

Key Points
-
•
We clinically characterized 3 additional, recurring KMT2A-r groups and identified specific ACAs of independent prognostic significance.
-
•
We present an optimized, fusion-based risk-group stratification of KMT2A-r pediatric AML.
Visual Abstract
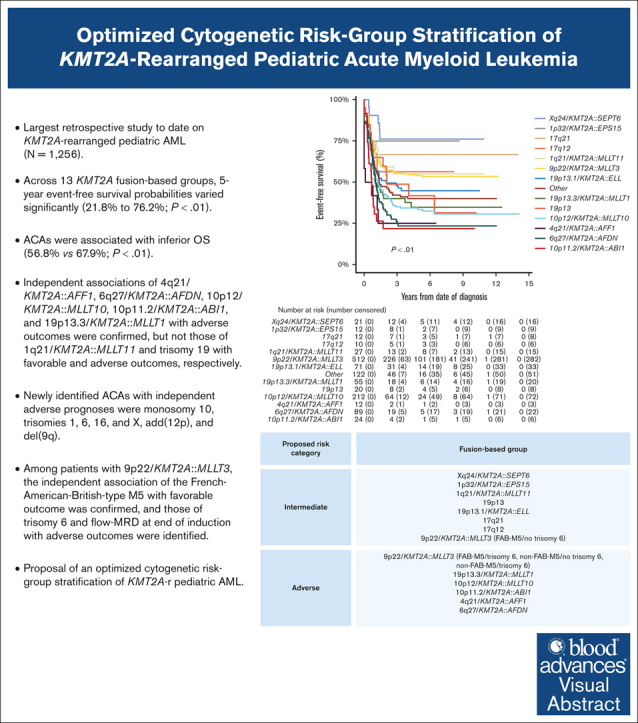
Abstract
A comprehensive international consensus on the cytogenetic risk-group stratification of KMT2A-rearranged (KMT2A-r) pediatric acute myeloid leukemia (AML) is lacking. This retrospective (2005-2016) International Berlin-Frankfurt-Münster Study Group study on 1256 children with KMT2A-r AML aims to validate the prognostic value of established recurring KMT2A fusions and additional cytogenetic aberrations (ACAs) and to define additional, recurring KMT2A fusions and ACAs, evaluating their prognostic relevance. Compared with our previous study, 3 additional, recurring KMT2A-r groups were defined: Xq24/KMT2A::SEPT6, 1p32/KMT2A::EPS15, and 17q12/t(11;17)(q23;q12). Across 13 KMT2A-r groups, 5-year event-free survival probabilities varied significantly (21.8%-76.2%; P < .01). ACAs occurred in 46.8% of 1200 patients with complete karyotypes, correlating with inferior overall survival (56.8% vs 67.9%; P < .01). Multivariable analyses confirmed independent associations of 4q21/KMT2A::AFF1, 6q27/KMT2A::AFDN, 10p12/KMT2A::MLLT10, 10p11.2/KMT2A::ABI1, and 19p13.3/KMT2A::MLLT1 with adverse outcomes, but not those of 1q21/KMT2A::MLLT11 and trisomy 19 with favorable and adverse outcomes, respectively. Newly identified ACAs with independent adverse prognoses were monosomy 10, trisomies 1, 6, 16, and X, add(12p), and del(9q). Among patients with 9p22/KMT2A::MLLT3, the independent association of French-American-British–type M5 with favorable outcomes was confirmed, and those of trisomy 6 and measurable residual disease at end of induction with adverse outcomes were identified. We provide evidence to incorporate 5 adverse-risk KMT2A fusions into the cytogenetic risk-group stratification of KMT2A-r pediatric AML, to revise the favorable-risk classification of 1q21/KMT2A::MLLT11 to intermediate risk, and to refine the risk-stratification of 9p22/KMT2A::MLLT3 AML. Future studies should validate the associations between the newly identified ACAs and outcomes and unravel the underlying biological pathogenesis of KMT2A fusions and ACAs.
Introduction
KMT2A-rearranged (KMT2A-r) acute myeloid leukemia (AML) is a heterogeneous pediatric AML subtype involving chromosomal rearrangement of the KMT2A (formerly known as MLL) gene located at chromosome 11q23.1 Outcome of this subtype is highly variable and related to the KMT2A fusion partner, as determined in our previous International Berlin-Frankfurt-Münster (I-BFM) Study Group (SG; I-BFM-SG) analysis (1993-2005).2 Our more recent (2005-2016) I-BFM-SG cohort demonstrated that the outcome of childhood KMT2A-r AML is also dependent on flow cytometry–based measurable residual disease (flow-MRD) at end of induction 2 (EOI2).3
The most frequently occurring KMT2A translocation, t(9;11)(p22;q23) (9p22/KMT2A::MLLT3 fusion), has been associated with an intermediate prognosis.2,4 However, among patients with 9p22/KMT2A::MLLT3, significantly better survival was repeatedly reported for those with the French-American-British (FAB)–type M5.2,5 The translocation t(1;11)(q21;q23) (1q21/KMT2A::MLLT11 fusion) has been associated with a favorable outcome,2 although not confirmed by others.4 Markedly inferior outcomes have been reported for the translocations t(4;11)(q21;q23) (4q21/KMT2A::AFF1 fusion), t(6;11)(q27;q23) (6q27/KMT2A::AFDN fusion), t(10;11)(p12;q23) (10p12/KMT2A::MLLT10 fusion), t(10;11)(p11.2;q23) (10p11.2/KMT2A::ABI1 fusion), and t(11;19)(q23;p13.3) (19p13.3/KMT2A::MLLT1 fusion),2,4 which are often considered as adverse risk. In our more recent I-BFM-SG analysis,3 these 5 fusions were clustered into an adverse-risk fusion-based group, which was independently associated with a poor outcome, like in the study by Pollard et al.4
The Children’s Oncology Group incorporated these 5 distinct adverse-risk KMT2A fusions as unfavorable prognostic markers into the treatment stratification algorithm of their ongoing AAML1831 trial (ClinicalTrials.gov identifier: NCT04293562).6,7 However, other SGs did not consider these fusions, incorporated only few, or relied more on flow-MRD as a prognostic factor because of the lack of a comprehensive international consensus on risk-group stratification of childhood KMT2A-r AML.8,9
Additional cytogenetic aberrations (ACAs) have been reported to be of prognostic value in childhood KMT2A-r AML.2,5 Trisomy 8 was associated with a favorable outcome, whereas trisomy 19 and structural aberrations were associated with adverse outcomes.5
The aims of this large I-BFM-SG study were to validate the prognostic value of previously defined recurring KMT2A fusions and reported ACAs and to define additional, recurring KMT2A fusions and ACAs, evaluating their prognostic relevance. The overall aim was to provide evidence to optimize the cytogenetic risk-group stratification of KMT2A-r pediatric AML.
Methods
Study design and patients
This retrospective study included patient data from 1256 children with KMT2A-r AML, assembled from 15 pediatric AML SGs/countries affiliated with the I-BFM-SG (supplemental Table 1). This same cohort was used in our previous study, analyzing the impact of flow-MRD and use of allogeneic stem cell transplantation (allo-SCT) in first complete remission (CR1) on the outcome of patients with KMT2A-r AML.3 Patients were treated according to national or SG pediatric AML clinical trials,10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23 approved by the institutional ethics committees of all collaborating centers. The study included patients aged<19 years with de novo KMT2A-r AML, diagnosed between 1 January 2005 and 31 December 2016. Patients with a diagnosis of acute promyelocytic leukemia, isolated myeloid sarcoma, myeloid leukemia of Down syndrome, and/or who had received previous anticancer treatment for diseases other than AML for >1 week were excluded a priori. Data were validated for accuracy and correctness.
Cytogenetic analysis
KMT2A rearrangements were detected by cytogenetics (G-, Q-, or R-banding according to local practice), with some confirmed by fluorescence in situ hybridization, or reverse transcription polymerase chain reaction within the local centers. Karyotypes were reviewed by 2 authors (R.E.v.W. and C.J.H.) and written according to the International System for Human Cytogenetic Nomenclature 2020.24 Patients were assigned to 1 of 10 individual, recurring fusion-based KMT2A-r groups or the KMT2A-other group, as previously described by Balgobind et al.2 Two authors (R.E.v.W. & C.J.H.) validated the group assignments and defined additional, recurring KMT2A-r groups when the same fusion was observed in at least 10 patients. Unknown fusions and those present in <10 patients remained assigned to the KMT2A-other group.
An ACA was defined as the presence of an acquired chromosomal abnormality in addition to the KMT2A rearrangement. Constitutional abnormalities did not constitute ACA. Patients with incomplete karyotypes were excluded from the ACA analysis. Ploidy changes were regarded as a single ACA. Patients with ACAs were categorized into having numerical, structural, or both numerical and structural aberrations. Numerical aberrations were defined as the loss or gain of whole chromosomes, whereas structural aberrations involved changes to chromosome short (p) or long (q) arms. Within the category of both numerical and structural aberrations, patients with separate numerical, and structural aberrations were included, as well as gains of structurally abnormal chromosomes, for example, +der(9p) and +i(8q). The gain of a marker chromosome was classified as a numerical aberration.
