

Abstract
Autophagy is a fundamental catabolic process whereby excessive or damaged cytoplasmic components are degraded through lysosomes to maintain cellular homeostasis. Studies of mTOR signaling have revealed that mTOR controls biomass generation and metabolism by modulating key cellular processes, including protein synthesis and autophagy. Primary cilia, the assembly of which depends on kinesin molecular motors, serve as sensory organelles and signaling platforms. Given these pathways’ central role in maintaining cellular and physiological homeostasis, a connection between mTOR and primary cilia signaling is starting to emerge in a variety of diseases. In this review, we highlight recent advances in our understanding of the complex crosstalk between the mTOR pathway and cilia and discuss its function in the context of related diseases.
1. Introduction
A major advance toward the discovery of mammalian Target of Rapamycin (mTOR) dates to 1964, when a Canadian expedition identified novel antimicrobial agents in the soil of the South Pacific Island of Rapa Nui. Streptomyces hygroscopicus produced a compound with remarkable immunosuppressive and antitumor properties (Eng, Sehgal, & Vézina, 1984; Martel, Klicius, & Galet, 1977; Vézina & Kudelski, 1975). The isolated compound was named sirolimus, but it is also known as rapamycin after its original site of discovery. The compound is now produced by fermentation of these bacteria and is indicated for the prophylaxis of organ rejection in renal transplantation and the treatment of patients with lymphangioleiomyomatosis.
The mechanisms by which rapamycin regulates immunosuppressive and antitumor properties remained elusive until 1992, when an initial study suggested that rapamycin could interact with a specific peptidyl-prolyl-isomerase named FKBP12, involved in regulating cell growth and proliferation (Chung, Kuo, Crabtree, & Blenis, 1992). In 1994, there was biochemical confirmation that the rapamycin-FKBP12 complex directly targets mTOR in mammals to inhibit the mTOR signaling pathway (Brown et al., 1994; Sabatini, Erdjument-Bromage, Lui, Tempst, & Snyder, 1994; Sabers et al., 1995). Since then, multiple studies have investigated this complex, and it has become apparent that the mTOR protein resides at the center of a vast network of key eukaryotic signaling pathways. These pathways regulate distinct functions, including cell growth and adaptation to environmental conditions, and play a fundamental role in cell organization and homeostasis. The importance of the TOR pathway is highlighted by its conservation among all eukaryotes, explaining the large success of TOR studies in Saccharomyces cerevisiae (Loewith & Hall, 2011). More recently, rapamycin and other mTOR inhibitors are being investigated for their potential to treat a wide variety of human diseases including cancer, diabetes, immune diseases, neurodegenerative diseases, and aging.
Although some of the key functions of mTOR have been elucidated recently, the range of processes in which mTOR is involved is very complex, and many questions about its exact mechanism in these processes remain unanswered.
2. Structure of mTOR
mTOR belongs to the PI3K-related kinase (PIKK) family. Specifically, it is a serine/threonine protein kinase which acts as the core component of two distinct complexes. These two complexes, conveniently named Complex 1 (mTORC1) and 2 (mTORC2), can be dynamically catalyzed by mTOR, depending on the cellular circumstances. Although the structure of both complexes is very similar, they are associated with their own sets of unique proteins, explaining their differentiated roles and relative sensitivity to rapamycin.
Both complexes contain mTOR and mLST8 (mammalian lethal with Sec13 protein 8, also known as GβL) (Hara et al., 2002; Kim et al., 2002; Kim et al., 2003) and the endogenous regulatory subunit DEPTOR (DEP domain containing mTOR interacting protein) (Peterson et al., 2009). However, mTORC1 has a unique Raptor (regulatory protein associated with mTOR) protein, specifically promoting substrate binding to mTORC1. The recruitment occurs through the TOR signaling (TOS) motif, which is a conserved domain on several canonical mTORC1 substrates (Nojima et al., 2003; Schalm, Fingar, Sabatini, & Blenis, 2003) and is crucial for the subcellular localization of mTORC1. In addition, the inhibitory effect of inhibitory subunit PRAS40 (proline rich AKT substrate of 40 kDa) may occur by direct competition with 4E-BP1 and S6K1 for raptor binding (Pearce et al., 2007; Schalm et al., 2003). mTORC2 does not have Raptor and is instead characterized by a different set of proteins, such as Rictor (Rapamycin insensitive companion of mTOR), regulatory subunits Protor1/2 (Pearce et al., 2007; Thedieck et al., 2007), and mSin1 (Frias et al., 2006; Yang, Inoki, Ikenoue, & Guan, 2006).
The mLST8 subunit is involved in growth and survival in response to a wide variety of nutrient, energy, and hormonal signals. While mLST8 is present in both mTORC1 and mTORC2, it is not entirely clear how its role is differentiated in the two complexes. Initial studies suggested that mLST8 does not play a significant role in mTORC1 function (Guertin et al., 2006), but more recent studies demonstrated its direct connection with the catalytic domain of mTORC1, highlighting a possible role in the kinase activation loop (Yang et al., 2006).
Cryo-EM structural studies of mammalian and yeast TORC1 revealed a primary diamond-shaped dimer with mTOR HEAT repeats and Raptor/mTOR as contact dimerization interfaces (Aylett et al., 2016; Baretić, Berndt, Ohashi, Johnson, & Williams, 2016). The rapamycin–FKBP12 complex directly inhibits mTORC1 by generating steric hindrance for substrate entry into the catalytic site of mTORC1. mTOR kinase domain bound to mLST8 showed that the rapamycin–FKBP12 complex binds to the FRB domain, rather than disrupting mTORC1 integrity as previously proposed (Yang et al., 2016). In comparison, the rapamycin insensitivity of mTORC2 is due to the masking of FKBP–rapamycin binding domain by the C-terminal domain of RICTOR (Fig. 1).
Fig. 1.
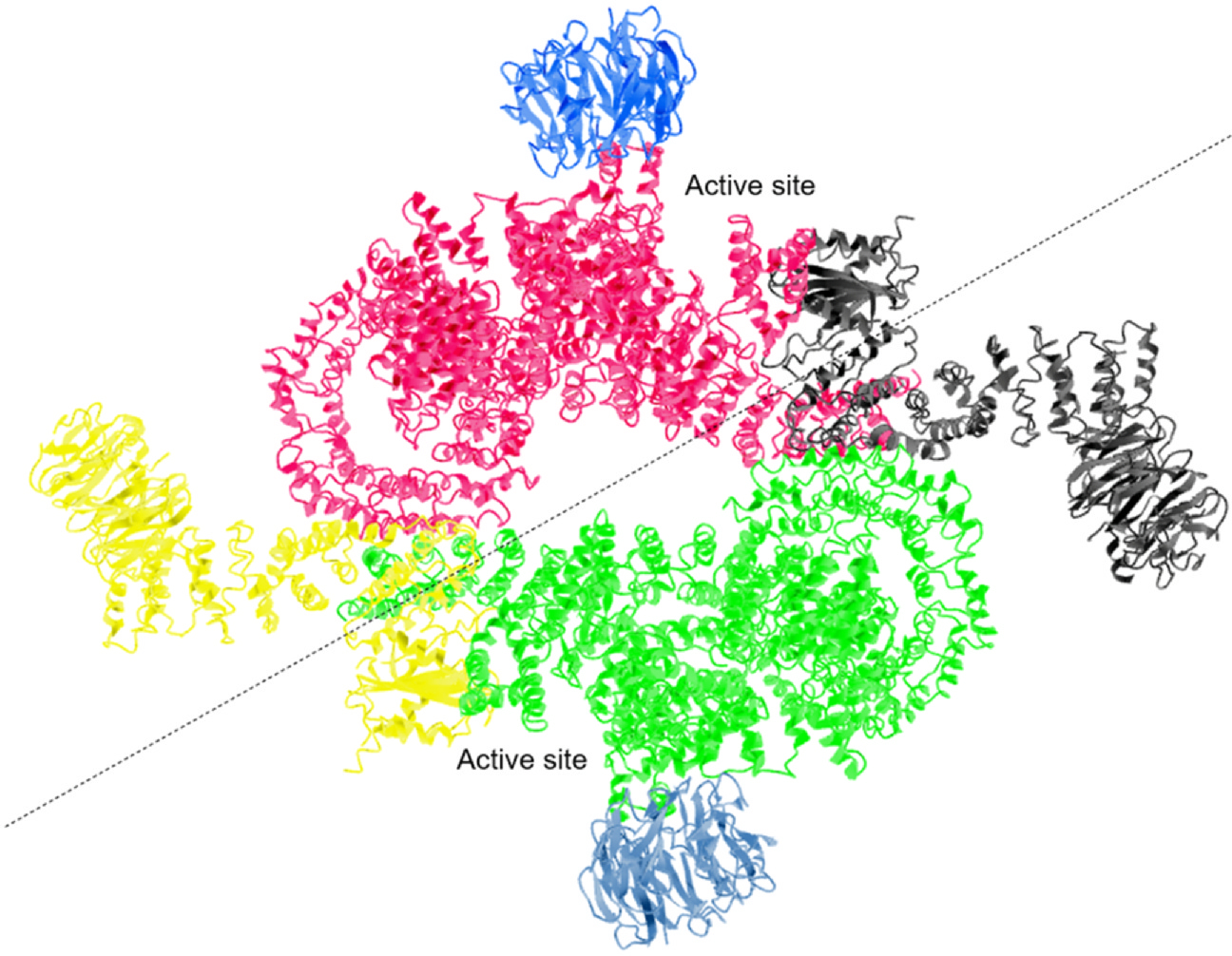
Cryo-EM structure of mTORC1. Color coded ribbon representation of mTORC1 structure showing the three essential components of human mTORC1. Serine/threonine-protein kinase mTOR (Red/Green), Regulatory-associated protein of mTOR RPTOR (yellow/black), and Target of rapamycin complex subunit LST8 (blue/celeste) are combined into a symmetric dimer of an heterotrimer consisting of mTOR, Raptor, and mLST8 itself. Adapted from Yang et al. (2016).
3. Nutrient sensing mTOR signaling pathways
3.1. Intricate upstream and downstream signaling dynamics of mTORC1 in cell growth and metabolism
In mammals during homeostasis, feeding promotes growth and energy storage in liver and muscle tissue. Growth signaling is directly initiated by diet and inhibited by fasting to conserve resources; mTOR is a central regulator of these processes. Upstream, mTORC1 is activated by a combination of pro-growth endocrine signals as well as molecular pathways that signal the presence of sufficient energy and chemical building blocks for macromolecular synthesis. Early studies of the mechanism showed that growth factor- and mitogen-dependent signaling pathways, such as those involving insulin/insulin-like growth factor-1 (IGF-1), can inhibit the Tuberous Sclerosis Complex (TSC) complex, which in turn negatively regulates mTORC1 signaling by binding to mTORC1 (Dibble et al., 2012; Long, Lin, Ortiz-Vega, Yonezawa, & Avruch, 2005; Sancak et al., 2007). Other upstream signals that depend on mTORC1 inhibition and converge on TSC1 include the inflammatory cytokines TNFα and Wnt. In addition to growth factors, mTORC1 is also sensitive to specific signaling pathways that serve as alarms to environmental stresses which are incompatible with cell growth, including hypoxia, low ATP levels, and others causing DNA damage. One example of these alarms is the metabolic regulator AMPK, which inhibits mTORC1 by phosphorylating TSC2 and Raptor on mTORC1 itself in response to glucose deprivation (Gwinn et al., 2008; Inoki, Li, Xu, & Guan, 2003) or hypoxia (Brugarolas et al., 2004). Moreover, the well-known DNA-damage cell cycle regulator p53 is also involved in AMPK and TSC regulation and has tight control over mTORC1 function (Feng et al., 2007).
