


Abstract
Small ubiquitin-like modifiers (SUMOs) are tiny but important protein regulators involved in orchestrating a broad spectrum of biological processes, either by covalently modifying protein substrates or by noncovalently interacting with other proteins. Here, we report an updated server, GPS-SUMO 2.0, for the prediction of SUMOylation sites and SUMO-interacting motifs (SIMs). For predictor training, we adopted three machine learning algorithms, penalized logistic regression (PLR), a deep neural network (DNN), and a transformer, and used 52 404 nonredundant SUMOylation sites in 8262 proteins and 163 SIMs in 102 proteins. To further increase the accuracy of predicting SUMOylation sites, a pretraining model was first constructed using 145 545 protein lysine modification sites, followed by transfer learning to fine-tune the model. GPS-SUMO 2.0 exhibited greater accuracy in predicting SUMOylation sites than did other existing tools. For users, one or multiple protein sequences or identifiers can be input, and the prediction results are shown in a tabular list. In addition to the basic statistics, we integrated knowledge from 35 public resources to annotate SUMOylation sites or SIMs. The GPS-SUMO 2.0 server is freely available at https://sumo.biocuckoo.cn/. We believe that GPS-SUMO 2.0 can serve as a useful tool for further analysis of SUMOylation and SUMO interactions.
Graphical Abstract
Graphical Abstract.

Introduction
Small ubiquitin-like modifier (SUMO) proteins are highly conserved ubiquitin-like (UBL) proteins that play essential roles in regulating a variety of biological processes, ranging from gene expression and chromatin remodelling to cellular dynamics and plasticity (1–3). In eukaryotic cells, SUMOs exhibit regulatory functions through covalently attaching to specific lysine residues in substrates or by noncovalently binding to proteins that consist of SUMO-interacting motifs (SIMs) (2–4). Previous studies have demonstrated that the lysine residues of SUMO acceptor sites are frequently embedded in the consensus motif ψ–K–X–E (ψ, a hydrophobic amino acid such as A, I, L, M, P, F, V or W; X, any amino acid residue) (5–10). Also, SUMOylation provides a noncovalent binding site for reader proteins comprising SIMs, which in turn shapes the function of SUMOylated substrates (2,9). The dysfunction of SUMOs is closely related to the occurrence of numerous types of human diseases, such as neurodegenerative diseases, autoimmune diseases, and cancers (11–13). Thus, the identification of SUMOylation sites and SIMs provides insight into the important roles of SUMOs in cellular, physiological and pathological processes (1,2), and facilitates the exploration of potential therapeutic targets for disease treatment (1,3).
In addition to the use of experimental screening and identification methods, a series of computational prediction tools have been constructed to provide useful information for efficiently identifying SUMOylation sites and SIMs. From 2006 to 2009, we developed the group-based prediction system (GPS) algorithm SUMOsp (14) and its update SUMOsp 2.0 (15) for the prediction SUMOylation sites. In 2014, we incorporated the SIM inference module and released the updated algorithm GPS-SUMO to predict both SUMOylation sites and SIMs (3). In addition, other reliable tools, such as MusiteDeep (16), ResSUMO (17), JASSA (18), and SUMOplot (http://www.abgent.com/sumoplot), have been developed for the study of lysine modification by SUMO conjugation (Supplementary Table S1). In particular, MusiteDeep and ResSUMO adopt machine learning approaches to directly learn informative features for in silico prediction of SUMOylation sites (16,17). With the progress of high-throughput proteomics, a large number of lysine-modified substrates and sites have been characterized (5–7,19,20). In our developed database CPLM 4.0, there are more than 590 000 lysine modification sites, which contain >53 000 SUMOylation sites (19). Recently, we pretrained a foundation model for general phosphorylation sites (p-sites) and fine-tuned each kinase-specific predictor for p-sites (21). Considering the large amount of accumulated protein lysine modification (PLM) data, it's not known whether pretraining a general model of lysine-modified sites followed by fine-tuning is helpful for computational identification of SUMOylation sites.
In this study, we released an updated online tool, GPS-SUMO 2.0, to predict SUMOylation sites and SIMs. We trained the model with a nonredundant dataset of 52 404 SUMOylation sites in 8262 proteins and 163 SIMs in 102 proteins. For the training of the predictor, three machine learning approaches, namely, the transformer, DNN and PLR approaches, were adopted, and contextual features, seven types of sequence features and three types of structural features were integrated. To further improve the accuracy of the computational prediction of SUMOylation sites, a foundation model was pretrained utilizing 145 545 nonredundant lysine modification sites in 38 069 proteins, followed by transfer learning to fine-tune the model. In comparison to other available tools, we carefully compiled an independent dataset not used in training. This dataset includes 6665 known SUMOylation sites as positive data and 71 248 negative sites. Our developed tool has demonstrated increased accuracy for SUMOylation prediction. In the web interface, all users can submit one or multiple protein sequences or identifiers, and the prediction results are presented as a tabular list. In addition, we further implemented 35 public resources for the annotation of SUMOylation sites or SIMs, including but not limited to experimental evidence, physical interactions, 3D structures, and disorder propensities. Overall, we anticipate that GPS-SUMO 2.0 could be useful and convenient for studying SUMOylation and SUMO interactions.
Materials and methods
Data collection and preparation
First, we downloaded 53495 experimentally identified SUMOylation sites in 11 705 proteins from CPLM 4.0 (19). In addition, 16 425 experimentally identified SUMOylation sites were collected from 12 public databases, including dbPTM (22), qPTM (23), iPTMnet (24), PhosphoSitePlus (25), ProteomeScout (26), mUbiSiDa (27), HPRD (28), ActiveDriverDB (29), VPTMdb (30), PTMcode v2 (31), UniProt (32) and BioGRID (33). Moreover, a series of keywords, including ‘SUMO’, ‘SUMOylation’, ‘(SIM or SBD or SBM) and SUMO’, were used to search the published literature from 2014 to 2022 in PubMed, and 5108 additional SUMOylation sites were obtained (Figure 1A). With the integration of the datasets in GPS-SUMO 1.0, a total of 74 927 SUMOylation sites and 176 SIMs in 14 809 proteins from 13 species were ultimately obtained. We employed the widely used clustering program CD-HIT (34) to avoid homology redundancy with a sequence similarity threshold of 40% (Figure 1B). Our nonredundant benchmark dataset included 59 069 SUMOylation sites and 163 SIMs (Supplementary Table S2).
