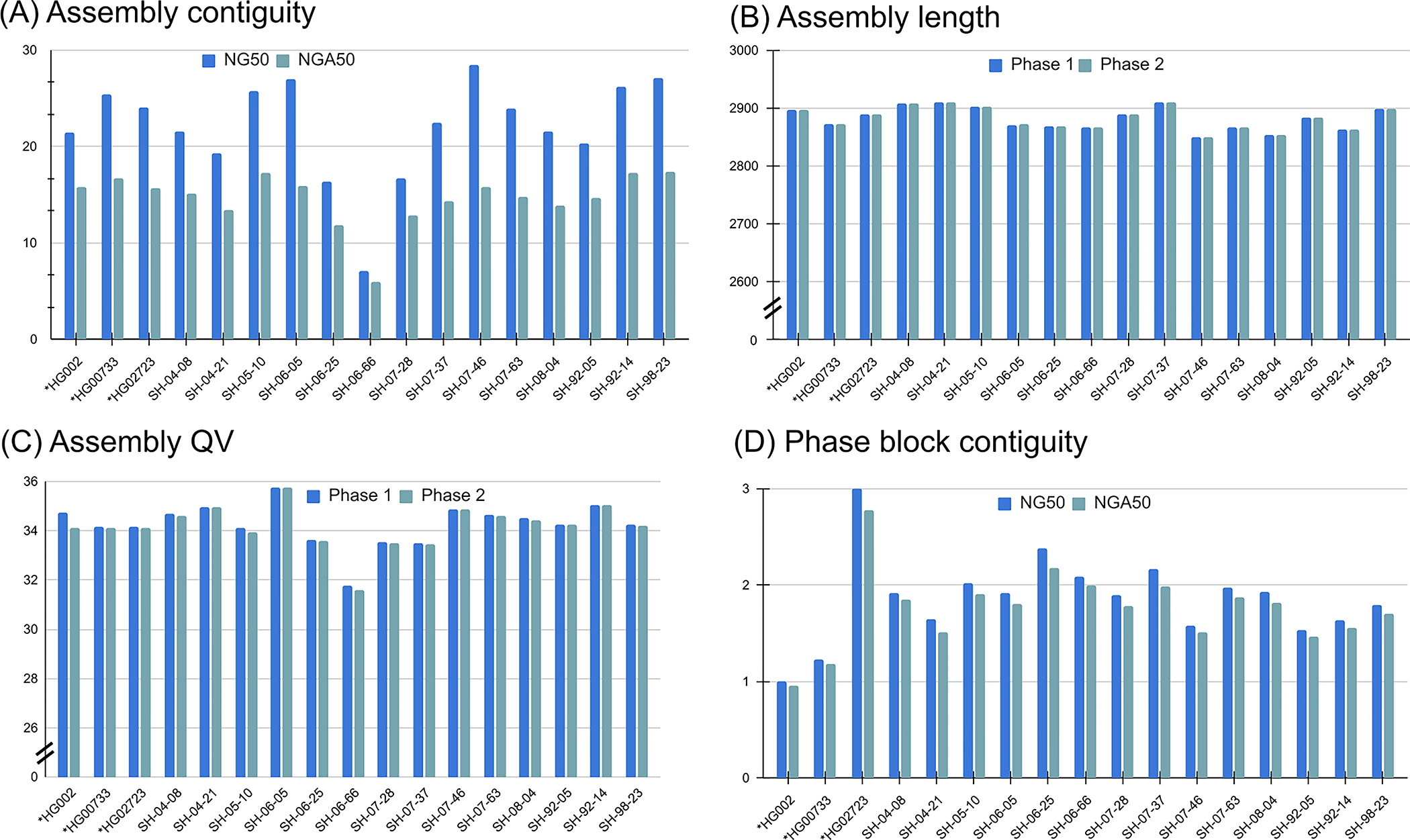
(A) NG50 and NGA50 contiguity measured using QUAST. Sample 06_66 had the lowest contiguity due to the decreased sequencing yield. (B) Assembly length. (C) Mean assemblies QV computed using yak. (D) Contiguity of phased blocks, broken at phase switches. An increased value for HG02723 suggests an increased heterozygosity rate. Cell lines marked with asterisks.
Extended Data Fig. 3. Assembly metrics comparison against HG002 assemblies produced in Jarvis et. al (2022).
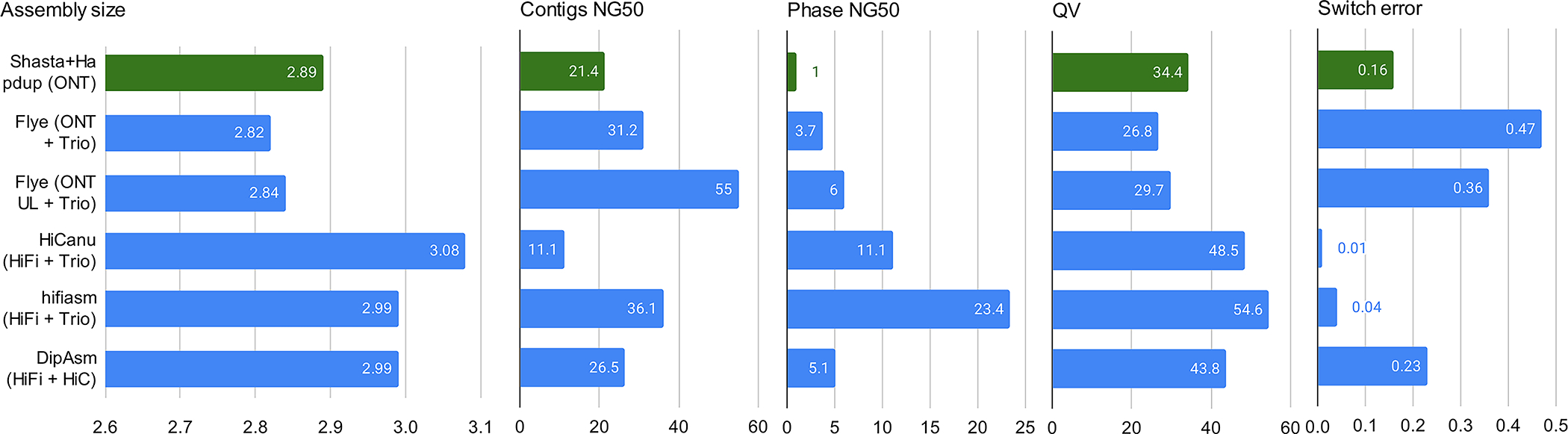
Our assemblies are highlighted in green. Flye (ONT+trio) were produced using standard ONT reads at 60x coverage and Illumina parental information; Flye (ONT UL + trio) is similar, but using ultra-long ONT extraction. HiCanu and hifiasm used 34x HiFi reads and Illumina parental sequencing. DipAsm used 34x HiFi reads and 60x Hi-C reads. Original evaluations from Jarvis et al. are shown. See Supplementary Table 5 for more detail.
