

Abstract
Background and Objectives
The complement system is known to play a role in multiple sclerosis (MS) pathogenesis. However, its contribution to disease progression remains elusive. The study investigated the role of the complement system in disability progression of patients with primary progressive MS (PPMS).
Methods
Sixty-eight patients with PPMS from 12 European MS centers were included in the study. Serum and CSF levels of a panel of complement components (CCs) were measured by multiplex enzyme-linked immunosorbent assay at a baseline time point (i.e., sampling). Mean (SD) follow-up time from baseline was 9.6 (4.8) years. Only one patient (1.5%) was treated during follow-up. Univariable and multivariable logistic regressions adjusted for age, sex, and albumin quotient were performed to assess the association between baseline CC levels and disability progression in short term (2 years), medium term (6 years), and long term (at the time of the last follow-up).
Results
In short term, CC played little or no role in disability progression. In medium term, an elevated serum C3a/C3 ratio was associated with a higher risk of disability progression (adjusted OR 2.30; 95% CI 1.17–6.03; p = 0.040). By contrast, increased CSF C1q levels were associated with a trend toward reduced risk of disability progression (adjusted OR 0.43; 95% CI 0.17–0.98; p = 0.054). Similarly, in long term, an elevated serum C3a/C3 ratio was associated with higher risk of disability progression (adjusted OR 1.81; 95% CI 1.09–3.40; p = 0.037), and increased CSF C1q levels predicted lower disability progression (adjusted OR 0.41; 95% CI 0.17–0.86; p = 0.025).
Discussion
Proteins involved in the activation of early complement cascades play a role in disability progression as risk (elevated serum C3a/C3 ratio) or protective (elevated CSF C1q) factors after 6 or more years of follow-up in patients with PPMS. The protective effects associated with C1q levels in CSF may be related to its neuroprotective and anti-inflammatory properties.
Introduction
Innate immune system components are known to play an important role in the immunopathology of multiple sclerosis (MS).1,2 The complement system is a key component of the innate immune system that consists of a network of circulating proteins and membrane-expressed proteins that recognize non–self-components and is activated by the classical (triggered by antigen-antibody complexes being IgM the most efficient isotype for complement activation), alternative, or lectin pathways (triggered by foreign surfaces and carbohydrates, respectively).3,4 Once activated, the complement system is critical for clearing pathogens as well as dead and apoptotic cells.5 Although the liver was originally considered to be the main site for the synthesis of complement factors,6 more recently it became apparent that most complement components (CCs) can also be synthesized within the CNS7 and they can play important roles in normal synaptic pruning and neurodevelopment.8,9
A large number of studies support a role of the complement system in MS pathogenesis. Immunohistochemical studies of postmortem MS brains have shown immunoreactivity for CCs, activation products, and complement regulators not only in plaque and periplaque areas but also in normal-appearing white matter and cortical regions.10,11 Several studies have also reported differences in the blood and CSF levels of CCs between patients with MS and healthy controls and between patients with different clinical forms of MS.12-14 However, few of these studies were performed in patients with progressive MS and none of them evaluated long-term outcomes. Of interest, immunoreactivity for CCs has been observed in chronic active and inactive plaques of patients with progressive MS in the absence of other inflammation markers such as blood immune cells, indicating that progression of inflammation in MS does not depend on infiltrating cells and can be driven by innate immune mechanisms including complement activation.10 Furthermore, complement was found to be activated in cortical gray matter lesions of patients with progressive MS, particularly in areas of increased numbers of complement receptor-positive microglia, suggesting that complement overactivation may contribute to the irreversible disease progression occurring in these patients.9 Altogether, these findings prompted us to investigate a potential relationship between the complement system and disability progression of patients with primary progressive MS (PPMS) after a long-term follow-up.
Methods
Patients
A multicentric cohort of 68 patients with PPMS recruited from 12 European MS centers (Madrid—Puerta de Hierro [n = 1], Madrid—Ramón y Cajal [n = 7], Madrid—Hospital Clínico San Carlos [n = 2], Barcelona—Cemcat [n = 11], Barcelona—Hospital de Bellvitge [n = 6], Barcelona—Hospital Clinic [n = 1], Valencia [n = 5], San Sebastián [n = 2], Sahlgrenska [n = 3], Innsbruck [n = 13], Ulm [n = 5], Poland [n = 12]) was included in the study. Selection of patients was performed based on (1) the availability of paired serum and CSF samples never thawed or thawed at most once and (2) follow-up of patients longer than 5 years preferably without receiving disease-modifying therapies.
Clinical Assessments and Definition of Disability Progression
Expanded Disability Status Scale (EDSS) scores were recorded at baseline (i.e., sampling time point), 2 and 6 years, and at the time of the last visit. Short-term disability progression was defined as an increase of at least 1 point in the EDSS if baseline EDSS ≤5.0 and 0.5 points if baseline EDSS ≥5.5 during the first 2 years. Taking into account that most of the patients would fulfill this progression criterion at medium and long term, to assess disability progression at these time points, progression rates were computed by dividing EDSS changes by the time on follow-up between baseline and 6 years for medium-term disability progression and between baseline and the time of the last visit for long-term disability progression. Then, medium and long-term progressors were defined as those patients displaying progression rates above the 75th percentile of disability progression.
CSF and Serum Processing and Analysis
CSF samples were collected by lumbar puncture for routine CSF diagnostics, centrifuged to remove cells, aliquoted, and stored frozen at −80°C until used. Peripheral blood was collected by standard venipuncture and allowed to clot spontaneously for 30 minutes. Serum was obtained by centrifugation and stored frozen at −80°C until used. CSF and serum concentrations of IgM and albumin were measured by nephelometry in a BN Prospect instrument (Siemens). The IgM index was calculated by using the following quotient: IgMCSF/IgMserum:AlbuminCSF/Albuminserum. The albumin CSF/serum ratio (Qalb) was also calculated to evaluate the relationship between CSF levels of CCs and blood-CSF barrier dysfunction.
Determination of CCs
CSF and serum samples were shipped to the University Hospital of Münster (Germany) on dry ice. For the quantification of serum and CSF levels of Ba, Bb, C3a, C4a, C5a, sC5b9, Factor H, Factor I, C1q, C3, C4, and C5, samples were thawed on ice and immediately processed. Multiplex enzyme-linked immunosorbent assays based on chemiluminesence were used according to the manufacturer's recommendations (Quidel, San Diego, CA; cat. number: A900, A917) to systematically profile protein concentrations (in serum and CSF samples). Each plate contained samples from all different groups to minimize consequences of potential interplate variation. Control samples provided by the manufacturer were included on each plate to ensure plate-to-plate consistency. For data points below the lower limit of quantification or above the upper limit of quantification, the respective threshold was used as value for analysis.
MRI Activity
To evaluate the effect of inflammation on blood and CSF levels of CCs, patients with PPMS were classified into inflammatory and noninflammatory according to the presence or absence of the following criteria: (1) contrast-enhancing lesions in a baseline MRI performed in proximity to sample collection (within a time window of ≤3 months) and (2) appearance of new T2 lesions or contrast-enhancing lesions in the follow-up MRI scans performed according to the standard clinical practice at each participating center. PPMS patients with radiologic disease activity at baseline and/or during follow-up were classified as inflammatory.