Figure 1.

The procedure of the development of GPS-SUMO 2.0, including data collection, data integration, feature encoding as well as model construction. (A) Data preparation of SUMOylation sites and SIMs curated from the literature and public databases. (B) Data integration. The collected SUMOylaiton sites and SIMs were merged, and the nonredundant datasets of modification sites and SIMs were generated using CD-HIT. (C) Feature encoding of the GPS-SUMO 2.0 algorithm. In total, 11 types of sequence-encoded features were used, including the contextual feature, 7 sequence features and 3 structural features. (D) The model construction methods used for GPS-SUMO 2.0. Three machine learning approaches, including transformer, DNN and PLR, were used for the construction of predictive models.
Next, we obtained 39 938 nonredundant sites exclusively SUMOylated under stress conditions, such as SUMO protease inhibition, proteasome inhibition and heat shock (5–7). This dataset was used to train an additional model for predicting SUMOylation sites under stress conditions. From previous studies (6,7), we obtained the relative abundance values of 40 765 SUMOylation sites, as well as their fractional protein intensity values under standard growth conditions. The relative abundance value is a SUMO site score, which has been calculated based on multiple parameters such as the Andromeda score, delta score, and localization delta score (6,7). The average SUMO site scores were determined for sites quantified in both studies, and 34 950 SUMOylation sites were reserved after redundancy clearance by CD-HIT (34). These SUMOylation sites were ranked based on SUMO site scores, and used for finetuning the default PLR model.
Then, we defined the SUMOylation site peptide SSP(m, n) as a lysine residue with m residues upstream and n residues downstream. We utilized SSP (30,30), a SUMOylation residue with 30 upstream residues and 30 downstream residues, for training. The SSP (30,30) items around experimentally identified SUMOylation sites were considered as positive data, while those around other lysine residues were treated as negative data. The training data includes 52404 positive sites in 8262 proteins (Supplementary Table S2A), and the independent test set that was not used during training includes 6665 positive sites (Supplementary Table S2B). As previously described (3), most of SIMs follow a hydrophobic motif of [IVL]{3, 5}, and a defined SIM(m, n) represents that a SIM core was flanked by m residues upstream and n residues downstream, respectively. Similarly, 163 known SIM (30,30) items were taken as positive data, and the remaining items around putative SIMs were taken as negative data (Supplementary Table S2C).
Performance evaluation measurements
To evaluate the accuracy of GPS-SUMO 2.0, true positive (TP), true negative (TN), false positive (FP), and false negative (FN) values were measured. Then, we calculated five commonly used measurements, sensitivity (Sn), specificity (Sp), accuracy (Ac), precision (Pr) and the Mathew correlation coefficient (MCC) separately, as follows:
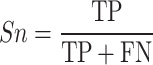 |
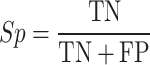 |
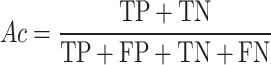 |
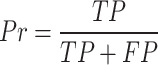 |
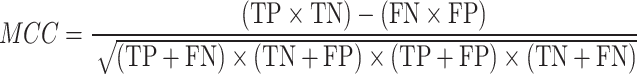 |
As we implemented 11 features in GPS-SUMO 2.0, 10-fold cross-validation was performed for each feature. In addition, 4-, 6-, 8- and 10-fold cross-validations were conducted to evaluate the accuracy and robustness of the final predictor. For the comparison of GPS-SUMO 2.0 with other existing tools, we used an independent test dataset with 6665 positive SUMOylation sites. The receiver operating characteristic (ROC) curve was generated based on the Sn and 1 − Sp scores, and the area under the curve (AUC) was calculated (Supplementary Table S3).
Feature encoding and model training
In previous studies, we designed and developed a series of hybrid learning frameworks to integrate multiple protein features to improve the prediction accuracy (21,35,36). Next, we utilized 11 features of SSP (30,30) to increase the prediction accuracy (Figure 1C). In this study, we first chose 145 545 nonredundant lysine modification sites in 38 069 proteins from CPLM 4.0 (19) to pretrain a transformer-based model. The model pretraining was carried on the ORISE Supercomputer. The HPC platform is based on a CPU+ Accelerator heterogeneous architecture. On each node, a 4-way 8-core X86 CPU with 128 GB memory is connected to four general-purpose GPU-like accelerators with 32 PCle buses while every accelerator accesses the CPU through direct memory access (DMA) and the nodes are linked by a high-speed network.
Next, the pretrained model was fine-tuned using SUMOylation site datasets. This transformer-based model was used for encoding the contextual information of SSP (30,30). Moreover, we encoded seven types of sequence-based features, namely, the GPS method (21), composition of k-spaced amino acid pairs (CKSAAPs), orthogonal binary coding (OBC), AAindex, position-specific scoring matrix (PSSM) and autocorrelation functions (ACFs), as well as three types of structural features, namely, secondary structure (SS), accessible surface area (ASA) and backbone torsion angles (BTAs), of SSP (30,30) items (Figure 1C). Then, for each individual feature, a 4-layer DNN model was constructed for training and prediction. The DNN model outputs a score to indicate the possibility of SUMOylation at lysine residues. Finally, we integrated the scores of the 11 features via the PLR model implemented in scikit-learn v1.0.2 with the ridge (L2) penalty and the ‘liblinear’ solver. The final score was calculated an indicator to estimate the probability of SUMOylation. Both transformer and DNN models were implemented in Keras 2.10.0 based on Tensorflow 2.10.0. For the prediction of SIMs, the same transformer framework and feature integration methods were adopted on SIM (30,30) for model training (Figure 1D).
