

Abstract
Introduction –
Arrhythmias are disturbances in the normal rhythm of the heart and account for significant cardiovascular morbidity and mortality worldwide. Historically, preclinical research has been anchored in animal models, though physiological differences between these models and humans have limited their clinical translation. The discovery of human induced pluripotent stem cells (iPSC) and subsequent differentiation into cardiomyocyte has led to the development of new in vitro models of arrhythmias with the hope of a new pathway for both exploration of pathogenic variants and novel therapeutic discovery.
Areas covered –
The authors describe the latest two-dimensional in vitro models of arrhythmias, several examples of the use of these models in drug development, and the role of gene editing when modeling diseases. They conclude by discussing the use of three-dimensional models in the study of arrythmias and the integration of computational technologies and machine learning with experimental technologies.
Expert opinion –
Human iPSC-derived cardiomyocytes models have significant potential to augment disease modeling, drug discovery, and toxicity studies in preclinical development. While there is initial success with modeling arrhythmias, the field is still in its nascency and requires advances in maturation, cellular diversity, and readouts to emulate arrhythmias more accurately.
Keywords: Arrythmia, Cardiomyocytes, Disease modeling, Drug development, Human induced pluripotent stem cells
1. Introduction
Proper mechanical function of the heart is dependent on the organized and coordinated electrical depolarization of atrial and ventricular muscle to move blood directionally forward into the pulmonary and systemic beds. Arrythmias are abnormal electrical depolarizations that can originate from any structure within the heart and lead to a loss of organized chamber depolarization such that mechanical function of the heart is impacted. These abnormalities can be triggered by a vast array of factors including fibrosis, ischemia, structural disease, and inherited mutations in ion channels. Arrythmias are a large clinical burden and can lead to sudden cardiac death, thrombosis, and heart failure. These disorders account for significant morbidity and mortality and carry a large financial burden to healthcare systems.
Model systems are key tools in cardiovascular research for investigating mechanisms of disease, genotype-phenotype relationships, and therapeutic development. Because of a historical lack of true cardiomyocyte culture systems, heterologous expression systems in cell lines like HEK293 cells have been successful not only in elucidating ion channel physiology but also drug mechanisms of action [1–3]. Genetically modified animal models, particularly the mouse, have been primarily utilized as a surrogate model system for both investigating disease pathogenesis and exploring new therapeutic agents. However, there are important and clinically relevant phenotypic differences between hearts from small animals and those from humans [4]. Ion channel currents, arrhythmia mechanisms, and responses to cardioactive drugs are species dependent, and extrapolating responses in one animal to a different species can prove to be an inaccurate assumption [5–9]. These differences are extremely relevant as we study mechanisms of disease pathophysiology and test the efficacy and safety of new therapeutic agents. There is a need for a physiologically relevant human model which accurately mimics in vitro the complexity of human pathophysiology and recapitulates the effects of known therapeutic agents to aid in the discovery of new cardioactive drugs.
The teams led by Shinya Yamanaka and James A. Thomson revolutionized the field of stem cell technology in 2007 with the generation of human induced pluripotent stem cells (iPSC) [10,11]. The ability to reprogram mature cells collected from peripheral blood or a skin biopsy into iPSC with the transduction of several transcription factors provided the field of cardiovascular research with a renewable resource for the in vitro study of human diseases. These cells have similar characteristics to embryonic stem cells (ESC), especially their potential to be differentiated into the three primary germ layers and respective derivative cells.
With exposure to the correct stimuli, these cells demonstrated the ability to be differentiated into functional cardiomyocytes [12,13]. Some of this work initially had been pursued with embryonic stem cells, which displayed the ability to form cardiomyocytes spontaneously in embryoid body aggregates stimulated with FBS [14]. Methods have expanded significantly in terms of complexity by incorporating a variety of mesodermal morphogens to direct differentiation in both two and three dimensions. Stimulation of specific pathways such as MAPK and WNT led eventually led to chemically defined media in which iPSC differentiation could predictably proceed through mesodermal development to generate cardiac progenitors and then cardiomyocytes [15–17]. For a overview of this evolution and all current methods for cardiac differentiation, we guide readers to this review [18]. Cardiomyocytes differentiated from iPSC resembled in vivo cardiomyocytes in terms of morphology, gene expression, and contraction behavior. However, there are significant differences between in vitro and in vivo cardiomyocytes, including mature gene expression, sarcomeric organization, and automaticity. One of the immediate advantages of iPSC-derived cardiomyocytes is their genetic recapitulation of the patient from whom they were derived, and thus their potential to model inherited disease states. With the simultaneous improvements in gene editing, specific genetic variations could also be introduced in apparently healthy wild-type cell lines or repaired in cells obtained from donors with genetic variants to create isogenic matched pairs (Figure 1) [19–21].
Figure 1. Study of arrhythmias using human iPSC-derived cardiomyocytes.
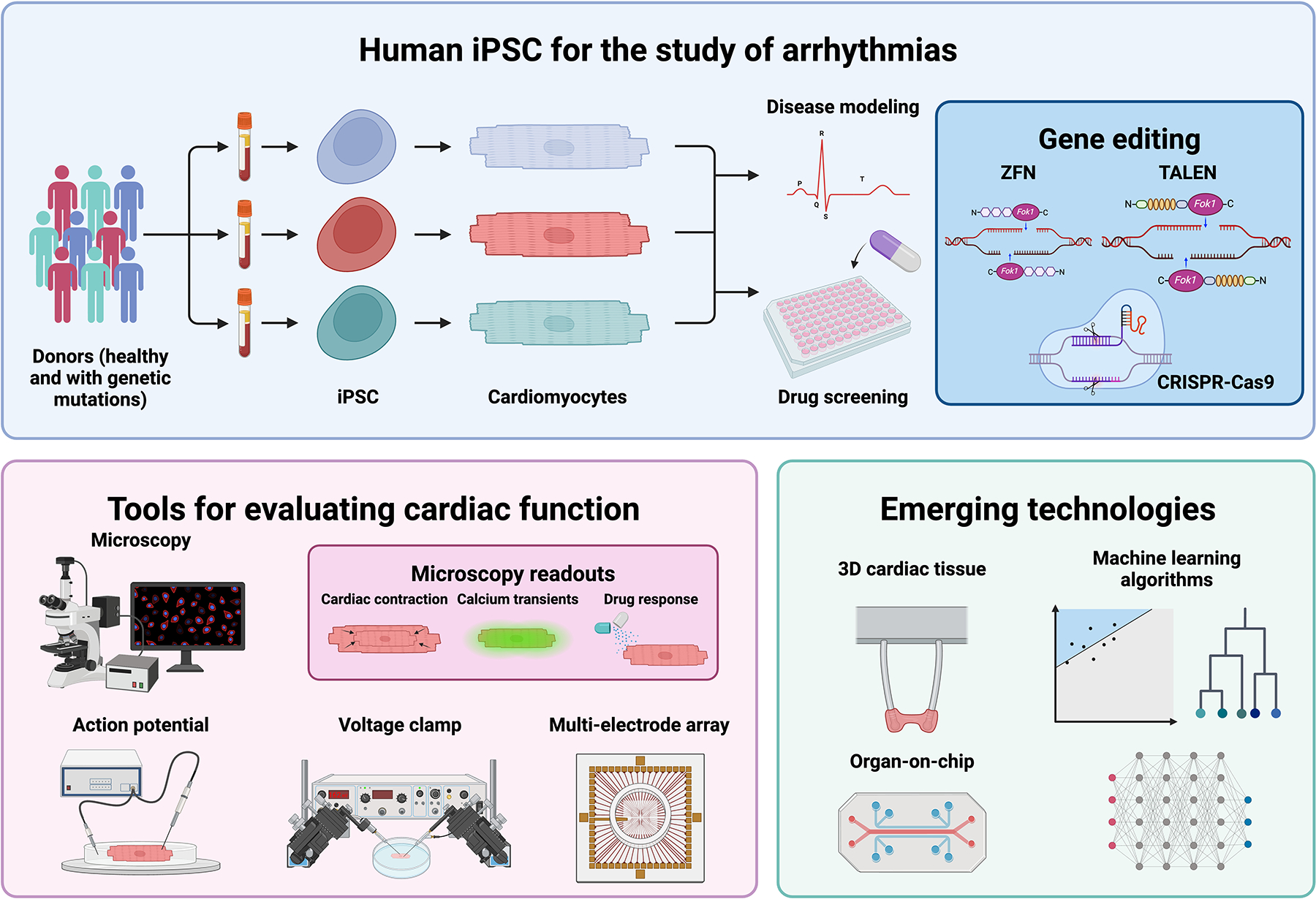
iPSCs from healthy donors and patients with genetic mutations can be differentiated into cardiomyocytes, which can then be studied for relevant in vitro phenotypes. These cells can be edited to correct inherited genetic mutations, or a specific mutation can be introduced to create isogenic pairs. There are numerous tools to evaluate the phenotype of iPSC-derived cardiomyocytes, from brightfield videos to assess cardiac contractility, calcium transients using dyes or fluorescent proteins, and several electrophysiological assays. Organ-on-chip, three-dimensional cardiac tissues, and machine learning algorithms are emerging technologies which are being incorporated with iPSC to improve the physiological complexity of these models and high-dimensional data analysis. Created with BioRender.com.