Statistical Methods
Mann-Whitney test was used to test for significant differences in serum and CSF CCs between progressors and nonprogressors in short, medium, and long term and also between patients with inflammatory and noninflammatory PPMS. Univariable linear models were fitted to evaluate the influence of age, sex, Qalb, duration of the disease, and EDSS on the complement system, using each CC as outcome. Univariable and multivariable logistic regressions adjusted by age, sex, and Qalb were built to evaluate the ability of baseline serum and CSF levels of CCs to predict disability progression in the short, medium, and long term. From each linear model and logistic regression, a diagnostic evaluation of the residuals was performed, and convenient transformations (log transformation, Box-Cox transformation, 1/x) were tested to accomplish the linear models' assumptions (normality and homoscedasticity of the residuals). Log transformation of the CCs were finally performed in all the models given the distribution of the data. Receiver-operating characteristic (ROC) curves were built to determine optimal cutoff points, and area under the ROC curve (AUC), sensitivity, and specificity were obtained to evaluate model performance. Correlations between CC and IgM index were performed using the Spearman rank correlation coefficient.
Data Availability
Anonymized data will be shared upon reasonable request.
Standard Protocol Approvals, Registrations, and Patient Consents
The study was approved by the corresponding Hospital Ethics Committee, and participants gave written informed consent.
Results
Patients
Demographic, clinical, and radiologic information of patients with PPMS is summarized in Table 1. At baseline (i.e., sampling), the mean (SD) age of patients was 52.2 (9.1) years, and 26 (38.0%) were men. Disease duration was 6.2 (6.9) years. Median (interquartile range) EDSS was 3.5 (2.5–5.5). None of the patients had a relapse in the 3 months before baseline sample collection.
Table 1.
Demographic, Clinical, and Radiologic Characteristics of Patients With PPMS
| Characteristics | Whole cohort |
| N | 68 |
| Age, ya,b | 52.2 (9.1) |
| Male/female, % men | 26/42 (38.0) |
| Disease duration, ya,c | 6.2 (6.9) |
| Follow-up time, ya,d | 9.6 (4.8) |
| EDSS at baseline | 3.5 (2.5–5.5) |
| EDSS at 2 y | 4.0 (3.0–6.5) |
| EDSS at 6 y | 6.0 (5.0–7.0) |
| EDSS at last visit | 6.3 (4.8–7.1) |
| Short-term progressors/nonprogressors, n (%)e | 21 (33.9)/41 (66.1) |
| Medium-term progressors/nonprogressors, n (%)e | 13 (22.4)/45 (77.6) |
| Long-term progressors/nonprogressors, n (%) | 17 (25.0)/51 (75.0) |
| Relapses in the previous 3 mo, n (%)f | 0 (0) |
| Treatment during follow-up, n (%)g | 1 (1.5) |
| MRI findings, n (%) | |
| Inflammatory | 24 (35.0) |
| Noninflammatory | 32 (47.0) |
| Unknown | 12 (18.0) |
| IgM index | 0.06 (0.03–0.14) |
| Qalb | 5.8 (4.2–7.2) |
Abbreviation: EDSS = Expanded Disability Status Scale.
Data are expressed as median (interquartile range) unless otherwise stated.
Data are expressed as mean (SD).
Refers to age at sample collection.
Refers to the time between disease onset and sample collection.
Refers to the time between sample collection and the time of the last visit.
Information missing in 6 (8.8%) and 10 (14.7%) patients, respectively.
Refers to the presence of relapses in the 3 mo before sample collection.
One patient was treated with interferon-beta for 2 y after lumbar puncture.
Associations Between CCs and Demographic and Clinical Variables at Baseline
Baseline median serum and CSF levels of CCs in the whole cohort of patients with PPMS are shown in eTable 1. eTables 2 and 3 show the results of the multivariable analysis at baseline of serum and CSF CCs, respectively. In serum, Bb, C5a, Factors H and I, SC5b9, and C5a/C5 were associated with age. Components Bb, C5a, and the C3a/C3 and C5a/C5 ratios were associated with disease duration. None of the serum components was associated with sex or with baseline EDSS and Qalb. In CSF, Ba, Bb, C3a, C5a, C4, C3a/C3, and C5a/C5 were associated with age. Bb, C3a, C1q, C4, C5, and the 3 ratios were associated with disease duration. All CCs were associated with Qalb, except Ba, C1q, C4, C4a/C4, and C5a/C5. None of the CSF components was associated with sex or with baseline EDSS.
Associations Between CSF and Serum Levels of CCs and Short-Term, Medium-Term, and Long-Term Disability Progression
Patients with PPMS were followed for a mean and median time of 9.6 (4.8) years and 8.0 (7.0–10.7) years, respectively. Median EDSS scores at 2 years, 6 years, and at the time of the last visit were 4.0 (3.0–6.5), 6.0 (5.0–7.0), and 6.3 (4.8–7.1), respectively (Table 1). Only one patient (1.5%) received treatment during follow-up (Table 1).
In the short term, 21 (33.9%) patients were classified as progressors. As shown in eTable 4, comparison of baseline serum levels of CCs between progressors and nonprogressors revealed significantly higher levels of C3a (p = 0.028), C4a (p = 0.039), and the C5a/C5 ratio (p = 0.028) in short-term progressors compared with nonprogressors. In CSF, levels of CCs were comparable between short-term progressors and nonprogressors (eTable 4). Serum C3a levels predicted short-term disability progression (OR 1.73; 95% CI 1.05–3.08; p = 0.044); however, significance was lost in the multivariable logistic regression (eTable 5). Elevated serum C5 levels were associated with lower risk of short-term disability progression only in the multivariable analysis (OR 0.12; 95% CI 0.01–0.68; p = 0.027). None of the CSF CCs predicted short-term disability progression in univariable or multivariable analyses (eTable 6).
In the medium term, 13 (22.4%) patients had progression rates above the 75th percentile and were classified as progressors. In serum, the C3a/C3 ratio at baseline was significantly higher in medium-term progressors compared with nonprogressors (p = 0.029; Figure 1A). A C3a/C3 ratio value of 2.38 was the best cutoff to classify medium-term progressors and nonprogressors with a sensitivity of 61.5% and specificity of 73% (Figure 1A). Serum levels of the remaining CCs were comparable between medium-term progressors and nonprogressors (eTable 7). As shown in Table 2, an increased C3a/C3 ratio was associated with a higher risk of medium-term disability progression both in univariable (OR 2.03; 95% CI 1.18–3.91; p = 0.018) and multivariable (OR 2.30; 95% CI 1.17–6.03; p = 0.040) analyses. Elevated levels of Bb were associated with increased risk of medium-term disability progression only in univariable analysis (OR 2.08; 95% CI 1.03–4.56; p = 0.049) (Table 2).
Figure 1. Serum C3a/C3 Ratio in Progressors and Nonprogressors.