For convenience, we also implemented the default model trained by PLR, and further finetuned by gene set enrichment analysis (GSEA) method (37,38), only using the GPS feature. First, the AUC value of the initial PLR model was calculated as 0.6930 using 10-fold cross-validation. Then, this model was used to score the 52404 positive sites in the training dataset, and the results were ranked by prediction values. By testing, 3495 (top 10%) sites with the highest SUMO site scores were taken as a gene set for enrichment analysis. Using a python package of GSEAPY (https://pypi.org/project/gseapy/) (37,38), the initial normalized enrichment score (NES) was calculated as 7.691. Next, weights of individual GPS features were randomly increased or decreased, and the manipulation was adopted if NES increased without decreasing the AUC value. Such a procedure was interactively performed with >10 000 times, until NES was not increased any longer. The final NES value was determined as 11.242, with an AUC value of 0.6963 (Supplementary Figure S1A).
Integrated annotations
We integrated several other tools to annotate our prediction results on the web site. A web tool, IceLogo (39) (http://iomics.ugent.be/icelogoserver/), was used for motif analysis. This tool shows enriched as well as depleted amino acids. We uploaded all positive SSP (30,30) and SIM (30,30) items to IceLogo, and sequence logos of SUMOylation sites and SIMs were generated, respectively. Moreover, we employed 3Dmol.js (40) to display the three-dimensional structure and predicted sites of the proteins. The disorder propensity values of the proteins were evaluated using IUPred (41). The ASA of the amino acids and the secondary structure were measured by NetSurfP 3.0 (42). Besides the basic statistics, the knowledge of 35 public resources was integrated (Supplementary Table S4).
Web server implementation
Up to 6 predictors were developed for the online service of GPS-SUMO 2.0: (i) PLR + GSEA, prediction based on PLR training and GSEA finetuning with the GPS feature; (ii) transformer, prediction based on transformer with the contextual information of sequences; (iii) all, prediction based on all models with all features; (iv) species-specific, species-specific prediction based on all models with all features; (v) comprehensive, prediction based on all models with all features and additional annotations of secondary structure and surface accessibility; (vi) stress conditions, prediction based on PLR, using 39 938 nonredundant SUMOylation sites identified under various stress conditions for training (Supplementary Figure S1B).
For each predictor, we defined high, medium and low thresholds using Sp values of 95%, 90% and 85%, respectively. The high threshold was selected as the default value. The web frontend was built with PHP 5.4 and JQuery 1.4.4, and the backend was built using Python 3.8. For visualization, charts were generated using JavaScript libraries. The online service GPS-SUMO 2.0 can be used on multiple platforms and browsers, including Google Chrome 107.0.5304.107, Mozilla Firefox 107.0.1, Safari 13.1.2 and Microsoft Edge 108.0.1462.46.
Results
Performance evaluation and comparison
Computational prediction of SUMOylation sites and SIMs in proteins provides a helpful means for studying the molecular mechanisms and regulatory roles of SUMOylation and SUMO interactions. Of note, the prediction performance heavily relies on the quantity and quality of experimentally identified SUMOylation sites or SIMs. Recent progresses in SUMO proteomic profiling have produced tens of thousands of SUMOylation sites, using the high-resolution tandem mass spectrometry (MS/MS) (5–7). For example, Hendriks et al. quantitatively identified up to 40 765 SUMOylation sites of 6747 proteins in human cell lines under standard or stress conditions (6). These newly identified SUMOylation sites will be highly valuable for improving the prediction performance. In GPS-SUMO, we only took 912 nonredundant SUMOylation sites for model training (3). For development of GPS-SUMO 2.0, we in total obtained 59069 nonredundant SUMOylation sites, with a 64.8-fold increase in the benchmark data set. A detailed comparison of GPS-SUMO 2.0 and our previous releases was shown in Supplementary Table S5.
In this study, three machine learning methods, including transformer, DNN and PLR, were employed for model training, and integrating the contextual information of sequences, seven types of sequence features and three types of structural features. To evaluate the prediction accuracy, the 10-fold cross-validation was individually conducted for each feature. The AUC values for the prediction of SUMOylation sites ranged from 0.5220 (ACFs) to 0.7621 (DNN) (Figure 2A), and the AUC values for predicting SIMs ranged from 0.5299 (ASA) to 0.9287 (Transformer) (Figure 2B). Moreover, n-fold cross-validations were adopted for the evaluation of the final prediction models. Using 4-, 6-, 8- and 10-fold cross-validations, AUC values for predicting SUMOylation sites ranged from 0.8933 to 0.8988 (Figure 2C), and AUC values for predicting SIMs ranged from 0.9563 to 0.9583 (Figure 2D). To further explore the performance of GPS-SUMO 2.0, a comparison was performed unbiasedly with other publicly available SUMOylation site predictors, including GPS-SUMO (3), MusiteDeep (16), ResSUMO (17), SUMOplot, SUMOsp 2.0 (15) and JASSA (18), using the independent dataset curated in this study. For JASSA, Sn and Sp values were calculated separately when high or low cut-off thresholds were selected (Figure 2E). Our analyses demonstrated that GPS-SUMO 2.0 exhibited better performance than did the other predictive tools (Figure 2E).
Figure 2.

Performance evaluation and comparison of GPS-SUMO 2.0. (A) The performance evaluation of the predictors of SUMOylation sites using various algorithms and features. The ROC curves and AUC values were separately presented for 13 predictive models. The GPS-SUMO 2.0 predictor was trained using 3 machine learning methods and 11 features. The PLR + GSEA model was constructed by PLR training and GSEA finetuning with the GPS feature. The other 11 models was individually trained by using DNN algorithm and single feature. (B) The performance measurement of SIM predictors utilizing different machine learning methods and features. The ROC curves were illustrated and AUC values were calculated for each predictive model. (C) The performance evaluation of SUMOylation predictor using n-fold cross-validations. For the evaluation of predictive models, 4-, 6-, 8-, 10-fold cross-validations were used, and the ROC curves and AUC values were presented. (D) The 4-, 6-, 8-, 10-fold cross-validations were utilized to evaluate the performance of constructed model for the prediction of SIMs. (E) The performance comparison between GPS-SUMO 2.0 and other existing predictors, including ResSUMO, MusiteDeep, SUMOplot, SUMOsp 2.0, GPS-SUMO and JASSA. (F) The evaluation of 11 types of features contributing to GPS-SUMO 2.0 by measuring the SHAP score for each feature.