Statistical analyses
CR was defined as <5% blasts in the bone marrow after 2 induction courses, with regeneration of peripheral blood cells and the absence of extramedullary disease and cells with Auer rods. Patients who did not achieve CR after induction therapy were considered refractory to treatment. Relapse was defined as ≥5% blasts in the bone marrow, reappearance of leukemic blasts in the peripheral blood, or the presence of extramedullary disease after initial CR. Event-free survival (EFS) was calculated from the date of diagnosis to the date of the first event or last follow-up. Events included induction failure (ie, death before the start of intended treatment, death within 42 days after the start of treatment [early death], death after >42 days but before CR assessment, or refractory disease), death in CR, relapse, and secondary malignancy. Induction failure was considered an event at time zero. Cumulative incidence of relapse (CIR) was defined as the time from end of induction 1 until relapse for patients in CR, with deaths without a relapse considered competing events. Overall survival (OS) was calculated from the date of diagnosis to the date of death or last follow-up.
The χ2 test was used to compare differences in proportions of clinical characteristics and groups. The Mann-Whitney U and Kruskal-Wallis tests were used to compare differences in medians of 2 or >2 groups, respectively. The prognostic impact of recurring ACAs, occurring in at least 10 patients, was explored. Probabilities of EFS and OS with 95% confidence intervals (CIs) were estimated with the Kaplan-Meier method and compared using the log-rank test. CIR estimates with 95% CIs were compared using the Gray test for competing risks. Cox proportional hazards models were used to calculate hazard ratios (HRs) with 95% CIs. Variables with a 2-sided P-value < .05 in univariable Cox regression analyses were included in multivariable Cox regression models. Subsequently, variables with a 2-sided P-value > .10 were omitted from the final multivariable models by stepwise backward elimination. To correct for multiple testing, 2-sided P-values ≤ .01 were considered statistically significant. SPSS version 28 and R version 4.1.2 were used.
Results
Patient characteristics and ACA distribution
A total of 1256 children with KMT2A-r AML were included, of whom 1130 (90.0%) were assigned to 1 of 13 KMT2A-r groups and 126 (10.0%) to the KMT2A-other group (Figure 1). 9p22/KMT2A::MLLT3 (n = 544, 43.3%), 10p12/KMT2A::MLLT10 (n = 218, 17.4%), 6q27/KMT2A::AFDN (n = 92, 7.3%), and t(11;19)(q23;p13.1) (19p13.1/KMT2A::ELL fusion) (n = 75, 6.0%) were most frequent, together accounting for around 75% of cases. As compared with Balgobind et al,2 3 additional, recurring KMT2A-r groups were defined: t(X;11)(q24;q23) (Xq24/KMT2A::SEPT6 fusion) (n = 22, 1.8%), t(1;11)(p32;q23) (1p32/KMT2A::EPS15 fusion) (n = 13, 1.0%), and t(11;17)(q23;q12) (17q12, variable at the molecular level) (n = 10, 0.8%).
Figure 1.

Distribution of the fusion-based groups and the presence and type of ACAs in our cohort of childhood KMT2A-r AML. 9p22/KMT2A::MLLT3 refers to t(9;11)(p22;q23) (n = 544), 10p12/KMT2A::MLLT10 to t(10;11)(p12;q23) (n = 218), 6q27/KMT2A::AFDN to t(6;11)(q27;q23) (n = 92), 19p13.1/KMT2A::ELL to t(11;19)(q23;p13.1) (n = 75), 19p13.3/KMT2A::MLLT1 to t(11;19)(q23;p13.3) (n = 56), 1q21/KMT2A::MLLT11 to t(1;11)(q21;q23) (n = 28), 10p11.2/KMT2A::ABI1 to t(10;11)(p11.2;q23) (n = 24), 19p13 to t(11;19)(q23;p13) without ascertained subband (n = 23), Xq24/KMT2A::SEPT6 to t(X;11)(q24;q23) (n = 22), 17q21 to t(11;17)(q23;q21) (n = 13), 1p32/KMT2A::EPS15 to t(1;11)(p32;q23) (n = 13), 4q21/KMT2A::AFF1 to t(4;11)(q21;q23) (n = 12), and 17q12 to t(11;17)(q23;q12) (n = 10). Of the 1200 patients with complete karyotypes, 638 had no ACAs and 562 had ACAs, of whom 211 had solely numerical ACAs, 159 solely structural aberrations, and 192 both numerical and structural ACAs. Figure created with BioRender.com.
Among the 1256 patients, only 56 (4.5%) had incomplete karyotypes. There were no statistically significant differences between patients with and without complete karyotypes in terms of sex, median age, white blood cell (WBC) count at diagnosis, central nervous system involvement, FAB-type, KMT2A-r groups, and clinical outcome (supplemental Table 2). Of the 1200 patients with complete karyotypes, 562 (46.8%) had ACAs (Figure 1). The number of ACAs ranged from 0 to 17. The median number of ACAs among patients with ACAs was 2 (interquartile range, 1.0-3.0).
Table 1 shows the clinical characteristics and outcomes of the total cohort, stratified by KMT2A-r group. There were statistically significant differences in median age and WBC count at diagnosis and in the proportions of FAB-type and type of ACAs between these groups (Table 1). In all KMT2A-r groups, the median age at diagnosis was <4 years except for the 19p13.3/KMT2A::MLLT1, t(11;19)(q23;p13) (19p13, subband unknown), and 6q27/KMT2A::AFDN groups, in which the median ages were 6.8, 9.2, and 10.5 years (P < .01), respectively. The median WBC count at diagnosis was higher in patients with 19p13 (50.8 × 109/L), 6q27/KMT2A::AFDN (65.8 × 109/L), and 4q21/KMT2A::AFF1 (114.3 × 109/L) than in other KMT2A-r groups (P < .01). Most patients were classified as FAB-M5 (n = 619, 71.5%) or FAB-M4 (n = 121, 14.0%), but these proportions were not similar across all groups (P < .01). For example, most children with 1q21/KMT2A::MLLT11 were classified as FAB-M4.
Table 1.
Clinical characteristics and outcomes of 1256 children with KMT2A-r AML and stratified by fusion-based group
| Total | 9p22/KMT2A::MLLT3 | 10p12/KMT2A::MLLT10 | 6q27/ KMT2A::AFDN | 19p13.1/KMT2A::ELL | 19p13.3/KMT2A::MLLT1 | 1q21/KMT2A::MLLT11 | 10p11.2/KMT2A::ABI1 | |
|---|---|---|---|---|---|---|---|---|
| No. (%) | 1256 (100) | 544 (43.3) | 218 (17.4) | 92 (7.3) | 75 (6.0) | 56 (4.5) | 28 (2.2) | 24 (1.9) |
| Sex, no. (%) (n = 1235) | ||||||||
| Male | 638 (51.7) | 276 (51.9) | 127 (58.5) | 44 (47.8) | 39 (52.7) | 30 (54.5) | 9 (32.1) | 14 (63.6) |
| Female | 597 (48.3) | 256 (48.1) | 90 (41.5) | 48 (52.2) | 35 (47.3) | 25 (45.5) | 19 (67.9) | 8 (36.4) |
| Age at diagnosis, y (n = 1256) | ||||||||
| Median (IQR) | 2.5 (1.0-10.0) | 3.0 (1.1-9.1) | 1.6 (0.7-7.7) | 10.5 (5.4-15.2) | 3.6 (0.6-12.3) | 6.8 (1.6-13.2) | 1.2 (0.5-3.0) | 1.5 (0.9-5.1) |
| WBC count, ×109/L, (n = 1186) | ||||||||
| Median (IQR) | 21.4 (5.7-87.8) | 12.3 (4.4-74.2) | 15.1 (5.2-57.7) | 65.8 (20.4-120.8) | 33.2 (11.0-93.7) | 28.1 (12.1-92.3) | 30.4 (9.0-63.0) | 30.9 (7.5-45.9) |
| CNS involvement, No. (%) (n = 722) | ||||||||
| Negative | 574 (79.5) | 274 (83.3) | 105 (80.8) | 41 (83.7) | 25 (69.4) | 19 (65.5) | 12 (80) | 7 (63.6) |
| Positive | 148 (20.5) | 55 (16.7) | 25 (19.2) | 8 (16.3) | 11 (30.6) | 10 (34.5) | 3 (20) | 4 (36.4) |