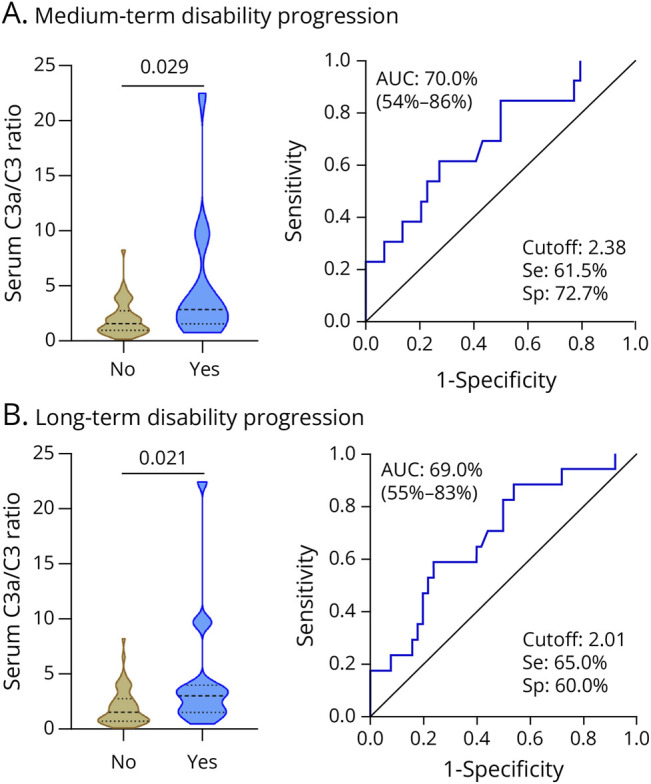
Violin plots showing the distribution of the serum C3a/C3 ratio at baseline in progressors and nonprogressors (left graphs), and performance of the serum C3a/C3 ratio to discriminate between progressors and nonprogressors in medium term (A) and long term (B) (right graphs). No: nonprogressors. Yes: progressors. AUC = area under the curve; SE = sensitivity; Sp = specificity.
Table 2.
Associations Between Serum and CSF Levels of Complement Components and Medium-Term Disability Progression
| Components | Univariable OR (95% CI); p value | Multivariable OR (95% CI); p value | Components | Univariable OR (95% CI); p value | Multivariable OR (95% CI); p value |
| Serum | CSF | ||||
| Ba (ng/mL) | 1.21 (0.48–3.47); 0.699 | 1.06 (0.36–3.43); 0.921 | Ba (ng/mL) | 1.45 (0.78–2.75); 0.241 | 1.17 (0.53–2.49); 0.679 |
| Bb (μg/mL) | 2.08 (1.03–4.56); 0.049 | 1.51 (0.68–3.59); 0.319 | Bb (μg/mL) | 1.48 (0.65–3.32); 0.329 | 0.67 (0.17–2.27); 0.538 |
| C3a (ng/mL) | 1.48 (0.83–2.89); 0.214 | 1.32 (0.69–2.78); 0.419 | C3a (ng/mL) | 1.26 (0.79–1.99); 0.322 | 1.12 (0.61–2.12); 0.706 |
| C4a (ng/mL) | 1.43 (0.92–2.30); 0.117 | 1.59 (0.92–2.97); 0.114 | C4a (ng/mL) | 0.90 (0.51–1.56); 0.714 | 1.08 (0.50–2.33); 0.835 |
| C5a (ng/mL) | 1.56 (0.96–2.65); 0.081 | 1.38 (0.76–2.59); 0.296 | C5a (ng/mL) | 1.42 (0.93–2.27); 0.113 | 1.05 (0.60–1.87); 0.856 |
| Factor H (μg/mL) | 0.47 (0.08–2.35); 0.360 | 1.2 (0.10–16.84); 0.886 | Factor H (μg/mL) | 0.24 (0.02–1.94); 0.189 | 0.10 (0.00–2.73); 0.191 |
| Factor I (ng/mL) | 0.86 (0.41–1.80); 0.686 | 1.25 (0.52–3.13); 0.621 | Factor I (ng/mL) | 1.08 (0.30–3.51); 0.901 | 1.23 (0.16–8.96); 0.839 |
| SC5b9 (ng/mL) | 1.23 (0.69–2.26); 0.487 | 1.06 (0.54–2.17); 0.863 | SC5b9 (ng/mL) | 0.97 (0.52–1.78); 0.912 | 0.88 (0.37–1.99); 0.762 |
| C1q (μg/mL) | 0.38 (0.04–3.12); 0.364 | 0.31 (0.02–5.03); 0.405 | C1q (μg/mL) | 0.44 (0.21–0.83); 0.015 | 0.43 (0.17–0.98); 0.054 |
| C3 (μg/mL) | 0.61 (0.34–1.08); 0.091 | 0.45 (0.19–0.93); 0.050 | C3 (μg/mL) | 0.23 (0.02–2.45); 0.222 | 0.08 (0.00–1.31); 0.076 |
| C4 (μg/mL) | 1.36 (0.96–1.97); 0.090 | 1.16 (0.74–1.83); 0.516 | C4 (μg/mL) | 0.73 (0.45–1.19); 0.186 | 0.70 (0.36–1.32); 0.265 |
| C5 (μg/mL) | 0.77 (0.15–3.91); 0.748 | 0.92 (0.13–6.07); 0.927 | C5 (μg/mL) | 0.70 (0.41–1.19); 0.189 | 0.65 (0.28–1.45); 0.295 |
| C3a/C3 ratio | 2.03 (1.18–3.91); 0.018 | 2.30 (1.17–6.03); 0.040 | C3a/C3 ratio | 1.32 (0.84–2.10); 0.224 | 1.24 (0.69–2.34); 0.483 |
| C4a/C4 ratio | 0.91 (0.60–1.30); 0.606 | 1.27 (0.74–2.39); 0.408 | C4a/C4 ratio | 1.09 (0.80–1.46); 0.553 | 1.17 (0.79–1.73); 0.424 |
| C5a/C5 ratio | 1.47 (0.95–2.33); 0.091 | 1.29 (0.77–2.23); 0.336 | C5a/C5 ratio | 1.34 (0.99–1.84); 0.063 | 1.12 (0.77–1.64); 0.541 |
OR represents multiplicative effects of doubling of each complement component. Multivariable analysis was adjusted by age, sex, and Qalb. Table 2 only shows the results for the complement components. Results for the remaining variables included in the multivariable analysis are shown in online eTables 8 and 9. OR = odds ratio.
In CSF, C1q levels were significantly higher in medium-term nonprogressors compared with progressors (p = 0.006; Figure 2A). C1q levels showed good performance to discriminate between medium-term progressors and nonprogressors, and a C1q value of 0.15 µg/mL resulted in the best cutoff to classify patients with a sensitivity of 77% and specificity of 71% (Figure 2A). No significant differences were observed for the remaining CSF CCs between medium-term progressors and nonprogressors (eTable 7). Elevated C1q levels were associated with a reduced risk of medium-term disability progression in univariable analysis (OR 0.44; 95% CI 0.21– 0.83; p = 0.015) and remained as a trend after adjustment (OR 0.43; 95% CI 0.17–0.98; p = 0.054) (Table 2).
Figure 2. CSF C1q Levels in Progressors and Nonprogressors.