Next, to investigate the differential contributions of the 11 features in GPS-SUMO 2.0, a well-characterized approach, namely, the SHapley Additive exPlanation (SHAP) approach (43,44), was adopted for the interpretation of the prediction model. The analysis revealed that all 11 features contributed to our developed predictors (Figure 2F). In particular, we observed that the feature encoded by the GPS algorithm had the highest SHAP score, indicating that obtaining information on sequence similarities is essential for in silico identification of modified sites. Notably, the transformer-encoded feature also had an important contribution to GPS-SUMO 2.0, suggesting that contextual information is useful for understanding the underlying mechanism of SUMOylation and SUMO interactions.
Usage of the GPS-SUMO 2.0 web server
In GPS-SUMO 2.0, we implemented 6 predictors that could be accessed at the advance page (https://sumo.biocuckoo.cn/advanced.php) (Supplementary Figure S1B). For convenience, the predictor based on PLR + GSEA with the GPS feature was provided at the home page, for its high speed and a still promising accuracy (Supplementary Figure S1C). For inputting, one or multiple protein sequences in FASTA format, or one or multiple UniProt accession numbers are acceptable. For all the predictive interfaces, there are two threshold panels for SUMOylation site and SIM prediction at the bottom of the web interface. The prediction results and additional annotations can be accessed after clicking the ‘Submit’ button (Figure 3A). After the predictions are generated, the results are illustrated in tabular form with 20 rows per page. Users are able to select the number of pages below the table when searching or browsing specific prediction results. The results table contains 9 types of informative resources, namely, ‘Name’, ‘Position’, ’Peptides’, ‘Score’, ‘Cut-off’, ‘Type’, ‘Source’, ‘PPI’ and ‘Logo’. The results presented in the table can be sorted by clicking on each of the column names (Figure 3B).
Figure 3.

The usage of the GPS-SUMO 2.0 web server. (A) The interface of sequence submission. The users can input the protein sequence in FASTA format or enter UniProt accession number, and select 3 different thresholds for the prediction of modification sites and SIMs. (B) The presentation of prediction results of the example. In tabular list, the predictive results contain the position of SUMOylation site or SIMs, the prediction score, cut-off value, identification by experimental or computational method, and PPI information. (C) The comprehensive annotations of the prediction results. The number of SUMO modification sites and SIMs predicted by GPS-SUMO 2.0 were presented, the location of SUMOylation sites were illustrated in 3D structure derived from PDB database, the ASA score and disorder score of SUMOylation sites were calculated.
In the ‘Source’ column, ‘Exp’ denotes that experimentally validated evidence of SUMOylation sites can be obtained by linking to the CPLM 4.0 database (19). Similarly, by integrating 13 public protein−protein interaction (PPI) databases (28,33,45–55), we provided detailed information on the predicted PPI network. For the prediction of SUMOylation sites, information regarding the interaction between a specific substrate and the SUMO E3 ligase was presented. If a reader protein contains a SIM pattern, we annotated it with SUMO proteins in the ‘PPI’ column. In default mode, the distribution of predictive results in the statistical chart is shown, and 3D structural information for potential modification sites of the substrate is visualized by 3Dmol.js if available (40). The disorder propensity scores of residues determined by IUPred (41) are also displayed in the form of a line chart. When selecting the comprehensive mode, the surface accessibility and secondary structures, including the α-helix, β-strand, and coil, are evaluated (Figure 3C). All the prediction results can be downloaded in .txt or .xlsx format. The ‘Export’ button beside the images allows the user to obtain a .png file. We also provided detailed descriptions on the ‘USER GUIDE’ page for all users to learn how to utilize the online services and interpret all the results.
An example of using GPS-SUMO 2.0
To demonstrate the usage of GPS-SUMO 2.0, the canonical sequence of human PTEN protein (UniProt ID: P60484) was selected as an example. PTEN has been characterized as a tumour suppressor gene and is involved in orchestrating various cellular physiological processes, including proliferation, survival, and energy metabolism. Previous studies revealed that post-translational modifications (PTMs), such as SUMOylation and ubiquitination, are essential for the structural and functional integrity of the PTEN protein (56,57), and crosstalk between PTEN SUMOylation and ubiquitination was discovered (58).
Here, we utilized the predictor based on all models with all features (The third option ‘All’ in the advanced page) to predict 12 potential SUMOylation sites in the PTEN protein, including 4 reported SUMOylation sites, K164, K254, K266 and K289 (Supplementary Table S2). The SUMOylation at K164 was identified from a SUMO proteomic profiling (6), whereas K266, K254 and K289 were identified as SUMOylation sites to regulate the nuclear localization and function of PTEN (58,59). In the results, 6 predicted SUMOylation sites had prediction scores greater than 0.9, including 3 reported sites, K266, K254 and K289 (Figure 4A, Supplementary Table S6). We retrieved the modification information regarding K66, K102 and K322 of PTEN from CPLM 4.0 (19), and found that only K66 was previously identified as a lysine modification site. Thus, our predictions indicated that this lysine residue might also be SUMOylated.
Figure 4.

The computational prediction of human PTEN protein. (A) The illustration of SUMOylation sites on PTEN predicted by using comprehensive predictors with all features. (B) The annotation of PTEN SUMOylation sites. The ASA score and disorder score were measured for the modification sites, including K66, K254, K266 and K289. (C) The calculated disorder score, ASA score and zoomed 3D structure of K66. (D) The database source information for annotating K66. The K66 site of human PTEN protein was identified to be modified by ubiquitination, according to the annotations in CPLM 4.0 database.
