

Abstract
SLC15A4 is an endolysosome-resident transporter linked with autoinflammation and autoimmunity. Specifically, SLC15A4 is critical for Toll-like receptor (TLR) 7–9 as well as the nucleotide-binding oligomerization domain-containing protein (NOD) signaling in several immune cell subsets. Notably, SLC15A4 is essential for the development of systemic lupus erythematosus in murine models and is associated with autoimmune conditions in humans. Despite its therapeutic potential, quality chemical probes targeting SLC15A4 functions remain limited. Here, we use an integrated chemical proteomics approach to develop a suite of chemical tools, including first-in-class functional inhibitors, for SLC15A4. We demonstrate the SLC15A4 inhibitors described herein suppress endolysosomal TLR and NOD functions in a variety of human and mouse immune cells, furnish evidence of their ability to suppress inflammation in vivo and in clinical settings, and provide insights into their mechanism of action. Our findings establish SLC15A4 as a druggable target for the treatment of autoimmune/autoinflammatory conditions.
Editor Summary:
Integrated chemoproteomic development of selective small molecule SLC15A4 inhibitors and their functional characterization establish SLC15A4 as a druggable target for autoimmune conditions.
Graphical Abstract
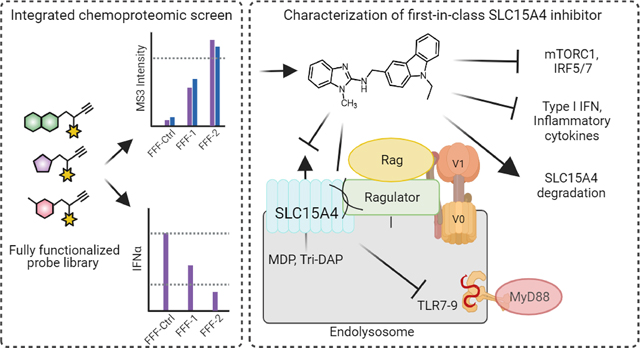
The innate immune system is the first line of defense against invading pathogens, and it intimately collaborates with the adaptive immune system to maintain physiological homeostasis. However, components of the immune response can sometimes become dysregulated, failing in this protective role, and even directly causing a variety of autoimmune diseases. Various innate immune sensors that are typically dedicated to protection against infection are sometimes usurped, and instead initiate and propagate autoimmune diseases, such as systemic lupus erythematosus (SLE) and Crohn’s disease 1. Specifically, self-induced signaling by nucleic acid-sensing endosomal Toll-like receptors (TLRs 7 and 9) and the unchecked production of pro-inflammatory cytokines (e.g. type I interferons; IFN-I) are central to the pathogenesis of numerous autoimmune conditions 1–3. Such autoimmune and inflammatory diseases affect millions of people worldwide; however, the mainstay treatments continue to be broad spectrum immune suppressant therapies that carry various risk factors, ranging from cardiovascular pathologies to increased susceptibility to infections 4,5. Therefore, the development of selective immune modulatory therapies is direly needed.
SLC15A4 is a 12-transmembrane (TM) domain protein with high gene expression in antigen presenting cells (APCs), such as plasmacytoid dendritic cells (pDCs) and B cells 6,7. SLC15A4 is a proton-coupled transporter of amino acids, such as histidine, and di- or tri-peptides, including the NOD1 ligand L-Ala-γ-D-Glu-mDAP (Tri-DAP) and the NOD2 ligand muramyl dipeptide (MDP) 8–10. SLC15A4 has also been intimately linked to TLR7, 8 and 9 mediated signaling as well as the production of IFN-I and other proinflammatory cytokines 6,7,10–15. Specifically, immune cells from Slc15a4 loss-of-function mutant (‘feeble’) and Slc15a4−/− mice are defective in IFN-α as well as TNFα, IL-6 and IL-12 production upon TLR 7–9 stimulation, but otherwise display normal development 6,10,15. Critically, Slc15a4 feeble and Slc15a4−/− mice show striking reductions in systemic lupus erythematosus (SLE)7,14,15, inflammatory bowel disease (IBD), and psoriasis manifestations 10,16. Finally, genome-wide association studies (GWAS) have revealed SLC15A4 to be associated with inflammatory diseases such as SLE in human populations 17–21.
Although these studies establish SLC15A4 as a critical modulator of inflammation and provide a strong therapeutic basis for the development of small molecule inhibitors, to our knowledge, none have yet been reported. Likely contributive to the dearth of chemical tools are general technical challenges posed by biochemical investigations of SLC family members 22, limited SLC15A4 structural information, and the absence of clear screenable functional readouts, stemming, in part, from an incomplete molecular understanding of how SLC15A4 mediates TLR-related signaling events. In this regard, a recent study has proposed that SLC15A4 mediates cytokine production via regulation of mTOR activity, which is required for TLR7- and TLR9 signaling and downstream IRF5 and IRF7 activation 13. Additionally, it was also found that SLC15A4 acts as a scaffold for the adaptor protein TASL, which was shown to support TLR7–9 mediated IRF5 signaling 11. More recently, it has been shown that SLC15A4 also affects the optimal trafficking of TLR9 and its ligands to the endolysosomes 7.
Considering these challenges, we pursued a mechanism-agnostic chemical proteomic strategy to identify small molecules that inhibit SLC15A4 inflammatory functions. Towards this end, we previously developed a method that unites fragment-based ligand discovery (FBLD) with chemical proteomics to broadly assess the ligandability of the proteome directly in live cells 23,24. Using a library of fully functionalized fragment (FFF) probes, we mapped thousands of reversible ligand–protein interactions in cells and showed that these interactions can be advanced to compounds capable of selectively modulating protein function. Here, we integrated this method with functional readouts, mainly the TLR9-mediated production of inflammatory cytokines, to identify small molecules capable of both binding to SLC15A4 and suppressing endolysosomal TLR functions directly in primary human immune cells (Fig. 1a). We demonstrate that optimized compounds stemming from this strategy block SLC15A4-mediated TLR 7–9 and NOD functions in a variety of human and mouse immune cells. We provide further evidence that these SLC15A4 inhibitors can suppress inflammatory cytokine production in lupus patient-derived cells and in vivo. Finally, mechanistic studies reveal that TLR 7–9 suppressive activity stems from, at least in part, compound-mediated disruption protein-protein interactions between SLC15A4 and components of the mTOR pathway, resulting in suppression of TLR-induced mTOR activation and downstream immune signaling. Notably, this disruption is followed by SLC15A4 destabilization and subsequent lysosomal-mediated degradation. Collectively, these studies shed new light on SLC15A4 functional roles in innate immune signaling and provide useful chemical tools that will enable investigations of SLC15A4 in basic and therapeutic contexts.
Fig. 1. Chemoproteomic discovery of SLC15A4 inhibitors.
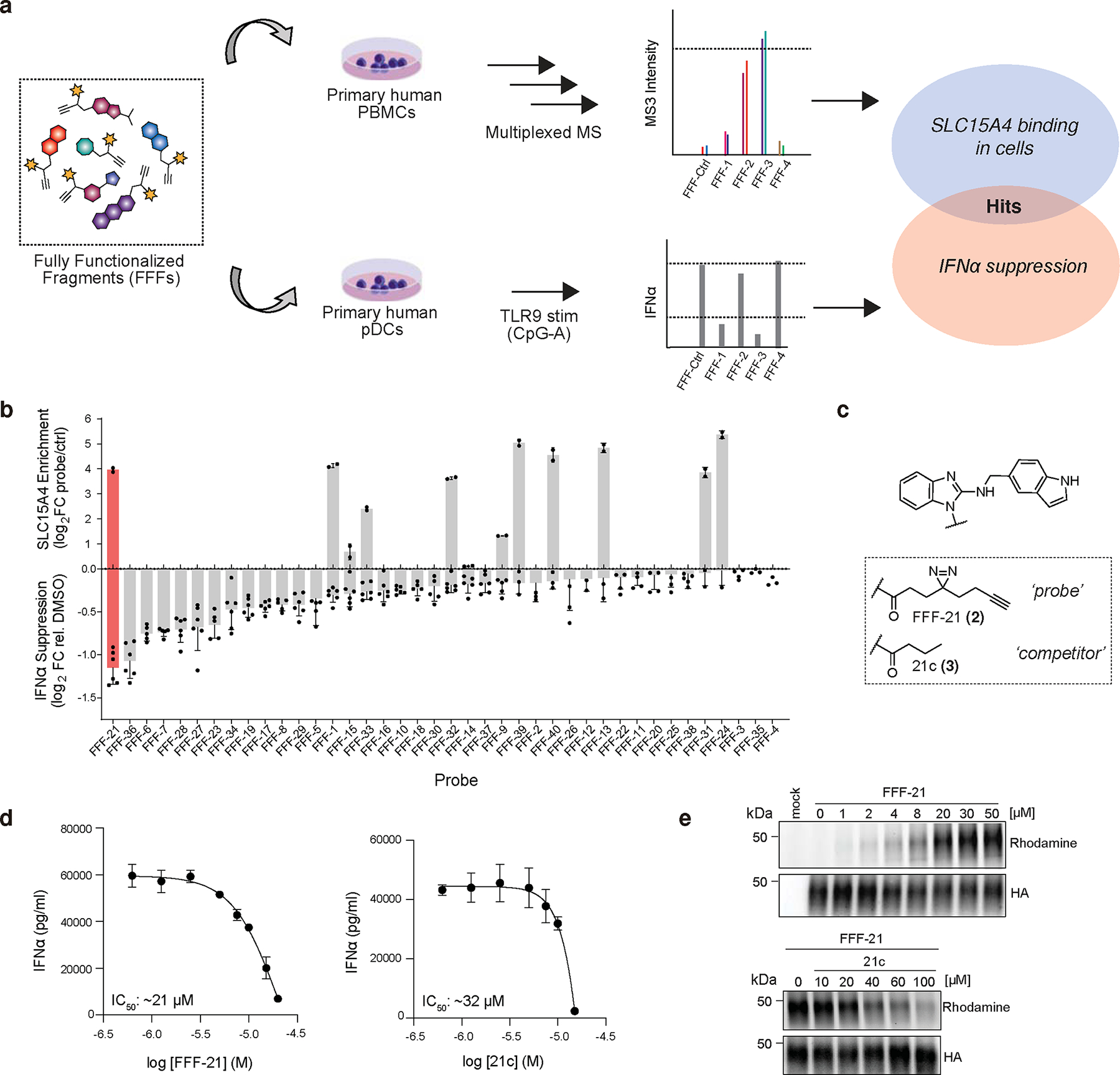
a, Schematic depiction of integrated chemoproteomic screen to identify fully functionalized fragment (FFF) probes capable of both binding SLC15A4 in human PBMCs and suppressing TLR9-mediated IFN-α production in primary human pDCs. See Methods for more details.
b, Overview of screening results. Upper x-axis displays SLC15A4 enrichment by FFF probes (10 μM) over control probe (10 μM) in human PBMCs via TMT-base proteomics. Lower x-axis displays corresponding IFN-α suppression in human pDCs after treatment with FFF probes (10 μM) and stimulation with CpG-A (1 μM, 24 h). Top hit, FFF21, is shaded in red. Data is presented as the mean ± s.d. (n = 2 independent biological replicates for proteomic enrichment analysis and n = 6 independent biological replicates for cytokine analysis).
c, Structures of hit FFF-21 probe and tag-free competitor analog 21c.
d, FFF-21 and 21c inhibit TLR9 mediated IFN-α production in primary pDCs. Primary human pDCs stimulated with CpG-A (1 μM) while being treated with escalating doses of compounds. Data is plotted as the mean ± s.d. (n = 4 independent biological replicates).
e, Gel profiling of HA-SLC15A4 engagement by FFF-21 and 21c in CAL-1 cells stably overexpressing HA-tagged SLC15A4. Cells were treated with DMSO or FFF-21 and binding was visualized by in-gel fluorescence scanning after PNGase treatment to reduce glycosylation (see Methods), click reaction with TAMRA-azide. For competition experiments (bottom), CAL-1 cells were co-treated with FFF-21 (10 μM) and 21c. Data are representative of two independent replicates.
