

Abstract
Polymorphous adenocarcinoma (PAC) is a common, usually low-grade salivary gland carcinoma. While conventional PACs are most associated with PRKD1 p.E710D hotspot mutations, the cribriform subtype is often associated with gene fusions in PRKD1, PRKD2, or PRKD3. These fusions have been primarily identified by fluorescence in situ hybridization (FISH) analysis, with a minority evaluated by next generation sequencing (NGS). Many of the reported fusions were detected by break apart FISH probes and therefore have unknown partners, or were negative by FISH altogether. In this study, we aimed to further characterize the fusions associated with PAC with NGS. 54 PACs (exclusively cribriform and mixed/intermediate types to enrich for fusion-positive cases) were identified and subjected to NGS. 51 cases were successfully sequenced, 28 of which demonstrated gene fusions involving PRKD1, PRKD2, or PRKD3. There were 10 cases with the PRKD1 p.E710D mutation. We identified a diverse group of fusion partners, including 13 novel partners, 3 of which were recurrent. The most common partners for the PRKD genes were ARID1A and ARID1B. The wide variety of involved genes is unlike other salivary gland malignancies and warrants a broader strategy of sequencing for molecular confirmation for particularly challenging cases, as our NGS study shows.
Introduction
The field of head and neck pathology, specifically salivary gland pathology, has witnessed a brisk expansion of novel tumor entities as well as detailed molecular classification of known tumor entities. Examples of this expansion include the recently described microsecretory adenocarcinoma as well as the related microcribriform adenocarcinoma (1)(2). These tumors are not only tied together by regionally similar morphologies, but also by a shared molecular underpinning, namely SS18 gene rearrangements. In a similar vein, polymorphous adenocarcinoma (PAC) is an entity with a unifying molecular pathogenesis. PAC predominantly affects the minor salivary glands, but has also been rarely identified in the parotid gland and other extraoral sites (3)(4)(5)(6)(7). PACs can demonstrate conventional fascicular or cribriform/glomerulopapillary patterns, or be mixed/indeterminate (3)(4)(5)(6)(7). Initially these tumors were described as low-grade adenocarcinomas with variable morphologies, which included papillary and cribriform patterns (3). Later, there was brief enthusiasm to separate tumors with prominent papillary and glomeruloid patterns into a separate tumor type known as cribriform adenocarcinoma (5)(6). Subsequent molecular analysis revealed that conventional PACs are most commonly associated with PRKD1 p.E710D hotspot mutations, whereas the cribriform adenocarcinoma subtype is generally associated with fusions of PRKD1, PRKD2, or PRKD3 (7)(8)(9). The segregation of molecular alteration category to histomorphologic subtype is modest, and interobserver reliability in characterizing histomorphologic subtypes is moderate, partly due to the high frequency of mixed/indeterminate tumors (9)(10). In the current WHO Classification of Head and Neck Tumors, classic/conventional and cribriform/papillary/glomeruloid tumors are regarded as subtypes of PAC. (11)
The PAC fusions have been primarily identified by fluorescence in situ hybridization (FISH) analysis, with only a small minority evaluated by next generation sequencing (NGS) (7). As a result, many of the reported fusions have unknown partners, or were negative by FISH altogether. In this study, we further characterize the genomic basis of the cribriform subtype of PAC, in addition to mixed/indeterminate tumors. If these specific molecular alterations ever contribute to prognosis and/or become targetable, it will become increasingly important for them to be sequenced and fully-characterized.
Materials/Subjects and Methods
Institutional research ethics board approval was obtained. Through a search of the laboratory information system (LIS), tumors diagnosed as PAC and classified as the cribriform subtype (n=35) or mixed/indeterminate (n=19) were reviewed for diagnosis confirmation by the contributing author according to the paper by Xu et al. (10). Tumors were classified as cribriform subtype if they demonstrated predominantly cribriform architecture, or as mixed/indeterminate if they demonstrated non-focal (>5%) conventional growth. Pure conventional PACs were excluded to enrich the study for fusion-positive cases. Tumors were classified as high-grade if they demonstrated abundant mitotic activity and comedo-type necrosis.