Next, we annotated the K66 residue, including the ASA score, disorder score, and zoomed 3D structure (Figure 4B, C). Additionally, experimental data from CPLM 4.0 were integrated (Figure 4D). According to the annotations, in comparison with K254 and K289, K66 exhibited a higher surface accessibility, suggesting that this residue might be easier to conjugate to SUMO modifiers. In particular, previous studies have revealed that K66 is ubiquitinated (60,61), and this ubiquitination event reduces the protein stability of PTEN in cancers (61,62). Thus, covalent conjugation of SUMO to K66 might dynamically compete with ubiquitination at the same residue, and regulate the PTEN stability at the protein level. Taken together, GPS-SUMO 2.0 predicts a highly potential SUMOylation site, K66, on PTEN, and provides useful clues for further experimental consideration.
To further demonstrate the usage of GPS-SUMO 2.0, 6 additionally reported SUMOylated substrates were scored using PLR + GSEA, transformer, and comprehensive models, respectively (Supplementary Table S6). Experimentally identified SUMOylation sites and/or SIMs were indicated for comparison.
Discussion
SUMO proteins are members of ubiquitin-like proteins that regulate cellular processes either by SUMOylation or SUMO interactions (1–4). Besides experimental identification, a variety of computational predictors have been developed for prioritization of potential SUMOylation sites and SIMs (Supplementary Table S1). Advances in both machine learning and SUMO proteomic profiling provided powerful methods and high-quality data sets for improving the prediction accuracy, and the predictions could provide helpful candidates for further experimental consideration.
In this study, we applied a pipeline of pretraining followed by transfer learning to develop 6 predictors for computationally identifying SUMOylation sites and SIMs, by combining much larger datasets with 3 machine learning algorithms and 11 features. We compiled a nonredundant benchmark data set containing 59069 SUMOylation sites and 163 SIMs (Supplementary Table S2). For training SUMOylation models, 52 404 SUMOylation sites were taken as the training data, while the remaining 6665 SUMOylation sites were taken as an independent test set. For each data set, lysine residues not reported to be SUMOylated in the same proteins were taken as negative data. It should be noted that a considerable proportion of the negative sites might be truly SUMOylated and yet to be identified in experiments. Thus, the prediction performance of predictive models might be considerably underestimated.
From previous studies (5–7), we collected 39938 nonredundant sites exclusively SUMOylated under stress conditions, occupying 67.6% of the 59 069 collected sites (Supplementary Figure S2A). To test whether the characteristics are different for SUMOylation sites under stress conditions and other conditions, we used the remaining 19 131 sites to train a model, and used the 39 938 stress-induced sites for testing. By comparison, the 10-fold cross-validation using the 19 131 sites achieved an AUC value of 0.7409, whereas the AUC value on the 39 938 stress-induced sites reduced to 0.6026 (Supplementary Figure S2B). We also trained a model using the 39938 stress-induced sites, and adopted the remaining 19 131 sites for testing. Again, the AUC value was decreased from 0.7294 (10-fold cross-validation) to 0.6692 (Independent testing) (Supplementary Figure S2C). Thus, our results demonstrated that stress-induced SUMOylation sites might be considerably different from SUMOylation sites under other conditions (usually the standard condition). In the advanced page, we implemented an additional predictor for identifying potentially stress-induced SUMOylation sites.
Previously, Hendriks et al. provided the information about the relative abundance of SUMOylation site usage per protein (6,7). It could be expected that the SUMOylation sites that are highly used might get a much higher score compared to the sites that are poorly used. To test this hypothesis, the 34 950 SUMOylation sites were equally separated into a high relative abundance group and a low relative abundance group, based on the median of SUMO site scores. These sites were scored using PLR + GSEA, transformer, and comprehensive models, and statistical analyses demonstrated that highly used SUMOylation sites got higher scores than poorly used sites (Supplementary Figure S3A–C, P< 0.001). In addition, we separated the 59069 SUMOylation sites into a consensus group with 7702 sites that follow the ψ–K–X–E motif, and a non-consensus group with 51 367 sites (Supplementary Table S2). Again, PLR + GSEA, transformer, and comprehensive models of GPS-SUMO 2.0 were used for scoring, respectively. The statistical results demonstrated that consensus sites generally achieved higher scores than non-consensus sites (Supplementary Figure S3D–F, P< 0.001). Taken together, GPS-SUMO 2.0 and following analyses might be helpful for biologists to further investigate the underlying mechanisms of SUMOylation and SUMO interactions.
In this study, although we have used three machine learning methods and large-scale datasets to train models for prediction of SUMOylation sites, it should be noted that there is still a partial mismatch between experimentally identified SUMOylation sites and SUMOylation sites predicted by GPS-SUMO 2.0, owing to the complexity of SUMO regulation. Thus, considering the limitation of our predictors, great efforts will be made to maintain and improve GPS-SUMO 2.0 for further increasing the accuracy of predicting SUMOylation sites as well as SIMs in the future. Currently, technologies for high-throughput identification of SUMOylation sites and SUMO interactions are being developed. Thus, we will expand and curate the training dataset as new SUMOylation sites and SIMs are discovered. In addition, more encoded features will be incorporated to improve the prediction performance. With the rapid development of cutting-edge AI algorithms, various novel state-of-the-art techniques will be adopted to test whether these approaches facilitate improvements in prediction accuracy. Moreover, the relative abundance of SUMOylation site usage per protein is an informative feature and orthogonal to sequence and structural features around SUMOylation sites (6,7). Indeed, incorporating this feature for model finetuning markedly increased the weight on experimentally validated sites with higher relative abundance values, as well as the weight to distinguish consensus SUMOylation sites following the ψ–K–X–E motif from non-consensus sites. In addition, given that the interplay between SUMOylation and other PTMs, especially ubiquitinated modifications, has been shown to play a role in regulating numerous cellular processes, the relationship between SUMOylation and other types of PTMs will be considered in the development of an updated version of the GPS-SUMO. In summary, we anticipate that GPS-SUMO 2.0 will be helpful for studying SUMOylation and SUMO interactions in signal transduction and cellular processes.
Supplementary Material
Acknowledgements
We thank all the users for their valuable comments and communications with us when using the GPS algorithm for the prediction of SUMOylation sites and SIMs. The manuscript was edited by Springer Nature Author Services (SNAS) prior to submission.