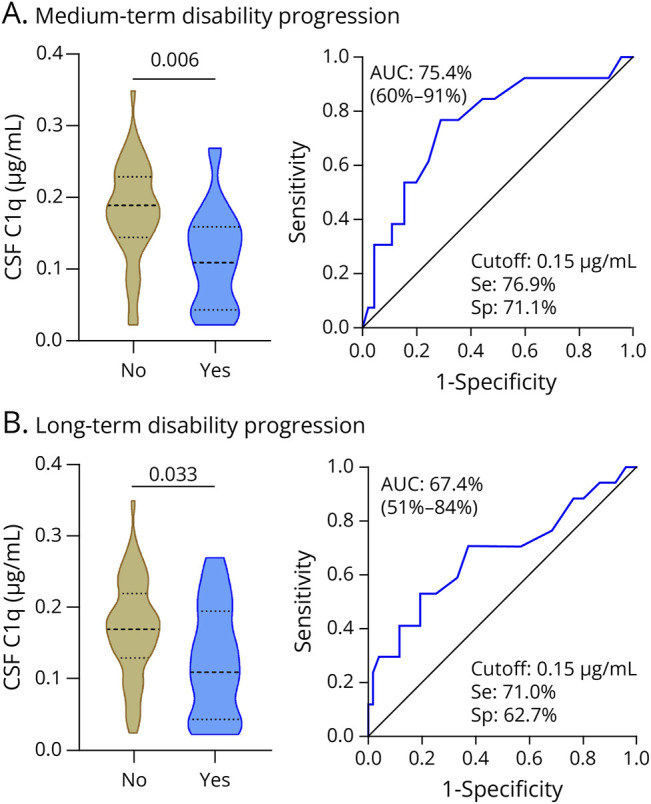
Violin plots showing the distribution of the CSF C1q levels in μg/mL at baseline in progressors and nonprogressors (left graphs), and performance of the CSF C1q levels to discriminate between progressors and nonprogressors in medium term (A) and long term (B) (right graphs). No: nonprogressors. Yes: progressors. AUC = area under the curve; SE = sensitivity; Sp = specificity.
In the long term, 17 (25.0%) patients had progression rates above the 75th percentile and were labeled as progressors. In serum, the C3a/C3 ratio at baseline was also significantly higher in long-term progressors vs nonprogressors (p = 0.021; Figure 1B). The best cutoff to classify patients was a C3a/C3 ratio value of 2.01, with a sensitivity of 65% and specificity of 60% (Figure 1B). No significant differences were observed for the remaining serum CCs between long-term progressors and nonprogressors (eTable 10). An elevated C3a/C3 ratio was associated with a higher risk of long-term disability progression both in univariable (OR 1.87; 95% CI 1.17–3.26; p = 0.015) and multivariable (OR 1.81; 95% CI 1.09–3.40; p = 0.037) analyses (Table 3).
Table 3.
Associations Between Serum and CSF Levels of Complement Components and Long-Term Disability Progression
| Components | Univariable OR (95% CI); p value | Multivariable OR (95% CI); p value | Components | Univariable OR (95% CI); p value | Multivariable OR (95% CI); p value |
| Serum | CSF | ||||
| Ba (ng/mL) | 1.05 (0.48–2.46); 0.896 | 0.87 (0.37–2.14); 0.751 | Ba (ng/mL) | 1.34 (0.79–2.30); 0.277 | 1.32 (0.71–2.45); 0.373 |
| Bb (μg/mL) | 1.59 (0.90–2.91); 0.120 | 1.28 (0.67–2.54); 0.455 | Bb (μg/mL) | 1.25 (0.65–2.33); 0.487 | 0.90 (0.36–2.07); 0.802 |
| C3a (ng/mL) | 1.51 (0.93–2.65); 0.120 | 1.36 (0.79–2.53); 0.289 | C3a (ng/mL) | 1.11 (0.75–1.61); 0.590 | 1.08 (0.66–1.73); 0.753 |
| C4a (ng/mL) | 1.25 (0.85–1.85); 0.258 | 1.24 (0.80–1.95); 0.340 | C4a (ng/mL) | 1.04 (0.64–1.69); 0.870 | 1.34 (0.74–2.49); 0.330 |
| C5a (ng/mL) | 1.34 (0.89–2.06); 0.161 | 1.25 (0.77–2.09); 0.373 | C5a (ng/mL) | 1.12 (0.78–1.62); 0.522 | 0.92 (0.57–1.46); 0.719 |
| Factor H (μg/mL) | 0.96 (0.20–4.46); 0.954 | 0.53 (0.05–4.83); 0.574 | Factor H (μg/mL) | 0.43 (0.06–2.71); 0.371 | 0.34 (0.02–4.87); 0.427 |
| Factor I (ng/mL) | 0.86 (0.44–1.67); 0.666 | 0.83 (0.37–1.80); 0.634 | Factor I (ng/mL) | 1.00 (0.32–2.92); 0.996 | 1.03 (0.18–5.24); 0.974 |
| SC5b9 (ng/mL) | 1.05 (0.65–1.73); 0.845 | 0.95 (0.55–1.67); 0.858 | SC5b9 (ng/mL) | 1.13 (0.65–1.98); 0.667 | 1.11 (0.56–2.21); 0.763 |
| C1q (μg/mL) | 0.49 (0.08–3.04); 0.438 | 0.39 (0.05–3.12); 0.367 | C1q (μg/mL) | 0.47 (0.25–0.85); 0.015 | 0.41 (0.17–0.86); 0.025 |
| C3 (μg/mL) | 0.68 (0.40–1.15); 0.150 | 0.61 (0.33–1.09); 0.105 | C3 (μg/mL) | 0.38 (0.05–3.06); 0.356 | 0.32 (0.03–3.18); 0.318 |
| C4 (μg/mL) | 1.24 (0.92–1.71); 0.168 | 1.19 (0.83–1.73); 0.334 | C4 (μg/mL) | 0.82 (0.57–1.19); 0.276 | 0.82 (0.54–1.24); 0.328 |
| C5 (μg/mL) | 0.81 (0.22–3.03); 0.754 | 0.67 (0.15–2.84); 0.590 | C5 (μg/mL) | 0.78 (0.49–1.22); 0.267 | 0.71 (0.37–1.34); 0.289 |
| C3a/C3 ratio | 1.87 (1.17–3.26); 0.015 | 1.81 (1.09–3.40); 0.037 | C3a/C3 ratio | 1.14 (0.78–1.63); 0.490 | 1.12 (0.71–1.74); 0.626 |
| C4a/C4 ratio | 0.91 (0.64–1.26); 0.599 | 0.96 (0.62–1.45); 0.838 | C4a/C4 ratio | 1.09 (0.86–1.38); 0.442 | 1.15 (0.88–1.50); 0.286 |
| C5a/C5 ratio | 1.26 (0.89–1.82); 0.195 | 1.21 (0.80–1.88); 0.367 | C5a/C5 ratio | 1.14 (0.89–1.46); 0.295 | 1.04 (0.77–1.41); 0.788 |
OR represents multiplicative effects of doubling of each complement component. Multivariable analysis was adjusted by age, sex, and Qalb. Table 3 only shows the results for the complement components. Results for the remaining variables included in the multivariable analysis are shown in online eTables 11 and 12. OR = odds ratio.
In CSF, C1q levels were significantly higher in long-term nonprogressors than in progressors (p = 0.033; Figure 2B). A C1q value of 0.15 µg/mL was again the best cutoff to discriminate between long-term progressors and nonprogressors, with a sensitivity of 71% and specificity of 63% (Figure 2B). CSF levels of the remaining CCs were comparable between both groups of patients (eTable 10). Similar to the medium term, higher C1q levels were also associated with a lower risk of long-term disability progression both at univariable (OR 0.47; 95% CI 0.25–0.85; p = 0.015) and multivariable (OR 0.41; 95% CI 0.17–0.86; p = 0.025) levels (Table 3).