RNA was isolated from formalin-fixed paraffin-embedded tissue, and complementary DNA (cDNA) synthesis was performed. In forty-seven cases, cDNA was captured from 1519 genes using IDT xGen hybrid capture probes and sequenced on a Nextseq 550 (Illumina) with at least 6 million reads per sample. Fusions were called by the StarFusion algorithm and manually confirmed in the Integrated Genomics Viewer (Broad Institute, Cambridge, USA) (12). The presence of PRKD1 p.E710D hotspot mutations was also evaluated in the RNA sequencing data. In four cases, targeted RNA sequencing was performed using an anchored multiplex polymerase chain reaction (PCR)-based clinical molecular diagnostic assay (MSK-Archer FusionPlex) in a CLIA-accredited laboratory, to detect oncogenic fusion transcripts including a panel of 123 genes as previously described (13). In one case, the Trusight RNA Fusion Panel targeting 507 known fusion-related gene targets was used to prepare the library and sequenced using an Illumina MiSeq with approximately 3 million reads per sample. Fusion gene analysis was performed using the STAR and BOWTIE2 aligners and the Manta and JAFFA fusion callers (14)(15). Where FISH data were available through prior study, the fusions were confirmed retrospectively.
Results
A total of 54 cases were identified through LIS review. The patient ages ranged from 34 to 93 years, with an average of 60 years. There was a 3.5:1 female predominance (42 females and 12 males). The tumor sites included palate (n=18), pharynx (n=12), lip and oral cavity (n=11), parotid gland (n=7), and sinonasal and skull base (n=6) (Figure 1). The histologic spectrum of tumors includes cribriform subtype (n=35) and mixed/indeterminate tumors (n=19), ranging from unequivocal cribriform morphology to mixed conventional and cribriform morphologies, some with the cribriform component comprising only a minority of the tumor. (Figure 2)
Figure 1:

Number of cases by anatomic site.
Figure 2:
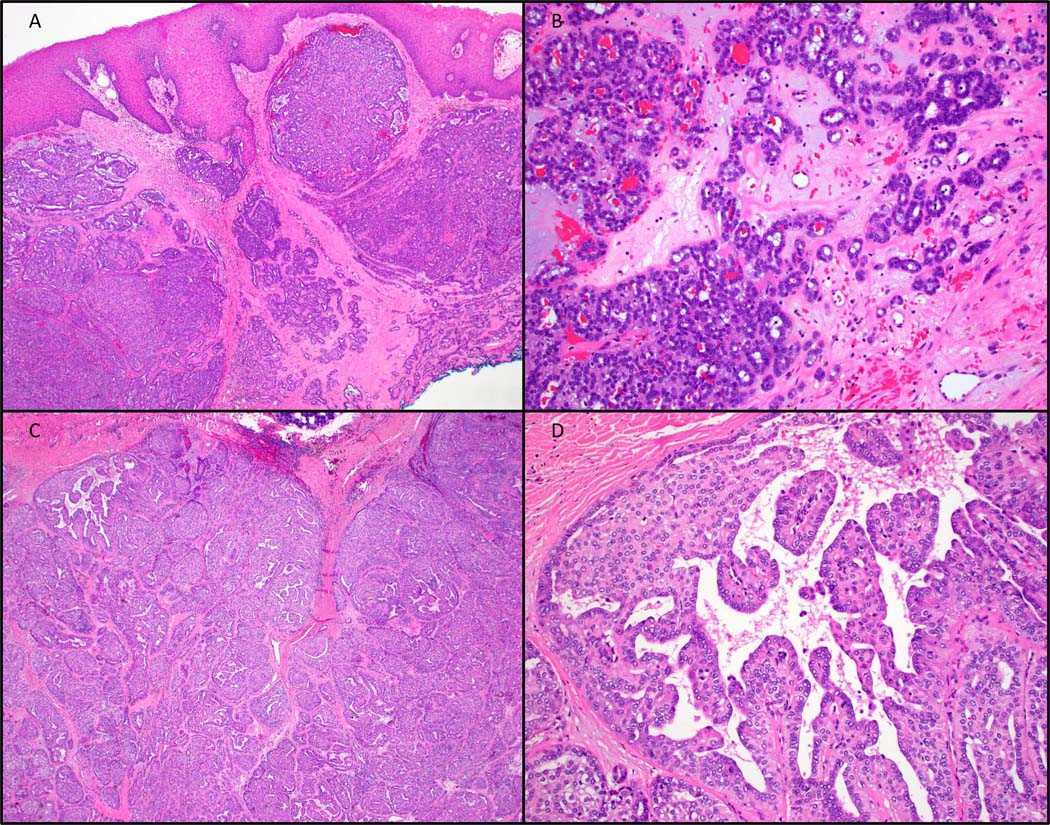
A. Low-power view of a mixed/indeterminate subtype tumour of the minor salivary glands with a PRKD1 E710D hotspot mutation. B. High-power view of a conventional focus. C. Low-power and D. high-power views of a cribriform subtype tumor of the parotid gland with a KTN1::PRKD1 fusion.