Extended Data Fig. 4. TT-Mars evaluation of Hapdup and Sniffles2 calls.
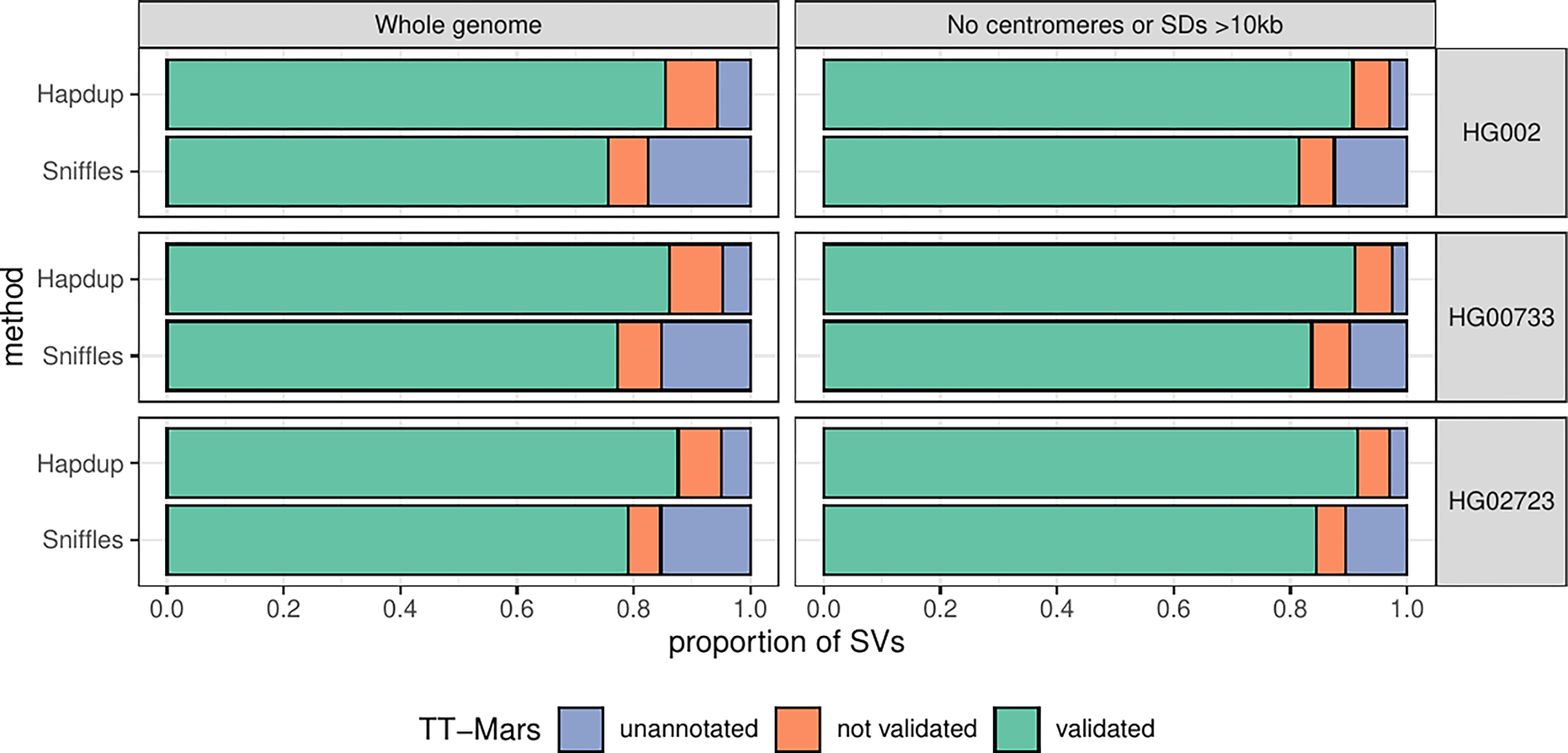
SV calls from Hapdup and Sniffles2 were compared to the assemblies from the HPRC for HG002 (top), HG00733 (middle), and HG02723 (bottom) with TT-Mars. The calls were either validated by the alignment (green), not validated (orange), or couldn’t be annotated by TT-Mars (blue). We evaluated all SVs across the genome (left), as well as the subset of SVs that don’t overlap centromeres or segmental duplications larger than 10 Kbp (right)
Extended Data Fig. 5. Flagger results based on HiFi alignments to cell line CARD and HPRC-Y1 assemblies.