Two-dimensional (2D) culture is straightforward for investigating molecular mechanisms, signal transduction pathways, and high-throughput drug screening. However, 2D cardiomyocyte systems alone lack the maturity and physiological complexity of the heart as an electrical-mechanical organ [22]. Tissue engineering has made considerable advances in three-dimensional (3D) systems through the inclusion of scaffold biomaterials, integration of different stromal populations, and application of external stimuli (electrical, mechanical, or biochemical) [23]. These developments have led to the generation of more nuanced and physiologically relevant models to study tissue function and mimic disease [24–27]. IPSC technology combined with tissue engineering now allows for establishing multiple isogenic tissue types from the same cell line which can be connected to emulate organ systems. This has proved particularly advantageous for studying drug metabolism and potential off-target toxicities [28–31].
In this review, we describe the use of iPSC-derived cardiomyocytes in the study of human arrhythmias and their application in drug development. We discuss several in vitro arrhythmia disease models, highlight examples of drug discovery and gene editing when modeling diseases, and conclude with our opinion on their current role and future applications.
2. In vitro disease modeling of arrhythmias
Shortly after the establishment of protocols for the differentiation of human cardiomyocytes from iPSC, somatic cells donated from patients with arrhythmias were reprogramed into iPSC and differentiated into cardiomyocytes to study if these cells still exhibited measurable cellular traits that mirrored electrophysiologic properties of clinical arrhythmias. These early studies of human arrhythmias in vitro using patient iPSC-derived cardiomyocytes confirmed the utility of this model system in studying a cardiovascular disease phenotype in vitro. Inherited arrhythmias are rare diseases, and the majority of disease-causing variants are near unique in terms of their individual prevalence. Thus, a critical question asked by both iPSC-based models and in vivo models more broadly, is whether using a patient-derived variant with inherited arrhythmia can generate physiologically relevant phenotype in vitro that more broadly represents the disease in question. This is a critical scientific assumption that is made frequently in animal models of disease and is a critical benchmark for iPS models broadly in order to demonstrate their utility for clinically relevant therapeutic discovery. Below, we summarize several in vitro models of arrhythmias (important details can be found in Table 1), discuss their applications, and examine how they have been used successfully to further expand our knowledge of these disorders by demonstrating key electrophysiological qualities linked to disease pathogenesis.
Table 1. In vitro models of arrhythmia using patient-derived cardiomyocytes.
Overview of several iPSC-based models of arrhythmias with a special focus on their phenotypes and drug response.
| Disease | Gene | Mutation | Cardiomyocytes phenotype | Drugs response | Ref. |
|---|---|---|---|---|---|
| LQT1 | KCNQ1 | c.569G>A | Prolonged APD. Reduction in IKs. Altered channel properties. | Higher susceptibility to catecholamine-induced tachyarrhythmia, which could be attenuated with β-blockers. | [34] |
| c.1893del | Increased FPD. Decreased IKs and tail current densities. | E-4031: prolonged FPD and induced arrhythmic events. Chromanol 293B: no response. Isoproterenol: increased the beating rate and induced EAD. Propranolol: decreased the number of arrhythmogenic events. Barium: prolonged the FPD. | [35] | ||
| c.922_1032del | Prolonged APD. Reduced IKs. | ML277: reversed the phenotype. | [36] | ||
| LQT2 | KCNH2 | c.1681G>A | Prolonged APD and FPD. | E-4031: prolonged APD and EAD. Isoprenaline: EAD. Propranolol: reverse isoprenaline-EAD. Nicorandil and PD118057: caused shortening of APD and abolished EAD. siRNA: normalized APD and potassium current and prevented isoprenaline or E-4031-induced arrhythmias. | [38,41] |
| c.821C>T | Reduction in IKr. Prolonged APD and FPD. Development of EAD. | E-4031: prolonged APD and FPD and increased arrhythmogenesis. Cisapride: prolonged FPD and arrhythmias. Nifedipine and pinacidil: shortening of APD and FPD and eliminated EAD. Ranolazine: arrhythmias with no effect on APD or FPD. | [39] | ||
| c.526C>T | Reduction in IKr. Prolonged APD. Shorter FPD. Rare EAD. | Isoprenaline: increased chronotropy. E4031 or sotalol: arryhtmias. Erythromycin or cisapride: no effect on arrythmias. | [40] | ||
| LQT3 | SCN5A | c.718G>A c.1604G>A |
Prolonged APD. Longer time to peak and to inactivation of Na+ channel. | –––– | [42] |
| c.5287G>A | Prolonged APD. | Increased tetrodotoxin-sensitive late or persistent Na+ current. Mexiletine: reduced APD and the late Na+ current. | [43] | ||
| LQT8 | CACNA1C | c.1216G>A | Slower and irregular contractions. Increased APD and number of DAD. Prolonged Ca2+ elevations. Reduction in IKr. | Several CDK5 inhibitors: reduced APD and rescued calcium mishandling. | [45–48] |
| Disease | Gene | Mutation | Cardiomyocytes phenotype | Drugs response | Ref. |
| CPVT | RYR2 | c.7447T>A | Arrythmias and DAD. Higher amplitudes and longer durations of spontaneous local calcium release events. | Isoproterenol: arrhythmias, DAD, and negative chronotropy. Caffeine: Ca2+ release. Foskolin and 8-Br-cAMP: steep rise in cytosolic Ca2+ and abolished Ca2+-induced Ca2+-release events after repolarization. | [50] |
| c.12056T>G | DAD. Abnormal calcium transients. | Isoproterenol or forskolin: increased DAD and induced calcium mishandling. Propranolol: rescued isoproterenol effect. Flecainide: eliminated or decreased afterdepolarizations. Thapsigargin: depressed postpacing afterdepolarizations. | [52] | ||
| c.6982C>T | Calcium mishandling. DAD and EAD. | Adrenaline: prolonged APD, calcium abnormalities, elevated diastolic Ca2+ levels, and decreased APD. | [53] | ||
| c.1217C>T | –––– | Isoproterenol: abnormal calcium handling. Dantrolene: restored calcium handling and rescued phenotype. | [54] | ||
| c.6933G>C | DAD. Calcium mishandling. Higher mean amplitude of AP. | Isoproterenol: increased frequency of DAD and exacerbated calcium mishandling. KN-93: stopped isoproterenol-induced DAD and stabilized calcium activation. | [55] | ||
| CASQ2 | c.919G>C | Prolonged APD. Pacing had a stabilizing effect. | Isoproterenol: increased Ca2+ abnormalities. DAD. |
[51] | |
| ARVC | PKP2 | c.1841T>C | –––– | Nifedipine: reduction of contraction amplitude. Isoproterenol: increased beat frequency. Caffeine: rapid and large release of Ca2+. | [58] |
| c.972insT/N | Prolonged FPD. | Isoproterenol or carbamylcholine: chronotropic response. | [59] | ||
| DSG2 | c.1912G>A | Reduced action potential amplitude and maximal depolarization velocity. EAD and DAD. Peak INa, INCX, Ito, ISK, and IKATP were reduced, while IKr was enhanced. Higher pacemaker activity. | Isoproterenol: shortened APD. Epinephrine: arrhythmias. PHP-1: reduced cell automaticity and arrhythmic events. | [60,62] | |
| DSP | c.1386del | Report of the generation of the iPSC line. | –––– | [61] |
AP, action potential. APD, action potential duration. ARVC, arrhythmogenic right ventricular cardiomyopathy. CPVT, catecholaminergic polymorphic ventricular tachycardia. DAD, delayed after depolarizations. EAD, early after depolarizations. FDP, field potential duration. LQT1, long QT syndrome 1. LQT2, long QT syndrome 2. LQT3, long QT syndrome 3. LQT8, long QT syndrome 8.
2.1. Long-QT syndrome 1
There are several subtypes of long QT (LQT) syndrome, each associated with variants in specific genes encoding ion channels or channel-associated proteins. These hereditary channelopathies are characterized by repolarization abnormalities, prolongation of the QT interval on the electrocardiogram, and the propensity to develop life-threatening cardiac arrhythmias and sudden cardiac death [32]. LQT syndrome 1 (LQT1) is a disorder of cardiac repolarization due to an abnormal potassium current. It is the most common LQT syndrome and is caused by mutations in KCNQ1, a gene that encodes a voltage-gated potassium channel [32,33]. In a seminal paper, somatic cells from a family affected by LQT1 were obtained, reprogrammed into iPSC, and differentiated into cardiomyocytes, which successfully demonstrated several canonical features of prolonged repolarization [34].
Several groups have generated patient-specific iPSC from members of families affected by LQT1 and recapitulated in vitro cardiac arrhythmias and drug responses as observed in patients. Cardiomyocytes with these mutations displayed prolonged action potential duration (APD) and corrected field potential duration (FPD), an analog to the QT interval on an electrocardiogram [34,35]. The slow delayed rectifier potassium current (IKs) was reduced with decreased peak and tail current densities as well as altered channel activation and deactivation properties [34,35]. Patients with LQT1 have an increased susceptibility to catecholamine-induced tachyarrhythmia, which was emulated in vitro when cardiomyocytes were exposed to the β1-adrenergic agonist isoproterenol. Conversely, this effect was successfully attenuated with β-adrenergic blockade [34,35]. Compounds with known ion channel activity demonstrated the utility of human iPSC-derived cardiomyocytes to study their impact on arrhythmias. ML277, an activator of KCNQ1, rescued the disease phenotype by increasing the amplitude and enhancing the activation of IKs [36]. Nifedipine, a calcium channel blocker, and pinacidil, an activator of ATP-sensitive potassium channels, also improved the phenotype of these cells through shortening the APD and calcium transient duration.