NGS was successful in 51 cases. Thirteen cases were negative for both PRKD1 p.E710D hotspot mutations and fusions involving PRKD1, PRKD2, or PRKD3 (See supplementary data). The remaining 38 cases had positive molecular results. These include 28 cases with gene fusions, all of which involved PRKD genes (PRKD1 n=23; PRKD2 n=3; PRKD3 n=2) (Table 1). There were 10 cases with the PRKD1 p.E710D mutation.
Table 1:
Demographic, histomorphologic, and molecular findings of the fusion-positive cases. Novel fusions are bolded. Not available listed as “-“.
| Age | Sex | Location | Histology | Grade | Mutation | Fusion |
|---|---|---|---|---|---|---|
| 53 | F | Parotid | Cribriform | High | – | ACTN1::PRKD1 |
| 48 | F | Palate | Mixed/indeterminate | Low | – | ANXA7::PRKD1 |
| 91 | F | Oral cavity | Mixed/indeterminate | Low | – | ANXA7::PRKD1 |
| 49 | M | Pharynx | Cribriform | Low | Negative | ARID1A::PRKD1 |
| 66 | F | Pharynx | Cribriform | Low | Negative | ARID1A::PRKD1 |
| 63 | F | Pharynx | Cribriform | Low | – | ARID1A::PRKD1 |
| 38 | F | Oral cavity | Cribriform | Low | – | ARID1A::PRKD1 |
| 37 | F | Pharynx | Cribriform | Low | – | ARID1A::PRKD1 |
| 57 | F | Sinonasal | Cribriform | High | Negative | ARID1A::PRKD1 and ATP5F1A::SS18 |
| 45 | F | Sinonasal | Cribriform | – | – | ARID1B::PRKD1 |
| 46 | F | Parotid | Cribriform | Low | – | ARID1B::PRKD1 |
| 46 | F | Parotid | Cribriform | Low | Negative | DDX3X::PRKD1 |
| 34 | F | Pharynx | Cribriform | Low | – | ERC1::PRKD1 |
| 38 | F | Sinonasal | Mixed/indeterminate | – | – | GPHN::PRKD1 |
| 47 | F | Palate | Cribriform | – | – | GPHN::PRKD1 |
| 46 | M | Sinonasal | Mixed/indeterminate | Low | – | KTN1::PRKD1 |
| 63 | F | Pharynx | Cribriform | Low | – | KTN1::PRKD1 |
| 78 | M | Parotid | Cribriform | – | – | KTN1::PRKD1 |
| 68 | F | Palate | Mixed/indeterminate | Low | – | NFIA::PRKD1 |
| 66 | F | Oral cavity | Cribriform | High | – | NOL4L::PRKD1 |
| 48 | F | Oral cavity | Mixed/indeterminate | – | – | RBM25::PRKD1 |
| 47 | F | Palate | Cribriform | – | – | TAX1BP1::PRKD1 |
| 43 | M | Palate | Cribriform | High | – | UBE2D3::PRKD1 |
| 65 | F | Pharynx | Cribriform | Low | – | CTNNB1::PRKD2 |
| 75 | M | Palate | Cribriform | Low | – | RBPMS::PRKD2 |
| 84 | F | Pharynx | Cribriform | Low | – | SS18::PRKD2 |
| 39 | F | Pharynx | Cribriform | Low | – | ATL2::PRKD3 |
| 59 | F | Palate | Mixed/indeterminate | Low | – | ATL2::PRKD3 |
A variety of novel fusion partners were identified (Table 1), with a recurrence of ANXA7::PRKD1, GPHN::PRKD1, and ARID1B::PRKD1. The latter of which, along with the previously recognized ARID1A::PRKD1, represented the most common fusion events in PAC (8 of 28 cases). Additionally, there was 1 high-grade cribriform variant that demonstrated 2 gene fusions, including ARID1A::PRKD1 and a novel gene fusion (ATP5F1A::SS18). Overall, there were 17 fusion partners to PRKD1, PRKD2, and PRKD3, including 13 novel partners. The fusions involved exons 11–13 of PRKD1, exons 10–11 of PRKD2, and exons 10–13 of PRKD3 (Table 2).
