

Summary
Background
FGFR alterations (alt) are reported across various malignancies and may act as oncogenic drivers in multiple histologies. Erdafitinib is an oral, selective pan-FGFR tyrosine kinase inhibitor with activity in FGFR-altered advanced urothelial carcinoma. We aimed to evaluate the safety and efficacy of erdafitinib in a phase 2 study of previously treated patients with FGFR-altered advanced solid tumours.
Methods
Patients were aged ≥12 years with advanced or metastatic tumours of any histology, except urothelial cancer, with predefined FGFR1–4alt (mutations/fusions per local/central testing) and disease progression on ≥1 prior line of systemic therapy and no alternative standard therapy and an Eastern Cooperative Oncology Group performance status of 0–1. Patients received once-daily oral erdafitinib (8 mg/day with provision for pharmacodynamically guided uptitration to 9 mg/day) on a continuous 21-day cycle until disease progression or intolerable toxicity. The primary endpoint was objective response rate (ORR) by independent review committee per RECIST version 1·1 and RANO. This primary analysis was conducted on the treated population of the Broad Panel Cohort. This ongoing study is registered with ClinicalTrials.gov, number NCT04083976.
Findings
RAGNAR initiated on Dec 5, 2019, and evaluated 217 erdafitinib-treated patients. 97 (45%) and 120 (55%) of the 217 patients were female and male, respectively. The data cutoff was Aug 15, 2022. At a median follow-up of 17·9 months (IQR 13·6–23·9), an ORR of 30% (95% CI 24–36) was observed in 64 of 217 patients across 16 distinct tumours. The most common grade 3–5 (all grade 3) treatment-emergent adverse events of any causality were stomatitis in 25 (12%), anaemia in 18 (8%), and palmar-plantar erythrodysaesthesia syndrome in 12 (6%) of 217 patients. The most commonly occurring serious treatment-related adverse events (grade 3 or higher) were stomatitis in 4 (2%) and diarrhoea in 2 (1%) of 217 patients. There were no treatment-related deaths.
Interpretation
RAGNAR results demonstrate clinical benefit for erdafitinib in the tumour-agnostic setting in patients with advanced solid tumours with susceptible FGFR alterations who have exhausted other treatment options. These results support the continued development of FGFR inhibitors in patients with advanced solid tumours.
Funding
Janssen Research & Development.
Introduction
The fibroblast growth factor receptor (FGFR) family plays important roles in normal cell proliferation, migration, differentiation, and survival. FGFR gene alterations, including activating mutations or fusions, can lead to constitutive activation of downstream cellular signalling and resultant oncogenesis.1–3 FGFR inhibitors are approved for the treatment of advanced or metastatic bladder cancer and cholangiocarcinoma with FGFR alterations.4–6 Erdafitinib is an oral, selective and potent pan-FGFR1–4 tyrosine kinase inhibitor, approved for the treatment of patients with locally advanced or metastatic urothelial carcinoma with susceptible FGFR2/3 alterations who progressed during or following at least one line of prior platinum-containing chemotherapy, including within 12 months of neoadjuvant or adjuvant platinum-containing chemotherapy.7 Erdafitinib was the first approved FGFR-targeted therapy and is the only FGFR therapy approved in metastatic urothelial carcinoma.6 In the phase 2 BLC2001 study of patients with locally advanced or metastatic urothelial carcinoma with selected FGFR alterations,8,9 treatment with erdafitinib resulted in an investigator-assessed objective response in 40 (40%; 95% CI 31–50) of 101 patients and a disease control rate (ie, the proportion of patients with complete response, partial response, and stable disease) in 81 (80%) of 101 patients.9 Clinical activity of erdafitinib has also been investigated in other tumours, including cholangiocarcinoma, in phase 1/2 trials.10–13 In the multicentre, single-arm phase 2a LUC2001 study in Asian patients with FGFR-altered cholangiocarcinoma, erdafitinib showed an objective response in 9 (41%) of 22 patients and a disease control rate of 82%.11 In a separately reported exploratory cohort of patients with FGFR-altered cholangiocarcinoma from the RAGNAR study, an objective response was observed in 21 (60%) of 35 patients with a disease control rate of 100%.13
Investigation of FGFR-targeted therapies in patients with FGFR alterations across tissue histologies has been limited and merits further evaluation. In the FIGHT-101 study, treatment with the FGFR1–3 inhibitor pemigatinib was associated with partial responses in 12 (9.4%) of 128 patients with advanced malignancies (of which five were in cholangiocarcinoma) with (62% FGFR+) and without FGFR alterations.14 Patients with advanced, unresectable, or metastatic solid tumours have a poor prognosis with almost universally incurable terminal conditions which are typically associated with short survival and significant impact on quality of life. Moreover, treatment options with standard therapies in the metastatic setting are limited further by potential toxicities or pre-existing comorbidities. For patients with advanced solid tumours who have exhausted standard therapies, few options remain outside of clinical trials. These patients often experience rapid progression and short survival. For example, glioblastoma has a median survival of 9 months with approved second-line therapies.15 There is a high unmet need for additional therapeutic options for patients with advanced solid tumours.
The RAGNAR study aimed to assess the antitumour activity and safety of erdafitinib in adult patients with advanced solid tumours with prespecified FGFR alterations. Here we report results from the primary analysis.
Methods
Study design and participants
The open-label, phase 2 RAGNAR study was conducted at 156 investigative centres in 15 countries across Asia, Europe, Oceania, South America, and the United States. Results reflect the primary cohort of the study, identified as the Broad Panel Cohort. Detailed description of all study cohorts is summarized in appendix pp 7, 12. Eligible patients (appendix pp 8, 10) were aged ≥12 years, with histological demonstration of an unresectable, locally advanced, or metastatic solid tumour malignancy harbouring a predefined FGFR1–4 mutation, or fusion with an intact kinase domain as determined by local or central laboratory screening, excluding FGFR gatekeeper and resistance alterations; had measurable disease according to Response Evaluation Criteria In Solid Tumors version 1·1 (RECIST v1·1) or Response Assessment In Neuro-Oncology (RANO) criteria for primary brain tumours; had disease progression on at least one prior line of systemic therapy and no alternative standard therapy; had an Eastern Cooperative Oncology Group (ECOG) performance status grade of 0 or 1 for adults, a Lansky Score of at least 80 for children and adolescents (aged ≥12 to <16 years), and a Karnofsky score of at least 80 for adolescents (aged ≥16 to <18 years). Patients with prior FGFR inhibitor treatment were excluded from the study (appendix p 9).
Central molecular eligibility screening (FoundationOne, Foundation Medicine, Inc, Cambridge, MA, USA, appendix p 11) was limited to the tumour types below, however, any solid tumour histology with evidence of an eligible FGFR alteration based on local testing was eligible for the study. Tumour samples were collected from either primary or metastatic sites. Central laboratory confirmation of local next-generation sequencing results was not required. Centrally screened tumour types included high-grade glioma (glioblastoma); low-grade glioma; squamous cell head and neck cancers; soft tissue sarcoma; cholangiocarcinoma; endometrial, cervical, and ovarian cancers; squamous non-small cell lung cancer; renal cell cancer; oesophageal cancer; gastric cancer; breast cancer; hepatocellular carcinoma; pancreatic cancer; salivary gland tumours; colorectal cancer; thymic cancer/thymoma; and paediatric patients with any solid tumour.