The use of iPSC-derived cardiomyocytes also allows for mechanistic studies of how mutations lead to disease beyond mere loss of function. For example, KCNQ1, which is normally found at the cell membrane as an ion channel, was instead observed in the cytoplasm and perinuclear areas of cells derived from several donors with several in-frame and missense heterozygous mutations [35,36]. Similar to defects observed in cystic fibrosis for chloride channels, these observations suggest a potassium channel trafficking abnormality plays a role in the pathogenesis of LQT1 due to this mutation and have opened the door to potential therapies that correct ion channel trafficking for patients with these mutations.
2.2. Long-QT Syndrome 2
LQT syndrome 2 (LQT2) involves mutations in KCNH2, which encodes a pore-forming subunit of the voltage-gated inward rectifying potassium channel. This subunit mediates the rapidly activating component of the delayed rectifying potassium current (IKr) [37]. Several iPSC lines have been generated from patients and the resulting differentiated cardiomyocytes emulated several arrhythmogenic features in vitro [38–41]. These cardiomyocytes displayed reduced IKr, prolonged APD and FPD, early after depolarizations (EAD or abnormal depolarization during phase 2 or 3 repolarization) and delayed after depolarizations (DAD or abnormal depolarizations during the phase 4 repolarization).
These electrical perturbations were then used to explore drug effects in vitro. EADs were exacerbated by β-adrenergic stimulation with isoproterenol, which could be reversed by β-blockers. The selective hERG blocker E-4031, as well as cisapride, prolonged APD and FDP and increasing arrhythmogenesis, while nifedipine and pinacidil led to significant APD and FPD attenutation and eliminated all arrhythmogenic events [38,39,41]. Nicorandil and PD-118057, experimental potassium channel enhancers, caused shortening of APD and reduced EAD, though this could be reversed by isoproterenol [38,41]. These findings demonstrated the power of human iPSC-derived cardiomyocytes to reconstitute multiple different ion channel and signaling pathways that impact both therapeutic utility and toxicity.
2.3. Long-QT Syndrome 3
LQT syndrome 3 (LQT3) is associated with gain of function mutations in the voltage gated sodium channel SCN5A [32]. These mutations lead to delayed sodium channel inactivation and consequently delayed repolarization of cardiomyocytes. The phenotype of several mutations observed in patients with LQT3 was successfully modeled in vitro with human iPSC-derived cardiomyocytes [42,43]. These cells exhibited prolonged APD, both when spontaneously beating or electrically paced, and a higher time to peak of sodium current. Exposing these cells to blockers of the sodium channels led to the reduction of APD and late sodium current.
2.4. Long-QT Syndrome 8
LQT syndrome 8 (LQT8), otherwise known as Timothy Syndrome, is a disease caused by mutations in the CACNA1C gene, which encodes a subunit of the voltage-gated calcium channel [44]. Cardiomyocytes from a patient-derived iPSC line demonstrated lower beat frequency, irregular contractions, increased APD, and slow and irregular calcium transients [45]. Roscovitine, a cyclin-dependent kinases (CDK) inhibitor, successfully rescued the calcium mishandling observed in the cells derived from patients [45,46]. To study the molecular mechanism underlying the positive effects of CDK inhibition in LQT8, iPSC-derived cardiomyocytes were used to test several roscovitine analogues and other CDK inhibitors [47]. Four compounds demonstrated beneficial effects increasing the spontaneous beating rate of these cardiomyocytes and decreasing the irregularity of their contractility. Three of these compounds were CDK5 inhibitors, supporting the hypothesis that inhibition of CDK5 had a positive effect on the phenotype of LQT8 and may be a potential therapeutic avenue in patients with this genetic mutation. In further work, the non-opioid intracellular receptor SIGMAR1 was shown to inhibit CDK5 activity. FDA-approved SIGMAR1 agonists fluvoxamine and dextromethorphan rescued the diseased phenotype, reverting the electrophysiological changes observed in these cardiomyocytes [48]. The effective dosages were shown to be safe in isogenic controls, and both in vitro efficacy and safety of dextromethorphan were confirmed in an animal model of LQT8.
2.5. Catecholaminergic polymorphic ventricular tachycardia
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is an arrhythmic disorder that leads to syncope or sudden cardiac death in states of high catecholamine surges such as physical activity or emotional stress [49]. This disease arises from abnormal calcium homeostasis caused by either mostly missense mutations in the ryanodine receptor (RYR2) or recessive mutations in calsequestrin (CASQ). In these situations, when the levels of catecholamines increase in the body, they can trigger abnormal calcium release from intracellular stores leading to the development of ventricular tachycardia.
Several iPSC lines have been derived from patients with CPVT and cardiomyocytes differentiated therefrom displayed several relevant molecular phenotypes: high arrhythmic activity, higher calcium amplitude, and longer durations of spontaneous calcium release events [50,51]. As expected, stimulation with either isoproterenol or adrenaline induced abnormal calcium handling, increased EAD’s and increased DAD’s [52,53]. Studies with different compounds were tested for managing arrhythmic activity in iPSC models of CPVT. KN-93 a Ca2+/calmodulin-dependent protein kinase II (CaMK-II) inhibitor with promising results in animal models, suppressed isoproterenol-induced DAD’s and stabilized calcium activation, while dantrolene was shown to improve RYR2 channel hyperactivity and reduce DAD’s [54,55].
2.6. Arrhythmogenic right ventricular cardiomyopathy
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is characterized by the replacement of normal heart muscle tissue with fatty or fibrous tissues, primarily in the right ventricle [56]. Mutations in desmosomal proteins such as plakophilin-2 (PKP2), desmoglein-2 (DSG2) and desmoplakin (DSP) account for most cases that lead to ARVC. As desmosomes are integral to the structural integrity of the myocardium, these mutations lead to disruption of cell-cell interactions followed by cell death and fibrofatty replacement. Cardiomyocytes differentiated from patient-specific iPSC lines with mutations in PKP2 display several features of ARVC and, interestingly, implicate abnormal metabolism as a contributing mechanism to the pathophysiology of this disease [57]. Several known compounds were applied to these models in order to validate iPSC cardiomyocyte physiological response[58,59]. For example, contraction amplitude was reduced after treatment with the calcium channel blocker nifedipine, while treatment with isoproterenol increased beat frequency. Caffeine, a potentiator of cAMP-mediated signal transduction pathways, evoked an instantaneous, rapid, and large release of calcium indicating that PKA modulation of calcium flux was intact in these cells.
iPSC obtained from donors with pathogenic variants in other genes associated with ARVC (DSP and DSG2) have demonstrated arrhythmogenicity and are potential models to identify novel therapeutics for treating arrhythmias in patients with these mutations [60,61]. As an example, cardiomyocytes derived from a patient with ARVC secondary to a mutation in the DSG2 gene displayed higher pacemaker activity, more arrhythmic events, and highly expressed NDPK-B and SK4 channels [62]. Treatment with PHP-1, an NDPK-B antagonist, suppressed cell beating, reduced SK4 channel activity, and diminished arrhythmic events, indicating a mechanistic role of NDPK-B and SK4 channels in the pathogenesis of ARVC. Future studies with other genotypes have similar potential to promote this precision medicine approach for mechanistic studies followed by therapeutic development tailored to specific genotypes.
3. Gene editing and the progression into precision medicine
The discovery of precise and facile gene editing techniques as zinc finger nuclease (ZFN) [63,64], transcription activator-like effector nuclease (TALEN) [65–68], and clustered regularly interspaced short palindromic repeats (CRISPR) [69–73] has accelerated iPSC disease modeling through both the introduction of variants in wild-type cell lines, as well as the correction of a mutation in a patient-derived cell line. These isogenic pairs are critical for studying genotype-phenotype relationships and making comparisons in readouts that can be potentially impacted by other genomic variations between cell lines. Isogenic controls for several diseases like LQT1 [74], LQT2 [75,74], LQT8 [47,48], and ARVC [57] have been generated using gene editing. By using these cells as controls for functional, electrophysiological, and calcium handling studies, the influence of the mutation in their phenotype can be investigated, confirming their validity for mechanistic and drug testing studies in the future.
Despite the in vitro nature of iPSC models, there have been reports that the degree of phenotypes observed in patient-derived cell lines can correlate to the severity of the disease of that patient. This has potential utility in classifying pathogenicity of variants and personalized drug response as an overall effort in precision medicine approaches. As an example, cells derived from LQT2 patients with milder phenotypes presented less frequent EAD and lower susceptibility to drugs known to increase arrhythmogenicity [40]. In another example, iPSC-derived cardiomyocytes were able to disentangle multiple mutations in a single patient with variants in both KCNH2 and SCN5A, genes associated with LQT2 and 3, respectively [76]. In vitro studies demonstrated arrhythmias due to aberrant sodium flux, but not influenced by the potassium flux from the polymorphism observed in the KCNH2 gene. These cardiomyocytes displayed an increased inhibition of their late sodium current when treated with mexiletine, a sodium channel blocker indicating potential drug response in that patient. These types of studies demonstrate promise for iPSC in both annotating genomic variants as well as in predicting possible therapeutic/toxicity interactions with specific variants.