RESULTS
Chemical Proteomic Discovery of SLC15A4 Inhibitors
Fully functionalized fragment (FFF) probes are composed of a: 1) a molecular recognition groups consisting of drug-like fragments intended to promote interactions with a subset of proteins; and (2) a photoactivatable retrieval tag consisting of a diazirine and alkyne handle, allowing for the UV-light-induced covalent capture, enrichment, and identification of interacting proteins (Supplementary Fig. 1)23,25. In order to identify compounds that directly bind to SLC15A4 in relevant immune cells, we screened an in-house library of FFFs (median fragment MW ~223 Da; Supplementary Fig. 1) in human peripheral blood mononuclear cells (PBMCs) using tandem mass tag (TMT) multiplexed proteomics, as previously described 24. In parallel, to prioritize SLC15A4 ligands that might perturb SLC15A4-mediated TLR functions, we evaluated FFFs for their ability to modulate IFN-α production in TLR9-stimulated human pDCs (Fig. 1a). Candidate hits were defined as FFFs that enriched SLC15A4 (>5-fold) over a control FFF (FFF-ctrl (1); Supplementary Fig. 1 and Supplementary Dataset) and induced >50% reduction in IFN-I production (10 μM). Among the nine FFFs that were found to substantially bind SLC15A4 in PBMCs, FFF-21 (2) suppressed IFN-α levels (Fig. 1b,c) to the highest extent, with an IC50 of approximately 21 μM whereas its tag-free analog, 21c (3), suppressed with a slightly lower IC50 of approximately 32 μM (Fig. 1d). We verified that FFF-21 engages SLC15A4 in CAL-1 cells, a pDC-like cell line 26, stably expressing lysosomal HA-SLC15A4 (Extended Data Fig. 1a) in a dose-dependent fashion which can be effectively blocked when co-treated with increasing concentrations of 21c (Fig. 1e). We further confirmed that FFF-21 and 21c can also block TLR 7/8 activation in monocyte-derived THP reporter cells (Extended Data Fig. 1b, c).
Recent studies have shown that SLC15A4 mediates the transport of bacterially derived components, such as the NOD2 ligand MDP, to the cytosol and is required for NOD responses to endosomal trafficked ligands 8,9,27,28. To assess whether FFF-21 also inhibits SLC15A4-mediated NOD activation, we generated a SLC15A4-dependent NF-κB reporter cell line utilizing a previously reported SLC15A4 mutant (L14A, L15A, L318A, V319A) that localizes to the plasma membrane 29. We confirmed production of luciferase was dependent upon exposure to MDP and the presence of these mutations (Extended Data Fig. 1d). Treatment with FFF-21 suppressed NF-κB activation in a concentration-dependent fashion, suggesting that it blocks SLC15A4-mediated transport of NOD2 ligand (Extended Data Fig. 1e).
SLC15A4 Inhibitor Optimization and Target Engagement Studies
Given the relatively low potency of FFF-21/21c and the limited structure-activity relationships (SAR) of compounds in our primary screen, we pursued the synthesis and functional evaluation of additional analogs. We synthesized 24 analogs of 21c, exploring modifications of the benzimidazole, indole, and alkyl groups and assessed their relative abilities to suppress IFN-α production in TLR9 - stimulated human pDCs and to block SLC15A4-mediated activation of NOD2 by MDP (Supplementary Table 1). We observed that alterations of the 2-aminobenzimidzole core were generally detrimental to activity, whereas modifications of the indole group were more tolerated. From these studies, we identified AJ2–30 (4) to be among the most potent in both assays (Fig. 2a), with an IFN-α suppression IC50 of 1.8 μM and a MDP transport inhibition IC50 of 2.6 μM (Fig. 2b–c). In addition, AJ2–30 showed no activity against a related family member SLC15A3 (Extended Data Fig. 2a), which also mediates NOD2 ligand transport and shares the highest sequence similarity to SLC15A4 9. Further, AJ2–30 displayed similar activity against murine Slc15a4-mediated NOD2 activation (Extended Data Fig. 2b) suggesting it also should be suitable for evaluation in mouse immune models. Critically, the AJ2–30 scaffold allowed for derivatization with a photoaffinity tag to enable cellular target engagment studies, with minimal effects on inhibitory potency (AJ2–32 (5); Fig 2b,c). We also identified structurally related analogs that displayed no observable activity in these assays and therefore could serve as negative control compounds (AJ2–18 (6) and corresponding photoaffinity probe AJ2–90 (7); Fig 2b,c). We confirmed that AJ2–30 also blocks R848-induced activation of TLR7/8 signalling (Extended Data Fig. 2c) as well as TriDAP- and MDP-induced activation of NOD1 and NOD2 signalling, respectively, in THP-1 reporter cells (Extended Data Fig. 2d).
Fig. 2. Functional valuation of SLC15A4 chemical probes and target engagement studies.
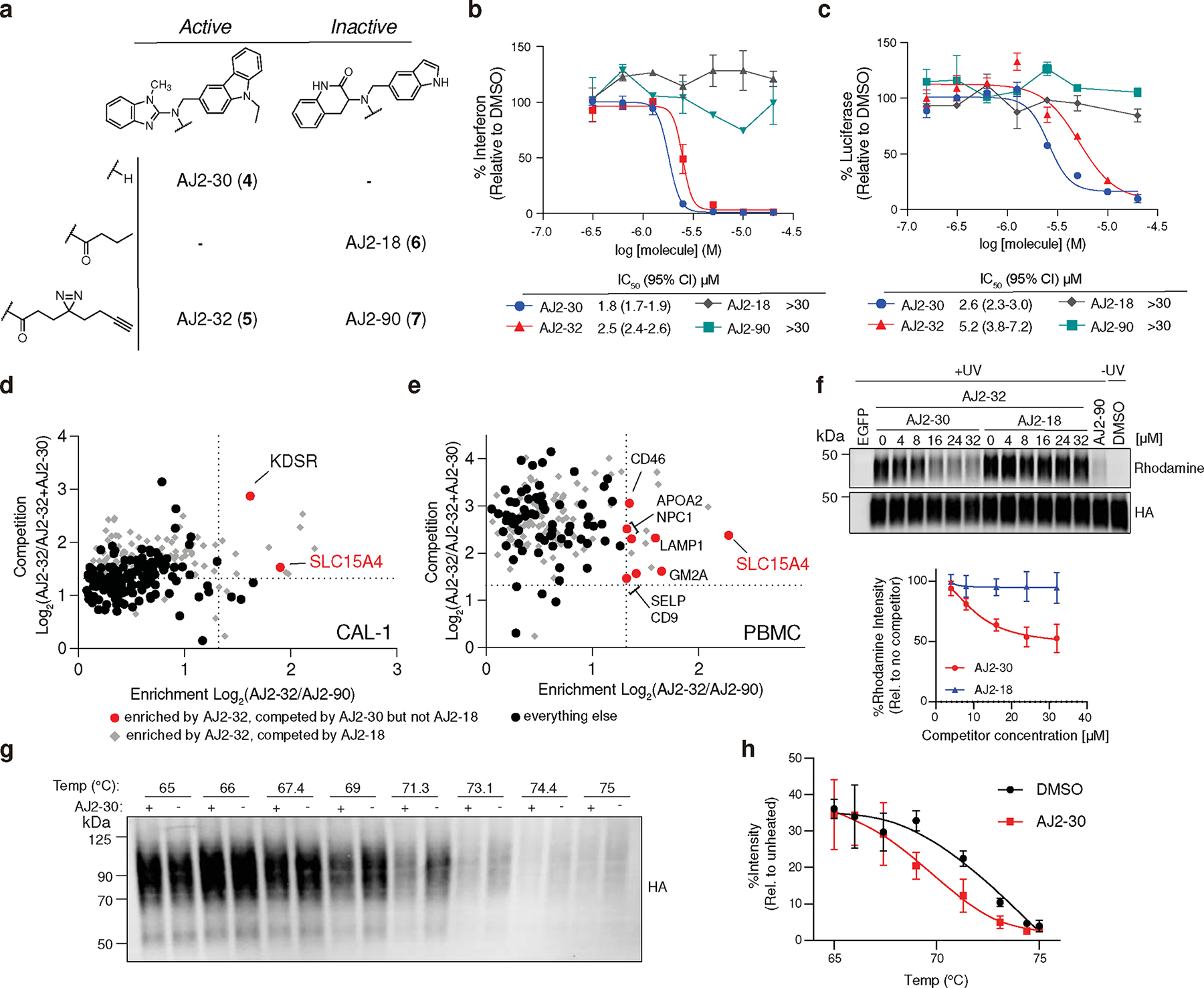
a, Structures of optimized active analog AJ2–30, inactive analog AJ2–18, and corresponding fully functionalized photoaffinity probes AJ2–32 and AJ2–90, respectively.
b, Concentration-dependent IFN-α suppression of CpG-A stimulated human pDCs by AJ2–30, AJ2–32, AJ2–18, and AJ2–90. Data is plotted as the mean ± s.d. (n = 3 independent biological replicates).
c, Concentration-dependent inhibition of MDP-mediated NOD2 signaling in A549 NF-κB reporter cells recombinantly expressing membrane-localized SLC15A4. Data is plotted as the mean ± s.d. (n = 3 independent biological replicates).
d, e, Chemical proteomic target engagement of AJ2–30 in (d) CAL-1 cells and € human PBMCs. The x-axis shows protein enrichment by AJ2–32 over AJ2–90 (4 μM), while the y-axis shows protein competition in cells treated with AJ2–32 (4 μM) and DMSO or AJ2–30 (24 μM). Dotted lines indicate threshold for proteins to be designated. Proteins preferentially enriched by AJ2–32 over AJ2–90 and competed by AJ2–30, but not competed by AJ2–18 are colored in red. Proteins plotted as gray diamonds are targets that were competed by AJ2–18 (24 μM). All other proteins plotted as black circles. Data is presented as the mean of replicated biological experiments (n = 2, p < 0.05; unpaired two tailed t test). Associated data set provided in Supplementary Dataset.
f, Immunoblot of HA-SLC15A4 engagement by AJ2–32 (4 μM) cotreated with indicated concentration of AJ2–30 and AJ2–18 as well as AJ2–90 (4 μM). Rhodamine signals were quantified and normalized to no competitor group samples. Data is presented as the mean ± s.d. of three independent replicates.
g, h, CETSA for AJ2–30 at varying temperatures. CAL-1 cells expressing HA-SLC15A4 were treated with AJ2–30 (10 μM), heated at indicated temperatures, and lysed. Soluble fractions were isolated by centrifugation and analyzed by immunoblot. Immunoblots were quantified and normalized to unheated (37°C) sample. Data is presented as the mean ± s.d. of three independent replicates.
