

Abstract
Genotype imputation is widely used in genome-wide association studies (GWAS). However, both the genotyping chips and imputation reference panels are dependent on next-generation sequencing (NGS). Due to the nature of NGS, some regions of the genome are inaccessible to sequencing. To date, there has been no complete evaluation of these regions and their impact on the identification of associations in GWAS remains unclear. In this study, we systematically assess the extent to which variants in inaccessible regions are underrepresented on genotyping chips and imputation reference panels, in GWAS results and in variant databases. We also determine the proportion of genes located in inaccessible regions and compare the results across variant masks defined by the 1000 Genomes Project and the TOPMed program. Overall, fewer variants were observed in inaccessible regions in all categories analyzed. Depending on the mask used and normalized for region size, only 4%–17% of the genotyped variants are located in inaccessible regions and 52 to 581 genes were almost completely inaccessible. From the Cooperative Health Research in South Tyrol (CHRIS) study, we present a case study of an association located in an inaccessible region that is driven by genotyped variants and cannot be reproduced by imputation in GRCh37. We conclude that genotyping, NGS, genotype imputation and downstream analyses such as GWAS and fine mapping are systematically biased in inaccessible regions, due to missed variants and spurious associations. To help researchers assess gene and variant accessibility, we provide an online application (https://gab.gm.eurac.edu).
Keywords: NGS, GWAS, accessibility, genotyping chips, web tool
Introduction
Genome-wide association studies (GWAS) have identified thousands of trait-associated loci, and have transformed our understanding of genetic susceptibility to common diseases [1]. Genotype imputation is frequently used to increase the number of variants available for association testing by estimating haplotypes in the genotyped individuals and matching them to reference panel haplotypes [2]. Variants that are present in the dense reference panel but not on the genotyping chip can thus be inferred for the study individuals. As the sample size of the reference panel increases, the quality of imputation improves, but the ability to impute variants is highly dependent on the variant quality and density, both of which are determined by next-generation sequencing (NGS) [3].
The quality of NGS based reference panels is impacted by the quality of the reference genome, as the sequenced reads must be mapped against it. Repetitive regions such as telomeres and GC-rich regions convolute the reference genome [4], which propagates to NGS [5]. To quantitatively assess this limitation, the 1000 Genomes Project [6] created two genome masks (“pilot” and “strict”) that defined regions of the genome as accessible to NGS, based on the low coverage whole genome sequencing of 2504 individuals initially mapped to GRCh37. A base was classified as inaccessible if [1] the nucleotide in the reference genome was N (no base assigned), [2] there was a significantly lower or higher than average depth of coverage [3], there were a high number of reads with zero or low mapping quality (Supplementary Table 1). Based on these criteria, 10.6% and 28.3% of bases were defined as inaccessible in the pilot and strict mask, respectively [7]. Following the release of GRCh38, the 1000 Genomes raw sequencing data were remapped to the new reference genome and the two inaccessibility masks were recalculated as for GRCh37 [8]. More recently, the TOPMed program performed deep whole genome sequencing on 53 581 individuals, and used their data mapped to GRCh38 to determine which regions “failed” sequencing using similar criteria to the 1000 Genomes Project (see Methods for details), finding 7.6% of bases inaccessible [9].
Few studies have investigated the effect of genome inaccessibility. Ebbert et al. [10] used whole-genome sequencing data from 10 unrelated individuals to assess which regions of the genome are inaccessible due to insufficient numbers of mappable reads (“dark by depth”) or ambiguous alignments caused by repetitive regions (“camouflaged”) [10]. While this study focused on analyzing the number of dark genes and proposed an algorithm to rescue camouflaged regions in sequencing studies, effect on downstream applications were not accessed. To follow up on these findings, Ryan et al. investigated whether these dark regions hindered the fine-mapping of exemplar GWAS loci for eight complex traits and the identification of disease-relevant genes and variants in two sequencing studies. As dark regions overlapped both the GWAS loci and disease genes, they concluded that GWAS fine mapping may fail due to dark regions, and that dark regions may contain disease-relevant variants that are missed by sequencing studies and likely contribute to missing heritability [11]. Despite these previous works examining dark regions exemplary, the full impact and extent of genome inaccessibility on NGS, genotyping, imputation and GWAS remains unclear, and researches may be unaware of the potential bias introduced by genome inaccessibility in their downstream analysis.
Here, we use five accessibility masks (1000 Genomes pilot GRCh37 and GRCh38, 1000 Genomes strict GRCh37 and GRCh38, and TOPMed GRCh38) to systematically assess the extent to which NGS inaccessibility affects the identification of trait-associated loci in GWAS and disease-relevant variants. Additionally, we present a case study from the Cooperative Health Research in South Tyrol (CHRIS) study, where a signal could not be reproduced in imputed data in GRCh37. Finally, we introduce an online application (https://gab.gm.eurac.edu) that enables users to verify whether a variant or gene of interest is located in an accessible or inaccessible region and how well it is covered on common genotyping chips.