Review boards at all participating institutions and countries approved the study and all the protocol amendments; the study was performed according to the principles of the Declaration of Helsinki and guidelines for Good Clinical Practice and applicable regulatory requirements. Patients or their legally acceptable representatives provided written consent before participation.
Procedures
Patients received erdafitinib orally once daily on a 21-day cycle until disease progression, intolerable toxicity, withdrawal of consent, or decision by the investigator to discontinue treatment. Dose interruptions and reductions were permitted for the management of toxicities. Adults (aged ≥18 years for dosing purposes) and adolescent patients aged ≥15 to <18 years started with erdafitinib 8 mg with possible up-titration to 9 mg based on cycle 1 day 14 serum phosphate levels. Adolescent patients aged ≥12 to <15 years started with erdafitinib 5 mg with possible up-titration to 6 mg or further to 8 mg based on cycle 1 day 14 and cycle 2 day 7 serum phosphate levels.8,9 If the investigator made an initial assessment of disease progression, and if the patient was clinically stable, treatment with erdafitinib could be continued. Following treatment discontinuation for any reason, all patients were followed for survival until death or withdrawal of consent.
For patients with non-central nervous system (CNS) tumours, efficacy was assessed using RECIST v1·1 by computed tomography or magnetic resonance imaging (MRI) of the chest, abdomen, and pelvis every 6 weeks for 12 months, and then every 12 weeks thereafter. Brain MRI was performed for all patients with a history of brain metastases or worsening neurological symptoms any time during the study. Efficacy was assessed by RANO for patients with primary brain tumours. Brain MRI was performed at baseline and every 6 weeks for disease assessment for the first 12 months and then every 12 weeks thereafter.
Safety assessments included adverse event reports and results of vital sign measurements, electrocardiograms, physical examinations, clinical laboratory tests, performance status assessment, and ophthalmologic examinations. Investigators assessed and graded adverse events and laboratory abnormalities according to the National Cancer Institute Common Terminology Criteria for Adverse Events (version 5·0) for the duration of the study.
Outcomes
The primary endpoint was the objective response rate (the proportion of patients with a confirmed complete response or a partial response) based on RECIST v1·1 or RANO as assessed by the independent review committee. Secondary endpoints were investigator-assessed objective response rate, duration of response (defined as the time of initial documentation of a response until the first documented evidence of progressive disease [or relapse for patients who had a complete response during the study] or death, whichever came first), disease control rate (the proportion of patients who achieved a best response of complete response, partial response, or stable disease), clinical benefit rate (the proportion of patients who achieved a best response of complete response, partial response, or durable stable disease [duration of at least 4 months]), progression-free survival (the time from the first dose of study drug until the first documented evidence of progressive disease [or relapse for patients who had a complete response during the study] or death, whichever came first), overall survival (the time from the first dose of study drug until death), safety, and health-related quality of life and pharmacokinetics (to be published elsewhere).
Statistical analysis
A sample size of approximately 200 response-evaluable patients was selected based on extensive simulations with various Bayesian Hierarchical Model designs. For enrolment in each tumour histology, the sample size was capped at approximately 30 patients to avoid enrichment from a specific tumour type.
The primary analysis was planned after the enrolment of approximately 200 response-evaluable patients in the Broad Panel Cohort 6 months after the first dose of erdafitinib was received by the last patient enrolled. The 200 response-evaluable patients, with a cap of 30 for each tumour histology, would achieve approximately 80% power to select at least 80% of tumour histologies (12/15) under the primary statistical null hypothesis of an objective response rate of 15% or less, with an alternative objective response rate being set at at least 35% for each tumour histology. Objective response rate by independent review committee assessment was calculated with 95% 2-sided exact CI. The analysis was based on the treated population in the Broad Panel Cohort.
Other secondary efficacy analyses that were focused on the Broad Panel Cohort include: the objective response rate assessed by the investigator calculated with 95% 2-sided exact CI; duration of response distributions summarised using Kaplan–Meier estimates based on responding patients with the estimated median reported with the 95% CI; disease control rate and clinical benefit rate calculated with 95% 2-sided exact CI; progression-free survival and overall survival, summarised using Kaplan–Meier methods.
The primary endpoint of objective response rate was calculated with 95% 2-sided exact CI. For duration of response and progression-free survival, data were censored at the last tumour assessment for patients who were progression-free and alive or had unknown status. For overall survival, if the patient was alive or the vital status was unknown, data were censored at the time the patient was last known to be alive.
The RAGNAR study protocol is provided in the appendix, together with the statistical analysis plan. SAS version 9·4 was used for all statistical analyses. This study is registered with ClinicalTrials.gov, NCT04083976.
Role of the funding source
Employees of the sponsor, Janssen Research & Development, were involved in the study’s conceptualisation and design and the collection, analysis, and interpretation of data.
Results
RAGNAR initiated on Dec 5, 2019, and remains ongoing. At the time of the clinical cutoff date of Aug 26, 2022, 217 patients were treated with erdafitinib, of which 134 completed the study due to death, with 67 patients ongoing (22 on treatment and 45 in the follow-up phase); 16 patients terminated participation prematurely (appendix p 13). Among the 217 treated patients, 174 (80%) were enrolled based on local next-generation sequencing reports and 43 (20%) were enrolled based on positive central molecular screening results. Overall, 11 353 patients underwent central screening and 333 patients had local screening submitted for FGFR alteration review. 144 (66%) of 217 patients had FGFR fusions and 75 (35%) of 217 patients had FGFR mutations (appendix p 19) across FGFR1 (10%), FGFR2 (47%), and FGFR3 (44%) genes (patients with ≥1 FGFR alterations were counted multiple times). No patients with FGFR4 alterations were enrolled, reflecting the low incidence of FGFR4 alterations in adult patients. The numbers of patients across tumour histologies and FGFR alterations are summarised in table 1 and appendix p 21, respectively.
Table 1:
Baseline characteristics
| Patients n=217 | |
|---|---|
|
| |
| Age, years | 57 (48–64) |
|
| |
| Sex | |
| Male | 120 (55%) |
| Female | 97 (45%) |
|
| |
| Region | |
| Europe | 94 (43%) |
| Asia | 53 (24%) |
| North America | 48 (22%) |
| Rest of world | 22 (10%) |
|
| |
| Race | |
| White | 112 (52%) |
| Asian | 57 (26%) |
| Black or African American | 6 (3%) |
| Other or not reported | 42 (19%) |
|
| |
| ECOG performance status* | n=215 |
| 0 | 65 (30%) |
| 1 | 149 (69%) |
| 2† | 1 (<1%) |
|
| |
| Time from progression/relapse on the last line of treatment to first dose, months‡ | n=214 1.25 (0·82–2·14) |
|
| |
| Metastatic sites | |
| Presence of metastases sites | 179 (83%) |
| Lymph node | 119 (55%) |
| Liver | 99 (46%) |
| Lung | 94 (43%) |
| Bone | 49 (23%) |
| Adrenal gland | 17 (8%) |
| Brain | 12 (6%) |
| Spinal cord | 5 (2%) |
| Other | 68 (31%) |
|
| |
| Number of body sites with metastatic disease§ | |
| Median | 2 (2–3) |
| 1 | 40 (22%) |
| 2 | 50 (28%) |
| ≥3 | 89 (50%) |
|
| |
| Prior systemic therapy in advanced/metastatic setting | |
| Chemotherapy | 215 (99%) |
| Immunotherapy | 67 (31%) |
| Other systemic therapy | 91 (42%) |
|
| |
| Number of prior lines of anticancer therapies | |
| Median | 2 (2–4) |
| 1 | 50 (23%) |
| 2 | 67 (31%) |
| ≥3 | 100 (46%) |
|
| |
| Tumour types | |
| Cholangiocarcinoma | 31 (14%) |
| High-grade glioma | 30 (14%) |
| Pancreatic cancer | 18 (8%) |
| Breast cancer | 16 (7%) |
| Squamous cell head and neck cancers | 15 (7%) |
| Squamous NSCLC | 14 (7%) |
| Non-squamous NSCLC | 9 (4%) |
| Carcinoma of unknown primary | 8 (4%) |
| Colorectal cancer | 8 (4%) |
| Endometrial cancer | 8 (4%) |
| Oesophageal cancer | 8 (4%) |
| Gastric cancer | 8 (4%) |
| Ovarian cancer | 8 (4%) |
| Low-grade glioma | 7 (3%) |
| Cervical cancer | 6 (3%) |
| Salivary gland cancer | 5 (2%) |
| Soft tissue sarcoma | 3 (1%) |
| Prostate cancer | 2 (1%) |
| Others | 13 (6%) |
Data are n (%) or median (IQR).
