

Abstract
The pathogenesis of epilepsy remains unclear; however, a prevailing hypothesis suggests that the primary underlying cause is an imbalance between neuronal excitability and inhibition. Glucose-6-phosphate dehydrogenase (G6PD) is a key enzyme in the pentose phosphate pathway, which is primarily involved in deoxynucleic acid synthesis and antioxidant defense mechanisms and exhibits increased expression during the chronic phase of epilepsy, predominantly colocalizing with neurons. G6PD overexpression significantly reduces the frequency and duration of spontaneous recurrent seizures. Furthermore, G6PD overexpression enhances signal transducer and activator of transcription 1 (STAT1) expression, thus influencing N-methyl-d-aspartic acid receptors expression, and subsequently affecting seizure activity. Importantly, the regulation of STAT1 by G6PD appears to be mediated primarily through reactive oxygen species signaling pathways. Collectively, our findings highlight the pivotal role of G6PD in modulating epileptogenesis, and suggest its potential as a therapeutic target for epilepsy.
Keywords: Epilepsy, G6PD, Reactive oxygen species, STAT1
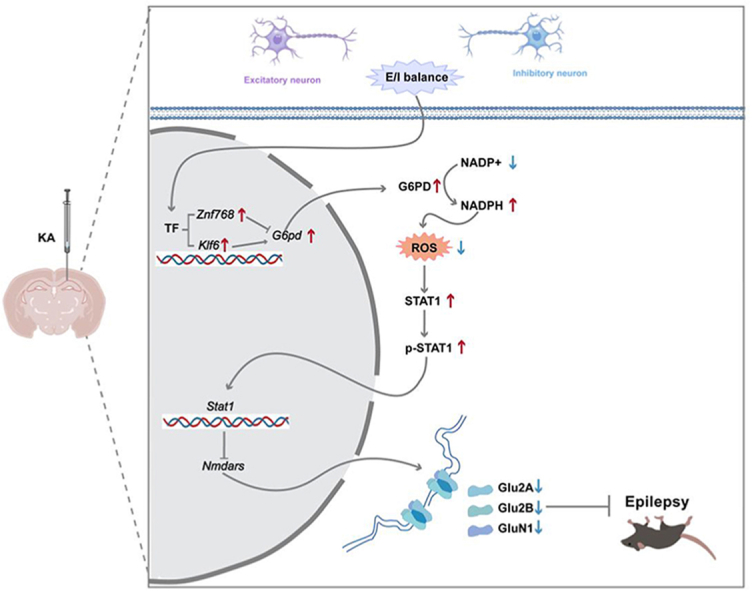
Graphical abstract
Schematic representation of the proposed G6PD-ROS-STAT1-NMDARs pathway that regulates epileptic seizures. The expression of STAT1 is facilitated by G6PD through suppression of ROS production, leading to an increase in the transcriptional inhibition of Nmdars (Grin1, Grin2a, and Grin2b), and a decrease in the expression of NMDARs, ultimately resulting in the alleviation of spontaneous seizures.
Abbreviations
- AAV
adeno-associated virus vectors
- ACSF
artificial cerebrospinal fluid
- AD
Alzheimer's disease
- AMPARs
α-amino-3-hydroxy-5-methyl-4-isoxazo-lepropionic acid receptors
- ASD
antiseizure drugs
- cDNA
complementary DNA
- DMSO
dimethyl sulfoxide
- DNA
deoxyribonucleic acid
- EEG
electroencephalogram
- G6PD
glucose-6-phosphate dehydrogenase
- KA
kainic acid
- NADPH
nicotinamide adenine dinucleotide phosphate
- NMDARs
N-methyl-d-aspartic acid receptors
- NOX-2
NADPH oxidase-2
- OS
oxidative stress
- PCR
polymerase chain reaction
- PD
Parkinson's disease
- PPP
pentose phosphate pathway
- PTZ
pentylenetetrazol
- qPCR
quantitative PCR
- RNA
ribonucleic acid
- ROI
region of interest
- ROS
reactive oxygen species
- SE
status epilepticus
- sEPSCs
spontaneous excitatory postsynaptic currents
- sIPSCs
spontaneous inhibitory postsynaptic currents
- SRSs
spontaneous recurrent seizures
- STAT1
signal transducer and activator of transcription 1
- WT
wild-type
1. Introduction
Epilepsy is a prevalent chronic neurological disorder, characterized by recurrent and long-term seizures, affecting approximately 70 million patients worldwide [1]. Approximately 30 % of patients exhibit clinical resistance to antiseizure drugs (ASD), necessitating urgent research on novel antiepileptic medications [2]. Neuronal excitatory-inhibitory imbalances resulting from genetic factors, trauma, infection, and metabolism can lead to hypersynchronous discharge, which is a crucial mechanism underlying epileptic seizures [3]. Some studies have further proposed that energy and metabolic abnormalities develop as the consequences of seizures, whereas others have suggested that energy and metabolic disorders contribute to the development of epilepsy and subsequent spontaneous recurrent seizures [4,5]. Energy changes in epilepsy depend on several metabolic pathways, most importantly glycolysis and the pentose phosphate pathway (PPP) [6].
Glucose-6-phosphate dehydrogenase (G6PD) is a pivotal enzyme in the PPP that is widely distribution and highly expressed in the brain [7]. Its primary roles include the regulation of ribose-5-phosphate for nucleotide synthesis, reduced nicotinamide adenine dinucleotide phosphate (NADPH) production for antioxidant defense against reactive oxygen species (ROS) [8]. A recent study revealed that patients with combined G6PD deficiency and Capicua transcriptional repressor mutations experienced more severe seizures [9]. Additionally, G6PD-deficient mice have been shown to exhibit increased oxidative damage in brain tissue [10]. Conversely, transgenic mice overexpressing G6PD and Drosophila models with elevated G6PD levels demonstrated enhanced NADPH concentrations that counteracted ROS-induced damage, resulting in improved resistance to age-related functional decline [11,12]. However, studies on the role of G6PD in epilepsy are lacking.
Oxidative Stress (OS) is characterized by the excessive production of ROS or a decrease in antioxidant capacity, both of which can result in cellular damage and death. OS further plays a crucial role in various neurological disorders such as stroke, epilepsy, Alzheimer's disease (AD), and Parkinson's disease (PD) [[13], [14], [15], [16], [17]]. A previous study by Prince Kumar Singh et al. showed that the upregulation of NADPH oxidase-2 (NOX2) expression as the main source of ROS in different epilepsy models [18]. Therefore, investigating the role of ROS is a pivotal step toward understanding the mechanisms underlying epilepsy.
In this study, we investigated the expression of G6PD during each phase following the establishment of an epileptic mouse model, examined its effect on epilepsy, and elucidated its underlying mechanism of action.
2. Materials and methods
2.1. Antibodies and reagents
The antibodies and reagents used in this study are listed in supplement Table 1-2.
2.2. Animals
Wild type (WT) C57BL/6J male mice (6-8-week-old, 20–25 g) were purchased from the Animal Center of the Chongqing Medical University. Map2-Cre-ERT2 mice were purchased from Shanghai Model Organisms Center, Inc. (Shanghai, China). Target mice were identified using a polymerase chain reaction (PCR). The required number of animals (sample size) was determined using a power calculation method, based on the prior experience. PCR analysis of the genomic DNA in the tail was performed using the following primers for Cre: (5′- GCATCACCCCGACGACTCAG - 3′), (5′- GTATTGGATAGCCTTCAGGCAC - 3′) and (5′- GGTGTTATAAGCAATCCCCAGAA - 3′). All PCR products were visualized on 1.2 % agarose gel.
All mice were housed in a specific-pathogen-free facility (five mice per cage) at 24 ± 2 °C and 60 % + 5 % relative humidity under a 12-h light-dark cycle (lights on from 09.00 to 21.00) with food and water available. All animal experiments were conducted under the protocols Institutional Animal Care and Use Committee-approved protocols at Chongqing University in accordance with the National Health Research Institute. Administration of 150 mg/kg of pentobarbital intraperitoneal was provided to deeply anesthetized the mice. Mice were then monitor until a lack of heart beat was noted for >60 s prior to tissue harvest. Rapid removal of the hippocampal tissue following cervical dislocation was followed by immediate placement on ice and collection of the hippocampal tissue on ice for further experimental requirements.