Results
First, we describe the general properties of inaccessible regions using the 1000 Genomes GRCh38 mask as an example. We then compare the 1000 Genomes strict masks with the pilot masks, the 1000 Genomes GRCh37 masks with the GRCh38 masks, and the mask derived from low coverage data (1000 Genomes GRCh38) against the mask derived from high coverage data (TOPMed). We present an online application that allows users to query the accessiblity status of a gene or variant of interest. Finally, we present a case study from the Cooperative Health Research in South Tyrol (CHRIS) study of a novel trait-associated locus in an inaccessible region that was not detected by imputation in GRCh37, but becomes accessible to sequencing and thus imputation in GRCh38.
Inaccessible regions defined by the GRCh38 pilot mask are depleted of variants
Here, we characterize inaccessible regions using the 1000 Genomes GRCh38 pilot mask exemplary. While approximately 10% of the reference autosome is classified as inaccessible by the GRCh38 pilot mask, only 1.4% of variants from the 1000G phase3 imputation reference panel, 1.5% of pathogenic ClinVar variants, 0.83% of common chip variants, 1.8% of known GWAS variants, 2.7% of genes and 4.4% of exons are located in these regions (Table 1).
Table 1.
Characteristics of inaccessible regions as defined by the five inaccessibility masks.
| Inaccessibility mask | Reference genome | % of autosomea | % of variants in the respective imputation reference panelb | % of ClinVar variants (all/pathogenic)b | % of chip variantsb | % of EBI GWAS hitsb | % of gene bodiesa | % of exomea |
|---|---|---|---|---|---|---|---|---|
| 1000G Pilot | GRCh37 | 10.6 | 2.6 | 2.8/1.3 | 0.65 | 1.3 | 3.1 | 4.9 |
| 1000G Pilot | GRCh38 | 9.9 | 1.4 | 3.0/1.5 | 0.83 | 1.8 | 2.7 | 4.4 |
| 1000G Strict | GRCh37 | 28.4 | 26.7 | 9.3/6.6 | 6.94 | 19.5 | 21.5 | 14.4 |
| 1000G Strict | GRCh38 | 23.8 | 20.6 | 6.7/4.4 | 3.78 | 14.3 | 17.9 | 11.8 |
| TOPMed | GRCh38 | 7.6 | 1.0 | 4.0/2.3 | 1.01 | 1.3 | 1.2 | 3.3 |
aPercent of the autosome that is located in inaccessible regions.
bPercent of the respective variant sets that are located in inaccessible regions.
Stratified by the size of the accessible and inaccessible regions (see Methods), only 12% of variants in the 1000 Genomes imputation reference panel fall into inaccessible regions, as do only 12% of pathogenic ClinVar variants, only 6% of variants on known genotyping chips, and only 14% of variants in the EBI GWAS Catalog (Fig. 1). Furthermore, only 20% of the gene bodies fall into inaccessible regions, as do only 30% of the exons.
Figure 1.

Characteristics of accessible and inaccessible regions. Proportion of variants (for panels “reference panel”, “pathogenic ClinVar”, “genotyping chips”, “EBI GWAS Catalog”) or proportion of base pairs (for panels “genes”, “exons”) that fall into accessible and inaccessible regions as defined by the five masks, stratified by the size of the inaccessible region in the different masks. TM = TOPMed.
The gene bodies of 167 (0.8%) genes were completely located in inaccessible regions, and for 837 genes (4.1%) at least 25% of the gene body was inaccessible (Fig. 2a). In total, 35.3 Mb of gene sequence was inaccessible. The exons of 197 genes (1.0%) were completely located in inaccessible regions, for 1017 genes (5.0%) at least 25% of the exonic region of the gene was inaccessible (Fig. 2b).
Figure 2.
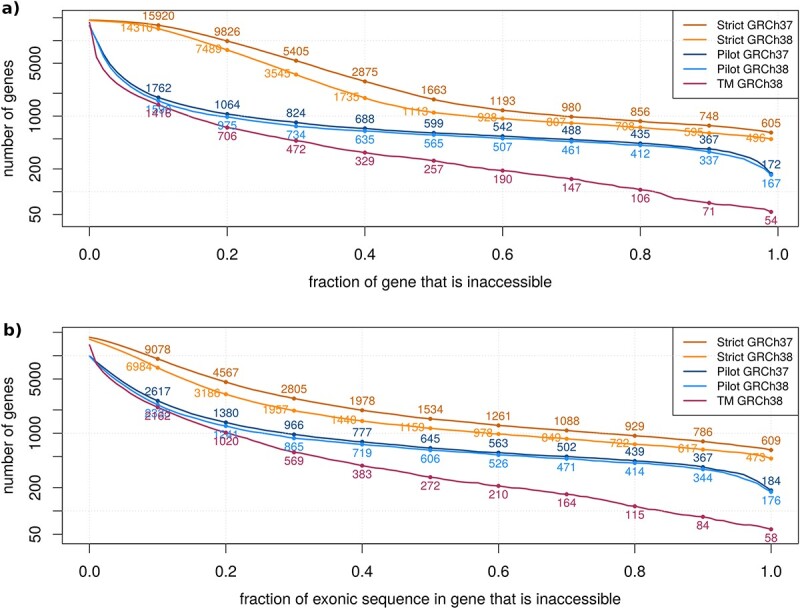
Number and proportion of genes and exons that are inaccessible. (a) Number of genes that are inaccessible by at least a certain proportion of the gene. (b) Number of genes by the proportion of exonic sequence within this gene that is inaccessible. TM = TOPMed.