Serum and CSF Levels of CCs in Patients With Inflammatory and Noninflammatory PPMS
As shown in Table 1, 24 (35.0%) patients with PPMS were classified as inflammatory and 32 (47.0%) patients as noninflammatory according to radiologic disease activity at baseline and/or during follow-up. In serum, levels of the CCs Bb, C3a, C4a, C5a, SC5b9, and the C5a/C5 ratio were significantly higher in inflammatory patients vs noninflammatory, whereas levels of Factor H and C5 were reduced in noninflammatory patients (eFigure 1). Serum levels of the remaining CCs were similar between inflammatory and noninflammatory patients (eTable 13). In CSF, levels of Ba, C3a, and the 3 ratios were significantly elevated in patients with inflammatory PPMS, whereas levels of C1q, C4, and C5 were decreased in noninflammatory patients (eFigure 1). No significant differences were observed for the CSF levels of the remaining CCs between both groups of patients (eTable 13).
Correlations Between Serum and CSF Levels of CCs and IgM Index
As shown in Table 4, strong correlations were observed between the IgM index and levels of the CCs C3a, C5a, and the ratio C5a/C5 both in serum and CSF; Bb only in serum; and the C3a/C5 ratio only in CSF. Moderate correlations were observed for the CC Ba both in serum and CSF and for SC5b9 and the C3a/C3 ratio only in serum. A moderate and inverse correlation was also observed for CSF levels of CC C4. Finally, serum and CSF levels of C4a weakly correlated with the IgM index.
Table 4.
Correlations Between Serum and CSF Levels of Complement Components and the IgM Index
| Serum components | ρ (p value) | CSF components | ρ (p value) |
| Ba (ng/mL) | 0.331 (0.010) | Ba (ng/mL) | 0.348 (0.006) |
| Bb (μg/mL) | 0.404 (0.001) | Bb (μg/mL) | 0.233 (0.070) |
| C3a (ng/mL) | 0.406 (0.001) | C3a (ng/mL) | 0.570 (0.000002) |
| C4a (ng/mL) | 0.283 (0.028) | C4a (ng/mL) | 0.270 (0.035) |
| C5a (ng/mL) | 0.408 (0.001) | C5a (ng/mL) | 0.421 (0.001) |
| Factor H (μg/mL) | −0.064 (0.626) | Factor H (μg/mL) | −0.128 (0.326) |
| Factor I (ng/mL) | 0.001 (0.994) | Factor I (ng/mL) | −0.118 (0.363) |
| SC5b9 (ng/mL) | 0.330 (0.010) | SC5b9 (ng/mL) | −0.090 (0.492) |
| C1q (μg/mL) | −0.119 (0.366) | C1q (μg/mL) | −0.195 (0.133) |
| C3 (μg/mL) | 0.036 (0.787) | C3 (μg/mL) | −0.097 (0.455) |
| C4 (μg/mL) | 0.224 (0.085) | C4 (μg/mL) | −0.389 (0.002) |
| C5 (μg/mL) | −0.179 (0.171) | C5 (μg/mL) | −0.230 (0.074) |
| C3a/C3 ratio | 0.341 (0.008) | C3a/C3 ratio | 0.581 (0.000001) |
| C4a/C4 ratio | −0.087 (0.510) | C4a/C4 ratio | 0.353 (0.005) |
| C5a/C5 ratio | 0.428 (0.001) | C5a/C5 ratio | 0.414 (0.001) |
Data show Spearman correlation coefficients (p values).
Discussion
Studies evaluating the relationship between the complement system and long-term outcomes of patients with progressive MS are lacking. In this study, we aimed to explore the role of the complement system in disability progression in a cohort of patients with PPMS by measuring the levels of a panel of CCs (C3, C4, C5, C1q), complement activation products (C3a, C4a, C5a, Ba, Bb, SC5b9), and complement regulators (Factor H and Factor I) in paired serum and CSF samples and analyzing their potential to predict disability progression in the short term (2 years), medium term (6 years), and long term (9.6 years).
In our study, the serum or CSF levels of the native CCs C3, C4, and C5 did not seem to play an important role in disability progression at any of the time points. Only elevated serum C5 levels were associated with decreased risk of short-term disability progression in multivariable analysis. Previous cross-sectional studies have shown increased levels of the CCs C3 and C4 in plasma15 and increased levels of C3 in the CSF16,17 of patients with MS compared with healthy controls or patients with noninflammatory neurologic disorders. Whereas in one of these studies, plasma C3 and C4 levels did not correlate with EDSS,15 in another study, CSF C3 levels were particularly higher in the group of patients with PPMS and correlated with EDSS in the whole group of patients with MS.16 However, these studies were cross-sectional and did not evaluate follow-up disability progression outcomes.
Regarding the complement activation products, the serum C3a/C3 ratio, which is indicative for general activation of the complement system, played an important role as risk factor of disability progression in patients with PPMS. Although it played no role in the short term, an elevated ratio increased the risk of disability progression in the medium and long term in univariable and multivariable analyses. Of interest, serum levels of the C3a/C3 ratio could also be used in clinical practice to identify patients with PPMS at high risk of disability progression after 6 or more years of follow-up with good sensitivity and specificity. These results indicate that complement activation of early complement activation products is present in serum of patients with PPMS and is higher in patients with higher risk of disability progression in the medium and long term. The reasons why these findings are restricted to serum and are not observed in paired CSF samples from the same patients are unknown.
C4a, which is generated during activation of the classical or lectin pathways, and the C4a/C4 ratio were not associated with disability progression in patients with PPMS. Previous cross-sectional studies found increased plasma and CSF levels of C4a in patients with MS compared with controls, particularly in patients with relapsing-remitting MS (RRMS) at the time of the acute relapses.15,18 Patients with progressive forms of MS had comparable plasma levels of C4a with patients with RRMS and controls, and a weak correlation with EDSS was also observed in patients with MS that was lost after adjustment.18
Other complement activation products of the alternative complement pathway, such as Bb and Ba, as well as later CC indicative of general activation of the complement system such as C5a (and the C5a/C5 ratio) and SC5b9, played little or no role in disability progression. Only elevated serum levels of Bb were associated with an increased risk of disability progression in the medium term in univariable analysis, but significance was lost after adjustment. Although previous studies observed increased CSF levels of the terminal complement complex in patients with MS compared with controls19,20 and CSF levels were found to correlate with EDSS in patients with RRMS,21 in our study neither serum nor CSF levels of SC5b9 played a role in disability progression during follow-up of patients with PPMS. Altogether, the lack of strong association between activation of C5 and SC5b9 in serum or CSF and disability progression indicates that disease progression in MS does not seem dependent on the effector mechanisms related with the terminal pathway activation and membrane attack complex formation, but rather with the chemotactic properties and local and systemic inflammatory responses induced by early blood CCs such as C3a.22
In this context, B-cell maturation and presence of germinal center (GC)-like lymphoid follicular structures in the cerebral meninges are believed to contribute to disease progression and disability accumulation in MS.23-25 Complement has a long-standing association with GC formation and function. Mice with GC B cells lacking complement receptor 2 (CR2/CD21), which binds C3 cleavage products, show deficient GC responses, indicating that normal GC processes are fundamentally perturbed by B-cell intrinsic changes to complement signaling.26,27 More recently, it was demonstrated that C3 cleavage on GC B-cell surfaces and activated CR2-receptor signals are required for positive B-cell selection.28 Thus, C3 cleavage products have a role in regulating B-cell maturation and amplifying GC B-cell responses in the absence of terminal complement complex formation, and such functions might contribute to aberrant immune responses driving disease progression in MS.
Concerning the complement regulators Factors H and I, none of them were associated with disability progression at any of the study time points. Plasma, serum, and CSF levels of Factor H, a major regulator of the alternative pathway with the ability to bind C3b and regulate the formation of the C3 and C5 convertases, have been reported to be increased in patients with MS, particularly in those with progressive forms of the disease.15,29,30 Furthermore, serum levels of complement Factor H were found to correlate with EDSS, although only at the univariable level.29 These findings are not in line with our results, insomuch as serum or CSF levels of factor H did not seem to influence disability progression in patients with progressive MS.