2.3. Epilepsy model
Mice were anesthetized with pentobarbital and fixed on a stereotaxic apparatus (RWD Life Science Co. Ltd., Shenzhen, China). Subsequently, 1.0 nmol of kainic acid (KA) (487-79-6; Sigma-Aldrich, USA) dissolved in 50 nL of saline was injected into the right hippocampus (anteroposterior, −1.5 mm; mediolateral, −1.5 mm; dorsoventral, −1.5 mm) to induced status epilepticus (SE) 2 h after the injection. SE was later terminated with diazepam (10 mg/kg). Mice (adCtrl, adG6PD, and adG6PD + fludarabine groups) were video monitored to quantify the number of spontaneous recurrent seizures (SRSs) over 7 consecutive days. Seizures (Racine's stages III–V) were quantified during an offline video review by two independent observers, one of whom was blinded to the treatment.
2.4. Adeno-associated virus vectors (AAV) construction and hippocampal injection
The AAV9 vectors carrying mouse full-length G6PD (adG6PD) or eGFP alone (adCtrl) were constructed by GeneChem Co., Ltd. (Shanghai, China). For adG6PD packaging, a rAAV‐CMV-DIO‐EGFP-mir155 (G6pdx) -WPRE vector was used. Empty AAV9 vectors coding eGFP were used as controls. Under anesthesia, AAV9 vectors (adCtrl and adG6PD groups) were injected bilaterally into the hippocampi of Map2-Cre-ERT2 mice. All AAV deliveries were performed with a volume of 0.6 μL (0.2 μL/min). The needle was kept in place for 5 min after each injection. To induce conditional overexpression of G6PD in the neurons, 75 mg/kg tamoxifen (10540-29-1; Sigma-Aldrich, UAS) was intraperitoneally administered daily for 5 d.
2.5. Pentylenetetrazol (PTZ)-induced seizures
As previously described, the convulsant PTZ (54-95-5; Sigma-Aldrich, UAS) was administered to induce acute seizures in mice that had received AG1 (or corn oil) and the 6-aminonicotinamide (or corn oil). As mentioned previously, seizures were triggered by repetitive intraperitoneal administration of PTZ every 10 min at a dose of 12.5 mg/kg, until generalized seizures were observed, or the cumulative dose reached 100 mg/kg. Seizures were monitored and assessed using the Racine scale, and mice that reached Racine scores of IV or V were considered to have epileptic kindling.
2.6. Electroencephalogram (EEG) recordings
Mice (adCtrl and adG6PD groups) that received intrahippocampal KA injections were used subjected to EEG recordings. After anesthetization, the mice were mounted on a stereotaxic apparatus, and electrodes were implanted in the brain cortex using a fixed head-mounted device. After surgery, the mice were returned to their home cage for 1 week. Then, the electrodes were then connected to the data conditioning and acquisition system (Pinnacle Technology) with an electrode wire, and the EEG activities were recorded continuously for 7 d. The Sirenia Seizure Pro software (Pinnacle Technology) was used for EEG signal analysis. Electrographic seizure events were defined as high amplitudes at two-folds of the baseline and durations lasting at least 10 s.
2.7. Drug administration
For G6PD activator administration, the mice received bilateral intrahippocampal injections of AG1 (HY-123962; MCE, USA) at 4 μg or corn oil before intraperitoneal injection PTZ.
Before the intraperitoneal injection of PTZ, mice were acclimatized to intraperitoneal injections of either the vehicle (saline) or the 6-aminonicotinamide (6-An) (HY–W010342; MCE, USA) for 5 d. The doses were based on a previous study [19].
For treatment with the STAT1 inhibitor fludarabine, mice received 5 mg/kg fludarabine (HY–B0069; MCE, USA) for 5 d [20].
2.8. Western blotting (WB)
Nuclear (R0050; Solarbio, China) and membrane (C500049; Sango Biotech, China) proteins were extracted in accordance with manufacturer's instructions provided with the commercial kit. Tissue samples were lysed for 30 min at 4 °C with radio immunoprecipitation assay lysis buffer lysate and centrifuged for 16000 g, at 4 °C, for 30 min to obtain the supernatant. Proteins were then is separated in sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel (8 % and 10 %), and transferred to a 0.45 μm polyvinylidene fluoride membrane (IPFL00010; Sigma-Aldrich, UAS). After transfer, the membranes were blocked for 15 min in protein free rapid blocking buffer (PS108P; Epizyme Biomedical Technology, China), after which they were incubated with primary antibody at 4 °C for 12–16 h. The next day, the membrane was washed three times with tris-buffered saline containing 0.1 % tween-20 for 5 min each. The membranes were then incubated with the secondary antibody for 1 h, after which they were washed for 5 min/three times. Membranes were imaged using the Fusion Imaging System and quantified using the ImageJ software. The antibodies used in this study and their dilution ratios are listed in Supplementary Table 1.
2.9. Quantitative PCR (qPCR)
Total ribonucleic acid (RNA) was extracted from epileptic or control hippocampal tissue samples using a Simple Total RNA Kit (DP451; TIANGEN, China), according to the manufacturer's instructions, after which complementary DNA (cDNA) was generated using a reverse transcriptase kit (R223-01; Vazyme, China). qPCR was performed using the Cham Q SYBR qPCR Master Mix kit (P071-01; Vazyme, China). β-actin was used as a standard control. The primers used in this study are listed in Supplementary Table 3.
2.10. Immunofluorescence staining
Frozen sections were prepared using previously reported methods [21]. The sections were equilibrated to room temperature, washed with phosphate buffer saline (PBS), permeabilized with PBS containing 0.4 % Triton X-100 (9036-19-5; Sigma-Aldrich, USA) for 30 min, and blocked with 10 % goat serum in PBS for 30 min. The samples were then incubated with primary antibody overnight at 4 °C, after which they were washed thrice with PBS and incubated with fluorophore-labeled secondary antibodies at room temperature for 1 h. Images were captured using a microscope (Nikon; Tokyo, Japan). Using Image Pro Plus software, each cell of the brain slices was segmented as an individual region of interest (ROI), and the mean pixel density of the ROI was measured [22,23]. The antibodies and dilution ratios used in this experiment are shown in Supplementary Table 2.
2.11. Oxidative stress parameters
To assess levels of ROS, including NADPH/NADP+ and GSH/GSSH detection in the control, 6-An, and 6-An and N-acetylcysteine (NAC) groups, HT22 cells in each well plate were first exposed to 100 μM KA for 12 h24, and then treated with 6-An (10 μM) or dimethyl sulfoxide (DMSO) for 12h [24,25]. The cells were treated with KA and 6-An prior treatment with DMSO or NAC (5 mM) for another 12h, after which they were collected for detection [26]. The levels of ROS, NADPH/NADP+, and GSH/GSSH levels were measured in the control, AG1, and dimethyl fumarate (DMF) groups. Twelve hours after the initial KA treatment, HT22 cells were treated with DMSO or AG1 (1 μM) for another 12 h, and cells were finally tested for ROS 12 h after receiving DMSO or DMF (100 μM) [27,28]. The ROS levels in HT22 cells were measured using 2′, 7′-dichlorofluorescein diacetates (DCFH-DA) (S0033S; Beyotime, China) and Dihydroethidium (DHE) (S0063; Beyotime, China) according to the manufacturer's protocol. The NADPH/NADP+ levels in HT22 cells were determined by using the WST-8 method according to the manufacturer's protocol (S0179; Beyotime, China). The GSH/GSSH levels in HT22 cells were determined by using the WST-8 method in accordance with the manufacturer's protocol (S0053; Beyotime, China).
2.12. Cell culture of HT22 and, HEK 293 cell
HT22 and HEK293 cells (Chinese Academy of Sciences) were maintained at 37 °C with 5 % CO2 and cultured in Dulbecco's modified Eagle's medium containing 10 % fetal bovine serum and 1 % penicillin-streptomycin.