Uncertainty of imputation is comparable between accessible and inaccessible regions in GRCh37
To compare imputation quality between accessible and inaccessible regions, we utilized HRC imputed data in GRCh37 as an example. The squared correlation of imputed dosages with sequenced WES hard calls (R2) in the CHRIS study revealed a lower R2 for variants in the 1000 Genomes inaccessible regions compared to the accessible regions for both the pilot and strict mask, particularly for the lower MAF range (see Supplementary Fig. 1a). The imputation quality estimated by the imputation software (rsq) and the uncertainty of imputation, calculated by computing the absolute difference in R2, are comparable between accessible and inaccessible regions (Supplementary Fig. 1b). This suggests that the rsq statistic for each imputed variant is equally reliable in both accessible and inaccessible regions.
Inaccessible regions defined by the strict masks contain more variants than inaccessible regions defined by the pilot masks
In the 1000 Genomes project using the phase 3 data, 10.6% and 9.9% of autosome base pairs were classified as inaccessible in the pilot masks on GRCh37 and GRCh38, respectively, and 28.4% and 23.8%, respectively in the strict masks. Stratified by region length, we observed fewer variants in the imputation reference panels and ClinVar, on common chips and among known GWAS signals in the inaccessible regions defined by the pilot masks compared to the inaccessible regions defined by the strict masks. The difference was greatest for the number of variants in the imputation reference panels. While only 12%–18% of variants were in inaccessible regions as defined by the pilot masks, 44%–48% of variants were in inaccessible regions as defined by the strict masks (Fig. 1). A similar trend was observed for variants in the EBI GWAS catalog and almost the same proportion of variants were in inaccessible regions between the pilot and strict masks for pathogenic ClinVar variants. Also on the common genotyping chips, we observed a higher proportion of variants located in inaccessible regions as defined by the strict masks compared to the pilot masks (Supplementary Fig. 2).
More genes and larger proportions of genes and exonic regions were inaccessible in the strict masks compared to the pilot masks (Fig. 2). 37.2 Mb of gene sequence was inaccessible according to the pilot masks, while 254.2 Mb of gene sequence was inaccessible according to the strict masks. For the pilot masks, only 20% of the gene body was located in inaccessible regions, whereas for the strict masks, 40% of the gene body was located in inaccessible regions.
GRCh38 is more accessible than GRCh37
Using the same rules to define inaccessible regions in GRCh38 as in GRCh37 resulted in 0.7% and 4.6% fewer autosomal bases being labeled as inaccessible in GRCh38 for pilot and strict, respectively. Stratified by region length, we observed more variants in the imputation reference panels in inaccessible regions as defined by the GRCh37 masks compared to the GRCh38 masks, but for the other categories we could not observe a clear trend.
More genes and a larger proportion of genes and exonic regions were inaccessible in GRCh37 compared to GRCh38, especially in the strict masks (Fig. 2). In the pilot and strict masks, 3.8 and 40.8 Mb of gene sequence were accessible in GRCh38 that were inaccessible in GRCh37. Despite this general trend, 268 and 305 genes were mostly (≥90%) accessible in GRCh37 and GRCh38, respectively, but mostly (≥90%) inaccessible in the other reference genome using the pilot mask.
Fewer genes are located in the inaccessible region defined by the TOPMed mask compared to the 1000 genomes GRCh38 masks
The TOPMed program has used a more stringent method to classify bases as inaccessible, resulting in only 7.6% of the autosome being labeled as inaccessible. Briefly, bases were labeled as inaccessible if the reference base was N, the base or mapping quality was too low, or the summary statistics were at the extremes of the distribution (see Methods). More genes and a larger proportion of genes and exonic regions were inaccessible in the 1000 Genomes masks (especially the strict mask) compared to the TOPMed mask (Fig. 2). In the TOPMed mask, only 16.0 Mb of gene sequence was inaccessible, while 35.3 Mb and 233.8 Mb of gene sequences was inaccessible in the pilot and strict masks, respectively.
Variants are depleted from telomeres and centromeres
To investigate whether telomeres and centromeres have different accessibility properties compared to the rest of the genome, we repeated the above analysis restricted to genomic regions that are not located in telomeres and centromeres. Less than half (42%–46%) of the inaccessible regions defined by the 1000 Genomes pilot masks and the TOPMed mask are located outside telomeres and centromeres, whereas the majority (77%–79%) of inaccessible regions defined by the strict masks are outside centromeres and telomeres (Supplementary Table 2). However, the total number of variants in the respective imputation reference panels, in ClinVar and the EBI GWAS catalog and on common genotyping chips are the same in all inaccessible regions and those outside of telomeres and centromeres (Table 1), indicating that essentially none of these variants are present in telomeres and centromeres. For example, only 804 of the ~47 million variants in the 1000 Genomes Phase 3 reference panel are in the 1000 Genomes GRCh37 pilot inaccessible regions inside the centromers or telomers, while the remaining ~1.2 million variants are in the inaccessible regions outside of the centromers and telomers.