| FAB-type, no. (%) (n = 866) | ||||||||
| FAB-M0 | 18 (2.1) | 7 (1.8) | 0 (0) | 3 (5.2) | 0 (0) | 2 (6.1) | 1 (5.3) | 0 (0) |
| FAB-M1 | 33 (3.8) | 9 (2.3) | 1 (0.7) | 12 (20.7) | 3 (6.5) | 0 (0) | 1 (5.3) | 0 (0) |
| FAB-M2 | 19 (2.2) | 6 (1.5) | 1 (0.7) | 1 (1.7) | 4 (8.7) | 1 (3.0) | 4 (21.1) | 0 (0) |
| FAB-M4 | 121 (14.0) | 29 (7.4) | 11 (7.2) | 15 (25.9) | 17 (37.0) | 6 (18.2) | 9 (47.4) | 1 (6.7) |
| FAB-M5 | 619 (71.5) | 308 (78.6) | 132 (86.3) | 26 (44.8) | 20 (43.5) | 22 (66.7) | 3 (15.8) | 13 (86.7) |
| FAB-M7 | 31 (3.6) | 21 (5.4) | 6 (3.9) | 0 (0) | 0 (0) | 2 (6.1) | 0 (0) | 0 (0) |
| FAB unspecified | 25 (2.9) | 12 (3.1) | 2 (1.3) | 1 (1.7) | 2 (4.3) | 0 (0) | 1 (5.3) | 1 (6.7) |
| ACA, no. (%) (n = 1200) | ||||||||
| No | 638 (53.2) | 271 (52.5) | 100 (47.6) | 60 (67.4) | 44 (61.1) | 26 (47.3) | 20 (71.4) | 14 (58.3) |
| Yes | 562 (46.8) | 245 (47.5) | 110 (52.4) | 29 (32.6) | 28 (38.9) | 29 (52.7) | 8 (28.6) | 10 (41.7) |
| ACA type, no. (%) (n = 562) | ||||||||
| Structural | 159 (28.3) | 53 (21.6) | 44 (40.0) | 6 (20.7) | 4 (14.3) | 9 (31.0) | 3 (37.5) | 7 (70) |
| Numerical | 211 (37.5) | 110 (44.9) | 30 (27.3) | 9 (31.0) | 11 (39.3) | 11 (37.9) | 2 (25) | 2 (20) |
| Both∗ | 192 (34.2) | 82 (33.5) | 36 (32.7) | 14 (48.3) | 13 (46.4) | 9 (31.0) | 3 (37.5) | 1 (10) |
| CR, no. (%) (n = 1219) | 1066 (87.5) | 464 (89.9) | 187 (87.8) | 77 (85.6) | 62 (87.3) | 50 (90.9) | 22 (81.5) | 22 (91.7) |
| Clinical outcome | No. | % (95% CI) | No. | % (95% CI) | No. | % (95% CI) | No. | % (95% CI) | No. | % (95% CI) | No. | % (95% CI) | No. | % (95% CI) | No. | % (95% CI) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5-y pEFS | 1199 | 44.8 (41.9-47.7) | 512 | 54.0 (49.5-58.5) | 212 | 33.4 (26.7-40.1) | 89 | 23.3 (14.3-32.3) | 71 | 44.8 (33.0-56.6) | 55 | 34.7 (21.6-47.8) | 27 | 55.0 (36.0-74.0) | 24 | 21.8 (4.9-38.7) |
| 5-y pCIR | 1057 | 44.8 (41.7-47.9) | 459 | 36.2 (31.6-40.7) | 186 | 57.4 (49.6-64.4) | 77 | 66.3 (54.0-76.0) | 61 | 41.9 (29.2-54.2) | 50 | 54.1 (39.1-66.9) | 22 | 27.9 (10.9-47.9) | 22 | 71.4 (45.4-86.6) |
| 5-y pOS | 1217 | 62.6 (59.9-65.3) | 522 | 70.2 (66.1-74.3) | 214 | 54.0 (47.1-60.9) | 90 | 39.0 (27.8-50.2) | 72 | 67.8 (56.6-79.0) | 55 | 51.7 (37.6-65.8) | 27 | 73.3 (56.3-90.4) | 24 | 50.9 (29.9-71.9) |
| 19p13 | Xq24/KMT2A::SEPT6 | 17q21 | 1p32/KMT2A::EPS15 | 4q21/KMT2A::AFF1 | 17q12 | Other | P value | |
|---|---|---|---|---|---|---|---|---|
| No. (%) | 23 (1.8) | 22 (1.8) | 13 (1.0) | 13 (1.0) | 12 (1.0) | 10 (0.8) | 126 (10.0) | |
| Sex, no. (%) (n = 1235) | ||||||||
| Male | 8 (38.1) | 9 (42.9) | 7 (53.8) | 8 (61.5) | 5 (41.7) | 6 (60) | 56 (44.8) | .24 |
| Female | 13 (61.9) | 12 (57.1) | 6 (46.2) | 5 (38.5) | 7 (58.3) | 4 (40) | 69 (55.2) | |
| Age at diagnosis, y (n = 1256) | ||||||||
| Median (IQR) | 9.2 (1.0-13.0) | 1.4 (0.9-5.7) | 1.0 (0.3-3.8) | 0.6 (0.3-1.6) | 0.6 (0.1-1.9) | 1.7 (0.6-13.9) | 1.5 (0.6-7.7) | <.01 |
| WBC count, ×109/L, (n = 1186) | ||||||||
| Median (IQR) | 50.8 (11.4-103.0) | 18.0 (5.4-48.1) | 39.2 (8.4-127.4) | 39.4 (13.5-56.9) | 114.3 (24.3-401.0) | 41.0 (8.8-102.3) | 30.0 (8.2-145.8) | <.01 |
| CNS involvement, no. (%) (n = 722) | ||||||||
| Negative | 17 (94.4) | 9 (75) | 8 (72.7) | 8 (80) | 4 (66.7) | 3 (75) | 42 (67.7) | .10 |
| Positive | 1 (5.6) | 3 (25) | 3 (27.3) | 2 (20) | 2 (33.3) | 1 (25) | 20 (32.3) | |
| FAB-type, no. (%) (n = 866) | ||||||||
| FAB-M0 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (11.1) | 0 (0) | 4 (4.5) | <.01 |
| FAB-M1 | 0 (0) | 2 (15.4) | 0 (0) | 0 (0) | 2 (22.2) | 0 (0) | 3 (3.4) | |
| FAB-M2 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (16.7) | 1 (1.1) | |
| FAB-M4 | 4 (28.6) | 3 (23.1) | 6 (60) | 1 (11.1) | 0 (0) | 2 (33.3) | 17 (19.1) | |
| FAB-M5 | 10 (71.4) | 8 (61.5) | 4 (40) | 8 (88.9) | 4 (44.4) | 3 (50) | 58 (65.2) | |
| FAB-M7 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 2 (2.2) | |
| FAB unspecified | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 2 (22.2) | 0 (0) | 4 (4.5) | |
| ACA, no. (%) (n = 1200) | ||||||||
| No | 12 (52.2) | 16 (72.7) | 5 (45.5) | 7 (53.8) | 3 (25) | 4 (40) | 56 (48.7) | .02 |
| Yes | 11 (47.8) | 6 (27.3) | 6 (54.5) | 6 (46.2) | 9 (75) | 6 (60) | 59 (51.3) | |
| ACA type, no. (%) (n = 562) | ||||||||
| Structural | 0 (0) | 0 (0) | 0 (0) | 3 (50) | 3 (33.3) | 2 (33.3) | 25 (42.4) | <.01 |
| Numerical | 7 (63.6) | 4 (66.7) | 5 (83.3) | 2 (33.3) | 2 (22.2) | 3 (50) | 13 (22.0) | |
| Both∗ | 4 (36.4) | 2 (33.3) | 1 (16.7) | 1 (16.7) | 4 (44.4) | 1 (16.7) | 21 (35.6) | |
| CR, no. (%) (n = 1219) | 20 (95.2) | 21 (100) | 12 (92.3) | 10 (83.3) | 6 (50) | 9 (90) | 104 (85.2) | .02 |
| Clinical outcome | No. | % (95% CI) | No. | % (95% CI) | No. | % (95% CI) | No. | % (95% CI) | No. | % (95% CI) | No. | % (95% CI) | No. | % (95% CI) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5-y pEFS | 20 | 41.8 (18.9-64.7) | 21 | 76.2 (58.0-94.4) | 12 | 66.7 (40.0-93.4) | 12 | 75.0 (50.5-99.5) | 12 | 25.0 (0.5-49.5) | 10 | 56.3 (24.0-88.6) | 122 | 39.9 (30.9-48.9) | <.01 |
| 5-y pCIR | 19 | 43.4 (20.1-64.8) | 21 | 19.0 (5.7-38.3) | 11 | 9.1 (0.4-35.0) | 10 | 10.0 (0.5-37.4) | 6 | 50.0 (7.7-82.9) | 9 | 37.5 (7.2-69.4) | 104 | 52.2 (41.8-61.7) | <.01 |
| 5-y pOS | 20 | 62.4 (39.9-84.9) | 21 | 90.5 (78.0-100) | 13 | 69.2 (44.1-94.3) | 13 | 92.3 (78.0-100) | 12 | 25.0 (0.5-49.5) | 10 | 50.0 (13.5-86.5) | 124 | 60.8 (52.0-69.6) | <.01 |
Values in boldface indicate statistical significance. Fusion-based groups are displayed in descending order of occurrence. 9p22/KMT2A::MLLT3 refers to t(9;11)(p22;q23), 10p12/KMT2A::MLLT10 to t(10;11)(p12;q23), 6q27/KMT2A::AFDN to t(6;11)(q27;q23), 19p13 to t(11;19)(q23;p13) without ascertained subband, 19p13.1/KMT2A::ELL to t(11;19)(q23;p13.1), 19p13.3/KMT2A::MLLT1 to t(11;19)(q23;p13.3), 1q21/KMT2A::MLLT11 to t(1;11)(q21;q23), 10p11.2/KMT2A::ABI1 to t(10;11)(p11.2;q23), Xq24/KMT2A::SEPT6 to t(X;11)(q24;q23), 17q21 to t(11;17)(q23;q21), 4q21/KMT2A::AFF1 to t(4;11)(q21;q23), 1p32/KMT2A::EPS15 to t(1;11)(p32;q23), and 17q12 to t(11;17)(q23;q12).
CNS, central nervous system; No., number of patients; pCIR, probability of CIR; pEFS, probability of EFS; pOS, probability of OS.
Including patients who had separate numerical and structural aberrations, as well as patients with gain of a chromosome with a structural aberration, for example, +der(9p) and +i(8q).
Supplemental Tables 3 and 4 show the frequency distributions of all numerical and structural ACAs, respectively, in total and stratified by KMT2A-r group. There were 28 recurring ACAs (supplemental Table 5), of which trisomy 8 (n = 210, 37.4% of all ACA cases) was the most common. Analyses to test for associations between specific ACAs and KMT2A-r groups were not possible because of the small numbers.