ECOG performance status is only applicable to adults.
One patient was enrolled with an ECOG performance status score of 2, which did not meet protocol eligibility criteria.
Applicable only to patients with non-missing values for progression/relapse date of last line of treatment.
Metastatic site with other will be considered as one body site.
ECOG=Eastern Cooperative Oncology Group. NSCLC=non-small cell lung cancer.
At the clinical cutoff date, median follow-up for efficacy was 17·9 months (IQR 13·6– 23·9). Median treatment duration was 4·3 months (IQR 2·1–9·2). The most common reasons for treatment discontinuation were progressive disease (73%) and adverse events (10%). One participant who was enrolled but not treated was not included in the primary analysis. All 217 treated patients had received a minimum of one prior line of systemic therapy, with a median of two lines of systemic therapy (IQR 2–4); 46% had received three or more prior treatments (table 1). Only 22 (10%) of 217 patients had a response (partial response) on the last line of therapy prior to enrolment as reported by the investigator.
159 (73%) of 217 patients had a reduction in tumour burden (figure 1A). The number of patients with an objective response per independent review committee assessment was 64 (30%; 95% CI 24–36) of 217 patients (figure 2). The study met its primary endpoint by rejecting the null hypothesis of an objective response rate of 15%. Among the 64 responding patients, 6 (3%) of 217 had a complete response and 58 (27%) had a partial response (figure 3; a description of patients with a complete response is in the appendix, p 29). Subgroup analysis of the overall objective response demonstrated consistent clinical benefit across prespecified subgroups (appendix p 16). Responses to erdafitinib were observed across 16 tumour types, including CNS, head and neck, thoracic, and gastrointestinal and gynaecological malignancies, as well as rare tumours such as salivary gland cancer and low-grade glioma. In the pancreatic cancer subgroup, the number of patients with an objective response was 10 (56%) of 18 patients. In high- and low-grade gliomas, the number of patients with an objective response rate was 3 (10%) of 30 patients and 2 (29%) of 7 patients, respectively. In adolescent patients (n=2), one patient (high-grade glioma) had confirmed response based on both independent review committee and investigator assessment, and the other (low-grade glioma) had confirmed response based on independent review committee assessment but not investigator assessment (appendix p 31).
Figure 1: Maximal percentage reduction of target lesion from baseline by tumour histology and FGFRalt by IRC assessment.

The best overall response was recorded from the start of study treatment to end of study, prior to progressive disease and subsequent anticancer therapy (subsequent surgery/procedure, subsequent radiotherapy, and subsequent systemic therapy), taking into account any requirement for confirmation. Disease evaluations were based on RECIST v1·1, maximum percentage reduction from baseline was calculated in sum of target lesion diameters; for disease evaluations based on RANO, maximum percentage reduction from baseline was calculated in sum of product of perpendicular dimension. Maximum percentage increase of target lesion from baseline greater than 100% was set to 100%.
alt=alterations. DNET=dysembryoplastic neuroepithelial tumour. IRC=independent review committee. MRI=magnetic resonance imaging. NSCLC=non-small cell lung cancer. RANO=Response Assessment in Neuro-Oncology. RECIST=Response Evaluation Criteria in Solid Tumors.
Figure 2: Confirmed responses across tumour types and FGFRalt.
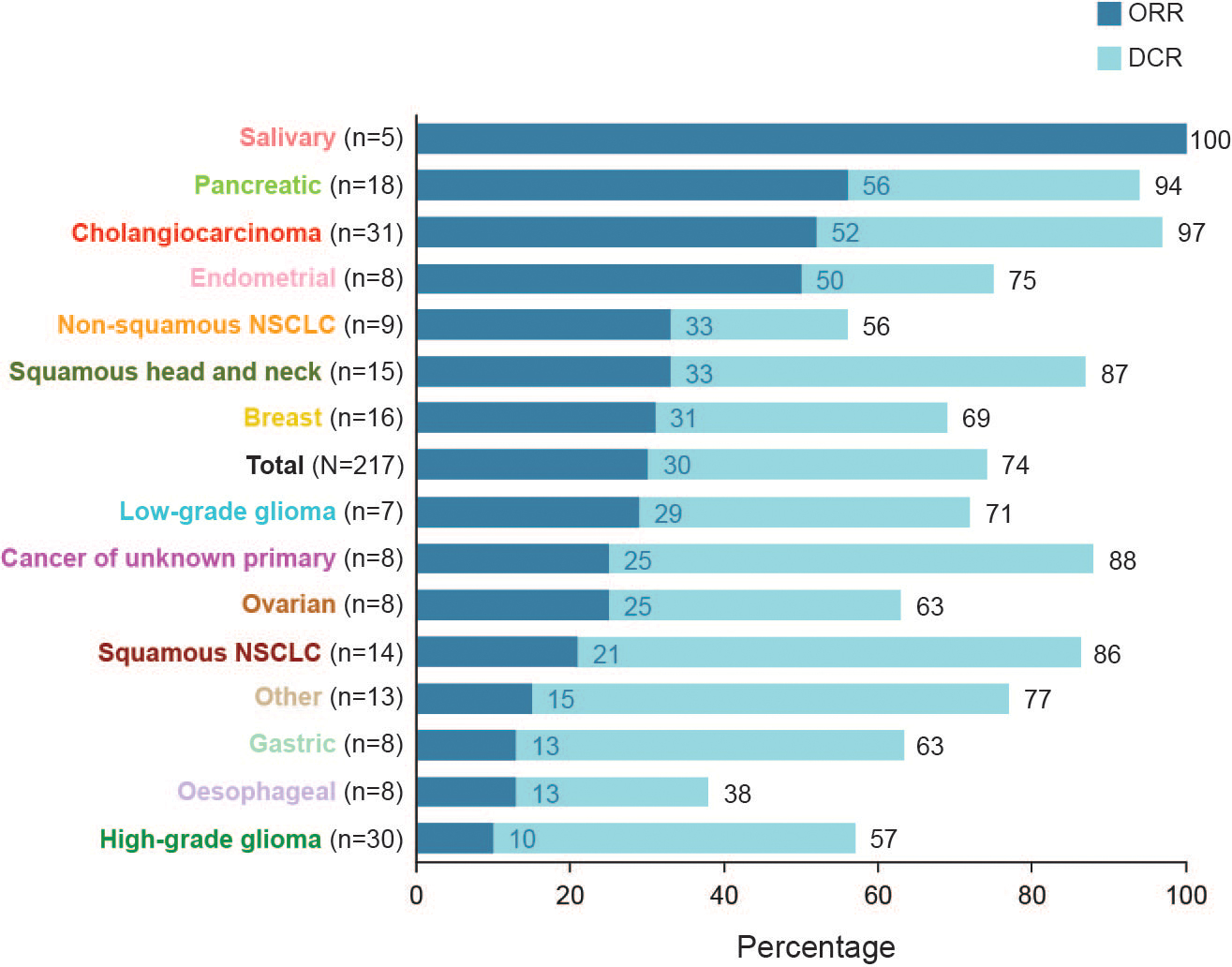
Only tumour types with at least one complete or partial response are shown. The objective response rate and disease control rate were equivalent for the salivary gland tumour type. alt=alterations. DCR=disease control rate. NSCLC=non-small cell lung cancer. ORR=objective response rate.