Case report of a false negative association in an inaccessible region
In a GWAS on multiple blood parameters in the CHRIS study, we observed an example where poor imputation in inaccessible regions resulted in a missed association. We found that the chip-genotyped variant rs2477642 at the locus 10p12.33 was significantly associated (P = 8.21 × 10−9) with aspartate aminotransferase (AST) in GRCh37 (Fig. 3).However, none of the 1000 Genomes Phase 3 or HRC imputed variants at this locus reached genome-wide significance. The pilot and strict masks added to the LocusZoom regional plot clearly show that the locus falls in an inaccessible region with a low density of both genotyped and imputed variants (Fig. 3a and b). Strikingly, rs2477642 has an allele frequency of 0.58 in the CHRIS sample, where it was directly genotyped, and 0.01 in the 1000 Genomes Project Phase 3 Europeans, where it was called from NGS on GRCh37. Re-imputation of the CHRIS genotype data with rs2477642 removed showed that this SNP was poorly imputed using the 1000 Genomes Phase 3 reference panel in GRCh37 (r2 = 0.13). The allele frequency of the imputed rs2477642 in the CHRIS sample decreased from 0.58 to 0.045 and the p-value increased to 0.06. Thus, without direct genotyping of the rs2477642 SNP, the association at 10p12.33 with AST would not have been detected in GRCh37 with neither 1000 Genomes Phase 3 nor the HRC imputed data (Supplementary Fig. 3). This locus has previously been shown to be associated with AST in a study of a Japanese population [12] and in a study of an Australian population [13]. In both cases, the reported SNPs (rs2477664 and rs2437528 respectively) were not found to be in LD with the most significantly associated variant in our study (rs2477642). Conditioning on rs2477642 showed that there were no additional signals in the region (Supplementary Fig. 4).
Figure 3.

Regional association plots (LocusZoom) of the aspartate aminotransferase genome-wide association results 500 kb around reference SNP rs2477642. The linkage disequilibrium between rs2477462 and all other variants is displayed as r2 values calculated from the 1000 genome Europeans. Genotyped variants are represented as filled circles, imputed variants as filled triangles. The genome wide significance line is indicated at 5 × 10−8. The regions of the genome defined as accessible according to the different masks are shown in black, inaccessible regions are shown in white. (a) 1000 genomes phase 3 (GRCh37), (b) HRC (GRCh37), (c) TOPMed (GRCh38), (d) 1000 genomes deep (GRCh38).
In GRCh38, however, this locus becomes accessible, regardless of the accessibility mask used, with many variants located in this region (Fig. 3c and d). The most significant variant in the TOPMed imputed CHRIS data was no longer rs2477642, but the imputed variant rs2477633, which is absent in the 1000 Genomes and HRC panel (P = 2.2 × 10−11, r2 = 0.94, 3 kb distance to rs2477642 with LD r2 = 0.82), which was not identified in 1000 Genomes Phase 3. In addition to two typed variants (rs2477642 and rs2436680), 25 imputed variants reached genome-wide significance at this locus, all with good imputation quality scores (r2 > 0.9). In GRCh37, the associated locus maps to the MRC1L1 gene (ENSG00000183748), of which 93.7% and 100.0% are inaccessible in the pilot and strict mask, respectively. In GRCh38, this gene model has been deprecated and replaced by MRC1 (ENSG00000260314) of which only 1.1%, 16.0%, 0.1% are inaccessible in the pilot, strict, and TOPMed masks, respectively. In GRCh37, MRC1 maps to the primary assembly at locus 10p12.33, less than 150 kb from MRC1L1 (ENSG00000120586) and to the patch HG544_PATCH (ENSG00000260314).
Online tool shows accessibility status of variants and genes
To allow users to query accessibility information for a specific gene or variant in both GRCh37 and GRCh38 on the autosomes and sex chromosomes, we have developed an online application. In the gene-based query, one table shows the proportion of the gene and its exonic region that is inaccessible according to the different masks. The second table shows all variants present in this gene on a genotyping chip of interest and shows inaccessibility information for all positions. The variant-based query shows whether the position is in an inaccessible region according to the different masks and on which common genotyping chips it is present. The application is freely available without registration at https://gab.gm.eurac.edu.
Discussion
Although individuals are not typically whole genome sequenced for GWAS, the impact of sequencing inaccessibility is evident in the use of genotyping chips and imputation. Genotyping chips are designed to measure variants at pre-specified sites. These sites are chosen based on prior knowledge of genetic variation within a population, which is typically determined by NGS. Therefore, as we have shown here, regions that are inaccessible to NGS, as defined by the 1000 Genomes Project, are underrepresented on genotyping chips. Indeed, across common genotyping chips and normalised for the differences in size between the inaccessible and accessible regions, only 4%–17% of genotyped variants are located in inaccessible regions as defined by the five masks (Fig. 1). The under-representation of inaccessible regions on genotyping chips is exacerbated by imputation. While imputation has proven extremely useful in increasing the number of variants available for association testing, the reference panels also have a sparsity of variants in inaccessible regions, as they are based on NGS. For the 1000 Genomes pilot masks and the TOPMed mask, only 11%–18% of variants in the imputation reference panel fall into inaccessible regions (normalized for region size), whereas for the 1000 Genomes strict masks there is no difference between accessible and inaccessible regions. An obvious consequence of the sparsity and low quality of known variants in inaccessible regions are false negative trait associations in GWAS. We tested this hypothesis using the ClinVar database and EBI GWAS Catalog. In both datasets we found that only 10%–22% of variants were in inaccessible regions as defined by the pilot and the TOPMed masks, normalized for region length.