Contributor Information
Yujie Gou, Department of Bioinformatics and Systems Biology, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, 430074, China; Key Laboratory of Molecular Biophysics of Ministry of Education, Hubei Bioinformatics and Molecular Imaging Key Laboratory, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, 430074, China.
Dan Liu, Department of Bioinformatics and Systems Biology, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, 430074, China; Key Laboratory of Molecular Biophysics of Ministry of Education, Hubei Bioinformatics and Molecular Imaging Key Laboratory, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, 430074, China.
Miaomiao Chen, Department of Bioinformatics and Systems Biology, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, 430074, China; Key Laboratory of Molecular Biophysics of Ministry of Education, Hubei Bioinformatics and Molecular Imaging Key Laboratory, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, 430074, China.
Yuxiang Wei, Department of Bioinformatics and Systems Biology, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, 430074, China; Key Laboratory of Molecular Biophysics of Ministry of Education, Hubei Bioinformatics and Molecular Imaging Key Laboratory, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, 430074, China.
Xinhe Huang, Department of Bioinformatics and Systems Biology, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, 430074, China; Key Laboratory of Molecular Biophysics of Ministry of Education, Hubei Bioinformatics and Molecular Imaging Key Laboratory, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, 430074, China.
Cheng Han, Department of Bioinformatics and Systems Biology, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, 430074, China; Key Laboratory of Molecular Biophysics of Ministry of Education, Hubei Bioinformatics and Molecular Imaging Key Laboratory, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, 430074, China.
Zihao Feng, Department of Bioinformatics and Systems Biology, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, 430074, China; Key Laboratory of Molecular Biophysics of Ministry of Education, Hubei Bioinformatics and Molecular Imaging Key Laboratory, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, 430074, China.
Chi Zhang, Department of Bioinformatics and Systems Biology, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, 430074, China; Key Laboratory of Molecular Biophysics of Ministry of Education, Hubei Bioinformatics and Molecular Imaging Key Laboratory, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, 430074, China.
Teng Lu, Computer Network Information Center, Chinese Academy of Sciences, Beijing, 100190, China.
Di Peng, Department of Bioinformatics and Systems Biology, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, 430074, China; Key Laboratory of Molecular Biophysics of Ministry of Education, Hubei Bioinformatics and Molecular Imaging Key Laboratory, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, 430074, China.
Yu Xue, Department of Bioinformatics and Systems Biology, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, 430074, China; Key Laboratory of Molecular Biophysics of Ministry of Education, Hubei Bioinformatics and Molecular Imaging Key Laboratory, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, 430074, China; Nanjing University Institute of Artificial Intelligence Biomedicine, Nanjing, 210031, China.
Data availability
The data underlying this article are available in the article and in its online supplementary material. GPS-SUMO 2.0 is freely available for all users at https://sumo.biocuckoo.cn/, and the related sources used in this study are available in Zenodo at https://dx.doi.org/10.5281/zenodo.10987312.
Supplementary data
Supplementary Data are available at NAR Online.
Funding
Funding for open access charge: National Key R&D Program of China [2022YFC2704304, 2021YFF0702000]; Natural Science Foundation of China [32341020, 32341021, 31930021]; Hubei Innovation Group Project [2021CFA005]; Hubei Province Postdoctoral Outstanding Talent Tracking Support Program, Strategic Priority Research Program of CAS [XDB0500101]; Research Core Facilities for Life Science (HUST).
Conflict of interest statement. None declared.
References
- 1. Geiss-Friedlander R., Melchior F. Concepts in sumoylation: a decade on. Nat. Rev. Mol. Cell Biol. 2007; 8:947–956. [DOI] [PubMed] [Google Scholar]
- 2. Vertegaal A.C.O. Signalling mechanisms and cellular functions of SUMO. Nature reviews. Mol. Cell Biol. 2022; 23:715–731. [DOI] [PubMed] [Google Scholar]
- 3. Zhao Q., Xie Y., Zheng Y., Jiang S., Liu W., Mu W., Liu Z., Zhao Y., Xue Y., Ren J. GPS-SUMO: a tool for the prediction of sumoylation sites and SUMO-interaction motifs. Nucleic Acids Res. 2014; 42:W325–W330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. González-Prieto R., Eifler-Olivi K., Claessens L.A., Willemstein E., Xiao Z., Talavera Ormeno C.M.P., Ovaa H., Ulrich H.D., Vertegaal A.C.O. Global non-covalent SUMO interaction networks reveal SUMO-dependent stabilization of the non-homologous end joining complex. Cell Rep. 2021; 34:108691. [DOI] [PubMed] [Google Scholar]
- 5. Hendriks I.A., D'Souza R.C., Yang B., Vries V.-d., M. M., Vertegaal A.C Uncovering global SUMOylation signaling networks in a site-specific manner. Nat. Struct. Mol. Biol. 2014; 21:927–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hendriks I.A., Lyon D., Young C., Jensen L.J., Vertegaal A.C., Nielsen M.L. Site-specific mapping of the human SUMO proteome reveals co-modification with phosphorylation. Nat. Struct. Mol. Biol. 2017; 24:325–336. [DOI] [PubMed] [Google Scholar]
- 7. Hendriks I.A., Lyon D., Su D., Skotte N.H., Daniel J.A., Jensen L.J., Nielsen M.L. Site-specific characterization of endogenous SUMOylation across species and organs. Nat. Commun. 2018; 9:2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Matic I., Schimmel J., Hendriks I.A., van Santen M.A., van de Rijke F., van Dam H., Gnad F., Mann M., Vertegaal A.C. Site-specific identification of SUMO-2 targets in cells reveals an inverted SUMOylation motif and a hydrophobic cluster SUMOylation motif. Mol. Cell. 2010; 39:641–652. [DOI] [PubMed] [Google Scholar]
- 9. Flotho A., Melchior F. Sumoylation: a regulatory protein modification in health and disease. Annu. Rev. Biochem. 2013; 82:357–385. [DOI] [PubMed] [Google Scholar]