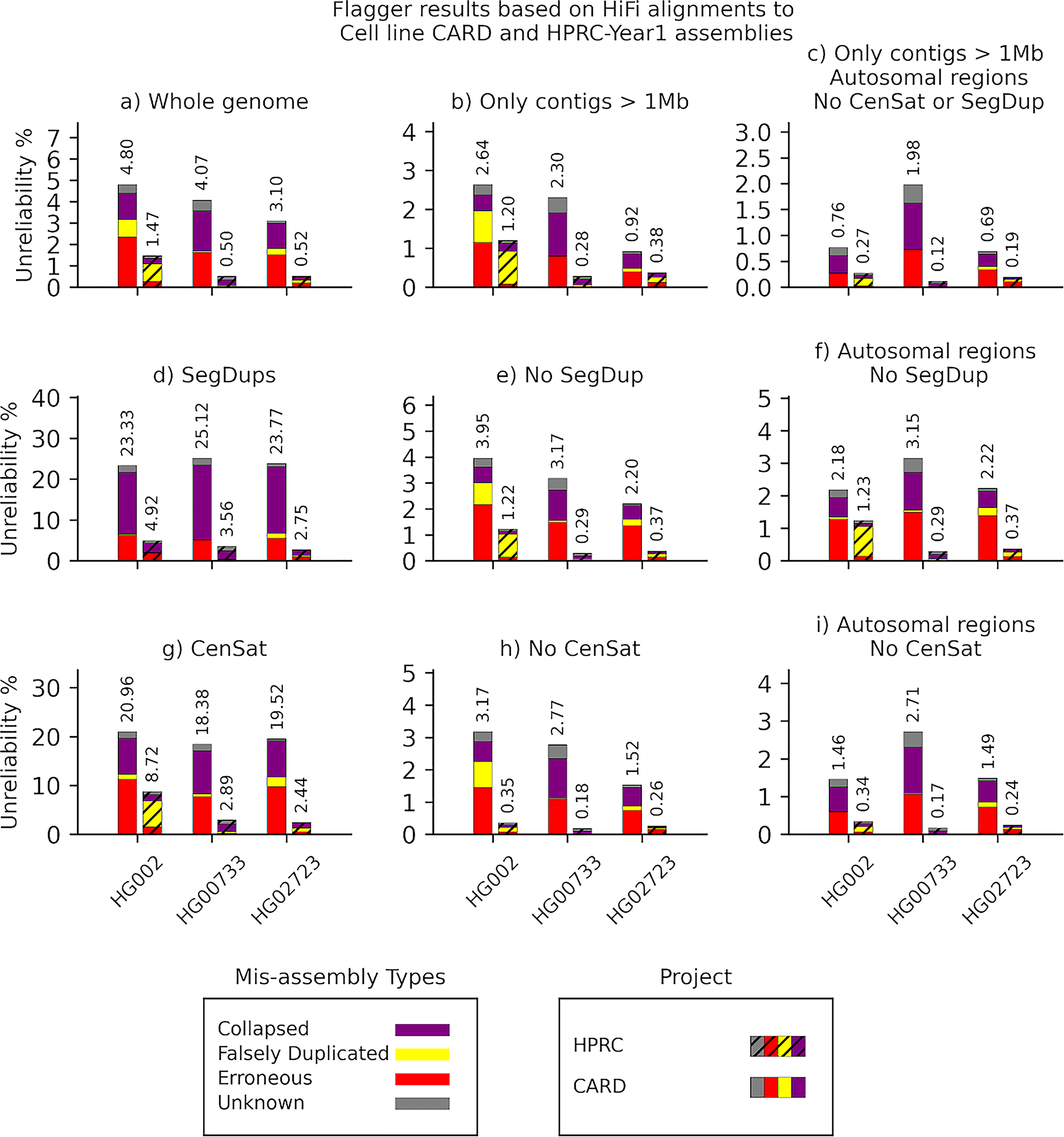
The y-axis of each panel indicates the unreliability percentages which are the total number of bases flagged as misassembly divided by the total assembly length and multiplied by one hundred.
Extended Data Fig. 6. Flagger results based on ONT alignments to cell line CARD and HPRC-Y1 assemblies.
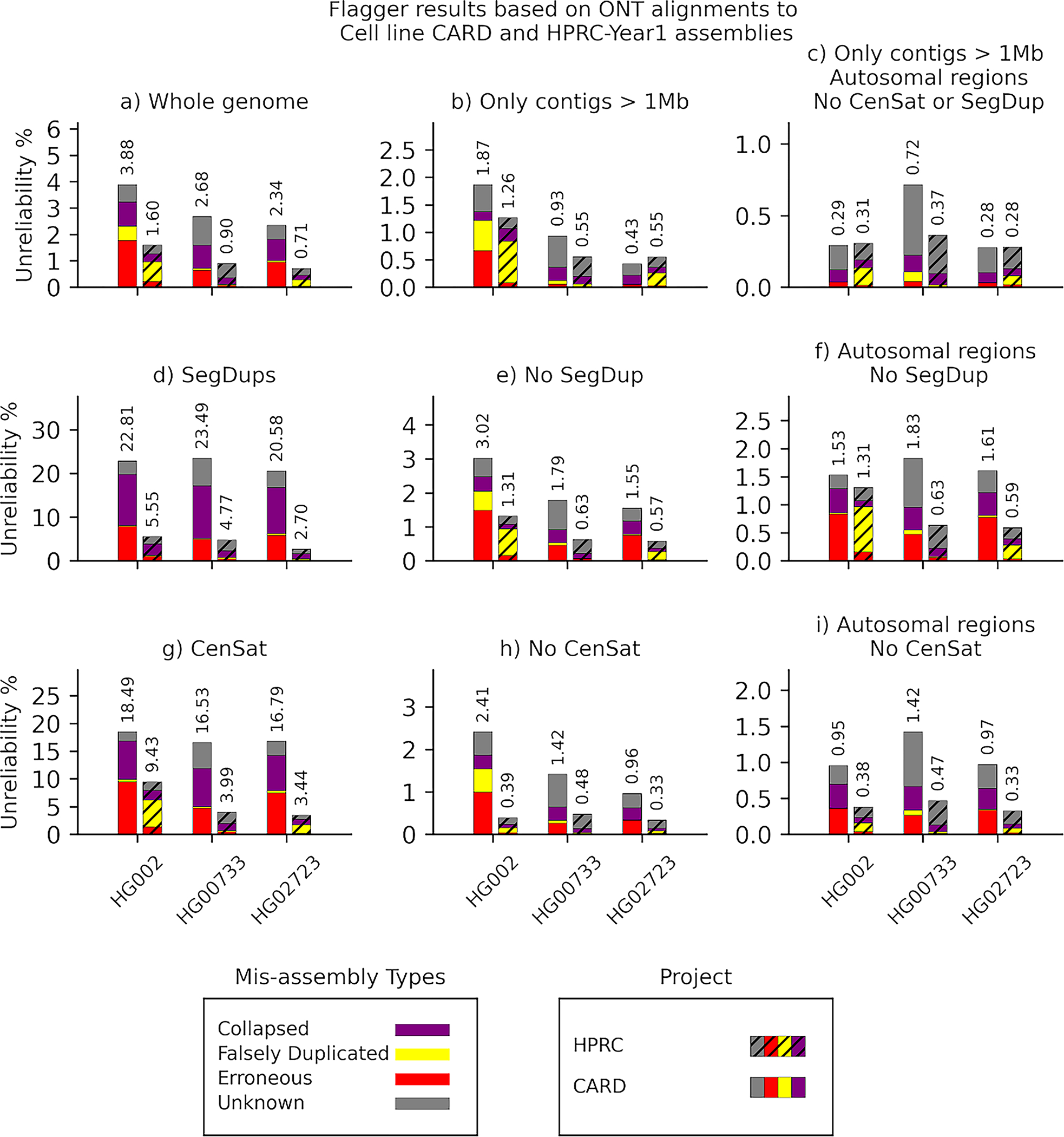
The y-axis of each panel indicates the unreliability percentages which are the total number of bases flagged as misassembly divided by the total assembly length and multiplied by one hundred.
Extended Data Fig. 7. Lenient SV catalog.
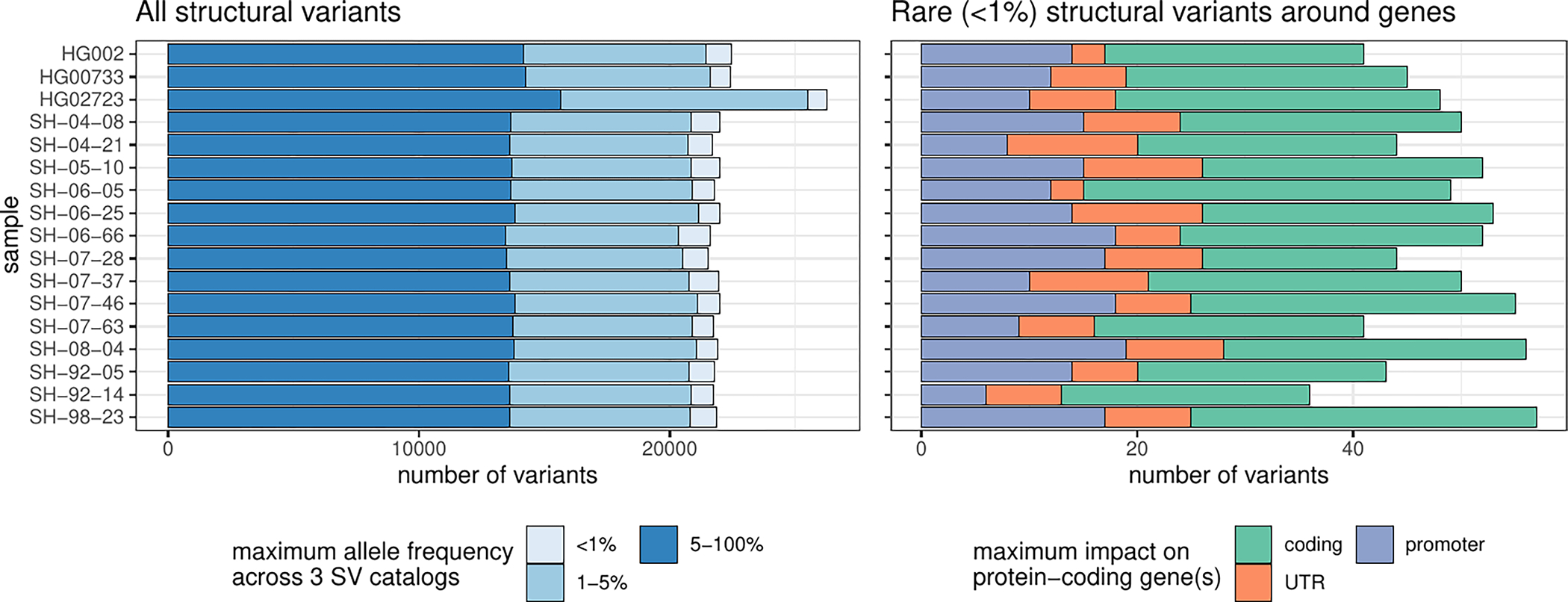
Similar to Fig. 5a but including SVs close to centromeres, telomeres, or within segmental duplications were removed. Number of SVs across samples. In the left panel, SVs were annotated with three SV catalogs (the gnomAD-SV database, a long-read-based SV catalog, and the HPRC v1.0 SV catalog). SVs are matched if they have at least 10% genomic overlap. The colors highlight the maximum frequency across these catalogs, the lighter blue showing ‘rare’ SVs (with an allele frequency below 1%) in the catalogs, or unmatched. SVs may be unmatched, either because they are novel or due to the difficulties in the database comparison. The right panel shows the number of rare SVs in protein-coding genes, grouped by their impact on the gene structure.