We have observed striking differences between the different inaccessibility masks, especially between the strict masks on the one side and the pilot and TOPMed masks on the other side. The 1000 Genomes strict masks classify about a quarter of the autosomes as inaccessible. However, the proportion of variants from the imputation reference panel and the EBI GWAS Catalog, as well as the proportion of genes in inaccessible regions as defined by the strict masks, is almost the same as the proportion in accessible regions, suggesting that a large proportion of bases classified as “inaccessible” by the strict masks have the same properties as the accessible bases and are therefore not truly inaccessible to sequencing. In general, GRCh38 is more accessible than GRCh37. A meaningful and informative genomic unit to compare different assemblies are gene definitions, especially since 97% of genes could be mapped from GRCh37 to GRCh38 (http://www.ensembl.info/2014/03/11/grch38-assembly-mapping-updating-coordinates-in-the-new-human-genome/) and Ensembl gene ids remained stable (http://www.ensembl.info/2014/07/08/maintaining-stable-ids-between-grch37-and-grch38/). While the general variant properties shown in Table 1 are similar for the GRCh37 and GRCh38 masks, genes become more accessible in GRCh38 (Fig. 2), suggesting that genetic studies would benefit from using GRCh38 gene models over GRCh37 gene models.
To consider the potential impact of inaccessibility on a particular target, a researcher can use our web tool to query the target and determine the best experimental setup. If the target region is indeed inaccessible, the researcher should be aware that genotyping followed by imputation, as well as next-generation sequencing, may not adequately cover the target. Other methods may be more appropriate for investigating this locus. Furthermore, it is recommended to retain all genotyped SNPs in the imputation output, including those that did not match SNPs in the reference panel (typed only). This is because they may be more accurate in inaccessible regions than imputed variants. However, some imputation programs do not enable the retention of genotyped SNPs by default. It has been demonstrated that imputation quality is inferior in inaccessible regions compared to accessible regions, particularly for very rare variants. However, the imputation quality statistic rsq provided by the imputation software is equally reliable or unreliable in inaccessible regions as in accessible regions. This suggests that a researcher can have the same level of confidence in an imputed variant and a GWAS signal based on it in an inaccessible or accessible region. In practical terms, the effect of inaccessibility on imputation and GWAS is confounded by the depletion of variants from inaccessible regions, which could result in missed associations.
We found a robust example of a trait-associated locus in the CHRIS study that cannot be detected by 1000 Genomes imputation in GRCh37. The novel association with aspartate aminotransferase at 10p12.33 was dependent on the significant SNP rs2477642 being directly genotyped. When we imputed this SNP in the CHRIS study and repeated the association analysis, we found that it was incorrectly imputed, and the association signal was lost. Low imputation quality does not only affect the discovery of trait associations in GWAS. As the AF of rs2477642 was considerably higher in the genotyped CHRIS sample (0.58) than in the imputed 1000 Genomes population (0.01), we might have mistakenly assumed that this variant was overrepresented in our local population. In GRCh38, however, the region changes from inaccessible to accessible (Fig. 3), which resolves the issue. In fact, our example represents one of the few genes, where the gene model could not be mapped directly between the assemblies, as the variants map to MRC1L1 in GRCh37, which has been deprecated and replaced with MRC1 in GRCh38.
To our knowledge, few other studies have systematically assessed the inaccessible or dark genome. Ebbert et al. analyzed whole genome sequencing of 10 individuals to identify regions inaccessible to sequencing and found 15 megabases (Mb) inaccessible in 6054 genes [10]. This is similar to the TOPMed mask, which identifies 16 Mb of gene regions as inaccessible. The pilot masks even classify 35–40 Mb and the strict masks 234–274 Mb as inaccessible in gene regions. Disregarding the strict masks, more genes are completely dark according to the results of Ebbert et al. (527) compared to the 1000 Genomes pilot masks and the TOPMed mask (54–172). Also considering genes that are at least 25% dark, Ebbert et al. identified 1608 genes, while only 573–923 genes are inaccessible according to the 1000 Genomes pilot masks and the TOPMed mask. Interestingly, they identified more than twice as many dark nucleotides in GRCh38 (excluding alternate contigs) than in GRCh37, while the 1000 Genomes masks contain fewer inaccessible regions in GRCh38 than in GRCh37. This discrepancy might be due to the differences in determining the “inaccessible” or “dark” regions or the quality of the underlying sequencing data. Ryan et al. followed up on these results and examined the overlap of the identified dark regions with GWAS loci and disease genes, from which they concluded that the causal variants in GWAS or sequencing studies may be located in dark regions and missed in fine-mapping studies [11].
