

Abstract
Splicing modulation is a promising treatment strategy pursued to date only in splicing factor-mutant cancers; however, its therapeutic potential is poorly understood outside of this context. Like splicing factors, genes encoding components of the cohesin complex are frequently mutated in cancer, including myelodysplastic syndromes (MDS) and secondary acute myeloid leukemia (AML), where they are associated with poor outcomes. Here, we showed that cohesin mutations are biomarkers of sensitivity to drugs targeting the splicing factor 3B subunit 1 (SF3B1) H3B-8800 and E-7107. We identified drug-induced alterations in splicing, and corresponding reduced gene expression, of a number of DNA repair genes, including BRCA1 and BRCA2, as the mechanism underlying this sensitivity in cell line models, primary patient samples and patient-derived xenograft (PDX) models of AML. We found that DNA damage repair genes are particularly sensitive to exon skipping induced by SF3B1 modulators due to their long length and large number of exons per transcript. Furthermore, we demonstrated that treatment of cohesin-mutant cells with SF3B1 modulators not only resulted in impaired DNA damage response and accumulation of DNA damage, but it sensitized cells to subsequent killing by poly(ADP-ribose) polymerase (PARP) inhibitors and chemotherapy and led to improved overall survival of PDX models of cohesin-mutant AML in vivo. Our findings expand the potential therapeutic benefits of SF3B1 splicing modulators to include cohesin-mutant MDS and AML.
INTRODUCTION
Cohesin is a multisubunit protein complex that is essential for sister chromatid cohesion, three-dimensional chromosome organization, and gene regulation (1). It forms a ring around DNA, with three structural subunits, structural maintenance of chromosomes 1A (SMC1A), structural maintenance of chromosomes 3 (SMC3), and RAD21 cohesin complex component (RAD21) bound to either STAG1 cohesin complex component (STAG1) or STAG2 cohesin complex component (STAG2) proteins. Cohesin is one of the most frequently mutated protein complexes in cancer, and mutations in genes encoding components of the cohesin ring and its modulators are frequent and recurrent drivers in myeloid malignancies. These include myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML), where cohesin mutations are associated with poor overall survival (2–5). Loss-of-function mutations in STAG2 are the most common mutations found in cohesin subunits and result in altered cellular functions including longer loop extrusion, altered gene expression, replication fork stress, and accumulation of DNA damage (6–10). There are currently no targeted therapies available for patients with cohesin-mutant cancers. However, our recent work describing DNA damage accumulation and the genetic dependency on DNA repair proteins in cohesin-mutant MDS and AML (6) has led to the development of a pilot proof-of-concept clinical trial of the poly(ADP-ribose) polymerase (PARP) inhibitor talazoparib treatment as monotherapy and in combination with decitabine (ClinicalTrials.gov identifier NCT03974217).
Splicing factor 3B subunit 1 (SF3B1) is the most frequently mutated gene in MDS, and splicing factor mutations in general comprise the most common class of genetic alterations found in MDS and secondary AML (11–14). Widespread alternative splicing and reliance on proper splicing function is a common feature of cancer cells that has been implicated in disease progression and exploited for therapeutic targeting (15–18). Cancer cells accumulate mis-spliced RNAs through a variety of mechanisms, including the acquisition of hotspot point mutations or alterations in the expression of splicing factor proteins (19–21). Therapeutic targeting of the spliceosome is of particular interest in myeloid malignancies such as MDS, AML, and chronic myelomonocytic leukemia (CMML), where splicing factor mutations are common (22). H3B-8800 and E-7107 are first-in-class compounds that modulate splicing function through targeting of SF3B1, which is a component of the U2 small nuclear ribonucleoprotein involved in branch point recognition (23). These compounds have shown promise in preclinical studies and phase 1/2 clinical trials where treatment with H3B-8800 resulted in transfusion independence in a subset of MDS patients with splicing factor mutations (24–26). However, the mechanisms mediating response to SF3B1 modulators are not well understood, and it remains unclear whether these drugs will provide therapeutic benefit beyond splicing factor-mutant cancers.
In this work, we identified widespread mis-splicing of DNA repair genes in cells treated with SF3B1 modulators, leading to an accumulation of DNA damage. Further, given the intrinsically elevated amount of DNA damage that occurs in cohesin-mutant cancer cells, treatment of these cells with H3B-8800 or E-7107 resulted in an increased therapeutic vulnerability to chemotherapeutic agents targeting DNA repair. We used human cell–derived in vitro and in vivo models of cohesin-mutant AML and patient samples from splicing factor-mutant patients enrolled in the H3B-8800 clinical trial to demonstrate that mis-splicing of DNA damage repair genes results in reduced expression of DNA damage response proteins and accumulation of DNA damage in cells. Although the splicing changes caused by SF3B1 modulators are genotype agnostic, we found that cohesin-mutant cells are specifically targeted for killing because of their dependence on proper DNA damage repair for survival (6, 27). We leveraged this vulnerability to develop a new therapeutic strategy in which treatment of cohesin-mutant cancer cells with low-dose splicing modulation markedly increased sensitivity to compounds that induce DNA damage or inhibit proper DNA damage repair, including PARP inhibitors and chemotherapeutic agents. We propose that splicing modulation in combination with inhibition of DNA damage repair may be an effective therapeutic strategy to target cohesin-mutant MDS and AML, as well as other cancers that exhibit an accumulation of DNA damage or selective reliance on the DNA repair machinery.
RESULTS
Cohesin-mutant cells are sensitive to SF3B1-targeting compounds
To generate cell line models of cohesin-mutant AML, we used CRISPR-Cas9 editing to establish a panel of isogenic single-cell–derived clones from parental U937 and K562 cells, which are wild type for all cohesin complex subunits and modulators (6). We engineered these cells to contain loss-of-function mutations in STAG2 and SMC3 commonly found in patients (Fig. 1A). We had previously used these cell lines to perform genome-wide CRISPR screens with the goal of identifying mutant-specific genetic dependencies and potential therapeutic vulnerabilities (6). In addition to a preferential dependency of STAG2-mutant cells on STAG1 and DNA damage and replication machinery, we identified STAG2-mutant cells to be more sensitive to knockout of genes involved in mRNA processing. To further explore the interaction of these mutations in patients, we analyzed comutation patterns in patients in our clinical MDS cohort and publicly available datasets (11, 12). We identified that mutations in STAG2 tend not to co-occur with SF3B1 mutations, suggestive of synthetic lethality (Fig. 1B and fig. S1A). To further probe this possibility, we tested whether loss-of-function cohesin mutations were tolerated in the background of the most frequently mutated SF3B1 codon K700E. We performed CRISPR-mediated depletion of STAG2 or SMC3 in isogenic SF3B1-K700E and SF3B1–wild-type (SF3B1-K700K) K562 cells and found that loss of any of the cohesin subunits reduced cell viability of SF3B1-K700E mutant relative to wild-type cells (fig. S1B). These data were consistent with our observation that STAG2 and SF3B1 mutations co-occur less frequently than predicted by chance and raised the possibility that cohesin-mutant cells may be vulnerable to splicing modulation.
Fig. 1. Cohesin-mutant cells are sensitive to SF3B1-targeting compounds.
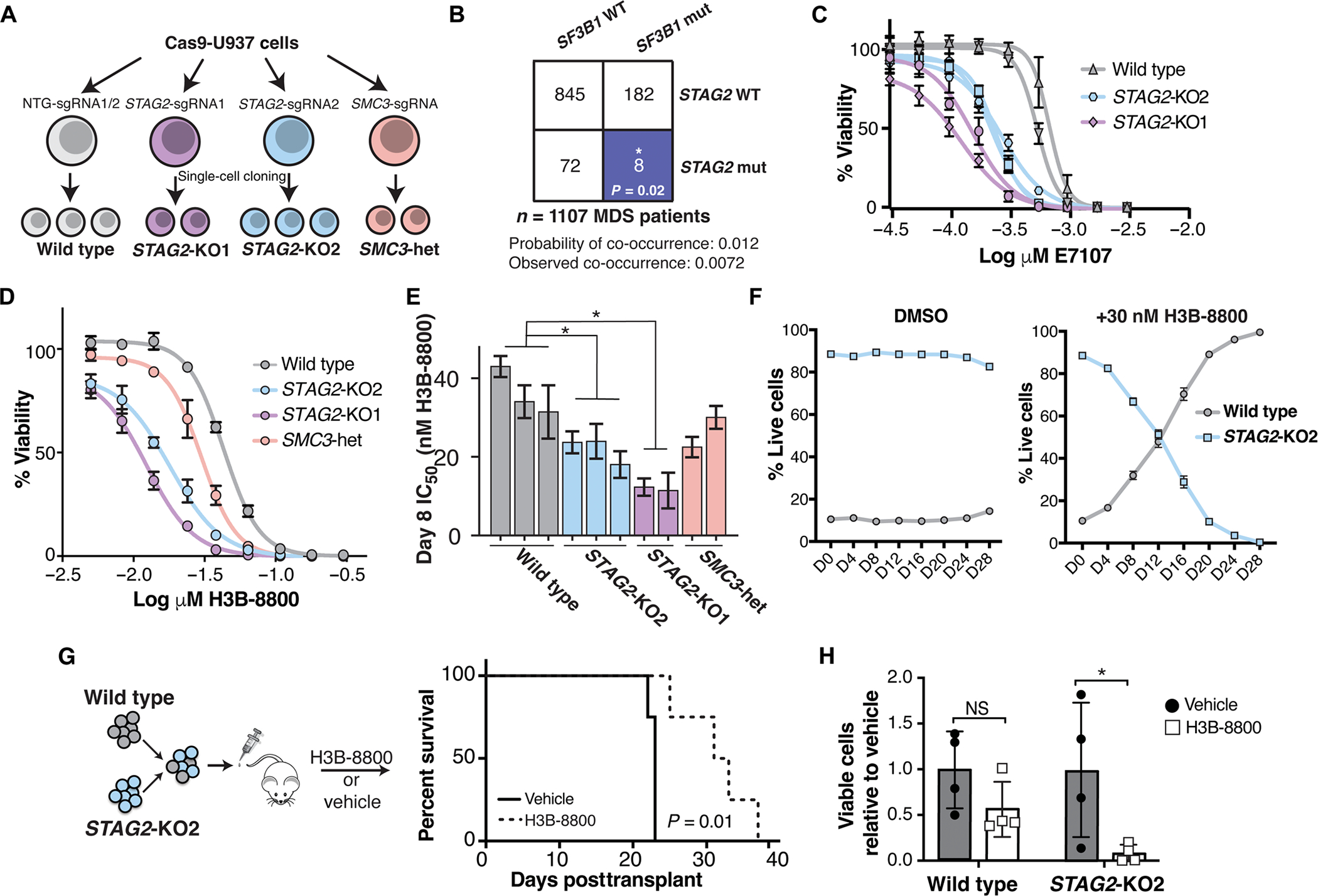
(A) Schematic of isogenic AML cell lines used in this study. U937 cells expressing Cas9 were nucleofected with single-guide RNAs (sgRNAs) targeting STAG2, SMC3, or nontargeting (NTG) sgRNAs. Two independent sgRNAs were used for STAG2 (KO1 and KO2) and NTG, and a single sgRNA was used to target SMC3. Independent single-cell–derived clones were used as biological replicates in this study. (B) Co-occurrence of SF3B1 and STAG2 mutations in a cohort of patients with MDS from the Dana-Farber Cancer Institute. Expected and observed probability of co-occurrence is listed. Blue color indicates a significant mutually exclusive relationship between SF3B1 and STAG2 mutations. *P < 0.05 (Z test). WT, wild type; mut, mutant. (C) Drug dose-response curves of E-7107–treated WT and STAG2-KO U937 clones on day 12 of treatment. Error bars represent SD of measurements of three technical replicates. (D) Drug dose-response curves of a representative set of wild-type and cohesin-mutant U937 cells treated with H3B-8800 for 8 days. Error bars represent SD of measurements in technical triplicates. (E) Quantification of IC50 among biological replicates (n = 2 or 3) of wild-type and cohesin-mutant U937 cells on day 8 of treatment with H3B-8800 tested in technical triplicates. STAG2-KO1 and STAG2-KO2 clones have a significantly lower IC50 than wild-type cells with H3B-8800 treatment (Kruskal-Wallis with post hoc test, P = 0.05). Error bars represent the 95% confidence interval of the IC50 calculated from technical triplicates of each cell line. (F) Competition assay with wild-type (mCherry) and STAG2-KO2 (GFP) U937 cells mixed in a 1:10 ratio in the presence of DMSO or H3B-8800 (30 nM) in vitro. % Live GFP+ or mcherry+ cells were determined using flow cytometry. error bars represent Sd of measurements of technical triplicates. (G) Schematic of the in vivo drug treatment of NSGS mice injected with wild-type (mcherry) and STAG2-Ko2 (GFP+) U937 cells mixed in a 1:1 ratio. Treatment with h3B-8800 or vehicle control was started 7 days posttransplant. Kaplan-Meier survival analysis was performed; P = 0.01. n = 4 mice per group. (H) leukemia burden in mice treated with h3B-8800 or vehicle was assessed in the spleens of animals at the time of sacrifice. % Live GFP+ or mcherry+ cells were determined using flow cytometry. Mean ± Sd is shown. P < 0.05 (Student’s t test). n = 4 mice per group.