Table 2:
Exonic breakpoints of a subset of fusion-positive cases.
| Fusion | Gene A | Gene A exon | Gene B | Gene B exon |
|---|---|---|---|---|
| ARID1B::PRKD1 | ARID1B | 4 | PRKD1 | 11 |
| KTN1::PRKD1 | KTN1 | 41 | PRKD1 | 11 |
| NOL4L::PRKD1 | NOL4L | 2 | PRKD1 | 11 |
| RBM25::PRKD1 | RBM25 | 2 | PRKD1 | 11 |
| ACTN1::PRKD1 | ACTN1 | 1 | PRKD1 | 12 |
| ANXA7::PRKD1 | ANXA7 | 5 | PRKD1 | 12 |
| ARID1A::PRKD1 | ARID1A | 1 | PRKD1 | 12 |
| UBE2D3::PRKD1 | UBE2D3 | 4 | PRKD1 | 12 |
| ANXA7::PRKD1 | ANXA7 | 5 | PRKD1 | 13 |
| ARID1A::PRKD1 | ARD1A | 1 | PRKD1 | 13 |
| ARID1A::PRKD1 | ARID1A | 1 | PRKD1 | 13 |
| ARID1B::PRKD1 | ARID1B | 4 | PRKD1 | 13 |
| ERC1::PRKD1 | ERC1 | 13 | PRKD1 | 13 |
| GPHN::PRKD1 | GPHN | 8 | PRKD1 | 13 |
| GPHN::PRKD1 | GPHN | 8 | PRKD1 | 13 |
| KTN1::PRKD1 | KTN1 | 33 | PRKD1 | 13 |
| KTN1::PRKD1 | KTN1 | 33 | PRKD1 | 13 |
| NFIA::PRKD1 | NFIA | 11 | PRKD1 | 13 |
| TAX1BP1::PRKD1 | TAX1BP1 | 8 | PRKD1 | 13 |
| CTNNB1::PRKD2 | CTNNB1 | 1 | PRKD2 | 10 |
| RBPMS::PRKD2 | RBPMS | 1 | PRKD2 | 11 |
| SS18::PRKD2 | SS18 | 10 | PRKD2 | 11 |
| ATL2::PRKD3 | ATL2 | 12 | PRKD3 | 10 |
| ATL2::PRKD3 | ATL2 | 11 | PRKD3 | 13 |
Of the 35 tumors with cribriform patterns, 21 demonstrated gene fusions (60%), 6 were positive for the hotspot mutation (17%), and there were 8 negative cases (23%) (Figure 3). Of the 16 indeterminate cases that were successfully sequenced, 7 demonstrated gene fusions (44%), 4 demonstrated the PRKD1 p.E710D hotspot mutation (25%), and 5 cases were negative (31%) (Figure 3).
Figure 3:

Molecular alterations arranged by histomorphologic subtype.
Discussion
The discovery of the PRKD1 p.E710D hotspot mutation in conventional PAC, followed by the discovery of fusions affecting PRKD1, PRKD2, and PRKD3 in cribriform PAC, provided a molecular link between what was previously thought by some to represent two entities (8)(9). Indeed, both types are included under the umbrella diagnosis of PAC in the current WHO Classification of Head and Neck Tumors (11). Our results highlight the molecular diversity of PAC, comprising different molecular mechanisms of tumorigenesis (point mutations versus gene fusions) affecting a number of different genes, both within the PRKD family and their partners. Importantly, we describe multiple novel recurrent gene fusions in PACs. These include, ANXA7::PRKD1, GPHN::PRKD1, and ARID1B::PRKD1 fusions, as detailed above.