Extended Data Fig. 8. IGV view of a 4.2 Kbp heterozygous deletion of a transcription start site and exon of RBFOX1.
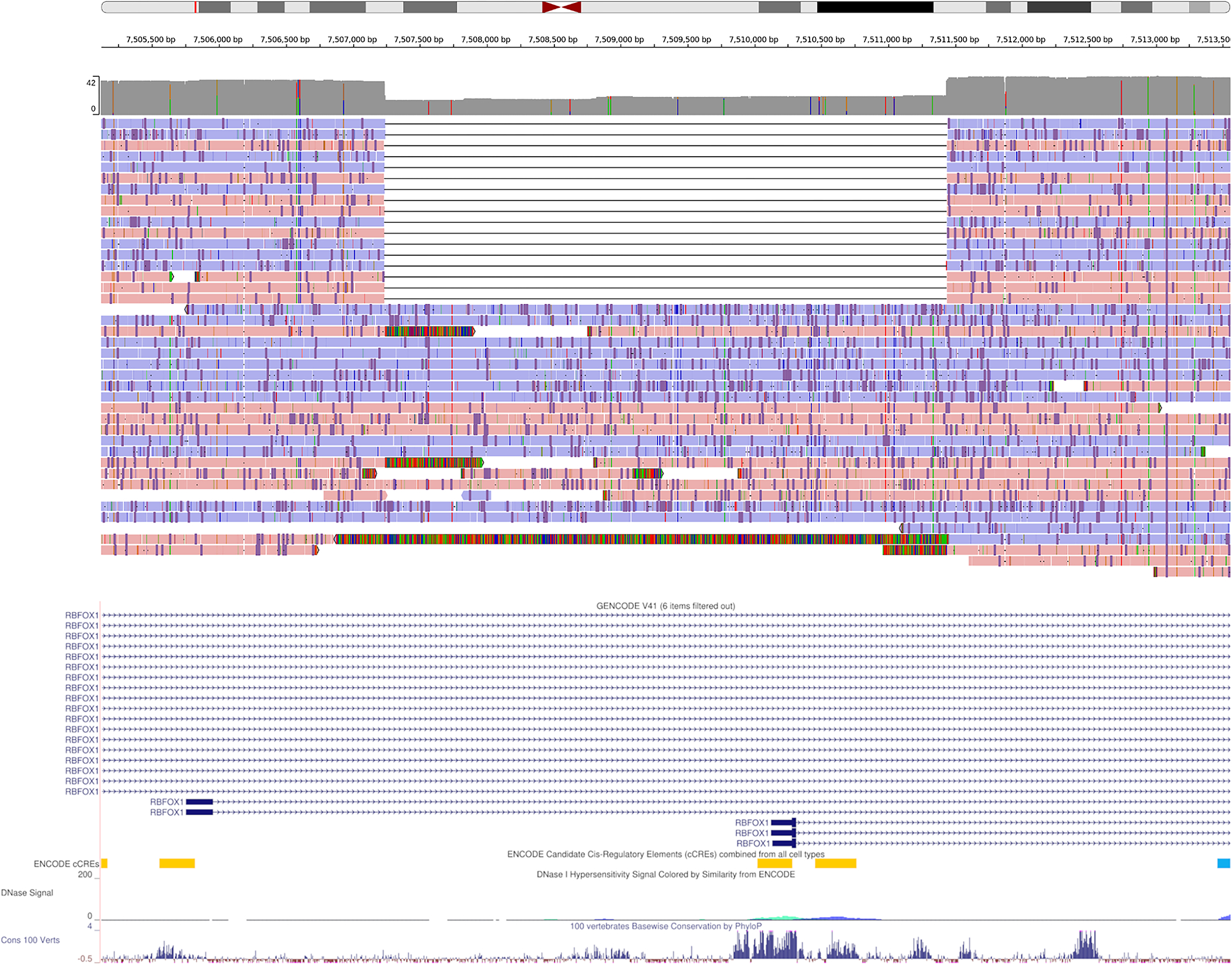
The coverage histogram (dark grey) shows the drop in read coverage. The alignment of about half of the reads, labelled by strand (red/blue), support the deletion. The GENCODE track, ENCODE candidate cis-regulatory elements, and conservation tracks are shown at the bottom.
Extended Data Fig. 9. Comparison of R9 and R10 sequencing runs using three cell lines.