Other groups have used gene editing techniques with iPSC to evaluate variant pathogenicity by interrogating serial mutations in a wild-type line [77,78]. Several mutations in KCNH2 were introduced into a single wild-type iPSC line, and mechanistic studies revealed that mutation proximity to the pore-forming domains was directly related to higher susceptibility to drug-induced arrhythmias [77]. These types of experiments demonstrate how this approach can reveal mechanistic insight into genotype-phenotype relationships, leading to enhanced risk stratification for patients.
These technologies also offer the potential to explore gene therapy modalities that can either suppress, correct, or replace mutant alleles with the ultimate goal of curative therapy. Animal alleles contain polymorphisms and critical sequence differences that can interfere with gene targeting beyond proof of principle studies. Approaches that specifically target human genes can ultimately require genomic humanization in animals to demonstrate safety and efficacy in preclinical studies. Human iPSC-derived cardiomyocytes, however, provide a model system in which the human gene therapy approaches can be safely optimized and validated in vitro. Several groups have tested this approach by delivering suppression and replacement gene therapy to rescue cells derived from donors with LQT syndromes [79,80]. For example, a short hairpin RNA (shRNA) (KCNQ1 for LQT1 or KCNH2 for LQT2) was used to suppress both alleles of the gene of interest, followed by gene replacement expression of an shRNA-resistant wild-type allele. Human iPSC-derived cardiomyocytes from several donor cell lines with genetic mutations were successfully rescued with the shortening of their APD. This approach is mutation agnostic and can be implemented in a range of pathogenic variants of different genetic diseases. Previous approval of similar genetic replacement therapies by the US Food and Drug Administration (FDA) in cases of retinal dystrophy and spinal muscular atrophy support this promising strategy to potentially cure genetic arrhythmogenic diseases by acting directly on the genetic level [81,82].
4. Screening of arrhythmic drugs and discovery of new antiarrhythmic agents
Drug toxicity is one of the most common reasons for drug development failure. Prolongation of the QT interval and inhibition of the hERG potassium channel are two major safety concerns as these can lead to a fatal ventricular tachycardia called Torsades de Pointes in humans. However, predicting toxicity in humans using in vitro models has been very difficult. The Comprehensive in vitro Proarrhythmia Assay (CiPA) collaboration, an international team comprised of regulatory officials, non-profits, academics, and drug developers, was assembled to evaluate the proarrhythmic risk of novel therapeutic agents [83]. This collaboration is focused on improving the prediction by integrating ion current studies, EKG analysis, in silico predictive modeling and, importantly, studies using iPSC-derived cardiomyocyte models. This was predicated on several published studies demonstrating iPSC cardiomyocytes display APD and FPD prolongation as well as changes in ion channel currents in response to known QT-prolonging drugs [84–86]. The goal of this initiative is to identify the risk of malignant arrhythmia prior to human testing in order to avoid costly late-stage drug failure [87], and the incorporation of iPSC-derived cardiomyocytes provides a scalable tool to quickly screen for new potential toxicities early in hit-to-lead campaigns (Figure 2).
Figure 2. Applications of iPSC-derived cardiomyocytes in drug discovery.

iPSC cardiomyocytes can be applied in three major scenarios to help improve the development of new therapeutic compounds or regimens. Healthy cardiomyocytes can be used to test arrhythmia potentials of new drug to identify potential limiting toxicities early in preclinical development (Top Panel). By using diseased cells, either from patients or genetically edited, new compounds can be screened and tested for efficacy. Promising compounds that revert diseased phenotypes can then move forward to further preclinical studies before an investigational new drug (IND) application (Middle Panel). Patients with genetic arrythmias can donated cells for the generation of patient-specific cardiomyocytes. These cells can be used for in vitro screening of new therapeutic regimens and identify specific therapies tailored to that patient (Bottom Panel). Created with BioRender.com.
Beyond assessing for potential arrhythmia toxicity, iPSC-based assays can help identify therapeutic potential in compounds using in vitro readouts that have some physiological relevance to the studied disease state. For example, iPSC-derived cardiomyocytes derived from a patient with CPVT were treated with several drugs in clinically relevant concentrations, and these findings were compared with the same drugs’ response in the same patient [88]. Treatment with the sodium channel blocker flecainide reduced exercise-induced ventricular tachycardia in the patient while simultaneously reducin adrenergic-induced arrhythmia in vitro. While treatment with labetalol had no effect in both settings, propranolol prevented the development of arrhythmias in vitro and successfully predicted decreased exercise-induced VT in the patient clinically.
Lumacaftor is an FDA approved drug that works as a chaperone to rescue trafficking defects in mutant CFTR chloride channels in a subset of patients with cystic fibrosis. This drug was tested in two patients with LQT2 who had mutations that were associated with defective membrane trafficking of hERG [89]. Both patients had their QT interval shortened significantly after one day of therapy, which rebounded after a wash-out recording [90]. Although these studies were only performed in a small number of patients, it is encouraging to observe a positive correlation between in vitro drug effects and clinical observations in patients, supporting the further use of human iPSC-derived cardiomyocytes to screen new or already approved drugs before testing them in large clinical trial settings.
iPSC-derived cardiomyocytes also play a role in the development of new drugs, especially in hit-to-lead campaigns. Novel therapeutic agents can be tested in vitro and can undergo several cycles of chemical refinement based on preclinical data before being tested in patients. Mexiletine is an antiarrhythmic drug used to reduce the QT interval in patients with LQT3, even though the mechanism of its therapeutic properties is not fully understood. Cardiomyocytes were differentiated from cells obtained from patients with LQT3 and were treated with mexiletine analogues [91]. Based on the analogues’ effects on APD shortening, arrhythmic activity, and ion current, the compounds were further engineered for optimized APD shortening and anti-arrhythmic activity. The resulting lead compounds from this drug development approach presented greater on-target potency and selectivity than the original drug. The efficacy of one of the new analogues was further tested in cells with different mutations in the same gene (SCN5A) and in cells which a LQT3-like phenotype generated pharmacologically. The new analogues shortened the APD in all models, while other treatments showed mutation-specific efficacy, and suppressed the emergence of arrythmia in healthy cells.
5. Integrating in silico technologies with experimental methodologies
Machine learning involves the development of algorithms that allow computers to make predictions and decisions based on pattern learning. Tools that have arisen from machine learning have enhanced our ability to decipher high-dimensional and complex data sets in biomedical research. Clinically, this has led to several advances in the interpretation of several areas including electrocardiograms, pathology sections, and radiographic images. This has also proved fruitful in translational research, assisting in the identification of disease phenotypes in vitro, the classification of cardioactive drugs, and predicting drug response [92].
The integration of machine learning algorithms with iPSC models allows for the facile analysis of high-dimensional data to identify phenotypes and drug response in vitro using novel classifiers in unsupervised or supervised manners. Several groups have reported the use of machine learning to classify iPSC-derived cardiomyocytes in healthy or diseased phenotypes based on different parameters of cardiac function [93–96]. Cells obtained from donors with different arrhythmias were successfully classified when comparing cells obtained from healthy donors. Using a noninvasive label-free approach, machine learning algorithms successfully discriminated iPSC-derived cardiomyocytes derived from patients with LQT8 as diseased cells when compared to healthy controls [97].
Machine learning has also been used to identify drug-induced arrhythmias in toxicity studies and to classify cardioactive drugs. Cardiomyocytes treated with drugs with different mechanisms were properly identified by algorithms based on changes in the cardiac functionality [98–100]. With elegant integration of in silico technologies and experimental methodologies, complex platforms can help identify the proarrhythmic risk of new drugs early in the development process [101].
In silico-based drug discovery has been proven particularly impactful, rapidly accelerating drug design campaigns by incorporating computational tools for drug binding optimization through molecular modeling and quantum mechanics [102]. Computational molecular dynamics and docking have been used to generate structure-activity relationships quickly. The integration of structural and ligand-based approaches for virtual screening allows for large lead compound screens with reduced labor costs, improved efficiency, and increased specificity of hits [103]. Moving forward, there is potential for synergy in drug development by combining computer-aided drug design with disease modeling with iPSC to rapidly accelerate validation of hits, identification of promising leads with high throughput functional assays, and early identification of toxicity.
6. Modeling 3D cardiac tissues for studies of arrythmia
Moving beyond 2D culture systems is crucial for better model systems of the heart that appreciate the complexity of a 3D mechanical organ and can also consist of a large number of non-cardiomyocytes that play a role in disease, as well as a complex extracellular matrix. The development of organs-on-chip technologies have allowed the formation of 3D cardiac tissues that incorporate different stromal cell types, extracellular matrices, and even vasculature for infusing external stimuli [23].
The power of these models is bolstered by the plasticity of iPSC and their ability to generate multiple isogenic cell types. By genetically editing iPSC-derived cardiomyocytes and fibroblasts to present a point mutation in the KCNH2 gene and integrating both cell types in a collagen-based hydrogel, a team of researchers developed a LQT2 model [78]. These 3D tissues formed anisotropic sarcomeres as well as recapitulated key features of LQT2, including prolonged APD and calcium mishandling. Exposure to E-4031, a hERG channel blocker, rescued the phenotype, demonstrating the applicability of these models to test novel therapeutic agents for this arrhythmic disease. Additional models have been successfully generated and emulated the pathophysiology and drug response of LQT1 [104], ARVC [105], and CPVT [106]. In addition to the modeling of human diseases in vitro, 3D cardiac tissues can also been used to study the molecular mechanisms behind arrhythmias, identifying the role of TFAP2A in the fibro-fatty remodeling observed in ARVC [107], and the mechanism behind drug-induced tachyarrhythmias with characteristics similar to Torsade de Pointes [108].