Interestingly, the discovery that heterodimeric Rag GTPases are also involved in these pathways showed that the activation of mTORC1 is coupled to diet-induced alterations in serum amino acid concentrations (Kim, Goraksha-Hicks, Li, Neufeld, & Guan, 2008; Sancak et al., 1979). Rag GPTases are bound to the lysosomal membrane, and the activation and conformational change by amino acid stimulation allow them to recruit mTORC1 to the lysosomal surface through binding to Raptor. Distinct mechanisms seem to be involved in the ability of mTORC1 to sense intra-lysosomal and cytosolic amino acids. One such mechanism is a possible direct effect of intralysosomal v-ATPases and the formation of a Rag-Ragulator-v-ATPase complex, which is necessary for arginine to activate mTORC1 (Jung, Genau, & Behrends, 2015; Rebsamen et al., 2015; Wang et al., 2015a). On the cytosolic side, the arginine signal to mTORC1 is in turn regulated by GATOR1 and GATOR2 complexes (Bar-Peled et al., 2013). Specifically, GATOR2 acts as a positive mTORC1 regulator, which interacts at the lysosomal membrane with the actual inhibitor of mTORC1, GATOR1 (Bar-Peled et al., 2013). GATOR2 also influences mTORC1 activation by directly binding the recently identified Cellular Arginine Sensor for mTORC1 (CASTOR1), supporting the picture of GATOR2 as a key player in the amino acid-dependent regulation of mTORC1 (Chantranupong et al., 2016). However, other mechanisms independent of Rag GTPases could play an equal role in these mechanisms, as shown by studies of the amino acid glutamine (Jewell et al., 1979).
mTOR plays a crucial role in regulating a wide variety of downstream signaling pathways. Specific signaling networks activated by mTOR depend on the downstream activation of either the mTORC1 or mTORC2 subunits. The mTORC1 pathway is involved in all these processes directly linked to cell growth and division. By regulating the suppression of catabolic pathways (autophagy) and the production of the various lipids, proteins, and nucleotides necessary for cell division, mTORC1 acts as a master switch between anabolism and catabolism in response to environmental conditions. The synthesis of proteins is regulated by the phosphorylation of the key effectors, eIF4E Binding Protein (4EBP) and p70S6 Kinase 1 (S6K1). Both of these kinases can activate or inhibit substrates that promote mRNA translation initiation (Gingras et al., 1999; Holz, Ballif, Gygi, & Blenis, 2021), and subsequent studies of acute mTOR inhibition revealed that only mRNAs involved in protein synthesis are primarily affected (Hsieh et al., 2012), supporting a role for mTOR in cell growth. However, the growth of cells also requires lipid synthesis, which mTORC1 facilitates through a transcription factor, the sterol-responsive element binding protein (SREBP), that regulates the expression of metabolic genes involved in fatty acid and cholesterol biosynthesis (Porstmann et al., 2008). Interestingly, mTORC1 also increases the translation of the master regulator of cellular and systemic homeostatic response to hypoxia (HIF-1) (Düvel et al., 2010), emphasizing its influence on pathways related to cell survival and angiogenesis.
A vital role of mTORC1 is also the regulation of catabolic mechanisms such as autophagy. Depletion of oxygen or nutrients, or other stress mechanism leads to the formation of the autophagosome via different kinases, such as the ULK1 kinase. mTORC1 can phosphorylate ULK1, preventing its activation through the AMP-activated protein kinase (AMPK) (Kim, Kundu, Viollet, & Guan, 2011). Another important component of catabolic processes is the transcription factor TFEB, which is involved not only in regulating genes for lysosomal biogenesis but also in the autophagy machinery (Martina, Chen, Gucek, & Puertollano, 2012; Roczniak-Ferguson et al., 2012; Settembre et al., 2012).
3.2. Regulation and signaling pathways of mTORC2 in proliferation and survival
mTORC2 participates in pathways that are primarily related to proliferation and survival. Insulin/PI3K signaling is one of the main upstream effectors of mTORC2. The mSin1 subunit, which is specific to mTORC2, has a phosphoinositide-binding PH domain that plays a crucial role in regulating insulin-dependent control of mTORC2. Briefly, in the absence of insulin, the PH domain inhibits the catalytic activity of insulin; this inhibition can be reversed by PI3K-generated phosphatidylinositol (3,4,5)-trisphosphate, (PIP3) at the plasma membrane (Liu et al., 2015). mTORC2 activity is also regulated by mTORC1 (Hsu et al., 1979; Yu et al., 1979) through a negative feedback loop between mTORC1 and insulin/PI3K signaling. This interaction between mTORC1 and mTORC2 complicates the pharmacological targeting of mTOR in disease.
The actin cytoskeleton regulator PKCα was one of the first kinases shown to be phosphorylated by mTORC2 (Jacinto et al., 2004; Sarbassov, Guertin, Ali, & Sabatini, 1979). Other PKC family members functioning as substrates for mTORC2 were subsequently shown to be involved in various aspects of cell migration and cytoskeletal organization. Direct phosphorylation of protein kinase Cζ (PKCζ) by the mTORC2 complex, for example, changes the kinase activity and stability of PKCζ and consequently its ability to influence actin cytoskeleton organization (Li & Gao, 2014) Phosphorylation of AKT, a key effector of insulin/PI3K signaling, is another critical action of mTORC2 (Sarbassov et al., 1979). The activation of ATK promotes proliferation and inhibits survival through several key substrates, including the aforementioned mTORC1 inhibitor TSC2 and the metabolic regulator GSK3β (Guertin et al., 2006; Jacinto et al., 2006).
3.3. Physiological roles of mTOR
Studies in mice have shown that mTORC1 plays a vital role in the response of the liver to diet. Under normal conditions, the liver activates alternative energy sources, such as ketones, when blood glucose levels drop to a critical level. Mice with TSC1 deletion in the kidney are unable to engage ketone metabolism during fasting, causing ectopic activation of mTORC1, which in turn inhibits the induction of autophagy (Efeyan et al., 2013; Sengupta, Peterson, Laplante, Oh, & Sabatini, 2010).
mTORC1 has also been shown to influence glucose homeostasis through the regulation of β-cell function and the subsequent effect on insulin production. Studies showing that mTORC1 hyperactivation results in insulin resistance initially raised hopes that mTORC1 inhibitors such as rapamycin might offer a novel treatment for type 2 diabetes. However, long-term rapamycin-dependent pharmacological inhibition of mTORC1 paradoxically also impairs glucose homeostasis and increases insulin resistance, possibly because chronic administration of Rapamycin can inhibit mTORC2 in some cell lines or tissues (Cunningham et al., 2007; Lamming et al., 2012).
Investigations on the biological properties of Rapamycin also revealed a role of mTORC1 in regulating immune function, especially by promoting the switch towards anabolic metabolism, which is a prerequisite for T-cell activation and expansion (Powell, Pollizzi, Heikamp, & Horton, 2012). Interestingly, the induction of T-cells is one of the mechanisms employed by cancers in immune evasion, underlining the potential of mTOR in cancer therapy. Consistent with involvement of the mTOR pathway in so many crucial pathways, mTORC1 is a downstream effector or a regulatory target for a wide variety of mutated oncogenic pathways, including the Ras/Raf/Mek/Erk (MAPK) and PI3K/AKT pathways, and known tumor suppressors such as TP53. Signaling related to mTORC2 has also been associated with tumor progression, especially due to its involvement in regulating pro-proliferative processes such as glycolysis and glucose uptake, which increase cell proliferation when upregulated in cancer (Guertin et al., 2009; Hietakangas & Cohen, 2008). Alterations of mTOR signaling in cancer extend to mutations in mTOR itself, which are found in various cancer subtypes. Although rapalogs, mTOR inhibitors approved for cancer treatment, showed initial promise, they have produced only moderate clinical responses overall. Rapalogs were effective in some cases that harbored mutations directly affecting the mTOR pathway. Their efficacy slowly declined, however, as additional mutations led to resistance and evasion via alternative pathways, and adverse effects, such as autophagy induced by the mTORC1 inhibition, promoted tumor progression (Iyer et al., 2012; Wagle et al., 2014a, 2014b).
4. Primary cilia overview
Primary cilia are conserved antenna-like sensors found in virtually all mammalian cell types. Although the presence of primary cilia has been known for more than one hundred years, these protrusions were dismissed as vestigial organelles for many decades. More recently, however, these antennas have attracted significant attention due to their involvement in key cellular homeostatic and developmental processes and their role in ciliopathies and cancer. They provide a unique microdomain characterized by an independent membrane distribution of receptors and signaling molecules. This location extends receptors far into the extracellular space, where they are exposed to widely diverse signals, including for chemical, mechanical, and static forces.
Primary cilia studies show that ciliogenesis is tightly linked with the cell cycle. Generally, primary cilia assemble when cells have exited the cell cycle, stopped proliferating, and entered a quiescent stage of differentiation and specialization while remaining ready to proliferate if necessary (Nachury, Seeley, & Jin, 2010). Quiescence requires significant metabolic changes from when the cell is actively participating in the cell cycle. The metabolic changes required for this switch are related to mTOR, especially the mTORC1 pathway.
mTORC1 activity is reduced in quiescent cells, and maintenance of a quiescent state directly depends on low activity of the protein complex. Investigation of mechanisms contributing to the downregulation of mTORC1 in quiescent cells is an established field (Leontieva, Demidenko, & Blagosklonny, 2014; Valcourt et al., 2012). Some studies suggest that primary cilia play an active role in restricting mTORC1 signaling activity (Singla & Reiter, 2006). The unique ability of primary cilia to sense extracellular signals and convert them into signaling by intracellular pathways, makes them appropriate sensors for signals that regulate mTOR activity. The link between primary cilia and mTOR activity is supported by reports that alterations in mTORC1 activity directly affect primary cilia formation and stability, and that, conversely, defects in primary cilia produce abnormal mTORC1 activation (Singla & Reiter, 2006). To understand how mTOR is involved in processes affecting cilia, we must first understand how these organelles are assembled and disassembled and which pathways regulate these processes.
As a result of cell division, each cell inherits two centrosomes, which are surrounded by pericentriolar material. Basal bodies develop from the centrosomes, move to the cell surface, and dock beneath the plasma membrane, where they serve as a nucleating hub for proteins involved in ciliogenesis (Prosser & Pelletier, 2020). Nine microtubule doublets emanate from the basal body, which functions as a microtubule organization center, causing the axoneme to elongate and establish the skeleton of the cilium (Sánchez & Dynlacht, 2016). As it elongates, the axoneme is simultaneously encased in a lipid bilayer which extends from the plasma membrane and creates a ciliary compartment. This area is completely separated from the cytoplasm and only connected by a specific boundary or transition zone, which regulates the influx and outflux from the ciliary shaft (Satir, Pedersen, & Christensen, 2010; Valente, Rosti, Gibbs, & Gleeson, 2014).
Assembly and disassembly of the primary cilium are subsequently regulated by a specific set of proteins that travel bi-directionally along the axoneme and are called intraflagellar transport (IFT) proteins. There are about 20 of these specialized transport proteins, subdivided into two sub-complexes A (IFT-A) and B (IFT-B), which can move ciliary building blocks - proteins and tubulins - across the transition zone (Lechtreck, 2015).
Membrane proteins use this intraflagellar transport system to transit into the ciliary compartment (Hsiao, Tuz, & Ferland, 2012; Lechtreck, 2015), but it is not clear if soluble proteins such as those involved in signal transduction are transported via the same system. Some evidence suggests that these proteins move in conjunction with membrane proteins (Garcia-Gonzalo & Reiter, 2012), but the possibility of simple diffusion through the transition zone has also been suggested (Breslow, Koslover, Seydel, Spakowitz, & Nachury, 2013; Kee et al., 2012). The numerous signaling proteins associated with the ciliary shaft have been studied extensively. The most widely known to play a critical role in primary cilia function are the Sonic Hedgehog (Shh), Wnt, and Notch and its receptors. Other important ciliary signaling proteins include various G-protein-coupled receptors (GPCR), platelet-derived growth factor (PDGF), and the mTORC1 pathway (Basten & Giles, 2013; Christensen, Clement, Satir, & Pedersen, 2012) (Fig. 2).
Fig. 2.