Figure 3: Treatment duration and response to erdafitinib.
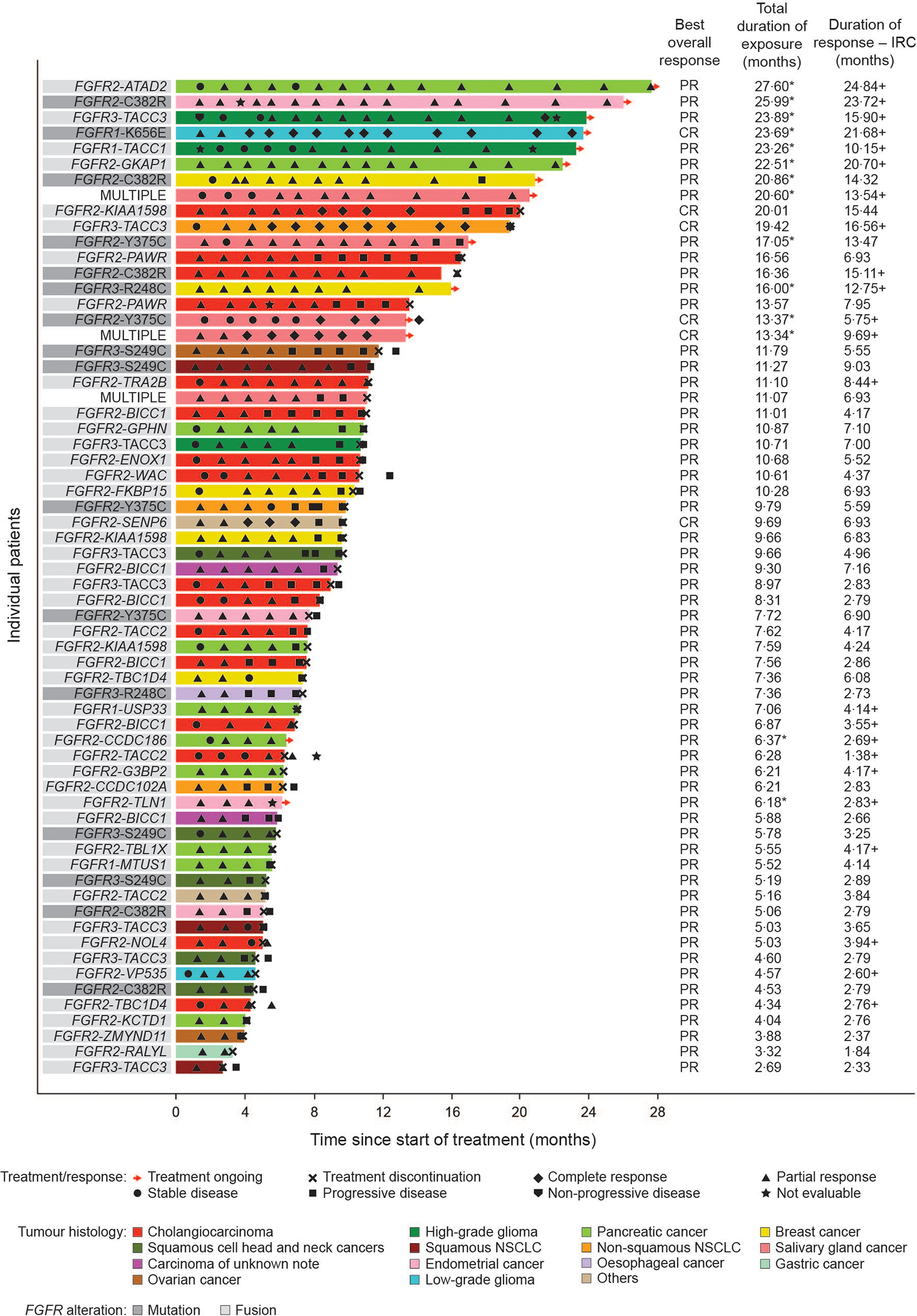
*Indicates patient remains on-treatment. +Indicates that the duration of response for a patient is currently censored. MULTIPLE signifies multiple alterations (mutations, fusions, or both).
CR=complete response. IRC=independent review committee. PR=partial response.
The median duration of response was 6·9 months (95% CI 4·4–7·1). 14 patients had ongoing responses at the time of the clinical cutoff. At the time of the clinical cutoff, 36% of confirmed responders were permanently censored (most commonly [11%] for receiving subsequent anticancer therapy). The median time to response was 1·4 months (IQR 1·4–2·7) after initiation of erdafitinib treatment. 57 patients continued treatment for at least 4 weeks beyond progressive disease (median 41·5 days [IQR 18–83]). The disease control rate was 74% (95% CI 67–80) and the clinical benefit rate was 46% (95% CI 39–53). Median progression-free survival and overall survival was 4·2 months (95% CI 4·1–5·5) and 10·7 months (95% CI 8·7–12·1) in the 217 patients, respectively (appendix p 22).
Efficacy was observed across patients with FGFR1–3 mutations and fusions (appendix p 23). The overall response rate was 25% (95% CI 16–37) in the prespecified subgroup analysis of patients with FGFR mutations compared with 33% (95% CI 25–41) in patients with FGFR fusions. Efficacy measures, including the objective response rate and the median duration of response per investigator assessment, demonstrated consistent results compared with that determined by the independent review committee data, with an investigator objective response rate of 25% (95% CI 20–32) and a median duration of response of 7 months (95% CI 5·5–8·5; appendix p 27). 82 (38%) of 217 patients received subsequent anticancer therapies (appendix p 30).
At the median treatment exposure of 4·3 months (IQR 2·1–9·2), 216 (99·5%) of 217 patients experienced at least one treatment-emergent adverse event (overall safety by sex, appendix p 33). Grade 3–5 treatment-emergent adverse events of any cause occurred in 152 (70%) patients (table 2, appendix p 34); the most common was stomatitis in 25 (12%), anaemia in 18 (8%), and palmar-plantar erythrodysaesthesia in 12 (6%) of 217 patients (appendix p 33; by sex, appendix p 33). Cumulative incidence of first onset of treatment-emergent adverse events is shown in the appendix, p 17. Treatment-emergent adverse events leading to dose reduction, dose interruption, or treatment discontinuation were observed in 136 (63%), 162 (75%), and 22 (10%) of 217 patients, respectively. Treatment-emergent adverse events considered to be related to erdafitinib (occurring in ≥30% of 217 patients) were hyperphosphataemia (in 154 [71%] patients), stomatitis (in 118 [54%] patients), diarrhoea (in 108 [50%] patients), dry mouth (in 103 [48%] patients), dry skin (in 76 [35%] patients), and palmar-plantar erythrodysaesthesia syndrome (in 73 [34%] patients; appendix p 37). Grade 3 or higher treatment-emergent adverse events related to erdafitinib occurred in 100 (46%) of 217 patients. Serious treatment-emergent adverse events occurred in 85 (39%) patients (appendix p 38). The most commonly occurring serious treatment-related adverse events of grade 3 or higher were stomatitis in 4 (2%) patients and diarrhoea in 2 (1%) of 217 patients (appendix p 39). The most common treatment-related adverse events that led to discontinuation were palmar-plantar erythrodysaesthesia syndrome and stomatitis in 3 (2%) of 217 patients each (appendix p 40).