7. Conclusion
There has been significant progress in the development of human iPSC-derived cardiomyocyte-based models of arrhythmias. Genetic studies through either derivation from patients with pathogenic variants or engineering mutations in healthy cell lines have captured several important features underlying arrhythmias. These have led to mechanistic insights into disease pathogenesis and clearly demonstrated clinically relevant drug responses. Advances in 3D tissue engineering have improved model fidelity and the application of machine learning has shown significant promise for high-dimensional data analysis and the discovery of new classifiers. All of this has set the stage for iPSC-based systems to provide both efficacy and safety data in preclinical drug development. This is timely as the FDA Modernization Act 2.0, passed in 2022, allows FDA regulators to consider preclinical data obtained from sources other than animal models when evaluating new therapeutic agents, empowering in silico computational-based models, artificial intelligence, and human iPSC culture systems. By integrating these models into the drug development pipeline, new leads can be studied in potentially more physiologically relevant models. These types of studies may give us more reliable safety and toxicity information before moving forward into clinical trials. With access to iPSC obtained from donors, in vitro clinical trials can be established to study the pharmacokinetic and pharmacodynamic profiles of new therapeutic agents and identify and predict risk for drug-induced toxicity. This will hopefully improve hit-to-lead/lead optimization campaigns and result in more effective and safe treatments for patients with arrhythmogenic disorders.
8. Expert opinion
One of the biggest impediments in cardiovascular research has been a lack of available human disease models. This stems in part from both a paucity of available tissue and an inability to sustain or easily manipulate primary culture systems from human heart tissue. This has led to a near exclusive reliance on rodent models of heart disease throughout the literature. These models have been bulwarked by genomic studies that have allowed us to validate human genetic drivers of disease as well as larger animal studies such as those in the pig, which more closely resembles human physiology. However, the rodent model has remained a poor predictor of therapeutic translation and is a potential culprit of many early human trial failures for therapies that progressed optimistically through preclinical investigational new drug (IND) application-enabling studies. This has presented an opportunity for iPSC to fill in a crucial gap in cardiovascular research as a potential parallel pipeline of both validation as well as discovery. The promise of this system cannot be overstated and the ability of iPSC-derived cardiomyocytes to recapitulate genetic diversity, electromechanical coupling, and calcium handling is encouraging, opening new avenues of research into the pathophysiology and treatment of a variety of cardiomyopathies. However, several key issues remain to be addressed to fully realize the potential of this system to model cardiovascular disease, specifically when it comes to appreciating the pathophysiology of arrhythmias (Figure 3). Though there has been success in applying genetic variant models, iPSC-derived cardiomyocytes will require a significant leap forward to tackle the far more common arrhythmias of the atria and ventricles such as atrial fibrillation and scar-related ventricular tachycardia.
Figure 3. Developing more physiological relevant models of arrhythmia.
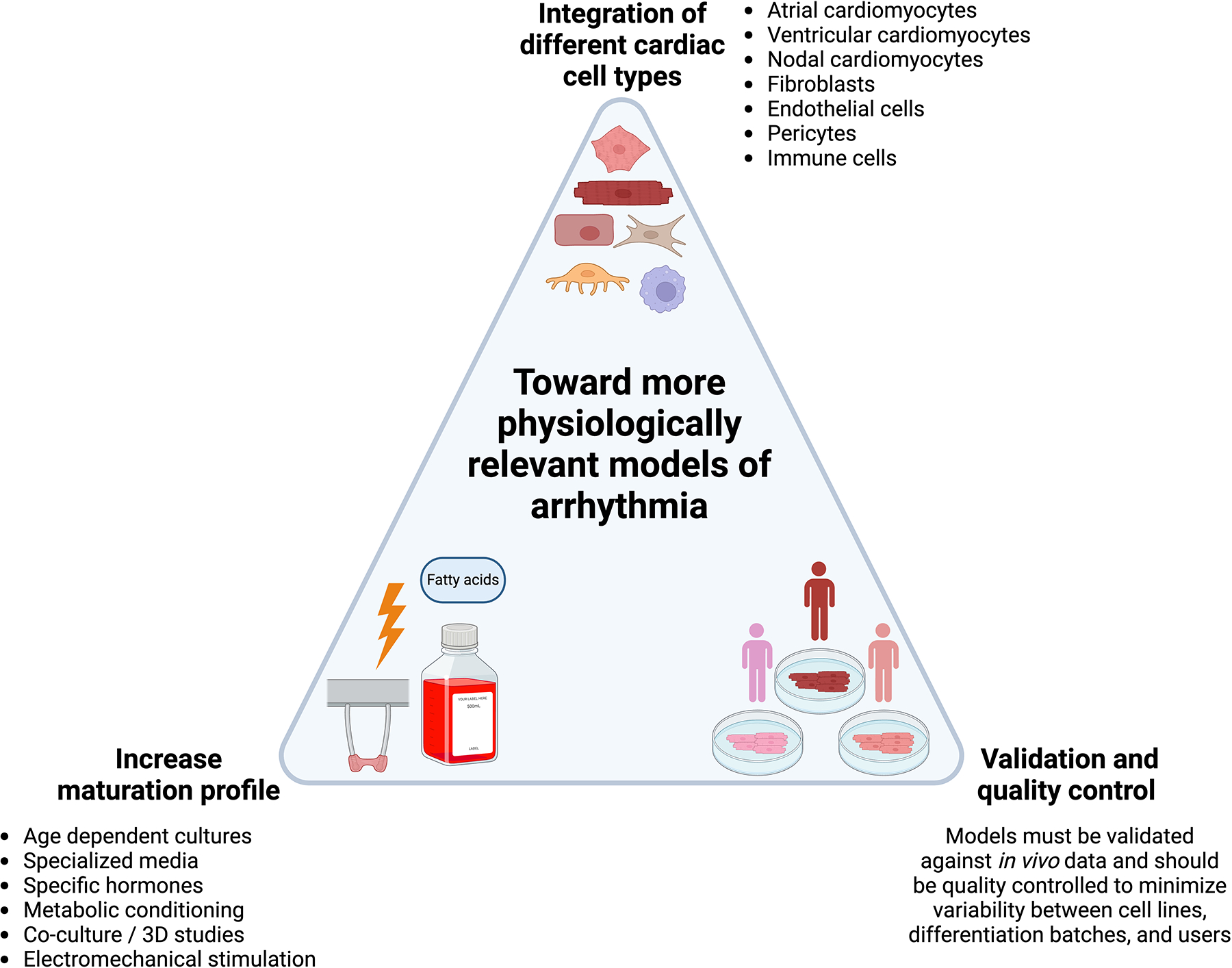
Current models can be further improved by integrating different types of cardiac cells, maturing models to a more adult-like phenotype, and validating and supervising their quality. Created with BioRender.com.
Probably one of the most frequently raised concerns about cardiomyocytes derived from iPSC is their level of maturation. At the end of standard differentiation protocols, cardiomyocytes have been mostly characterized as neonatal [109]. This has significant impact on recapitulating both genetic variants and acquired pathological arrhythmias that depend on adrenergic signaling, mature ion channel constituency, cell to cell communication, and calcium homeostasis. Advances have been made to improve maturation of these cells after differentiation through a variety of methods such as age dependent cultures, specialized media, exposure to specific hormones, altering the metabolic source of energy, co-culture studies, and electromechanical stimulation in 3D culture [26,110–114]. This latter method has proved particularly impressive in terms of gene expression and force generation [26]. Maturity though remains a critical impediment and creating models that more closely resemble adult cardiomyocytes will be needed to boost the fidelity and translational potential of arrhythmia models.
Furthermore, not all cardiomyocytes are the same. There are significant differences between atria and ventricle and between left and right sided structures in terms of their developmental origins, gene expression profiles, electromechanical properties, and ion channel constitution. Arrhythmias can arise in nearly any part of the heart and to model these accurately, we will need to improve our ability to direct differentiation toward specific sublineages. Standard differentiation protocols produce a heterogeneous population that initially displays multiple foci of autologous depolarization with varied action potential morphologies. While there has been significant progress toward both improving discrimination of differentiation between subpopulations of cardiomyocytes and increasing population purity using commonly found expression markers, we are still far from having well-defined labels and in vitro characteristics to properly differentiate all the subpopulations in the native heart [115]. We believe initiatives to develop criteria that different subclasses of in vitro differentiated cardiomyocytes will improve the use of different cell subtypes in disease modeling [116] Whether these can enhance clinical translation significance remains to be tested. However, this improved specification will allow more nuanced models that can both investigate chamber-specific contributions to arrhythmogenesis as well as those in which chamber-specific cardiomyocytes are electrically connected to mimic actual human physiology.