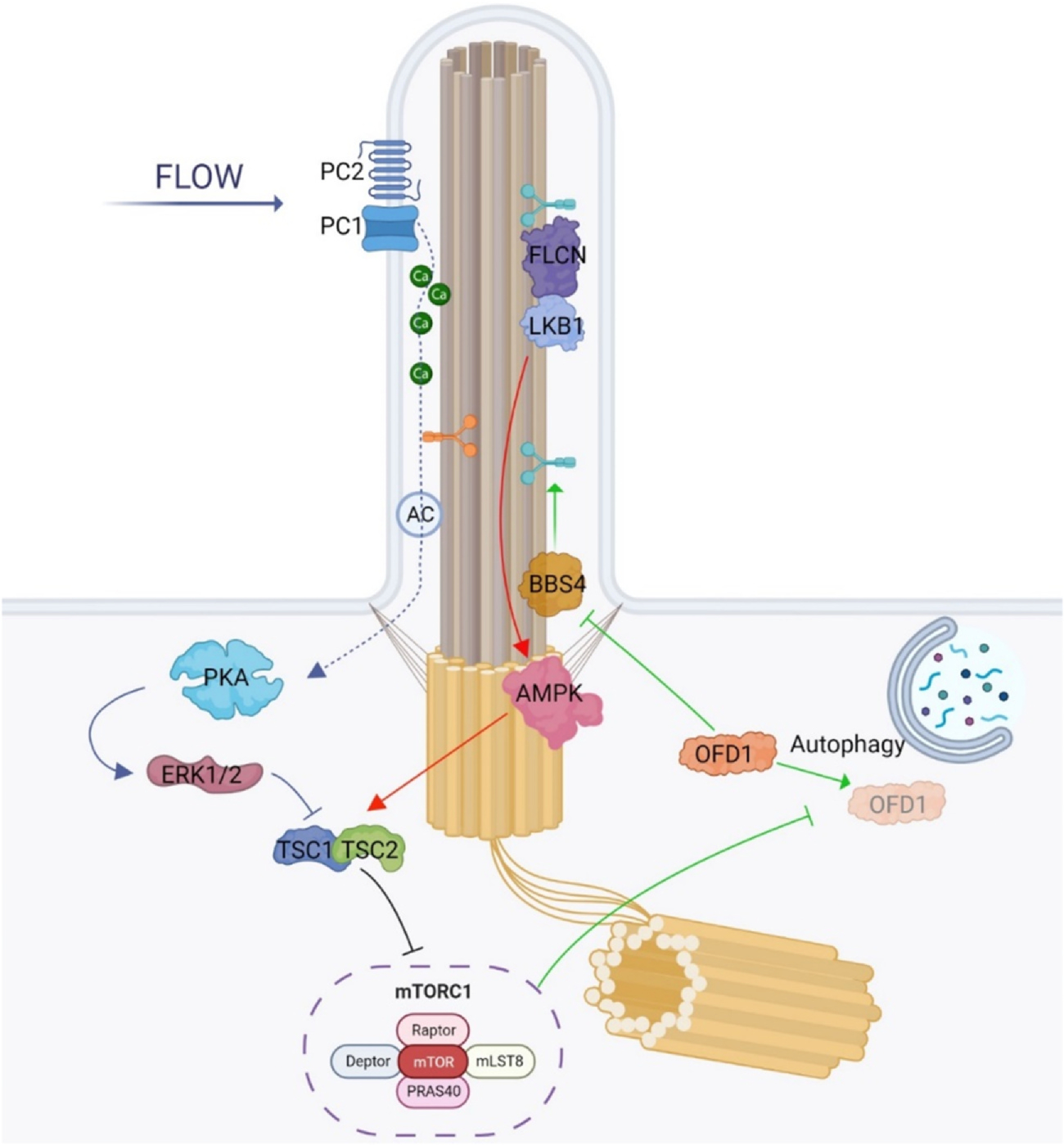
mTORC1 pathway in primary cilia: GREEN: Autophagy plays a role in regulating ciliogenesis. When there is a lack of nutrients and starvation conditions prevail, there is an increase in autophagy. This increase in autophagy promotes ciliogenesis by selectively degrading oral-facial-digital syndrome type 1 (OFD1), which is an inhibitor of BBSome formation. However, the autophagy-mediated degradation of OFD1 is negatively regulated by mechanistic target of Rapamycin complex 1 (mTORC1). The other two main mTOR/cilia related pathways rely on the flow stress-induced regulation of mechanistic target of Rapamycin complex 1 (mTORC1) as depicted in RED: Under flow stress conditions that cause primary cilia to deflect, mTORC1 activity reduces. This reduction is brought about by the accumulation of liver kinase B1 (LKB1) at the cilium through FLCN-mediated mechanisms. The accumulated LKB1 triggers the activation of AMP-activated protein kinase (AMPK) at the basal body, which subsequently phosphorylates TSC2, leading to the downregulation of mTORC1. BLUE: Polycystin-1 (PC-1) plays an important role in regulating mechanistic target of Rapamycin complex 1 (mTORC1). One of the main functions attributed to PC-1 is as a mechanosensor in primary cilia. When cilia are subjected to flow stress which results in bending, PC-1 is activated, which subsequently stimulates the ion channel PC-2, leading to calcium influx into the ciliary compartment. The ciliary calcium level increase in turn inhibits adenylyl cyclases (AC) and reduces the cyclic adenosine monophosphate (cAMP) level within the ciliary compartment. Conversely, when the flow stress is removed or there is dysfunction in PC-1, the calcium entry into the cilia is blocked, leading to a decrease in the ciliary calcium level. Thus, adenylyl cyclase activity increases, driving the ciliary cAMP level higher, which in turn activates PKA leading to the downregulation of mTORC1 through the ERK1/2 and TSC2.
5. Crosstalk between mTOR and Cilia
5.1. mTORC1 regulation by flow stress
Clear evidence of a link between primary cilia function and mTOR was provided through kidney epithelial cell studies, which showed that a deficiency in primary cilia function or structure was generally associated with noticeable alterations in cell size. Cells with defective cilia were abnormally enlarged, and this increase in size appeared to be mediated by mTORC1-dependent signaling (Boehlke et al., 2010). Kidney epithelial cells express primary cilia that extend into the lumen of the renal tubule and collecting duct and detect extracellular fluid flowing through the kidney filtering system. Under normal conditions, the cilia-dependent fluid sensation which is activated by cilia bending initiates downstream signaling pathways that restrict mTORC1 activity. In this setting, primary cilia can be regarded as a brake for mTOR signaling; as long as normal fluid flow is detected, cells remain differentiated, resulting in cell and tissue homeostasis. However, when the ability of the primary cilia to transduce these signals is impaired by defects in ciliary proteins or cilia formation, then the cell becomes unresponsive to flow stress and mTORC1 activity significantly increases. This may serve as an adaptive response to enable possible repair mechanisms. Detailed studies have revealed that the regulation of mTORC1 through the primary cilium requires the function of the Serine/threonine kinase 11 (STK11), here referred to as liver kinase B1 (LKB1). Although LKB1 actively shuttles between the nucleus and cytoplasm and has been shown to accumulate at the cell-cell junctions (Sebbagh, Santoni, Hall, Borg, & Schwartz, 2009), it has also been shown to concentrate in both primary cilia and the basal bodies (Maurin et al., 2015). Since ciliary levels of LKB1 are negligible in the absence of flow stress and only increased flow stress leads to LKB1 accumulation within the cilium, the association of LKB1 and primary cilia was not recognized immediately. When cells detect flow through the cilium, the accumulation of LKB1 activates one of the master regulators of cell metabolism, the 5′ AMP-activated protein kinase or AMPK (Boehlke et al., 2010). This enzyme plays a role in cellular energy homeostasis and through phosphorylation of TSC2 can keep mTORC1 signaling inhibited and unable to propagate (Zhong et al., 2016). One protein involved in flow induced accumulation of LKB1 within primary cilia is Folliculin (FLCN), a tumor suppressor with structural similarities to regulators of small GTPases (Nookala et al., 2012; Shen et al., 2019; Tsun et al., 2013) that also resides within the primary cilium (Luijten et al., 2013; Zhong et al., 2016). Tumors with mutations in FLCN demonstrate mTORC1 hyperactivity which is suggested to be one of the main drivers of disease pathogenesis (Baba et al., 2008; Hasumi et al., 2009). Although it is not entirely clear how FLCN controls the presence of LKB1 in cilia, mutations affecting its activity or causing its loss prevent the accumulation of LKB1 in the ciliary compartment, ultimately resulting in an inactive AMPK state and making mTORC1 unresponsive under flow conditions (Zhong et al., 2016).
5.2. mTORC1 and polycystin-1
Another link between primary cilia and mTOR was revealed by studies of one of the best-known ciliopathies. Autosomal dominant polycystic kidney disease (ADPKD) is among the most common, life-threatening inherited human kidney diseases and is characterized by the formation of small fluid-filled cysts in both kidneys. Cysts form and expand at the expense of normal renal tissue, which causes a progressive loss of renal function and ultimately kidney failure. Many studies have recognized that the mTORC1 pathway plays an important role in driving disease progression: mTORC1 levels are abnormal in renal cyst tissues from multiple sources, including animal models of PKD and patients with ADPKD (Shillingford et al., 2006; Weimbs, 2006). There are also numerous reports that administration of mTORC1 inhibitors mitigates disease progression and markedly reduces cyst formation and kidney size (Perico et al., 2010; Shillingford et al., 2006; Walz et al., 2010). Although the genes responsible for ADPKD were identified decades ago and the disease has been extensively studied, the precise mechanisms of pathogenesis have been elusive and treatment options consequently remain unsatisfactory. The genes responsible for the disease are PKD1 and PKD2, which encode two proteins, polycystin-1 (PC-1) and polycystin-2 (PC-2) (Streets, Prosseda, & Ong, 2020). PC1 is a large surface membrane receptor of 4302 amino acids, which partially explains the difficulty in studying its function biochemically (Harris, Germino, Klinger, Landes, & van Adelsberg, 1995; Wilson, 2001).
The relatively large N-terminal extracellular domain is suggested to have sensor-like properties, tuned to respond to extracellular stimuli such as force by flow or mechanical bending (Forman, Qamar, Paci, Sandford, & Clarke, 2005). These signals are then transmitted via the C-terminal cytoplasmic tail of the protein and transduced by its interactions with diverse proteins. In addition to the nonselective cation channel PC-2 (also known as TRPP2) (Qian et al., 1997; Tsiokas, Kim, Arnould, Sukhatme, & Walz, 1997), the most important C-terminal interacting proteins include members of the β-catenin family (Lal et al., 2008) and TSC2, which is known for its ability to block mTOR target phosphorylation (Dere, Wilson, Sandford, & Walker, 2010).
PC-1 is mainly found in primary cilia and cell adhesion structures. The localization in primary cilia is essential for normal kidney epithelial cell function, as mutations affecting this positioning worsen disease progression (Cai et al., 2014). The localization of PC2 to cilia is also important for PC1-PC2 complex function within the primary cilium (Geng et al., 2006; Yoder, Hou, & Guay-Woodford, 2002), which is required for upholding the signaling pathways involved in cell homeostasis and suppression of renal cystogenesis. Although both proteins are directly or indirectly associated to some extent with the mTOR pathway, one of the most promising targets in mTOR regulation appears to be a PC2/calcium-dependent pathway. In general, PC2 acts primarily as an ion/calcium channel within renal cilia. Mechanical bending due to extracellular flow initiates ciliary calcium influx which produces a cascade of events leading to a further rise in cytoplasmic calcium levels (Praetorius & Spring, 2001). Because the ability of PC2 to regulate intracellular calcium levels appears to be intrinsically dependent on its interaction with PC1, PC1 and PC2 can be seen as a complex where mechanosensory impulses are conveyed to PC2 via PC1 (Ibraghimov-Beskrovnaya & Natoli, 2011). Abundant evidence has confirmed that this increase in intracellular calcium levels subsequently reduces intracellular cyclic adenosine monophosphate (cAMP) (Chabardès, Imbert-Teboul, & Elalouf, 1999; Cooper, Yoshimura, Zhang, Chiono, & Mahey, 1994). Although some studies showed that cAMP inhibits mTORC1 via the activation of PKA, the actual regulation appears to be more complex; cAMP can either inhibit or stimulate the formation of mTOR complexes depending on the cell type studied (Distefano et al., 2009; Xie et al., 2011).
PC1 is indirectly involved in abnormally activating mTORC1 through its effect on calcium regulation of PC2, but the model is subject to some caveats. For example, there are studies that point out that ciliary calcium changes do not seem to affect cytoplasmic calcium levels to the degree necessary to induce the required signaling cascade (Delling et al., 2016).
PC1 has been suggested to act in an alternative way in conjunction with the transcription factor STAT6 and its coactivator P100, both also found within the primary cilium (Low et al., 2006). In renal epithelial cells, the C-terminal cytoplasmic tail of PC-1 undergoes proteolytic cleavage when no flow bends the primary cilium. The cleaved protein translocates into the nucleus in conjunction with STAT6 and P100 and stimulates expression of the cytokine receptor Interleukin 13 (IL13), which subsequently produces PI3K- and AKT-dependent mTORC1 activation (Olsan et al., 2011).
Interestingly, the C-terminal region of PC1 directly interacts with TSC2 (Dere et al., 2010), anchoring it at the plasma membrane where it is found as a complex with TSC1. Here it can interact with and inhibit the membrane-bound Ras homolog Rheb, a protein involved in the mTOR pathway and cell cycle regulation. Release of TSC2 from the plasma membrane is enabled by its phosphorylation through AKT, which consequently inhibits its attenuation of the small GTPase. However, convincing evidence that this specific effect and the involvement of PC-1 or PC-2 need the primary cilium or mechanosensation to influence mTORC1 is lacking, and some of these mechanisms appear to be cilia-independent. Suggestive evidence was provided in a recent study showing that flow-dependent mTORC1 regulation relies on the mechanosensory function of PC-1, but not PC-2, suggesting a calcium- but not cilia-independent pathway, and that the ability of PC-1 to transmit a mechanical signal to mTORC1 involves LKB1 and its target AMPK (Viau et al., 2018, 2020).