To understand whether splice-modulating compounds may provide therapeutic benefit in cohesin-mutant MDS and AML, we treated cohesin-mutant U937 and K562 AML cell lines with the SF3B1-targeting compounds H3B-8800 and E-7 107 at increasing concentrations in vitro. Cohesin-mutant cells were more sensitive to both splicing modulators as compared with the isogenic, wild-type controls (Fig. 1, C to E, and fig. S1, C to E). In competition assays, STAG2-mutant cells were outcompeted by wild-type cells in the presence of low-dose H3B-8800 even when present at a 10-fold excess (Fig. 1F). We also performed in vivo competition experiments using mouse xenograft models injected with a mixture of wild-type and STAG2-mutant AML cells and observed both selective killing of the STAG2-mutant cells by H3B-8800 and a survival benefit in animals treated with the drug (Fig. 1, G and H). These results demonstrated that cohesin-mutant AML cell lines exhibit sensitivity to SF3B1-targeted splicing modulation and led us to further interrogate the mechanism driving this response.
To determine whether the sensitivity to SF3B1-modulating compounds in cohesin-mutant cells is driven by common splicing alterations in cohesin-mutant and SF3B1 K700E-mutant cells at baseline, we compared the splicing events altered upon mutation in each of these models. First, we compared RNA sequencing (RNA-seq) from U937 cell clones of each cohesin-mutant genotype with wild-type control cells (Fig. 1A). We quantified a total of 3740 splicing events that were significantly altered in any of the three cohesin-mutant lines [false discovery rate (FDR) < 0.05, delta percent spliced in (ΔPSI) > 5%]. K-means clustering of these events revealed highly similar patterns of splicing among the cohesin-mutant cells, with many exon skipping and exon inclusion events in common (fig. S1F). Next, we analyzed publicly available RNA-seq data from K562 cells with SF3B1 K700E mutations (26) or with STAG2-KO (28) and identified 2874 splicing events that were significantly altered in either genotype as compared with wild-type K562 cells (FDR < 0.05, ΔPSI > 5%). K-means clustering performed on these events demonstrated little overlap between the splicing changes observed in the SF3B1 K700E-mutant versus STAG2-KO cells (fig. S1G). These results suggested that the reliance of cohesin-mutant cells on proper splicing does not reflect a common set of disrupted splicing events in cohesin-mutant and SF3B1 K700E-mutant cells. Instead, we speculated that disruption of splicing with SF3B1 inhibitors targets cancer cells through a shared therapeutic vulnerability that exists downstream of splicing regulation in both cohesin-mutant and SF3B1 K700E-mutant cells.
H3B-8800 treatment induces mis-splicing and down-regulation of DNA damage repair genes
To understand the mechanism by which splicing modulating drugs target cohesin-mutant cells, we treated our panel of 10 isogenic U937 cell lines with increasing concentrations of the SF3B1 modulator H3B-8800 and quantified splicing and gene expression changes relative to dimethyl sulfoxide (DMSO) treatment using RNA-seq. We hypothesized that treatment with H3B-8800 may target cohesin-mutant cells by either (i) selectively modulating splicing of essential genes in cohesin-mutant but not wild-type cells or (ii) leading to similar splicing changes in all genotypes but targeting genes that are selective dependencies for the survival of cohesin-mutant cells. We identified more than 13,000 splicing changes in H3B-8800–treated cells relative to DMSO, the majority of which were exon skipping events that were alternatively spliced in a dose-dependent manner (Fig. 2A and fig. S2, A and B). We used k-means clustering to group splicing events on the basis of the percent change in splicing and observed that H3B-8800–induced splicing changes were largely conserved in all cells, including both wild type and cohesin-mutant (Fig. 2B). In general, splicing changes induced by H3B-8800 occur in exons that are normally fully included (PSI = 1) and are spliced out uniquely in the presence of the drug (Fig. 2C, top). Conversely, we found that introns retained upon H3B-8800 treatment were usually fully spliced out (PSI = 0) (Fig. 2C, bottom). Thus, rather than reflecting alternative splicing, the changes observed in H3B-8800–treated cells may represent mis-splicing and the formation of aberrant mRNA products. In agreement with this model, the median gene expression of H3B-8800 target genes decreased in a dose-dependent manner, suggesting that drug-induced alterations in splicing result in impaired gene expression (fig. S2C). These results suggested that H3B-8800 causes similar splicing changes in all genotypes and that the preferential killing of cohesin-mutant cells by H3B-8800 is driven by mis-splicing of genes that are selectively essential for survival in cohesin-mutant but not wild-type cells.
Fig. 2. H3B-8800 treatment induces mis-splicing and down-regulation of DNA damage repair genes.
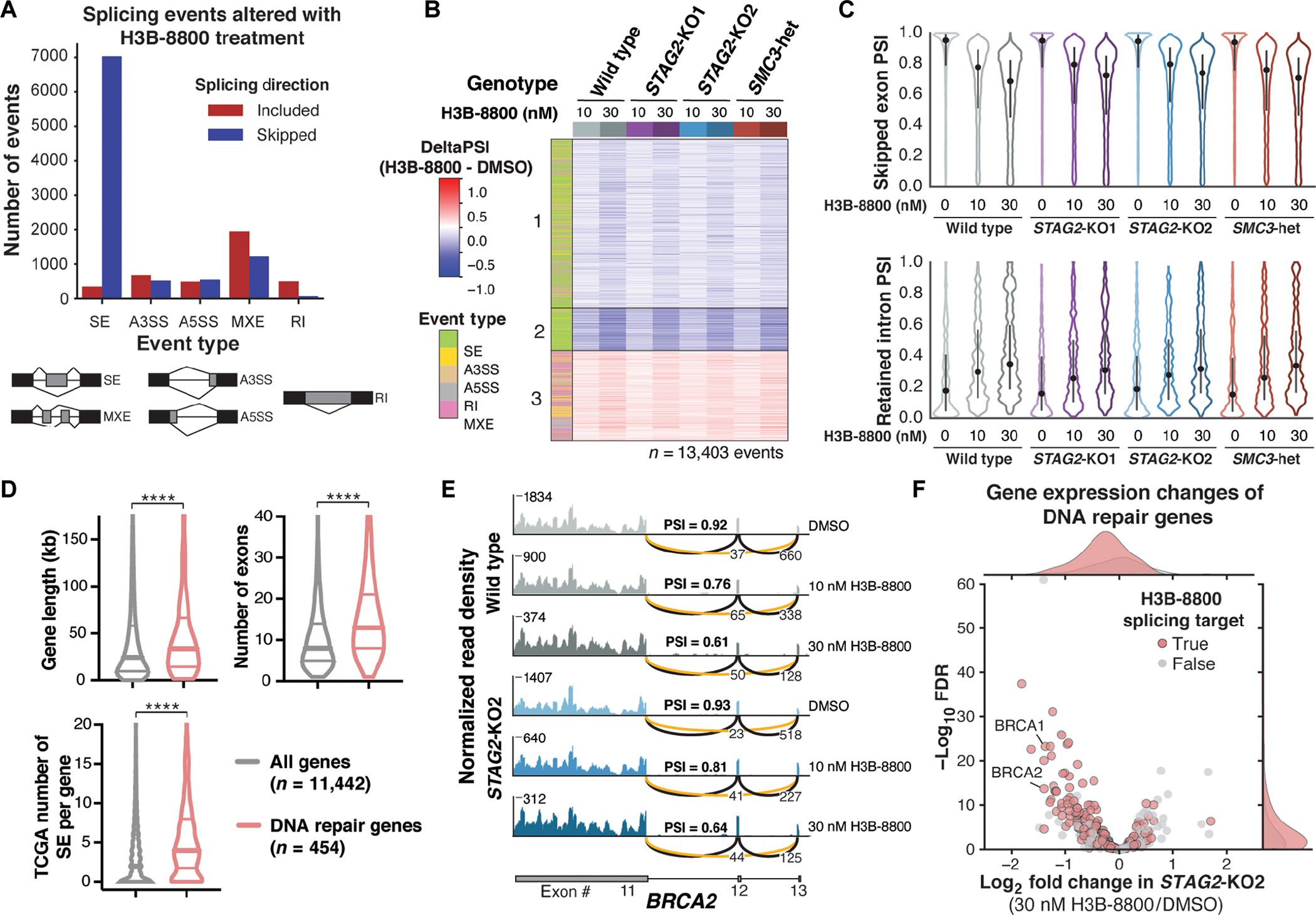
(A) Total number and directionality of splicing alterations induced by 6-hour H3B-8800 treatment in all U937 cell lines. Events are categorized by event type and direction of regulation in H3B-8800–treated relative to DMSO-treated cells. SE, skipped exon; A3SS, alternative 3′ splice site; A5SS, alternative 5′ splice site; MXE, mutually exclusive exon; RI, retained intron. (B) Heatmap of ΔPSI scores for H3B-8800–regulated exons [from (A)] across all conditions separated into three k-means clusters. Each comparison consists of two (STAG2-KO1 and SMC3-heterozygous) or three (STAG2-KO2 and wild type) independent single-cell clones for each concentration of drug compared with DMSO controls within the same genotype. Columns are organized by genotype and concentration of H3B-8800. Color bar on the left indicates the type of splicing event that was called. PSI, percent spliced in. (C) Violin plots showing the distribution of PSI scores for H3B-8800–regulated skipped exons (top) and retained introns (bottom) under each treatment condition tested. Dot represents the median, and bars extend from the first to third quartile range. (D) Violin plots depicting the gene length, number of exons, and number of skipped exons in TCGA comparing DNA repair genes (n = 454) with all expressed protein-coding genes (n = 11,442). Pan-cancer TCGA exon skipping events collated from ExonSkipDB (29). Horizontal lines in violin plots depict the median and first and third quartiles. ****P < 0.0001, Mann-Whitney test. (E) RNA-seq–normalized read density and splice junction track of exon skipping in BRCA2 exon12 from one representative replicate of wild-type and STAG2-KO2 cells treated with DMSO and 10 or 30 nM H3B-8800 for 6 hours. Average PSI scores from three biological replicate samples of exon12 are shown. Average number of reads supporting exon skipping (orange line) and exon inclusion (black line) are reported. (F) Volcano plot of gene expression changes in DNA repair genes in STAG2-KO2 cells treated with 30 nM H3B-8800 relative to DMSO-treated controls. Average log2 fold change of three biological replicates of STAG2-KO2 versus wild-type U937 cells is shown. DNA repair genes that contain H3B-8800–regulated splicing changes are highlighted in red.