To assess engagement of SLC15A4 by these new analogs, we profiled the protein targets of AJ2–32 in CAL-1 cells and human PBMCs using TMT isobaric labeling and mass spectrometry (MS). We found SLC15A4 to be the top target of AJ2–32 and AJ2–30 in both cell types, as it is substantially enriched over inactive probe AJ2–90 and this binding could be blocked upon co-incubation with AJ2–30 but not with the inactive analog AJ2–18 (Fig. 2d,e and Supplementary Dataset). Though additional targets of AJ2–30 were identified (e.g. KDSR in CAL-1 cells and APOA2, GM2A, NPC1 in PMBCs), they were completely non-overlapping between the cell types and do not possess biological functions associated with endolysosomal TLR signaling. We confirmed that recombinant SLC15A4 stably expressed in CAL-1 cells was strongly labeled and this labeling was effectively blocked in a dose-dependent fashion with increasing concentrations of AJ2–30 (Fig. 2f; Supplementary Fig. 2), but not AJ2–18, indicative of a stoichiometric interaction. We sought to further validate AJ2–30 engagement of SLC15A4 in cells by monitoring the thermal stability of SLC15A4 in the presence of AJ2–30 via a cellular thermal shift assay (CETSA) 30. Interestingly, when we applied a temperature gradient, SLC15A4 was found to be markedly destabilized by treatment with AJ2–30 (Fig. 2g–h). In addition, this decrease in thermal stability was found to be concentration-dependent and was not observed with the inactive control AJ2–18 (Extended Data Fig. 2e, f).
To further characterize AJ2–30, we also pursued cell free profiling (at 10 μM) across a panel of 468 kinases, revealing only three kinases (MAPKAPK2, PHKG2, and ERBB3) identified as minimally interacting partners (Supplementary Fig. 3a), further supporting the selectivity of the compound. Additionally, these compounds showed no evidence of toxicity in multiple primary immune cells, including pDCs, B cells, PBMCs, as well as in the immune cell lines CAL-1 and THP-1 (Supplementary Fig. 3b–e). Finally, AJ2–30 was found to possess suitable mouse pharmacokinetic properties for evaluation in short-term in vivo models of inflammation (Supplementary Fig. 3f). Collectively, our analyses point to SLC15A4 as being a primary target of AJ2–30.
AJ2–30 Inhibits SLC15A4 Functions in Primary Immune Cells
We next evaluated the ability of AJ2–30 to suppress additional innate signaling pathways in multiple immune cell subsets. In human pDCs, AJ2–30, but not control analog AJ2–18, suppressed IFN-α production following stimulation with Class A (CpG2216) and Class B (CpG2006) TLR9 agonists, the TLR7/8 stimulus R848, the TLR7-specific agonist R837 and TLR9 agonistic LL37:DNA complexes 31 (Fig. 3a; Supplementary Fig. 4a). Further, when human pDCs were challenged with influenza virus, IFN-α production was significantly inhibited by AJ2–30 but not AJ2–18 (Supplementary Fig. 4b). Additional pDC-derived cytokines and chemokines, including IL-6, TNF-α, G-CSF, CCL3, and CCL4, were also significantly reduced after treatment with AJ2–30 after TLR9 stimulation (Supplementary Fig. 5a), and not after TLR7/8 stimulation (Supplementary Fig. 5b). SLC15A4 is also significantly expressed in monocytes, macrophages, and B cells, and genetic disruption of Slc15a4 in these cell types interferes with their activation following endosomal TLR and NOD signaling 8,9,13,14. We found AJ2–30, but not AJ2–18 inhibited TLR7/8-induced production of TNFα in human monocytes (Fig. 3b; Supplementary Fig. 4c). Consistent with our observations in NOD reporter assays described above, treatment with AJ2–30 also suppressed MDP- and TriDAP-mediated TNFα production in human macrophages (Fig. 3c). Flow cytometry analysis of cell surface activation markers showed that AJ2–30 also suppresses B cell activation following stimulation with TLR 7/8 or TLR9 agonists (Fig. 3d, e). In addition, AJ2–30 but not AJ2–18 reduced total IgG, IL-6, TNFα and IL-10 production from B cells upon TLR9 (Fig. 3f,g and Supplementary Fig. 4c) or TLR7/8 (Supplementary Fig. 4e–f, 5d) stimulation. Finally, AJ2–30 also displays cross-species reactivity, suppressing IFN-α production in mouse pDCs (Supplementary Fig. 5e) as well as IL-6 (Supplementary Fig. 5f) and IgG production (Supplementary Fig. 5g) in mouse B cells with TLR7–9 stimulation.
Fig. 3. Pharmacological inhibition of SLC15A4 suppresses multiple innate signaling pathways in human immune cells.
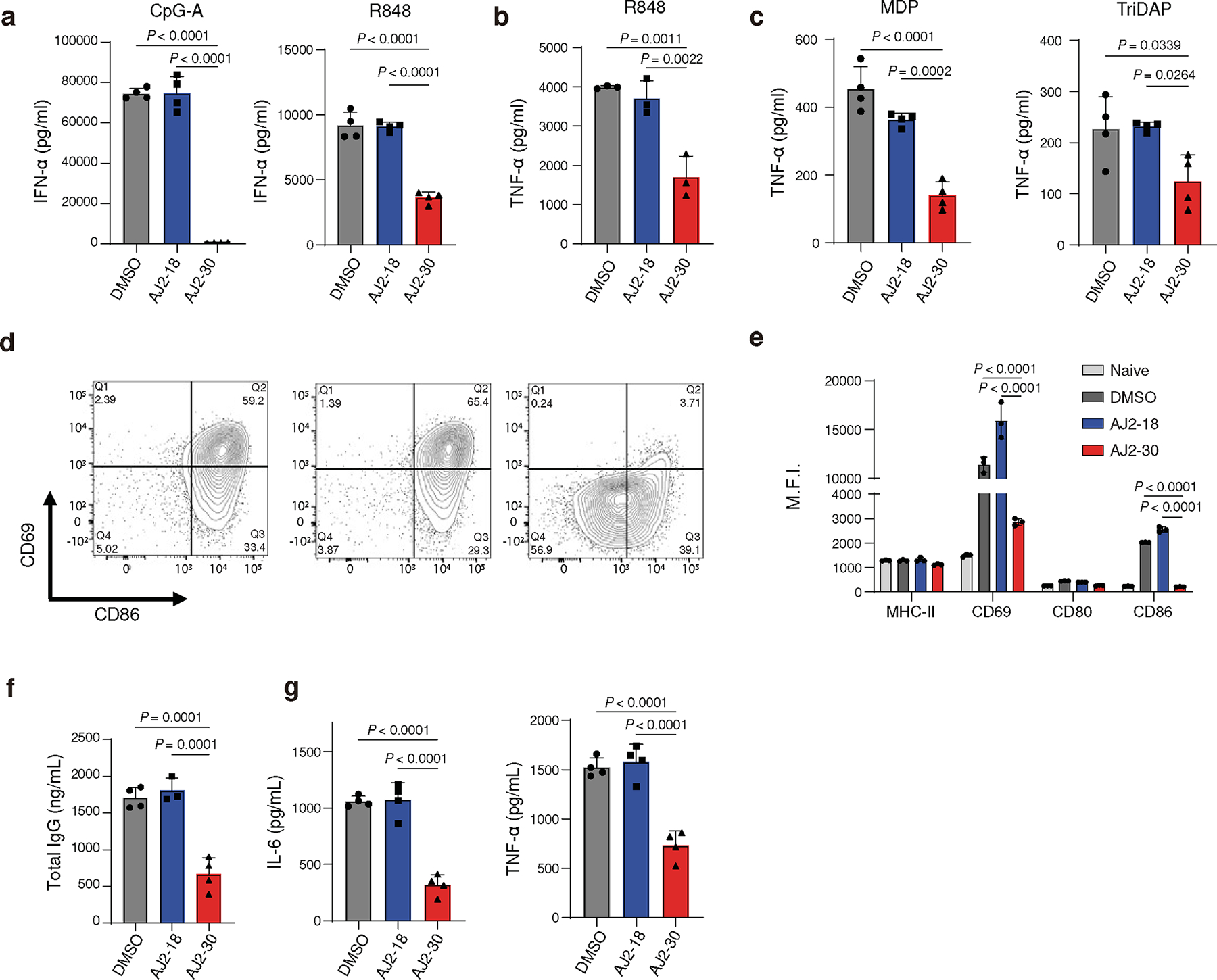
a, AJ2–30 inhibits TLR7, TLR7/8, and TLR9-induced pDC IFN-α production. pDCs were isolated from human PBMCs and treated with AJ2–18 or AJ2–30 (5 μM, 24 h) along with CpG-A (1 μM) or R848 (5 μg/mL).
b, AJ2–30 inhibits TLR7/8 mediated production of TNF-α in primary human monocytes. Monocytes were isolated from PBMCs and treated with AJ2–18 or AJ2–30 (5 μM, 24 h) along with R848 (5 μg/mL).
c, AJ2–30 inhibits bacterial dipeptides MDP and TriDAP mediated NOD1 and NOD2 signaling. Primary human derived macrophages were treated with AJ2–18 or AJ2–30 (5 μM), polarized with IFN-γ, and then challenged with the bacterial dipeptides MDP (150 ng/ml) or TriDAP (2000 ng/mL) for 24 h.
d, e, Primary human B cells isolated from PBMCs were treated with AJ2–18 or AJ2–30 (5 μM, 24 h) while stimulated with CpG-B (1 μM). B cell activation was assessed by measuring the surface expression of CD69, CD80, CD86, and MHC-II on live cells.
f, In vitro IgG secretion from primary human B cells stimulated with CpG-B for 6 days in the presence of either AJ2–18 and AJ2–30 (5 μM). The compounds were not replenished during the treatment.
g, In vitro secretion of IL-6 and TNFα from isolated primary human B cells in the presence of either AJ2–18 or AJ2–30 (5 μM) when stimulated by CpG-B after 24 h.
Results are presented as mean ± s.d. of n = 3 independent biological replicates and at least 3 independent experiments. Statistical analysis was performed using ANOVA analysis followed by multiple comparisons test. P-values are shown.
AJ2–30 Anti-Inflammatory Activity is Dependent on SLC15A4
We next sought to confirm that AJ2–30 activity is specific to endolysosomal TLR signaling. Towards this end, AJ2–30 did not suppress production of TNFα with non-endolysosomal TLR stimulation, including the TLR2 agonist Pam3CSK4 and the TLR4 agonist LPS (Fig. 4a), or IFN-β production upon STING stimulation (Fig. 4b) in primary human monocytes. As was previously described in Slc15a4−/− derived macrophages 14, we observe no suppressive effects of AJ2–30 in human macrophages upon TLR7/8 stimulation (Extended Data Fig. 3a). AJ2–30 also did not affect IgG production or CD86 expression in LPS stimulated B cells, and consistent with B cells derived from Slc15a4−/− mice (Extended Data Fig. 3b) 13, did not suppress IgM production upon TLR7–9 stimulation (Extended Data Fig. 3c), confirming that AJ2–30 is not broadly immunosuppressive.
Fig. 4. AJ2–30 suppresses TLR7–9 and NOD activation of mouse immune cells in a Slc15a4-dependent manner.