5.3. Cilia length regulation by mTORC1 pathway
Interestingly, although we discussed the possible role of cilia in regulating mTORC1 pathways, it is likely that there are multiple feedback mechanisms and that, conversely, the mTOR pathway is also able to influence primary cilia function. This notion is supported by studies of mouse embryonic fibroblasts (MEFs) showing that an impairment of TSC1 reduces levels of PC1 in the cilium. This reduction can be restored by treatment with Rapamycin, suggesting that mTORC1 is directly involved in controlling the expression of PC-1 (Lai & Jiang, 2020).
mTORC1 activity is also linked directly to ciliogenesis, and there is evidence that mTORC1 regulates changes in both cilium length and overall ciliation frequency: increased mTORC1 activity has been reported to be associated with an increase in ciliary length (Jin, Liu, & Zhong, 2015; Takahashi, Nagai, Chiba, Nakayama, & Mizuno, 2018), and, conversely, mTORC1 inhibition via rapamycin has been found to reduce ciliary length (Rosengren, Larsen, Pedersen, Christensen, & Møller, 2018), although rapamycin has also been observed to increase ciliary length in a dose-dependent manner (Sherpa, Atkinson, Ferreira, & Nauli, 2016). The ability of mTORC1 to influence cilium length regulation has been suggested to be related to its ability to regulate protein synthesis (Yuan et al., 2012), but how this stability may be regulated is unknown. It is, then, evident, that present models include discrepancies, possibly because different cells have been studied. Nevertheless, although cell-to-cell variations may prevent a unifying view of the pathway’s function, the involvement of mTORC1 in controlling protein synthesis in metabolic pathways suggests a definite contribution to direct or indirect regulation of primary cilia assembly and stability.
5.4. Regulation of ciliogenesis by mTORC1 pathway
We have seen that ciliogenesis is tightly coupled with the cell cycle. In cell culture, ciliation occurs when cells are forced to exit the cell cycle by nutrient or growth factor starvation or contact inhibition in fully confluent conditions. These observations affirm that cells are constantly sensing their extracellular environment for external growth factor availability, and that mTORC1-dependent autophagy plays a key role in this activity. In general, autophagy is always active at a low basal level where intracellular components are turning over and recycling, which allows the cell to function under optimal conditions. Nevertheless, as a fundamental cell survival mechanism, it is also highly adaptive and ready to respond to metabolic stress by becoming significantly more active when growth factor or nutrient signaling is diminished (Mizushima, 2007). Studies of Oral-facial-digital syndrome type I (OFD1) indicate that autophagy can specifically degrade multiple proteins which inhibit the initiation of ciliogenesis. Mutations in the gene for the OFD1 protein cause a disease with many of the typical features of ciliopathies (Ferrante et al., 2001). The OFD1 protein localizes to centrosomes and aids docking of the basal body to the plasma membrane. It also influences the ability of one of the core components of the IFT retrograde complex B, IFT88, to maintain the bidirectional motility of ciliary proteins along the axonemes (Singla, Romaguera-Ros, Garcia-Verdugo, & Reiter, 2010). Induction of autophagy via starvation produces a concomitant decrease of OFD1, which allows it to bind to BBS4, a member of the BBSome that plays a central role in primary cilia homeostasis and is involved in trafficking cargos to the primary cilium. Conversely, blocking autophagy leads to the accumulation of OFD1, preventing recruitment and function of BBS4 and leading to impaired cilia formation (Tang et al., 2013).
Under normal conditions, low-level basal autophagy seems to balance ciliogenesis, primarily by selective degradation of cilia and the Golgi complex resident IFT B protein IFT20, which prevents transport of building blocks from the Golgi apparatus to the ciliary shaft (Follit, Xu, Keady, & Pazour, 2009; Pampliega et al., 2013).
5.5. Primary cilia in autophagy and cancer
Since primary cilia present as antennas, they are placed advantageously to detect extracellular cues, including those which influence metabolic pathways. Consistent with this surveillance function, cells with defective primary cilia have significant alterations in autophagy activity. Many of the proteins involved in autophagy, such as Vps15, AMBRA-1, GABARAP, LC3, and several Atg protein members, are associated with either the primary cilium or basal bodies (Pampliega et al., 2013), suggesting that the primary cilium is an important hub, where multiple components of the autophagy machinery are connected. Removal of this hub by disrupted ciliogenesis, therefore, significantly impairs the ability to form autophagosomes upon serum deprivation (Pampliega et al., 2013).
Interestingly, increasing cilia-associated Sonic Hedgehog (Shh) signaling, but not inhibiting mTORC1 with Rapamycin, rescues autophagy under starvation condition (Pampliega et al., 2013), suggesting involvement of the Shh pathway in regulating autophagy. The report that Shh signaling is also associated with an increased expression of autophagy-related genes is consistent with this suggestion (Petralia et al., 2013).
Additional evidence that primary cilia represent a critical hub for signaling molecules that regulate autophagy includes studies of mechanotransduction showing that flow induces a reduction of cell size and increased autophagy in kidney epithelial cells, which is mediated by the LKB1–AMPK signaling axis (Orhon et al., 2016). Also supporting the notion is the finding in mouse embryonic fibroblasts that removing Retinitis pigmentosa GTPase regulator-interacting protein-1-like (RPGRIP1L) from its normal localization site at the ciliary transition zone significantly reduces autophagy by activating mTORC1 activation (Struchtrup, Wiegering, Stork, Rüther, & Gerhardt, 2018).
Available evidence also supports the idea that primary cilia play an essential role in mechanical stress mediated ERK/mTOR signaling and autophagy expression in chondrocytes (Xiang et al., 2019). Chondrocytes, a specialized cell type responsible for cartilage formation that produces the cartilage matrix and maintains the extracellular matrix (ECM), are very sensitive to mechanical stress. Recent studies reveal an interaction between primary cilia and autophagy that is involved in regulating mechanical stress-mediated cartilage development via the ERK/mTOR axis (Xiang et al., 2019). These studies found that application of cyclic tensile strain can influence primary cilia elongation and autophagy in an intensity-dependent manner.
Because mTOR signaling is often upregulated and contributes to increased cell growth and proliferation in cancer, mTOR inhibitors have been explored as potential targets for treating various forms of cancer. Clinical studies showed the effectiveness of mTOR inhibitors in treating certain types of cancers, such as renal cell carcinoma and certain forms of breast cancer. However, in other cases of malignancy, these inhibitors have proved to be only partially effective as a component of combination therapy. The complexity of tumor signaling, the potential that alternative signaling pathways will be activated in response to inhibition, and the relatively frequent side effects demonstrated in clinical trials (Zou, Tao, Li, & Zhu, 2020), show that a general mTOR inhibition of multiple diverse cell types is not a feasible treatment strategy, and that a more targeted approach is required. Primary cilia are associated with the cell cycle and disrupted primary cilia function has been implicated in the development of cancer (Streets et al., 2020), but the relationship between cilia and the mTOR pathway, which is also dysregulated in a variety of cancer types, is not clear (Zou et al., 2020).
Autophagy and ciliogenesis play crucial roles in maintaining the intracellular energy balance, and during conditions of serum deprivation, they occur simultaneously within the cell. This observation leads to the hypothesis that these processes are correlated and that they act synergistically to maintain the energy balance within the cell. Reports that primary cilia have a positive effect on autophagy regulation suggest that cells lacking primary cilia will exhibit reduced autophagic activity (Shim & Dixon, 2023; Wang, Deng, Chong, & Wang, 2018). This suggestion is supported by studies showing that renal cell carcinoma (RCC) cells with decreased ciliation frequency also demonstrate suppressed autophagy (Pampliega et al., 2013; Wang, Livingston, Su, & Dong, 2015b). However, autophagy in cilia-deficient cancer models presents a complex picture: cells differ in autophagic activity, and autophagy has both tumor-suppressing and tumor-promoting effects depending on the type of cancer cell examined and the stage of the disease when they are derived (Ko, Lee, & Park, 2019; White, 2015).
Research on pancreatic ductal adenocarcinoma (PDAC) cell lines revealed increased autophagic activity despite the absence of primary cilia, suggesting a cilia-independent mechanism for autophagy regulation in this case. Inhibiting autophagy in PDAC cell lines reduced growth and tumorigenesis, indicating potential anti-cancer effects (Yang et al., 2011). Similar findings were also observed in thyroid cancer cells (Lee et al., 2016). These observations point to the difficulty in elucidating a clear pathway accounting for the bidirectional interaction between primary cilia and autophagy.
Crosstalk between Hedgehog (Hh) and mTOR signaling has also been observed in several different types of tumor cells (Buonamici et al., 2010; Hao, Tian, Qin, Xie, & Yang, 2013; Kaylani et al., 2013; Yoo et al., 2011). Because primary cilia have an essential role in Hh signaling in mammals, the connection to mTOR suggests strongly that these cells have an additional connection to cilia. In cells that lack the TSC1 gene, this crosstalk seems to be mediated via the transforming growth factor beta (TGF-β) pathway (Larsen & Møller, 2020). However, a mutually positive relationship between Hh and mTORC1 signaling has also been suggested, in which increased activity in one pathway leads to increased activity in the other. Moreover, the mTORC1 complex and the PI3K/AKT/mTOR pathway can increase Hh signaling through GLI1/2/3 transcription factors which in turn activate mTORC1 by reducing negative regulators of mTOR through upregulation of NKX2.2. That GSK3 and AMPK, which are members of both pathways, can inhibit Hh signaling by phosphorylating GLI1 and inhibit mTORC1 activity by activating phosphorylation of TSC2 (Larsen & Møller, 2020) highlights the intricacy of this crosstalk and the likelihood that there are also complex interrelationships among additional pathways remaining to be elucidated.
6. Role of mTOR and cilia signaling in kidney disease
The widespread expression of mTORC1 and mTORC2 in the kidney makes it challenging to determine their roles in renal physiology. However, pharmacological inhibition and targeted genetic deletion have allowed researchers to analyze mTOR function in rodents and humans. While most of the current understanding of mTOR function relates to the podocyte or tubular epithelial cells, recent research suggests that renal endothelial and immune cells also play significant roles in regulating renal homeostasis and metabolism (Basso, Andrade-Oliveira, & Câmara, 2021). The current dogma associates dysregulation of mTOR signaling in the kidney with disruption of the normal functioning of renal cells and subsequently to the development of kidney diseases such as AKI, kidney fibrosis, glomerular disease, PKD, and renal cancer.
The most common type of kidney disease is chronic kidney disease (CKD), which affects more than 10% of the worldwide population (Kovesdy, 2022). The disease is characterized by excessive deposition of extracellular matrix in the interstitial space of the kidney and fibrosis. All resident renal cells, including fibroblasts, tubular epithelial cells, pericytes, and endothelial cells, contribute to these conditions. Interestingly, Rapamycin mitigates interstitial inflammation and fibrosis in various types of kidney disease, including ischemia-reperfusion injury (IRI), transplantation, and glomerulopathy (Bonegio et al., 2005; Wu et al., 2006). Other forms of CKD include Autosomal dominant polycystic kidney disease (ADPKD), which is caused by genetic mutations in either PKD1 or PKD2 and is one of the most prevalent human monogenic diseases. In ADPKD, mTOR signaling plays a critical role in cyst formation and enlargement. The cytoplasmic tail of PC1 interacts with tuberin to create a complex that acts as an endogenous inhibitor of Rheb, a constitutive activator of mTORC1. Mutations in PC1 also result in inappropriate activation of mTORC1 and increased cyst formation in both animal models and patients (Shillingford et al., 2006). PC1 control of mTORC1 activation relies on ERK-mediated TSC2 phosphorylation. When PC1 is absent, mTORC1 signaling is activated by ERK phosphorylation of tuberin at Ser664 (Distefano et al., 2009; Shillingford et al., 2006).
In mouse models of ADPKD, metformin reduces cyst growth by stimulating AMPK and inhibiting mTORC1 (Takiar et al., 2011). Direct phosphorylation by S6K1 of Afadin, a component of cell adhesion systems, has also recently been reported to alter cell division orientation and increase kidney cyst formation in TSC1-mutant mice (Bonucci et al., 2020). Rapamycin (Sirolimus) reduces cell proliferation in tubules with and without cysts, retards progression of cystogenesis, and protects kidney function in rats with ADPKD (Holditch et al., 2019; Li et al., 2017). Moreover, high doses of mTOR inhibitor can decrease progression of cystogenesis, halt cyst growth, and increase parenchymal volume in patients with ADPKD. These effects were also observed in other studies where Rapamycin treatment significantly reduced polycystic kidney size in transplant recipients with ADPKD (Shillingford et al., 2006). However, clinical trials for mTOR inhibitors in ADPKD have yielded mixed results: some showed no significant decrease in total kidney volume or progression of renal impairment compared to placebo groups (Braun, Schold, Stephany, Spirko, & Herts, 2014; Walz et al., 2010). These results suggest that despite promising results, the timing and dosage of mTOR inhibitors that will provide the most effect treatment for patients with ADPKD remain to be determined.