In our study, findings in the CSF with the recognition component of the classical complement pathway C1q were interesting and at the same time surprising. We observed a clear protective role of elevated C1q levels in the CSF lowering the risk of disability progression in the medium and long term both in univariable analyses and after adjustment. Of note, the CSF C1q levels showed good potential to discriminate between progressors and nonprogressors and could be used in clinical practice to identify those patients with PPMS with lower risk of developing disability progression in the medium and long term. Studies in MS have shown that C1q staining is present in MS lesions,31 and CSF levels of C1q are found to be elevated in patients with MS and early phases of the disease compared with controls.17,32 However, C1q without the presence of C1r and C1s has neuroprotective and anti-inflammatory properties that foster resilience and resolution of injury.33,34 To name a few, C1q increased the uptake of apoptotic neurons by microglia, thus preventing the release of toxic intracellular contents and excitotoxic damage to neighboring neurons35; it downregulated the expression of proinflammatory cytokines by microglia in response to LPS;35 in vitro C1q induced the expression of genes important for neuronal survival and neurite outgrowth and neurotrophic factors.36,37 Furthermore, C1q was also found to inhibit NLRP3 inflammasome activation in human macrophages,38 a multiprotein complex proposed by our group as a therapeutic target in patients with PPMS.39
Stratification of patients with PPMS into inflammatory and noninflammatory according to radiologic activity overall revealed increased serum or CSF levels of complement activation products and decreased levels of intact CCs, the latter probably due to compound consumption associated with an inflammatory environment. Some of our findings are supported by previous studies showing correlations between CSF C3a levels and the total number of T2 lesions and the number of gadolinium-enhancing lesions32 or increased plasma C4a levels during acute relapses.18
Finally, because the IgM is the most efficient immunoglobulin isotype for complement activation, we also investigated the relationship between CCs and the IgM index. As expected, most of the complement activation products in serum or the CSF correlated more or less strongly with the IgM index, whereas levels of native CCs and complement regulators did not correlate, except for the CC of the classical pathway C4, which inversely correlated with the IgM index, most likely in the context of compound consumption associated with a higher IgM index–related complement activation.
In conclusion, early CCs such as the C3a/C3 ratio in serum and C1q in the CSF seem to play a role in disability progression of patients with PPMS in the medium and long term, and their levels may be used in clinical practice to identify patients with PPMS at higher (elevated serum C3a/C3 ratio) or lower (elevated CSF C1q) risk of disability progression during follow-up. The protective findings observed for C1q in the CSF are probably related to the neuroprotective and anti-inflammatory properties attributed to this component of the classical complement pathway. However, considering the size of our PPMS cohort, further studies are needed to confirm the role of the complement system in disability progression of patients with MS with this clinical form.
Acknowledgment
The authors particularly acknowledge patients and the Biobank HUB-ICO-IDIBELL (PT20/00171) integrated in the ISCIII Biobanks and Biomodels Platform and Xarxa Banc de Tumors de Catalunya (XBTC) for their collaboration. The authors also thank the Hospital Universitario Puerta de Hierro Majadahonda (HUPHM)/Instituto de Investigación Sanitaria Puerta de Hierro-Segovia de Arana (IDIPHISA) Biobank (Carlos III Health Institute Biobanks and Biomodels Platform) for the human specimens used in this study. Samples and data of patients included in this study from the Biobank HUB-ICO- IDIBELL and the HUPHM/IDIPHISA were processed following standard operating procedures with the appropriate approval of the Ethics and Scientific Committees.
Glossary
- CC
complement components
- EDSS
Expanded Disability Status Scale
- MS
multiple sclerosis
- PPMS
primary progressive MS
- ROC
receiver operating characteristic
- RRMS
relapsing-remitting MS
Appendix. Authors
| Name | Location | Contribution |
| Jan D. Lunemann, MD | Department of Neurology with Institute of Translational Neurology, University Hospital Münster, Germany | Major role in the acquisition of data; study concept or design; analysis or interpretation of data |
| Harald Hegen, MD, PhD | Department of Neurology, Medical University of Innsbruck, Austria | Major role in the acquisition of data; analysis or interpretation of data |
| Luisa María Villar, MD, PhD | Departments of Neurology and Immunology, Hospital Universitario Ramón y Cajal, Instituto Ramón y Cajal de Investigacion Sanitaria | Major role in the acquisition of data; analysis or interpretation of data |
| Konrad Rejdak, MD | Department of Neurology, Medical University of Lublin, Poland | Major role in the acquisition of data; analysis or interpretation of data |
| Augusto Sao-Aviles, PhD | Servei de Neurologia, Centre d'Esclerosi Múltiple de Catalunya (Cemcat), Institut de Recerca Vall d'Hebron (VHIR), Hospital Universitari Vall d'Hebron, Universitat Autònoma de Barcelona, Spain | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; analysis or interpretation of data |
| Pere Carbonell-Mirabent, PhD | Servei de Neurologia, Centre d'Esclerosi Múltiple de Catalunya (Cemcat), Institut de Recerca Vall d'Hebron (VHIR), Hospital Universitari Vall d'Hebron, Universitat Autònoma de Barcelona, Spain | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; analysis or interpretation of data |
| Jaume Sastre-Garriga, MD, PhD | Servei de Neurologia, Centre d'Esclerosi Múltiple de Catalunya (Cemcat), Institut de Recerca Vall d'Hebron (VHIR), Hospital Universitari Vall d'Hebron, Universitat Autònoma de Barcelona, Spain | Major role in the acquisition of data; analysis or interpretation of data |
| Neus Mongay-Ochoa, MD | Servei de Neurologia, Centre d'Esclerosi Múltiple de Catalunya (Cemcat), Institut de Recerca Vall d'Hebron (VHIR), Hospital Universitari Vall d'Hebron, Universitat Autònoma de Barcelona, Spain | Major role in the acquisition of data; analysis or interpretation of data |
| Klaus Berek, MD | Department of Neurology, Medical University of Innsbruck, Austria | Major role in the acquisition of data; analysis or interpretation of data |
| Sergio Martínez-Yélamos, MD, PhD | Department of Neurology, Bellvitge University Hospital, Barcelona, Spain | Major role in the acquisition of data; analysis or interpretation of data |
| Francisco Pérez-Miralles, MD, PhD | Neuroimmunology Unit, València University and Polytechnic Hospital La Fe, Spain | Major role in the acquisition of data; analysis or interpretation of data |
| Ahmed Abdelhak, MD | Department of Neurology, Ulm University, Germany; Division of Neuroinflammation and Glial Biology, Department of Neurology, University of California, San Francisco | Major role in the acquisition of data; analysis or interpretation of data |