Where available, the exonic breakpoints of each gene involved in the fusions were assessed (Table 2). The PRKD genes demonstrated consistent break points upstream of the protein kinase domain, the same domain affected by the activating E710D point mutation characteristic of conventional PAC. The PRKD genes share a common structure consisting of a 5’ regulatory cysteine-rich zinc-finger motif, an autoregulatory pleckstrin homology (PH) domain, and a protein kinase domain (Figure 4) (16)(17)(18)(19)(20)(21). The PRKD gene fusions recurrently occur in regions that preserve the protein kinase domain while removing the cysteine rich regulatory domain and either disrupting or removing the PH domain (exons 11–13 of PRKD1, exons 10–11 of PRKD2, exons 10–13 of PRKD3) (Figure 4). Further, deletion of all or part of the PH domain has been shown to increase basal kinase activity in PRKD1, and likely acts similarly in PRKD2 and PRKD3 (16). Additionally, a specific amino acid residue (T513) in PKD1, that when mutated loses its autoregulatory function, is lost in all of our PRKD1 fusion-positive cases where exon-level data was available (22).
Figure 4:

Structure of the PRKD genes. Cysteine-rich regulatory domains (red; C); pleckstrinhomology domain (purple; PH); protein kinase domain (green; PKD). Exonic breakpoints found in our cohort denoted by hashed black bars (exons 11, 12 and 13 in PRKD1; exons 10 and 11 in PRKD2; exons 10 and 13 in PRKD3). Terminal codons and E710 hot-spot indicated in gray (21)(22).
The diverse gene partners included genes that have a wide variety of cellular functions. ARID1A, the most common recurrent fusion identified, only had fusion breakpoints observed in the first intron. This region is far upstream of any functional domains and commonly occurring deactivating mutations in cancers (23)(24)(25). The breakpoints described on ARID1B are also upstream of the ARID domain (26). A number of other gene partners in our cohort featured intron 1 fusion breakpoints (ACTN1, CTNNB1, and RBPMS). It is unlikely that any biochemical activity is gained from these 5’ fusion partners. In other instances, however, more of the functional domains are included in the fusions (ATL2, ERC1, NFIA, SS18, TAX1BP1, and KTN1). Considering the diversity of gene partners and the most commonly recurrent fusion contributing only one exon, the fusions likely function by the contribution of a promoter or enhancer from the first gene partner to the protein kinase domain of the PRKD gene, in addition to removing the cysteine-rich zinc-finger regulatory domain and inactivating the auto regulatory PH domain of the PKRD gene, through complete removal or disruption of its function. Beyond this, the contribution of the 5’ partner, if any, is unknown.
We identified 2 cases with a novel ARID1B::PRKD1 gene fusion. When taken in the context of the well described ARID1A::PRKD1 fusions in these tumors, fusion events involving this family of genes appear to characterize the most common rearrangements in PAC. We also describe the first fusions partners for PRKD2 (CTNNB1, RBPMS, and SS18). Additionally, in our study, we identified 2 cases with fusions involving the PRKD3 gene, both of which have ATL2 as the fusion partner. This finding has been described before, and perhaps characterizes the extent of PRKD3 fusions seen in PAC (27).
Interestingly, we identified 2 tumors with fusions involving the SS18 gene. One of these tumors was high-grade, and demonstrated 2 fusions: the canonical ARID1A::PRKD1 fusion, as well as an ATP5F1A::SS18 fusion. Morphologically, the tumor had high-grade areas in addition to areas that were similar to microsecretory adenocarcinoma (Figure 5A). The possible explanations for this case include a single tumor with 2 gene fusions and high-grade transformation, or alternatively, a collision tumor between a high-grade polymorphous adenocarcinoma and a microsecretory adenocarcinoma. The other tumor demonstrated a single fusion between SS18 and PRKD2, a novel finding in our study. Within this tumor there were foci that demonstrated luminal secretions reminiscent of microsecretory adenocarcinoma (Figure 5B). This case raises the possibility that the distinction between these two tumors may not be as clear as initially described. Both entities are frequently located in the oral cavity, and demonstrate bland cytology and infiltrative, occasionally single-filed, growth, with a shared immunophenotype (1). This case also highlights a potential pitfall of relying solely on FISH analysis for molecular diagnosis.