Beside cyst formation, scarring and fibrosis are major characteristics of kidney damage, and here the mTOR pathway is involved. Activation of mTOR signaling leads to kidney fibrosis through various routes. One route is via glomerular mesangial cells, where TGFβ1 induces mesangial cell hypertrophy and matrix expansion through the PI3K-AKT-mTORC1 axis (Dey, Ghosh-Choudhury, Kasinath, & Choudhury, 2012). In tubular epithelial cells, Rapamycin prevents the mTOR-promoted epithelial-to-mesenchymal transition. Studies in kidney fibroblasts have also shown that inhibition of mTOR with Rapamycin prevents TGFβ1-induced fibroblast activation, which has been used to stimulate mTORC signaling in a dose- and time-dependent manner (Lee et al., 2011; Li et al., 2015). In addition, the progression of fibrosis in cases of severe kidney injury has been associated with increased formation of the TOR-autophagy spatial coupling compartment along with secretion of profibrotic factors in tubular cells (Canaud et al., 2019); both processes act to downregulate mTORC1 activation (Narita et al., 1979).
mTOR also plays a role in acute kidney injury (AKI), which represents a major public health concern because of its high morbidity and mortality. A large body of evidence suggests that mTOR signaling contributes to maintaining renal tubular homeostasis and thus preventing tubular cell death following acute injury (Grahammer et al., 2014). This evidence includes investigations of the activation of the mTOR signaling pathway in tubular cells that show protection against AKI. Activating both mTORC1 and mTORC2 in kidney fibroblasts, for example, protects against tubular cell death and AKI (Gui et al., 2019). Consistent with these findings, in vivo studies in mice show that inhibition of mTORC1 not only prevents tubular cell regeneration and delays renal function recovery following AKI, but also enhances the immunosuppressive activity of regulatory T cells and myeloid-derived suppressor cells in ischemic kidneys (Zhang et al., 2017). However, other studies in rat have pointed out that Rapamycin administration significantly reduces ischemia-reperfusion injury (IRI), decreases inflammatory mediators in situ, and improves graft function after kidney transplantation (Cicora et al., 2012). Interestingly, a recent report of an association between shortening of primary cilia and enhanced sensitivity of epithelial cells to injury cues (Wang & Dong, 2013) also suggests a possible link to the mTOR pathway and the need for more studies of these interrelationships.
Podocytes are specialized cells responsible for maintaining the selective permeability of the glomerular filtration barrier and for preserving cellular morphology through mTOR (Gödel et al., 2011). Their loss can yield proteinuria, glomerular sclerosis, and progression of kidney dysfunction, ultimately leading to ESKD. Interestingly, deletion of mTOR-related genes selectively from podocytes also produced proteinuria (Cinà et al., 2012), progressive glomerulosclerosis, and eventually end-stage renal failure. This outcome underscores the indispensable role of mTORCs in maintaining glomerular homeostasis. Inappropriate activation of mTORC1 in glomerular diseases has been shown to be responsible for suppressing podocyte autophagy, damaging podocyte function, and exacerbating diabetic kidney disease (Zhao et al., 2018). However, treatment in animal models with mTOR inhibitors yielded conflicting results, ranging from remission to deterioration of kidney function (Clark, 2009; Liern, De Reyes, Fayad, & Vallejo, 2012), while in patients, Rapamycin treatment frequently causes podocyte apoptosis and proteinuria. Although a connection to the mTOR pathway is likely, the observation that expression of primary cilia is transient and that they gradually disappear in rat podocytes during glomerular development (Ichimura, Kurihara, & Sakai, 2010) poses a challenge to determining the involvement of mTOR and cilia in kidney diseases and to answering the many open questions about their function in podocytes.
One of the hallmarks of renal cell carcinoma (RCC) is loss of primary cilia, and several studies have investigated mTOR signaling inhibitors as a treatment. RCC, a rare cancer that originates in the renal cortex and is caused by mutations in the VHL gene that upregulate factors promoting tumor angiogenesis and proliferation. The activation of the PI3K–AKT–mTORC1 pathway and the positive feedback loop between VHL and mTOR in RCC suggest mTOR signaling as a promising therapeutic target (Clark, 2009; Pal & Quinn, 2013). Various mTOR signaling inhibitors have been tested as treatments, including mTOR kinase domain inhibitors (Zhang et al., 2013), mTOR/PI3K dual inhibitors (Roulin et al., 2011), PI3K-selective inhibitors, and programmed cell death 6, a proapoptotic protein (Rho et al., 2012). Results have been mediocre, however, and these agents do not currently represent a front-line treatment for RCC.
7. Role of mTOR and cilia in brain development and diseases
Primary cilia in the brain are well known to play significant roles in such processes as early patterning, neurogenesis, neuronal maturation and survival, and tumorigenesis (Alvarado et al., 2021). These roles are largely accomplished by the ability of cilia to regulate cell cycle progression and WNT and Hh signaling. Evidence for the importance of primary cilia in transducing Wnt signaling includes a recent study showing that a hyperactivating mutation in mTOR causes defective neuronal ciliogenesis, which leads to abnormal activation of canonical Wnt signaling and inactivation of non-canonical Wnt signaling. This abnormal signaling disrupts neuronal migration due to disturbed neuronal polarization (Park et al., 2018a). Recent evidence has revealed a mTOR-dependent role of primary cilia on radial glia cells in determining the size of the surface of their ventricular apical domain through regulation of the mTORC1 pathway. Radial glia cells are progenitor cells that bear a primary cilium and give rise to neurons and glia. Interestingly, mutant mice with defective cilia show dramatic increases in mTOR signaling during brain development, leading to enlarged apical domains of radial glia cells and subsequent dilatation of brain ventricles (Foerster et al., 2017). The study showed that primary cilia regulate ventricular morphogenesis by acting as a brake on the mTORC1 pathway, and that treatment with the mTORC1 inhibitor rapamycin rescues the observed phenotypes. This research may open new avenues for the diagnosis and treatment of hydrocephalus (Foerster et al., 2017).
A recent study has demonstrated that mitochondrial stress caused by respiratory complex inhibitors and excessive fission can promote ciliogenesis in various cell types, including neurons. This process is facilitated by the generation of reactive oxygen species (ROS), which activate AMPK and autophagy. The findings suggest that primary cilia play an essential role in promoting pro-survival responses during mitochondrial stress (Bae et al., 2019).
Other evidence of a link between primary cilia and mTOR in the brain derives from focal malformations of cortical developments (FMCDs), which are a group of brain disorders that include conditions such as hemimegalencephaly and focal cortical dysplasia (FCD). In brain tissues of FMCD patients, somatic activating mutations in the mTOR gene can block autophagy, the body’s natural process of removing cellular waste, leading to the accumulation of cellular components, such as OFD1, which can cause defective neuronal ciliogenesis (Park et al., 2018a). Because normal neuronal ciliogenesis is essential for proper brain development, defective ciliogenesis can contribute to the accumulation of damaged cellular components in brain tissues of FMCDs patients. This accumulation of cellular waste can decrease the quality of cellular components necessary for cilia formation, which can further disrupt neuronal ciliogenesis. Consequently, FMCD patients may present with abnormal cilia and subsequent alterations in the brain’s development that are responsible for the intractable epilepsy and autism-spectrum disorders (Lim & Crino, 2013; Park et al., 2018a; Wegiel et al., 2010) often associated with these conditions. While defective autophagy has been implicated in a range of neurodevelopmental and neurodegenerative disorders, further studies are needed to fully understand the role of primary cilia in the pathophysiology of these disorders.
8. mTOR and cilia in eye development and disease
Normal interaction between primary cilia and the mTOR pathway is essential for proper eye development and function. Here, we describe the (i) mechanisms of regulation of the mTOR pathway and primary cilia during normal eye development (ii) dysregulations of the mTOR pathway and primary cilia in the pathogenesis of ocular diseases and (iii) potential therapeutic interventions for eye diseases aimed at correcting primary cilia dysfunction and mTOR dysregulation.
The mTOR pathway is involved in the process of neuronal differentiation in the eye, and precise spatiotemporal regulation of its activity is central to the normal development of the retina and optic nerve (Avet-Rochex et al., 2014). Photoreceptor development in animal models constitutes one of the first areas to illuminate the mechanisms of control of neurogenesis by the mTOR pathway. For example, studies of mutants of upstream and downstream components of the mTOR pathway have demonstrated that activation of the pathway causes precocious differentiation of photoreceptors, whereas inhibition delays differentiation (Avet-Rochex et al., 2014; Bateman & McNeill, 2004). Several gene mutations in Drosophila affect the temporal control of photoreceptor differentiation through the mTOR pathway. For example, the loss of TSC1 causes precocious expression of neuronal markers in photoreceptors (Bateman & McNeill, 2004). Usually, TSC1 forms a complex with TSC2 upstream to negatively regulate the mTOR pathway. Conversely, loss-of-function clones of downstream elements S6k and Rheb demonstrate that inhibition of mTOR signaling delays, but does not block, differentiation (McNeill, Craig, & Bateman, 2008).
Other gene mutations in mouse models have emphasized the importance of the mTOR signaling pathway in photoreceptor development (Ma et al., 2015). Loss of either mTORC1 or mTORC2 impacts inner and outer segment structure through their accessory proteins Raptor and Rictor, respectively. Specifically, retina-specific knockout of Raptor or Rictor causes abnormally thickened cone inner segments and abnormally thinned outer segments, without affecting long-term cone survival or function. However, simultaneous loss of mTORC1 and mTORC2 reduces cone function without affecting cone viability (Ma et al., 2015).
Retinal ganglion cell (RGC) differentiation is temporally correlated with mTOR signaling in rodent and human retinas, suggesting a conserved relationship between the pathway and retinal development (Jones, Hägglund, & Carlsson, 2019; Teotia, Van Hook, Fischer, & Ahmad, 2019). Specifically, the differentiation of human induced pluripotent stem cells (iPSCs) into RGCs increases when the mTOR pathway is activated by lentivirus-mediated transduction of TSC2-shRNA and decreases when cells are exposed to rapamycin (Teotia et al., 2019). Recent studies suggest that balanced mTORC1 activity is essential for regulating the coordinated process of retinal progenitor cell (RPC) differentiation during mouse eye development (Fort et al., 2022; Jones et al., 2019). A relatively high level of protein synthesis in neural tissues facilitates the synthesis of cell cycle regulator proteins and the projection of RGC axons out of the eye to form the optic nerve. Additionally, mTORC1 may promote the degradation of proteins by activating protein turnover through a mechanism involving the immunoproteasome (Choi et al., 2018). The activity of mTORC1 in human iPSC-derived retinal organoids (RO) also closely resembles that found during human retinal development (Kim et al., 2002). For example, hyperactivation of mTORC1 during early RO development increases RGC proliferation and retinal thickness, whereas inhibition of mTORC1 decreases retinal thickness (Lee et al., 2022).
The mTOR signaling pathway has a crucial role in promoting proper neurodevelopment, and its dysregulation is involved in a range of neurodegenerative diseases of the retina and optic nerve, including glaucoma (Yao & van Wijngaarden, 2020). Several studies have revealed expression of mTOR components at relatively high levels in the ganglion cell layer and RGCs of rodent and human retinas (Losiewicz et al., 2020; Teotia et al., 2019). For example, immunoprecipitation studies have demonstrated that mTORC1 is primarily localized in RGCs and their axons, while mTORC2 is predominantly localized in RGCs (Losiewicz et al., 2020). The mTOR pathway has also been investigated in models of ocular hypertension; results have varied depending on the approach used to establish injury (Park, Kim, & Park, 2018b; Zhou, Chen, Yan, & Xia, 2021). In rats, chronically elevated intraocular pressure (IOP) induced by laser burns of the trabecular meshwork (TM) and episcleral veins, amplified the levels of mTOR and its downstream pathways S6K1 and 4E-BP1 in the retina, and upregulated the levels of Hypoxia inducible factor-1 (HIF-1α) and vascular endothelial growth factors (VEGF). These results suggest a possible role for mTOR in regulating ischemic hypoxia in glaucoma (Zhou et al., 2021). In contrast, elevated IOP induced by episcleral vein cauterization is associated with downregulation of the mTOR pathway in glaucomatous retinas (Park et al., 2018b).
Degeneration of RGC axons in the optic nerve is a hallmark of glaucoma and is almost inevitably followed by apoptotic RGC death. Interestingly, rapamycin does not significantly affect IOP in laser-induced hypertensive glaucoma models. However, treatment with rapamycin appears to promote RGC survival in laser-induced ocular hypertension models (Kitaoka et al., 2013; Su, Li, Jia, & Zhuo, 2019). Rapamycin is an enhancer of autophagy, a cellular pathway involved in protein and organelle degradation that has been shown to provide neuroprotection in rodent and primate chronic hypertensive glaucoma models (Deng et al., 2013; Koch & Lingor, 2016). However, the role of autophagy in glaucoma remains controversial as several studies have suggested that activation of autophagy contributes to axonal degeneration (Knöferle et al., 2010; Piras, Gianetto, Conte, Bosone, & Vercelli, 2011). For example, acute axonal degeneration following optic nerve crush is significantly delayed when autophagy is inhibited by 3-MA in the rat optic nerve (Knöferle et al., 2010).