- 10. Impens F., Radoshevich L., Cossart P., Ribet D Mapping of SUMO sites and analysis of SUMOylation changes induced by external stimuli. Proc. Nat. Acad. Sci. U.S.A. 2014; 111:12432–12437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chang H.M., Yeh E.T.H. SUMO: from bench to bedside. Physiol. Rev. 2020; 100:1599–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Eifler K., Vertegaal A.C.O. SUMOylation-mediated regulation of cell cycle progression and cancer. Trends Biochem. Sci. 2015; 40:779–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Demel U.M., Boger M., Yousefian S., Grunert C., Zhang L., Hotz P.W., Gottschlich A., Kose H., Isaakidis K., Vonficht D. et al. Activated SUMOylation restricts MHC class I antigen presentation to confer immune evasion in cancer. J. Clin. Invest. 2022; 132:e152383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xue Y., Zhou F., Fu C., Xu Y., Yao X. SUMOsp: a web server for sumoylation site prediction. Nucleic Acids Res. 2006; 34:W254–W257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ren J., Gao X., Jin C., Zhu M., Wang X., Shaw A., Wen L., Yao X., Xue Y. Systematic study of protein sumoylation: development of a site-specific predictor of SUMOsp 2.0. Proteomics. 2009; 9:3409–3412. [DOI] [PubMed] [Google Scholar]
- 16. Wang D., Liu D., Yuchi J., He F., Jiang Y., Cai S., Li J., Xu D MusiteDeep: a deep-learning based webserver for protein post-translational modification site prediction and visualization. Nucleic Acids Res. 2020; 48:W140–W146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhu Y., Liu Y., Chen Y., Li L. ResSUMO: a deep learning architecture based on residual structure for prediction of lysine SUMOylation sites. Cells. 2022; 11:2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Beauclair G., Bridier-Nahmias A., Zagury J.F., Saïb A., Zamborlini A. JASSA: a comprehensive tool for prediction of SUMOylation sites and SIMs. Bioinformatics. 2015; 31:3483–3491. [DOI] [PubMed] [Google Scholar]
- 19. Zhang W., Tan X., Lin S., Gou Y., Han C., Zhang C., Ning W., Wang C., Xue Y. CPLM 4.0: an updated database with rich annotations for protein lysine modifications. Nucleic Acids Res. 2022; 50:D451–D459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hendriks I.A., Vertegaal A.C. A comprehensive compilation of SUMO proteomics. Nat. Rev. Mol. Cell Biol. 2016; 17:581–595. [DOI] [PubMed] [Google Scholar]
- 21. Chen M., Zhang W., Gou Y., Xu D., Wei Y., Liu D., Han C., Huang X., Li C., Ning W. et al. GPS 6.0: an updated server for prediction of kinase-specific phosphorylation sites in proteins. Nucleic Acids Res. 2023; 51:W243–W250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huang K.Y., Lee T.Y., Kao H.J., Ma C.T., Lee C.C., Lin T.H., Chang W.C., Huang H.D. dbPTM in 2019: exploring disease association and cross-talk of post-translational modifications. Nucleic Acids Res. 2019; 47:D298–D308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yu K., Wang Y., Zheng Y., Liu Z., Zhang Q., Wang S., Zhao Q., Zhang X., Li X., Xu R.H. et al. qPTM: an updated database for PTM dynamics in human, mouse, rat and yeast. Nucleic Acids Res. 2023; 51:D479–D487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Huang H., Arighi C.N., Ross K.E., Ren J., Li G., Chen S.C., Wang Q., Cowart J., Vijay-Shanker K., Wu C.H. iPTMnet: an integrated resource for protein post-translational modification network discovery. Nucleic Acids Res. 2018; 46:D542–D550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hornbeck P.V., Kornhauser J.M., Latham V., Murray B., Nandhikonda V., Nord A., Skrzypek E., Wheeler T., Zhang B., Gnad F. 15 years of PhosphoSitePlus®: integrating post-translationally modified sites, disease variants and isoforms. Nucleic Acids Res. 2019; 47:D433–D441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Matlock M.K., Holehouse A.S., Naegle K.M. ProteomeScout: a repository and analysis resource for post-translational modifications and proteins. Nucleic Acids Res. 2015; 43:D521–D530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen T., Zhou T., He B., Yu H., Guo X., Song X., Sha J. mUbiSiDa: a comprehensive database for protein ubiquitination sites in mammals. PLoS One. 2014; 9:e85744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Goel R., Harsha H.C., Pandey A., Prasad T.S. Human Protein Reference Database and Human Proteinpedia as resources for phosphoproteome analysis. Mol. Biosyst. 2012; 8:453–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Krassowski M., Pellegrina D., Mee M.W., Fradet-Turcotte A., Bhat M., Reimand J. ActiveDriverDB: interpreting genetic variation in human and cancer genomes using post-translational modification sites and signaling networks (2021 update). Front. Cell Dev. Biol. 2021; 9:626821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xiang Y., Zou Q., Zhao L. VPTMdb: a viral posttranslational modification database. Brief Bioinform. 2021; 22:bbaa251. [DOI] [PubMed] [Google Scholar]
- 31. Minguez P., Letunic I., Parca L., Garcia-Alonso L., Dopazo J., Huerta-Cepas J., Bork P. PTMcode v2: a resource for functional associations of post-translational modifications within and between proteins. Nucleic Acids Res. 2015; 43:D494–D502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. UniProt C. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019; 47:D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Oughtred R., Stark C., Breitkreutz B.J., Rust J., Boucher L., Chang C., Kolas N., O’Donnell L., Leung G., McAdam R. et al. The BioGRID interaction database: 2019 update. Nucleic Acids Res. 2019; 47:D529–D541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fu L., Niu B., Zhu Z., Wu S., Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012; 28:3150–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ning W., Xu H., Jiang P., Cheng H., Deng W., Guo Y., Xue Y. HybridSucc: a hybrid-learning architecture for general and species-specific succinylation site prediction. Genomics Proteomics Bioinformatics. 2020; 18:194–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang C., Tan X., Tang D., Gou Y., Han C., Ning W., Lin S., Zhang W., Chen M., Peng D. et al. GPS-Uber: a hybrid-learning framework for prediction of general and E3-specific lysine ubiquitination sites. Brief Bioinform. 2022; 23:bbab574. [DOI] [PubMed] [Google Scholar]