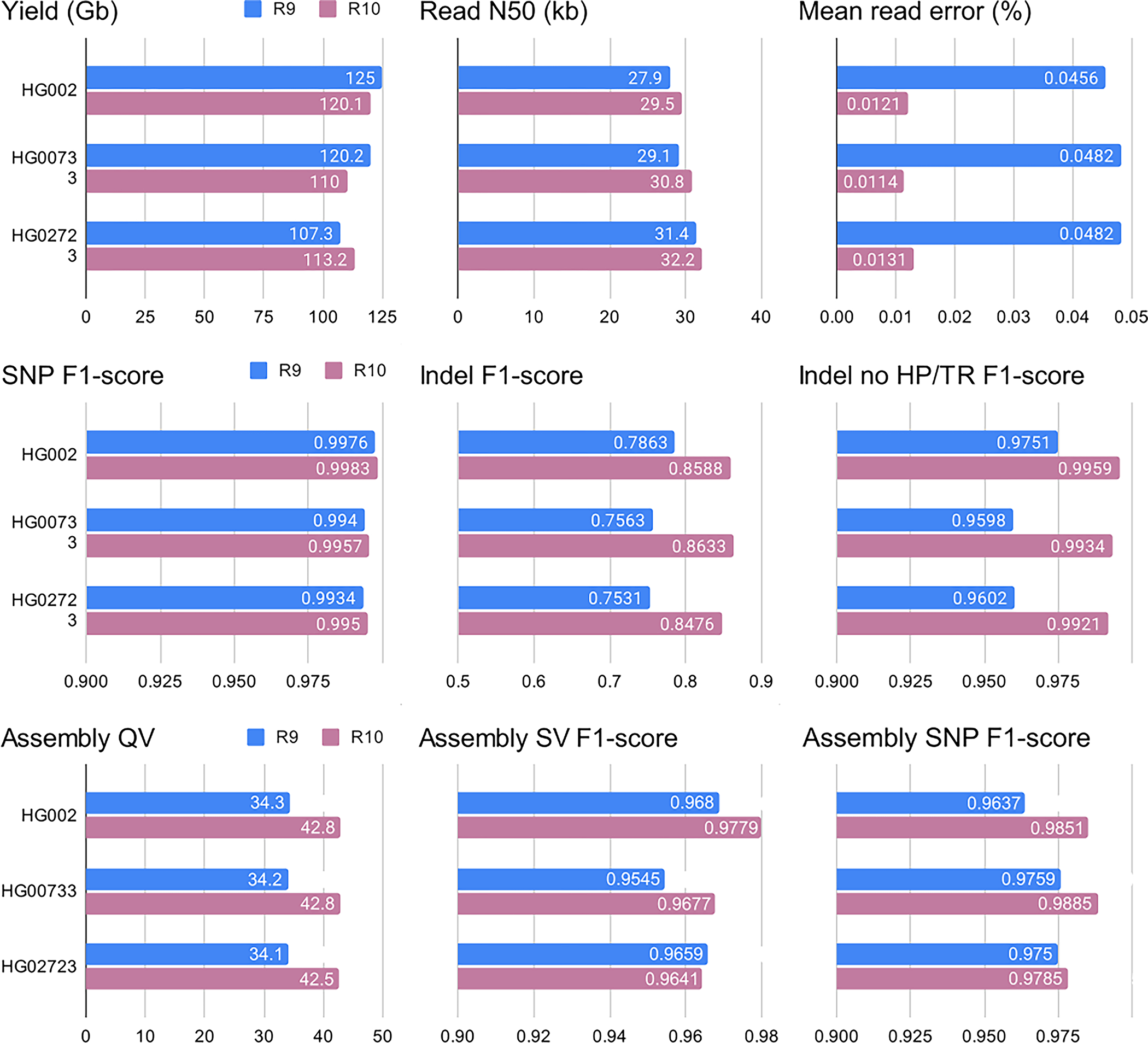
Benchmarks were performed similarly to those described in Figs. 2–4. ‘Indel no HP/TD’ corresponds to indels outside of homopolymers and tandem repeats. Assembly SV F1 scores were computed outside of centromeres and segmental duplications. Additional statistics are given in Supplementary Table 13.
Extended Data Fig. 10. F1-score for SV inside clusters of different sizes.
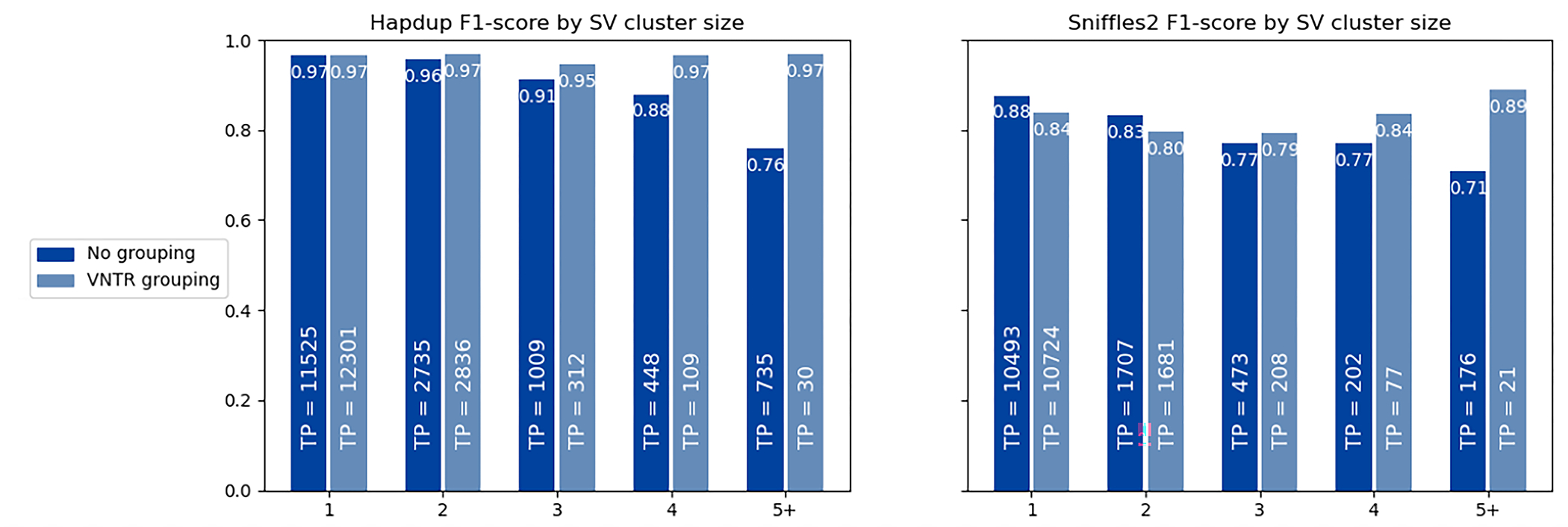
The HiFi calls for HG002 genome were used as reference, and calls within 2 kbp were clustered using single linkage clustering. The number of true positive calls in each category is shown as text. When VNTR grouping is enabled, all insertions and deletions within the same haplotype in a single VNTR are combined into a single call. A substantial portion of the reduced Sniffles2 concordance is explained by the differences in representation of SV clusters by the assembly-based and mapping-based approaches.
Supplementary Material
Acknowledgements.