The strength of our work lies in the systematic evaluation and comparison of inaccessible regions defined by different masks on the GRCh37 and GRCh38 genome assemblies, shedding light on the limitations of genotyping and genotype imputation that have received little attention to date. Our online application allows users to assess the level of accessibility of a region of interest to help evaluate the results of genotyping, NGS or GWAS. With the CHRIS case study, we demonstrate a real-world example of a missed association in GWAS. The work described also has some limitations that were beyond the scope of this article and warrant further research. First, our analysis focuses on short-read technologies and their downstream applications, as these are the current state of the art, and we have not evaluated whether a bias also exists in long-read technolgies. Second, we did not investigate whether the technical inaccessibility of genomic regions is related to biological function, e.g. whether genes in inaccessible regions have lower expression levels or are transcriptionally repressed due to histone methylation [14].
Overall, our work shows that sequencing inaccessibility remains a problem regardless of genome assembly and sequencing data quality. In recent years, however, resources have been invested in exploring the dark or inaccessible genome in search of variants that may explain the missing heritability for diseases such as Alzheimer’s [15]. Variants located in regions with ambiguous mapping could be rescued by masking all but one highly similar region in the reference genome, realigning the extracted reads, and then calling variants in these remapped regions [10]. In addition, the alignment of multi-mapping reads can be improved by extending the standard fragment size in short-read sequencing, which should improve the quality of variant calling in these repetitive regions [16]. Finally, linked- and long-read sequencing technologies can largely overcome the problem of genome inaccessibility and facilitate the detection of structural variation [15, 16]. Indeed, a nearly complete genome (T2T-CHM13), adding five full chromosome arms and 8% of previously missed sequence covering 229 Mbp and 207 protein-coding genes, has recently been assembled using long-read sequencing technologies [17, 18]. Remapping of the 1000 Genomes short-read samples against T2T-CHM13 revealed 2 million variants within previously unresolved and thereby inaccessible regions. Furthermore, the mismatch rate was reduced by 20%–25% for the short-read data, and by 5%–40% for 17 aligned long-read samples [18], which further reduces the problem of inacessibility, since insufficient mapping quality was the second most prevalent reason for base inaccessibility in the 1000 Genomes masks (Supplementary Table 1). However, until this newly assembled genome becomes the de facto reference and long-read sequencing technologies are more widely used, the problem of genome inaccessibility will remain in practice.
In summary, we have provided a comprehensive characterization of inaccessible regions in GRCh37 and GRCh38 and have shown that inaccessible regions are systematically underrepresented on genotyping chips and in imputation reference panels, resulting in fewer GWAS associations and pathogenic variants identified in these regions. While missed variation due to insufficient target sequence coverage is a well-recognized limitation of NGS, its impact on genotyping, genotype imputation and GWAS has been neglected. Genome inaccessibility is likely to explain a proportion of the missing heritability in GWAS and will remain a limitation of genomic analyses that are based on the GRCh37 and GRCh38 reference assemblies using currently established methods. However, our interactive tool (https://gab.gm.eurac.edu) will allow researchers to investigate the impact on their genes and regions of interest.
Materials and methods
All analyses described in this manuscript were restricted to the autosomes.
Accessibility masks
The 1000 Genomes Project assigned each base in the GRCh37 and GRCh38 reference genomes to one of seven classes according to two different criteria (“pilot” and “strict”), creating two different accessibility masks for each genome assembly (Supplementary Table 1). The inaccessible bases in the pilot masks are a subset of those in the strict masks. The primary difference between the masks is the mapping quality requirements. The pilot masks have more lenient thresholds for classifying a base as accessible. The pilot and strict inaccessibility masks were downloaded as bed files in the GRCh37 and GRCh38 reference assemblies (http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/supporting/accessible_genome_masks, http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000_genomes_project/working/20160622_genome_mask_GRCh38/README.accessible_genome_mask.20160622).
The TOPMed program also defined an accessibility mask based on the whole-genome sequencing data of 1000 randomly selected individuals of the data freeze 5 [9]. For each individual, base coverage was calculated using reads with mapping quality greater than 20 and base quality greater than 20. For each base pair, the coverage was then aggregated across all individuals. Base-pairs with N as reference allele, low base quality, or where all reads had low mapping quality were not considered for aggregation. Finally, a base-pair was declared inaccessible if: i) the mapped reference allele was N; ii) the base mapping quality was below a threshold or all mapping qualities were below a threshold across all individuals; iii) the calculated mapping summary statistics did not fall between the 1 and 99 percentiles for autosomal chromosomes and pseudoautosomal regions on chromosome X, and between the 1 and 99.9 percentiles for non-pseudoautosomal regions on chromosome X. The TOPMed “failed regions” mask was obtained directly from the authors of the study upon request.
Imputation reference panel comparison
1000 Genomes Project Phase 3 genetic variants were downloaded as vcf files in genome assemblies GRCh37 (http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/) and GRCh38 (http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000_genomes_project/release/20190312_biallelic_SNV_and_INDEL/ALL.wgs.shapeit2_integrated_snvindels_v2a.GRCh38.27022019.sites.vcf.gz). Genetic variants from the TOPMed project were downloaded using the TOPMed imputation server (https://imputation.biodatacatalyst.nhlbi.nih.gov/). Singletons were removed. The number of variants in inaccessible regions was determined with vcftools and their proportion compared to the proportion of the autosome that was inaccessible according the different masks.