2.13. Luciferase activity assay
HEK293 cells were seeded in black 96-well plates and transfected with luciferase reporter plasmids for 24 h. Luciferase activity was measured using a GloMax 96-microplate luminometer (Promega) and a Dual-Lumi Luciferase Reporter Gene Assay Kit (MA0520-1; Meilunbio, China).The control vector 3xflag-pcDNA3.1, expression plasmid G6pd promoter pDualuc-Basic, shuttle plasmid Znf768 (NM_146202)-pcDNA 3.1-3xFlag-C and Klf6 (NM_011803)-pcDNA 3.1-3xFlag-C were purchased from YouBio (Changsha, China)
2.14. RNA-Seq
RNA extraction, library building, sequencing, and bioinformatics analysis using RNA-Seq were conducted by LC Bio Technology Co., Ltd., Hangzhou., China.
2.15. Whole-cell patch-clamp recordings
Whole-cell patch-clamp recordings were conducted, as described in a previous study from our laboratory [29]. After anesthetization, mice in the adCtrl and adG6PD groups were sacrificed for the preparation of brain slices. Transverse hippocampal slices (300 μM) were prepared in a cold sterile slice solution (2 mM MgCl2, 2 mM CaCl2, 2.5 mM KCl, 1.25 mM KH2PO4, 26 mM NaHCO3, 220 mM sucrose, and 10 mM glucose [pH 7.4], bubbled with 95 % O2/5 % CO2). Slices were then allowed to recover in a storage chamber containing Mg2+-free artificial cerebrospinal fluid (ACSF) (2.5 mM KCl, 125 mM NaCl, 2 mM CaCl2, 1.25 mM KH2PO4, 26 mM NaHCO3, and 25 mM glucose [pH 7.4] bubbled with 95 % O2/5 % CO2) at 34 °C for 1 h. The slices were then fully submerged in flowing Mg2+-free ACSF (4 mL/min) at room temperature for recording. Spontaneous excitatory postsynaptic currents (sEPSCs) and spontaneous inhibitory postsynaptic currents (sIPSCs) were recorded. In the voltage clamp mode, the pyramidal neurons were clamped at −70 mV, and picrotoxin (100 μM) was added into the electrode fluid to record sEPSCs. In addition, 6,7-dinitroquinoxaline-2,3-dione (20 μM) and D-(−)-2-amino-5-phosphonovalerate (D-APV) (50 μM) were added to record the sIPSC.
All patch-clamp data were obtained using a MultiClamp 700 B patch-clamp amplifier (Axon Instruments, Burlingame, CA, USA) with a 2 kHz low-pass filter and 10 kHz sample rate and were recorded using pClamp 10 software (Molecular Devices; Sunnyvale, CA, USA). The data were analyzed using the Clampfit software (Molecular Devices).
2.16. Statistical analysis
Statistical analyses were performed using GraphPad Prism software (version 8.0; GraphPad Software, La Jolla, CA, USA). Differences in variables between two groups were assessed using an unpaired two-tailed Student's t-test. Differences in variables between more than two groups were assessed using one-way analysis of variance (ANOVA), followed by Tukey's post-hoc test. Differences between groups were analyzed using one- or two-way ANOVA. The sample size was selected based on a previously reported method. Data are presented as mean ± SEM. Significant differences are indicated as follows: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, while nonsignificant differences are indicated by the abbreviation “ns.”
3. Results
3.1. G6PD increases in the epileptic chronic phase
We conducted a comprehensive analysis of metabolic enzymes involved in glycolysis and PPP in epileptic and sham mice to investigate the correlation between metabolic enzymes and epilepsy. WB results demonstrated a significant increase in G6PD levels in the cortical and hippocampal tissues of epileptic mice compared to those in the sham group (Fig. 1a–d). To investigate the potential role of G6PD in temporal lobe epilepsy (TLE), we assessed the transcript levels and enzymatic activity of G6PD at various time points following KA-induced SE using qPCR and the WST-8 method. At 28 d, both the transcription level and enzymatic activity of G6PD were significantly higher than those in the sham control group (Fig. 1e and f).
Fig. 1.

Expression and functionality of pivotal enzymes in the glycolytic and pentose phosphate pathways. (a–d) Representative immunoblots (a, c) and quantification (b, d) of metabolism enzymes in the glycolysis and pentose phosphate pathways from the cortex and hippocampus of sham and chronic epileptic mice (n = 6). Data are presented as means ± SEM. **P < 0.01, ***P < 0.001, unpaired two-tailed Student's t-test. (e) Quantitative real-time polymerase chain reaction analysis of the expression of G6pd mRNA in the hippocampus at different time points after induction of status epilepticus (SE) (n = 5). Data are presented as means ± SEM. **P < 0.01, ***P < 0.001, one-way ANOVA with Tukey's post hoc test. (f) Relative enzyme activity of G6PD in the hippocampus at different time points after induction of SE (n = 3). Data are presented as means ± SEM. **P < 0.01, ***P < 0.001, one-way ANOVA with Tukey's post hoc test.
3.2. G6PD co-localized with neurons
The expression of G6PD in the central nervous system has previously been confirmed; however, its localization in the hippocampus has not yet been identified [30]. To address this gap, double-label immunofluorescence labeling was performed to investigate the specific cellular localization of endogenous G6PD. The results showed that G6PD colocalized with neuron-specific nucleoprotein (NeuN, a marker of neurons) in the hippocampus, but not with glial fibrillary acidic protein (GFAP, a marker of astrocytes) or ionized calcium-binding adapter molecule 1 (Iba-1, a marker of microglia) (Fig. 2a, b, and S1a-c). Consistent with the increased total expression of G6PD in the hippocampus, although the number of neurons decreased after KA modeling and the number of activated glial cells increased (Fig. 2c), the fluorescence intensity of G6PD increased (Fig. 2d). A similar outcome was observed in the temporal cortex alongside the hippocampal CA3 region, where G6PD colocalized with NeuN (Figs. S2a–b).
Fig. 2.

Localization of G6PD in epileptic brain tissues. (a–b) Immunostaining for G6PD with neuron-specific nucleoprotein (NeuN), glial fibrillary acidic protein (GFAP), and ionized calcium-binding adapter molecule 1 (Iba-1) in the sham and kainic acid (KA) groups at 28 d. Scale bars: 50 μm. (b–c) Quantitative analysis for the relative intensity of G6PD, NeuN, GFAP, and Iba-1 in single cells in the hippocampus CA3 region of epileptic mice at 28 d after KA injection (n = 50 cells from nine sections of each group, from three mice). Data are presented as means ± SEM. ***P < 0.001, unpaired two-tailed Student's t-test.
3.3. G6PD negatively modulates seizures
We further observed an upregulation of G6PD in neurons during the chronic phase of epilepsy; however, its role in promoting seizures or exerting an antiepileptic effect remains unclear. G6PD plays a neuroprotective role in cerebral ischemia by facilitating the PPP, suggesting that increased expression of G6PD during the chronic phase of epilepsy may exert antiepileptic properties. Considering that the intervention of the acute phase and/or latency will not only affect the severity of seizures during this period, but also affect the formation of chronic spontaneous epilepsy, we used Map2-Cre-ERT2 mice to overexpress G6PD at 28 days after KA modeling to detect the effect of exogenous addition of G6PD on chronic spontaneous seizures of epilepsy [31]. The efficiency of G6PD overexpression was confirmed after 3 weeks by immunofluorescence and WB (Fig. 3a–c). We further investigated the impact of enhanced G6PD expression on epileptic behavior in a KA-induced TLE model 40 day after SE (Fig. 3d), and found that G6PD overexpression decreased the daily number of SRSs, as well as the duration of seizure-like events in chronic epileptic mice (Fig. 3e–g). These results demonstrated that G6PD decreased seizure severity in chronic epilepsy models. We further assessed the role of G6PD in excitatory/inhibitory (E/I) balance, and analyzed the occurrence of spontaneous postsynaptic currents. Our findings suggest that upregulation of G6PD expression did not influence the frequency or amplitude of spontaneous inhibitory postsynaptic currents (Fig. 3h–j). However, it reduced the frequency and amplitude of spontaneous excitatory postsynaptic currents (Fig. 3k-m). These results indicated that increased G6PD expression leads to decreased excitatory signaling in the hippocampus.
Fig. 3.