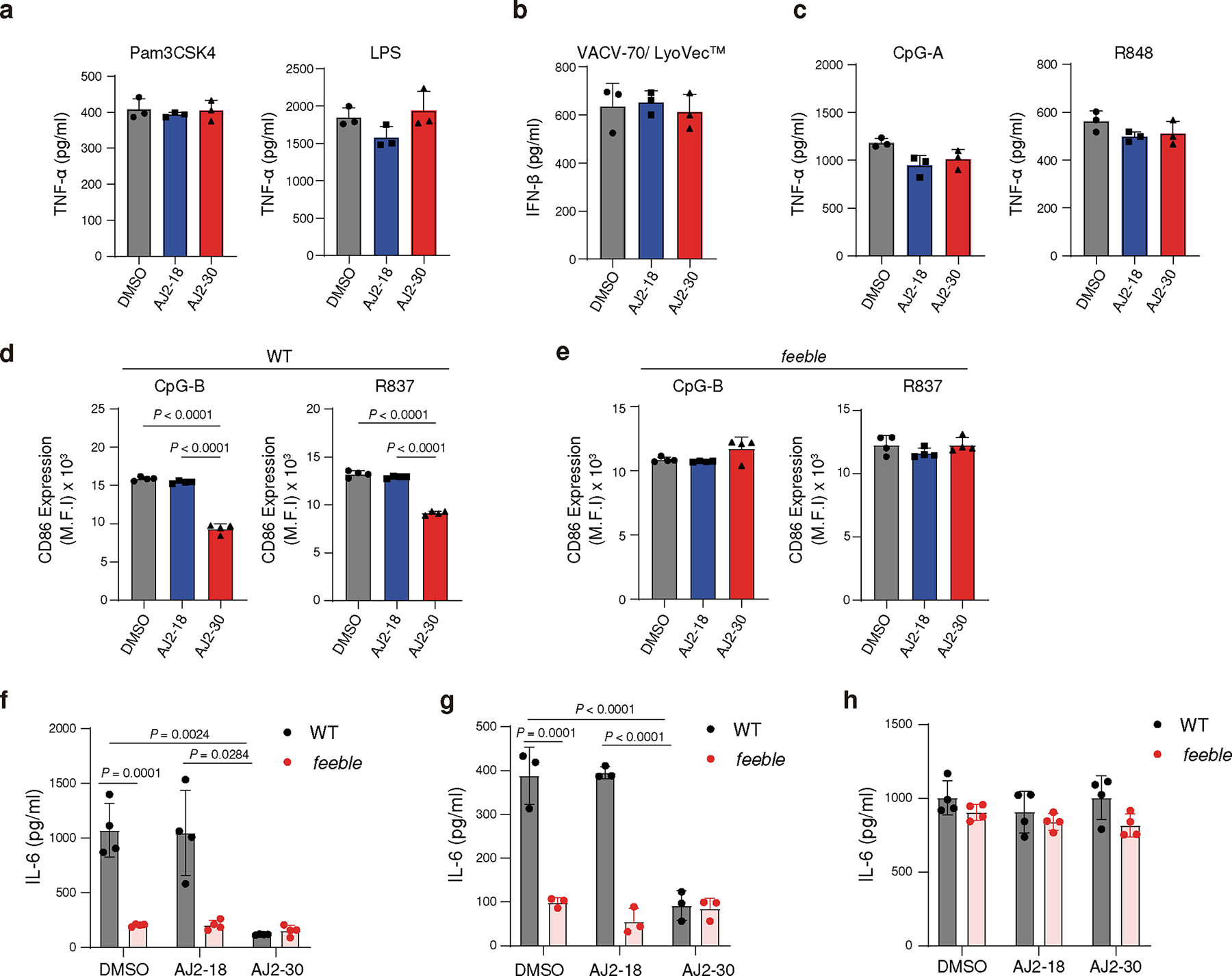
a, b, Secretion of cytokines from primary monocytes is not inhibited by treatment of AJ2–30 (5 μM) when stimulated by non-endosomal TLR (a, Pam3CSK4, LPS) and STING (b, VACV-70/LyoVec) agonists for 24 h.
c, AJ2–30 does not inhibit TNFα production from GM-CSF-differentiated dendritic cells. Bone marrow cells were harvested from wild-type C57BL/6J (WT) mice and differentiated into conventional dendritic cells with GM-CSF for 10 days. Dendritic cells were then stimulated with CpG-A or R848 for 24 h in the presence of DMSO, AJ2–18 or AJ2–30 (5 μM).
d, e, AJ2–30 suppresses B cell activation in WT but not feeble B cells. CD86 expression was detected by flow cytometry on WT (d) or feeble (e) B cells treated with either AJ2–18 or AJ2–30 (5 μM) 24 h after TLR7 and TLR9 stimulation.
f,g AJ2–30 (5 μM) inhibits production of IL-6 induced by R837 in mouse WT but not feeble B cells (f), or following stimulation with the bacterial dipeptide MDP in BMDMs (g) 24 h after stimulation.
h, AJ2–30 does not inhibit TLR7/8 signaling in mouse macrophages. BMDMs, generated from both WT and feeble mice, were treated with either AJ2–18 or AJ2–30 (5 μM) and then stimulated with R848 for 24 h.
Results in this figure are presented mean ± s.d. of 3 independent experiments (n = 3 independent biological replicates for a-e and g, n = 4 independent biological replicates for f and h). Statistical analysis was performed using ANOVA analysis followed by multiple comparisons test. P-values are shown.
We next explored whether AJ2–30 suppressive effects were dependent on SLC15A4. We first evaluated AJ2–30 in GM-CSF derived conventional dendritic cells (cDCs), which in contrast to pDCs, have significantly lower expression of Slc15a4 6. We observed no suppressive effects of AJ2–30 on TLR-induced TNFα production in cDCs (Fig. 4c). We also evaluated AJ2–30 in primary immune cells derived from feeble mice, which lack Slc15a4 expression. B cells from Slc15a4−/− mice display reduced CD86 upregulation following R837 stimulation compared to wild-type B cells13, however we observed AJ2–30 treatment did not further suppress activation following TLR7/8 or 9 stimulations (Fig. 4d,e and Extended Data Fig. 3d), nor did it further affect IL-6 production in these cells (Fig. 4f). Similarly, bone marrow derived macrophages (BMDMs) from feeble mice exhibited reduced IL-6 production following MDP stimulation and AJ2–30 did not have additional suppressive effects (Fig. 4g). It was recently reported that cytokine levels from Slc15a4−/− BMDMs are seemingly unaffected upon TLR7/8 stimulation 14, similarly, we observed that feeble BMDMs produce comparable IL-6 levels compared to WT controls and critically, AJ2–30 did not have any suppressive effects (Fig. 4h). Taken together, these data demonstrate that AJ2–30 suppressive effects are specific to TLR 7–9 as well as NOD activation and dependent on SLC15A4.
Molecular Characterization of SLC15A4 Inhibition with AJ2–30
We next sought to characterize how SLC15A4 inhibition with AJ2–30 disrupts TLR7–9-mediated cytokine production. Previously, it was shown that although SLC15A4 loss suppresses TLR7/8-mediated cytokine production, it does not disrupt proximal TLR7/8 signaling in Slc15a4−/− B cells 14. Consistent with these observations, AJ2–30 did not disrupt normal IRAK1, IKKα/β, and IκBα activation upon stimulation with R848, confirming that SLC15A4 inhibition by AJ2–30 does not disrupt proximal TLR7/8 signaling (Supplementary Fig. 6a). In contrast, we observed that AJ2–30, but not inactive AJ2–18, disrupts proximal TLR9 signaling upon stimulation with CpG in human and mouse B cells (Supplementary Fig. 6a, b). Interestingly, we observed no significant changes in TLR9 proximal signaling in feeble B cells (Supplementary Fig. 6c), suggestive of differences between genetic depletion and pharmacological disruption mediated by AJ2–30. Next, we sought to assess the relative kinetics of AJ2–30 suppressive activity. Here we observed that AJ2–30 suppresses the induction of IFNβ transcripts and IFN-induced STAT1 phosphorylation within 1–2hrs of TLR stimulation (Extended Data Fig. 4a, b), demonstrating that AJ2–30 effects on TLR signaling occur rapidly. It was recently shown that Slc15a4 is required for optimal trafficking of TLR9 and TLR9 ligands to endolysosomes7, raising the possibility that AJ2–30 may impose similar effects. Indeed, we observe that AJ2–30 reduces TLR9 localization to LAMP1-positive lysosomes in mouse B cells (Extended Data Fig. 4c, d).
Numerous studies have shown that SLC15A4 loss is disruptive to the mTOR signaling pathway, leading to failure of the IRF7-IFN-I regulatory circuit 12,13,32,33. Specifically, it was shown that SLC15A4 loss was associated with a decrease in lysosomal acidity and impairment of the vacuolar H+-ATPase (v-ATPase) activity 13, the primary regulator of acidification of the endocytic pathway 34 and a regulator of mTOR activity35. Interestingly, we observed a modest increase in lysosomal acidity when B cells were treated with AJ2–30 (Extended Data Fig. 4e) and no effects on ATPase activity was detected in lysosomal enriched fractions of mouse B cells treated with AJ2–30, despite strong inhibition with the ATPase inhibitor bafilomycin A1 (BafA, Extended Data Fig. 4f). One possible explanation for these observed differences is that AJ2–30 inhibits SLC15A4-mediated proton export from the lysosome36 while congenital depletion disrupts other unrealized lysosomal maintenance functions of SLC15A4. We next examined whether AJ2–30 interferes with mTOR signaling in immune cells. We observed a strong, dose-dependent impairment of mTOR pathway activation in human B cells stimulated with either TLR7/8 or TLR9 agonists by AJ2–30, but not with the inactive control AJ2–18 (Fig. 5a). Similar inhibitory effects on mTOR signaling were also observed in human pDCs (Extended Data Fig. 5a). Notably, AJ2–30 does not block activation of the mTOR pathway upon stimulation with the TLR2 agonist Pam3CSK4, suggesting it does not directly target mTOR itself and inhibition is restricted to endolysosomal TLR7–9-mediated mTOR activation (Fig. 5a).
Fig. 5. Mechanistic characterization of SLC15A4 inhibition by AJ2–30.