Table 2:
Treatment-emergent adverse events (≥10% any grade) by worst toxicity grade in the safety population (n=217)
| Grade 1–2 | Grade 3 | Grade 4 | Grade 5 | |
|---|---|---|---|---|
| All treatment-emergent events | 64 (29%) | 124 (57%) | 20 (9%) | 8 (4%) |
| Hyperphosphataemia | 143 (66%) | 11 (5%) | 0 | 0 |
| Diarrhoea | 119 (55%) | 9 (4%) | 0 | 0 |
| Stomatitis | 95 (44%) | 25 (12%) | 0 | 0 |
| Dry mouth | 105 (48%) | 1 (<1%) | 0 | 0 |
| Dry skin | 73 (34%) | 4 (2%) | 0 | 0 |
| Palmar-plantar erythrodysaesthesia syndrome | 61 (28%) | 12 (6%) | 0 | 0 |
| Constipation | 64 (29%) | 2 (1%) | 0 | 0 |
| Fatigue | 56 (26%) | 7 (3%) | 0 | 0 |
| Alanine aminotransferase increased | 51 (24%) | 11 (5%) | 0 | 0 |
| Aspartate aminotransferase increased | 53 (24%) | 5 (2%) | 0 | 0 |
| Decreased appetite | 55 (25%) | 3 (1%) | 0 | 0 |
| Anaemia | 39 (18%) | 18 (8%) | 0 | 0 |
| Dry eye | 48 (22%) | 0 | 0 | 0 |
| Alopecia | 44 (20%) | 0 | 0 | 0 |
| Nausea | 41 (19%) | 2 (1%) | 0 | 0 |
| Paronychia | 37 (17%) | 6 (3%) | 0 | 0 |
| Nail disorder | 36 (17%) | 4 (2%) | 0 | 0 |
| Onycholysis | 34 (16%) | 6 (3%) | 0 | 0 |
| Arthralgia | 36 (17%) | 3 (1%) | 0 | 0 |
| Epistaxis | 38 (18%) | 0 | 0 | 0 |
| Vomiting | 33 (15%) | 5 (2%) | 0 | 0 |
| Dysgeusia | 37 (17%) | 0 | 0 | 0 |
| Abdominal pain | 24 (11%) | 10 (5%) | 0 | 0 |
| Blood alkaline phosphatase increased | 32 (15%) | 2 (1%) | 0 | 0 |
| Nail discolouration | 32 (15%) | 0 | 0 | 0 |
| Weight decreased | 26 (12%) | 3 (1%) | 0 | 0 |
| Asthenia | 21 (10%) | 7 (3%) | 0 | 0 |
| Vision blurred | 25 (12%) | 2 (1%) | 0 | 0 |
| Pyrexia | 23 (11%) | 2 (1%) | 0 | 1 (<1%) |
| Hyponatraemia | 17 (8%) | 6 (3%) | 2 (<1%) | 0 |
| Pain in extremity | 24 (11%) | 1 (<1%) | 0 | 0 |
| Nail dystrophy | 20 (9%) | 3 (1%) | 0 | 0 |
| Back pain | 20 (9%) | 2 (1%) | 0 | 0 |
| Headache | 20 (9%) | 2 (1%) | 0 | 0 |
| Myalgia | 22 (10%) | 0 | 0 | 0 |
| Thrombocytopenia | 19 (9%) | 3 (1%) | 0 | 0 |
Data are n (%). Patients with one or more related treatment-emergent adverse events were counted only once for each adverse event and worst adverse event grade reported. Treatment-emergent adverse events that occurred in at least 10% of patients are shown ordered relative to the descending frequency of any-grade treatment-emergent adverse event. All treatment-emergent adverse events with the outcome of death were considered by the investigator to be unrelated to erdafitinib, and most events (126/140) occurred in the context of progressive disease. Hyperphosphataemia was graded on the basis of protocol-defined criteria: 5·5–6·9 mg/dL as grade 1; 7·0–8·9 mg/dL as grade 2; 9·0–10·0 mg/dL as grade 3; >10·0 mg/dL as grade 4.
Central serous retinopathy, a treatment-emergent adverse event of special interest, occurred in 31 (14%) of 217 patients. Central serous retinopathy represents a grouped term comprising specific preferred terms; the most frequently reported central serous retinopathy-specific preferred terms (occurring in ≥2% of 217 patients) were chorioretinopathy in 8 (4%) patients, detachment of retinal pigment epithelium in 7 (3%) patients, and retinal detachment in 6 (3%) patients. All central serous retinopathy events were grade 1–2 except for one (<1%) of 217 patients with a serious grade 3 treatment-emergent adverse event (retinal oedema). Central serous retinopathy led to an erdafitinib dose reduction in 19 (9%) of 217 patients, dose interruption in 17 (8%) of 217 patients, and treatment discontinuation in one (<1%) of 217 patients (appendix p 40). Central serous retinopathy events resolved in 20 (65%) of 31 patients. Unresolved central serous retinopathy was grade 1 in 10 of 11 patients and grade 2 in the remaining patient.
Treatment-emergent adverse events led to death in 8 (4%) of 217 patients (multiple organ dysfunction syndrome, pyrexia, COVID-19, sepsis, pulmonary embolism, respiratory failure, cardiac arrest, and subdural haematoma, n=1 each); all were considered unrelated to treatment with erdafitinib by investigator assessment.
Discussion
Primary analysis data from RAGNAR confirm antitumour activity of erdafitinib in patients with prespecified FGFR alterations across histologies. Efficacy data based on independent review committee assessment show a robust objective response rate of 30% and a clinically meaningful duration of response of 6·9 months across 16 distinct tumour histologies in patients with advanced solid tumours who had no effective alternative treatments. The consistency of observed response and clinical benefit across tumour histologies and FGFR alterations supports the tumour-agnostic activity of erdafitinib and the validity of the biomarker panel used to determine eligibility.
A key strength of RAGNAR is that, to our knowledge, this study represents the first pivotal phase 2 tumour-agnostic trial of a FGFR-targeted therapy reported to date. The study enrolled patients with a wide variety of tumour histologies, such as cholangiocarcinomas, high-grade gliomas, non-small cell lung cancer, and pancreatic cancer. An important element of the study design was limiting the sample size by tumour type to prevent a subset of histologies from dominating enrolment, resulting in improved representation of rarer tumour types based on 32 distinct tumour types enrolled.