Figure 5:
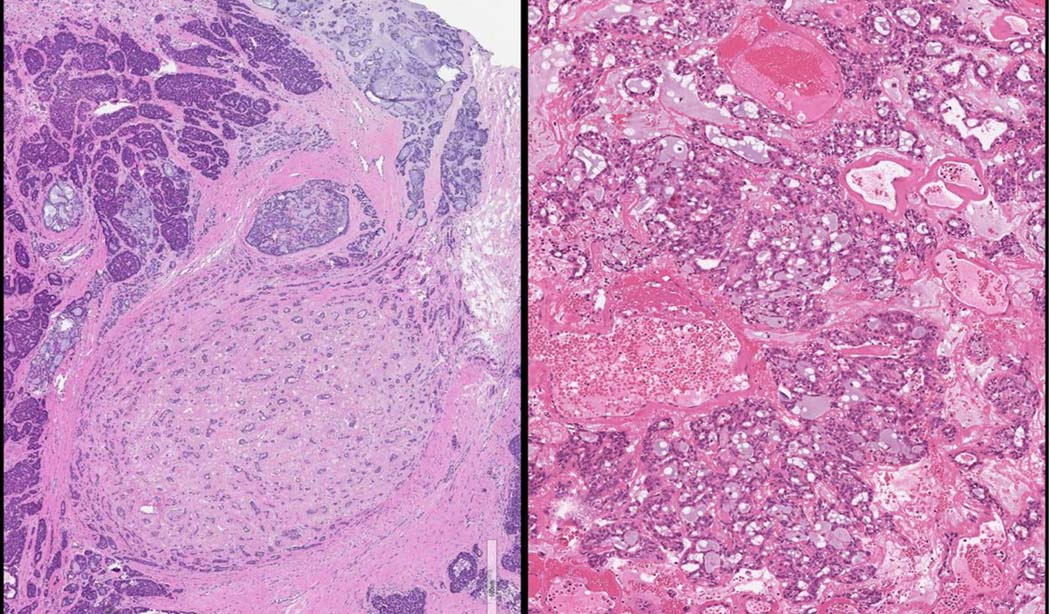
A. Tumor with ARID1A::PRKD1 and ATP5F1A::SS18 fusions, demonstrating both high-grade areas and areas morphologically reminiscent of microsecretory adenocarcinoma. B. SS18::PRKD2 fusion-positive tumor, also with foci reminiscent of microsecretory adenocarcinoma.
Histomorphologic characterization segregates tumors by molecular alteration with modest reliability, with conventional/fascicular tumors typically harboring PRKD1 hotspot mutations, and those with cribriform morphologies harboring fusions involving PRKD1, PRKD2, and PRKD3 (7). There are frequent exceptions to this rule, however, as is demonstrated by our data, which further brings into question the value of routine histologic subtyping. Interobserver reliability has been demonstrated to be moderate in histomorphologically characterizing these tumors (10). In the current clinical workflow, the use of histomorphology alone is generally sufficient for diagnostic accuracy as it pertains to clinical management. Laboratories with NGS panels that do not include PRKD1, PRKD2, or PRKD3, may be able to target one of the above described fusion partners to confirm a diagnosis of PAC in difficult cases. Should one of the molecular alterations be proven targetable, histomorphology alone will be insufficient, and molecular testing of all PAC cases will be necessary in the identification of these alterations.
Conclusion
NGS of cribriform and mixed/indeterminate PAC has elucidated a wide variety of gene fusions, including entirely novel fusions, and multiple PRKD gene partners, in addition to a non-negligible number of PRKD1 p.E710D hotspot-mutated tumors. The most common partners for PRKD1 are ARID1A and ARID1B, while PRKD3 seems to preferentially associate with ATL2. Two PAC cases showed SS18 fusions highlighting that this gene is not specific for microsecretory or microcribriform adenocarcinomas. The wide variety of involved genes is unlike other salivary gland malignancies that are typically characterized by one or two canonical fusions, and warrants a broader strategy of sequencing for diagnostic fusion confirmation. As we trudge further into the era of personalized medicine and targeted therapies, our results highlight the need for more comprehensive molecular analysis to fully elucidate pathogenic fusions in these tumors.