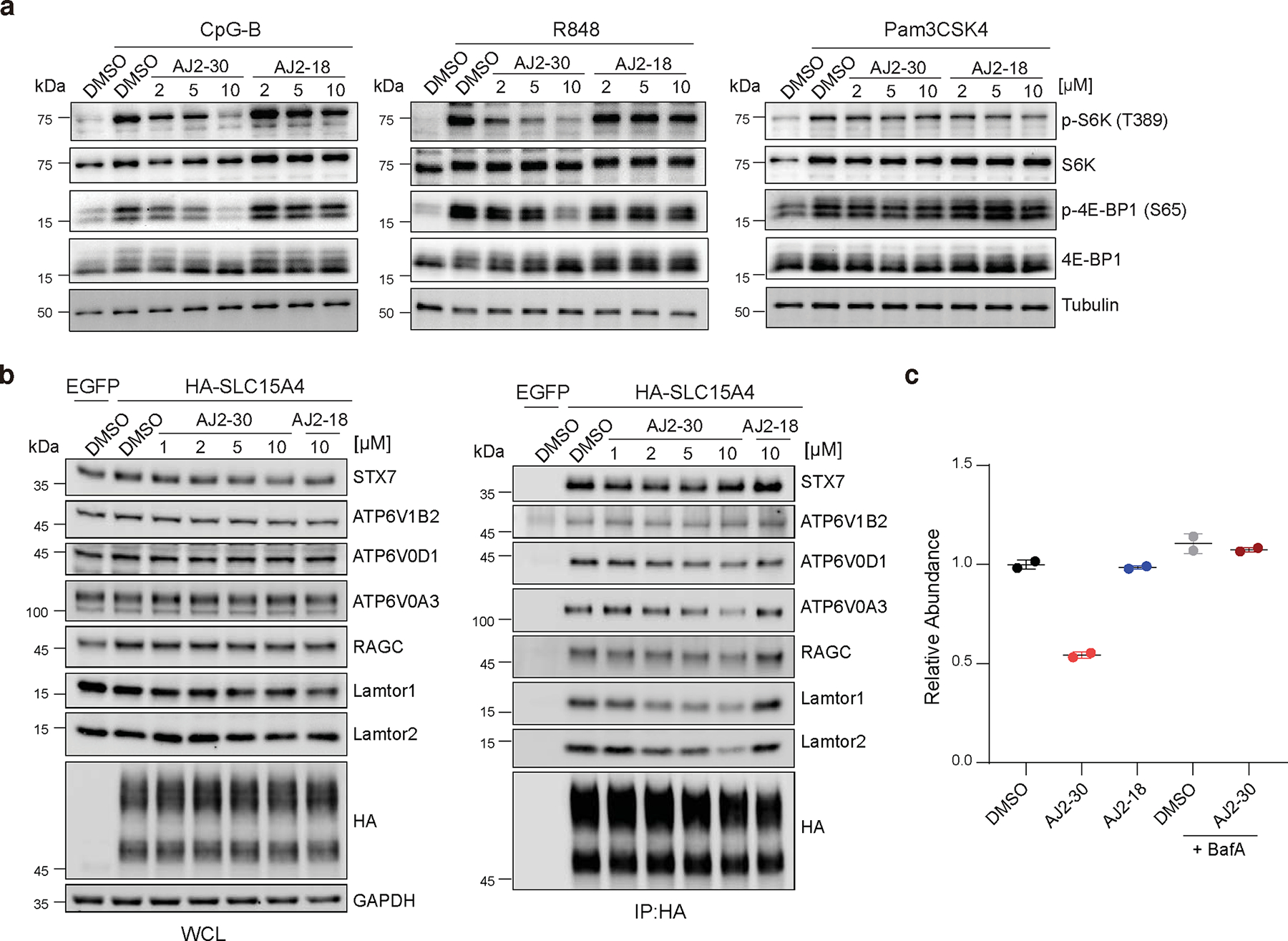
a, Immunoblot of mTOR substrates in B cells isolated from human PBMCs. Cells were co-treated with compounds or DMSO and stimuli (1 μM CpG-B, 5 μg/mL R848, 1 μg/mL Pam3CSK4) for 4 h. Data are representative of three independent experiments.
b, Immunoprecipitation of HA-SLC15A4 in CAL-1 cells reveal interactions with mTOR signaling components. CAL-1 cells expressing EGFP or HA-SLC15A4 were treated with compounds or DMSO for 1h. Whole cell lysates (WCL) from each condition were immunoprecipitated (IP) with anti-HA beads. WCL and immunoprecipitates were analyzed by immunoblot with indicated antibodies. Data are representative of three independent experiments.
c, AJ2–30 induces lysosomal degradation of endogenous SLC15A4 in human B cells. Human B cells were pretreated with or without BafA (500 nM) for 1 h and co-incubated with indicated compounds (10 μM) or DMSO for 16 h. Relative abundances of SLC15A4 was determined by TMT-based quantitative proteomics and normalized to DMSO treated samples. Data presented as mean of n = 2 independent biological replicates. Associated data set provided in Supplementary Dataset.
The observation that AJ2–30 destabilizes SLC15A4 thermal stability (Fig. 2g–h; Extended Data Fig. 2e-f), coupled to recent reports suggesting that SLC15A4 is proximal, or interacts directly, with mTOR signaling components12,32, raised the possibility that AJ2–30 may be disrupting SLC15A4 interactions critical for mTOR activation. To explore this further, we first generated a CAL-1 cell line stably expressing SLC15A4 fused to the peroxidase APEX232,37 to identify proteins within proximity to SLC15A4. We identified 269 proteins that were substantially enriched in APEX2-SLC15A4 CAL-1 cells relative to control cells, among which were numerous proteins involved in the mTOR signaling, including Ragulator-Rag complex members Lamtor 1–3 and the GTPases RagB and C, as well as multiple v-ATPase subunits (Extended Data Fig. 5b-c, Supplementary Dataset). We noted that Lamtor1 in particular, was the most enriched protein in these studies. We subsequently confirmed several of these proteins are co-enriched when HA-tagged SLC15A4 is immunoprecipitated, including Lamtor1, Lamtor2, RagC, and vATPase subunits ATP6V1B2, ATP6V0A3, ATP6V0D1, as well as SNARE complex protein STX7, suggestive of direct interactions (Fig. 5b). Notably, we observe that AJ2–30, but not inactive AJ2–18, strongly disrupts the interactions with Lamtor1 and Lamtor2, and to a lesser extent, v-ATPase subunits ATP6V0A3 and ATP6V0D1, and RagC and no disruption for other highly enriched proteins (e.g. STX7, ATP6V1B2) identified in our proximity labeling studies (Fig. 5b, Extended Data Fig 5d). Recently, it has been shown that SLC15A4 also interacts with the adaptor protein TASL, which serves as a scaffold for IRF5 activation11. Though we did not detect TASL in our proximity labeling studies, we confirmed HA-SLC15A4 engagement with endogenous as well as recombinant TASL via immunoprecipitation, however, AJ2–30 did not disrupt this interaction in either case (Supplementary Fig. 7).
The Ragulator complex is a pentamer containing Lamtor1–5, where Lamtor1 serves an anchor holding the complex to the lysosomal membrane, which in turn, tethers Rag GTPases enabling recruitment of mTORC1 and subsequent activation 38–45. Given that AJ2–30 disrupts interactions between SLC15A4 and Ragulator-Rag complex members, primarily Lamtor1/2, and blocks TLR-induced mTORC1 signaling, we wondered whether it would also affect mTORC1 recruitment to the lysosome. Indeed, we observe substantial blockade of mTORC1 localization to LAMP1-positive lysosomes in AJ2–30 treated primary human B cells stimulated either with TLR7/8 or TLR9 agonists (Extended Data Fig. 6a-b). Recent loss-of-function studies have associated SLC15A4 with autophagy12,46, a conserved lysosomal degradation pathway tightly linked to mTORC144, raising the possibility that SLC15A4 pharmacological inhibition may also affect this pathway. Consistent with this hypothesis, AJ2–30, but not inactive AJ2–18, was found to induce LC3B-II levels and trigger LC3 puncta in human B cells (Supplementary Fig. 8a–c).
mTOR is critical for TLR-mediated expression of type I IFNs and other proinflammatory cytokines in various immune cells47, and pharmacological inhibition of mTORC1 impairs IRF5 activation as well as phosphorylation, translation, and nuclear translocation of IRF7 48,49. Consistent with a mechanism that involves AJ2–30 mediated suppression of mTOR activation upon endolysosomal TLR activation, we observed that mTORC1 inhibitor Torin suppressed both TLR7/8 and TLR9-induced IFN-α production, but only TLR 9-induced TNF-α in pDCs, phenocopying AJ2–30 activity in these cells (Supplementary Fig. 8d–e). Additionally, when human pDCs were stimulated with either CpG or R848, we observed a strong blockade of IRF7 phosphorylation (Extended Data Fig. 6c) as well as IRF5 and IRF7 nuclear translocation with AJ2–30, but not with inactive control AJ2–18 (Extended Data Fig. 6d-e). Finally, we observe that AJ2–30, but not AJ2–18, reduced both TLR7/8 and TLR9-induced IRF5 and IRF7 protein expression in human B cells (Extended Data Fig. 6f). Together, these data demonstrate that engagement of SLC15A4 by AJ2–30 impairs endolysosomal TLR7–9-mediated mTOR activation, IRF5 and 7 activation, and subsequent cytokine production.
AJ2–30 Induces Lysosomal-Mediated Degradation of SLC15A4
Having observed that AJ2–30 destabilizes the thermal stability of SLC15A4 (Fig. 2g–h; Extended Data Fig. 2e-f), we wondered what the consequences of such effects on SLC15A4 stability in cells over longer exposure times could be. To investigate this further, we monitored HA-SLC15A4 levels in CAL-1 cells for extended time periods and observed that levels significantly decreased with treatment of AJ2–30 beginning at 4–6h of incubation in CAL-1 cells (Extended Data Fig. 7a). This effect was concentration dependent and was not observed with the inactive analog AJ2–18 (Extended Data Fig. 7a, b). Notably, we also observed substantial decreases of endogenous SLC15A4 levels in primary human B cells from multiple donors (Fig. 5c; Extended Data Fig. 7c), primary mouse B cells (Extended Data Fig. 7d), and CAL-1 cells (Extended Data Fig. 7e) by MS-based proteomics (Supplementary Dataset) upon treatment with AJ2–30, suggesting a conserved mechanism across multiple cell types. Given that SLC15A4 is localized to the membranes of endolysosomes and that SLC15A4 remains localized to LAMP1-positive lysosomes after AJ2–30 treatment but prior to abundance decreases (Extended Data Fig. 7f-g), we hypothesized that the observed SLC15A4 loss was the result of lysosome-mediated degradation. In line with this hypothesis, we observed that SLC15A4 loss can be rescued by pretreatment with BafA, which disrupts lysosomal function via inhibition of v-ATPase activity, resulting in inhibition of acidification and lysosome-mediated protein degradation50, both in primary human B cells and CAL-1 cells (Fig. 5c; Extended Data Fig. 7h; Supplementary Fig. 9a and Supplementary Dataset). We note that in addition to SLC15A4, we observe decreased abundance (>2-fold) for ~60 other proteins (~1.4% of total proteins quantified), though the vast majority are lysosome-resident (37 proteins), and interestingly, include mTOR components we observed to interact with SLC15A4, Lamtor 1–2, RagB, and RagC, and which were also rescued upon treatment with BafA (Supplementary Fig. 9b–d, Supplementary Dataset). Collectively, these data suggest that AJ2–30 engages SLC15A4, resulting in protein destabilization and subsequent lysosome-mediated degradation.