The type of ACAs differed significantly across KMT2A-r groups (P < .01; Table 1). For example, numerical aberrations were more common in patients with 9p22/KMT2A::MLLT3 (44.9%), 17q12 (50%), 19p13 (63.6%), Xq24/KMT2A::SEPT6 (66.7%), and t(11;17)(q23;q21) (17q21, fusion unknown) (83.3%) than in other KMT2A-r groups. Associations between the presence and type of ACAs and clinical characteristics are shown in supplemental Table 6. The median WBC count at diagnosis was lower in patients with ACAs than in patients without ACAs, whereas ACAs in general were more likely to occur in patients with FAB-M7 and FAB-M0.
Outcome
Of the 1219 patients known to have commenced chemotherapy, 1066 (87.5%) achieved CR, with no significant differences in CR rates among KMT2A-r groups (P = .02; Table 1). However, EFS, CIR, and OS estimates of the KMT2A-r groups differed significantly (all P < .01; Table 1; Figure 2). Good outcomes were observed in patients with Xq24/KMT2A::SEPT6 and 1p32/KMT2A::EPS15, with EFS and OS rates exceeding 75% and 90%, respectively, and CIR rates below 20%. Most patients that achieved CR in these 2 groups were treated with chemotherapy only and did not receive allo-SCT in CR1 (16/21 patients with Xq24/KMT2A::SEPT6; 9/10 patients with 1p32/KMT2A::EPS15). Relapses in these groups occurred only in patients who did not undergo transplantation, and all but 1 patient were salvaged. Patients with 10p11.2/KMT2A::ABI1, 6q27/KMT2A::AFDN, and 4q21/KMT2A::AFF1 had very poor outcomes with EFS rates of 21.8% (95% CI, 4.9-38.7), 23.3% (95% CI, 14.3-32.3), and 25.0% (95% CI, 0.5-49.5), respectively. Patients with 10p12/KMT2A::MLLT10 and 19p13.3/KMT2A::MLLT1 also had poor outcomes, with EFS rates <40%. In these 5 KMT2A-r groups with poor outcomes, CIR rates were ≥50% (Table 1).
Figure 2.

Survival curves for pediatric patients with KMT2A-rearranged AML, stratified by fusion-based group. Kaplan-Meier estimates of (A) EFS, (B) CIR, and (C) OS of KMT2A fusion-based groups. KMT2A::MLLT3 refers to t(9;11)(p22;q23) (n = 544), KMT2A::MLLT10 to t(10;11)(p12;q23) (n = 218), KMT2A::AFDN to t(6;11)(q27;q23) (n = 92), KMT2A::ELL to t(11;19)(q23;p13.1) (n = 75), KMT2A::MLLT1 to t(11;19)(q23;p13.3) (n = 56), KMT2A::MLLT11 to t(1;11)(q21;q23) (n = 28), KMT2A::ABI1 to t(10;11)(p11.2;q23) (n = 24), 19p13 to t(11;19)(q23;p13) without ascertained subband (n = 23), KMT2A::SEPT6 to t(X;11)(q24;q23) (n = 22), 17q21 to t(11;17)(q23;q21) (n = 13), KMT2A::EPS15 to t(1;11)(p32;q23) (n = 13), KMT2A::AFF1 to t(4;11)(q21;q23) (n = 12), and 17q12 to t(11;17)(q23;q12) (n = 10).
Patients with ACAs had inferior OS compared with patients without ACAs (56.8% [95% CI, 52.5-61.1] vs 67.9% [95% CI, 64.2-71.6]; P < .01), but EFS and CIR rates were not statistically significantly different (supplemental Table 7; supplemental Figure 1). Patients with numerical ACAs showed better outcomes than patients with structural or both numerical and structural ACAs (EFS, 52.0% [95% CI, 44.9-59.1] vs 33.7% [95% CI, 25.9-41.5] vs 37.3% [95% CI, 30.1-44.6]; P < .01; CIR, 34.5% [95% CI, 27.5-41.6] vs 57.0% [95% CI, 47.5-65.4] vs 51.7% [95% CI, 43.2-59.6]; P < .01; OS, 64.0% [95% CI, 57.1-70.9] vs 53.9% [95% CI, 47.7-62.1] vs 50.8% [95% CI, 43.0-58.6]; P < .01; supplemental Table 7). EFS curves for patients with and without recurring ACAs that were included in multivariable Cox regression analyses on the basis of entry criterion 2-sided P value < .05 in univariable Cox regression analyses (supplemental Table 5) are shown in Figure 3, and the CIR and OS curves of these patients are shown in supplemental Figure 2. Patients with trisomy 8 had statistically significantly superior EFS and a lower CIR than patients without trisomy 8. Patients with monosomy 10 and del(9q) had statistically significantly inferior EFS and OS and a higher CIR than patients without these ACAs. Patients with trisomies 4, 6, 12, 16, and X, and add(12p) had statistically significantly inferior EFS and/or higher CIR than patients without these ACAs. Patients with trisomy 1 had a statistically significantly inferior OS than patients without trisomy 1.
Figure 3.

Survival curves for pediatric patients with KMT2A-rearranged AML with and without specific, recurring ACAs. Kaplan-Meier estimates of EFS of patients with and without (A) add (12p), (B) del(9q), (C) monosomy 10, (D) trisomy 1, (E) trisomy 4, (F) trisomy 6, (G) trisomy 8, (H) trisomy 12, (I) trisomy 16, (J) trisomy 17, and (K) trisomy X. Patients with specific ACAs are compared with patients with other ACAs.
Multivariable analyses
In multivariable analyses (Table 2), age >10 years was independently associated with inferior OS (HR, 1.8; P < .01), and WBC count >100 × 109/L with inferior EFS (HR, 1.3; P < .01) and OS (HR, 1.5; P < .01). The 10p12/KMT2A::MLLT10, 6q27/KMT2A::AFDN, and 10p11.2/KMT2A::ABI1 groups were independently associated with inferior EFS ([HR, 1.7; P < .01], [HR, 1.8; P < .01], and [HR, 2.6; P < .01], respectively) and OS ([HR, 1.8; P < .01], [HR, 1.8; P < .01], and [HR, 2.4; P < .01], respectively), and higher CIR ([HR, 1.7; P < .01], [HR, 2.0; P < .01], and [HR, 3.0; P < .01], respectively). The 19p13.3/KMT2A::MLLT1 group was independently associated with inferior EFS (HR, 1.6; P = .01) and higher CIR (HR, 1.8; P < .01). The 4q21/KMT2A::AFF1 group was independently associated with inferior EFS (HR, 2.7; P < .01) and OS (HR, 4.5; P < .01). Among ACAs, trisomy 6 was independently associated with inferior EFS (HR, 1.6; P = .01) and OS (HR, 1.7; P = .01) and higher CIR (HR, 2.0; P < .01). Add(12p) was independently associated with inferior EFS (HR, 2.2; P = .01) and trisomy 16 with inferior EFS (HR, 2.5; P < .01) and higher CIR (HR, 4.5; P < .01). Monosomy 10, trisomy 1, trisomy X, and del(9q) were independently associated with inferior OS ([HR, 2.8; P < .01], [HR, 3.9; P < .01], [HR, 2.8; P < .01], and [HR, 2.8; P < .01], respectively).
Table 2.