In the RAGNAR study, clinical benefit was observed in patients with advanced solid tumours with limited therapeutic options, such as pancreatic cancer with an objective response rate of 56%. While limited data exist regarding the ability of FGFR inhibitors to cross the blood–brain barrier, encouraging activity was also observed in CNS tumours with an objective response rate of 29% in low-grade gliomas (14% by investigator) and 10% (20% by investigator) in high-grade gliomas. Notably, 46% of patients had three or more prior lines of systemic therapy, with only 10% of patients achieving a response on the last line of therapy prior to enrolment, as reported by the investigator. Responses to erdafitinib included complete and partial responses observed in the tumour-agnostic setting across FGFR1–3 mutations and fusions. The observed activity with erdafitinib in patients with a wide variety of histologies and FGFR alterations is notable given the apparent limited efficacy reported for other FGFR inhibitors in similar patient populations.14
Results of secondary endpoints showed a disease control rate of 74%, a median progression-free survival of 4.2 months, and a median overall survival of 10.7 months. These findings are clinically impactful in the context of a last-line therapy population as observed for tumour types examined in RAGNAR. For example, glioblastoma has a median survival of 9 months with approved second-line therapies,15 whereas recurrent or metastatic squamous cell carcinoma of the head and neck has a median survival of 7 to 8 months with second-line programmed death-ligand 1 therapies.18 For patients with metastatic pancreatic cancer previously treated with gemcitabine-based therapies, treatment with nanoliposomal irinotecan with fluorouracil and folinic acid led to a median overall survival of approximately 6 months.19 The clinical benefit demonstrated in the RAGNAR study confirms that a targeted therapy can have tumour-agnostic efficacy in a biomarker-selected population.
Limited or no responses were observed in some tumour types, such as colorectal cancer, but these findings are difficult to interpret given the small sample size of select tumour types. Metastatic solid tumours that failed multiple prior therapies are expected to harbour a variety of genomic alterations, which may limit responsiveness to FGFR inhibitor treatment. Notably, 70% of patients in RAGNAR did not achieve a response, highlighting the need to better understand the biology of solid tumours with FGFR alterations, the type of FGFR alterations, and the role of concurrent genomic alterations in conferring resistance to FGFR-targeted therapies. For example, further analysis is warranted to understand erdafitinib activity in tumours with FGFR kinase domain alterations. However, the robust clinical response observed across 16 tumour types with FGFR1–3 mutations and fusions underscores the dependency of multiple tumour types on the FGFR oncogenic axis for growth, progression, and survival. Data from adolescent patients were relatively limited in this analysis; however, a specific paediatric cohort is currently ongoing and will be reported on separately.
Observed toxicities, including central serous retinopathy, were consistent with the known safety profile of erdafitinib and other FGFR inhibitors.8,9,16,17 The erdafitinib dose personalisation strategy was based on serum phosphate levels and intended to maximise the proportion of patients within the target phosphate range for efficacy while maintaining tolerability.20 Additionally, treatment-emergent adverse events related to erdafitinib were managed by dose interruptions, dose reductions, and supportive care, minimising discontinuations due to treatment-emergent adverse events. Dose interruptions and dose reductions were observed in the majority of patients, including 10% who discontinued due to adverse events, which may have compromised efficacy. These challenges highlight the need for further investigation to explore pathogenetic mechanisms, which may lead to the development of novel FGFR-targeted therapies with improved tolerability.
The open-label, single-arm study design limits the ability to interpret RAGNAR study results. However, in this patient population, a randomised trial design would not be feasible due to the rarity of FGFR-altered solid tumours and the absence of equipoise in settings without available standard therapies. In addition, combining multiple tumour types and histologies in a single randomised trial may be methodologically inappropriate given the variability of prognoses and natural histories. Therefore, in this setting, real-world data may provide additional information that would serve to contextualise the study results by exploring the prognostic significance of FGFR alterations and, potentially, real-world outcomes with non–FGFR-targeted therapies.
In conclusion, efficacy and safety results from the RAGNAR primary analysis confirm that erdafitinib has a favourable benefit–risk profile in the tumour-agnostic setting and provides meaningful clinical benefit in patients with advanced solid tumours who have susceptible FGFR alterations and have exhausted other treatment options.
Supplementary Material
Research in Context.
Evidence before this study
In a phase 1 study, erdafitinib showed preliminary clinical activity in patients with fibroblast growth factor receptor (FGFR)-altered urothelial carcinoma, cholangiocarcinoma, and other advanced solid tumours. In the global phase 2 BLC2001 study, erdafitinib demonstrated antitumor efficacy in patients with locally advanced or metastatic urothelial carcinoma with prespecified FGFR alterations. Antitumour activity of erdafitinib was also observed in patients with FGFR-altered cholangiocarcinoma in the phase 2a LUC2001 study.
We searched the literature for clinical trials of FGFR inhibitors used to treat patients in the tumour-agnostic setting. We found a paucity of investigations examining FGFR inhibitors in this setting.
Added value of this study
To our knowledge, RAGNAR is the first tumour-agnostic study demonstrating antitumour activity of erdafitinib across histologies and FGFR1–3 alterations. Our findings demonstrate that erdafitinib treatment leads to a robust objective response rate and a clinically meaningful duration of response and clinical benefit across a wide variety of tumour histologies in patients with FGFR-altered advanced solid tumours who had progressed on or after at least one line of systemic therapy and had no effective alternative treatments for their underlying disease.
Implications of all the available evidence
Prior phase 2 or 3 studies of FGFR inhibitors were limited to advanced or metastatic urothelial carcinoma and cholangiocarcinoma. Results from the RAGNAR study demonstrate cross-histology activity of erdafitinib in patients with FGFR-altered advanced or metastatic solid tumours who have exhausted available therapies. The RAGNAR findings validate selected FGFR alterations as a therapeutic target across solid tumour histologies. They also highlight a potential role for erdafitinib to benefit patients with prespecified FGFR alterations who have exhausted standard treatment options. Further, these results may help inform the design of future studies with FGFR inhibitors.
Acknowledgements
Erdafitinib (JNJ-42756493) was discovered in collaboration with Astex Pharmaceuticals. This study was funded by Janssen Research & Development. We thank Christopher Moy and Mingxuan Xia for computational biology and preclinical support in the defining of Broad Panel Cohort-eligible FGFR alterations, respectively. The authors would like to thank the patients who participated in this trial, their families, the investigators, study coordinators, study teams, and nurses.
Role of the funding source
Writing assistance was provided by Ira Mills, PhD, of Parexel, and was funded by Janssen Global Services, LLC.