G6PD modulates epileptic seizure activity. (a) Immunofluorescence labeling of enhanced green fluorescent proteins in the hippocampal CA1 region transfected with AAV-eGFP-adG6PD (n = 1). (b–c) Immunoblotting images (b) and quantification (c) of G6PD protein levels in the hippocampus of mice infected with the indicated AAVs (n = 6). Data are presented as the means ± SEM, ***P < 0.001, unpaired two-tailed Student's t-test. (d, e) Representative baseline and chronic epileptic electroencephalogram recordings from the cortex of chronic epileptic (adCtrl group) and G6PD overexpression after chronic epilepsy modeling (adG6PD group). (f, g) Quantitative analysis of the daily number of spontaneous recurrent seizures and duration of seizure-like events in the KA-induced epilepsy model in the adCtrl (n = 11) and adG6PD (n = 8) groups. Data are presented as means ± SEM. **P < 0.01, ***P < 0.001, unpaired two-tailed Student's t-test. (h–j) Representative traces and a summary of the amplitudes and frequencies of spontaneous excitatory postsynaptic currents in the hippocampal CA1 region in the adCtrl (n = 3, 6 cells) and adG6PD (n = 3, 8 cells) groups. (k–m) Representative traces and summary of the amplitude and frequency of spontaneous inhibitory postsynaptic currents in the hippocampal CA1 region of the adCtrl (n = 3, 8 cells) and adG6PD (n = 3, 8 cells) groups. Data are presented as means ± SEM.**P < 0.01, ***P < 0.001, unpaired two-tailed Student's t-test. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.4. G6PD inhibited NMDARs expression by promoting STAT1 expression
To further explore the molecular mechanism of G6PD in epilepsy, we used RNA-Sequence (RNA-Seq) technology, which revealed increased expression of the following genes: MGP_C57BL6NJ_G0016503, Usp18, MGP_C57BL6NJ_G0025477, Etnppl, Rnf213, Oasl2, Ifit1, Zc3hav1, MGP_C57BL6NJ_G0011418, Stat1 (Fig. 4a). STAT1, a member of the STAT protein family, has been associated with seizures [[32], [33], [34]]. We first demonstrated that the overexpression of G6PD after KA only increased the total protein expression of STAT1, rather than that of STAT3 (Fig. 4b and c). Phosphorylated STAT1 is known to dimerize, which in turn leads to ectopic activation of the nucleus [35]. Nuclear STAT1 activation can inhibit the expression of the NMDARs: GluN1, Glu2A, and Glu2B, by directly binding to the promoters of GluN1, GluN2A, and GluN2B [36]. Therefore, we speculate that G6PD plays a role in inhibiting NMDARs expression by promoting STAT1 expression. G6PD increased the expression of phosphorylated STAT1 (Fig. 4b and c) and nuclear STAT1 (Fig. 4d and e) during the chronic phase of epilepsy. Finally, we found that G6PD inhibited the total protein expression of NMDARs rather than α-amino-3-hydroxy-5-methyl-4-isoxazo-lepropionic acid receptors (AMPARs) (Fig. 4f and g). Similarly, NMDARs membrane protein levels decreased, while AMPARs (GluA1 and GluA2) were not affected in the adG6PD group (Fig. 4h and i). To determine whether G6PD regulates NMDARs expression via STAT1. The STAT1 inhibitor fludarabine also effectively reversed the observed decline in daily SRSs and the reduction in total NMDARs induced by G6PD (Fig. 4j-l). These results suggested that G6PD reduced NMDARs expression by promoting STAT1 expression.
Fig. 4.

G6PD negatively regulates the expression of N-methyl-d-aspartic acid receptors (NMDARs) through increasing STAT1 (a) RNA sequencing revealed that G6PD increased the transcription of Stat1 in epileptic mice. (b–e) Representative immunoblots (b, d) and quantification (c, e) of total STAT1 (n = 6), STAT3 (n = 6), p-STAT1 (n = 3), and nuclear STAT1 (n = 6) expression in the hippocampi of mice from the adCtrl and adG6PD groups. (f–i) Representative immunoblots (f, h) and quantification (g, i) of total and surface protein expression of NMDARs in the mouse hippocampus from the adCtrl and adG6PD groups (n = 6). Data are presented as means ± SEM. **P < 0.01, ***P < 0.001, unpaired two-tailed Student's t-test. (j) Effect of G6PD overexpression on the number of daily spontaneous recurrent seizures in the adCtrl, adG6PD, and adG6PD + fludarabine groups (n = 8). Data are presented as means ± SEM. ***P < 0.001, one-way ANOVA with Tukey's post-hoc test. (k–l) Representative immunoblots (b) and quantification (c) of NMDARs (Glu2A, Glu2B, and GluN1) in the hippocampi of mice in the adCtrl, adG6PD, and adG6PD + fludarabine groups (n = 6). Data are presented as means ± SEM. *P < 0.05, ***P < 0.001, one-way ANOVA with Tukey's post-hoc test.
3.5. G6PD promotes STAT1 expression by scavenging ROS
Our results show that G6PD plays an antiepileptic role in spontaneous seizures by affecting the expression of NMDARs through STAT1; however, the mechanism by which G6PD affects the expression of STAT1 is not clear. NOX2, an enzyme downstream of G6PD, promotes the expression of STAT3 by inhibiting ROS production [37]. Considering that STAT1 and STAT3 both belong to the STAT family and are related to epilepsy, we speculated that G6PD could promote the expression of STAT1 by affecting ROS [32,33]. In vitro, treatment with the G6PD agonist AG1 significantly reduced the levels ROS and increased the NADPH/NADP+ and GSH/GSSH ratio; these changes caused by G6PD activation were reversed by the ROS agonist DMF (Fig. 5a–d). In vitro, treatment with the G6PD inhibitor 6-An significantly increased ROS and decreased the NADPH/NADP+ and GSH/GSSH ratios; these changes in ROS and NADPH/NADP+ and GSH/GSSH levels caused by G6PD activation could be rescued by the ROS scavenger NAC (Fig. 5e–h). To verify whether G6PD promotes STAT1 expression by reducing ROS production, we pretreated HT22 cells with G6PD agonists and inhibitors and determined whether the changes in STAT1 expression caused by G6PD intervention could be rescued by ROS agonists or scavengers. As predicted, the ROS agonist DMF significantly attenuated the increase in STAT1 expression induced by G6PD activation, whereas the ROS scavenger NAC significantly reversed the decrease in STAT1 expression induced by the G6PD inhibitor, 6-An (Fig. 5i and j). These results indicate that G6PD may promote the expression of STAT1 by limiting ROS production, thereby limiting the synthesis of NMDARs that exert antiepileptic effects.
Fig. 5.

G6PD promotes the expression of STAT1 by decreasing reactive oxygen species (ROS) (a–d) Quantification of ROS, NADPH/NADP+, and GSH/GSSH in the control, AG1, AG1+DMF, 6-An, and 6-An + NAC groups after kainic acid (KA) treatment of HT22 cells. Data are presented as means ± SEM. ****P < 0.0001, one-way ANOVA with Tukey's post-hoc test. (e–h) Quantification of ROS, NADPH/NADP+, and GSH/GSSH in the control, 6-An, 6-An, 6-An, and 6-An + NAC groups after kainic acid (KA) treatment of HT22 cells. Data are presented as means ± SEM. ****P < 0.0001, one-way ANOVA with Tukey's post-hoc test. (i–i) Representative immunoblots (i) and quantification (j) of STAT1 in agonist (Con, AG1, and AG1+DMF groups) and inhibitor (Con, 6-An, and 6-An + NAC groups) groups after KA treatment of HT22 cells. Data are presented as means ± SEM. **P < 0.01, ***P < 0.001, one-way ANOVA with Tukey's post-hoc test.