Multivariable analyses of EFS, CIR, and OS in childhood KMT2A-r AML
| pEFS |
pCIR |
pOS |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. | HR | 95% CI | P value | No. | HR | 95% CI | P value | No. | HR | 95% CI | P value | |
| Age at diagnosis, y | ||||||||||||
| ≤10 | 837 | 1.0 | 751 | 1.0 | 845 | 1.0 | ||||||
| >10 | 277 | 1.3 | 1.0-1.5 | .02 | 262 | 1.3 | 1.0-1.6 | .03 | 279 | 1.8 | 1.4-2.2 | <.01 |
| WBC count, ×109/L | ||||||||||||
| ≤100 | 865 | 1.0 | NA | 872 | 1.0 | |||||||
| >100 | 249 | 1.3 | 1.1-1.6 | <.01 | NA | NA | NA | NA | 252 | 1.5 | 1.2-1.8 | <.01 |
| Fusion-based group | ||||||||||||
| 9p22/KMT2A::MLLT3 | 479 | 1.0 | 440 | 1.0 | 484 | 1.0 | ||||||
| Xq24/KMT2A::SEPT6 | 21 | 0.5 | 0.2-1.1 | .08 | 21 | 0.5 | 0.2-1.3 | .14 | 21 | 0.3 | 0.1-1.4 | .13 |
| 1p32/KMT2A::EPS15 | 12 | 0.5 | 0.2-1.5 | .22 | 10 | 0.2 | 0.0-1.6 | .13 | 13 | 0.3 | 0.0-1.9 | .19 |
| 17q21 | 10 | 0.6 | 0.2-1.8 | .40 | 10 | 0.2 | 0.0-1.5 | .12 | 10 | 0.9 | 0.3-2.6 | .84 |
| 17q12 | 10 | 0.9 | 0.4-2.5 | .90 | 9 | 1.0 | 0.3-3.3 | .94 | 10 | 1.3 | 0.5-3.6 | .61 |
| 1q21/KMT2A::MLLT11 | 26 | 1.1 | 0.6-2.0 | .78 | 22 | 0.7 | 0.3-1.6 | .40 | 26 | 1.1 | 0.5-2.4 | .76 |
| 19p13.1/KMT2A::ELL | 67 | 1.3 | 0.9-1.9 | .13 | 58 | 1.3 | 0.8-2.0 | .24 | 67 | 1.1 | 0.7-1.8 | .65 |
| 19p13 | 14 | 1.1 | 0.5-2.3 | .85 | 19 | 1.3 | 0.6-2.6 | .51 | 14 | 1.4 | 0.6-3.3 | .38 |
| 19p13.3/KMT2A::MLLT1 | 52 | 1.6 | 1.1-2.3 | .01 | 49 | 1.8 | 1.2-2.8 | <.01 | 52 | 1.5 | 0.9-2.3 | .10 |
| 10p12/KMT2A::MLLT10 | 198 | 1.7 | 1.3-2.1 | <.01 | 178 | 1.7 | 1.3-2.2 | <.01 | 200 | 1.8 | 1.4-2.4 | <.01 |
| 4q21/KMT2A::AFF1 | 11 | 2.7 | 1.3-5.5 | <.01 | 6 | 1.6 | 0.5-5.1 | .40 | 11 | 4.5 | 2.2-9.4 | <.01 |
| 6q27/KMT2A::AFDN | 81 | 1.8 | 1.3-2.4 | <.01 | 74 | 2.0 | 1.4-2.8 | <.01 | 82 | 1.8 | 1.3-2.5 | <.01 |
| 10p11.2/KMT2A::ABI1 | 22 | 2.6 | 1.6-4.4 | <.01 | 22 | 3.0 | 1.7-5.2 | <.01 | 22 | 2.4 | 1.3-4.5 | <.01 |
| Other | 111 | 1.3 | 1.0-1.8 | .06 | 95 | 1.5 | 1.1-2.1 | .02 | 112 | 1.3 | 0.9-1.9 | .11 |
| Recurring ACAs | Yes/no | Yes/no | Yes/no | |||||||||
| Monosomy 10 | 13/1101 | 2.0 | 1.1-3.8 | .03 | 10/1003 | 2.1 | 1.0-4.5 | .04 | 13/1111 | 2.8 | 1.5-5.5 | <.01 |
| Trisomy 1 | NA | NA | NA | NA | NA | NA | NA | NA | 13/1111 | 3.9 | 2.0-7.4 | <.01 |
| Trisomy 4 | ∗ | ∗ | ∗ | ∗ | ∗ | ∗ | ∗ | ∗ | ∗ | ∗ | ∗ | ∗ |
| Trisomy 6 | 60/1054 | 1.6 | 1.1-2.3 | .01 | 47/966 | 2.0 | 1.4-3.0 | <.01 | 60/1064 | 1.7 | 1.1-2.5 | .01 |
| Trisomy 8 | 194/920 | 0.8 | 0.6-1.0 | .05 | 172/841 | 0.7 | 0.5-1.0 | .03 | ∗ | ∗ | ∗ | ∗ |
| Trisomy 12 | ∗ | ∗ | ∗ | ∗ | ∗ | ∗ | ∗ | ∗ | ∗ | ∗ | ∗ | ∗ |
| Trisomy 16 | 12/1102 | 2.5 | 1.3-4.8 | <.01 | 9/1004 | 4.5 | 2.1-9.5 | <.01 | NA | NA | NA | NA |
| Trisomy 17 | 8/1106 | 2.1 | 0.9-4.7 | .07 | ∗ | ∗ | ∗ | ∗ | 8/1116 | 2.2 | 0.9-5.1 | .08 |
| Trisomy X | 12/1102 | 2.2 | 1.1-4.3 | .03 | NA | NA | NA | NA | 12/1112 | 2.8 | 1.3-5.9 | <.01 |
| Add(12p) | 13/1101 | 2.2 | 1.2-3.9 | .01 | ∗ | ∗ | ∗ | ∗ | NA | NA | NA | NA |
| Del(9q) | 16/1098 | 2.0 | 1.1-3.5 | .03 | 14/999 | 2.2 | 1.1-4.5 | .02 | 16/1108 | 2.8 | 1.5-5.1 | <.01 |
Values in boldface indicate statistical significance. ACA type was excluded from multivariable analyses, as the specific ACAs and the ACA type variables are related. 9p22/KMT2A::MLLT3 refers to t(9;11)(p22;q23), 10p12/KMT2A::MLLT10 to t(10;11)(p12;q23), 6q27/KMT2A::AFDN to t(6;11)(q27;q23), 19p13 to t(11;19)(q23;p13) without ascertained subband, 19p13.1/KMT2A::ELL to t(11;19)(q23;p13.1), 19p13.3/KMT2A::MLLT1 to t(11;19)(q23;p13.3), 1q21/KMT2A::MLLT11 to t(1;11)(q21;q23), 10p11.2/KMT2A::ABI1 to t(10;11)(p11.2;q23), Xq24/KMT2A::SEPT6 to t(X;11)(q24;q23), 17q21 to t(11;17)(q23;q21), 4q21/KMT2A::AFF1 to t(4;11)(q21;q23), 1p32/KMT2A::EPS15 to t(1;11)(p32;q23), and 17q12 to t(11;17)(q23;q12).
NA, not applied (not significant variables in univariable Cox regression analyses, see supplemental Table 5); abbreviations are explained in Table 1.
Omitted from the final multivariable model of EFS/CIR/OS because of a P-value > .10 in stepwise backward elimination.
The 9p22/KMT2A::MLLT3 group
Within this group, patients with FAB-M5 had superior EFS (65.9% [95% CI, 60.2-71.6] vs 37.9% [95% CI, 26.9-48.9]; P < .01) and OS (80.3% [95% CI, 75.6-85.0] vs 52.3% [95% CI, 42.0-63.5]; P < .01), and lower CIR (24.5% [95% CI, 19.4-29.9] vs 48.6% [95% CI, 35.9-60.1]; P < .01) than patients with non–FAB-M5 (supplemental Table 7). In multivariable analyses (supplemental Table 8), FAB-M5 was independently associated with superior EFS (HR, 0.5; P < .01) and OS (HR, 0.4; P < .01) and lower CIR (HR, 0.5; P < .01), whereas trisomy 6 was independently associated with inferior EFS (HR, 2.2; P < .01) and OS (HR, 2.3; P = .01) and higher CIR (HR, 2.7; P < .01). Subsequently, multivariable analyses were performed including a combination variable of FAB-type and trisomy 6, showing that, in reference to FAB-M5/no trisomy 6, the combinations of FAB-M5/trisomy 6, non–FAB-M5/no trisomy 6, or non–FAB-M5/trisomy 6 were all independently associated with inferior EFS ([HR, 3.9; P < .01], [HR, 2.3; P < .01], and [HR, 3.1; P < .01], respectively) and OS ([HR, 4.3; P < .01], HR, 2.8; P < .01], and [HR, 4.1; P < .01], respectively) and higher CIR ([HR, 3.3; P = .02], [HR, 2.1; P < .01], and [HR, 5.0; P < .01], respectively). The prognostic value of recurring ACAs within other KMT2A-r groups was explored, but none were significantly associated with EFS, CIR, or OS (data not shown).
Having previously demonstrated the independent prognostic significance of flow-MRD at EOI2 in childhood KMT2A-r AML (with detailed analysis described previously),3 we explored its prognostic significance in the 9p22/KMT2A::MLLT3 group within the context of FAB-type (M5 vs non-M5). It was not feasible to assess this in both the context of FAB-type and recurring ACAs because of the lack of flow-MRD at EOI2 data in 60% of patients with 9p22/KMT2A::MLLT3. Patients with EOI2 MRD negativity had superior EFS (54.2% [95% CI, 47.1-61.3] vs 35.3% [95% CI, 12.6-58.0]; P = .02) and OS (73.6% [95% CI, 67.3-79.9] vs 47.1% [95% CI, 23.4-70.8]; P < .01), but CIR was not statistically significantly different. Among patients with FAB-M5, as well as among patients with non–FAB-M5, EFS did not statistically significantly differ between patients with EOI2 MRD negativity and MRD positivity, but subgroup numbers were small (supplemental Table 7). In multivariable analyses (supplemental Table 8), FAB-M5 was independently associated with superior EFS (HR, 0.5; P = .02), whereas flow-MRD at EOI2 was independently associated with inferior EFS (HR, 2.4; P = .04) and OS (HR, 2.5; P = .05). The addition of the variable flow-MRD at EOI2 did not change the effect of FAB-type on survival, and vice versa, as confirmed by the nonsignificant interaction terms (P = .52 for EFS; P = .63 for OS).
Discussion
This largest study on childhood KMT2A-r AML confirmed the independent adverse prognostic significance of the previously defined recurring, adverse-risk KMT2A-r groups (ie, 4q21/KMT2A::AFF1, 6q27/KMT2A::AFDN, 10p12/KMT2A::MLLT10, 4q21/KMT2A::ABI1, and 19p13.3/KMT2A::MLLT1; together representing about 30% of pediatric KMT2A-r AML cases), defined and clinically characterized 3 additional, recurring KMT2A-r groups (ie, Xq24/KMT2A::SEPT6, 1p32/KMT2A::EPS15, and 17q12), the former 2 with good outcomes, and refined risk-group stratification of the most frequently occurring KMT2A-r group, 9p22/KMT2A::MLLT3, based on FAB-type and the presence/absence of trisomy 6. Flow-MRD at EOI2 was also identified as an independent adverse prognosticator in this group. Furthermore, we identified novel ACAs that were independently associated with inferior EFS (ie, add(12p) and trisomies 6 and 16), higher CIR (ie, trisomies 6 and 16), or inferior OS (ie, monosomy 10, trisomies 1, 6, and X, and del(9q)).