Having established that H3B-8800–induced splicing changes are shared by cohesin wild-type and mutant cells, we next investigated candidate genes whose mis-splicing could lead to preferential killing of cohesin-mutant cells. We started by quantifying enrichment of gene ontology (GO) terms among the genes in each cluster of H3B-8800–induced splicing changes from Fig. 2B. DNA repair was the only GO category among the top five enriched terms in all three clusters (fig. S2D and table S1). Cohesin-mutant cells are known to accumulate DNA damage and rely on proper DNA repair for survival (6, 27), making this category of genes particularly interesting for further study.
DNA damage repair genes tend to be significantly longer, contain more exons per transcript, and are more often found to have skipped exons compared with all other expressed genes in the Cancer Genome Atlas (TCGA) dataset (P < 0.0001) (Fig. 2D and fig. S2, E to H) (29). We proposed that these features make DNA damage repair genes highly dependent on accurate splicing for their expression, rendering them more sensitive to splicing changes induced by H3B-8800. An example of H3B-8800–induced exon skipping is shown for exon12 of BRCA2, which is skipped in 40 to 45% of transcripts in wild-type and STAG2-KO2 cells treated with 30 nM H3B-8800 for 6 hours (Fig. 2E). Among the 454 annotated DNA damage repair genes (30) expressed in U937 cells, 70% contained H3B-8800–induced splicing changes, and ~40% of those contained a reduction in gene expression (Fig. 2F and fig. S2I). Therefore, the splicing changes observed in DNA damage repair genes in cells treated with H3B-8800 may lead to loss of proper DNA repair, a process that is shared between wild-type and mutant cells but is particularly critical for survival of cohesin-mutant cells.
Mis-splicing of DNA repair genes alters protein function and results in accumulation of DNA damage
Among the top mis-spliced and down-regulated genes with H3B-8800 treatment were BRCA1 DNA repair associated (BRCA1) and BRCA2 DNA repair associated (BRCA2) (Fig. 3A and fig. S3A), proteins that play a critical role in homology-directed DNA damage repair and whose loss of function is known to correlate with sensitivity to PARP inhibitors (31–33). We confirmed that cells treated with H3B-8800 nearly completely lost protein expression of both BRCA1 and BRCA2 72 hours after treatment (Fig. 3B). Mis-splicing in other DNA damage repair genes, such as checkpoint kinase 2 (CHEK2), did not lead to major changes in RNA expression but resulted in splicing out of amino acid residues in key functional domains (Fig. 3C and fig. S3A), which has been previously shown to alter the protein activity (34). H3B-8800–induced skipping of CHEK2 exon2 spliced out the T68 amino acid targeted for phosphorylation and initiation of downstream signaling, as well as exon10, a portion of the kinase domain required for phosphorylation of its substrates (Fig. 3C). H3B-8800 treatment partially depleted total CHK2 and pCHK2 protein abundance (Fig. 3D and fig. S3B), thus impairing the ability to initiate proper DNA repair through signaling of the CHK2 DNA damage checkpoint. As evidence of accumulating DNA damage after H3B-8800 treatment, Western blotting for the marker of DNA damage gamma H2A.X variant histone (γH2AX) showed a substantial increase in signal in both cohesin-mutant and wild-type U937 cells, with the greatest accumulation of γH2AX in cohesin-mutant cells treated with H3B-8800 (Fig. 3E and fig. S3C). Furthermore, we observed a functional defect in homology-directed repair (HDR) using the RAD51 foci formation assay. At baseline, STAG2-mutant cells exhibited a significantly reduced ability to form RAD51 foci in response to γ-irradiation as compared with wild-type cells (P < 0.05) (Fig. 3F and fig. S3D), consistent with their increased baseline sensitivity to PARP inhibition (6, 27). Treatment with H3B-8800 reduced the ability of wild-type and STAG2-KO cells to form RAD51 foci, rendering STAG2 KO cells HDR deficient (Fig. 3F and fig. S3C). These results supported a model in which H3B-8800 inhibits DNA repair through mis-splicing of key DNA repair genes, resulting in the strongest accumulation of DNA damage and defect in HDR in cells with inherent dysfunction in DNA repair.
Fig. 3. Mis-splicing of DNA repair genes alters protein function and results in accumulation of DNA damage.
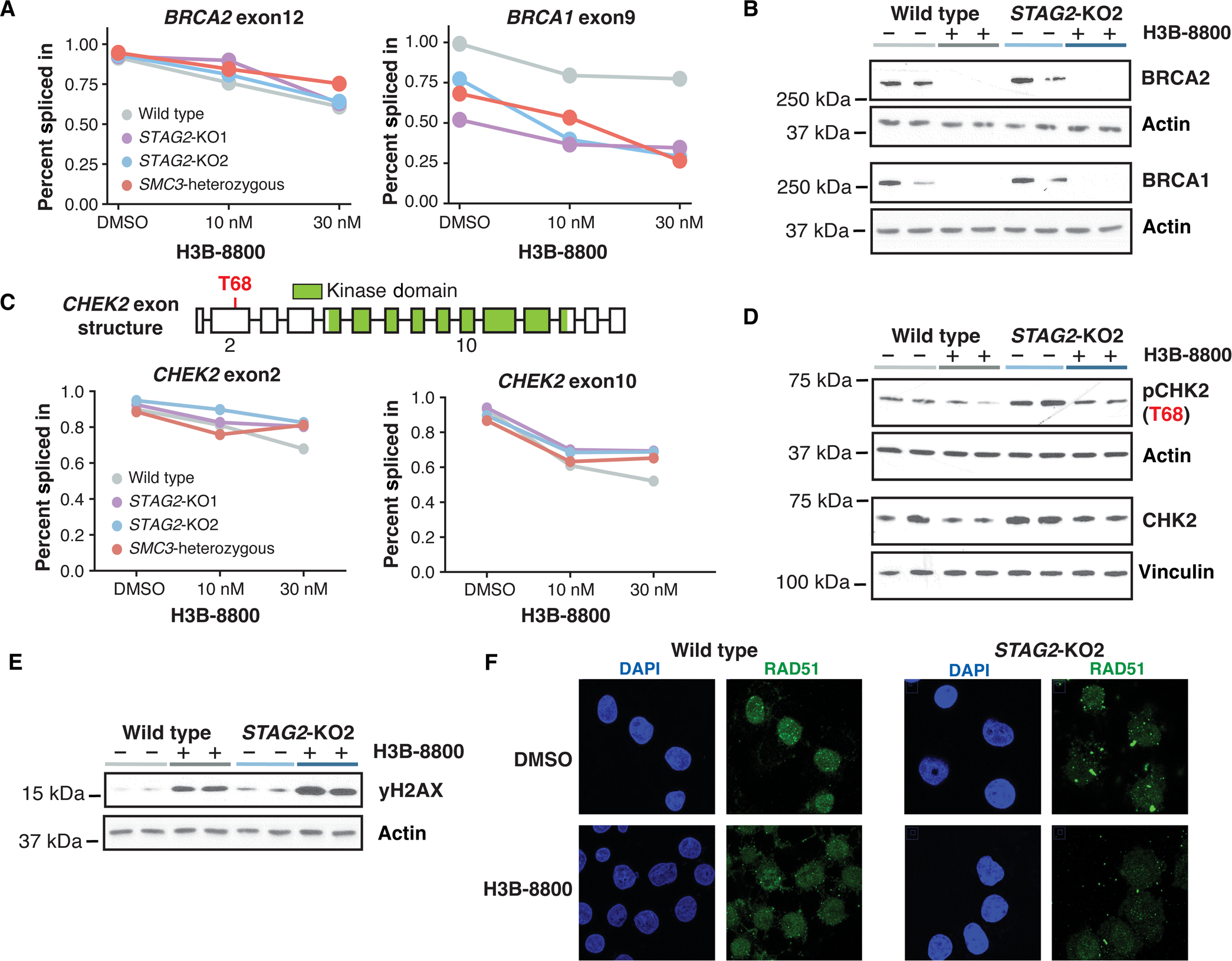
(A) average PSI scores of BRCA2 exon12 and BRCA1 exon9 are plotted for each genotype and drug condition. data points represent the mean of biological triplicates (wild type and STAG2-KO2) or duplicates (STAG2-KO1 and SMC3-heterozygous) for each treatment condition. (B) Western blot analysis of BCRA1 and BRCA2 protein expression in wild-type and STAG2-KO2 cells treated with 50 nM H3B-8800 or DMSO for 3 days. each lane represents an independent single-cell clone of U937 cells transduced with a nontargeting sgRNA (wild type) or sgRNA targeting STAG2. actin was used as a loading control. (C) Schematic of CHEK2 exon structure (top) with the Thr68 phosphorylation residue highlighted in red and the annotated kinase domain shown in green. Percent spliced in scores of exon2 and exon10 are shown for each treatment condition. data points represent the mean of biological triplicates (wild type and STAG2-KO2) or duplicates (STAG2-KO1 and SMC3-heterozygous) for each treatment condition. (D) Western blot analysis of pCHK2 and total CHK2 protein in wild-type and STAG2-KO2 cells treated for 3 days with 50 nM H3B-8800 or DMSO. each lane represents an independent single-cell clone of U937 cells transduced with a nontargeting sgRNA (wild type) or sgRNA targeting STAG2. Vinculin and actin were used as loading controls. (E) Western blot analysis of γh2aX protein in wild-type and STAG2-KO2 cells treated for 3 days with 50 nM H3B-8800 or DMSO. each lane represents an independent single-cell clone of U937 cells transduced with a nontargeting sgRNA (wild type) or sgRNA targeting STAG2. actin was used as a loading control. (F) representative images of cells treated with 50 nM H3B-8800 or DMSO control for 24 hours and stained for DNA/nuclei (DAPI, blue) and RAD51 (green).
Splicing modulation sensitizes cohesin-mutant AML cell lines to killing by talazoparib and chemotherapy
One prediction of this model is that treatment of cells with H3B-8800 or E-7107 impairs DNA damage repair and therefore renders cells more sensitive to subsequent treatment with DNA damage repair inhibitors or DNA damage–inducing agents. Having established drug-induced mis-splicing and loss of expression of BRCA1 and BRCA2, two key regulators of HDR and biomarkers of PARP inhibitor sensitivity, we then tested whether sequential treatment of cells with an SF3B1 modulator followed by the PARP inhibitor talazoparib potentiates killing of cohesin-mutant cells. In agreement with our previous work (6), STAG2-KO U937 cells were more sensitive than wild-type cells to treatment with talazoparib (Fig. 4A, shown here after pretreatment with DMSO). Pretreatment of cohesin wild-type U937 cells with either of the two available SF3B1 modulators, H3B-8800 or E-7107, followed by treatment with talazoparib, resulted in a modest decrease in cell viability (Fig. 4, A and B). Pretreatment of STAG2-KO U937 cells with DMSO, followed by treatment with talazoparib, recapitulated increased sensitivity of STAG2-KO versus wild-type cells to talazoparib, as we have previously reported (6). However, pretreatment of STAG2-KO U937 cells with either of the two splicing modulators markedly increased their sensitivity to talazoparib with up to a 10-fold increase in killing (Fig. 4, A and B, and fig. S4A). Furthermore, cells accumulated more DNA damage when subjected to sequential treatment of H3B-8800 followed by talazoparib than with either drug alone, as assessed by γH2AX Western blot (fig. S4, B and C).