Footnotes
Declaration of interests
SP has received consulting fees from Ipsen, Janssen, Novartis, and Zymeworks; and institutional research funding from 4D Pharma, Arcus Biosciences, Astellas Pharma, Boehringer Ingelheim, Bristol Myers Squibb, Elicio Therapeutics, Janssen, Lilly, Mirati Therapeutics, NGM Biopharmaceuticals, Novartis, Purple Biotech, Rgenix, and Xencor. MS has received consulting fees from Amgen, AstraZeneca, Boehringer Ingelheim, Bristol Myers Squibb, GlaxoSmithKline, Janssen Oncology, Merck Serono, Novartis, Roche, Sanofi, and Takeda; honoraria from Amgen, Boehringer Ingelheim, Bristol Myers Squibb, Janssen-Cilag, and Novartis; institutional research funding from AstraZeneca and Bristol Myers Squibb; and has institutional patents, royalties, or other intellectual property for a highly sensitive method for mutation detection by PCR. GI has received consulting fees from Basilea, Bayer, Flare Therapeutics, Janssen, Loxo/Lilly, and Mirati Therapeutics; speakers’ bureau fees from Gilead Sciences and Lynx Group; and institutional research funding from Bayer, Debiopharm Group, Janssen, Mirati Therapeutics, Novartis, and Seagen. OW has received consulting fees from Bayer US, LCC, Bristol Myers Squibb, Day One Therapeutics, Novartis, and Roche; and research funding from AstraZeneca, Bayer US, LLC, Blueprint Medicines, Day One Therapeutics, GlaxoSmithKline, Janssen, Lilly, Loxo, Novartis, and Roche. TD has received consulting fees from AbbVie, Amgen, Boehringer Ingelheim, Chugai Pharma, Daiichi Sankyo, Janssen, Kaken Pharmaceutical, Kyowa Kirin, MSD, Otsuka, PRA Health Science, Rakuten Medical, Shionogi, Sumitomo Dainippon Pharma, Taiho Pharmaceutical, and Takeda; honoraria from AstraZeneca, Bristol Myers Squibb Japan, Chugai Pharma, Daicchi Sankyo, Ono Pharmaceutical, Rakuten Medical Japan, and Taiho Pharmaceutical; and institutional research funding from AbbVie, Boehringer Ingelheim, Bristol Myers Squibb, Chugai Pharma, Daiichi Sankyo, Eisai, IQVIA, Janssen, MSD, Ono Pharmaceutical, Novartis, Pfizer, PRA Health Sciences, Sumitomo Dainippon Pharma, and Taiho Pharmaceutical. SQ does not declare any interests. JT has received consulting fees from Array BioPharma, AstraZeneca, Bayer, Boehringer Ingelheim, Cardiff Oncology, Chugai Pharma, Daiichi Sankyo, F. Hoffman LaRoche, Genentech, HalioDx, Ikena Oncology, Inspirna, IQVIA, Lilly, Menarini, Merck Serono, Merus, Mirati Therapeutics, MSD, NeoPhore, Ona Therapeutics, Orion Biotechnology, Hutchison MediPharma, Novartis, Peptomyc, Pfizer, Pierre Fabre, Samsung Bioepis, Sanofi, Scandion Oncology, Scorpion Therapeutics, Servier, Sotio, Taiho Pharmaceutical, Tessa Therapeutics, TheraMyc, and Tolremo; other financial interests from Imedex/HMP, Medscape, MJH Life Sciences, Peerview, Physicians’ Education Resource; and stock and other ownership interests from Oniria Therapeutics. DAR has received consulting fees from Advantagene, Agenus, Agios, AnHeart Therapeutics, Bayer, Bristol Myers Squibb, Delmar Pharmaceuticals, Ellipses Pharma, EMD Serono, Genenta Science, Imvax, Kintara Therapeutics, Kiyatec, Medicenna, Merck, Merck KGaA, Novocure, Oncorus, Regeneron, Taiho Pharmaceutical, and Vivacitas Oncology; honoraria from Advantagene, Agenus, AnHeart Therapeutics, Bayer, Bristol Myers Squibb, Deciphera, DelMar Pharmaceuticals, Ellipses Pharma, EMD Serono, Genenta Science, Imvax, Inovio Pharmaceuticals, Kintara Therapeutics, Kiyatec, Medicenna, Merck, Merck KGaA, Neuvogen, Oncorus, Novocure, Regeneron, Sumitomo Dainippon Pharma, Taiho Pharmaceutical, Vivacitas Oncology, and Y-mAbs Therapeutics; and institutional research funding from Acerta Pharma, Agenus, Celldex, EMD Serono, Enterome, Incyte, and Omniox. CM has received consulting fees from Amgen, Astellas Pharma, AstraZeneca, Bayer, BeiGene, Blueprint Medicines, Bristol Myers Squibb, Celgene, Debiopharm Group, Faron Pharmaceuticals, Genentech/Roche, Innate Pharma, Ipsen, Janssen, Lilly, MSD, Novartis, Orion, Pfizer, PharmaMar, Sanofi, and Taiho Pharmaceutical. AM has received consulting fees from Bayer, Chugai, GlaxoSmithKline, Janssen, Merck, and Novartis Oncology; has been reimbursed for travel, accommodations, or expenses from Amgen; and has received fees for participating in a data safety monitoring board or advisory board from Faron, Genmab, Janssen, Merck, and Takeda. IL has received consulting fees from Boehringer Ingelheim; honoraria from Agenus, Amgen, AstraZeneca, BeiGene, Bristol Myers Squibb, Celon, Cullinan Oncology, Jacobio, Janssen, Loxo, Macrogenics, Menarini, MSD, Pfizer, Rhizen, Roche, Sanofi, and Takeda; institutional research funding from Agenus and Roche; and has been reimbursed for travel, accommodations, or expenses from Bristol Myers Squibb. OC has received consulting fees from Bayer US, LLC, and Janssen; honoraria from Bayer US, LLC, Janssen, Merck, Novartis, and Pfizer; and speakers’ bureau fees from Merck and Pfizer. DA has received consulting fees from AstraZeneca, Boston Scientific, Bristol Myers Squibb, CRA International, Gilead, Janssen Cilag, MSD, onkowissen, Pierre Fabre Pharma, Seagen, and Terumo; honoraria from Amgen, Aptitude Health, art tempi media, AstraZeneca, Boehringer Ingelheim, Boston Scientific, Bristol Myers Squibb, Clinical Care Options, Eisai, From Research to Practice, GlaxoSmithKline, Imedex, Ipsen, mci, MedAhead (Austria), Merck Serono, MSD, Pierre Fabre Pharma, PRMA Consulting, Roche, Sanofi (Genzyme), Seagen, Servier, Streamitup Germany, Tactics MD LLC, Terumo, Viatris, and WebMD; fees for editorial roles with Elsevier; fees and an institutional education grant from AbbVie; institutional trial support from Bristol Myers Squibb and Oncolytics; and has served in a leadership role with EORTC. MG has received consulting fees from Celularity and Guardant. HW has received honoraria from Bristol Myers Squibb, GlaxoSmithKline, and Novartis; and has been reimbursed for travel, accommodations, or expenses from Janssen. KS, LC, SN, CH, ST, AS-W, ST, and HS received personal fees from Janssen during the conduct of the study. YL has received consulting fees from Astellas Pharma, AstraZeneca, Bristol Myers Squibb, Immunomedics, Janssen (and institutional), MSD Oncology (and institutional), Loxo/Lilly, Pfizer/EMD Serono, Roche, and Taiho Pharmaceutical; has been reimbursed for travel, accommodations, or expenses from Astellas, AstraZeneca, Janssen Oncology, MSD Oncology, and Roche; and has received institutional research funding from Astellas Pharma, AstraZeneca, Basilea, Bristol Myers Squibb, Exelixis, Gilead Sciences, Incyte, Janssen Oncology, Merck KGaA, MSD Oncology, Nektar, Pfizer, Roche, Sanofi, and Taiho Pharmaceutical.
To be presented at the 2023 ASCO Annual Meeting, June 2–5, 2023, Chicago, IL, & Online.
Contributor Information
Shubham Pant, Department of Gastrointestinal Medical Oncology, The University of Texas MD Anderson Cancer Center, Houston, TX, USA.
Martin Schuler, Department of Medical Oncology, West German Cancer Center, University Hospital Essen, University Duisburg-Essen, Essen, Germany.
Gopa Iyer, Department of Medicine, Memorial Sloan Kettering Cancer Center and Weill Cornell Medical College, New York, NY, USA.
Olaf Witt, Hopp Children’s Cancer Center (KiTZ), Heidelberg University Hospital, German Cancer Research Center and National Center for Tumor Diseases (NCT), Heidelberg, Germany.
Toshihiko Doi, National Cancer Center Hospital East, Kashiwa, Japan.