Autophagy is activated by mechanical stretch acting on primary cilia in TM cells located in the anterior segment of the eye. For example, application of cyclic mechanical stretch has been shown to induce autophagy and activate AKT1 and SMAD2/3 as critical regulatory components of IOP homeostasis (Shim, Nettesheim, Dixon, & Liton, 2021). Although the mTOR pathway acts as a core sensor in autophagic regulation, the induction of autophagy in response to mechanical stretch appears to occur in an mTOR-independent manner. Thus, the role of mTOR and primary cilia in the pathogenesis of glaucoma appears to be complex (Prosseda et al., 2017).
9. Conclusion
Primary cilia clearly play a key role as sensory organs in regulating various pathways of the mTOR network, and much of the available evidence emphasizes their function as inhibitors of mTORC1 activity. Studies of the relationships between primary cilia and mTORC1 pathways have yielded many discrepancies and numerous questions about how these pathways are regulated remain unanswered. Current data points to the LKB1/AMPK-dependent pathways as one promising candidate to contribute to this regulation. This pathway was shown to directly transduce mechanical flow stimulation into mTORC1 activity; other studies provide evidence for regulation by this pathway based on correlations between PC-1 deficiency and defective mTORC1 activation. The exact mechanisms by which cilia bending signaling -mediated by PC-1- activates LKB1 are still under investigation, but this information may offer novel targets for treating ciliopathies and metabolic diseases. Recent research has provided new insights into mTOR function and regulation, and studies in mice have increased our understanding of how mTOR dysfunction contributes to disease. However, there is still much to learn about the importance of different signals that activate mTOR and the contexts in which they are meaningful. For example, it is unclear which inputs to the TSC complex are most significant and how the relative weight of these inputs varies depending on the physiological context. Similar questions exist for nutrient sensing by the Rag GTPases, including the purpose of sensing different types of amino acids and which tissues rely on certain nutrient sensors. To answer these questions, more in-depth biochemical studies of these complexes in vitro and better mouse models that allow for more nuanced perturbation and monitoring of these sensors in vivo are necessary.
Future mTOR research will focus on improving its therapeutic targeting in the clinic. Although current treatments have been successful in certain contexts, they are limited by the essential functions of mTOR in human tissues. Complete inhibition of mTOR causes severe dose-limiting toxicities, and because rapalogs lack tissue specificity, they disrupt mTORC2. Future research may focus on developing new mTOR-targeting therapeutics, such as mTORC1-specific inhibitors for use in diabetes and neurodegeneration, and to prolong lifespan, or tissue-specific mTORC1 agonists for use in muscle wasting diseases and immunotherapy. These approaches may involve the connection between mTOR and primary cilia, targeting tissue-specific receptors or signaling molecules upstream of mTOR, like the amino acid sensors Sestrin2 and CASTOR1, which specifically regulate mTORC1. Ultimately, these insights could enable rational targeting of mTOR signaling for better therapeutic outcomes.
Acknowledgements
This work was supported by R01-EY025295 (Y.S.), R01-EY032159 (Y.S.) VA CX001298 (Y.S.) VA CX 001481 (YS), R01-EY026885 (PBL), R01-EY-033600 (PBL), Children’s Health Research Institute Award (Y.S.), an unrestricted grant from Research to Prevent Blindness, New York, NY., Research for Prevention of Blindness Unrestricted grant (Stanford Ophthalmology); NIH NEI P30-EY026877 grant (Stanford Ophthalmology).
Footnotes
Competing interests
The authors declare no financial/non-financial competing interest.
References
- Alvarado JA, et al. (2021). Developmental distribution of primary cilia in the retinofugal visual pathway. Journal of Comparative Neurology, 529, 1442–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avet-Rochex A, et al. (2014). Unkempt is negatively regulated by mTOR and uncouples neuronal differentiation from growth control. PLoS Genetics, 10, e1004624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylett CHS, et al. (2016). Architecture of human mTOR complex 1. Science (1979), 351, 48–52. [DOI] [PubMed] [Google Scholar]
- Baba M, et al. (2008). Kidney-targeted Birt-Hogg-Dubé gene inactivation in a mouse model: Erk1/2 and Akt-mTOR activation, cell hyperproliferation, and polycystic kidneys. Journal of the National Cancer Institute, 100, 140–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae JE, et al. (2019). Primary cilia mediate mitochondrial stress responses to promote dopamine neuron survival in a Parkinson’s disease model. Cell Death & Disease, 10, 952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baretić D, Berndt A, Ohashi Y, Johnson CM, & Williams RL (2016). Tor forms a dimer through an N-terminal helical solenoid with a complex topology. Nature Communications, 7, 11016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Peled L, et al. (2013). A tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science (1979), 340, 1100–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso PJ, Andrade-Oliveira V, & Câmara NOS (2021). Targeting immune cell metabolism in kidney diseases. Nature Reviews Nephrology, 17, 465–480. 10.1038/s41581-021-00413-7. [DOI] [PubMed] [Google Scholar]
- Basten SG, & Giles RH (2013). Functional aspects of primary cilia in signaling, cell cycle and tumorigenesis. Cilia, 2, 6. 10.1186/2046-2530-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman JM, & McNeill H (2004). Temporal control of differentiation by the insulin receptor/tor pathway in Drosophila. Cell, 119, 87–96. [DOI] [PubMed] [Google Scholar]
- Boehlke C, et al. (2010). Primary cilia regulate mTORC1 activity and cell size through Lkb1. Nature Cell Biology, 12, 1115–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonegio RGB, et al. (2005). Rapamycin ameliorates proteinuria-associated tubulointerstitial inflammation and fibrosis in experimental membranous nephropathy. Journal of the American Society of Nephrology, 16, 2063–2072. [DOI] [PubMed] [Google Scholar]
- Bonucci M, et al. (2020). mTOR and S6K1 drive polycystic kidney by the control of Afadin-dependent oriented cell division. Nature Communications, 11, 3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun WE, Schold JD, Stephany BR, Spirko RA, & Herts BR (2014). Low-dose rapamycin (Sirolimus) effects in autosomal dominant polycystic kidney disease: An open-label randomized controlled pilot study. Clinical Journal of the American Society of Nephrology, 9, 881–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslow DK, Koslover EF, Seydel F, Spakowitz AJ, & Nachury MV (2013). An in vitro assay for entry into cilia reveals unique properties of the soluble diffusion barrier. Journal of Cell Biology, 203, 129–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EJ, et al. (1994). A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature, 369, 756–758. [DOI] [PubMed] [Google Scholar]
- Brugarolas J, et al. (2004). Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes & Development, 18, 2893–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buonamici S, et al. (2010). Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Science Translational Medicine, 2, 51ra70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, et al. (2014). Altered trafficking and stability of polycystins underlie polycystic kidney disease. Journal of Clinical Investigation, 124, 5129–5144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canaud G, et al. (2019). Cyclin G1 and TASCC regulate kidney epithelial cell G 2 -M arrest and fibrotic maladaptive repair. Science Translational Medicine, 11, eaav4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabardès D, Imbert-Teboul M, & Elalouf JM (1999). Functional properties of Ca2+-inhibitable type 5 and type 6 adenylyl cyclases and role of Ca2+ increase in the inhibition of intracellular cAMP content. Cellular Signalling, 11, 651–663. 10.1016/S0898-6568(99)00031-5. [DOI] [PubMed] [Google Scholar]
- Chantranupong L, et al. (2016). The CASTOR proteins are arginine sensors for the mTORC1 pathway. Cell, 165, 153–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JH, et al. (2018). MTORC1 accelerates retinal development via the immunoproteasome. Nature Communications, 9, 2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen ST, Clement CA, Satir P, & Pedersen LB (2012). Primary cilia and coordination of receptor tyrosine kinase (RTK) signalling. Journal of Pathology, 226, 172–184. 10.1002/path.3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung J, Kuo CJ, Crabtree GR, & Blenis J (1992). Rapamycin-FKBP specifically blocks growth-dependent activation of and signaling by the 70 kd S6 protein kinases. Cell, 69, 1227–1236. [DOI] [PubMed] [Google Scholar]
- Cicora F, et al. (2012). Preconditioning donor with a combination of tacrolimus and rapamycin to decrease ischaemia-reperfusion injury in a rat syngenic kidney transplantation model. Clinical and Experimental Immunology, 167, 169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinà DP, et al. (2012). Inhibition of MTOR disrupts autophagic flux in podocytes. Journal of the American Society of Nephrology, 23, 412–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark PE (2009). The role of VHL in clear-cell renal cell carcinoma and its relation to targeted therapy. Kidney International, 76, 939–945. 10.1038/ki.2009.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper DMF, Yoshimura M, Zhang Y, Chiono M, & Mahey R (1994). Capacitative Ca2+ entry regulates Ca2+-sensitive adenylyl cyclases. Biochemical Journal, 297, 437–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham JT, et al. (2007). mTOR controls mitochondrial oxidative function through a YY1-PGC-1α transcriptional complex. Nature, 450, 736–740. [DOI] [PubMed] [Google Scholar]
- Delling M, et al. (2016). Primary cilia are not calcium-responsive mechanosensors. Nature, 531, 656–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng S, et al. (2013). Autophagy in retinal ganglion cells in a rhesus monkey chronic hypertensive glaucoma model. PLoS One, 8, e77100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dere R, Wilson PD, Sandford RN, & Walker CL (2010). Carboxy terminal tail of polycystin-1 regulates localization of TSC2 to repress mTOR. PLoS One, 5, e9239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey N, Ghosh-Choudhury N, Kasinath BS, & Choudhury GG (2012). TGFβ-stimulated microRNA-21 utilizes PTEN to orchestrate AKT/mTORC1 signaling for mesangial cell hypertrophy and matrix expansion. PLoS One, 7, e42316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibble CC, et al. (2012). TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Molecular Cell, 47, 535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Distefano G, et al. (2009). Polycystin-1 regulates extracellular signal-regulated kinase-dependent phosphorylation of tuberin to control cell size through mTOR and its downstream effectors S6K and 4EBP1. Molecular and Cellular Biology, 29, 2359–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Düvel K, et al. (2010). Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Molecular Cell, 39, 171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efeyan A, et al. (2013). Regulation of mTORC1 by the Rag GTPases is necessary for neonatal autophagy and survival. Nature, 493, 679–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng CP, Sehgal SN, & Vézina C (1984). Activity of rapamycin (ay-22,989) against transplanted tumors. The Journal of Antibiotics, 37, 1231–1237. [DOI] [PubMed] [Google Scholar]
- Feng Z, et al. (2007). The regulation of AMPK β1, TSC2, and PTEN expression by p53: Stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Research, 67, 3043–3053. [DOI] [PubMed] [Google Scholar]
- Ferrante MI, et al. (2001). Identification of the gene for oral-facial-digital type 1 syndrome. American Journal of Human Genetics, 68, 569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foerster P, et al. (2017). MTORC1 signaling and primary cilia are required for brain ventricle morphogenesis. Development (Cambridge), 144, 201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follit JA, Xu F, Keady BT, & Pazour GJ (2009). Characterization of mouse IFT complex B. Cell Motility and the Cytoskeleton, 66, 457–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman JR, Qamar S, Paci E, Sandford RN, & Clarke J (2005). The remarkable mechanical strength of polycystin-1 supports a direct role in mechanotransduction. Journal of Molecular Biology, 349, 861–871. [DOI] [PubMed] [Google Scholar]
- Fort PE, et al. (2022). mTORC1 regulates high levels of protein synthesis in retinal ganglion cells of adult mice. Journal of Biological Chemistry, 298, 101944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frias MA, et al. (2006). mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Current Biology, 16, 1865–1870. [DOI] [PubMed] [Google Scholar]
- Garcia-Gonzalo FR, & Reiter JF (2012). Scoring a backstage pass: Mechanisms of ciliogenesis and ciliary access. Journal of Cell Biology, 197, 697–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng L, et al. (2006). Polycystin-2 traffics to cilia independently of polycystin-1 by using an N-terminal RVxP motif. Journal of Cell Science, 119, 1383–1395. [DOI] [PubMed] [Google Scholar]
- Gingras AC, et al. (1999). Regulation of 4E-BP1 phosphorylation: A novel two step mechanism. Genes & Development, 13, 1422–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gödel M, et al. (2011). Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. Journal of Clinical Investigation, 121, 2197–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grahammer F, et al. (2014). MTORC1 maintains renal tubular homeostasis and is essential in response to ischemic stress. Proceedings of the National Academy of Sciences of the United States of America, 111, E2817–E2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin DA, et al. (2006). Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCα, but not S6K1. Developmental Cell, 11, 859–871. [DOI] [PubMed] [Google Scholar]