This work was supported in part by the Intramural Research Program of the National Cancer Institute (M.K.), the National Human Genome Research Institute (A.M.P.), National Institute on Aging (B.J.T.), and the Center for Alzheimer’s and Related Dementias (C.B.), within the Intramural Research Program of the NIA and the National Institute of Neurological Disorders and Stroke (ZIANS003154, ZIAAG000538), National Institutes of Health (AG000538). The Brain and Body Donation Program has been supported by the National Institute of Neurological Disorders and Stroke (U24 NS072026 National Brain and Tissue Resource for Parkinson’s Disease and Related Disorders), the National Institute on Aging (P30 AG19610 and P30AG072980, Arizona Alzheimer’s Disease Center), the Arizona Department of Health Services (contract 211002, Arizona Alzheimer’s Research Center), the Arizona Biomedical Research Commission (contracts 4001, 0011, 05–901 and 1001 to the Arizona Parkinson’s Disease Consortium) and the Michael J. Fox Foundation for Parkinson’s Research. B.P. was partly supported by NIH grants: R01HG010485, U24HG010262, U24HG011853, OT3HL142481, U01HG010961, and OT2OD033761. M.M. was supported by NIH grant T32HG012344. K.D was supported by JSPS Research Fellowship for Japanese Biomedical and Behavioral Researchers at NIH. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript. We acknowledge the support of Oxford Nanopore Technologies staff in generating this data set, in particular A. Markham. We acknowledge the support of the Circulomics Inc team in generating this protocol, in particular K. Liu, J. Burke, M. Kim & D. Kilburn. We also acknowledge the Terra support team for their help with the data storage and cloud computing solutions. This work utilized the computational resources of the NIH HPC Biowulf cluster (https://hpc.nih.gov). We thank members of the North American Brain Expression Consortium (NABEC) for providing samples derived from brain tissue. We are grateful to the Banner Sun Health Research Institute Brain and Body Donation Program of Sun City, Arizona for the provision of human biological materials.
Footnotes
Competing interests. K.S. is an employee of Google LLC and owns Alphabet stock as part of the standard compensation package; authors from Google LLC did not have access to the cell line and brain tissue sample data. WT has two patents (8,748,091 and 8,394,584) licensed to Oxford Nanopore Technologies. F.J.S. received research support from Illumina, Pacific Biosciences and Oxford Nanopore Technologies. S.W.S. serves on the Scientific Advisory Council of the Lewy Body Dementia Association and the Multiple System Atrophy Coalition. S.W.S. and B.J.T. receive research support from Cerevel Therapeutics. B.J.T. holds patents on the clinical testing and therapeutic implications of the C9orf72 repeat expansion. The remaining authors declare no competing interests.
Code availability.
The Napu implementation in WDL is available at https://github.com/nanoporegenomics/card_nanopore_wf. Hapdup is available as a standalone tool at: https://github.com/KolmogorovLab/hapdup. Hapdiff is available at: https://github.com/KolmogorovLab/hapdiff.
Data availability.
The cell line data (HG002, HG0073 and HG02723) and openly available through the AnVIL workspace: https://anvil.terra.bio/#workspaces/anvil-datastorage/ANVIL_NIA_CARD_Coriell_Cell_Lines_Open. Human brain sequencing datasets are under controlled access and require a dbGap application (phs001300.v4). Afterwards, the data will be available through the restricted AnVIL workspace: https://anvil.terra.bio/#workspaces/anvil-datastorage/ANVIL_NIA_CARD_LR_WGS_NABEC_GRU. Matching Illumina data used for cell lines evaluations is available at: https://www.internationalgenome.org/data-portal/data-collection/30x-grch38. HPRC assemblies are available at: https://github.com/human-pangenomics/HPP_Year1_Data_Freeze_v1.0. GIAB benchmarks are available at: https://www.nist.gov/programs-projects/genome-bottle.
References
- 1.DePristo MA et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.1000 Genomes Project Consortium et al. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.100,000 Genomes Project Pilot Investigators et al. 100,000 Genomes Pilot on Rare-Disease Diagnosis in Health Care - Preliminary Report. N. Engl. J. Med. 385, 1868–1880 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karczewski KJ et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang K-L et al. Pathogenic Germline Variants in 10,389 Adult Cancers. Cell 173, 355–370.e14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 578, 82–93 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sedlazeck FJ, Lee H, Darby CA & Schatz MC Piercing the dark matter: bioinformatics of long-range sequencing and mapping. Nat. Rev. Genet. 19, 329–346 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Chen X et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32, 1220–1222 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Mahmoud M et al. Structural variant calling: the long and the short of it. Genome Biol. 20, 246 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zarate S et al. Parliament2: Accurate structural variant calling at scale. Gigascience 9, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zook JM et al. A robust benchmark for detection of germline large deletions and insertions. Nat. Biotechnol. 38, 1347–1355 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wagner J et al. Curated variation benchmarks for challenging medically relevant autosomal genes. Nat. Biotechnol. 40, 672–680 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wagner J et al. Benchmarking challenging small variants with linked and long reads. Cell Genom 2, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee H & Schatz MC Genomic dark matter: the reliability of short read mapping illustrated by the genome mappability score. Bioinformatics 28, 2097–2105 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin M et al. WhatsHap: fast and accurate read-based phasing. bioRxiv 085050 (2016) doi: 10.1101/085050. [DOI] [Google Scholar]
- 16.Loh P-R et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat. Genet. 48, 1443–1448 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sedlazeck FJ et al. Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 15, 461–468 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang T et al. Long-read-based human genomic structural variation detection with cuteSV. Genome Biol. 21, 189 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shafin K et al. Haplotype-aware variant calling with PEPPER-Margin-DeepVariant enables high accuracy in nanopore long-reads. Nat. Methods 18, 1322–1332 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin J-H, Chen L-C, Yu S-C & Huang Y-T LongPhase: an ultra-fast chromosome-scale phasing algorithm for small and large variants. Bioinformatics (2022) doi: 10.1093/bioinformatics/btac058. [DOI] [PubMed] [Google Scholar]