ClinVar comparison
The ClinVar database version 2020-06-16 was downloaded in GRCh37 and GRCh38. High confidence pathogenic and likely pathogenic variants were selected by restricting to variants with the review status (CLNREVSTAT) “reviewed by expert panel” or “criteria provided, multiple submitters, no conflicts” and the clinical significance (CLNSIG) “pathogenic” or “likely pathogenic”. The number of variants in inaccessible regions was determined using vcftools and their proportion compared to the proportion of the autosome that was inaccessible according to the different masks.
Comparison of genotyping chips
Variants genotyped by common genotyping chips (Illumina and Affymetrix) were downloaded from (https://www.well.ox.ac.uk/~wrayner/strand/sourceStrand/index.html), resulting in 130 available chips for GRCh37 and 140 available chips for GRCh38 (http://www.well.ox.ac.uk/~wrayner/strand/index.html). The number of chip variants in inaccessible regions was determined and averaged across all chips.
Comparison of GWAS catalog
The NHGRI-EBI Catalog version e98 of March 8th 2020 (https://www.ebi.ac.uk/gwas/) of published GWAS was downloaded, lifted over to GRCh37, and both the original and the lifted catalog were filtered to include only genome-wide significant associations (P-value < 5 × 10−8). The number of catalog variants in inaccessible regions was determined.
Calculation of gene and exon inaccessibility
The genomic start and end coordinates of all autosomal protein-coding genes and their exons for GRCh37 and GRCh38 were downloaded from Ensembl Biomart for Ensembl release 100. For each gene in both genome assemblies, the proportion of the gene located in an inaccessible region was determined for all five masks using the Ensembl stable id definition to map genes between GRCh37 and GRCh38, as the HGNC gene name may change between assemblies. We refer to the genomic region of a gene from the start coordinate to the end coordinate as the gene body. Accordingly, the proportion of exons in an inaccessible region was determined for each gene.
Telomeres and centromeres
Features of accessible and inaccessible regions were also analyzed, restricting the analysis to genomic regions outside of telomeres and centromeres. For GRCh37 centromere and telomere coordinates were obtained from the USCS genome browser (http://genome.ucsc.edu/cgi-bin/hgTables) using the ‘gap’ table. For GRCh38, the gap table contains only telomeres, not centromeres. Therefore, the coordinates were extracted from the ‘cytoband’ table (gimsa stain acen).
Stratification of the masks
The five accessibility masks are of different sizes, making it difficult to directly compare their characteristics (see Supplementary Fig. 5 for an example). To determine the relative proportions of imputation reference panel variants, pathogenic ClinVar variants, variants on common genotyping chips, variants in the EBI GWAS catalog, exonic and genomic base pairs in accessible and inaccessible regions stratified by the mask size (Fig. 1), the following procedure was used. Let Va and Vi be the number of variants (SNPs and indels) in the accessible and inaccessible region of a category (i.e. the imputation reference panel), respectively. Let Ba and Bi be the number of base pairs in the accessible and inaccessible regions of a mask, respectively. First, the number of variants or base pairs was normalized to the region size, i.e. Va_rel = Va/Ba and Vi_rel = Vi/Bi. Then, the relative proportion was calculated as Fa = Va_rel/(Va_rel + Vi_rel) and Fi = Vi_rel/(Va_rel + Vi_rel). That is, if stratified by region size, the same number of variants would fall into accessible and inaccessible regions, Fa = Fi = 0.5.
Genotyping, imputation, and GWAS in the CHRIS study
The Cooperative Health Research in South Tyrol (CHRIS) study [19] is a population-based study of more than 13 000 participants recruited between 2011 and 2018 from the Vinschgau/Val Venosta, South Tyrol, Italy. Participants underwent blood and urine sampling, anthropometric measurements, a 20 min electrocardiogram (ECG), and blood pressure measurements. Here, we analyze the first 4639 CHRIS participants, for which genotyping was performed on an Illumina HumanOmniExpress chip. Genotypes were called using GenomeStudio on GRCh37, resulting in 653 660 genotyped variants. Variants with GenTrain score < 0.6, cluster separation score < 0.4, or call rate < 80% were considered technical failures in the genotyping laboratory and were deleted before data release. Samples with call rate < 98%, singletons, and variants with Hardy–Weinberg equilibrium P < 10−6 were removed.
Imputation of CHRIS genotypes into the 1000 Genomes phase 3 reference panel (GRCh37 and GRCh38), the HRC reference panel (GRCh37), the TOPMed r1 panel (GRCh38), and the 1000 Genomes deep panel (GRCh38) was performed using the Michigan Imputation Server (https://imputationserver.sph.umich.edu). Genome-wide association testing was performed on a range of numeric biochemical traits using the epacts software version 3.2.6 with the q.emmax test. Residuals were generated from a linear mixed model of the natural log transformed trait with sex, age and machine change as fixed effects and week of examination as a random effect. In the evaluation of the trait aspartate aminotransferase (AST), the significantly associated variant rs2477642, which had a much higher minor allele frequency in CHRIS, was examined in more detail (see Results).