- Guertin DA, et al. (2009). mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell, 15, 148–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui Y, et al. (2019). Fibroblast mTOR/PPARγ/HGF axis protects against tubular cell death and acute kidney injury. Cell Death and Differentiation, 26, 2774–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwinn DM, et al. (2008). AMPK phosphorylation of raptor mediates a metabolic checkpoint. Molecular Cell, 30, 214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao K, Tian XD, Qin CF, Xie XH, & Yang YM (2013). Hedgehog signaling pathway regulates human pancreatic cancer cell proliferation and metastasis. Oncology Reports, 29, 1124–1132. [DOI] [PubMed] [Google Scholar]
- Hara K, et al. (2002). Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell, 110, 177–189. [DOI] [PubMed] [Google Scholar]
- Harris PC, Germino G, Klinger K, Landes G, & van Adelsberg J (1995). The PKD1 gene product. Nature Medicine, 49, 1, 493. 10.1038/nm0695-493a. [DOI] [PubMed] [Google Scholar]
- Hasumi Y, et al. (2009). Homozygous loss of BHD causes early embryonic lethality and kidney tumor development with activation of mTORC1 and mTORC2. Proceedings of the National Academy of Sciences of the United States of America, 106, 18722–18727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hietakangas V, & Cohen SM (2008). TOR complex 2 is needed for cell cycle progression and anchorage-independent growth of MCF7 and PC3 tumor cells. BMC Cancer, 8, 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holditch SJ, et al. (2019). A study of sirolimus and mtor kinase inhibitor in a hypomorphic pkd1 mouse model of autosomal dominant polycystic kidney disease. American Journal of Physiology. Renal Physiology, 317, F187–F196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holz MK, Ballif BA, Gygi SP, & Blenis J (2021). mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell, 184, 2255. 10.1016/j.cell.2021.03.060. [DOI] [PubMed] [Google Scholar]
- Hsiao YC, Tuz K, & Ferland RJ (2012). Trafficking in and to the primary cilium. Cilia, 1, 4. 10.1186/2046-2530-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh AC, et al. (2012). The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature, 485, 55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PP, et al. (2011). The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science (New York, N. Y.) (1979), 332, 1317–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibraghimov-Beskrovnaya O, & Natoli TA (2011). MTOR signaling in polycystic kidney disease. Trends in Molecular Medicine, 17, 625–633. 10.1016/j.molmed.2011.06.003. [DOI] [PubMed] [Google Scholar]
- Ichimura K, Kurihara H, & Sakai T (2010). Primary cilia disappear in rat podocytes during glomerular development. Cell and Tissue Research, 341, 197–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Li Y, Xu T, & Guan KL (2003). Rheb GTpase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes & Development, 17, 1829–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer G, et al. (2012). Genome sequencing identifies a basis for everolimus sensitivity. Science (New York, N. Y.), 338, 221. 10.1126/science.1226344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacinto E, et al. (2004). Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nature Cell Biology, 6, 1122–1128. [DOI] [PubMed] [Google Scholar]
- Jacinto E, et al. (2006). SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell, 127, 125–137. [DOI] [PubMed] [Google Scholar]
- Jewell JL, et al. (2015). Differential regulation of mTORC1 by leucine and glutamine. Science (New York, N. Y.) (1979), 347, 194–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin D, Liu P, & Zhong TP (2015). Prostaglandin signaling in ciliogenesis during development. Cell Cycle (Georgetown, Tex.), 14, 2632–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones I, Hägglund AC, & Carlsson L (2019). Reduced mTORC1-signalling in retinal progenitor cells leads to visual pathway dysfunction. Biology Open, 8, bio044370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J, Genau HM, & Behrends C (2015). Amino acid-dependent mTORC1 regulation by the lysosomal membrane protein SLC38A9. Molecular and Cellular Biology, 35, 2479–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaylani SZ, et al. (2013). Rapamycin targeting mTOR and hedgehog signaling pathways blocks human rhabdomyosarcoma growth in xenograft murine model. Biochemical and Biophysical Research Communications, 435, 557–561. [DOI] [PubMed] [Google Scholar]
- Kee HL, et al. (2012). A size-exclusion permeability barrier and nucleoporins characterize a ciliary pore complex that regulates transport into cilia. Nature Cell Biology, 14, 431–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, et al. (2003). GβL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Molecular Cell, 11, 895–904. [DOI] [PubMed] [Google Scholar]
- Kim DH, et al. (2002). mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell, 110, 163–175. [DOI] [PubMed] [Google Scholar]
- Kim E, Goraksha-Hicks P, Li L, Neufeld TP, & Guan KL (2008). Regulation of TORC1 by Rag GTPases in nutrient response. Nature Cell Biology, 10, 935–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, & Guan KL (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature Cell Biology, 13, 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitaoka Y, et al. (2013). Axonal protection by Nmnat3 overexpression with involvement of autophagy in optic nerve degeneration. Cell Death & Disease, 4, e860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knöferle J, et al. (2010). Mechanisms of acute axonal degeneration in the optic nerve in vivo. Proceedings of the National Academy of Sciences of the United States of America, 107, 6064–6069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko JY, Lee EJ, & Park JH (2019). Interplay between primary cilia and autophagy and its controversial roles in cancer. Biomolecules and Therapeutics, 27, 337–341. 10.4062/biomolther.2019.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch JC, & Lingor P (2016). The role of autophagy in axonal degeneration of the optic nerve. Experimental Eye Research, 144, 81–89. 10.1016/j.exer.2015.08.016. [DOI] [PubMed] [Google Scholar]
- Kovesdy CP (2022). Epidemiology of chronic kidney disease: An update 2022. Kidney International Supplements, 12, 7–11. 10.1016/j.kisu.2021.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y, & Jiang Y (2020). Reciprocal regulation between primary cilia and mTORC1. Genes (Basel), 11, 711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal M, et al. (2008). Polycystin-1 C-terminal tail associates with β-catenin and inhibits canonical Wnt signaling. Human Molecular Genetics, 17, 3105–3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamming DW, et al. (2012). Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science (New York, N. Y.), 335, 1638–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen LJ, & Møller LB (2020). Crosstalk of Hedgehog and mTORC1 pathways. Cells, 9, 2316. 10.3390/cells9102316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechtreck KF (2015). IFT-cargo interactions and protein transport in cilia. Trends in Biochemical Sciences, 40, 765–778. 10.1016/j.tibs.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, et al. (2016). Defective ciliogenesis in thyroid hürthle cell tumors is associated with increased autophagy. Oncotarget, 7, 79117–79130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, et al. (2011). Albumin-induced epithelial-mesenchymal transition and ER stress are regulated through a common ROS-c-Src kinase-mTOR pathway: Effect of imatinib mesylate. American Journal of, 300, F1214–F1222. [DOI] [PubMed] [Google Scholar]
- Lee SH, et al. (2022). Role of mTORC1 activity during early retinal development and lamination in human-induced pluripotent stem cell-derived retinal organoids. Cell Death Discovery, 8, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leontieva OV, Demidenko ZN, & Blagosklonny MV (2014). Contact inhibition and high cell density deactivate the mammalian target of rapamycin pathway, thus suppressing the senescence program. Proceedings of the National Academy of Sciences of the United States of America, 111, 8832–8837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, et al. (2017). Rapamycin treatment dose-dependently improves the cystic kidney in a new ADPKD mouse model via the mTORC1 and cell-cycle-associated CDK1/cyclin axis. Journal of Cellular and Molecular Medicine, 21, 1619–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, et al. (2015). Rictor/mTORC2 signaling mediates TGFβ1-induced fibroblast activation and kidney fibrosis. Kidney International, 88, 515–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, & Gao T (2014). MTORC2 phosphorylates protein kinase Cζ to regulate its stability and activity. EMBO Reports, 15, 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liern M, De Reyes V, Fayad A, & Vallejo G (2012). Use of sirolimus in patients with primary steroid-resistant nephrotic syndrome. Nefrologia: Publicacion Oficial de la Sociedad Espanola Nefrologia, 32, 321–328. [DOI] [PubMed] [Google Scholar]
- Lim KC, & Crino PB (2013). Focal malformations of cortical development: New vistas for molecular pathogenesis. Neuroscience, 252, 262–276. 10.1016/j.neuroscience.2013.07.037. [DOI] [PubMed] [Google Scholar]
- Liu P, et al. (2015). Ptdins(3,4,5) P3-dependent activation of the mTORC2 kinase complex. Cancer Discovery, 5, 1194–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewith R, & Hall MN (2011). Target of rapamycin (TOR) in nutrient signaling and growth control. Genetics, 189, 1177–1201. 10.1534/genetics.111.133363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long X, Lin Y, Ortiz-Vega S, Yonezawa K, & Avruch J (2005). Rheb binds and regulates the mTOR kinase. Current Biology, 15, 702–713. [DOI] [PubMed] [Google Scholar]
- Losiewicz MK, et al. (2020). mTORC1 and mTORC2 expression in inner retinal neurons and glial cells. Experimental Eye Research, 197, 108131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low SH, et al. (2006). Polycystin-1, STAT6, and P100 function in a pathway that transduces ciliary mechanosensation and is activated in polycystic kidney disease. Developmental Cell, 10, 57–69. [DOI] [PubMed] [Google Scholar]
- Luijten MNH, et al. (2013). Birt-hogg-dubé syndrome is a novel ciliopathy. Human Molecular Genetics, 22, 4383–4397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S, et al. (2015). Loss of mTOR signaling affects cone function, cone structure and expression of cone specific proteins without affecting cone survival. Experimental Eye Research, 135, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martel RR, Klicius J, & Galet S (1977). Inhibition of the immune response by rapamycin, a new antifungal antibiotic. Canadian Journal of Physiology and Pharmacology, 55, 48–51. [DOI] [PubMed] [Google Scholar]
- Martina JA, Chen Y, Gucek M, & Puertollano R (2012). MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy, 8, 903–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurin P, et al. (2015). New LKB1 function in the primary cilium. Cilia, 4, P71. 10.1186/2046-2530-4-S1-P71. [DOI] [Google Scholar]
- McNeill H, Craig GM, & Bateman JM (2008). Regulation of neurogenesis and epidermal growth factor receptor signaling by the insulin receptor/target of rapamycin pathway in drosophila. Genetics, 179, 843–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N (2007). Autophagy: Process and function. Genes and Development, 21, 2861–2873. 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- Nachury M. v, Seeley ES, & Jin H. (2010). Trafficking to the ciliary membrane: How to get across the periciliary diffusion barrier. Annual Review of Cell and Developmental Biology, 26, 59–87. 10.1146/annurev.cellbio.042308.113337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita M, et al. (2011). Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science (New York, N. Y.) (1979), 332, 966–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nojima H, et al. (2003). The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. Journal of Biological Chemistry, 278, 15461–15464. [DOI] [PubMed] [Google Scholar]
- Nookala RK, et al. (2012). Crystal structure of folliculin reveals a hidDENN function in genetically inherited renal cancer. Open Biology, 2, 120071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsan EE, et al. (2011). Signal transducer and activator of transcription-6 (STAT6) inhibition suppresses renal cyst growth in polycystic kidney disease. Proceedings of the National Academy of Sciences of the United States of America, 108, 18067–18072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orhon I, et al. (2016). Primary-cilium-dependent autophagy controls epithelial cell volume in response to fluid flow. Nature Cell Biology, 18, 657–667. [DOI] [PubMed] [Google Scholar]
- Pal SK, & Quinn DI (2013). Differentiating mTOR inhibitors in renal cell carcinoma. Cancer Treatment Reviews, 39, 709–719. 10.1016/j.ctrv.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pampliega O, et al. (2013). Functional interaction between autophagy and ciliogenesis. Nature, 502, 194–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HYL, Kim JH, & Park CK (2018b). Different contributions of autophagy to retinal ganglion cell death in the diabetic and glaucomatous retinas. Scientific Reports, 8, 13321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SM, et al. (2018a). Brain somatic mutations in MTOR disrupt neuronal ciliogenesis, leading to focal cortical dyslamination. Neuron, 99, 83–97. [DOI] [PubMed] [Google Scholar]
- Pearce LR, et al. (2007). Identification of protor as a novel Rictor-binding component of mTOR complex-2. Biochemical Journal, 405, 513–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perico N, et al. (2010). Sirolimus therapy to halt the progression of ADPKD. Journal of the American Society of Nephrology, 21, 1031–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson TR, et al. (2009). DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell, 137, 873–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petralia RS, et al. (2013). Sonic hedgehog promotes autophagy in hippocampal neurons. Biology Open, 2, 499–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Activation of autophagy in a rat model of retinal ischemia following high intraocular pressure Piras A, Gianetto D, Conte D, Bosone A, & Vercelli A. (2011). Activation of autophagy in a rat model of retinal ischemia following high intraocular pressure. PLoS One, 6, e22514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porstmann T, et al. (2008). SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metabolism, 8, 224–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell JD, Pollizzi KN, Heikamp EB, & Horton MR (2012). Regulation of immune responses by mTOR. Annual Review of Immunology, 30, 39–68. 10.1146/annurev-immunol-020711-075024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praetorius HA, & Spring KR (2001). Bending the MDCK cell primary cilium increases intracellular calcium. Journal of Membrane Biology, 184, 71–79. [DOI] [PubMed] [Google Scholar]