3.6. G6PD expression is regulated by the transcription factors Znf768 and Klf6
It has previously been shown that G6PD overexpression can alleviate the severity of seizures, while its downstream mechanisms have been explored. Therefore, we investigated whether G6PD is regulated in chronic epilepsy. Our previous results suggested that the transcription level of G6PD is increased in the chronic phase (Fig. 1e). Therefore, we sought to explain the possible reasons for the increase in G6PD levels at the transcriptional level during the chronic phase of epilepsy. We used qPCR to examine the presence of transcriptional regulators in the top five promoter regions that may control G6PD expression, as listed on the JASPER website. We found that compared to that of ETS Transcription Factor4 (Elk4), Kruppel-like transcription factor 2 (Klf2), and Kruppel-like transcription factor11 (Klf11), the transcriptional levels of zinc finger protein 768 (Znf768) and Kruppel-like transcription factor 6 (Klf6) were significantly increased (Fig. 6a). Next, we performed WB to further examine the protein expression levels of ZNF768 and KLF6 in the hippocampus of mice with epilepsy, confirming the observed increases in the expression of these factors (Fig. 6b and c). Furthermore, we verified the regulation of these transcription factors by the G6PD promoter. We co-transferred a plasmid containing the upstream transcription factors Znf768 or Klf6 with a plasmid containing the target gene G6pd into HEK293 cells. Sual-luciferase reporter assay subsequently showed that Znf768 exerted a suppressive effect on the expression of the G6pd gene, whereas Klf6 had a stimulatory effect on the expression of the G6pd gene (Fig. 6d). The above experiments indicate that the transcription factors Znf768 and Klf6 regulate the expression of G6PD at the transcriptional level.
Fig. 6.

Transcription factors Znf768 and Klf6 regulate G6PD expression. (a) Quantitative real-time polymerase chain reaction analysis of predicted transcription factors (n = 4). Data are presented as means ± SEM, ns P > 0.05, **P < 0.01, nonsignificant unpaired two-tailed Student's t-test. (b–c) Representative immunoblots (b) and quantification (c) of ZNF768 and KLF6 from the hippocampus of sham and chronic epileptic mice (n = 6). Data are presented as means ± SEM, **P < 0.01, ***P < 0.001, unpaired two-tailed Student's t-test. (d) Relative luciferase activities in HEK293 cells. Cells were transiently co-transfected G6pd with vector or Znf768 or Klf6 (n = 6). Data are presented as means ± SEM, **P < 0.001, unpaired two-tailed Student's t-test.
3.7. The G6PD agonist AG1 alleviates the epileptic susceptibility
Previous behavioral results suggested that increased G6PD levels in the chronic phase could alleviate the number and duration of seizures (Fig. 3e–g). We wanted to further explored whether existing agonists and inhibitors of G6PD could be used as new drugs to treat epilepsy. PTZ is a γ-Aminobutyric acid type A (GABAA) receptors antagonist, for which previous research has shown that continuous low-dose intraperitoneal administration can induce severe tonic clonic seizures [38]. Neuronal damage observed in this model is consistent with histological changes in the brain of epileptic patients [39]. Therefore, this epilepsy model is commonly used to screen for antiepileptic drugs and epilepsy-related genes [40]. We chose the PTZ model to verify the effects of the G6PD agonist AG1 and inhibitor 6-An on susceptibility of epilepsy. Initial results demonstrated that the intraperitoneal injection of PTZ caused seizures and electrophysiological changes (Fig. 7a). We subsequently intraperitoneally injected mice with 6-An for five consecutive days to inhibit G6PD enzyme activity. After 2 days of rest, the mice were intraperitoneally injected with PTZ to facilitate behavioral observation [19]. The results showed that 6-An reduced the time of the first induction of excitatory toxicity, shortened the latency, and triggered all mice in a shorter period (Fig. 7c–e). These findings suggest that 6-An promotes seizures. As the role of AG1 in the central nervous system has not previously been reported, we directly injected AG1 into the hippocampus (excluding the effect of the blood-brain barrier) to activate G6PD. Overall, we found that a dosage of 4 μg exerts a significant excitation effect, and is more appropriate (Fig. 7f and g). Therefore, we intraperitoneally injected PTZ 24 h after 4 μg of AG1 was injected into the hippocampus and observed the behavioral changes in the mice. We found that, compared with the 100 % kindling rate of the control group, the AG1 group had a kindling rate of only 37.5 % (Fig. 7h). Further, the mice were kindled later, with some not showing any kindling until the end of the induction protocol (10 intraperitoneal injections) (Fig. 7i). Overall, these results suggest that the G6PD agonist, AG1, may relieve seizures and could therefore represent a new target for the selection of clinical drugs.
Fig. 7.

The G6PD activator AG1 alleviates seizure susceptibility (a) Example traces matching the minimal and tonic-clonic seizures of PTZ-injected mice. Scale bars, 0.3 mV and 1 s. (b) Timeline of 6-An and AG1 injection before the administration of pentylenetetrazol (PTZ) (12.5 mg/kg PTZ was injected into C57BL/6 mice once every 10 min, up to 10 times). (c) The seizure susceptibility in the sham and 6-An group following the injection of PTZ (n = 8). Data are presented as means ± SEM, **P < 0.01, unpaired two-tailed Student's t-test. (f, g) Immunoblot images and statistical analysis of hippocampal G6PD after AG1 administration (n = 4). Data are presented as means ± SEM. *P < 0.05, one-way ANOVA with Tukey's post-hoc test. (h, i) The seizure susceptibility in the sham and AG1 groups following the injection of PTZ (n = 8). Data are presented as means ± SEM, *P < 0.05, unpaired two-tailed Student's t-test.
4. Discussion
The brain is an organ with a robust energy metabolism that relies heavily on glycolysis, the tricarboxylic acid cycle, and oxidative phosphorylation [41]. In the brain, glucose is converted into glucose-6-phosphate by hexokinase 1/2 (HK1/2), which produces lactic acid through the action of glycolytic enzymes (fructose-2,6-biphosphatase 3, PFKFB3, phosphofructokinase, PFK1) and pyruvate kinase m2 (PKM2). Additionally, 5-phosphate ribose and NADPH are generated by the PPP enzymes G6PD, 6-phosphogluconate dehydrogenase (6PGD), and transketolase (TKT). The inhibition of the glycolysis pathway using 2-dehydroglucose may promote seizures [42]. Furthermore, during the onset of spontaneous recurrent epilepsy, the increased accumulation of lactic acid produced by glucose metabolism exacerbates seizures, while inhibition of lactic acid production leads to more severe seizures. NOX2/4 metabolizes the NADPH produced by the PPP, thereby promoting ROS production. Research has shown increases in NOX2/4 in epilepsy, while inhibition of NOX2/4 has been shown to play a neuroprotective role in epilepsy [43,44]. Interventions involving HK2 and PKM2 affect the learning and memory abilities of AD mice [45,46]. Additionally, G6PD protects against ischemic brain injury by enhancing the PPP [30]. Furthermore, the inhibition of TKT in PD leads to dopaminergic neuron toxicity [47]. In the present study, we investigated the alterations in the expression of glycolytic and PPP-related enzymes in epilepsy, and only G6PD expression was elevated in the hippocampal neurons of epileptic mice.
The imbalance between excitatory and inhibitory signals within neural networks is a direct cause of TLE [40]. The development of chronic spontaneous epilepsy is associated with long-term progressive changes in these networks [48]. In the present study, we found that changes in G6PD levels were most significant in mice with chronic epilepsy. G6PD, a crucial enzyme in the PPP, is involved in both physiological and pathological processes [7]. Further, it regulates the production of 5-ribose, DNA synthesis required for nucleotide synthesis, and NADPH required for antioxidant production, thus affecting rapid cell growth [49]. Furthermore, compared to normal mice, G6PD deficient mice are more susceptible to oxidative stress injury with aging, whereas overexpression of G6PD can potentially extend the lifespan by combating oxidative stress injury [11,50]. Based on these findings, we hypothesized that G6PD plays a protective role against epilepsy. We validated this hypothesis by overexpressing G6PD in the Map2-Cre-ERT2 mice with chronic epilepsy.