Though cardiomyocytes by volume occupy the bulk of heart tissue, they numerically actually comprise a minority of cells in the heart. The contribution of other cell types to the development of cardiovascular disease is becoming increasingly appreciated and these cells need to be incorporated into model systems. Fibroblasts, for instance, play a key role in the maintenance of structural integrity after myocardial injury through a fibrotic response to the permanent loss of cardiomyocytes. The result, however, can be scarring heterogeneity with islands of viable cardiomyocytes creating short tracks of conduction that lead to scar-mediated ventricular tachycardia. Immune cells have also been recently implicated in exciting research demonstrating that recruited macrophages are key to the development of atrial fibrillation in inflammatory states [117]. Advances in iPSC differentiation now allow for directed differentiation of isogenic fibroblasts and immune cells including macrophages whose incorporation allows for more complex multicellular models. As a recent example of this power, improved atrial fibrillation models in 3D tissue organoids was recently demonstrated through incorporation of isogenic iPSC-derived atrial specific fibroblasts [118].
Modeling organotypic function in a cell or tissue-based culture systems requires a dimensional reduction of physiological phenotypes to laboratory-based readouts which we rationalize have a relationship to the original disease being studied. As an example, cardiomyocyte contractile properties by video microscopy can be correlated to sarcomeric dysfunction from cardiomyopathic iPSC lines. The danger of this approach is that most of these laboratory readouts can be confounded by small changes in differentiation efficiency, batch to batch variation, maturity level, and cell heterogeneity. Biological replicates and the use of several different cell lines can help control for both these phenotypic variations as well as potential unknown genetic interactions in the case of using just one genetic background. However, the fact that we utilize the same quality benchmarks for the generation of iPSC-derived cardiomyocytes and the surrogates for therapeutic response will remain a significant weakness of this system and modeling arrhythmogenesis is no exception. Cellular measurements such as patch clamp, action potentials, and calcium flux using various reporters are all subject to significant variation during the generation of iPSC-derived cardiomyocytes. Consistency and reduction of variability in the manufacturing of iPSC-derived cardiomyocytes are necessary to have confidence that model systems can measure a pathologically relevant change to a readout that is not the result of the variance of iPSC’s differentiation.
Overall, there have been significant progress in modeling arrhythmias using cardiomyocytes differentiated from iPSC. Advances in iPSC methodologies, gene editing, tissue engineering, and in silico technologies portend an optimistic future where these models will become established components of drug development pathways.
Article highlights box.
Induced pluripotent stem cells (iPSC) provide a new source of human cardiomyocytes for studying cardiovascular disease in vitro.
Models of arrhythmias have been successfully generated with both iPSC and gene editing technologies.
Important readouts of arrhythmias such as early and delayed depolarizations have been captured by iPSC-derived cardiomyocytes.
Early drug studies focusing on toxicity and efficacy have shown promise with several inherited arrhythmias.
Tissue engineering has improved the physiological complexity of these models and machine learning is being applied to these systems to improve high-dimensional data analysis.
Funding:
The authors are supported by the National Institutes of Health through a grant from the National Heart, Lung, and Blood Institute (grant no. R01HL166387) and the National Institute of Biomedical Imaging and Bioengineering (grant no. P41EB027062).
Footnotes
Declaration of Interest:
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer Disclosures:
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Bibliography
- [1].Pérez-García MT, Kamp TJ, Marbán E. Functional properties of cardiac L-type calcium channels transiently expressed in HEK293 cells. Roles of alpha 1 and beta subunits. J Gen Physiol. 1995;105:289–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Cribbs LL, Lee JH, Yang J, et al. Cloning and characterization of alpha1H from human heart, a member of the T-type Ca2+ channel gene family. Circ Res. 1998;83:103–109. [DOI] [PubMed] [Google Scholar]
- [3].Anderson CL, Delisle BP, Anson BD, et al. Most LQT2 mutations reduce Kv11.1 (hERG) current by a class 2 (trafficking-deficient) mechanism. Circulation. 2006;113:365–373. [DOI] [PubMed] [Google Scholar]
- [4].Blackwell DJ, Schmeckpeper J, Knollmann BC. Animal models to study cardiac arrhythmias. Circ Res. 2022;130:1926–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Wang ZG, Pelletier LC, Talajic M, et al. Effects of flecainide and quinidine on human atrial action potentials. Role of rate-dependence and comparison with guinea pig, rabbit, and dog tissues. Circulation. 1990;82:274–283. [DOI] [PubMed] [Google Scholar]
- [6].Zicha S, Moss I, Allen B, et al. Molecular basis of species-specific expression of repolarizing K+ currents in the heart. Am J Physiol Heart Circ Physiol. 2003;285:H1641–1649. [DOI] [PubMed] [Google Scholar]
- [7].Akar FG, Wu RC, Deschenes I, et al. Phenotypic differences in transient outward K+ current of human and canine ventricular myocytes: insights into molecular composition of ventricular Ito. Am J Physiol Heart Circ Physiol. 2004;286:H602–609. [DOI] [PubMed] [Google Scholar]
- [8].Blechschmidt S, Haufe V, Benndorf K, et al. Voltage-gated Na+ channel transcript patterns in the mammalian heart are species-dependent. Prog Biophys Mol Biol. 2008;98:309–318. [DOI] [PubMed] [Google Scholar]
- [9].O’Hara T, Rudy Y. Quantitative comparison of cardiac ventricular myocyte electrophysiology and response to drugs in human and nonhuman species. Am J Physiol Heart Circ Physiol. 2012;302:H1023–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. [DOI] [PubMed] [Google Scholar]
- [11].Yu J, Vodyanik MA, Smuga-Otto K, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. [DOI] [PubMed] [Google Scholar]
- [12].Gai H, Leung EL-H, Costantino PD, et al. Generation and characterization of functional cardiomyocytes using induced pluripotent stem cells derived from human fibroblasts. Cell Biol Int. 2009;33:1184–1193. [DOI] [PubMed] [Google Scholar]
- [13].Zhang J, Wilson GF, Soerens AG, et al. Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ Res. 2009;104:e30–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kehat I, Kenyagin-Karsenti D, Snir M, et al. Human embryonic stem cells can differentiate into myocytes with structural and functional properties of cardiomyocytes. J Clin Invest. 2001;108:407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Graichen R, Xu X, Braam SR, et al. Enhanced cardiomyogenesis of human embryonic stem cells by a small molecular inhibitor of p38 MAPK. Differentiation. 2008;76:357–370. [DOI] [PubMed] [Google Scholar]
- [16].Xu XQ, Graichen R, Soo SY, et al. Chemically defined medium supporting cardiomyocyte differentiation of human embryonic stem cells. Differentiation. 2008;76:958–970. [DOI] [PubMed] [Google Scholar]
- [17].Yang L, Soonpaa MH, Adler ED, et al. Human cardiovascular progenitor cells develop from a KDR+ embryonic-stem-cell-derived population. Nature. 2008;453:524–528. [DOI] [PubMed] [Google Scholar]
- [18]**.Lyra-Leite DM, Gutiérrez-Gutiérrez Ó, Wang M, et al. A review of protocols for human iPSC culture, cardiac differentiation, subtype-specification, maturation, and direct reprogramming. STAR Protoc. 2022;3:101560. [DOI] [PMC free article] [PubMed] [Google Scholar]; Comprehensive review of differentiation protocols of human iPSC to cardiomyocytes.
- [19].Urnov FD, Rebar EJ, Holmes MC, et al. Genome editing with engineered zinc finger nucleases. Nat Rev Genet. 2010;11:636–646. [DOI] [PubMed] [Google Scholar]
- [20].Joung JK, Sander JD. TALENs: A widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol. 2013;14:49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Knott GJ, Doudna JA. CRISPR-Cas guides the future of genetic engineering. Science. 2018;361:866–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].van den Berg CW, Okawa S, Chuva de Sousa Lopes SM, et al. Transcriptome of human foetal heart compared with cardiomyocytes from pluripotent stem cells. Development. 2015;142:3231–3238. [DOI] [PubMed] [Google Scholar]
- [23].Hirt MN, Hansen A, Eschenhagen T. Cardiac tissue engineering: State of the art. Circ Res. 2014;114:354–367. [DOI] [PubMed] [Google Scholar]
- [24].Tiburcy M, Hudson JE, Balfanz P, et al. Defined engineered human myocardium with advanced maturation for applications in heart failure modeling and repair. Circulation. 2017;135:1832–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lemoine MD, Mannhardt I, Breckwoldt K, et al. Human iPSC-derived cardiomyocytes cultured in 3D engineered heart tissue show physiological upstroke velocity and sodium current density. Sci Rep. 2017;7:5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26]*.Ronaldson-Bouchard K, Ma SP, Yeager K, et al. Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature. 2018;556:239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]; Major advance in tissue engineering for generating three dimensional cardiac tissue for disease modeling and drug discovery
- [27].Zhao Y, Rafatian N, Feric NT, et al. A platform for generation of chamber-specific cardiac tissues and disease modeling. Cell. 2019;176:913–927.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Herland A, Maoz BM, Das D, et al. Quantitative prediction of human pharmacokinetic responses to drugs via fluidically coupled vascularized organ chips. Nat Biomed Eng. 2020;4:421–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Novak R, Ingram M, Marquez S, et al. Robotic fluidic coupling and interrogation of multiple vascularized organ chips. Nat Biomed Eng. 2020;4:407–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chramiec A, Teles D, Yeager K, et al. Integrated human organ-on-a-chip model for predictive studies of anti-tumor drug efficacy and cardiac safety. Lab Chip. 2020;20:4357–4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ronaldson-Bouchard K, Teles D, Yeager K, et al. A multi-organ chip with matured tissue niches linked by vascular flow. Nat Biomed Eng. 2022;6:351–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Schwartz PJ, Crotti L, Insolia R. Long-QT syndrome: From genetics to management. Circ Arrhythm Electrophysiol. 2012;5:868–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Sanguinetti MC. Long QT syndrome: Ionic basis and arrhythmia mechanism in long QT syndrome type 1. J Cardiovasc Electrophysiol. 2000;11:710–712. [DOI] [PubMed] [Google Scholar]
- [34]**.Moretti A, Bellin M, Welling A, et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med. 2010;363:1397–1409. [DOI] [PubMed] [Google Scholar]; First manuscript demonstrating the recapitulation of LQT syndrome in vitro using human iPSC-derived cardiomyocytes.