Fig. 4. Splicing modulation sensitizes cohesin-mutant AML cell lines to killing by talazoparib and chemotherapy.
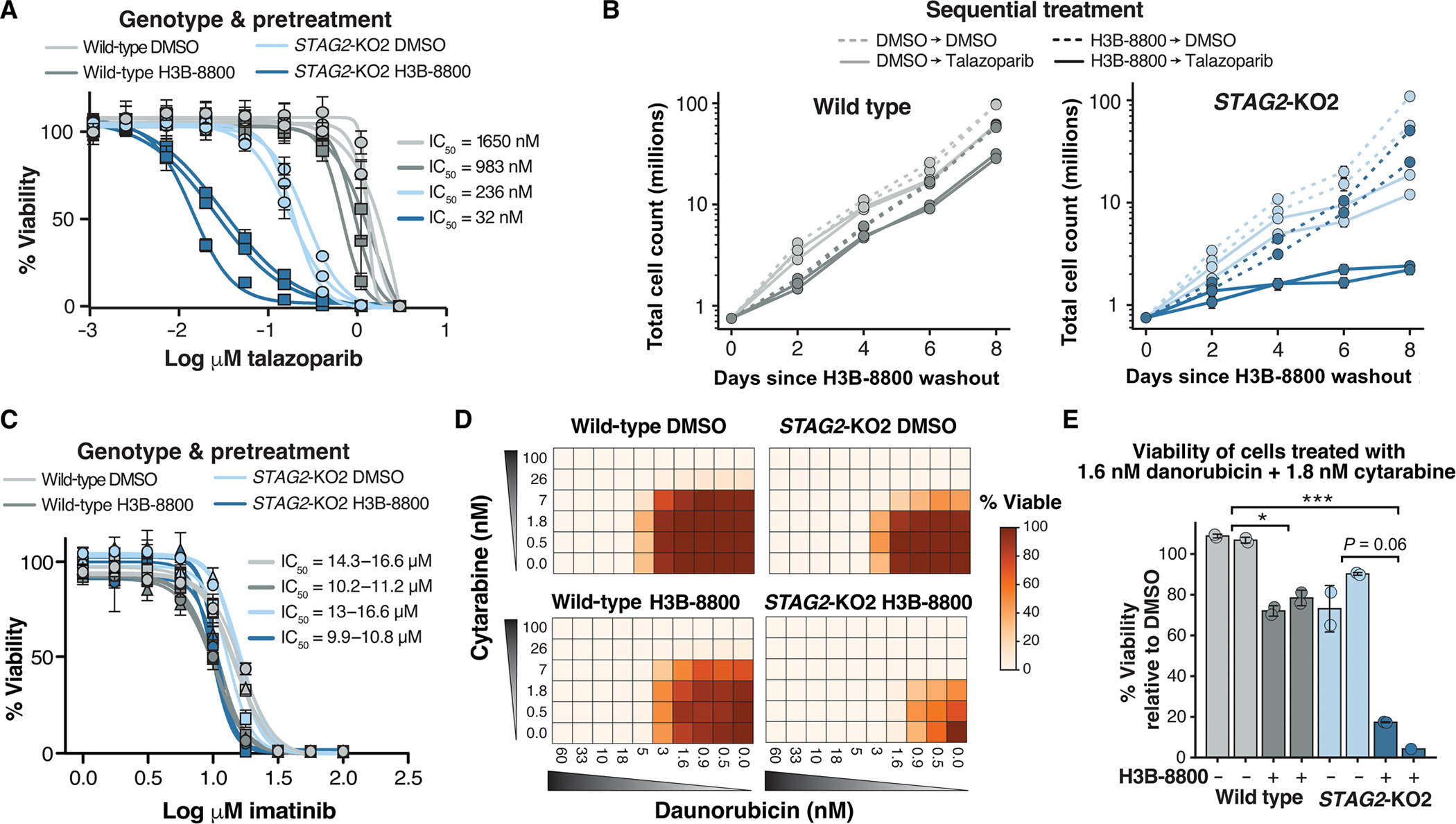
(A) drug dose-response curves of wild-type and STAG2-KO2 cells pretreated with 50 nM H3B-8800 or DMSO for 3 days, followed by drug washout and 8 days of treatment with talazoparib. error bars represent Sd of technical triplicate measurements for each biological triplicate sample (n = 3 per genotype and condition). (B) Growth curves depicting total number of wild type (left) or STAG2-KO2 cells (right) pretreated with DMSO or H3B-8800 (50 nM) for 3 days, followed by treatment with talazoparib (50 nM) or DMSO for 8 days. error bars represent Sd of technical duplicate measurements for each biological replicate sample (n = 2 per genotype and condition). (C) drug dose-response curves of wild-type and STAG2-KO2 cells pretreated with 50 nM H3B-8800 or DMSO for 3 days, followed by drug washout and 8 days of treatment with imatinib. error bars represent SD of technical triplicate measurements for each biological replicate sample (n = 2 per genotype and condition). (D) heatmap of cell viabilities for wild-type and STAG2-KO2 cells pretreated with DMSO (top) or 50 nM H3B-8800 (bottom) for 3 days, followed by treatment with a combination of daunorubicin and cytarabine for 8 days. cell viabilities are normalized to DMSO-treated controls on each plate (0 nM cytarabine and 0 nM daunorubicin). Values shown are the average of two technical replicate samples for one representative biological replicate sample. (E) Bar plot of percent viable cells after combination treatment with 1.6 nM daunorubicin and 1.8 nM cytarabine relative to DMSO-treated controls. cells received either 3 days of 50 nM H3B-8800 (dark bars) or DMSO control (light bars) before chemotherapy. data points show technical replicates n = 2 per condition (except biological replicate 2 of H3B-8800 treated STAG2-KO2 cells n = 1). each bar represents an independent biological replicate sample. P = 0.008 (Kruskal-Wallis), *P < 0.05, ***P < 0.001 in post hoc analysis using the Dunn’s test.
We next determined whether treatment of cells with an SF3B1 splicing modulator could sensitize cells to additional DNA damage–inducing agents, such as chemotherapy. We tested a panel of chemotherapeutic drugs, including single-agent and combination treatment of cytarabine and daunorubicin commonly used in the treatment of AML, as well as etoposide, cisplatin, and mitomycin C. Pretreatment of cells with H3B-8800 followed by treatment with any of these chemotherapeutic agents resulted in increased killing of both cohesin-mutant and wild-type cells, with preferential sensitivity of cohesin-mutant cells over wild-type cells (fig. S4D). In contrast, pretreatment with H3B-8800 had no effect on viability of cells treated with imatinib, an ABL proto-oncogene 1 (ABL1) kinase inhibitor used to treat chronic myeloid leukemia (CML) and CML blast crisis, which does not induce DNA damage (Fig. 4C). Furthermore, treatment of cells with a combination of daunorubicin and cytarabine, the standard-of-care regimen for AML, resulted in 80 to 90% depletion of cohesin-mutant cells after a single dose pretreatment with H3B-8800 compared with only 25 to 30% killing in wild-type cells (Fig. 4, D and E, and fig. S4E). Therefore, short-term treatment with a splicing modulator may be an effective method to increase the therapeutic window of many DNA damage–inducing chemotherapeutic agents, particularly in subsets of cancer with an underlying DNA damage repair defect, such as cohesin-mutant AML.
Low-dose splicing modulation combined with talazoparib or chemotherapy targets PDX AML in vivo
To test whether treatment with splicing modulators sensitizes primary patient samples to DNA damage repair inhibitors in vivo, we generated four serially transplantable patient-derived xenograft (PDX) models of AML from two different human STAG2-mutant and two cohesin wild-type AML samples (Fig. 5A and fig. S5A) (6). We first tested whether treatment with E-7107 could reproduce the effect on splicing and down-regulation of DNA damage repair genes in vivo. To do this, we performed RNA-seq on human bone marrow cells isolated from one of the STAG2-mutant and one cohesin wild-type AML PDX model after 3 or 5 days of E-7107 treatment. Consistent with our cell line results, we observed widespread exon skipping and splicing changes that were conserved at the event-level among genes that were detected in the PDX cells (Fig. 5, B and C, and fig. S5, B and C). DNA repair genes, including BRCA1 and BRCA2, were down-regulated in both PDX models tested (Fig. 5D and fig. S5, D to G). These data supported that our initial observations from AML cell lines were conserved in primary patient cell–derived xenograft models.
Fig. 5. Low-dose splicing modulation combined with talazoparib or chemotherapy targets PDX AML in vivo.
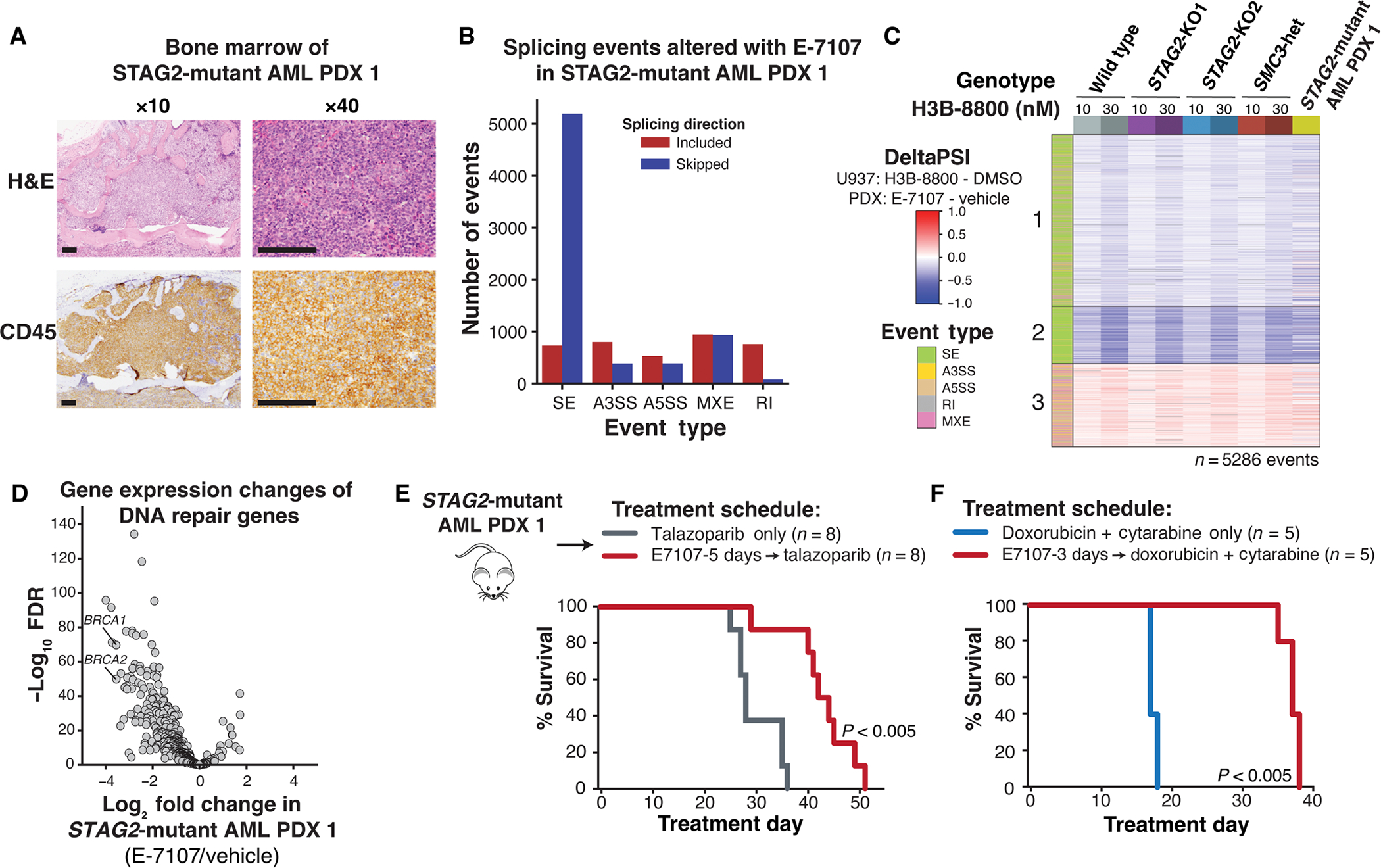
(A) Morphologic evaluation of bone marrow of STAG2-mutant AMl1 patient–derived xenograft shows infiltration with human leukemia blasts. images were stained using hematoxylin and eosin (H&E) (top) and hCD45-targeting antibody (bottom) and imaged at ×10 and ×40 (scale bars, 0.125 mm) original magnification. (B) Total number and directionality of significant (FDR < 0.05, ΔPSI >5%) splicing alterations differentially called in STAG2-mutant human AML1 PDX cells isolated from bone marrow of NSGS mice treated with E-7107 compared with vehicle for 5 days in vivo. Splicing events are categorized by event type and direction of regulation in E-7107 versus vehicle-treated mice. SE, skipped exon; A3SS, alternative 3′ splice site; A5SS, alternative 5′ splice site; MXE, mutually exclusive exon; RI, retained intron. N = 3 mice per condition. (C) heatmap of ΔPSI scores for H3B-8800–regulated exons called from U937 cells (Fig. 2B) that are expressed in STAG2-mutant AML PdX1 treated with E-7107 in vivo. each comparison consists of two (STAG2-KO1 and SMC3-heterozygous) or three (STAG2-KO2, wild type, and STAG2-mutant PDX) independent biological replicates compared with either DMSO or vehicle-treated controls. color bar on the left indicates the type of splicing event that was called, and column colors are labeled by genotype and drug treatment on the right. (D) Volcano plot depicting differential gene expression of DNA repair genes in STAG2-mutant human AML1 PDX cells isolated from the bone marrow of NSGS mice treated with E-7107 versus vehicle for 5 days in vivo. N = 3 mice per condition. (E) Schematic of in vivo E-7107 drug treatment and survival analysis of STAG2-mutant AML1 PDX model (other mutations include BCOR/RUNX1/U2AF1/DNMT3A). Treatment of mice assigned to two treatment arms was initiated 3 weeks after bone marrow transplantation: talazoparib only (n = 8) or E-7107 for 5 days followed by talazoparib (n = 8). Survival data were combined from two independent experiments. P < 0.005 (log-rank test). (F) Survival analysis of STAG2-mutant AML1 PDX mice treated with 3 days of E-7107 followed by combination chemotherapy (5 + 3 doxorubicin + cytarabine) or combination chemotherapy alone (n = 5 mice per arm). P < 0.005 (log-rank test).