In our mice, we injected KA into the hippocampus, resulting in epileptiform discharges originating from the hippocampus that subsequently spreading to the cortex [51,52]. Previous studies on chronic spontaneous epilepsy typically employed KA modeling after intervention, followed by behavioral assessments and mechanism investigation [53,54]. However, a limitation of this approach is its susceptibility to acute seizure severity, which affects the development of chronic spontaneous epilepsy [31]. Consequently, employing viral intervention proteins prior to KA epilepsy modeling fails to account for potential alterations in acute-phase-related proteins that may influence chronic seizures. In this study, we used a Map2-Cre-ERT2 mouse model combined with viral injection into the hippocampus. Furthermore, Map2-Cre-ERT2 mice combined with viral hippocampal injections were used to induce the specific overexpression of G6PD in neurons for behavioral evaluation and mechanistic exploration during the chronic stage of epileptic seizures. Specifically, KA was modeled in Map2-Cre-ERT2 mice pre-infected with the virus (adCtrl or adG6PD group), while tamoxifen was injected intraperitoneally at 28 days after KA modeling to induce the specific overexpression of G6PD in neurons for behavioral evaluation and mechanistic exploration during the chronic stage of epileptic seizures [23]. This method effectively avoids the influence of changes in acute phase-related proteins on chronic seizures, and facilitates the intervention of specific cell types in specific brain regions at specific time points. The findings of this study demonstrated that increased G6PD expression can mitigate both the number and duration of spontaneous seizures in mice with chronic epilepsy.
In the rodent brain, neuronal excitability can be increased by activating glutamate (an excitatory neurotransmitter), which acts on the AMPARs, NMDARs, and kainic acid receptors [55]. NMDARs are composed of two GluN1 subunits and two identical GluN2 subunits or two different GluN2 subunits [56]. These receptors play crucial roles in the regulation of neuronal plasticity, excitatory neurotransmission, and neurotoxicity regulation [57,58]. Maintaining the activity of NMDARs within a stable range is essential to ensure normal physiological function and the prevention of disease development [58]. In patients with refractory temporal lobe epilepsy, elevated levels of extracellular glutamate during seizures activate NMDARs, leading to neuroexcitatory toxicity [59,60]. Excessive NMDARs excitation has also been associated with seizure occurrence [61]. As such, direct targeting of NMDARs can affect seizure control. Our study further confirmed that STAT1 acts as a transcription factor that inhibits NMDARs [36,62]. Additionally, G6PD overexpression suppressed NMDARs expression, which was reversed by STAT1 intervention. Thus, we propose that the G6PD-STAT1-NMDARs axis plays a significant role in antiepileptic mechanisms. As part of the STAT family of proteins, combining STAT1 with STAT3 has shown potential for alleviating seizures by suppressing NMDARs expression; this avenue warrants further investigation [63,64].
G6PD plays a role in oxidative stress damage, such as ROS production by providing NADPH [65]. Prior research has shown that a reduction in oxidative stress can reduce seizures [66]. Our results suggest that G6PD reduces the ROS levels in epilepsy. For the first time, we proposed the existence of a G6PD-ROS-STAT1 axis. The impact of ROS on STAT1 regulation remains a topic of debate. ROS can enhance STAT1 expression; however, our experiment revealed that ROS inhibits STAT1 expression by ROS [67]. We speculate that the possible reasons for this are as follows: 1) our study specifically focused on investigating the pathogenesis of epilepsy, which differs from the previous models examined; 2) after overexpressing G6PD in our study, we confirmed its potential involvement in seizures through the G6PD-ROS-STAT1 axis; however, considering the complex pathogenic environment in mice, G6PD may also alleviate seizures through other mechanisms; and 3) notably, ROS also inhibits STAT3 expression [37]. STAT1 and STAT3 belong to the same family and have similar functions; therefore, it is plausible that ROS may also inhibit STAT1 expression [68].
Currently, more than 30 types of ASD are used in clinical practice [69]. Most drugs, such as sodium phenytoin, ethyl ketamine (which regulates ion channels), and clonazepam (which regulates E/I imbalance), primarily function as symptomatic treatments for seizure control [70,71]. However, some patients remain unresponsive to these medications, necessitating the development of novel anti-recurrent spontaneous epilepsy drugs [2]. Animal models used for antiepileptic drug screening must have an etiology, clinical manifestations, and pathological features consistent with those of patients with epilepsy and can show the characteristics of drug resistance to antiepileptic drugs [72,73]. PTZ is a nonselective GABA inhibitor with attributes such as high utilization rates, strong permeability, short incubation periods, and rapid distribution throughout all organs of the body [38,74]. The PTZ-induced model is widely recognized both domestically and internationally, and has been utilized as a preclinical drug screening models [40]. In our study, which used the PTZ epilepsy model, we discovered that the G6PD agonist AG1 significantly reduced the susceptibility to seizures. Consequently, we hypothesized that AG1 could potentially represent a new small-molecule drug with clinical anti-seizure applications. Further investigation is required to validate the effects of AG1 on seizures in different epilepsy models.
In conclusion, our study showed that the overexpression of G6PD reduced the severity of SRSs and exerted antiepileptic effects through action on the G6PD-ROS-STAT1-NMDARs axis. The administration of a G6PD agonist significantly alleviated seizure susceptibility in the PTZ model, providing a novel approach for the clinical management of seizures. Overall, our results revealed a promising novel therapeutic target for epilepsy.
Funding information
This work was supported by grants from the National Natural Science Foundation of China (82271496, 82001378), National Natural Science Foundation of Chongqing (CSTB2022NSCQ-MSX0747, CSTB2023NSCQ-JQX0035, CSTB2022NSCQ-LZX0038, cstc2021ycjh-bgzxm0035), Future Medical Youth Innovation Team of Chongqing Medical University (W0043), Young and Middle-aged Medical Excellence Team Program of Chongqing (2022), and Chongqing Chief Expert Studio program (2022), Key Basic Research Project of the Discipline Climbing Plan of the First Affiliated Hospital of Chongqing Medical University (cyyy-xkdfjh-jcyj-202306), China Postdoctoral Science Foundation (2023M730443), the Joint Project of Chongqing Health Commission and Science and Technology Bureau (2023QNXM009), Science and Technology Research Program of Chongqing Education Commission (KJQN202200435).
Ethical approval
All animal experiments were approved by the Ethics Committee of Chongqing Medical University.
CRediT authorship contribution statement
Liqin Hu: Writing – review & editing, Writing – original draft, Project administration, Methodology, Investigation, Data curation. Yan Liu: Writing – review & editing, Methodology, Data curation, Conceptualization. Ziwei Yuan: Software, Resources, Formal analysis. Haokun Guo: Resources, Investigation, Data curation. Ran Duan: Supervision, Project administration, Conceptualization. Pingyang Ke: Supervision, Software, Data curation. Yuan Meng: Visualization, Formal analysis, Conceptualization. Xin Tian: Software, Funding acquisition. Fei Xiao: Writing – review & editing, Funding acquisition, Conceptualization.
Declaration of competing interest
The authors declare no competing interests.
Acknowledgments
None.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2024.103236.
Contributor Information
Xin Tian, Email: xintian@cqmu.edu.cn.
Fei Xiao, Email: feixiao81@126.com.
Appendix A. Supplementary data
The following are the Supplementary data to this article.
Data availability
Data will be made available on request.