| Franziska Bachhuber, MD | Department of Neurology, Ulm University, Germany | Major role in the acquisition of data; analysis or interpretation of data |
| Hayrettin Tumani, MD, PhD | Department of Neurology, Ulm University, Germany | Major role in the acquisition of data; analysis or interpretation of data |
| Jan N. Lycke, MD, PhD | Department of Clinical Neuroscience, Institute of Neuroscience and Physiology at Sahlgrenska Academy, University of Gothenburg, Sweden | Major role in the acquisition of data; analysis or interpretation of data |
| Igal Rosenstein, MD, PhD | Department of Clinical Neuroscience, Institute of Neuroscience and Physiology at Sahlgrenska Academy, University of Gothenburg, Sweden | Major role in the acquisition of data; analysis or interpretation of data |
| Roberto Alvarez-Lafuente, PhD | Environmental Factors in Degenerative Diseases Research Group, Hospital Clínico San Carlos, Instituto de Investigación Sanitaria del Hospital Clínico San Carlos (IdISSC), Madrid, Spain | Major role in the acquisition of data; analysis or interpretation of data |
| Tamara Castillo-Trivino, MD, PhD | Neurology Department, Hospital Universitario Donostia, San Sebastián; Center for Networked Biomedical Research on Neurodegenerative Diseases (CIBERNED) - ISCIII, Madrid, Spain | Major role in the acquisition of data; analysis or interpretation of data |
| David Otaegui, PhD | Center for Networked Biomedical Research on Neurodegenerative Diseases (CIBERNED) - ISCIII, Madrid; Multiple Sclerosis Unit. Biodonostia Health Research Institute, San Sebastián, Spain | Major role in the acquisition of data; analysis or interpretation of data |
| Sara Llufriu, MD, PhD | Center of Neuroimmunology, Service of Neurology, Hospital Clinic and Institut d'Investigacions Biomèdiques August Pi Sunyer (IDIBAPS), University of Barcelona, Spain | Major role in the acquisition of data; analysis or interpretation of data |
| Yolanda Blanco, MD, PhD | Center of Neuroimmunology, Service of Neurology, Hospital Clinic and Institut d'Investigacions Biomèdiques August Pi Sunyer (IDIBAPS), University of Barcelona, Spain | Major role in the acquisition of data; analysis or interpretation of data |
| Antonio J. Sánchez López, PhD | Neuroimmunology Unit; Biobank, Puerta de Hierro-Segovia de Arana Health Research Institute, Madrid, Spain | Major role in the acquisition of data; analysis or interpretation of data |
| Juan Antonio Garcia Merino, MD, PhD | Neuroimmunology Unit, Puerta de Hierro-Segovia de Arana Health Research Institute, Madrid, Spain | Major role in the acquisition of data; analysis or interpretation of data |
| Nicolas Fissolo, PhD | Center for Networked Biomedical Research on Neurodegenerative Diseases (CIBERNED) - ISCIII, Madrid; Servei de Neurologia, Centre d'Esclerosi Múltiple de Catalunya (Cemcat), Institut de Recerca Vall d'Hebron (VHIR), Hospital Universitari Vall d'Hebron, Universitat Autònoma de Barcelona, Spain | Major role in the acquisition of data; analysis or interpretation of data |
| Lucia Gutierrez, BSc | Servei de Neurologia, Centre d'Esclerosi Múltiple de Catalunya (Cemcat), Institut de Recerca Vall d'Hebron (VHIR), Hospital Universitari Vall d'Hebron. Universitat Autònoma de Barcelona, Spain | Major role in the acquisition of data; analysis or interpretation of data |
| Javier Villacieros-Álvarez, MD | Servei de Neurologia, Centre d'Esclerosi Múltiple de Catalunya (Cemcat), Institut de Recerca Vall d'Hebron (VHIR), Hospital Universitari Vall d'Hebron. Universitat Autònoma de Barcelona, Spain | Major role in the acquisition of data; analysis or interpretation of data |
| Enric Monreal, MD, PhD | Department of Neurology, Hospital Universitario Ramón y Cajal, REEM, IRYCIS, Universidad de Alcalá, Madrid, Spain | Major role in the acquisition of data; analysis or interpretation of data |
| Adrián Valls-Carbó, MD | Fundación INCE (Iniciativa para las Neurociencias), Madrid, Spain | Drafting/revision of the manuscript for content, including medical writing for content |
| Heinz Wiendl, MD | Department of Neurology with Institute of Translational Neurology, University Hospital Münster, Germany | Major role in the acquisition of data; analysis or interpretation of data |
| Xavier Montalban, MD, PhD | Servei de Neurologia, Centre d'Esclerosi Múltiple de Catalunya (Cemcat), Institut de Recerca Vall d'Hebron (VHIR), Hospital Universitari Vall d'Hebron, Universitat Autònoma de Barcelona; Center for Networked Biomedical Research on Neurodegenerative Diseases (CIBERNED) - ISCIII, Madrid, Spain | Major role in the acquisition of data; study concept or design; analysis or interpretation of data |
| Manuel Comabella, MD, PhD | Servei de Neurologia, Centre d'Esclerosi Múltiple de Catalunya (Cemcat), Institut de Recerca Vall d'Hebron (VHIR), Hospital Universitari Vall d'Hebron, Universitat Autònoma de Barcelona; Center for Networked Biomedical Research on Neurodegenerative Diseases (CIBERNED) - ISCIII, Madrid, Spain | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; study concept or design; analysis or interpretation of data |
Study Funding
The authors report no targeted funding.
Disclosure
The authors report no relevant disclosures. Go to Neurology.org/NN for full disclosures.
References
- 1.Weiner HL. A shift from adaptive to innate immunity: a potential mechanism of disease progression in multiple sclerosis. J Neurol. 2008;255(suppl 1):3-11. doi: 10.1007/s00415-008-1002-8 [DOI] [PubMed] [Google Scholar]
- 2.Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23:683-747. doi: 10.1146/annurev.immunol.23.021704.115707 [DOI] [PubMed] [Google Scholar]
- 3.Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344(14):1058-1066. doi: 10.1056/NEJM200104053441406 [DOI] [PubMed] [Google Scholar]
- 4.Walport MJ. Complement. Second of two parts. N Engl J Med. 2001;344(15):1140-1144. doi: 10.1056/NEJM200104123441506 [DOI] [PubMed] [Google Scholar]
- 5.Sarma JV, Ward PA. The complement system. Cell Tissue Res. 2011;343(1):227-235. doi: 10.1007/s00441-010-1034-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perlmutter DH, Colten HR. Molecular immunobiology of complement biosynthesis: a model of single-cell control of effector-inhibitor balance. Annu Rev Immunol. 1986;4:231-251. doi: 10.1146/annurev.iy.04.040186.001311 [DOI] [PubMed] [Google Scholar]
- 7.Veerhuis R, Nielsen HM, Tenner AJ. Complement in the brain. Mol Immunol. 2011;48(14):1592-1603. doi: 10.1016/j.molimm.2011.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stevens B, Allen NJ, Vazquez LE, et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131(6):1164-1178. doi: 10.1016/j.cell.2007.10.036 [DOI] [PubMed] [Google Scholar]