To determine the efficacy of single-agent splicing modulation using these PDX models, we treated two STAG2-mutant and two cohesin and splicing factor wild-type AML PDX models with H3B-8800 or E-7107 in vivo for two weeks (fig. S5H). We assessed leukemia burden by quantifying the total number of human leukemia cells in the bone marrow relative to vehicle-treated control mice. Consistent with the cell line data, treatment with either of the two splicing modulators reduced the leukemia burden in both STAG2-mutant PDX models but not in the cohesin wild-type PDX model 1, characterized by mutations in FLT3-ITD, NPM1, DNMT3A, and PTPN11 (fig. S5H). The cohesin wild-type PDX model 2, characterized by the lysine methyltransferase 2A (MLL)/calmodulin (CALM) fusion and NRAS proto-oncogene NRAS(Q61L) mutation, also demonstrated reduction of the leukemia burden with E-7107 treatment. This prompted us to explore potential biomarkers of sensitivity to SF3B1 splicing modulators. On the basis of our observations that SF3B1 modulator induced mis-splicing and subsequent loss of expression of DNA damage repair genes, including BRCA1 and BRCA2, involved in homologous recombination, we sought to examine whether the RAD51 foci formation assay, a functional readout of homologous recombination efficiency in a setting of γ-irradiation–induced dsDNA breaks, could (i) demonstrate the functional consequence of SF3B1 splicing modulation on homologous recombination activity and (ii) predict sensitivity to treatment with SF3B1 inhibitors. We quantified γ-irradiation–induced RAD51 foci formation across all four PDX models and found reduced RAD51 foci formation in the three models that were sensitive to splicing modulation, including the two STAG2-mutant, and cohesin wild-type PDX model 2, but not in the cohesin wild-type PDX model 1, which was resistant to splicing modulation (fig. S5, I and J). These data further support that the increased sensitivity of cohesin-mutant cells to SF3B1 splicing modulation is driven by their underlying defects in DNA damage repair, and we propose the use of the RAD51 foci formation assay as a marker of sensitivity to SF3B1 modulation.
We next sequentially treated our STAG2-mutant and cohesin wild-type AML PDX models with splicing modulation and talazoparib or chemotherapy to determine whether the addition of splicing modulation could improve overall survival. First, we treated two separate cohorts of STAG2-mutant PDX mice with 3 or 5 days of E-7107, followed by talazoparib, talazoparib alone, or vehicle control. We observed a significant benefit in overall survival in both E-7107–containing treatment arms, with the longer splicing modulator pretreatment time contributing to the strongest benefit in overall survival (Fig. 5E and fig. S6, A and B) (P < 0.005, P < 0.05, P < 0.05, and P < 0.005, as presented in the figures). Pretreatment with E-7107 also reduced the disease-associated thrombocytopenia, circulating human peripheral blood cells, total human leukemia cells in the bone marrow, and disease infiltration into spleen and liver (fig. S6, C to G). Treatment of this model with E-7107 and combination chemotherapy of daunorubicin and cytarabine also increased overall survival and reduced disease-associated thrombocytopenia (Fig. 5F and fig. S6H). We treated a second STAG2-mutant model (PDX 2) with 5 days of E-7107 followed by talazoparib, talazoparib alone, or vehicle control. After 6 weeks of treatment, we euthanized all animals and observed a reduction in the total number of human cells in the bone marrow and spleens of animals treated with E-7107 and talazoparib compared with vehicle controls (fig. S6I). These results confirm that low-dose SF3B1 splicing modulation provides a therapeutic benefit in combination with PARP inhibition or chemotherapy in vivo and could be considered as a potential treatment strategy for cohesin-mutant cancers.
To test the specificity of this approach to cohesin-mutant models, we performed sequential treatment of splicing modulation and PARP inhibition in the two cohesin wild-type models that we had tested with single-agent splicing modulation (fig. S5H). The cohesin wild-type PDX model 1 (FLT3-ITD, NPM1, DNMT3A, and PTPN11-mutant) showed no difference in overall survival between animals treated with combination chemotherapy or E-7107 followed by combination chemotherapy (fig. S6J). The cohesin wild-type PDX model 2 (MLL/CALM and NRAS-mutant) showed a trend toward increased survival with H3B-8800 and talazoparib compared with talazoparib alone (fig. S6K). Furthermore, we tested the effect and toxicity of our combination treatment in healthy primary human hematopoietic stem and progenitor cells, which would be expected to have normal baseline DNA damage repair capacity. We engrafted CD34+ hematopoietic stem and progenitor cells isolated from mobilized peripheral blood of healthy donors into NSG-SGM3 (NSGS) mice and tested the effect of sequential treatment with the splicing modulator E-7107 followed by talazoparib after establishing engraftment. After 4 weeks of treatment, we noted no effect on the number of engrafted human cells in the bone marrow or spleen, with only a mild reduction of circulating human cells in peripheral blood (fig. S6L). We then examined the effect on differentiation and quantified numbers of mature and immature cells in the bone marrow. We observed a mild increase in the number of CD34+CD38− hematopoietic stem and progenitor cells and CD33+ myeloid cells, with no significant effect on the CD34−CD38+ progenitor compartment, CD11B+ myeloid, or CD71+ erythroid compartments (fig. S6L) with sequential treatment compared to vehicle (P > 0.05). These results suggested that healthy human hematopoiesis or PDX models of AML without an underlying defect in DNA damage repair are not affected by sequential treatment of low-dose short-term splicing modulation followed by PARP inhibition.
Splicing changes and down-regulation of DNA repair genes are conserved in patients with MDS/AML
Having established that SF3B1 modulator treatment leads to mis-splicing of DNA damage repair genes and sensitization of cohesin-mutant cells and PDX models to DNA damage–inducing drugs, we next sought to confirm that these effects were conserved in patient samples. We first interrogated splicing and gene expression changes among the 11 genes that were assessed using a custom nanostring panel as on-target biomarkers in the phase 1 clinical trial of H3B-8800 in splicing factor-mutant MDS and AML (25). Ten of 11 genes were expressed in U937 cells, and eight contained H3B-8800–regulated splicing changes that met our stringent threshold for significance (fig. S7A). Moreover, seven of the eight mis-spliced genes showed a dose-dependent effect on gene expression (fig. S7B). The remaining two genes, F-box and WD repeat domain containing 5 (FBXW5) and dynein light chain Tctex-type 1 (DYNLT1), contained multiple introns that were retained in a dose-dependent manner throughout the gene body but did not reach statistical significance (FDR > 0.05) (fig. S7, C and D). These data confirm on-target activity of H3B-8800 in our in vitro AML cell line models.
Next, we addressed whether H3B-8800 treatment in patients led to mis-splicing of DNA damage repair genes. Because comprehensive RNA-seq analysis on patients from the H3B-8800 clinical trial has not been reported, we performed total RNA-seq on peripheral blood samples collected from patients pretreatment and 2 to 4 hours after receiving their first oral dose of H3B-8800 (Fig. 6A and table S2). Splicing changes relative to the pretreatment sample were quantified for three patients independently, given the variable doses administered. Consistent with our in vitro data, exon skipping was the most common alternative splicing event detected after H3B-8800 treatment, and the number of events increased in a dose-dependent manner among the three patients (Fig. 6B). We then asked whether the drug-induced splicing changes identified in U937 cells (Fig. 2B) were conserved in patient samples. We observed a dose-dependent increase in splicing alterations that mirrored the splicing changes observed in U937 cells (Fig. 6C and fig. S7E). In the patient who received the highest dose of H3B-8800, 35% of expressed DNA repair genes contained at least one drug-induced splicing change, including the same event that was detected in BRCA1 in U937 cells (Fig. 6D). To examine whether splicing changes that occurred within 2 to 4 hours of H3B-8800 administration could affect gene expression, we performed a paired differential expression analysis comparing each patient pre- and posttreatment. As expected from the short time frame of treatment, changes in gene expression were generally small in magnitude. However, a trend toward down-regulation of DNA repair genes was observed in all patients after treatment with H3B-8800 (Fig. 6E). These results support that there is conservation at the splicing event-level with H3B-8800 treatment in vivo and in vitro and that DNA repair genes are targeted in patient samples.
Fig. 6. Splicing changes and down-regulation of DNA repair genes are conserved in patients with MDS and AML.