- [35].Egashira T, Yuasa S, Suzuki T, et al. Disease characterization using LQTS-specific induced pluripotent stem cells. Cardiovasc Res. 2012;95:419–429. [DOI] [PubMed] [Google Scholar]
- [36].Ma D, Wei H, Lu J, et al. Characterization of a novel KCNQ1 mutation for type 1 long QT syndrome and assessment of the therapeutic potential of a novel IKs activator using patient-specific induced pluripotent stem cell-derived cardiomyocytes. Stem Cell Res Ther. 2015;6:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].January CT, Gong Q, Zhou Z. Long QT syndrome: Cellular basis and arrhythmia mechanism in LQT2. J Cardiovasc Electrophysiol. 2000;11:1413–1418. [DOI] [PubMed] [Google Scholar]
- [38].Matsa E, Rajamohan D, Dick E, et al. Drug evaluation in cardiomyocytes derived from human induced pluripotent stem cells carrying a long QT syndrome type 2 mutation. Eur Heart J. 2011;32:952–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Itzhaki I, Maizels L, Huber I, et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature. 2011;471:225–229. [DOI] [PubMed] [Google Scholar]
- [40].Lahti AL, Kujala VJ, Chapman H, et al. Model for long QT syndrome type 2 using human iPS cells demonstrates arrhythmogenic characteristics in cell culture. Dis Model Mech. 2012;5:220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Matsa E, Dixon JE, Medway C, et al. Allele-specific RNA interference rescues the long-QT syndrome phenotype in human-induced pluripotency stem cell cardiomyocytes. Eur Heart J. 2014;35:1078–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Fatima A, Kaifeng S, Dittmann S, et al. The disease-specific phenotype in cardiomyocytes derived from induced pluripotent stem cells of two long QT syndrome type 3 patients. PLoS One. 2013;8:e83005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ma D, Wei H, Zhao Y, et al. Modeling type 3 long QT syndrome with cardiomyocytes derived from patient-specific induced pluripotent stem cells. Int J Cardiol. 2013;168:5277–5286. [DOI] [PubMed] [Google Scholar]
- [44].Splawski I, Timothy KW, Sharpe LM, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119:19–31. [DOI] [PubMed] [Google Scholar]
- [45].Yazawa M, Hsueh B, Jia X, et al. Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature. 2011;471:230–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Song L, Awari DW, Han EY, et al. Dual optical recordings for action potentials and calcium handling in induced pluripotent stem cell models of cardiac arrhythmias using genetically encoded fluorescent indicators. Stem Cells Transl Med. 2015;4:468–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Song L, Park S-HE, Isseroff Y, et al. Inhibition of CDK5 alleviates the cardiac phenotypes in Timothy Syndrome. Stem Cell Reports. 2017;9:50–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48]**.Song L, Bekdash R, Morikawa K, et al. Sigma non-opioid receptor 1 is a potential therapeutic target for long QT syndrome. Nat Cardiovasc Res. 2022;1:142–156. [DOI] [PMC free article] [PubMed] [Google Scholar]; Evaluation of a potential therapeutic agents using human iPSC-derived cardiomyocytes and its validation in an animal model.
- [49].Leenhardt A, Denjoy I, Guicheney P. Catecholaminergic polymorphic ventricular tachycardia. Circ Arrhythm Electrophysiol. 2012;5:1044–1052. [DOI] [PubMed] [Google Scholar]
- [50].Fatima A, Xu G, Shao K, et al. In vitro modeling of ryanodine receptor 2 dysfunction using human induced pluripotent stem cells. Cell Physiol Biochem. 2011;28:579–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Novak A, Barad L, Zeevi-Levin N, et al. Cardiomyocytes generated from CPVTD307H patients are arrhythmogenic in response to β-adrenergic stimulation. J Cell Mol Med. 2012;16:468–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Itzhaki I, Maizels L, Huber I, et al. Modeling of catecholaminergic polymorphic ventricular tachycardia with patient-specific human-induced pluripotent stem cells. J Am Coll Cardiol. 2012;60:990–1000. [DOI] [PubMed] [Google Scholar]
- [53].Kujala K, Paavola J, Lahti A, et al. Cell model of catecholaminergic polymorphic ventricular tachycardia reveals early and delayed afterdepolarizations. PLoS One. 2012;7:e44660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Jung CB, Moretti A, Mederos y Schnitzler M, et al. Dantrolene rescues arrhythmogenic RYR2 defect in a patient-specific stem cell model of catecholaminergic polymorphic ventricular tachycardia. EMBO Mol Med. 2012;4:180–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Di Pasquale E, Lodola F, Miragoli M, et al. CaMKII inhibition rectifies arrhythmic phenotype in a patient-specific model of catecholaminergic polymorphic ventricular tachycardia. Cell Death Dis. 2013;4:e843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Corrado D, Basso C, Judge DP. Arrhythmogenic cardiomyopathy. Circulation Research. 2017;121:784–802. [DOI] [PubMed] [Google Scholar]
- [57].Kim C, Wong J, Wen J, et al. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature. 2013;494:105–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Ma D, Wei H, Lu J, et al. Generation of patient-specific induced pluripotent stem cell-derived cardiomyocytes as a cellular model of arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2013;34:1122–1133. [DOI] [PubMed] [Google Scholar]
- [59].Caspi O, Huber I, Gepstein A, et al. Modeling of arrhythmogenic right ventricular cardiomyopathy with human induced pluripotent stem cells. Circ Cardiovasc Genet. 2013;6:557–568. [DOI] [PubMed] [Google Scholar]
- [60].El-Battrawy I, Zhao Z, Lan H, et al. Electrical dysfunctions in human-induced pluripotent stem cell-derived cardiomyocytes from a patient with an arrhythmogenic right ventricular cardiomyopathy. Europace. 2018;20:f46–f56. [DOI] [PubMed] [Google Scholar]
- [61].Loiben A, Friedman CE, Chien W-M, et al. Generation of human iPSC line from an arrhythmogenic cardiomyopathy patient with a DSP protein-truncating variant. Stem Cell Res. 2023;66:102987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Buljubasic F, El-Battrawy I, Lan H, et al. Nucleoside diphosphate kinase B contributes to arrhythmogenesis in human-induced pluripotent stem cell-derived cardiomyocytes from a patient with arrhythmogenic right ventricular cardiomyopathy. J Clin Med. 2020;9:486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Hockemeyer D, Soldner F, Beard C, et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat Biotechnol. 2009;27:851–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Zou J, Maeder ML, Mali P, et al. Gene targeting of a disease-related gene in human induced pluripotent stem and embryonic stem cells. Cell Stem Cell. 2009;5:97–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Miller JC, Tan S, Qiao G, et al. A TALE nuclease architecture for efficient genome editing. Nat Biotechnol. 2011;29:143–148. [DOI] [PubMed] [Google Scholar]
- [66].Cermak T, Doyle EL, Christian M, et al. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011;39:e82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Hockemeyer D, Wang H, Kiani S, et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nat Biotechnol. 2011;29:731–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Reyon D, Tsai SQ, Khayter C, et al. FLASH assembly of TALENs for high-throughput genome editing. Nat Biotechnol. 2012;30:460–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Jinek M, Chylinski K, Fonfara I, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Gasiunas G, Barrangou R, Horvath P, et al. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci U S A. 2012;109:E2579–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Hou Z, Zhang Y, Propson NE, et al. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc Natl Acad Sci U S A. 2013;110:15644–15649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Mali P, Yang L, Esvelt KM, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Wang Y, Liang P, Lan F, et al. Genome editing of isogenic human induced pluripotent stem cells recapitulates long QT phenotype for drug testing. J Am Coll Cardiol. 2014;64:451–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Bellin M, Casini S, Davis RP, et al. Isogenic human pluripotent stem cell pairs reveal the role of a KCNH2 mutation in long-QT syndrome. EMBO J. 2013;32:3161–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Terrenoire C, Wang K, Tung KWC, et al. Induced pluripotent stem cells used to reveal drug actions in a long QT syndrome family with complex genetics. J Gen Physiol. 2013;141:61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Brandão KO, van den Brink L, Miller DC, et al. Isogenic Sets of hiPSC-CMs Harboring Distinct KCNH2 Mutations Differ Functionally and in Susceptibility to Drug-Induced Arrhythmias. Stem Cell Reports. 2020;15:1127–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Veldhuizen J, Mann HF, Karamanova N, et al. Modeling long QT syndrome type 2 on-a-chip via in-depth assessment of isogenic gene-edited 3D cardiac tissues. Sci Adv. 2022;8:eabq6720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79]*.Dotzler SM, Kim CSJ, Gendron WAC, et al. Suppression-replacement KCNQ1 gene therapy for type 1 long QT syndrome. Circulation. 2021;143:1411–1425. [DOI] [PubMed] [Google Scholar]; Rescue of cells from patients with LQT syndrome with gene therapy.