This and our previous studies2,3 provide strong evidence for the inclusion of the previously defined adverse-risk KMT2A-r groups into the cytogenetic risk-group stratification algorithm of childhood KMT2A-r AML. With EFS rates <40%, patients with adverse-risk KMT2A fusions may benefit from high-risk–adapted treatment. However, we and others have previously shown that high-risk–adapted treatment approaches superior to allo-SCT in CR1 are urgently needed in this disease.2, 3, 4 In this regard, studies to elucidate the biological role of KMT2A fusions and identify novel therapeutic targets are needed to improve survival. Ongoing phase 1/2 studies including patients with KMT2A-r acute leukemia have shown encouraging clinical responses with the menin inhibitors SNDX-5613 (revumenib)25 and KO-539 (ziftomenib),26 which target and disrupt the KMT2A fusion protein complex. These inhibitors constitute a novel, promising class of targeted therapeutics for this disease. However, a recent study suggests that mutations in menin are acquired with the use of SNDX-5613, thereby mediating clinical resistance.27 Hopefully, such mutations can be avoided when menin inhibitors are combined with chemotherapy. Furthermore, the incorporation of the CD33-targeting immunoconjugate gemtuzumab ozogamicin into induction therapy in the AAML0531 trial improved EFS and reduced relapse risk in children with KMT2A-r AML.4
The outcome of the 1q21/KMT2A::MLLT11 group was not statistically significantly superior compared with that of the 9p22/KMT2A::MLLT3 group, which is in contrast to Balgobind et al2 but in agreement with Pollard et al.4 Therefore, we recommend revision of the previous favorable-risk classification of 1q21/KMT2A::MLLT11 to intermediate risk. The underlying biological function of 1q21/KMT2A::MLLT11 remains unclear, although high MLLT11 expression has been shown to be an independent adverse prognosticator in pediatric AML.28 Another study showed that MIR29B directly regulates MLLT11 expression in vitro and that low MIR29B expression corresponded to high MLLT11 expression in patients with AML, resulting in poor survival.29 In pediatric patients with 1q21/KMT2A::MLLT11 AML, it remains unknown how MLLT11 expression is regulated.
We defined and clinically characterized Xq24/KMT2A::SEPT6 (n = 22) and 1p32/KMT2A::EPS15 (n = 13) as 2 additional, recurring KMT2A-r groups with good outcomes, although not statistically significantly superior. Our findings need to be validated in future large cohort studies, which may include more patients with these fusions as screening methods have improved over the years. For now, it seems justified to consider these fusions as intermediate risk.
Our study confirms the independent favorable prognostic significance of FAB-M5 in the most common KMT2A-r group, 9p22/KMT2A::MLLT3, which is a notable repeated finding.2,5 It remains to be determined how FAB-M5 is associated with favorable outcomes in these children. Potentially, high sensitivity to chemotherapy,30 or overexpression of specific genes, such as IGSF4, may be related to differences in outcome. IGSF4 has been identified as a discriminative, epigenetically, upregulated gene in children with 9p22/KMT2A::MLLT3 AML with FAB-M5 morphology.31 We propose to consider patients who are non–FAB-M5 as adverse risk, as their EFS rate was <40% (supplemental Table 7). Although the use of FAB morphology has dwindled in clinical pediatric AML practice, the determination of FAB-type in these patients thus remains relevant. Furthermore, although the number of cases in specific subgroups may be limited, we propose to consider children with 9p22/KMT2A::MLLT3 AML with FAB-M5/no trisomy 6 as intermediate risk and those with FAB-M5/trisomy 6, non–FAB-M5/no trisomy 6, or non–FAB-M5/trisomy 6 as adverse risk, as their EFS rates were <40% and on par with those of adverse-risk KMT2A fusions (supplemental Table 7).
Compared with Coenen et al (supplemental Table 9),5 our study identified different recurring ACAs to have a prognostic impact. This may be explained by the larger number of patients, allowing us to identify ACAs of greater independent prognostic significance, conduction of our study over a different period with the application of different treatment protocols, the co-occurrence of ACAs, or concurrent gene mutation profiles. Trisomy 8 has been previously reported to be an independent prognosticator for improved survival among children with KMT2A-r AML.5 In our study, trisomy 8 was significantly associated with a superior outcome in univariable analyses only. Regarding structural ACAs in general, they have been previously reported as independent adverse prognostic indicators of EFS in childhood KMT2A-r AML.5 To our knowledge, we are the first to specifically identify add(12p) and del(9q) to be independently associated with inferior EFS and OS, respectively. Abnormalities of 12p have also been associated with an adverse outcome in pediatric AML in general,32, 33, 34 and del(9q) has been previously reported to be associated with lower CR rates among children with t(8;21)(q22;q22)/RUNX1::RUNX1T1 AML.35 Regarding numerical ACAs, an independent association of trisomy 6 with higher CIR was found in our study, whereas in the study of Coenen et al,5 it was associated with inferior OS in univariable analysis only. Furthermore, independent associations of monosomy 10 and trisomies 1, 16, and X with inferior outcomes were found, whereas the former 3 ACAs had not been previously described as recurring ACAs in pediatric AML. To date, biological indicators of the prognostic relevance of the ACAs identified in our study remain to be elucidated.
Limitations of our study include its retrospective nature, the diverse treatment regimens used across SGs/countries, although with similar chemotherapeutic backbones, and no inclusion of data on allo-SCT in CR1 and flow-MRD response at EOI2 in the entire cohort analysis. Inclusion of the latter was precluded because subgroups became too small because of the large number of KMT2A-r groups and the low number of patients with specific ACAs, as well as the overall low transplantation rate and lack of flow-MRD data, which was also discussed in our previous study analyzing the impact of flow-MRD and use of allo-SCT in CR1 on outcome in this disease.3 Furthermore, 10% of the cohort were assigned to the KMT2A-other group, and molecular genetic data were not available to analyze the mutational landscape. With the increasing use of flow-MRD assays, quantitative polymerase chain reaction, and next-generation sequencing, future studies will likely allow for accurate detection of MRD in all patients and identification of all (cryptic) fusion genes, as well as gene mutations, which may affect the prognosis of this disease.
In conclusion, from this study, we can propose an optimized cytogenetic risk-group stratification of KMT2A-r pediatric AML in the pre-menin inhibitor era. Table 3 highlights how the risk of fusion-based groups has evolved over time and shows our proposal. Children with KMT2A-r AML may be stratified into an intermediate-risk or adverse-risk group based on cytogenetics, or both cytogenetics and FAB morphology in 9p22/KMT2A::MLLT3 cases. Regarding the 3 additional, recurring KMT2A-r groups and newly identified ACAs, future studies should validate the associations found in this study. In addition, once a large cohort becomes available with more flow-MRD data, it would be of great interest to evaluate whether individual KMT2A-r groups and ACAs retain their independent prognostic value if flow-MRD response is considered in multivariable analyses. Moreover, future studies should validate the role of allo-SCT in CR1, especially among the adverse-risk KMT2A-r groups, elucidate the mutational landscape, and further unravel the underlying biological pathogenesis of KMT2A fusions and ACAs. Together with ongoing discoveries and encouraging results from new targeted therapeutics, it will likely lead to improved risk-group stratification and risk-adapted treatment, as well as enhanced survival of childhood KMT2A-r AML.
Table 3.