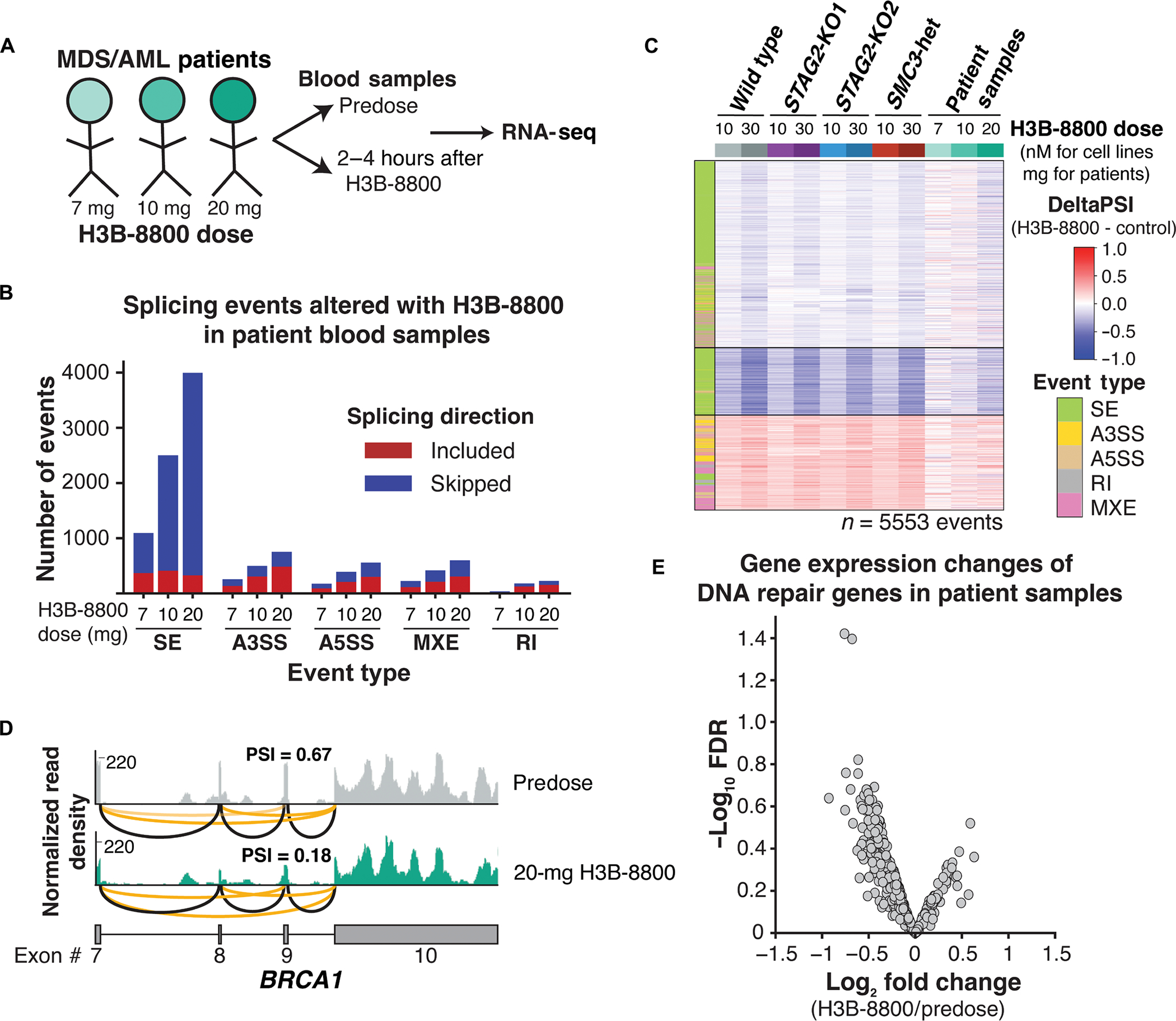
(A) Schematic of samples collected from patients with MDS and AML treated with three different doses of H3B-8800 on clinical trial (clinicalTrials.gov identifier NCT02841540). (B) Total number and directionality of significant (FDR < 0.05, ΔPSI >5%) splicing alterations differentially called in each patient sample pre– and post–H3B-8800. Patients are sorted on the x axis according to increasing doses of H3B-8800. Splicing events are categorized by event type and direction of regulation in H3B-8800 versus pretreatment sample. SE, skipped exon; A3SS, alternative 3′ splice site; A5SS, alternative 5′ splice site; MXE, mutually exclusive exon; RI, retained intron. (C) heatmap of ΔPSI scores for H3B-8800–regulated splicing changes called from U937 cells (Fig. 3a) that are expressed in patient samples. Patient samples are sorted by increasing dose of H3B-8800 received. color bar on the left indicates the type of splicing event that was called, and column colors are labeled by genotype and drug treatment. (D) RNA-seq–normalized read density and splice junction track of exon skipping in BRCA1 exon9 from the pre- and posttreatment sample in the patient who received 20 mg of H3B-8800. Black lines indicate constitutive splicing junctions, and orange lines indicate splice junctions that contain exon skipping. (E) Volcano plot depicting differential gene expression of DNA repair genes from a paired analysis of all patients pre– and post–H3B-8800 treatment.
DISCUSSION
Our studies established a role for RNA splicing modulation in the therapeutic targeting of cohesin-mutant MDS and AML. We demonstrated increased sensitivity of cohesin-mutant cells to splice-modulating drugs that target the SF3B complex and proposed to include SF3B1 modulation in combination with chemotherapy or PARP inhibition to target cohesin-mutant cells. Although E-7107 and H3B-8800 were first designed to treat MDS and AML patients harboring splicing factor mutations, this work suggests that patients with MDS and AML with cohesin mutations may also derive therapeutic benefit from this class of drugs.
We characterized a mechanism by which H3B-8800 and E-7107 induce cell death through mis-splicing of DNA repair proteins, a class of genes that are enriched for long genes with many exons. We showed that treatment with H3B-8800 results in accumulation of γH2AX and reduced function and expression of key DNA damage response genes including CHEK2, BRCA1, and BRCA2, which we have previously described as an important genetic dependency in cohesin-mutant cells (6). Although there are currently no targeted therapies approved for cohesin-mutant patients, clinical testing of single-agent PARP inhibition and combination with the hypomethylating agent decitabine in cohesin-mutant MDS and AML is ongoing (ClinicalTrials.gov identifier NCT03974217). We propose to expand this effort by using a dual-agent strategy in which low-dose splicing modulation is used to further sensitize cohesin-mutant cells to PARP inhibition or standard chemotherapy. We showed that this approach increases survival in primary patient-derived STAG2-mutant AML xenografts.
Although splicing modulators that target the SF3B complex were first developed to treat patients with splicing factor mutations, current research suggests that they may have widespread utility beyond this subset of patients with cancer. Other cancer types, including MYC-driven triple-negative breast cancer, T cell acute lymphoblastic leukemia (T-ALL) with up-regulation of SRSF7, DNMT3A-mutant AML, CLL irrespective of SF3B1 mutation status, and aggressive prostate cancer, have shown efficacy of SF3B1-targeting drugs in preclinical models (35–39). Our findings suggest that cohesin-mutant cancers may similarly benefit from splice-modulating therapies, particularly those targeting the SF3B complex. Furthermore, we identified the RAD51 foci formation assay as a potential marker to predict sensitivity to SF3B1 splicing modulation of additional DNA damage repair–deficient cancer types. We therefore suggest that the ongoing single-agent study with H3B-8800, which is currently limited to lower-risk transfusion-dependent MDS may benefit from additional evaluation in higher risk MDS and AML subtypes with underlying DNA damage repair defects in combination with PARP inhibition or chemotherapy. Our data suggest that the addition of short-term splicing modulation to PARP inhibition does not lead to major toxicity to healthy human hematopoietic stem and progenitor cells. It remains to be seen whether this sensitivity will extend to other types of splice-modulating drugs that are undergoing clinical development, including SR protein kinase inhibitors, RNA binding motif protein 39 degraders, and protein arginine methyltransferase (PRMT) inhibitors (22, 40). RNA processing defects have been previously shown to lead to genomic instability as a consequence of R loop formation (41). Because our study did not focus on R loop formation, we cannot rule out that an additional mechanism by which SF3B splicing modulators lead to accumulation of DNA damage is due to accumulation of R loops.
Our study has some limitations. None of the patient samples available for analysis from the H3B-8800 clinical trial contained mutations in STAG2 or other cohesin genes. However, we did observe a strong conservation of on-target splicing changes and down-regulation of DNA repair genes in cohesin wild-type and mutant PDX models and cell line models. These results highlight the reliance on inherent vulnerabilities of proper DNA repair in cancer cells as a critical determinant in our proposed model of SF3B1 splicing modulator–induced selective killing (fig. S8). More broadly, we propose that any cancer cells that are addicted to proper DNA repair for survival may be selectively targeted with a treatment strategy of splicing modulation followed by PARP inhibition or chemotherapy (42). These include BRCA-mutant breast and ovarian cancers characterized by a “BRCA-ness” phenotype of dysfunctional HDR and resulting sensitivity to PARP inhibition (43, 44). For example, treatment with pladienolide B has been previously shown to sensitize ovarian and breast cancer cell lines to cisplatin (45). Recent work has suggested that spliceosome-mutant MDS cell line models are also sensitive to PARP inhibition, making this class of cancers another candidate for dual splicing and PARP inhibition therapy (46).
A major challenge with single-agent PARP inhibitor treatment is acquired resistance through escape mechanisms in which cells repair HDR defects (44). However, targeting cancer dependency pathways with multiple, non-overlapping agents is a proven strategy to prevent acquisition of resistance. Our study identifies a critical connection between cohesin mutations and splicing modulation in AML, creating a therapeutic strategy for cohesin-mutant patients with very limited treatment options and poor outcomes. In summary, we propose low-dose splicing modulation followed by PARP inhibition or standard chemotherapy as a therapeutic strategy to be considered in cohesin-mutant MDS and AML and suggest a potential benefit of this strategy in other cancer types that are deficient in proper DNA damage repair.
MATERIALS AND METHODS
Study design
The objective of this study was to determine the role of RNA splicing modulation in the survival of cohesin-mutant MDS/AML. Isogenic cell line models with multiple independent single-cell clones per genotype were used for all in vitro work. Two or three independent biological replicates were used for each individual experiment, many of which also included additional technical replicates. For in vivo studies, the primary endpoint was overall survival, and the secondary endpoint was depletion of cohesin-mutant cells. Animals were euthanized according to the predetermined endpoints of 20% weight loss from the beginning of treatment or a body condition score of 2 or lower. For animal studies, treatment groups were assigned to normalize blood counts per group before treatment. The studies were not performed in a blind fashion.
U937 and K562 cell lines and culture
U937 and K562 cell lines used in this study were previously published (6). Cells were grown in RMPI + 10% fetal bovine serum + 1% penicillin/streptomycin/glutamine at 37°C with 5% CO2.
U937 cell transplantation mouse model
Eight- to 10-week-old female NSGS mice [NOD-S CID; IL2Rγ null; Tg(IL3, CSF2, KITL)] were obtained from the Jackson Laboratory (strain 013062). Recipient mice were sublethally irradiated using a Gamma cell irradiator (Best Theratronics) at a dose of 250 rads and injected by tail vein with 500,000 green fluorescent protein–positive (GFP+) or mCherry+ STAG2-mutant or WT U937 cells. Mice were dosed daily with H3B-8800 (8 mg/kg) or an equivalent volume of vehicle in 0.5% methylcellulose solution daily by oral gavage, starting on day 7 after transplantation. Treatment continued for 15 to 30 days until the mice were euthanized upon disease development. All mice were housed in a pathogen-free animal facility in microisolator cages, and experiments were conducted according to an institutional animal care and use committe (IACUC)–approved protocol 19–020 at the Dana-Farber Cancer Institute.
PDX mouse models
The PDX models used in this study were generated using serial transplantation of human AML samples in adult female NSGS mice [NOD-SCID; IL2Rγ null; Tg(IL3, CSF2, KITL), the Jackson Laboratory, strain 013062]. Details of the PDX mouse models and in vivo drug treatments used in the study are included in the Supplementary Materials and Methods.