- 21.Mahmoud M, Doddapaneni H, Timp W & Sedlazeck FJ PRINCESS: comprehensive detection of haplotype resolved SNVs, SVs, and methylation. Genome Biol. 22, 268 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Logsdon GA, Vollger MR & Eichler EE Long-read human genome sequencing and its applications. Nat. Rev. Genet. 21, 597–614 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rhie A et al. Towards complete and error-free genome assemblies of all vertebrate species. Nature 592, 737–746 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nurk S et al. The complete sequence of a human genome. Science 376, 44–53 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liao W-W et al. A Draft Human Pangenome Reference. bioRxiv 2022.07.09.499321 (2022) doi: 10.1101/2022.07.09.499321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jarvis ED et al. Automated assembly of high-quality diploid human reference genomes. bioRxiv 2022.03.06.483034 (2022) doi: 10.1101/2022.03.06.483034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koren S et al. Canu: scalable and accurate long-read assembly via adaptive -mer weighting and repeat separation. Genome Res. 27, 722–736 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jain M et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 36, 338–345 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kolmogorov M, Yuan J, Lin Y & Pevzner PA Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Shafin K et al. Nanopore sequencing and the Shasta toolkit enable efficient de novo assembly of eleven human genomes. Nat. Biotechnol. 38, 1044–1053 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rautiainen M et al. Verkko: telomere-to-telomere assembly of diploid chromosomes. bioRxiv 2022.06.24.497523 (2022) doi: 10.1101/2022.06.24.497523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.J Billingsley K et al. Processing human frontal cortex brain tissue for population-scale Oxford Nanopore long-read DNA sequencing SOP v2. (2022) doi: 10.17504/protocols.io.kxygxzmmov8j/v2. [DOI] [Google Scholar]
- 33.J Billingsley K Processing frozen human blood samples for population-scale Oxford Nanopore long-read DNA sequencing SOP v1. (2022) doi: 10.17504/protocols.io.ewov1n93ygr2/v1. [DOI] [Google Scholar]
- 34.Gibbs JR et al. Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet. 6, e1000952 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schatz MC et al. Inverting the model of genomics data sharing with the NHGRI Genomic Data Science Analysis, Visualization, and Informatics Lab-space. Cell Genom 2, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li H yak: Yet another k-mer analyzer. (Github; ). [Google Scholar]
- 37.Smolka M et al. Comprehensive Structural Variant Detection: From Mosaic to Population-Level. bioRxiv 2022.04.04.487055 (2022) doi: 10.1101/2022.04.04.487055. [DOI] [Google Scholar]
- 38.English AC, Menon VK, Gibbs RA, Metcalf GA & Sedlazeck FJ Truvari: refined structural variant comparison preserves allelic diversity. Genome Biol. 23, 271 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang J & Chaisson MJP TT-Mars: structural variants assessment based on haplotype-resolved assemblies. Genome Biol. 23, 110 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vollger MR et al. Long-read sequence and assembly of segmental duplications. Nat. Methods 16, 88–94 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kirsche M et al. Jasmine: Population-scale structural variant comparison and analysis. bioRxiv 2021.05.27.445886 (2021) doi: 10.1101/2021.05.27.445886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chowdhury M, Pedersen BS, Sedlazeck FJ, Quinlan AR & Layer RM Searching thousands of genomes to classify somatic and novel structural variants using STIX. Nat. Methods 19, 445–448 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li H et al. A synthetic-diploid benchmark for accurate variant-calling evaluation. Nat. Methods 15, 595–597 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Byrska-Bishop M et al. High-coverage whole-genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. Cell 185, 3426–3440.e19 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin Y et al. Assembly of long error-prone reads using de Bruijn graphs. Proc. Natl. Acad. Sci. U. S. A. 113, E8396–E8405 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mikheenko A, Prjibelski A, Saveliev V, Antipov D & Gurevich A Versatile genome assembly evaluation with QUAST-LG. Bioinformatics vol. 34 i142–i150 Preprint at 10.1093/bioinformatics/bty266 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheng H, Concepcion GT, Feng X, Zhang H & Li H Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cheng H et al. Haplotype-resolved assembly of diploid genomes without parental data. Nat. Biotechnol. 40, 1332–1335 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li H, Feng X & Chu C The design and construction of reference pangenome graphs with minigraph. Genome Biol. 21, 265 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wick RR, Schultz MB, Zobel J & Holt KE Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Heller D & Vingron M SVIM-asm: Structural variant detection from haploid and diploid genome assemblies. Bioinformatics 36, 5519–5521 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Razaghi R et al. Modbamtools: Analysis of single-molecule epigenetic data for long-range profiling, heterogeneity, and clustering. bioRxiv 2022.07.07.499188 (2022) doi: 10.1101/2022.07.07.499188. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The cell line data (HG002, HG0073 and HG02723) and openly available through the AnVIL workspace: https://anvil.terra.bio/#workspaces/anvil-datastorage/ANVIL_NIA_CARD_Coriell_Cell_Lines_Open. Human brain sequencing datasets are under controlled access and require a dbGap application (phs001300.v4). Afterwards, the data will be available through the restricted AnVIL workspace: https://anvil.terra.bio/#workspaces/anvil-datastorage/ANVIL_NIA_CARD_LR_WGS_NABEC_GRU. Matching Illumina data used for cell lines evaluations is available at: https://www.internationalgenome.org/data-portal/data-collection/30x-grch38. HPRC assemblies are available at: https://github.com/human-pangenomics/HPP_Year1_Data_Freeze_v1.0. GIAB benchmarks are available at: https://www.nist.gov/programs-projects/genome-bottle.