- [80].Bains S, Zhou W, Dotzler SM, et al. Suppression and replacement gene therapy for KCNH2-mediated arrhythmias. Circ Genom Precis Med. 2022;15:e003719. [DOI] [PubMed] [Google Scholar]
- [81].Russell S, Bennett J, Wellman JA, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet. 2017;390:849–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Lowes LP, Alfano LN, Arnold WD, et al. Impact of age and motor function in a Phase 1/2A study of infants with SMA type 1 receiving single-dose gene replacement therapy. Pediatr Neurol. 2019;98:39–45. [DOI] [PubMed] [Google Scholar]
- [83].Sager PT, Gintant G, Turner JR, et al. Rechanneling the cardiac proarrhythmia safety paradigm: A meeting report from the Cardiac Safety Research Consortium. Am Heart J. 2014;167:292–300. [DOI] [PubMed] [Google Scholar]
- [84].Blinova K, Stohlman J, Vicente J, et al. Comprehensive translational assessment of human-induced pluripotent stem cell derived cardiomyocytes for evaluating drug-induced arrhythmias. Toxicol Sci. 2017;155:234–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Millard D, Dang Q, Shi H, et al. Cross-site reliability of human induced pluripotent stem cell-derived cardiomyocyte based safety assays using microelectrode arrays: Results from a blinded CiPA pilot study. Toxicol Sci. 2018;164:550–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Honda Y, Li J, Hino A, et al. High-throughput drug screening system based on human induced pluripotent stem cell-derived atrial myocytes ~ A novel platform to detect cardiac toxicity for atrial arrhythmias. Front Pharmacol. 2021;12:680618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Gintant G, Sager PT, Stockbridge N. Evolution of strategies to improve preclinical cardiac safety testing. Nat Rev Drug Discov. 2016;15:457–471. [DOI] [PubMed] [Google Scholar]
- [88].Maizels L, Huber I, Arbel G, et al. Patient-specific drug screening using a human induced pluripotent stem cell model of catecholaminergic polymorphic ventricular tachycardia type 2. Circ Arrhythm Electrophysiol. 2017;10:e004725. [DOI] [PubMed] [Google Scholar]
- [89].Mehta A, Ramachandra CJA, Singh P, et al. Identification of a targeted and testable antiarrhythmic therapy for long-QT syndrome type 2 using a patient-specific cellular model. Eur Heart J. 2018;39:1446–1455. [DOI] [PubMed] [Google Scholar]
- [90].Schwartz PJ, Gnecchi M, Dagradi F, et al. From patient-specific induced pluripotent stem cells to clinical translation in long QT syndrome Type 2. Eur Heart J. 2019;40:1832–1836. [DOI] [PubMed] [Google Scholar]
- [91].McKeithan WL, Feyen DAM, Bruyneel AAN, et al. Reengineering an antiarrhythmic drug using patient hiPSC cardiomyocytes to improve therapeutic potential and reduce toxicity. Cell Stem Cell. 2020;27:813–821.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92]**.Trayanova NA, Popescu DM, Shade JK. Machine learning in arrhythmia and electrophysiology. Circ Res. 2021;128:544–566. [DOI] [PMC free article] [PubMed] [Google Scholar]; Review on machine learning, its use on arrhythmia and electrophysiology studies, and main challenges.
- [93].Juhola M, Joutsijoki H, Penttinen K, et al. Detection of genetic cardiac diseases by Ca2+ transient profiles using machine learning methods. Sci Rep. 2018;8:9355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Joutsijoki H, Penttinen K, Juhola M, et al. Separation of HCM and LQT cardiac diseases with machine learning of Ca2+ transient profiles. Methods Inf Med. 2019;58:167–178. [DOI] [PubMed] [Google Scholar]
- [95].Juhola M, Joutsijoki H, Penttinen K, et al. Machine learning to differentiate diseased cardiomyocytes from healthy control cells. Informatics in Medicine Unlocked. 2019;14:15–22. [Google Scholar]
- [96].Hwang H, Liu R, Maxwell JT, et al. Machine learning identifies abnormal Ca2+ transients in human induced pluripotent stem cell-derived cardiomyocytes. Sci Rep. 2020;10:16977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Teles D, Kim Y, Ronaldson-Bouchard K, et al. Machine learning techniques to classify healthy and diseased cardiomyocytes by contractility profile. ACS Biomater Sci Eng. 2021;7:3043–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Lee EK, Kurokawa YK, Tu R, et al. Machine learning plus optical flow: a simple and sensitive method to detect cardioactive drugs. Sci Rep. 2015;5:11817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Heylman C, Datta R, Sobrino A, et al. Supervised machine learning for classification of the electrophysiological effects of chronotropic drugs on human induced pluripotent stem cell-derived cardiomyocytes. PLoS One. 2015;10:e0144572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Juhola M, Penttinen K, Joutsijoki H, et al. Analysis of drug effects on iPSC cardiomyocytes with machine learning. Ann Biomed Eng. 2021;49:129–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Serrano R, Feyen DAM, Bruyneel AAN, et al. A deep learning platform to assess drug proarrhythmia risk. Cell Stem Cell. 2023;30:86–95.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Sadybekov AV, Katritch V. Computational approaches streamlining drug discovery. Nature. 2023;616:673–685. [DOI] [PubMed] [Google Scholar]
- [103].Shaker B, Ahmad S, Lee J, et al. In silico methods and tools for drug discovery. Comput Biol Med. 2021;137:104851. [DOI] [PubMed] [Google Scholar]
- [104].Giacomelli E, Sala L, Oostwaard DW, et al. Cardiac microtissues from human pluripotent stem cells recapitulate the phenotype of long-QT syndrome. Biochem Biophys Res Commun. 2021;572:118–124. [DOI] [PubMed] [Google Scholar]
- [105].Bliley JM, Vermeer MCSC, Duffy RM, et al. Dynamic loading of human engineered heart tissue enhances contractile function and drives a desmosome-linked disease phenotype. Sci Transl Med. 2021;13:eabd1817. [DOI] [PubMed] [Google Scholar]
- [106].Goldfracht I, Efraim Y, Shinnawi R, et al. Engineered heart tissue models from hiPSC-derived cardiomyocytes and cardiac ECM for disease modeling and drug testing applications. Acta Biomater. 2019;92:145–159. [DOI] [PubMed] [Google Scholar]
- [107].Kohela A, van Kampen SJ, Moens T, et al. Epicardial differentiation drives fibro-fatty remodeling in arrhythmogenic cardiomyopathy. Sci Transl Med. 2021;13:eabf2750. [DOI] [PubMed] [Google Scholar]
- [108].Kawatou M, Masumoto H, Fukushima H, et al. Modelling Torsade de Pointes arrhythmias in vitro in 3D human iPS cell-engineered heart tissue. Nat Commun. 2017;8:1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Karakikes I, Ameen M, Termglinchan V, et al. Human induced pluripotent stem cell-derived cardiomyocytes: Insights into molecular, cellular, and functional phenotypes. Circ Res. 2015;117:80–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Tulloch NL, Muskheli V, Razumova MV, et al. Growth of engineered human myocardium with mechanical loading and vascular coculture. Circ Res. 2011;109:47–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Kamakura T, Makiyama T, Sasaki K, et al. Ultrastructural maturation of human-induced pluripotent stem cell-derived cardiomyocytes in a long-term culture. Circ J. 2013;77:1307–1314. [DOI] [PubMed] [Google Scholar]
- [112].Ruan J-L, Tulloch NL, Razumova MV, et al. Mechanical stress conditioning and electrical stimulation promote contractility and force maturation of induced pluripotent stem cell-derived human cardiac tissue. Circulation. 2016;134:1557–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Parikh SS, Blackwell DJ, Gomez-Hurtado N, et al. Thyroid and glucocorticoid hormones promote functional T-tubule development in human-induced pluripotent stem cell-derived cardiomyocytes. Circ Res. 2017;121:1323–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Yang X, Rodriguez ML, Leonard A, et al. Fatty acids enhance the maturation of cardiomyocytes derived from human pluripotent stem cells. Stem Cell Reports. 2019;13:657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Lee JH, Protze SI, Laksman Z, et al. Human pluripotent stem cell-derived atrial and ventricular cardiomyocytes develop from distinct mesoderm populations. Cell Stem Cell. 2017;21:179–194.e4. [DOI] [PubMed] [Google Scholar]
- [116].Kane C, Terracciano CMN. Concise review: Criteria for chamber-specific categorization of human cardiac myocytes derived from pluripotent stem cells. Stem Cells. 2017;35:1881–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Hulsmans M, Schloss MJ, Lee I-H, et al. Recruited macrophages elicit atrial fibrillation. Science. 2023;381:231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Brown GE, Han YD, Michell AR, et al. Engineered cocultures of iPSC-derived atrial cardiomyocytes and atrial fibroblasts for modeling atrial fibrillation. Sci Adv. 2024;10:eadg1222. [DOI] [PMC free article] [PubMed] [Google Scholar]