Shukui Qin, Jinling Hospital, Nanjing University of Chinese Medicine, Nanjing, China.
Josep Tabernero, Vall d’Hebron Hospital Campus and Institute of Oncology (VHIO), IOB-Quiron, Barcelona, Spain.
David A Reardon, Center for Neuro-Oncology, Dana-Farber Cancer Institute and Harvard Medical School, Boston, MA, USA.
Christophe Massard, Université Paris-Saclay, Le Kremlin Bicêtre – France INSERM U1030, Molecular Radiotherapy, Gustave Roussy, Université Paris-Saclay, Paris, France.
Anna Minchom, The Royal Marsden NHS Foundation Trust, Sutton, UK.
Iwona Lugowska, Department of Soft Tissue/Bone Sarcoma and Melanoma, Narodowy Instytut Onkologii im. Marii Sklodowskiej-Curie – Panstwowy Instytut Badawczy, Warszawa, Poland.
Omar Carranza, Hospital Privado de Comunidad de Mar del Plata, Mar del Plata, Argentina.
Dirk Arnold, Department of Oncology, AK Altona, Asklepios Tumourzentrum Hamburg, Hamburg, Germany.
Martin Gutierrez, John Theurer Cancer Center, Hackensack University Medical Center, Hackensack, NJ, USA.
Helen Winter, Bristol Haematology and Oncology Centre, Bristol, UK.
Kim Stuyckens, Janssen Research & Development, Beerse, Belgium.
Lauren Crow, Janssen Research & Development, Spring House, PA, USA.
Saltanat Najmi, Janssen Research & Development, Spring House, PA, USA.
Constance Hammond, Janssen Research & Development, Spring House, PA, USA.
Shibu Thomas, Janssen Research & Development, Spring House, PA, USA.
Ademi Santiago-Walker, Janssen Research & Development, Spring House, PA, USA.
Spyros Triantos, Janssen Research & Development, Spring House, PA, USA.
Hussein Sweiti, Janssen Research & Development, Spring House, PA, USA.
Yohann Loriot, Department of Cancer Medicine, INSERM U981, Gustave Roussy, Université Paris-Saclay, Villejuif, France.
Data sharing
Janssen Pharmaceutical Companies of Johnson & Johnson’s data sharing policy is available at https://www.janssen.com/clinical-trials/transparency. As noted on this site, requests for study data access can be submitted through the Yale Open Data Access (YODA) Project site at http://yoda.yale.edu.
References
- 1.Dienstmann R, Rodon J, Prat A, et al. Genomic aberrations in the FGFR pathway: opportunities for targeted therapies in solid tumors. Ann Oncol 2014; 25: 552–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.AACR Project GENIE Consortium. AACR Project GENIE: powering precision medicine through an international consortium. Cancer Discov 2017; 7: 818–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Katoh M Fibroblast growth factor receptors as treatment targets in clinical oncology. Nat Rev Clin Oncol 2019; 16: 105–22. [DOI] [PubMed] [Google Scholar]
- 4.US Food & Drug Administration. FDA grants accelerated approval to pemigatinib for cholangiocarcinoma with an FGFR2 rearrangement or fusion. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-pemigatinib-cholangiocarcinoma-fgfr2-rearrangement-or-fusion (accessed February 28, 2023).
- 5.US Food & Drug Administration. FDA grants accelerated approval to futibatinib for cholangiocarcinoma. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-futibatinib-cholangiocarcinoma (accessed February 28, 2022).
- 6.US Food & Drug Administration. FDA grants accelerated approval to erdaftinib for metastatic urothelial carcinoma. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-erdafitinib-metastatic-urothelial-carcinoma (accessed February 1, 2023).
- 7.BALVERSA® [prescribing information]. Janssen Products, LP, Horsham, PA; 2023. [Google Scholar]
- 8.Loriot Y, Necchi A, Park SH, et al. Erdafitinib in locally advanced or metastatic urothelial carcinoma. N Engl J Med 2019; 381: 338–48. [DOI] [PubMed] [Google Scholar]
- 9.Siefker-Radtke AO, Necchi A, Park SH, et al. Efficacy and safety of erdafitinib in patients with locally advanced or metastatic urothelial carcinoma: long-term follow-up of a phase 2 study. Lancet Oncol 2022; 23: 248–58. [DOI] [PubMed] [Google Scholar]
- 10.Bahleda R, Italiano A, Hierro C, et al. Multicenter phase I study of erdafitinib (JNJ-42756493), oral pan-fibroblast growth factor receptor inhibitor, in patients with advanced or refractory solid tumors. Clin Cancer Res 2019; 25: 4888–97. [DOI] [PubMed] [Google Scholar]
- 11.Feng Y-H, Su W-C, Oh D-Y, et al. Updated analysis with longer follow up of a phase 2a study evaluating erdfitinib in Asian patients (pts) with advanced cholangiocarcinoma (CCA) and fibroblast growth factor receptor (FGFR) alterations. J Clin Oncol 2022; 40: 430.34882498 [Google Scholar]
- 12.Tabernero J, Bahleda R, Dienstmann R, et al. Phase I dose-escalation study of JNJ-42756493, an oral pan-fibroblast growth factor receptor inhibitor, in patients with advanced solid tumors. J Clin Oncol 2015; 33: 3401–8. [DOI] [PubMed] [Google Scholar]
- 13.Pant S, Schuler M, Iyer G, et al. Efficacy and safety of erdafitinib in adults with cholangiocarcinoma (CCA) with prespecified fibroblast growth factor receptor alterations (FGFRalt) in the phase 2 open-label, single-arm RAGNAR trial: Expansion cohort results. J Clin Oncol 2023; 41: 610. [Google Scholar]
- 14.Subbiah V, Iannotti NO, Gutierrez M, et al. FIGHT-101, a first-in-human study of potent and selective FGFR 1–3 inhibitor pemigatinib in pan-cancer patients with FGF/FGFR alterations and advanced malignancies. Ann Oncol 2022; 33: 522–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Friedman HS, Prados MD, Wen PY, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol 2009; 27: 4733–40. [DOI] [PubMed] [Google Scholar]
- 16.Abou-Alfa GK, Sahai V, Hollebecque A, et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol 2020; 21: 671–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meric-Bernstam F, Bahleda R, Hierro C, et al. Futibatinib, an Irreversible FGFR1–4 Inhibitor, in Patients with Advanced Solid Tumors Harboring FGF/FGFR Aberrations: A Phase I Dose-Expansion Study. Cancer Discov 2022; 12: 402–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferris RL, Blumenschein G Jr., Fayette J, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med 2016; 375: 1856–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang-Gillam A, Li CP, Bodoky G, et al. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): a global, randomised, open-label, phase 3 trial. Lancet 2016; 387: 545–57. [DOI] [PubMed] [Google Scholar]
- 20.Dosne AG, Valade E, Stuyckens K, et al. Erdafitinib’s effect on serum phosphate justifies its pharmacodynamically guided dosing in patients with cancer. CPT Pharmacometrics Syst Pharmacol 2022; 11: 569–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Touat M, Ileana E, Postel-Vinay S, Andre F, Soria JC. Targeting FGFR signaling in cancer. Clin Cancer Res 2015; 21: 2684–94. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Janssen Pharmaceutical Companies of Johnson & Johnson’s data sharing policy is available at https://www.janssen.com/clinical-trials/transparency. As noted on this site, requests for study data access can be submitted through the Yale Open Data Access (YODA) Project site at http://yoda.yale.edu.