AJ2–30 Reduces Inflammation in Lupus Patient Cells and Mice
Finally, we sought to assess the therapeutic potential of small molecule SLC15A4 functional inhibition. We first investigated immunosuppressive effects of AJ2–30 in PBMCs isolated from a cohort of lupus patients both with and without a therapeutic regime for treatment of their disease (Supplementary Table 2). As expected, stimulation of PBMCs from these patients with TLR7/8 or TLR9 agonists produced significant levels of IFN-α and other inflammatory cytokines, which were substantially blunted by AJ2–30 treatment (Fig. 6a–b and Supplementary Fig. 10a–b). Additionally, AJ2–30 significantly reduced IgG levels (Fig. 6c) as well as the expression of the costimulatory molecules CD80, CD86, and MHC-II on B cells following TLR7/8 or TLR9 stimulation (Extended Data Fig. 8a,b). Elevated IFN-I signatures, cytokine production and cellular activation is observed in lupus patients 51–53. Treatment of unstimulated lupus patient-derived PBMCs with AJ2–30 significantly suppressed production of IFN-γ, IL-6, and IL-10 (Fig. 6d and Supplementary Fig. 10c). Moreover, the elevated levels of IgG and B cell co-stimulatory molecules were reduced following treatment with AJ2–30 (Fig. 6e and Extended Data Fig. 8c), demonstrating that the heightened activation state of lupus PBMCs can be suppressed by AJ2–30. Finally, we sought to obtain preliminary in vivo evidence that SLC15A4 inhibition results in disruption of type 1 and 2 IFN production. Towards this end, IFN production was stimulated with DOTAP-complexed CpG-A, as previously described 14, and serum cytokine levels were measured following treatment with AJ2–30. Consistent with findings in Slc15a4-deficient mice 6,14,15 and our cellular studies, AJ2–30 significantly reduced IFN-α, IFN-β, and IFN-γ (Fig. 6f). Critically, we observed a concatenate suppression of early p-mTOR and p-4E-BP1 (Fig. 6g), consistent with inhibition of SLC15A4-mediated mTOR signaling as observed in cellulo. Collectively, these data support that AJ2–30 can mimic SLC15A4 functional loss in vivo and may be therapeutically effective against lupus and other autoimmune conditions.
Fig. 6. Pharmacological inhibition of SLC15A4 suppresses inflammatory cytokine production in lupus patient derived PBMCs.
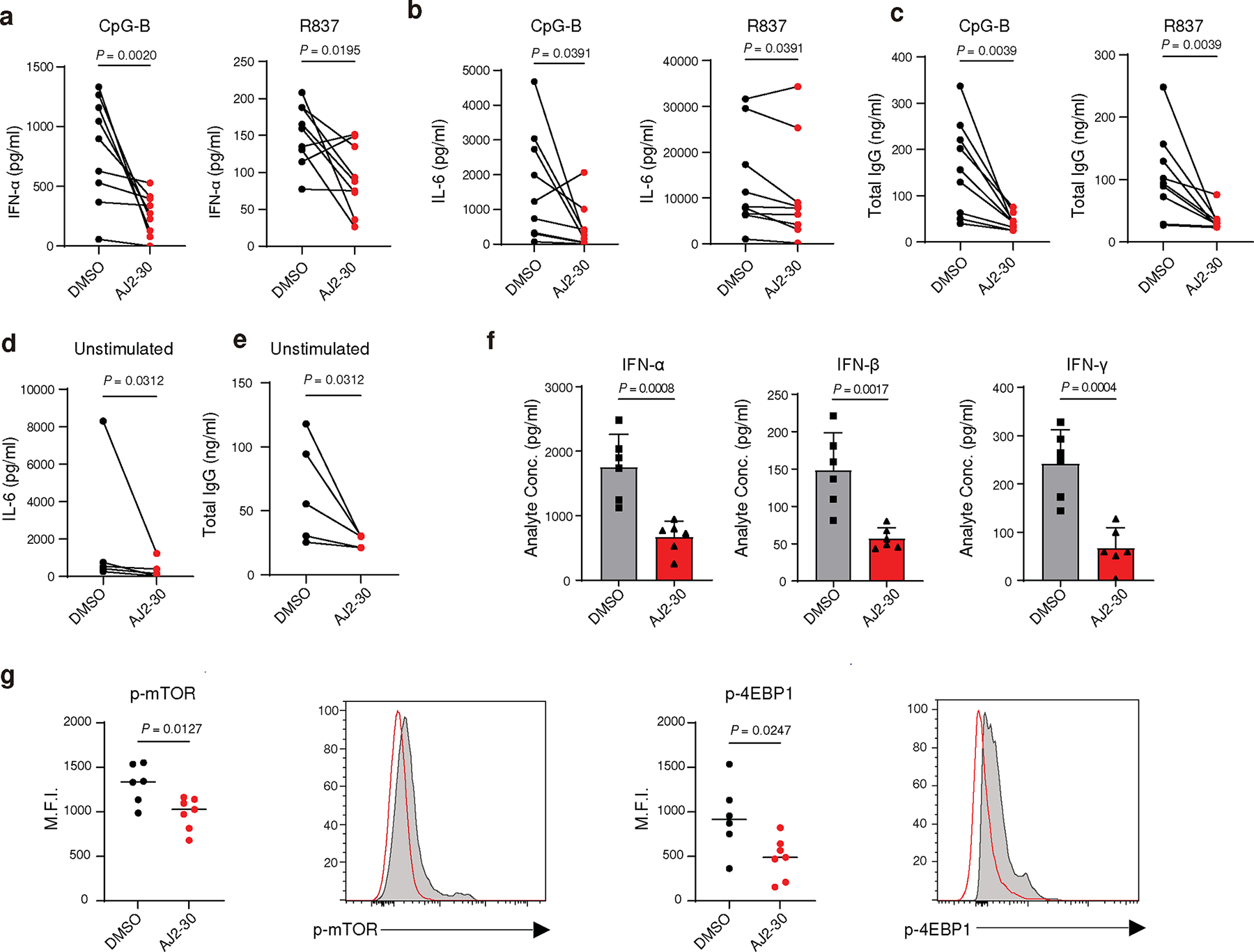
a, Secretion of IFN-α from PBMCs of lupus patients in the presence of either DMSO or AJ2–30 (5 μM) when stimulated by either CpG-A (1 μM) or R837 (10 μg/mL) after 24 h.
b, Secretion of IFN-γ, IL-6 and IL-10 from PBMCs of lupus patients in the presence of either DMSO or AJ2–30 (5 μM) when stimulated by either CpG-B (1 μM) or R837 (10 μg/mL) after 24 h.
c, Total IgG from PBMCs stimulated with CpG-B or R837 in the presence of AJ2–30 (5 μM) or DMSO was measured by ELISA after 6 days of stimulation.
d, The levels of IL-6 were measured in the supernatants of unstimulated PBMCs from lupus patients in the presence of either AJ2–30 (5 μM) or DMSO for 24 h.
e, Total IgG levels were measured by ELISA to assess the effect of AJ2–30 on suppression of antibody production from unstimulated PBMCs from lupus patients after 6 days of stimulation.
f, AJ2–30 reduced proinflammatory cytokine levels in mice. Mice were treated with compound AJ2–30 (50 mpk) or vehicle by intraperitoneal injection and then challenged with the TLR9 ligand CpG-A complexed with DOTAP (2 μg CpG-A per mouse), administered via tail vein injection. 6 h post injection, blood was harvested, plasma collected, and cytokine levels were measured from plasma samples.
g, AJ2–30 inhibits the mTOR signaling pathway in splenic mouse pDCs. C57BL/6 mice, treated with either vehicle or compound AJ2–30 via intraperitoneal injection (50 mpk), were challenged with the TLR9 ligand CpG-A complexed with DOTAP (2 μg CpG-A per mouse). 2 hours post-injection, spleens from these challenged mice were isolated. From these splenocyte population, CD11c+ cells were enriched and the p-mTOR and p-4EBP1 levels of the splenic pDCs, defined as B220+, CD11cmid, was assessed by flow cytometry.
Results are presented n = 9 (a-c), n = 5 (d and e) from independent donors, and n = 6 (f and g) independent mice. Statistical significance was determined using a Wilcoxon matched-pairs signed rank two tailed test (a-e) or unpaired two tailed t-test (f and g; mean ± s.d.). P-values are shown.
DISCUSSION
Despite a concerted effort to identify safe and effective targeted therapies to ameliorate autoimmune disease and autoinflammatory conditions, the mainstay treatments continue to be broad spectrum immune suppressants that carry considerable risk factors. Several genetic studies have identified the solute transport protein SLC15A4 as a promising therapeutic target to treat inflammation and autoimmune conditions. Experimental studies in mice where Slc15a4 is mutated or deleted demonstrate the importance of this protein in driving various autoimmune pathologies 6,13–15. Moreover, multiple GWAS studies have also genetically linked SLC15A4 to several autoimmune conditions 17–21. Given the restricted expression of SLC15A4 in immune cell populations critical for disease propagation coupled to its requirement for the function of major innate signaling pathways, SLC15A4 is an ideal therapeutic target to curb autoimmune pathologies.
Unlike many other SLCs that have proven more straightforward for chemical probe development 22, SLC15A4 poses unique challenges, including restricted expression to specific immune cell subsets, limited understanding of endogenous or exogenous transport substrates and assays, and localization to the endolysosomal compartment, where SLC15A4 is engages in numerous protein-protein interactions and is subject to intricate post-translational modifications (e.g. glycosylation) 11,32. Moreover, though some progress has been made, our understanding of how SLC15A4 regulates endolysosomal TLR signaling remains incomplete, underscoring the potential value of chemical probes for elucidating the mechanism of action of this critical immune modulatory protein.
To overcome these obstacles, we pursued an integrated chemical proteomic strategy to identify small molecules capable of both liganding SLC15A4 directly in relevant human immune cells and suppressing down-stream inflammatory cytokine production. We demonstrate that our tool compound, AJ2–30, directly engages SLC15A4 in multiple cell types, and inhibits functions regulated by SLC15A4, including the activation of TLR7–9 signaling, inflammatory cytokine production and SLC15A4-mediated NOD 1/2 activation both in primary human and mouse immune cells 6,7,10,13–15. Critically, we establish that the AJ2–30 suppressive effects largely phenocopy profiles of SLC15A4-deficient immune cells, are dependent on SLC15A4, and are specific to endolysosomal TLR stimulation. We also demonstrate that pharmacological inhibition of SLC15A4 results in disruption of an mTOR circuit essential for IFN-I and other inflammatory cytokine production, supporting recent genetic studies suggesting SLC15A4 plays a central role in this pathway13,46. Finally, we demonstrate that AJ2–30 substantially blunted inflammatory cytokine production both in an in vivo mouse model of inflammation and in lupus patient derived immune cells, underscoring the therapeutic potential of SLC15A4 inhibitors. Though, further optimization of the potency and pharmacokinetic properties of the molecules described herein are likely needed for evaluation in advanced autoimmune disease models. Such optimization endeavors will likely be facilitated by determination of the AJ2–30 binding site as well as recent structural elucidation of human SLC15A4 published while this manuscript was under review 54.
Mechanistically, AJ2–30 was found to directly induce destabilization of SLC15A4 (Fig. 2g–h; Extended Data Fig. 2e-f), which we hypothesize may is a consequence of native stabilizing protein-protein interactions (PPIs). Though we observe AJ2–30 does not directly disrupt interactions with recently reported TASL, it does disrupt interactions of SLC15A4 with components of mTORC1 signaling, including components of the Ragulator-Rag complex, notably Lamtor1/2. AJ2–30 disrupts mTORC1 lysosomal localization and selectively suppresses TLR7–9 induced mTOR activation, potentially stemming from disruption of the aforementioned PPIs. While the necessity of mTOR signaling in endolysosomal TLR mediate cytokine production is well-recognized 48,49,55, how TLR stimulation modulates mTOR activation is less established. In this regard, our data intimates at a previously unrealized scaffolding-like role of SLC15A4 for mTORC1 recruitment to the lysosome. It is tempting to speculate that SLC15A4 may serve as a signal relay for endolysosomal TLR ligand binding, resulting in mTOR activation, though further investigation is needed.