- 9.Watkins LM, Neal JW, Loveless S, et al. Complement is activated in progressive multiple sclerosis cortical grey matter lesions. J Neuroinflammation. 2016;13(1):161. doi: 10.1186/s12974-016-0611-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ingram G, Loveless S, Howell OW, et al. Complement activation in multiple sclerosis plaques: an immunohistochemical analysis. Acta Neuropathol Commun. 2014;2:53. doi: 10.1186/2051-5960-2-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saez-Calveras N, Brewster AL, Stuve O. The validity of animal models to explore the pathogenic role of the complement system in multiple sclerosis: a review. Front Mol Neurosci. 2022;15:1017484. doi: 10.3389/fnmol.2022.1017484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ingram G, Hakobyan S, Robertson NP, Morgan BP. Complement in multiple sclerosis: its role in disease and potential as a biomarker. Clin Exp Immunol. 2009;155(2):128-139. doi: 10.1111/j.1365-2249.2008.03830.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tatomir A, Talpos-Caia A, Anselmo F, et al. The complement system as a biomarker of disease activity and response to treatment in multiple sclerosis. Immunol Res. 2017;65(6):1103-1109. doi: 10.1007/s12026-017-8961-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saez-Calveras N, Stuve O. The role of the complement system in multiple sclerosis: a review. Front Immunol. 2022;13:970486. doi: 10.3389/fimmu.2022.970486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ingram G, Hakobyan S, Hirst CL, et al. Systemic complement profiling in multiple sclerosis as a biomarker of disease state. Mult Scler. 2012;18(10):1401-1411. doi: 10.1177/1352458512438238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aeinehband S, Lindblom RP, Al Nimer F, et al. Complement component C3 and butyrylcholinesterase activity are associated with neurodegeneration and clinical disability in multiple sclerosis. PLoS One. 2015;10(4):e0122048. doi: 10.1371/journal.pone.0122048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lindblom RP, Aeinehband S, Ström M, et al. Complement receptor 2 is increased in cerebrospinal fluid of multiple sclerosis patients and regulates C3 function. Clin Immunol. 2016;166-167:89-95. doi: 10.1016/j.clim.2016.04.003 [DOI] [PubMed] [Google Scholar]
- 18.Ingram G, Hakobyan S, Robertson NP, Morgan BP. Elevated plasma C4a levels in multiple sclerosis correlate with disease activity. J Neuroimmunol. 2010;223(1-2):124-127. doi: 10.1016/j.jneuroim.2010.03.014 [DOI] [PubMed] [Google Scholar]
- 19.Mollnes TE, Vandvik B, Lea T, Vartdal F. Intrathecal complement activation in neurological diseases evaluated by analysis of the terminal complement complex. J Neurol Sci. 1987;78(1):17-28. doi: 10.1016/0022-510x(87)90074-8 [DOI] [PubMed] [Google Scholar]
- 20.Sanders ME, Koski CL, Robbins D, Shin ML, Frank MM, Joiner KA. Activated terminal complement in cerebrospinal fluid in Guillain-Barré syndrome and multiple sclerosis. J Immunol. 1986;136(12):4456-4459. doi: 10.4049/jimmunol.136.12.4456 [DOI] [PubMed] [Google Scholar]
- 21.Sellebjerg F, Jaliashvili I, Christiansen M, Garred P. Intrathecal activation of the complement system and disability in multiple sclerosis. J Neurol Sci. 1998;157(2):168-174. doi: 10.1016/s0022-510x(98)00086-0 [DOI] [PubMed] [Google Scholar]
- 22.Schanzenbacher J, Köhl J, Karsten CM. Anaphylatoxins spark the flame in early autoimmunity. Front Immunol. 2022;13:958392. doi: 10.3389/fimmu.2022.958392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Magliozzi R, Howell O, Vora A, et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain. 2007;130(Pt 4):1089-1104. doi: 10.1093/brain/awm038 [DOI] [PubMed] [Google Scholar]
- 24.Magliozzi R, Howell OW, Reeves C, et al. A Gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann Neurol. 2010;68(4):477-493. doi: 10.1002/ana.22230 [DOI] [PubMed] [Google Scholar]
- 25.Howell OW, Reeves CA, Nicholas R, et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain. 2011;134(Pt 9):2755-2771. doi: 10.1093/brain/awr182 [DOI] [PubMed] [Google Scholar]
- 26.Ahearn JM, Fischer MB, Croix D, et al. Disruption of the Cr2 locus results in a reduction in B-1a cells and in an impaired B cell response to T-dependent antigen. Immunity. 1996;4(3):251-262. doi: 10.1016/s1074-7613(00)80433-1 [DOI] [PubMed] [Google Scholar]
- 27.Croix DA, Ahearn JM, Rosengard AM, et al. Antibody response to a T-dependent antigen requires B cell expression of complement receptors. J Exp Med. 1996;183(4):1857-1864. doi: 10.1084/jem.183.4.1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cumpelik A, Heja D, Hu Y, et al. Dynamic regulation of B cell complement signaling is integral to germinal center responses. Nat Immunol. 2021;22(6):757-768. doi: 10.1038/s41590-021-00926-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ingram G, Hakobyan S, Hirst CL, et al. Complement regulator factor H as a serum biomarker of multiple sclerosis disease state. Brain. 2010;133(Pt 6):1602-1611. doi: 10.1093/brain/awq085 [DOI] [PubMed] [Google Scholar]
- 30.Ingram G, Hakobyan S, Loveless S, Robertson N, Morgan BP. Complement regulator factor H in multiple sclerosis. J Cell Biochem. 2011;112(10):2653-2654. doi: 10.1002/jcb.23204 [DOI] [PubMed] [Google Scholar]
- 31.Breij EC, Brink BP, Veerhuis R, et al. Homogeneity of active demyelinating lesions in established multiple sclerosis. Ann Neurol. 2008;63(1):16-25. doi: 10.1002/ana.21311 [DOI] [PubMed] [Google Scholar]
- 32.Håkansson I, Ernerudh J, Vrethem M, Dahle C, Ekdahl KN. Complement activation in cerebrospinal fluid in clinically isolated syndrome and early stages of relapsing remitting multiple sclerosis. J Neuroimmunol. 2020;340:577147. doi: 10.1016/j.jneuroim.2020.577147 [DOI] [PubMed] [Google Scholar]
- 33.Thielens NM, Tedesco F, Bohlson SS, Gaboriaud C, Tenner AJ. C1q: a fresh look upon an old molecule. Mol Immunol. 2017;89:73-83. doi: 10.1016/j.molimm.2017.05.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bohlson SS, Tenner AJ. Complement in the brain: contributions to neuroprotection, neuronal plasticity, and neuroinflammation. Annu Rev Immunol. 2023;41:431-452. doi: 10.1146/annurev-immunol-101921-035639 [DOI] [PubMed] [Google Scholar]
- 35.Fraser DA, Pisalyaput K, Tenner AJ. C1q enhances microglial clearance of apoptotic neurons and neuronal blebs, and modulates subsequent inflammatory cytokine production. J Neurochem. 2010;112(3):733-743. doi: 10.1111/j.1471-4159.2009.06494.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Benoit ME, Tenner AJ. Complement protein C1q–mediated neuroprotection is correlated with regulation of neuronal gene and microRNA expression. J Neurosci. 2011;31(9):3459-3469. doi: 10.1523/JNEUROSCI.3932-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pisalyaput K, Tenner AJ. Complement component C1q inhibits beta-amyloid- and serum amyloid P-induced neurotoxicity via caspase- and calpain-independent mechanisms. J Neurochem. 2008;104(3):696-707. doi: 10.1111/j.1471-4159.2007.05012.x [DOI] [PubMed] [Google Scholar]
- 38.Benoit ME, Clarke EV, Morgado P, Fraser DA, Tenner AJ. Complement protein C1q directs macrophage polarization and limits inflammasome activity during the uptake of apoptotic cells. J Immunol. 2012;188(11):5682-5693. doi: 10.4049/jimmunol.1103760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Malhotra S, Costa C, Eixarch H, et al. NLRP3 inflammasome as prognostic factor and therapeutic target in primary progressive multiple sclerosis patients. Brain. 2020;143(5):1414-1430. doi: 10.1093/brain/awaa084 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data will be shared upon reasonable request.