Supplementary Material
Funding:
Research reported in this publication was supported in part by the Cancer Center Support Grant of the National Institutes of Health/National Cancer Institute under award number P30 CA008748 (BX, NK, SD).
Footnotes
Ethics Approval and Consent to Participate:
Institutional research ethics board approval was obtained from the represented institutions.
Conflict of Interest: None to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data availability statement:
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request. The majority of the data generated or analyzed during this study are included in this published article and its supplementary information files.
References
- 1.Bishop JA, Weinreb I, Swanson D, Westra WH, Qureshi HS, Sciubba J, et al. Microsecretory Adenocarcinoma: A Novel Salivary Gland Tumor Characterized by a Recurrent MEF2C-SS18 Fusion. Am J Surg Pathol. 2019. Aug;43(8):1023–32. [DOI] [PubMed] [Google Scholar]
- 2.Weinreb I, Hahn E, Dickson BC, Rooper LM, Rupp NJ, Freiberger SN, et al. Microcribriform Adenocarcinoma of Salivary Glands: A Unique Tumor Entity Characterized by an SS18::ZBTB7A Fusion. Am J Surg Pathol. 2023. Feb 1;47(2):194–201. [DOI] [PubMed] [Google Scholar]
- 3.Evans HL, Batsakis JG. Polymorphous low-grade adenocarcinoma of minor salivary glands. A study of 14 cases of a distinctive neoplasm. Cancer. 1984. Feb 15;53(4):935–42. [DOI] [PubMed] [Google Scholar]
- 4.Evans HL, Luna MA. Polymorphous low-grade adenocarcinoma: a study of 40 cases with long-term follow up and an evaluation of the importance of papillary areas. Am J Surg Pathol. 2000. Oct;24(10):1319–28. [DOI] [PubMed] [Google Scholar]
- 5.Michal M, Skálová A, Simpson RH, Raslan WF, Curík R, Leivo I, et al. Cribriform adenocarcinoma of the tongue: a hitherto unrecognized type of adenocarcinoma characteristically occurring in the tongue. Histopathology. 1999. Dec;35(6):495–501. [DOI] [PubMed] [Google Scholar]
- 6.Skalova A, Sima R, Kaspirkova-Nemcova J, Simpson RHW, Elmberger G, Leivo I, et al. Cribriform adenocarcinoma of minor salivary gland origin principally affecting the tongue: characterization of new entity. Am J Surg Pathol. 2011. Aug;35(8):1168–76. [DOI] [PubMed] [Google Scholar]
- 7.Sebastiao APM, Xu B, Lozada JR, Pareja F, Geyer FC, Da Cruz Paula A, et al. Histologic spectrum of polymorphous adenocarcinoma of the salivary gland harbor genetic alterations affecting PRKD genes. Mod Pathol Off J U S Can Acad Pathol Inc. 2020. Jan;33(1):65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weinreb I, Zhang L, Tirunagari LMS, Sung YS, Chen CL, Perez-Ordonez B, et al. Novel PRKD gene rearrangements and variant fusions in cribriform adenocarcinoma of salivary gland origin. Genes Chromosomes Cancer. 2014. Oct;53(10):845–56. [DOI] [PubMed] [Google Scholar]
- 9.Weinreb I, Piscuoglio S, Martelotto LG, Waggott D, Ng CKY, Perez-Ordonez B, et al. Hotspot activating PRKD1 somatic mutations in polymorphous low-grade adenocarcinomas of the salivary glands. Nat Genet. 2014. Nov;46(11):1166–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu B, Barbieri AL, Bishop JA, Chiosea SI, Dogan S, Di Palma S, et al. Histologic Classification and Molecular Signature of Polymorphous Adenocarcinoma (PAC) and Cribriform Adenocarcinoma of Salivary Gland (CASG): An International Interobserver Study. Am J Surg Pathol. 2020. Apr;44(4):545–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Skalova A, Thompson LDR, Bishop JA, Mehrotra R, Hyrcza MD. Salivary gland tumours. In: Board WCTE, editor. Head and neck tumours. 5th ed. Lyon: International Agency for Research on Cancer; 2022. [Google Scholar]