RNA sequencing
U937 cells were treated with H3B-8800 at 10 and 30 nM or DMSO for 6 hours. One million cells were collected in 350 μl of TRizol and flash-frozen. RNA was extracted with Zymogen Direct-Zol columns. One hundred nanograms of input RNA was used for library preparation with the KAPA RNA hyper prep kit with RiboErase treatment according to the manufacturer’s instructions. Libraries were sequenced on the Illumina NOVASeq in PE100 mode to a depth of 100 M reads per sample at the Dana-Farber Cancer Institute. For patient sample analysis, blood samples were collected from patients immediately before and 2 to 4 hours after their first dose of H3B-8800. RNA was extracted from whole blood and quantified, and 100 ng was used as the input for total RNA-seq. Details of the RNA-seq and splicing analyses are included in the Supplementary Materials and Methods.
In vitro drug treatment and in vitro competition assays
All drug dose response assays were conducted in 96-w ell plates. For U937 and K562 cell lines, 10,000 cells were plated per well at 0.05 × 106 cells/ml. For pretreatment with DMSO, H3B-8800 (50 nM), or E-7107 (1.14 nM), cells were cultured in bulk for 3 days before plating in 96-well format for secondary drug dosing. Viability was measured every 4 days using the CellTiter-Glo luminescent cell viability assay (Promega, G7573). Drug dose-response curves were fitted, and median inhibitory concentrations (IC50) were calculated using GraphPad Prism. For competition experiments, mCherry-labeled wild-type and GFP-labeled STAG2-KO2 U937 cells were mixed in a 1:10 ratio and plated in 96-well plates with three technical replicates. Cells were stained for viability with 4′,6-diamidino-2-phenylindole (DAPI), and % mCherry+DAPI− and GFP+DAPI− cells were determined by flow cytometry. Additional details are provided in the Supplementary Materials and Methods.
Western blotting
Primary antibodies used in our studies include the following: vinculin (V9131 Sigma-Aldrich, 1:1000), CHK2 (MP 05–649, 1:5000), pCHK2 [Cell Signaling Technology (CST) 2661, 1:1000], y-H2AX (CST9718, 1:1000), actin (CST8H10D10, 1:10,000), BRCA2 (EMD Milipore OP95, 1:500), and BRCA1 (gift from D. Livingston at the Dana-Farber Cancer Institute, MS110, 1:1000). Details of the Western blotting protocol used are included in the Supplementary Materials and Methods.
RAD51 foci detection
U937 and PDX cells were treated in culture with 50 nM H3B-8800 or DMSO for 24 hours. Cells were then collected and treated with irradiation (5 Gy), cultured for an additional 4 hours, and collected and spun onto coverslips for immunofluorescence staining. Cells were fixed in 4% paraformaldehyde, permeabilized, and stained for RAD51 (ab63801). Slides were mounted with DAPI mounting medium and imaged on a confocal microscope. Data were analyzed using a Cell Profiler pipeline.
Statistical analysis
Primary data are presented in data file S1. Statistical analysis was performed in GraphPad Prism 8 and with the Scipy Stats module in Python (v1.5.0). Data with two groups were analyzed using unpaired Student’s t test, Mann-Whitney test, or Welch’s t test. Data with multiple groups were analyzed using one-way analysis of variance (ANOVA) or Kruskal-Wallis test with Dunn’s post hoc test. Co-occurrence and mutual exclusivity of STAG2 and SF3B1 mutations were analyzed using a Z test. Kaplan-Meier analysis and the log-rank test were used to determine overall survival differences. Data are shown as mean ± SD. Significance is shown as *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.001, and nonsignificant (NS). Additional details of statistical tests used, sample size, and technical replicates for each experiment are described in the figure legends. No outliers were removed from data analysis.
Supplementary Material
Acknowledgments:
We would like to thank the members of the Tothova and Adelman labs for helpful discussion and feedback on this work. We thank S. Lazo, M. Buonopane, and J. Marcucella at the Dana-Farber Flow Cytometry core for help with cell sorting and analysis; M. Berkeley and Z. Hebert from the Molecular Biology Core Facility at Dana-Farber for preparation of RNA-seq libraries and sequencing and acknowledge support from the Ted and Eileen Pasquarello Tissue Bank in Hematologic Malignancies at the Dana-Farber Cancer Institute. We thank E. Hatchi and A. Idelevich for providing BRCA1 and BRCA2 antibodies and helpful discussion on this work.
Funding:
The work of Z.T. and J.T. was supported by the Doris Duke Charitable Foundation. The work of Z.T. was supported by the Burroughs Welcome Fund and the Edward P. Evans Foundation. The work of Z.T. and K.A. was supported by the Ludwig Center at Harvard and the Quadrangle Fund for Advancing and Seeding Translational Research (QFASTR) at Harvard Medical School. The work of J.S.G. was supported by the NCI K08CA245209 award. The work of Y.P. was supported by the St. Baldricks consortium grant and and Hannah’s Heroes. E.C.W. was supported by the NIH Ruth L. Kirschstein F32 postdoctoral award (F32HL159905). B.J.E.M. is supported by CIHR Banting Postdoctoral Fellowship.
Footnotes
Competing interests: M.S. and S.B. were employees of H3 Biomedicine Inc. at the time the research was conducted. O.A.-W. has served as a consultant for H3B Biomedicine, Foundation Medicine Inc., Merck, Prelude Therapeutics, and Janssen and is on the Scientific Advisory Board of Codify, Envisagenics Inc., AIChemy, Harmonic Discovery Inc., and Pfizer Boulder; O.A.-W. has received prior research funding from H3B Biomedicine, Nurix Therapeutics, Minovia Therapeutics, and LOXO Oncology unrelated to the current manuscript. J.S.G. is on the Scientific Advisory Board of AbbVie, Astellas, BMS, Genentech, and Servier and has received institutional funding support for trials from AbbVie, AstraZeneca, Pfizer, Prelude, and Genentech, all unrelated to the current manuscript. K.A. is a consultant for Syros Pharmaceuticals and is on the SAB of CAMP4 Therapeutics. K.A. and Z.T. received research funding from Novartis not related to this work. The other authors declare that they have no competing interests.
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary Materials. All of the raw and processed RNA-seq data are available on GEO (accession GSE 198518). Original code has been deposited at Zenodo (54, 55). H3B-8800 and E-7107 were obtained from H3B Biosciences and Hemavant Sciences under institutional materials transfer agreements.
REFERENCES AND NOTES
- 1.Losada A, Cohesin in cancer: Chromosome segregation and beyond. Nat. Rev. Cancer 14, 389–393 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Jann JC, Tothova Z, Cohesin mutations in myeloid malignancies. Blood 138, 649–661 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Antony J, Chin CV, Horsfield JA, Cohesin mutations in cancer: Emerging therapeutic targets. Int. J. Mol. Sci. 22, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heimbruch KE, Meyer AE, Agrawal P, Viny AD, Rao S, A cohesive look at leukemogenesis: The cohesin complex and other driving mutations in AML. Neoplasia 23, 337–347 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kon A, Shih LY, Minamino M, Sanada M, Shiraishi Y, Nagata Y, Yoshida K, Okuno Y, Bando M, Nakato R, Ishikawa S, Sato-Otsubo A, Nagae G, Nishimoto A, Haferlach C, Nowak D, Sato Y, Alpermann T, Nagasaki M, Shimamura T, Tanaka H, Chiba K, Yamamoto R, Yamaguchi T, Otsu M, Obara N, Sakata-Yanagimoto M, Nakamaki T, Ishiyama K, Nolte F, Hofmann WK, Miyawaki S, Chiba S, Mori H, Nakauchi H, Koeffler HP, Aburatani H, Haferlach T, Shirahige K, Miyano S, Ogawa S, Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat. Genet. 45, 1232–1237 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Tothova Z, Valton AL, Gorelov RA, Vallurupalli M, Krill-Burger JM, Holmes A, Landers CC, Haydu JE, Malolepsza E, Hartigan C, Donahue M, Popova KD, Koochaki S, Venev SV, Rivera J, Chen E, Lage K, Schenone M, D’Andrea AD, Carr SA, Morgan EA, Dekker J, Ebert BL, Cohesin mutations alter DNA damage repair and chromatin structure and create therapeutic vulnerabilities in MDS/AML. JCI Insight 6, e142149 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cuadrado A, Losada A, Specialized functions of cohesins STAG1 and STAG2 in 3D genome architecture. Curr. Opin. Genet. Dev. 61, 9–16 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Viny AD, Bowman RL, Liu Y, Lavallee VP, Eisman SE, Xiao W, Durham BH, Navitski A, Park J, Braunstein S, Alija B, Karzai A, Csete IS, Witkin M, Azizi E, Baslan T, Ott CJ, Pe’er D, Dekker J, Koche R, Levine RL, Cohesin members Stag1 and Stag2 display distinct roles in chromatin accessibility and topological control of HSC self-renewal and differentiation. Cell Stem Cell 25, 682–696.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mazumdar C, Shen Y, Xavy S, Zhao F, Reinisch A, Li R, Corces MR, Flynn RA, Buenrostro JD, Chan SM, Thomas D, Koenig JL, Hong WJ, Chang HY, Majeti R, Leukemia-associated cohesin mutants dominantly enforce stem cell programs and impair human hematopoietic progenitor differentiation. Cell Stem Cell 17, 675–688 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mullenders J, Aranda-Orgilles B, Lhoumaud P, Keller M, Pae J, Wang K, Kayembe C, Rocha PP, Raviram R, Gong Y, Premsrirut PK, Tsirigos A, Bonneau R, Skok JA, Cimmino L, Hoehn D, Aifantis I, Cohesin loss alters adult hematopoietic stem cell homeostasis, leading to myeloproliferative neoplasms. J. Exp. Med. 212, 1833–1850 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, Yoon CJ, Ellis P, Wedge DC, Pellagatti A, Shlien A, Groves MJ, Forbes SA, Raine K, Hinton J, Mudie LJ, McLaren S, Hardy C, Latimer C, Della Porta MG, O’Meara S, Ambaglio I, Galli A, Butler AP, Walldin G, Teague JW, Quek L, Sternberg A, Gambacorti-Passerini C, Cross NC, Green AR, Boultwood J, Vyas P, Hellstrom-Lindberg E, Bowen D, Cazzola M, Stratton MR, Campbell PJ, C., Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 122, 3616–3627 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, Schnittger S, Sanada M, Kon A, Alpermann T, Yoshida K, Roller A, Nadarajah N, Shiraishi Y, Shiozawa Y, Chiba K, Tanaka H, Koeffler HP, Klein HU, Dugas M, Aburatani H, Kohlmann A, Miyano S, Haferlach C, Kern W, Ogawa S, Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 28, 241–247 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lindsley RC, Saber W, Mar BG, Redd R, Wang T, Haagenson MD, Grauman PV, Hu ZH, Spellman SR, Lee SJ, Verneris MR, Hsu K, Fleischhauer K, Cutler C, Antin JH, Neuberg D, Ebert BL, Prognostic mutations in myelodysplastic syndrome after stem-cell transplantation. N. Engl. J. Med. 376, 536–547 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lindsley RC, Mar BG, Mazzola E, Grauman PV, Shareef S, Allen SL, Pigneux A, Wetzler M, Stuart RK, Erba HP, Damon LE, Powell BL, Lindeman N, Steensma DP, Wadleigh M, DeAngelo DJ, Neuberg D, Stone RM, Ebert BL, Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 125, 1367–1376 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li J, Pan T, Chen L, Wang Q, Chang Z, Zhou W, Li X, Xu G, Li X, Li Y, Zhang Y, Alternative splicing perturbation landscape identifies RNA binding proteins as potential therapeutic targets in cancer. Mol. Ther. Nucleic Acids 24, 792–806 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kahles A, Lehmann KV, Toussaint NC, Huser M, Stark SG, Sachsenberg T, Stegle O, Kohlbacher O, Sander C, Ratsch G, Comprehensive analysis of alternative splicing across tumors from 8,705 patients. Cancer Cell 34, 211–224 e216 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Climente-Gonzalez H, Porta-Pardo E, Godzik A, Eyras E, The functional impact of alternative splicing in cancer. Cell Rep. 20, 2215–2226 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Oltean S, Bates DO, Hallmarks of alternative splicing in cancer. Oncogene 33, 5311–5318 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, Chalkidis G, Suzuki Y, Shiosaka M, Kawahata R, Yamaguchi T, Otsu M, Obara N, Sakata-Yanagimoto M, Ishiyama K, Mori H, Nolte F, Hofmann WK, Miyawaki S, Sugano S, Haferlach C, Koeffler HP, Shih LY, Haferlach T, Chiba S, Nakauchi H, Miyano S, Ogawa S, Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478, 64–69 (2011). [DOI] [PubMed] [Google Scholar]