AJ2–30 was also found, after longer exposures (4–6hrs), to induce lysosomal-mediated degradation of SLC15A4. However, our data demonstrates that inhibition of TLR signaling initiates relatively earlier, as AJ2–30 suppression of IFNβ gene induction occurs rapidly (< 1hr), followed by inhibition of STAT1 phosphorylation (1–2hrs), suggesting functional SLC15A4 inhibition proceeds degradation. The cascade of molecular events leading to, and the functional consequences of this degradation, are not yet completely clear. It is possible AJ2–30 either perturbs SLC15A4 structure directly or disrupts native stabilizing interactions (e.g. Lamtor1/2), leading to degradation through a number of potential pathways, such as endo-lysosome protein quality control 56 or microautophagy-like processes 57. In this regard, we do observe induction of autophagic activation markers (Supplementary Fig. 8a–c), though, it is not yet evident if this autophagic state is triggered by AJ2–30-mediated mTOR inhibition, leading to non-specific degradation of SLC15A4 consequently or, selective towards SLC15A4 and proximal proteins, a result of AJ2–30 mediated destabilization of a lysosomal membrane complex. Finally, the differential requirement for SLC15A4 to promote TLR signaling in different cell types, coupled to the ability of mTOR to modulate innate immune signaling in a cell-type specific manner, may explain some of the differences observed with AJ2–30 suppressive abilities following TLR7/8 versus TLR9 signaling58. Further refinement of AJ2–30’s mechanism of action will be the subject of future studies, which will be enabled by concurrent elucidation of SLC15A4 at the interface of endolysosomal TLR and mTOR signaling, as inferred by the observations herein and by others12,13,32,46.
In summary, we have developed, to our knowledge, the first inhibitors that disrupt all functions of SLC15A4, and validated this protein as a druggable target with promising potential for the treatment of inflammatory and autoimmune conditions. Projecting forward, we envision that the integration of targeted chemical proteomic ligand screening with phenotypical readouts will facilitate the discovery of useful chemical probes for additional SLC members as well as other technically challenging, but therapeutically compelling targets.
METHODS
All antibodies and reagents are listed in the Supplementary Table 3.
Human cell lines
HEK293T and A549 cells were purchased from ATCC. THP1-Dual™ Cells were purchased from InvivoGen. CAL-1 cells were obtained from S. Kamihira, Nagasaki University26. HEK293T cells were maintained in DMEM media supplemented with 10% (v/v) fetal bovine serum (FBS), penicillin (100 U ml−1), streptomycin (100 μg ml−1) and L-glutamine (2 mM). A549 cells were maintained in DMEM supplemented with 2 mM L-glutamine, 25 mM HEPES, 10% (v/v) heat-inactivated FBS, 1× non-essential amino acids, and 55 μM β-Mercaptoethanol. CAL-1 cells were maintained in RPMI1640 media supplemented with 10% (v/v) FBS, penicillin (100 U ml−1), streptomycin (100 μg ml−1) and L-glutamine (2 mM). THP1-Dual™ Cells were cultured according to manufacturer’s instructions. All cell lines were grown at 37 °C in a humidified 5% CO2 atmosphere.
Generation of CAL-1 cells stably overexpressing EGFP or HA-SLC15A4
Lentiviral particles were produced by co-transfecting HEK293T cells with pMD2.G (Addgene, #12259), psPAX2 (Addgene, #12260), and pLEX305 vectors carrying EGFP, HA tagged SLC15A4, or V5-APEX2 tagged SLC15A4 using PEI MAX (Polysciences Inc). The growth medium was changed 24 h later, and the viral supernatants were collected 48 h and 72 h after transfection and filtered through a 0.2-μm PES membrane. CAL-1 cells were replaced with viral supernatants containing 8 μg ml-1 polybrene and then centrifuged for 30 min at 800 ×g at room temperature to enhance transduction. Transduced cells were selected with 2 μg ml−1 puromycin and cultured in RPMI1640 complete media (same as parental CAL-1 cell line) containing 1 μg ml-1 puromycin.
Mouse models
All procedures involving handling and treatment of mice were approved by the Institutional Animal Care and Use Committees (IACUC) of The Scripps Research Institute. All mice were bred and maintained within The Scripps Research Institute vivarium, housed under a 12 h light/dark cycle (light from 6 AM to 6 PM local time) in a temperature-controlled room (24°C) with free access to water and food. A C57BL/6J breeding colony maintained by the Teijaro Lab provided all experimental C57BL/6J mice for this study. Feeble (Slc15a4m1Btlr) mice (MGI:4835997) were provided as a gift from Dr. Bruce Beutler 6. C57BL/6JFeeble/Feeble mice were generated by backcrossing Feeble mice with C57BL/6J mice for 10 generations. This colony was maintained using C57BL/6JFeeble/Feeble x C57BL/6JFeeble/Feeble breeding. 8 to 12-week-old mice were used for experiments with age and sex matched C57BL/6J mice used as controls for the C57BL/6JFeeble/Feeble mice.
Isolation of peripheral blood mononuclear cells (PBMCs) and primary cells
Mouse cells
Mouse cells were isolated from a single cell suspension of mouse splenocytes, coming from either Wild-type C57BL/6J or feeble mice 6 which had been back-crossed onto the C57BL/6J line. Mouse immune cells were isolated using corresponding isolation kits from StemCell Technologies. Isolated B cells were cultured in complete medium consisting of RPMI 1640, 2 mM L-glutamine, 25 mM HEPES, 10% heat-inactivated FBS, 1× non-essential amino acids, and 55 μM β-Mercaptoethanol.
Bone marrow was harvested from murine femurs and tibias from both Wild-type C57BL/6J or feeble mice. Briefly, bones were flushed with RPMI-1640, erythrocytes were lysed with ACK lysing buffer (Quality Biological), and subsets were obtained through various differentiation protocols. Dendritic cells were generated by culturing bone marrow cells in DMEM supplemented with 2 mM L-glutamine, 25 mM HEPES, 10% heat-inactivated FBS, 1× non-essential amino acids, and 55 μM β-Mercaptoethanol. 10 ng ml−1 granulocytes macrophage colony-stimulating factor (GM-CSF) (Fisher Scientific) was added to the culture and cells were allowed to differentiate for 7 days, with GM-CSF containing medium being replenished on Day 3 and Day 6 to derive mouse dendritic cells. Bone marrow-derived macrophages (BMDMs) were generated from bone marrow cells derived in the manner described above in growth medium consisting of RPMI 1640, 2 mM L-glutamine, 25 mM HEPES, 10% heat-inactivated FBS, 1× non-essential amino acids, and 55 μM β-Mercaptoethanol, supplemented with M-CSF (50 ng ml−1) (Fisher Scientific) for 6 days, with the M-CSF containing medium being replaced on Day 3. Flt3-generated pDCs were differentiated from bone marrow cells as follows. Bone marrow cells were cultured at a concentration of 1.5× 106 cells ml−1 in growth medium consisting of DMEM, 2 mM L-glutamine, 25 mM HEPES, 10% heat-inactivated FBS, 1× non-essential amino acids, and 55 μM β-Mercaptoethanol, supplemented with Flt-3L-Ig (200 ng ml−1) (BioXcell) for 8 days respectively.
Healthy donors
For unenriched proteomic analysis, B cells were isolated from three fresh human peripheral blood Leukopaks (half-size) obtained from StemCell Technologies. Donor information is as follows: 24-year-old female, O+ blood; 44-year-old male, O+ blood. Remaining red blood cells (RBCs) were eliminated through lysis (ACK Lysing Buffer, Quality Biological Inc.) and primary human B cells were isolated using EasySep Human B Cell Isolation Kit (StemCell Technologies, negative selection).
All other studies with non-diseased primary human cells were performed on samples obtained from Scripps Research’s Normal Blood Donor Service following protocols approved by The Scripps Research Institute Institutional Review Board (IRB). Peripheral blood mononuclear cells (PBMCs) were isolated from heparinized whole blood using a Lymphoprep (StemCell Technologies) gradient and washed twice with fluorescence-activated cell sorting (FACS) buffer (Phosphate-buffered saline (PBS), 1× without calcium and magnesium; 2% FBS, and 1 mM EDTA).
Plasmacytoid dendritic cells (pDCs) were isolated via positive selection using the CD304 (BDCA-4/Neuropilin-1) MicroBead Kit (Miltenyi Biotec) following the manufacturers’ instructions. From the flow-through of the pDC isolation, primary human B cells or monocytes were isolated using EasySep Human B Cell Isolation Kit (StemCell Technologies, negative selection), or EasySep Human Monocyte Isolation Kit (StemCell Technologies, negative selection), respectively, according to manufacturer’s instructions.
Patient Samples
Lupus patient samples were obtained through StemCell Technologies Diseased Human Peripheral Blood Mononuclear Cells Catalog. Donor cells were collected following IRB-approved consent forms and protocols. Donors are screened for HIV-1, HIV-2, hepatitis B, and hepatitis C, and medical information of donors is available in Supplementary Table 2. PBMCs were stored in liquid nitrogen upon arrival and thawed with RPMI 1640, 2 mM L-glutamine, 25 mM HEPES, 10% heat-inactivated FBS, 1× non-essential amino acids, and 55 μM β-mercaptoethanol. Once thawed, cells are cultured at a concentration of 1×106 per ml.
Culturing and stimulation of immune cells
Synthesized oligodeoxynucleotides (ODNs) and toll-like receptor (TLR) agonists for stimulation of immune cells were purchased from InvivoGen (See Reagents Table). Isolated cells were maintained in complete RPMI growth medium consisting of RPMI 1640, 2 mM L-glutamine, 25 mM HEPES, 10% heat-inactivated (FBS), 1× non-essential amino acids, and 55 μM β-mercaptoethanol. Human primary pDCs and Flt-3L-Ig derived mouse pDCs were stimulated with 1 μM CpG-A, 1 μM CpG-B, 5 μg ml−1 R837, or 5 μg ml−1 R848 for 24 hr. Human and mouse pDCs were stimulated with one multiplicity of infection of influenza for 24 hr. For LL37:DNA complex formation, whole cell genomic DNA was isolated from CAL-1 cells using Qiagen Blood and Tissue Kit. 150 μg of genomic DNA was incubated with 100 μg of LL37 protein and allowed to complex for 1 h in 250 μl complete RPMI growth medium. Complexed DNA was added to primary pDCs to obtain a final concentration of 10 μg ml−1 of LL37. Human primary monocytes were stimulated with the following concentrations of reagents: 5 μg ml−1 R848 or R837, 100 ng ml−1 LPS, 100 ng ml−1 Pam3CSK4, or 5 μg ml−1 VACV-70/ LyoVec for 24 h. Both mouse and human B-cells were stimulated at the following concentrations unless otherwise noted: R837, 10 μg ml−1; LPS, 10 μg ml−1; R848, 5 μg ml−1; CpG-B 1 μM; and Pam3CSK4, 1 μg ml−1. For quantitation of cytokines and analysis of B cell activation, cells were stimulated for 24 hrs. For quantitation of in vitro immunoglobin production, cells were stimulated for 6 days. Following treatment, cells were centrifuged (500g for 6 min) and the supernatant was transferred to new plates. Supernatants were stored at −80°C until cytokine quantitation was performed. To assess IFN-α production of PBMCs isolated from lupus patient samples were stimulated with 1 μM CpG-A, 1 μM CpG-B, or 10 μg ml−1 R837. 24 h post stimulation, supernatant was collected and stored at −80°C until cytokine quantitation was performed. To assess the effect of compounds on the B cell activation of the lupus patients, cells were stimulated with 10 μg ml−1 R837 or 1 μM CpG-B. 24 h post stimulation, activation markers were assessed by flow cytometry and supernatant was collected and stored at −80°C until cytokine quantitation was performed. To assess antibody production, PBMCs were cultured for 6 days, at which point supernatant was collected.