- 37. Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Nat. Acad. Sci. U.S.A. 2005; 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fang Z., Liu X., Peltz G. GSEApy: a comprehensive package for performing gene set enrichment analysis in Python. Bioinformatics. 2023; 39:btac757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Maddelein D., Colaert N., Buchanan I., Hulstaert N., Gevaert K., Martens L. The iceLogo web server and SOAP service for determining protein consensus sequences. Nucleic Acids Res. 2015; 43:W543–W546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rego N., Koes D 3Dmol.js: molecular visualization with WebGL. Bioinformatics. 2015; 31:1322–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Erdős G., Pajkos M., Dosztányi Z. IUPred3: prediction of protein disorder enhanced with unambiguous experimental annotation and visualization of evolutionary conservation. Nucleic Acids Res. 2021; 49:W297–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hoie M.H., Kiehl E.N., Petersen B., Nielsen M., Winther O., Nielsen H., Hallgren J., Marcatili P. NetSurfP-3.0: accurate and fast prediction of protein structural features by protein language models and deep learning. Nucleic. Acids. Res. 2022; 50:W510–W515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yuan G.H., Wang Y., Wang G.Z., Yang L. RNAlight: a machine learning model to identify nucleotide features determining RNA subcellular localization. Brief Bioinform. 2023; 24:bbac509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lundberg S., Lee S.-I. A Unified Approach to Interpreting Model Predictions. NIPS'17: Proceedings of the 31st International Conference on Neural Information Processing Systems. 2017; 4768–4777. [Google Scholar]
- 45. Razick S., Magklaras G., Donaldson I.M. iRefIndex: a consolidated protein interaction database with provenance. BMC Bioinf. 2008; 9:405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Calderone A., Castagnoli L., Cesareni G. mentha: a resource for browsing integrated protein-interaction networks. Nat. Methods. 2013; 10:690–691. [DOI] [PubMed] [Google Scholar]
- 47. Basha O., Shpringer R., Argov C.M., Yeger-Lotem E. The DifferentialNet database of differential protein-protein interactions in human tissues. Nucleic Acids Res. 2018; 46:D522–D526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Higueruelo A.P., Jubb H., Blundell T.L. TIMBAL v2: update of a database holding small molecules modulating protein-protein interactions. Database. 2013; 2013:bat039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hu Y., Vinayagam A., Nand A., Comjean A., Chung V., Hao T., Mohr S.E., Perrimon N. Molecular Interaction Search Tool (MIST): an integrated resource for mining gene and protein interaction data. Nucleic Acids Res. 2018; 46:D567–D574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kotlyar M., Pastrello C., Sheahan N., Jurisica I. Integrated interactions database: tissue-specific view of the human and model organism interactomes. Nucleic Acids Res. 2016; 44:D536–D541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Alanis-Lobato G., Andrade-Navarro M.A., Schaefer M.H. HIPPIE v2.0: enhancing meaningfulness and reliability of protein-protein interaction networks. Nucleic Acids Res. 2017; 45:D408–D414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cowley M.J., Pinese M., Kassahn K.S., Waddell N., Pearson J.V., Grimmond S.M., Biankin A.V., Hautaniemi S., Wu J. PINA v2.0: mining interactome modules. Nucleic Acids Res. 2012; 40:D862–D865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Das J., Yu H. HINT: high-quality protein interactomes and their applications in understanding human disease. BMC Syst. Biol. 2012; 6:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li T., Wernersson R., Hansen R.B., Horn H., Mercer J., Slodkowicz G., Workman C.T., Rigina O., Rapacki K., Staerfeldt H.H. et al. A scored human protein-protein interaction network to catalyze genomic interpretation. Nat. Methods. 2017; 14:61–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Szklarczyk D., Gable A.L., Lyon D., Junge A., Wyder S., Huerta-Cepas J., Simonovic M., Doncheva N.T., Morris J.H., Bork P. et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019; 47:D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rodríguez J.A. Interplay between nuclear transport and ubiquitin/SUMO modifications in the regulation of cancer-related proteins. Semin. Cancer Biol. 2014; 27:11–19. [DOI] [PubMed] [Google Scholar]
- 57. Bassi C., Ho J., Srikumar T., Dowling R.J., Gorrini C., Miller S.J., Mak T.W., Neel B.G., Raught B., Stambolic V. Nuclear PTEN controls DNA repair and sensitivity to genotoxic stress. Science. 2013; 341:395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. González-Santamaría J., Campagna M., Ortega-Molina A., Marcos-Villar L., de la Cruz-Herrera C.F., González D., Gallego P., Lopitz-Otsoa F., Esteban M., Rodríguez M.S. et al. Regulation of the tumor suppressor PTEN by SUMO. Cell Death. Dis. 2012; 3:e393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Huang J., Yan J., Zhang J., Zhu S., Wang Y., Shi T., Zhu C., Chen C., Liu X., Cheng J. et al. SUMO1 modification of PTEN regulates tumorigenesis by controlling its association with the plasma membrane. Nat. Commun. 2012; 3:911. [DOI] [PubMed] [Google Scholar]
- 60. Hu Q., Li C., Wang S., Li Y., Wen B., Zhang Y., Liang K., Yao J., Ye Y., Hsiao H. et al. LncRNAs-directed PTEN enzymatic switch governs epithelial-mesenchymal transition. Cell Res. 2019; 29:286–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gupta A., Leslie N.R. Controlling PTEN (phosphatase and tensin homolog) stability: a dominant role for lysine 66. J. Biol. Chem. 2016; 291:18465–18473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wang K., Liu J., Li Y.L., Li J.P., Zhang R. Ubiquitination/de-ubiquitination: a promising therapeutic target for PTEN reactivation in cancer. Biochim. Biophys. Acta Rev. Cancer. 2022; 1877:188723. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in the article and in its online supplementary material. GPS-SUMO 2.0 is freely available for all users at https://sumo.biocuckoo.cn/, and the related sources used in this study are available in Zenodo at https://dx.doi.org/10.5281/zenodo.10987312.