- 20.Pereira B, Billaud M, Almeida R, RNA-binding proteins in cancer: Old players and new actors. Trends Cancer 3, 506–528 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Saez B, Walter MJ, Graubert TA, Splicing factor gene mutations in hematologic malignancies. Blood 129, 1260–1269 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.El Marabti E, Abdel-Wahab O, Therapeutic modulation of RNA splicing in malignant and non-malignant disease. Trends Mol. Med. 27, 643–659 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Folco EG, Coil KE, Reed R, The anti-tumor drug E7107 reveals an essential role for SF3b in remodeling U2 snRNP to expose the branch point-binding region. Genes Dev. 25, 440–444 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Obeng EA, Chappell RJ, Seiler M, Chen MC, Campagna DR, Schmidt PJ, Schneider RK, Lord AM, Wang L, Gambe RG, McConkey ME, Ali AM, Raza A, Yu L, Buonamici S, Smith PG, Mullally A, Wu CJ, Fleming MD, Ebert BL, Physiologic expression of Sf3b1(K700E) causes impaired erythropoiesis, aberrant splicing, and sensitivity to therapeutic spliceosome modulation. Cancer Cell 30, 404–417 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steensma DP, Wermke M, Klimek VM, Greenberg PL, Font P, Komrokji RS, Yang J, Brunner AM, Carraway HE, Ades L, Al-Kali A, Alonso-Dominguez JM, Alfonso-Pierola A, Coombs CC, Deeg HJ, Flinn I, Foran JM, Garcia-Manero G, Maris MB, McMasters M, Micol JB, De Oteyza JP, Thol F, Wang ES, Watts JM, Taylor J, Stone R, Gourineni V, Marino AJ, Yao H, Destenaves B, Yuan X, Yu K, Dar S, Ohanjanian L, Kuida K, Xiao J, Scholz C, Gualberto A, Platzbecker U, Phase I first-in-human dose escalation study of the oral SF3B1 modulator H3B-8800 in myeloid neoplasms. Leukemia 35, 3542–3550 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seiler M, Yoshimi A, Darman R, Chan B, Keaney G, Thomas M, Agrawal AA, Caleb B, Csibi A, Sean E, Fekkes P, Karr C, Klimek V, Lai G, Lee L, Kumar P, Lee SC, Liu X, Mackenzie C, Meeske C, Mizui Y, Padron E, Park E, Pazolli E, Peng S, Prajapati S, Taylor J, Teng T, Wang J, Warmuth M, Yao H, Yu L, Zhu P, Abdel-Wahab O, Smith PG, Buonamici S, H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat. Med. 24, 497–504 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mondal G, Stevers M, Goode B, Ashworth A, Solomon DA, A requirement for STAG2 in replication fork progression creates a targetable synthetic lethality in cohesin-mutant cancers. Nat. Commun. 10, 1686 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Antony J, Gimenez G, Taylor T, Khatoon U, Day R, Morison IM, Horsfield JA, BET inhibition prevents aberrant RUNX1 and ERG transcription in STAG2 mutant leukaemia cells. J. Mol. Cell Biol. 12, 397–399 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim P, Yang M, Yiya K, Zhao W, Zhou X, ExonSkipDB: Functional annotation of exon skipping event in human. Nucleic Acids Res. 48, D896–D907 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hussmann JA, Ling J, Ravisankar P, Yan J, Cirincione A, Xu A, Simpson D, Yang D, Bothmer A, Cotta-Ramusino C, Weissman JS, Adamson B, Mapping the genetic landscape of DNA double-strand break repair. Cell 184, 5653–5669.e25 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Groelly FJ, Fawkes M, Dagg RA, Blackford AN, Tarsounas M, Targeting DNA damage response pathways in cancer. Nat. Rev. Cancer 23, 78–94 (2023). [DOI] [PubMed] [Google Scholar]
- 32.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T, Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A, Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Yang HW, Kim TM, Song SS, Shrinath N, Park R, Kalamarides M, Park PJ, Black PM, Carroll RS, Johnson MD, Alternative splicing of CHEK2 and codeletion with NF2 promote chromosomal instability in meningioma. Neoplasia 14, 20–28 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou Y, Han C, Wang E, Lorch AH, Serafin V, Cho BK, Gutierrez Diaz BT, Calvo J, Fang C, Khodadadi-Jamayran A, Tabaglio T, Marier C, Kuchmiy A, Sun L, Yacu G, Filip SK, Jin Q, Takahashi YH, Amici DR, Rendleman EJ, Rawat R, Bresolin S, Paganin M, Zhang C, Li H, Kandela I, Politanska Y, Abdala-Valencia H, Mendillo ML, Zhu P, Palhais B, Van Vlierberghe P, Taghon T, Aifantis I, Goo YA, Guccione E, Heguy A, Tsirigos A, Wee KB, Mishra RK, Pflumio F, Accordi B, Basso G, Ntziachristos P, Posttranslational regulation of the exon skipping machinery controls aberrant splicing in leukemia. Cancer Discov. 10, 1388–1409 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ten Hacken E, Valentin R, Regis FFD, Sun J, Yin S, Werner L, Deng J, Gruber M, Wong J, Zheng M, Gill AL, Seiler M, Smith P, Thomas M, Buonamici S, Ghia EM, Kim E, Rassenti LZ, Burger JA, Kipps TJ, Meyerson ML, Bachireddy P, Wang L, Reed R, Neuberg D, Carrasco RD, Brooks AN, Letai A, Davids MS, Wu CJ, Splicing modulation sensitizes chronic lymphocytic leukemia cells to venetoclax by remodeling mitochondrial apoptotic dependencies. JCI Insight 3, e121438 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang D, Hu Q, Liu X, Ji Y, Chao HP, Liu Y, Tracz A, Kirk J, Buonamici S, Zhu P, Wang J, Liu S, Tang DG, Intron retention is a hallmark and spliceosome represents a therapeutic vulnerability in aggressive prostate cancer. Nat. Commun. 11, 2089 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hsu TY, Simon LM, Neill NJ, Marcotte R, Sayad A, Bland CS, Echeverria GV, Sun T, Kurley SJ, Tyagi S, Karlin KL, Dominguez-Vidana R, Hartman JD, Renwick A, Scorsone K, Bernardi RJ, Skinner SO, Jain A, Orellana M, Lagisetti C, Golding I, Jung SY, Neilson JR, Zhang XH, Cooper TA, Webb TR, Neel BG, Shaw CA, Westbrook TF, The spliceosome is a therapeutic vulnerability in MYC-driven cancer. Nature 525, 384–388 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ramabadran R, Wang JH, Reyes JM, Guzman AG, Gupta S, Rosas C, Brunetti L, Gundry MC, Tovy A, Long H, Gu T, Cullen SM, Tyagi S, Rux D, Kim JJ, Kornblau SM, Kyba M, Stossi F, Rau RE, Takahashi K, Westbrook TF, Goodell MA, DNMT3A-coordinated splicing governs the stem state switch towards differentiation in embryonic and haematopoietic stem cells. Nat. Cell Biol. 25, 528–539 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eymin B, Targeting the spliceosome machinery: A new therapeutic axis in cancer? Biochem. Pharmacol. 189, 114039 (2021). [DOI] [PubMed] [Google Scholar]
- 41.Chan YA, Hieter P, Stirling PC, Mechanisms of genome instability induced by RNA-processing defects. Trends Genet. 30, 245–253 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krajewska M, Dries R, Grassetti AV, Dust S, Gao Y, Huang H, Sharma B, Day DS, Kwiatkowski N, Pomaville M, Dodd O, Chipumuro E, Zhang T, Greenleaf AL, Yuan GC, Gray NS, Young RA, Geyer M, Gerber SA, George RE, CDK12 loss in cancer cells affects DNA damage response genes through premature cleavage and polyadenylation. Nat. Commun. 10, 1757 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lord CJ, Ashworth A, PARP inhibitors: Synthetic lethality in the clinic. Science 355, 1152–1158 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Curtin NJ, Szabo C, Poly(ADP-ribose) polymerase inhibition: Past, present and future. Nat. Rev. Drug Discov. 19, 711–736 (2020). [DOI] [PubMed] [Google Scholar]
- 45.Anufrieva KS, Vcapital CSO, Arapidi GP, Pavlyukov MS, Shakhparonov MI, Shnaider PV, Butenko IO, Lagarkova MA, Govorun VM, Therapy-induced stress response is associated with downregulation of pre-mRNA splicing in cancer cells. Genome Med. 10, 49 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nguyen DH, Liu ZS, Sinha S, Bannister M, Arriaga-Gomez E, Song A, Zong D, Corral V, Leibson A, Rai S, Graubert TA, Lee SC, Spliceosome mytant myeloid malignancies are preferentially sensitive to PARP inhibition. ASH Meeting Abstract. biorxiv: 2022.09.26.509430 (2021). [Google Scholar]
- 47.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liao Y, Smyth GK, Shi W, The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 47, e47 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bray NL, Pimentel H, Melsted P, Pachter L, Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Shen S, Park JW, Lu ZX, Lin L, Henry MD, Wu YN, Zhou Q, Xing Y, rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. U. S. A. 111, E5593–E5601 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pikman Y, Tasian SK, Sulis ML, Stevenson K, Blonquist TM, Winger BA, Cooper TM, Pauly M, Maloney KW, Burke MJ, Brown PA, Gossai N, McNeer JL, Shukla NN, Cole PD, Kahn JM, Chen J, Barth MJ, Magee JA, Gennarini L, Adhav AA, Clinton CM, Ocasio-Martinez N, Gotti G, Li Y, Lin S, Imamovic A, Tognon CE, Patel T, Faust HL, Contreras CF, Cremer A, Cortopassi WA, Ruiz DG, Jacobson MP, Dharia NV, Su A, Robichaud AL, Conway AS, Tarlock K, Stieglitz E, Place AE, Puissant A, Hunger SP, Kim AS, Lindeman NI, Gore L, Janeway KA, Silverman LB, Tyner JW, Harris MH, Loh ML, Stegmaier K, Matched targeted therapy for pediatric patients with relapsed, refractory, or high-risk leukemias: A report from the LEAP consortium. Cancer Discov. 11, 1424–1439 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, Chanda SK, Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10, 1523 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martin BJ, Mimoso C, AdelmanLab/GetGeneAnnotation_GGA: AdelmanLab/GetGeneAnnotation_GGA/v1. Zenodo (2021). 10.5281/zenodo.5519928. [DOI] [Google Scholar]
- 55.Martin BJ, subset_rmats_junctioncountonly.py. Zenodo (2023). 10.5281/zenodo.10219272. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials. All of the raw and processed RNA-seq data are available on GEO (accession GSE 198518). Original code has been deposited at Zenodo (54, 55). H3B-8800 and E-7107 were obtained from H3B Biosciences and Hemavant Sciences under institutional materials transfer agreements.