- 12.Haas BJ, Dobin A, Li B, Stransky N, Pochet N, Regev A. Accuracy assessment of fusion transcript detection via read-mapping and de novo fusion transcript assembly-based methods. Genome Biol. 2019. Oct 21;20(1):213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu G, Benayed R, Ho C, Mullaney K, Sukhadia P, Rios K, et al. Diagnosis of known sarcoma fusions and novel fusion partners by targeted RNA sequencing with identification of a recurrent ACTB-FOSB fusion in pseudomyogenic hemangioendothelioma. Mod Pathol Off J U S Can Acad Pathol Inc. 2019. May;32(5):609–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu S, Tsai WH, Ding Y, Chen R, Fang Z, Huo Z, et al. Comprehensive evaluation of fusion transcript detection algorithms and a meta-caller to combine top performing methods in paired-end RNA-seq data. Nucleic Acids Res. 2016. Mar 18;44(5):e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Källberg M, et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinforma Oxf Engl. 2016. Apr 15;32(8):1220–2. [DOI] [PubMed] [Google Scholar]
- 16.Iglesias T, Rozengurt E. Protein kinase D activation by mutations within its pleckstrin homology domain. J Biol Chem. 1998. Jan 2;273(1):410–6. [DOI] [PubMed] [Google Scholar]
- 17.Sturany S, Van Lint J, Muller F, Wilda M, Hameister H, Hocker M, et al. Molecular cloning and characterization of the human protein kinase D2. A novel member of the protein kinase D family of serine threonine kinases. J Biol Chem. 2001. Feb 2;276(5):3310–8. [DOI] [PubMed] [Google Scholar]
- 18.Hayashi A, Seki N, Hattori A, Kozuma S, Saito T. PKCnu, a new member of the protein kinase C family, composes a fourth subfamily with PKCmu. Biochim Biophys Acta. 1999. May 6;1450(1):99–106. [DOI] [PubMed] [Google Scholar]
- 19.Steinberg SF. Regulation of protein kinase D1 activity. Mol Pharmacol. 2012. Mar;81(3):284–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou X, Edmonson MN, Wilkinson MR, Patel A, Wu G, Liu Y, et al. Exploring genomic alteration in pediatric cancer using ProteinPaint. Nat Genet. 2016. Jan;48(1):4–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.UniProt Consortium. UniProt: the Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023. Jan 6;51(D1):D523–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ziemba BP, Pilling C, Calleja V, Larijani B, Falke JJ. The PH domain of phosphoinositide-dependent kinase-1 exhibits a novel, phospho-regulated monomer-dimer equilibrium with important implications for kinase domain activation: single-molecule and ensemble studies. Biochemistry. 2013. Jul 16;52(28):4820–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu JN, Roberts CWM. ARID1A mutations in cancer: another epigenetic tumor suppressor? Cancer Discov. 2013. Jan;3(1):35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guan B, Mao TL, Panuganti PK, Kuhn E, Kurman RJ, Maeda D, et al. Mutation and loss of expression of ARID1A in uterine low-grade endometrioid carcinoma. Am J Surg Pathol. 2011. May;35(5):625–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cornen S, Adelaide J, Bertucci F, Finetti P, Guille A, Birnbaum DJ, et al. Mutations and deletions of ARID1A in breast tumors. Oncogene. 2012. Sep 20;31(38):4255–6. [DOI] [PubMed] [Google Scholar]
- 26.Hurlstone AFL, Olave IA, Barker N, van Noort M, Clevers H. Cloning and characterization of hELD/OSA1, a novel BRG1 interacting protein. Biochem J. 2002. May 15;364(Pt 1):255–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Freiberger SN, Brada M, Fritz C, Höller S, Vogetseder A, Horcic M, et al. SalvGlandDx - a comprehensive salivary gland neoplasm specific next generation sequencing panel to facilitate diagnosis and identify therapeutic targets. Neoplasia N Y N. 2021. May;23(5):473–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request. The majority of the data generated or analyzed during this study are included in this published article and its supplementary information files.