References
- 1.Thijs R.D., Surges R., O'Brien T.J., Sander J.W. Epilepsy in adults. Lancet. 2019;393:689–701. doi: 10.1016/s0140-6736(18)32596-0. [DOI] [PubMed] [Google Scholar]
- 2.Engel J., Jr., Pitkänen A. Biomarkers for epileptogenesis and its treatment. Neuropharmacology. 2020;167 doi: 10.1016/j.neuropharm.2019.107735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scheffer I.E., et al. ILAE classification of the epilepsies: position paper of the ILAE commission for classification and terminology. Epilepsia. 2017;58:512–521. doi: 10.1111/epi.13709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neal E.G., et al. A randomized trial of classical and medium-chain triglyceride ketogenic diets in the treatment of childhood epilepsy. Epilepsia. 2009;50:1109–1117. doi: 10.1111/j.1528-1167.2008.01870.x. [DOI] [PubMed] [Google Scholar]
- 5.Zsurka G., Kunz W.S. Mitochondrial dysfunction and seizures: the neuronal energy crisis. Lancet Neurol. 2015;14:956–966. doi: 10.1016/s1474-4422(15)00148-9. [DOI] [PubMed] [Google Scholar]
- 6.Goodman A.M., Szaflarski J.P. Recent advances in neuroimaging of epilepsy. Neurotherapeutics. 2021;18:811–826. doi: 10.1007/s13311-021-01049-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cappellini M.D., Fiorelli G. Glucose-6-phosphate dehydrogenase deficiency. Lancet. 2008;371:64–74. doi: 10.1016/s0140-6736(08)60073-2. [DOI] [PubMed] [Google Scholar]
- 8.Stanton R.C. Glucose-6-phosphate dehydrogenase, NADPH, and cell survival. IUBMB Life. 2012;64:362–369. doi: 10.1002/iub.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alagoz M., Kherad N., Gunger E., Kaymaz S., Yuksel A. The new CIC mutation associates with mental retardation and severity of seizure in Turkish child with a rare class I glucose-6-phosphate dehydrogenase deficiency. J. Mol. Neurosci. 2020;70:2077–2084. doi: 10.1007/s12031-020-01614-8. [DOI] [PubMed] [Google Scholar]
- 10.Jeng W., Loniewska M.M., Wells P.G. Brain glucose-6-phosphate dehydrogenase protects against endogenous oxidative DNA damage and neurodegeneration in aged mice. ACS Chem. Neurosci. 2013;4:1123–1132. doi: 10.1021/cn400079y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nóbrega-Pereira S., et al. G6PD protects from oxidative damage and improves healthspan in mice. Nat. Commun. 2016;7 doi: 10.1038/ncomms10894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Legan S.K., et al. Overexpression of glucose-6-phosphate dehydrogenase extends the life span of Drosophila melanogaster. J. Biol. Chem. 2008;283:32492–32499. doi: 10.1074/jbc.M805832200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Borowicz-Reutt K.K., Czuczwar S.J. Role of oxidative stress in epileptogenesis and potential implications for therapy. Pharmacol. Rep. 2020;72:1218–1226. doi: 10.1007/s43440-020-00143-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang R., et al. Nrf2-a promising therapeutic target for defensing against oxidative stress in stroke. Mol. Neurobiol. 2017;54:6006–6017. doi: 10.1007/s12035-016-0111-0. [DOI] [PubMed] [Google Scholar]
- 15.Pearson-Smith J.N., Patel M. Metabolic dysfunction and oxidative stress in epilepsy. Int. J. Mol. Sci. 2017;18 doi: 10.3390/ijms18112365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheignon C., et al. Oxidative stress and the amyloid beta peptide in Alzheimer's disease. Redox Biol. 2018;14:450–464. doi: 10.1016/j.redox.2017.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.A N.K., et al. ROS networks: designs, aging, Parkinson's disease and precision therapies. NPJ Syst Biol Appl. 2020;6:34. doi: 10.1038/s41540-020-00150-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saadi A., Sandouka S., Grad E., Singh P.K., Shekh-Ahmad T. Spatial, temporal, and cell-type-specific expression of NADPH Oxidase isoforms following seizure models in rats. Free Radic. Biol. Med. 2022;190:158–168. doi: 10.1016/j.freeradbiomed.2022.08.009. [DOI] [PubMed] [Google Scholar]
- 19.Politis M.J. 6-Aminonicotinamide selectively causes necrosis in reactive astroglia cells in vivo. Preliminary morphological observations. J. Neurol. Sci. 1989;92:71–79. doi: 10.1016/0022-510x(89)90176-7. [DOI] [PubMed] [Google Scholar]
- 20.Zhao Y., et al. STAT1 contributes to microglial/macrophage inflammation and neurological dysfunction in a mouse model of traumatic brain injury. J. Neurosci. 2022;42:7466–7481. doi: 10.1523/jneurosci.0682-22.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiong Y., et al. Expression of Glypican-4 in the brains of epileptic patients and epileptic animals and its effects on epileptic seizures. Biochem. Biophys. Res. Commun. 2016;478:241–246. doi: 10.1016/j.bbrc.2016.07.061. [DOI] [PubMed] [Google Scholar]
- 22.Zhang H., et al. TMEM25 modulates neuronal excitability and NMDA receptor subunit NR2B degradation. J. Clin. Invest. 2019;129:3864–3876. doi: 10.1172/jci122599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen Z.P., et al. Lipid-accumulated reactive astrocytes promote disease progression in epilepsy. Nat. Neurosci. 2023;26:542–554. doi: 10.1038/s41593-023-01288-6. [DOI] [PubMed] [Google Scholar]
- 24.Lin W., et al. Inhibition of miR-134-5p protects against kainic acid-induced excitotoxicity through Sirt3-mediated preservation of mitochondrial function. Epilepsy Res. 2021;176 doi: 10.1016/j.eplepsyres.2021.106722. [DOI] [PubMed] [Google Scholar]
- 25.Li Y., et al. Targeting glucose-6-phosphate dehydrogenase by 6-AN induces ROS-mediated autophagic cell death in breast cancer. FEBS J. 2023;290:763–779. doi: 10.1111/febs.16614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choi J.H., et al. Baicalein protects HT22 murine hippocampal neuronal cells against endoplasmic reticulum stress-induced apoptosis through inhibition of reactive oxygen species production and CHOP induction. Exp. Mol. Med. 2010;42:811–822. doi: 10.3858/emm.2010.42.12.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hwang S., et al. Correcting glucose-6-phosphate dehydrogenase deficiency with a small-molecule activator. Nat. Commun. 2018;9:4045. doi: 10.1038/s41467-018-06447-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xie X., et al. Dimethyl fumarate induces necroptosis in colon cancer cells through GSH depletion/ROS increase/MAPKs activation pathway. Br. J. Pharmacol. 2015;172:3929–3943. doi: 10.1111/bph.13184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gu J., et al. KCTD13-mediated ubiquitination and degradation of GluN1 regulates excitatory synaptic transmission and seizure susceptibility. Cell Death Differ. 2023;30:1726–1741. doi: 10.1038/s41418-023-01174-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cao L., et al. G6PD plays a neuroprotective role in brain ischemia through promoting pentose phosphate pathway. Free Radic. Biol. Med. 2017;112:433–444. doi: 10.1016/j.freeradbiomed.2017.08.011. [DOI] [PubMed] [Google Scholar]
- 31.Puttachary S., Sharma S., Thippeswamy A., Thippeswamy T. Immediate epileptogenesis: impact on brain in C57BL/6J mouse kainate model. Front. Biosci. 2016;8:390–411. doi: 10.2741/e775. [DOI] [PubMed] [Google Scholar]
- 32.Feng X., et al. Down-regulated microRNA-183 mediates the Jak/Stat signaling pathway to attenuate hippocampal neuron injury in epilepsy rats by targeting Foxp1. Cell Cycle. 2019;18:3206–3222. doi: 10.1080/15384101.2019.1671717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grabenstatter H.L., et al. The effect of STAT3 inhibition on status epilepticus and subsequent spontaneous seizures in the pilocarpine model of acquired epilepsy. Neurobiol. Dis. 2014;62:73–85. doi: 10.1016/j.nbd.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aaronson D.S., Horvath C.M. A road map for those who don't know JAK-STAT. Science. 2002;296:1653–1655. doi: 10.1126/science.1071545. [DOI] [PubMed] [Google Scholar]
- 35.Chen X., et al. Crystal structure of a tyrosine phosphorylated STAT-1 dimer bound to DNA. Cell. 1998;93:827–839. doi: 10.1016/s0092-8674(00)81443-9. [DOI] [PubMed] [Google Scholar]