- Prosseda PP, et al. (2017). Loss of OCRL increases ciliary PI(4,5)P2 in Lowe oculocerebrorenal syndrome. Journal of Cell Science, 130, 3447–3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser SL, & Pelletier L (2020). Centriolar satellite biogenesis and function in vertebrate cells. Journal of Cell Science, 133, jcs239566. 10.1242/jcs.239566. [DOI] [PubMed] [Google Scholar]
- Qian F, et al. (1997). PKD1 interacts with PKD2 through a probable coiled-coil domain. Nature Genetics, 16, 179–183. [DOI] [PubMed] [Google Scholar]
- Rebsamen M, et al. (2015). SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature, 519, 477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rho SB, et al. (2012). Programmed cell death 6 (PDCD6) inhibits angiogenesis through PI3K/mTOR/p70S6K pathway by interacting of VEGFR-2. Cellular Signalling, 24, 131–139. [DOI] [PubMed] [Google Scholar]
- Roczniak-Ferguson A, et al. (2012). The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Science Signaling, 5, ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosengren T, Larsen LJ, Pedersen LB, Christensen ST, & Møller LB (2018). TSC1 and TSC2 regulate cilia length and canonical Hedgehog signaling via different mechanisms. Cellular and Molecular Life Sciences, 75, 2663–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roulin D, et al. (2011). Targeting renal cell carcinoma with NVP-BEZ235, a dual PI3K/mTOR inhibitor, in combination with sorafenib. Molecular Cancer, 10, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P, & Snyder SH (1994). RAFT1: A mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell, 78, 35–43. [DOI] [PubMed] [Google Scholar]
- Sabers CJ, et al. (1995). Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. Journal of Biological Chemistry, 270, 815–822. [DOI] [PubMed] [Google Scholar]
- Sancak Y, et al. (2007). PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Molecular Cell, 25, 903–915. [DOI] [PubMed] [Google Scholar]
- Sancak Y, et al. (2008). The rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science (New York, N. Y.) (1979), 320, 1496–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez I, & Dynlacht BD (2016). Cilium assembly and disassembly. Nature Cell Biology, 18, 711–717. 10.1038/ncb3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, & Sabatini DM (2005). Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science (New York, N. Y.) (1979), 307, 1098–1101. [DOI] [PubMed] [Google Scholar]
- Satir P, Pedersen LB, & Christensen ST (2010). The primary cilium at a glance. Journal of Cell Science, 123, 499–503. 10.1242/jcs.050377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schalm SS, Fingar DC, Sabatini DM, & Blenis J (2003). TOS motif-mediated raptor binding regulates 4E-BP1 multisite phosphorylation and function. Current Biology, 13, 797–806. [DOI] [PubMed] [Google Scholar]
- Sebbagh M, Santoni MJ, Hall B, Borg JP, & Schwartz MA (2009). Regulation of LKB1/STRAD localization and function by E-cadherin. Current Biology, 19, 37–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta S, Peterson TR, Laplante M, Oh S, & Sabatini DM (2010). MTORC1controls fasting-induced ketogenesis and its modulation by ageing. Nature, 468, 1100–1104. [DOI] [PubMed] [Google Scholar]
- Settembre C, et al. (2012). A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO Journal, 31, 1095–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K, et al. (2019). Cryo-EM structure of the human FLCN-FNIP2-Rag-Ragulator complex. Cell, 179, 1319–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherpa RT, Atkinson KF, Ferreira VP, & Nauli SM (2016). Rapamycin increases length and mechanosensory function of primary cilia in renal epithelial and vascular endothelial cells. International Education and Research Journal, 2, 91–97. [PMC free article] [PubMed] [Google Scholar]
- Shillingford JM, et al. (2006). The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proceedings of the National Academy of Sciences of the United States of America, 103, 2236519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim MS, Dixon A, et al. (2023). Shear stress induces autophagy in Schlemm’s canal cells via primary cilia-mediated SMAD2/3 signaling pathway. Autophagy Reports, 2, 2236519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim MS, Nettesheim A, Dixon A, & Shim PB (2021). Primary cilia and the reciprocal activation of AKT and SMAD2/3 regulate stretch-induced autophagy in trabecular meshwork cells. Proceedings of the National Academy of Sciences of the United States of America, 118, e2021942118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singla V, & Reiter JF (2006). The primary cilium as the cell’s antenna: Signaling at a sensory organelle. Science (New York, N. Y.), 313, 629–633. 10.1126/science.1124534. [DOI] [PubMed] [Google Scholar]
- Singla V, Romaguera-Ros M, Garcia-Verdugo JM, & Reiter JF (2010). Ofd1, a human disease gene, regulates the length and distal structure of centrioles. Developmental Cell, 18, 410–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streets AJ, Prosseda PP, & Ong ACM (2020). Polycystin-1 regulates ARHGAP35-dependent centrosomal RhoA activation and ROCK signaling. JCI Insight, 5, e135385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struchtrup A, Wiegering A, Stork B, Rüther U, & Gerhardt C (2018). The ciliary protein RPGRIP1L governs autophagy independently of its proteasome-regulating function at the ciliary base in mouse embryonic fibroblasts. Autophagy, 14, 567–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su W, Li Z, Jia Y, & Zhuo Y (2019). Correction: Rapamycin is neuroprotective in a rat chronic hypertensive glaucoma model. PLoS One, 14, e0213489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Nagai T, Chiba S, Nakayama K, & Mizuno K (2018). Glucose deprivation induces primary cilium formation through mTORC1 inactivation. Journal of Cell Science, 131, jcs208769. [DOI] [PubMed] [Google Scholar]
- Takiar V, et al. (2011). Activating AMP-activated protein kinase (AMPK) slows renal cystogenesis. Proceedings of the National Academy of Sciences of the United States of America, 108, 2462–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, et al. (2013). Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites. Nature, 502, 254–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teotia P, Van Hook MJ, Fischer D, & Ahmad I (2019). Human retinal ganglion cell axon regeneration by recapitulating developmental mechanisms: Effects of recruitment of the mTOR pathway. Development (Cambridge), 146, dev178012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thedieck K, et al. (2007). PRAS40 and PRR5-like protein are new mTOR interactors that regulate apoptosis. PLoS One, 2, e1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsiokas L, Kim E, Arnould T, Sukhatme VP, & Walz G (1997). Homo- and heterodimeric interactions between the gene products of PKD1 and PKD2. Proceedings of the National Academy of Sciences of the United States of America, 94, 6965–6970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsun ZY, et al. (2013). The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Molecular Cell, 52, 495–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valcourt JR, et al. (2012). Staying alive: Metabolic adaptations to quiescence. Cell Cycle (Georgetown, Tex.), 11, 1680–1696. 10.4161/cc.19879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente EM, Rosti RO, Gibbs E, & Gleeson JG (2014). Primary cilia in neurodevelopmental disorders. Nature Reviews Neurology, 10, 27–36. 10.1038/nrneurol.2013.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vézina C, & Kudelski A (1975). Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. The Journal of Antibiotics, 28, 721–726. [DOI] [PubMed] [Google Scholar]
- Viau A, et al. (2018). Cilia-localized LKB 1 regulates chemokine signaling, macrophage recruitment, and tissue homeostasis in the kidney. The EMBO Journal, 37, e98615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viau A, et al. (2020). Divergent function of polycystin 1 and polycystin 2 in cell size regulation. Biochemical and Biophysical Research Communications, 521, 290–295. [DOI] [PubMed] [Google Scholar]
- Wagle N, et al. (2014b). Activating mTOR mutations in a patient with an extraordinary response on a phase I trial of everolimus and pazopanib. Cancer Discovery, 4, 546–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagle N, et al. (2014a). Response and acquired resistance to everolimus in anaplastic thyroid cancer. New England Journal of Medicine, 371, 1426–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walz G, et al. (2010). Everolimus in patients with autosomal dominant polycystic kidney disease. New England Journal of Medicine, 363, 830–840. [DOI] [PubMed] [Google Scholar]
- Wang S, & Dong Z (2013). Primary cilia and kidney injury: Current research status and future perspectives. American Journal of Physiology - Renal Physiology, 305, F1085–F1098. 10.1152/ajprenal.00399.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, et al. (2015a). Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science (1979), 347, 188–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Livingston MJ, Su Y, & Dong Z (2015b). Reciprocal regulation of cilia and autophagy via the MTOR and proteasome pathways. Autophagy, 11, 607–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZL, Deng Q, Chong T, & Wang ZM (2018). Autophagy suppresses the proliferation of renal carcinoma cell. European Review for Medical and Pharmacological Sciences, 22, 343–350. [DOI] [PubMed] [Google Scholar]
- Wegiel J, et al. (2010). The neuropathology of autism: Defects of neurogenesis and neuronal migration, and dysplastic changes. Acta Neuropathologica, 119, 755–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weimbs T (2006). Regulation of mTOR by polycystin-1: Is polycystic kidney disease a case of futile repair. Cell Cycle (Georgetown, Tex.), 5, 2425–2429. 10.4161/cc.5.21.3408. [DOI] [PubMed] [Google Scholar]
- White E (2015). The role for autophagy in cancer. Journal of Clinical Investigation, 46, 125, 42–46. 10.1172/JCI73941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson PD (2001). Polycystin: New aspects of structure, function, and regulation. Journal of the American Society of Nephrology, 12, 834–845. 10.1681/asn.v124834. [DOI] [PubMed] [Google Scholar]
- Wu MJ, et al. (2006). Rapamycin attenuates unilateral ureteral obstruction-induced renal fibrosis. Kidney International, 69, 2029–2036. [DOI] [PubMed] [Google Scholar]
- Xiang W, et al. (2019). Primary cilia and autophagy interaction is involved in mechanical stress mediated cartilage development via ERK/mTOR axis. Life Sciences, 218, 308–313. [DOI] [PubMed] [Google Scholar]
- Xie J, et al. (2011). CAMP inhibits mammalian target of rapamycin complex-1 and −2 (mTORC1 and 2) by promoting complex dissociation and inhibiting mTOR kinase activity. Cellular Signalling, 23, 1927–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, et al. (2016). 4.4 Å resolution cryo-EM structure of human mTOR complex 1. Protein Cell, 7, 878–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Inoki K, Ikenoue T, & Guan KL (2006). Identification of Sin1 as an essential TORC2 component required for complex formation and kinase activity. Genes & Development, 20, 2820–2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, et al. (2011). Pancreatic cancers require autophagy for tumor growth. Genes & Development, 25, 717–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao A, & van Wijngaarden P (2020). Metabolic pathways in context: mTOR signalling in the retina and optic nerve - A review. Clinical and Experimental Ophthalmology, 48, 1072–1084. 10.1111/ceo.13819. [DOI] [PubMed] [Google Scholar]
- Yoder BK, Hou X, & Guay-Woodford LM (2002). The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. Journal of the American Society of Nephrology, 13, 2508–2516. [DOI] [PubMed] [Google Scholar]
- Yoo YA, et al. (2011). Sonic hedgehog pathway promotes metastasis and lymphangiogenesis via activation of Akt, EMT, and MMP-9 pathway in gastric cancer. Cancer Research, 71, 7061–7070. [DOI] [PubMed] [Google Scholar]
- Yu Y, et al. (2011). Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science (New York, N. Y.) (1979), 332, 1322–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan S, et al. (2012). Target-of-rapamycin complex 1 (Torc1) signaling modulates cilia size and function through protein synthesis regulation. Proceedings of the National Academy of Sciences of the United States of America, 109, 2021–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, et al. (2017). The mtor signal regulates myeloid-derived suppressor cells differentiation and immunosuppressive function in acute kidney injury. Cell Death & Disease, 8, e2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, et al. (2013). A comparison of Ku0063794, a dual mTORC1 and mTORC2 inhibitor, and temsirolimus in preclinical renal cell carcinoma models. PLoS One, 8, e54918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, et al. (2018). Advanced glycation end-products suppress autophagic flux in podocytes by activating mammalian target of rapamycin and inhibiting nuclear translocation of transcription factor EB. Journal of Pathology, 245, 235–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong M, et al. (2016). Tumor suppressor folliculin regulates mTORC1 through primary cilia. Journal of Biological Chemistry, 291, 11689–11697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Chen F, Yan A, & Xia X (2021). Role of mammalian target of rapamycin in regulating HIF-1α and vascular endothelial growth factor signals in glaucoma. Archives of Physiology and Biochemistry, 127, 44–50. [DOI] [PubMed] [Google Scholar]
- Zou Z, Tao T, Li H, & Zhu X (2020). MTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell and Bioscience, 10, 31. 10.1186/s13578-020-00396-1. [DOI] [PMC free article] [PubMed] [Google Scholar]