HRC imputation accuracy was determined by computing the squared correlation of imputed dosages with WES hard calls (R2) for all variants that were both imputed and sequenced, but not genotyped. To obtain a measure for the uncertainty of imputation, the absolute difference of R2 and the estimated imputation quality estimate of the imputation software (rsq) was computed.
Supplementary Material
Acknowledgements
We thank Daniele di Domizio for IT support and Giacomo Antonello and David Emmert for feedback. The CHRIS study is a collaborative effort between the Eurac Research Institute for Biomedicine and the Healthcare System of the Autonomous Province of Bozen/Bolzano (Südtiroler Sanitätsbetrieb/Azienda Sanitaria dell’Alto Adige) [19]. Investigators thank all CHRIS study participants, the general practitioners, the study teams of the CHRIS center at the Hospital of Schlanders/Silandro and of the CHRIS Biobank for their support and collaboration. The CHRIS biobank was assigned the “Bioresource Research Impact Factor” (BRIF) code BRIF6107. The CHRIS study is funded by the Department of Innovation, Research and University of the Autonomous Province of Bozen/Bolzano. The authors thank the Department of Innovation, Research and University of the Autonomous Province of Bozen/Bolzano for covering the Open Access publication costs.
Conflict of interest statement: None declared.
Contributor Information
Eva König, Institute for Biomedicine (affiliated to the University of Lübeck), Eurac Research, Via Volta 21, Bolzano 39100, Italy.
Jonathan Stewart Mitchell, Institute for Biomedicine (affiliated to the University of Lübeck), Eurac Research, Via Volta 21, Bolzano 39100, Italy.
Michele Filosi, Institute for Biomedicine (affiliated to the University of Lübeck), Eurac Research, Via Volta 21, Bolzano 39100, Italy.
Christian Fuchsberger, Institute for Biomedicine (affiliated to the University of Lübeck), Eurac Research, Via Volta 21, Bolzano 39100, Italy.
References
- 1. Abdellaoui A, Yengo L, Verweij KJH. et al. 15 years of GWAS discovery: realizing the promise. Am J Hum Genet 2023;110:179–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Howie B, Fuchsberger C, Stephens M. et al. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet 2012;44:955–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Das S, Abecasis GR, Browning BL. Genotype imputation from large reference panels. Annu Rev Genomics Hum Genet 2018;19:73–96. [DOI] [PubMed] [Google Scholar]
- 4. Guo Y, Dai Y, Yu H. et al. Improvements and impacts of GRCh38 human reference on high throughput sequencing data analysis. Genomics 2017;109:83–90. [DOI] [PubMed] [Google Scholar]
- 5. Treangen TJ, Salzberg SL. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet 2012;13:36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Auton A, Abecasis GR, Altshuler DM. et al. A global reference for human genetic variation. Nature 2015;526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Altshuler DL, Durbin RM, Abecasis GR. et al. A map of human genome variation from population-scale sequencing. Nature 2010;467:1061–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zheng-Bradley X, Streeter I, Fairley S. et al. Alignment of 1000 genomes project reads to reference assembly GRCh38. Gigascience 2017;6:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Taliun D, Harris DN, Kessler MD. et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed program. Nature 2021;590:290–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ebbert MTW, Jensen TD, Jansen-West K. et al. Systematic analysis of dark and camouflaged genes reveals disease-relevant genes hiding in plain sight. Genome Biol 2019;20:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ryan NM, Corvin A. Investigating the dark-side of the genome: a barrier to human disease variant discovery? Biol Res 2023;56:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kamatani Y, Matsuda K, Okada Y. et al. Genome-wide association study of hematological and biochemical traits in a Japanese population. Nat Genet 2010;42:210–5. [DOI] [PubMed] [Google Scholar]
- 13. Middelberg RP, Ferreira MA, Henders AK. et al. Genetic variants in LPL, OASL and TOMM40/APOE-C1-C2-C4 genes are associated with multiple cardiovascular-related traits. BMC Med Genet 2011;12:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vaillant I, Paszkowski J. Role of histone and DNA methylation in gene regulation. Curr Opin Plant Biol 2007;10:528–33. [DOI] [PubMed] [Google Scholar]
- 15. Raybould R, Sims R. Searching the dark genome for Alzheimer’s disease risk variants. Brain Sci 2021;11:332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Iadarola B, Xumerle L, Lavezzari D. et al. Shedding light on dark genes: enhanced targeted resequencing by optimizing the combination of enrichment technology and DNA fragment length. Sci Rep 2020;10:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nurk S, Koren S, Rhie A. et al. The complete sequence of a human genome. Science 2022;376:44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Aganezov S, Yan SM, Soto DC. et al. A complete reference genome improves analysis of human genetic variation. Science 376:eabl3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pattaro C, Gögele M, Mascalzoni D. et al. The cooperative Health Research in South Tyrol (CHRIS) study: rationale, objectives, and preliminary results. J Transl Med 2015;13:348. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.