Evolution in risk of fusion-based groups over time and our proposed cytogenetic risk-group stratification of childhood KMT2A-r AML
| Fusion-based group | Balgobind et al 20092 |
Pollard et al 20214 |
van Weelderen et al 2024 |
||||||
|---|---|---|---|---|---|---|---|---|---|
| No. | 5-y pEFS (%) | Risk-group | No. | 5-y pEFS (%) | Risk-group | No. | 5-y pEFS (%) | Risk-group | |
| 1q21/KMT2A::MLLT11 | 24 | 92 | Favorable∗ | 5 | 60 | Intermediate | 27 | 55 | Intermediate |
| 9p22/KMT2A::MLLT3 | 321 | 50 | Intermediate | 82 | 49 | Intermediate | 512 | 54 | Intermediate |
| Non–FAB-M5 | 59 | 31 | Adverse | ND | ND | ND | 81 | 38 | Adverse |
| FAB-M5 | 254 | 59 | Intermediate† | ND | ND | ND | 298 | 66 | Intermediate† |
| FAB-M5/no trisomy 6 | ND | ND | ND | ND | ND | ND | 273 | 68 | Intermediate |
| FAB-M5/trisomy 6 | ND | ND | ND | ND | ND | ND | 11 | 29‡ | Adverse§ |
| Non–FAB-M5/no trisomy 6 | ND | ND | ND | ND | ND | ND | 66 | 39 | Adverse§ |
| Non–FAB-M5/trisomy 6 | ND | ND | ND | ND | ND | ND | 10 | 27 | Adverse§ |
| 19p13 | 31 | 49 | Intermediate | ND | ND | ND | 20 | 42 | Intermediate |
| 19p13.1/KMT2A::ELL | 33 | 46 | Intermediate | 15 | 65 | Intermediate | 71 | 45 | Intermediate |
| 19p13.3/KMT2A::MLLT1 | 23 | 46 | Intermediate | 7 | 14 | Adverse‖ | 55 | 35 | Adverse∗ |
| 17q21 | 12 | 42 | Intermediate | ND | ND | ND | 12 | 67 | Intermediate |
| 10p12/KMT2A::MLLT10 | 97 | 31 | Adverse∗ | 40 | 20 | Adverse‖ | 212 | 33 | Adverse∗ |
| 4q21/KMT2A::AFF1 | 13 | 29 | Adverse | 2 | 0 | Adverse‖ | 12 | 25 | Adverse∗ |
| 10p11.2/KMT2A::ABI1 | 12 | 17 | Adverse∗ | 6 | 17 | Adverse‖ | 24 | 22 | Adverse∗ |
| 6q27/KMT2A::AFDN | 35 | 11 | Adverse∗ | 15 | 15 | Adverse‖ | 89 | 23 | Adverse∗ |
| Xq24/KMT2A::SEPT6 | ND | ND | ND | 5 | 80 | Intermediate | 21 | 76 | Intermediate |
| 1p32/KMT2A::EPS15 | ND | ND | ND | ND | ND | ND | 12 | 75 | Intermediate |
| 17q12 | ND | ND | ND | ND | ND | ND | 10 | 56 | Intermediate |
Risk-group assignment was determined arbitrarily according to the EFS rate. Fusion-based groups with an EFS rate of <40% were classified as adverse-risk, whereas those with an EFS rate >40% were designated at intermediate risk. Furthermore, fusion-based groups with an EFS rate >75%, which demonstrated an independent association with superior EFS compared with patients with 9p22/KMT2A::MLLT3, were considered favorable risk. 9p22/KMT2A::MLLT3 refers to t(9;11)(p22;q23), 10p12/KMT2A::MLLT10 to t(10;11)(p12;q23), 6q27/KMT2A::AFDN to t(6;11)(q27;q23), 19p13.1/KMT2A::ELL to t(11;19)(q23;p13.1), 19p13.3/KMT2A::MLLT1 to t(11;19)(q23;p13.3), 1q21/KMT2A::MLLT11 to t(1;11)(q21;q23), 10p11.2/KMT2A::ABI1 to t(10;11)(p11.2;q23), 19p13 to t(11;19)(q23;p13) without ascertained subband, Xq24/KMT2A::SEPT6 to t(X;11)(q24;q23), 17q21 to t(11;17)(q23;q21), 1p32/KMT2A::EPS15 to t(1;11)(p32;q23), 4q21/KMT2A::AFF1 to t(4;11)(q21;q23), and 17q12 to t(11;17)(q23;q12).
Independently associated with superior/inferior EFS compared with patients with 9p22/KMT2A::MLLT3.
Independently associated with superior EFS compared with patients with 9p22/KMT2A::MLLT3 with non–FAB-M5 morphology.
For patients with FAB-M5 morphology with trisomy 6, the 3-year pEFS is shown because this estimate could not be extrapolated to 5 years.
Independently associated with inferior EFS compared with patients with 9p22/KMT2A::MLLT3 with FAB-M5 morphology and without trisomy 6.
In the study by Pollard et al,4 these 5 fusion-based groups were clustered into a high-risk cohort, which was independently associated with inferior EFS compared with the non–high-risk cohort.
Conflict-of-interest disclosure: B.B. reports being on the speakers' bureau of Beckman Coulter, Becton Dickinson, and Amgen, and received travel and accommodation expenses from Beckman Coulter, Becton Dickinson, and Amgen. B.D.M. received honoraria from Novartis, Gilead Sciences, Pfizer, and Daiichi Sankyo, and travel expenses from Jazz Pharmaceuticals. S.E. received honoraria from Novartis and Medison, and reports a consulting or advisory role with Amgen. E.G. holds stock and other ownership interests in Pfizer and Moderna Therapeutics; reports a consulting or advisory role with Syndax and Jazz Pharmaceuticals; and is on the speakers' bureau of Jazz Pharmaceuticals. H.H. reports a consulting or advisory role with Novartis. F.L. received honoraria from Bellicum Pharmaceuticals, Miltenyi Biotec, bluebird bio, medac, Sobi, and Amgen, and reports a consulting or advisory role with Amgen, Novartis, and Pfizer. T.M. received honoraria from Amgen, Novartis, Sumitomo Dainippon Pharma Oncology, and Chugai Pharma. J.E.R. reports a consulting or advisory role in Kura Oncology, Biomea Fusion, and Pinotbio, and received research funding from AbbVie. A.T. received honoraria from and is on the speakers' bureau of BD Biosciences. C.M.Z. reports a consulting or advisory role in Takeda, Pfizer, AbbVie, Jazz Pharmaceuticals, Incyte, Novartis, and Kura Oncology, and received research funding from Takeda, AbbVie/Genentech, Pfizer, Jazz Pharmaceuticals, Kura Oncology, and Daiichi Sankyo. D.T. received honoraria from Amgen, Novartis, Chugai Pharma, Nippon Shinyaku, Ohara Pharmaceutical, Takeda, and Taiho Pharmaceutical, and reports a consulting or advisory role in Novartis and Meiji Seika Kaisha. The remaining authors declare no competing financial interests.
Acknowledgments
This study was an initiative of the International Berlin-Frankfurt-Münster Study Group including 15 individual pediatric AML study groups/countries: Aghia Sophia Children’s Hospital (Department of Pediatric Hematology and Oncology, Greece), Associazione Italiana Ematologia Oncologia Pediatrica (Italy), Berlin-Frankfurt-Münster (Germany and Austria), Belgian Society of Pediatric Hematology Oncology (Belgium); Children’s Cancer and Leukaemia Group (United Kingdom), Children’s Oncology Group (United States), Czech Pediatric Hematology (Czech Republic), Dutch Childhood Oncology Group (The Netherlands), Hong Kong Pediatric Hematology and Oncology Study Group (Hong Kong), Israel National Study (Israel), Japanese Pediatric Leukemia/Lymphoma Study Group (Japan), Leucémie Aiguë Myéloblastique Enfant (France), Nordic Society for Pediatric Hematology and Oncology (Scandinavia), Spanish Society of Pediatric Hematology and Oncology (Spain), and St. Jude Children’s Research Hospital (United States).
The authors thank Amaya Fernández, Andy Kolb, Anna Castleton, Arnaud Petit, Betsy Hirsch, Brenda Gibson, Harm van Tinteren, Iveta Janotová, Joyce Le, Karin Nebral, Katharine Patrick, Kathy Jackson, Kilian Crespí, Majid Madni, Markéta Žaliová, Michelle Cummins, Nora Mühlegger, Oskar Haas, Patricio Ledesma, Riccardo Masetti, Rubén Fernández, Shannon Cawte, and Todd Cooper for their individual efforts and contributions to this manuscript.
The studies from which any Children’s Oncology Group data were used were supported by National Cancer Institute/National Institutes of Health National Clinical Trial Network Operations Center grant number U10CA180886 (E.G. and R.B.G.). The content of this work is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This work was also supported by Swedish Childhood Cancer Foundation grants KF-2017-0010 and PL2018-0007 (U.N.-N.).
Authorship
Contribution: R.E.v.W., K.K., and G.J.L.K. conceptualized and designed the study; G.J.L.K. provided financial support; C.J.H., J.A., T.A., R.A., N.A.-C., E.B.-D., B.D.M., M.N.D., J.M.F.N., A.G., R.B.G., E.G., S.-Y.H., H.H., C.K., H.L., F.L., S.P., J.E.R., J.S., D.T., C.M.Z., and G.J.L.K. provided study materials or patients; R.E.v.W., K.K., C.J.H., J.A., T.A., R.A., N.A.-C., E.B.-D., B.B., B.D.M., M.N.D., S.E., J.M.F.N., A.G., R.B.G., H.A.d.G.-K., E.G., S.-Y.H., H.H., C.K., H.L., G.L., F.L., U.N.-N., S.P., M.R., J.E.R., J.S., A.T., and D.T. collected and assembled data; R.E.v.W., K.K., C.J.H., Y.J., B.F.G., E.G., H.H., F.L., J.E.R., J.S., C.M.Z., and G.J.L.K. analyzed and interpreted the data; and all authors wrote the manuscript and approved and were accountable for all aspects of the work.
Footnotes
Presented in abstract form at the 62nd annual meeting of the American Society of Hematology, San Diego, CA, December 2020, and at the 63rd annual meeting of the American Society of Hematology, Atlanta, GA, December 2021.
Individual participant data are not available to share. Participating study groups/countries should be contacted directly for the original data. Any overlap in our data set has been published in https://doi.org/10.1200/JCO.22.02120.
The full-text version of this article contains a data supplement.