- 36.Li X.G., et al. Tau accumulation triggers STAT1-dependent memory deficits by suppressing NMDA receptor expression. EMBO Rep. 2019;20 doi: 10.15252/embr.201847202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu C., et al. Nox2 impairs VEGF-A-induced angiogenesis in placenta via mitochondrial ROS-STAT3 pathway. Redox Biol. 2021;45 doi: 10.1016/j.redox.2021.102051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shimada T., Yamagata K. Pentylenetetrazole-induced kindling mouse model. J. Vis. Exp. 2018 doi: 10.3791/56573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ueno H., et al. Alteration of extracellular matrix molecules and perineuronal nets in the Hippocampus of pentylenetetrazol-kindled mice. Neural Plast. 2019 doi: 10.1155/2019/8924634. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barker-Haliski M., Steve White H. Validated animal models for antiseizure drug (ASD) discovery: advantages and potential pitfalls in ASD screening. Neuropharmacology. 2020;167 doi: 10.1016/j.neuropharm.2019.107750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arnold S.E., et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat. Rev. Neurol. 2018;14:168–181. doi: 10.1038/nrneurol.2017.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stafstrom C.E., et al. Anticonvulsant and antiepileptic actions of 2-deoxy-D-glucose in epilepsy models. Ann. Neurol. 2009;65:435–447. doi: 10.1002/ana.21603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Skwarzynska D., Sun H., Williamson J., Kasprzak I., Kapur J. Glycolysis regulates neuronal excitability via lactate receptor, HCA1R. Brain. 2023;146:1888–1902. doi: 10.1093/brain/awac419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Singh P.K., Saadi A., Sheeni Y., Shekh-Ahmad T. Specific inhibition of NADPH oxidase 2 modifies chronic epilepsy. Redox Biol. 2022;58 doi: 10.1016/j.redox.2022.102549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bustamante M.F., et al. Hexokinase 2 as a novel selective metabolic target for rheumatoid arthritis. Ann. Rheum. Dis. 2018;77:1636–1643. doi: 10.1136/annrheumdis-2018-213103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pan R.Y., et al. Positive feedback regulation of microglial glucose metabolism by histone H4 lysine 12 lactylation in Alzheimer's disease. Cell Metabol. 2022;34:634–648.e636. doi: 10.1016/j.cmet.2022.02.013. [DOI] [PubMed] [Google Scholar]
- 47.Kang H., et al. PARIS reprograms glucose metabolism by HIF-1α induction in dopaminergic neurodegeneration. Biochem. Biophys. Res. Commun. 2018;495:2498–2504. doi: 10.1016/j.bbrc.2017.12.147. [DOI] [PubMed] [Google Scholar]
- 48.Rho J.M., Boison D. The metabolic basis of epilepsy. Nat. Rev. Neurol. 2022;18:333–347. doi: 10.1038/s41582-022-00651-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boros L.G., et al. Oxythiamine and dehydroepiandrosterone inhibit the nonoxidative synthesis of ribose and tumor cell proliferation. Cancer Res. 1997;57:4242–4248. [PubMed] [Google Scholar]
- 50.Bermúdez-Muñoz J.M., et al. G6PD overexpression protects from oxidative stress and age-related hearing loss. Aging Cell. 2020;19 doi: 10.1111/acel.13275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lévesque M., Avoli M. The kainic acid model of temporal lobe epilepsy. Neurosci. Biobehav. Rev. 2013;37:2887–2899. doi: 10.1016/j.neubiorev.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bielefeld P., et al. A standardized protocol for stereotaxic intrahippocampal administration of kainic acid combined with electroencephalographic seizure monitoring in mice. Front. Neurosci. 2017;11:160. doi: 10.3389/fnins.2017.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zheng F., et al. CD36 deficiency suppresses epileptic seizures. Neuroscience. 2017;367:110–120. doi: 10.1016/j.neuroscience.2017.10.024. [DOI] [PubMed] [Google Scholar]
- 54.Yang Y., et al. GPR40 modulates epileptic seizure and NMDA receptor function. Sci. Adv. 2018;4 doi: 10.1126/sciadv.aau2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Karakas E., Regan M.C., Furukawa H. Emerging structural insights into the function of ionotropic glutamate receptors. Trends Biochem. Sci. 2015;40:328–337. doi: 10.1016/j.tibs.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tu Y.C., Kuo C.C. The differential contribution of GluN1 and GluN2 to the gating operation of the NMDA receptor channel. Pflügers Archiv. 2015;467:1899–1917. doi: 10.1007/s00424-014-1630-z. [DOI] [PubMed] [Google Scholar]
- 57.Paoletti P., Bellone C., Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013;14:383–400. doi: 10.1038/nrn3504. [DOI] [PubMed] [Google Scholar]
- 58.Mony L., Paoletti P. Mechanisms of NMDA receptor regulation. Curr. Opin. Neurobiol. 2023;83 doi: 10.1016/j.conb.2023.102815. [DOI] [PubMed] [Google Scholar]
- 59.Hunt D.L., Castillo P.E. Synaptic plasticity of NMDA receptors: mechanisms and functional implications. Curr. Opin. Neurobiol. 2012;22:496–508. doi: 10.1016/j.conb.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Möddel G., et al. The NMDA receptor NR2B subunit contributes to epileptogenesis in human cortical dysplasia. Brain Res. 2005;1046:10–23. doi: 10.1016/j.brainres.2005.03.042. [DOI] [PubMed] [Google Scholar]
- 61.Xu X.X., Luo J.H. Mutations of N-Methyl-D-Aspartate receptor subunits in epilepsy. Neurosci. Bull. 2018;34:549–565. doi: 10.1007/s12264-017-0191-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tang F.L., et al. MBD5 regulates NMDA receptor expression and seizures by inhibiting Stat1 transcription. Neurobiol. Dis. 2023;181 doi: 10.1016/j.nbd.2023.106103. [DOI] [PubMed] [Google Scholar]
- 63.Hong X.Y., et al. STAT3 ameliorates cognitive deficits by positively regulating the expression of NMDARs in a mouse model of FTDP-17. Signal Transduct. Targeted Ther. 2020;5:295. doi: 10.1038/s41392-020-00290-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wan H.L., et al. STAT3 ameliorates cognitive deficits via regulation of NMDAR expression in an Alzheimer's disease animal model. Theranostics. 2021;11:5511–5524. doi: 10.7150/thno.56541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.TeSlaa T., Ralser M., Fan J., Rabinowitz J.D. The pentose phosphate pathway in health and disease. Nat. Metab. 2023;5:1275–1289. doi: 10.1038/s42255-023-00863-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kovac S., Domijan A.M., Walker M.C., Abramov A.Y. Seizure activity results in calcium- and mitochondria-independent ROS production via NADPH and xanthine oxidase activation. Cell Death Dis. 2014;5 doi: 10.1038/cddis.2014.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kotla S., Singh N.K., Traylor J.G., Jr., Orr A.W., Rao G.N. ROS-dependent Syk and Pyk2-mediated STAT1 activation is required for 15(S)-hydroxyeicosatetraenoic acid-induced CD36 expression and foam cell formation. Free Radic. Biol. Med. 2014;76:147–162. doi: 10.1016/j.freeradbiomed.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Verhoeven Y., et al. The potential and controversy of targeting STAT family members in cancer. Semin. Cancer Biol. 2020;60:41–56. doi: 10.1016/j.semcancer.2019.10.002. [DOI] [PubMed] [Google Scholar]
- 69.Perucca E., Perucca P., White H.S., Wirrell E.C. Drug resistance in epilepsy. Lancet Neurol. 2023;22:723–734. doi: 10.1016/s1474-4422(23)00151-5. [DOI] [PubMed] [Google Scholar]
- 70.Patocka J., Wu Q., Nepovimova E., Kuca K. Phenytoin - an anti-seizure drug: overview of its chemistry, pharmacology and toxicology. Food Chem. Toxicol. 2020;142 doi: 10.1016/j.fct.2020.111393. [DOI] [PubMed] [Google Scholar]
- 71.Sills G.J., Rogawski M.A. Mechanisms of action of currently used antiseizure drugs. Neuropharmacology. 2020;168 doi: 10.1016/j.neuropharm.2020.107966. [DOI] [PubMed] [Google Scholar]
- 72.Dlugos D., et al. Epilepsy benchmarks area III: improve treatment options for controlling seizures and epilepsy-related conditions without side effects. Epilepsy Curr. 2014;16:192–197. doi: 10.5698/1535-7511-16.3.192. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stables J.P., et al. Therapy discovery for pharmacoresistant epilepsy and for disease-modifying therapeutics: summary of the NIH/NINDS/AES models II workshop. Epilepsia. 2003;44:1472–1478. doi: 10.1111/j.0013-9580.2003.32803.x. [DOI] [PubMed] [Google Scholar]
- 74.Ramzan I.M., Levy G. Kinetics of drug action in disease states. XIV. Effect of infusion rate on pentylenetetrazol concentrations in serum, brain and cerebrospinal fluid of rats at onset of convulsions. J. Pharmacol. Exp. Therapeut. 1985;234:624–628. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.