Supplementary Material
References
- 1.Creutzig U, van den Heuvel-Eibrink MM, Gibson B, et al. Diagnosis and management of acute myeloid leukemia in children and adolescents: recommendations from an international expert panel. Blood. 2012;120(16):3187–3205. doi: 10.1182/blood-2012-03-362608. [DOI] [PubMed] [Google Scholar]
- 2.Balgobind BV, Raimondi SC, Harbott J, et al. Novel prognostic subgroups in childhood 11q23/MLL-rearranged acute myeloid leukemia: results of an international retrospective study. Blood. 2009;114(12):2489–2496. doi: 10.1182/blood-2009-04-215152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van Weelderen RE, Klein K, Harrison CJ, et al. Measurable residual disease and fusion partner independently predict survival and relapse risk in childhood KMT2A-rearranged acute myeloid leukemia: a study by the International Berlin-Frankfurt-Munster Study Group. J Clin Oncol. 2023;41(16):2963–2974. doi: 10.1200/JCO.22.02120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pollard JA, Guest E, Alonzo TA, et al. Gemtuzumab ozogamicin improves event-free survival and reduces relapse in pediatric KMT2A-rearranged AML: results from the phase III Children's Oncology Group Trial AAML0531. J Clin Oncol. 2021;39(28):3149–3160. doi: 10.1200/JCO.20.03048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coenen EA, Raimondi SC, Harbott J, et al. Prognostic significance of additional cytogenetic aberrations in 733 de novo pediatric 11q23/MLL-rearranged AML patients: results of an international study. Blood. 2011;117(26):7102–7111. doi: 10.1182/blood-2010-12-328302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cooper TM, Ries RE, Alonzo TA, et al. Revised risk stratification criteria for children with newly diagnosed acute myeloid leukemia: a report from the Children's Oncology Group. Blood. 2017;130(suppl 1):407. [Google Scholar]
- 7.Lamble AJ, Tasian SK. Opportunities for immunotherapy in childhood acute myeloid leukemia. Blood Adv. 2019;3(22):3750–3758. doi: 10.1182/bloodadvances.2019000357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klein K, de Haas V, Kaspers GJL. Clinical challenges in de novo pediatric acute myeloid leukemia. Expert Rev Anticancer Ther. 2018;18(3):277–293. doi: 10.1080/14737140.2018.1428091. [DOI] [PubMed] [Google Scholar]
- 9.Rubnitz JE, Kaspers GJL. How I treat pediatric acute myeloid leukemia. Blood. 2021;138(12):1009–1018. doi: 10.1182/blood.2021011694. [DOI] [PubMed] [Google Scholar]
- 10.Rubnitz JE, Inaba H, Dahl G, et al. Minimal residual disease-directed therapy for childhood acute myeloid leukaemia: results of the AML02 multicentre trial. Lancet Oncol. 2010;11(6):543–552. doi: 10.1016/S1470-2045(10)70090-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rubnitz JE, Lacayo NJ, Inaba H, et al. Clofarabine can replace anthracyclines and etoposide in remission induction therapy for childhood acute myeloid leukemia: the AML08 multicenter, randomized phase III trial. J Clin Oncol. 2019;37(23):2072–2081. doi: 10.1200/JCO.19.00327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tierens A, Bjørklund E, Siitonen S, et al. Residual disease detected by flow cytometry is an independent predictor of survival in childhood acute myeloid leukaemia; results of the NOPHO-AML 2004 study. Br J Haematol. 2016;174(4):600–609. doi: 10.1111/bjh.14093. [DOI] [PubMed] [Google Scholar]
- 13.Pession A, Masetti R, Rizzari C, et al. Results of the AIEOP AML 2002/01 multicenter prospective trial for the treatment of children with acute myeloid leukemia. Blood. 2013;122(2):170–178. doi: 10.1182/blood-2013-03-491621. [DOI] [PubMed] [Google Scholar]
- 14.Creutzig U, Zimmermann M, Bourquin JP, et al. Randomized trial comparing liposomal daunorubicin with idarubicin as induction for pediatric acute myeloid leukemia: results from study AML-BFM 2004. Blood. 2013;122(1):37–43. doi: 10.1182/blood-2013-02-484097. [DOI] [PubMed] [Google Scholar]
- 15.De Moerloose B, Reedijk A, de Bock GH, et al. Response-guided chemotherapy for pediatric acute myeloid leukemia without hematopoietic stem cell transplantation in first complete remission: results from protocol DB AML-01. Pediatr Blood Cancer. 2019;66(5) doi: 10.1002/pbc.27605. [DOI] [PubMed] [Google Scholar]
- 16.Gamis AS, Alonzo TA, Meshinchi S, et al. Gemtuzumab ozogamicin in children and adolescents with de novo acute myeloid leukemia improves event-free survival by reducing relapse risk: results from the randomized phase III Children's Oncology Group trial AAML0531. J Clin Oncol. 2014;32(27):3021–3032. doi: 10.1200/JCO.2014.55.3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aplenc R, Meshinchi S, Sung L, et al. Bortezomib with standard chemotherapy for children with acute myeloid leukemia does not improve treatment outcomes: a report from the Children's Oncology Group. Haematologica. 2020;105(7):1879–1886. doi: 10.3324/haematol.2019.220962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Creutzig U, Zimmermann M, Lehrnbecher T, et al. Less toxicity by optimizing chemotherapy, but not by addition of granulocyte colony-stimulating factor in children and adolescents with acute myeloid leukemia: results of AML-BFM 98. J Clin Oncol. 2006;24(27):4499–4506. doi: 10.1200/JCO.2006.06.5037. [DOI] [PubMed] [Google Scholar]
- 19.Petit A, Ducassou S, Leblanc T, et al. Maintenance therapy with interleukin-2 for childhood AML: results of ELAM02 phase III randomized trial. Hemasphere. 2018;2(6):e159. doi: 10.1097/HS9.0000000000000159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van der Velden VH, van der Sluijs-Geling A, Gibson BE, et al. Clinical significance of flowcytometric minimal residual disease detection in pediatric acute myeloid leukemia patients treated according to the DCOG ANLL97/MRC AML12 protocol. Leukemia. 2010;24(9):1599–1606. doi: 10.1038/leu.2010.153. [DOI] [PubMed] [Google Scholar]
- 21.Waack K, Schneider M, Walter C, et al. Improved outcome in pediatric AML - the AML-BFM 2012 study. Blood. 2020;136(suppl 1):12–14. [Google Scholar]
- 22.Tomizawa D, Tawa A, Watanabe T, et al. Excess treatment reduction including anthracyclines results in higher incidence of relapse in core binding factor acute myeloid leukemia in children. Leukemia. 2013;27(12):2413–2416. doi: 10.1038/leu.2013.153. [DOI] [PubMed] [Google Scholar]
- 23.Burnett AK, Russell NH, Hills RK, et al. Optimization of chemotherapy for younger patients with acute myeloid leukemia: results of the medical research council AML15 trial. J Clin Oncol. 2013;31(27):3360–3368. doi: 10.1200/JCO.2012.47.4874. [DOI] [PubMed] [Google Scholar]
- 24.McGowan-Jordan J, Hastings RJ, Moore S. Karger; 2020. ISCN 2020: An International System for human Cytogenomic Nomenclature. [DOI] [PubMed] [Google Scholar]
- 25.Issa GC, Aldoss I, DiPersio J, et al. The menin inhibitor revumenib in KMT2A-rearranged or NPM1-mutant leukaemia. Nature. 2023;615(7954):920–924. doi: 10.1038/s41586-023-05812-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Erba HP, Fathi AT, Issa GC, et al. Update on a phase 1/2 first-in-human study of the menin-KMT2A (MLL) inhibitor ziftomenib (KO-539) in patients with relapsed or refractory acute myeloid leukemia. Blood. 2022;140(suppl 1):153–156. [Google Scholar]
- 27.Perner F, Stein EM, Wenge DV, et al. MEN1 mutations mediate clinical resistance to menin inhibition. Nature. 2023;615(7954):913–919. doi: 10.1038/s41586-023-05755-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tse W, Meshinchi S, Alonzo TA, et al. Elevated expression of the AF1q gene, an MLL fusion partner, is an independent adverse prognostic factor in pediatric acute myeloid leukemia. Blood. 2004;104(10):3058–3063. doi: 10.1182/blood-2003-12-4347. [DOI] [PubMed] [Google Scholar]
- 29.Xiong Y, Li Z, Ji M, et al. MIR29B regulates expression of MLLT11 (AF1Q), an MLL fusion partner, and low MIR29B expression associates with adverse cytogenetics and poor overall survival in AML. Br J Haematol. 2011;153(6):753–757. doi: 10.1111/j.1365-2141.2011.08662.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zwaan CM, Kaspers GJ, Pieters R, et al. Cellular drug resistance profiles in childhood acute myeloid leukemia: differences between FAB types and comparison with acute lymphoblastic leukemia. Blood. 2000;96(8):2879–2886. [PubMed] [Google Scholar]
- 31.Kuipers JE, Coenen EA, Balgobind BV, et al. High IGSF4 expression in pediatric M5 acute myeloid leukemia with t(9;11)(p22;q23) Blood. 2011;117(3):928–935. doi: 10.1182/blood-2010-05-286138. [DOI] [PubMed] [Google Scholar]
- 32.Harrison CJ, Hills RK, Moorman AV, et al. Cytogenetics of childhood acute myeloid leukemia: United Kingdom Medical Research Council Treatment trials AML 10 and 12. J Clin Oncol. 2010;28(16):2674–2681. doi: 10.1200/JCO.2009.24.8997. [DOI] [PubMed] [Google Scholar]
- 33.Creutzig U, Zimmermann M, Reinhardt D, et al. Changes in cytogenetics and molecular genetics in acute myeloid leukemia from childhood to adult age groups. Cancer. 2016;122(24):3821–3830. doi: 10.1002/cncr.30220. [DOI] [PubMed] [Google Scholar]
- 34.Quessada J, Cuccuini W, Saultier P, Loosveld M, Harrison CJ, Lafage-Pochitaloff M. Cytogenetics of pediatric acute myeloid leukemia: a review of the current knowledge. Genes (Basel) 2021;12(6) doi: 10.3390/genes12060924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klein K, Kaspers G, Harrison CJ, et al. Clinical impact of additional cytogenetic aberrations, cKIT and RAS mutations, and treatment elements in pediatric t(8;21)-AML: results from an international retrospective study by the International Berlin-Frankfurt-Munster Study Group. J Clin Oncol. 2015;33(36):4247–4258. doi: 10.1200/JCO.2015.61.1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.