Differentiation and treatment of primary macrophages
Human monocytes were isolated using EasySep Human Monocyte Isolation Kit, 50,000 cells are plated in 200 μl differentiation culture media (RPMI with 2 mM L-Glutamine, 10% Heat Inactivated FBS, penicillin (100 U ml−1), streptomycin (100 μg ml−1), 10 ng ml−1 recombinant Human M-CSF Protein (StemCell Technologies)) in a 96 well, flat-bottom plate. Cells were allowed to differentiate for 5 days. On day 5, differentiation media was replaced, and cells were polarized using IFN-γ (100 ng ml−1) for 1 h, Polarized macrophages were then treated with 150 ng ml−1 MDP, 2000 ng ml−1 TriDAP, or 5 μg ml−1 R848. Macrophages were treated for 24 h at 37 °C and 5% CO2, after which supernatant was collected and stored at −80°C until cytokine quantitation.
Mouse bone marrow derived macrophages (BMDM) were derived as described above using the bone marrow of both Wild-type C57BL/6J and feeble mice. On day 6 of culture, 2×105 BMDMs were plated per well in 96-well flat bottom plate in in growth medium consisting of RPMI 1640, 2 mM L-glutamine, 25 mM HEPES, 10% heat-inactivated FBS, 1× non-essential amino acids, and 55 μM β-mercaptoethanol, supplemented with M-CSF (50 ng ml−1). The following day, growth medium was replaced and the BMDMs were stimulated with 5 μg ml−1 R848 or 10 μg ml−1 MDP with 5 ng ml−1 LPS. Cells were cultured at 37 °C for 24 h, and the culture media were collected. IL-6 cytokine levels were measured by ELISA (ELISA MAX Deluxe Set Mouse IL-6, Biolegend).
Quantitation of cytokines and immunoglobulin levels
Quantitation of interferon levels were determined by ELISAs using the Human and Mouse IFN-α and IFN-β kits according to manufacturer’s instructions. Broad cytokine analysis of cellular supernatants was performed using the Bio-Plex Pro Human Cytokine 27-plex Assay (Bio-Rad M500KCAF0Y) and the Bio-Plex Pro Mouse Cytokine 23-plex Assay (Bio-Rad M60009RDPD). Analysis of individual analytes and immunoglobulin levels was performed using individual ELISA kits following manufacturer’s instructions. ELISAs were read using the ClarioStar plate reader (BMG Labtech). Bio-plex cytokine assays were run on Bio-plex 200 System (Bio-Rad).
In situ labeling of living cells by FFF probes
CAL-1 cells or PBMCs were incubated with serum-free RPMI1640 media (1×107 cells/mL) containing indicated probes, and, if applicable, competitors or DMSO vehicle for 30 min at 37 °C under a 5% CO2 atmosphere. The cells were then irradiated with UV light (365 nm, Stratagene, UV Stratalinker 1800 with 5× Hitachi F8T5 UV bulbs) for 20 min, scraped, washed twice with ice-cold DPBS (1,400 ×g, 4 °C, 3 min), and collected into 15 ml Falcon tubes. Cell pellets were either directly processed or stored at −80 °C until the next stage of processing.
Proximity labeling experiments
For proximity labeling, CAL-1 cells expressing a V5-APEX2-SLC15A4 fusion were processed as previously described 59. 70 million cells were incubated with 500 μM biotin phenol (ApexBio, CAS No. 41994–02-9, Cat. No. A8011) for 30 min and then treated with 1 mM H2O2 for 1 min, and the reaction was quenched with quencher solution (5 mM Trolox, 10 mM sodium ascorbate, and 10 mM sodium azide in PBS). Cells were then harvested and washed twice with ice-cold PBS. Pellets were processed with the method described below.
Preparation of samples for chemoproteomic analysis
All chemoproteomic samples were prepared as previously described25. In brief, after cells were treated as described in “In situ labeling of living cells by FFF probes” section, cell pellets were then resuspended, lysed by sonication in cold DPBS buffer. Protein concentrations were normalized using a DC Protein Assay (Bio-Rad), followed by a click reaction to couple the biotin-PEG3-azide to the probe-labeled proteins. After the click reaction, excess reagents were removed by a protein precipitation assay (MeOH/CHCl3/DPBS, 4:1:2, v/v). Pellets were resuspended in 6M urea solution (500 μl in DPBS) with SDS (10 μl of 10% w/v), followed by the reduction and alkylation steps. Then samples were diluted to 5.5 ml with DPBS. 100 μl of streptavidin-agarose bead suspension (Thermo Fisher Scientific, 20349) was added to each sample for probe-labeled protein enrichment. After 90 min, the beads were washed completely, transferred to 1.5 ml LoBind microcentrifuge tubes, digested by trypsin (Promega, V5111) in TEAB buffer (100 μl, 100 mM, pH 8.5) with 100 μM CaCl2 at 37 °C overnight. The digest was labeled with respective tandem-mass-tags (TMT, Thermo Fisher Scientific) reagents, quenched by hydroxylamine followed by adding formic acid before drying under vacuum centrifugation. The samples were then combined and fractionated using the Pierce high pH Reversed-Phase Fractionation Kit (Thermo Fisher Scientific, 84868) according to manufacturer’s instructions into 6 final fractions, dried, and stored at −80 °C until ready for mass spectrometry analysis. All chemoproteomics data is generated from two independent biological replicates, unless otherwise noted.
Preparation of samples for unenriched proteomic analysis
Cell pellets were resuspended in 100 μL ice-cold DPBS containing 1× Halt protease inhibitor (Thermo Fisher Scientific), lysed via sonication and proteins were quantified as described above. 100 – 200 μg protein per sample was aliquoted to tubes containing urea (8 M final concentration) and proteins were reduced (with TCEP and K2CO3) and alkylated (with IAA) as described above. Proteins were then precipitated with 1.2 ml ice-cold 4:1 MeOH/CHCl3 followed by 600 μl ice-cold DPBS. Precipitated proteins were centrifuged (12,000 ×g, 4 °C, 10 min), forming a disk, and the supernatant was removed. Proteins were then washed with 600 μL ice-cold 4:1 MeOH/CHCl3, re-centrifuged and the supernatant was again removed. Protein pellets were resuspended in 160 μl 100 mM TEAB via sonication and endopeptidase LysC (NEB, P8109S) was added at a ratio of 1:100 with protein amount. The LysC digest was conducted for 2 h, after which sequencing-grade trypsin (Promega, 1:100 with protein amount), ProteaseMAX (Promega, 0.05% final concentration), and CaCl2 (1 mM final concentration) were added, and the digestion was allowed to proceed overnight. Following the digest, samples were centrifuged (12,000 ×g, 4 °C, 5 min) and digested peptides were quantified using the Pierce Quantitative Fluorometric Peptide Assay (Thermo Fisher Scientific) according to manufacturer’s instructions. 25 μg from each sample was aliquoted, desalted with C18 spin columns (See Reagents Table for Catalog Numbers), and labeled with TMT10plex reagents according to manufacturer’s instructions. TMT-labeled peptides were combined in 0.1% TFA as described above and fractionated with the Pierce high pH Reversed-Phase Fractionation Kit. The peptide fractions were eluted from the spin column with solutions of 0.1% triethylamine containing an increasing concentration of MeCN (7.5 – 75% MeCN; 18 fractions). The fractions were combined into 9 final fractions in a pairwise fashion (fraction 1 and fraction 7, fraction 2 and fraction 8, etc.), dried via vacuum centrifugation, and stored at −80 °C until ready for mass spectrometry analysis.
LC-MS analysis of TMT samples
TMT-labelled peptides were resuspended in MS sample buffer (0.1 % formic acid in water) before subjected to LC–MS analysis as previously described25. In general, samples were loaded and eluted using an UltiMate 3000 RSLCnano system (Thermo Fisher Scientific) with a 220-min gradient separation method. The eluents were then analyzed and quantified with a Thermo Fisher Scientific Orbitrap Fusion Lumos mass spectrometer. A list of TMT 10plex-labeled SLC15A4 peptide molecular weights (573.6666, 872.9958, 463.8029, 688,3862, 1239.1694, 1012.5444, 705.4477, 459.2593, 582.3318, 859.9966, 517.7844, 721.8893, 470.6345, 675.3653) were employed for parallel reaction monitoring (PRM). MS1 spectra were specified 375 to 1,500 m/z scan range with a resolution of 120,000; peptide isolated for MS2 spectra were fragmented by collision-induced dissociation (CID, 30% collision energy); synchronous precursor selection60 was used to isolate up to 10 MS2 ions for the MS3-based quantification through high-energy collision-induced dissociation (HCD, 65% collision energy).
Cloning and plasmids construction
A template for cloning human SLC15A4 was obtained from Addgene (pDONR221_SLC15A4, #131910). cDNA for TASL (Cxorf21:476835) was obtained from Genetic Perturbation Screening core in UF Scripps. For transient transfection, SLC15A4 or TASL cDNA was cloned to pRK5 backbone in frame with either an N-terminal HA tag or a C-terminal FLAG tag. For lentiviral transduction, SLC15A4 cDNA was cloned into pLEX305 backbone in frame with a N-terminal HA tag. To construct transport assay cell line, gBlocks containing the codon-optimized human SLC15A3, human SLC15A4, mouse SLC15A4, and human NOD2 sequences were ordered from Integrated DNA Technologies. Human SLC15A3, human SLC15A4, mouse SLC15A4 and human NOD2 sequences were cloned into the BamH1 site of pLPC retrovirus backbone containing an mCherry protein in frame with a Glycine-Serine bridge using the NEBuilder HiFi Assembly kit (New England Biolabs).
Mutational disruption of the two dileucine motifs to induce plasma membrane localization was performed in both the mouse and human SLC15A4 constructs as well as the human SLC15A3 contruct using Q5® Site-Directed Mutagenesis Kit (New England Biolabs), which lead to the generation of the following mutants: human SLC15A4 (L14A, L15A, L318A, V319A), mouse SLC15A4 (L8A, L9A, L319A, V320A), and human SLC15A3 (L19A, L20A). All primers were ordered from Integrated DNA Technologies.
Primers: SLC15A4 Mut1 F (5’-AAGGGCACCTgcggccGGCGCAAGACGGGCAGCCGC-3’); SLC15A4 Mut1 R (5’-TCCCCAGCCCCACCGCCG-3’) (Undercase represents site of mutation);
SLC15A4 Mut2 F (5’-TGTGAAAGCTgcggccAAGATTGTCC-3’); SLC15A4 Mut2 R (5’-TCTTCCACTTTCTCTTCTG-3’);
mouse SLC15A4 Mut1 F (5’-GAGAGCTCCTgcggccGGTAGTCGCCGACCC-3’); mouse SLC15A4 Mut1 R (5’-TCTCCCTCCATGGTTCTG-3’);
mouse SLC15A4 Mut2 F (5’-TGTAAAGGCTgcggccAAAATCGTAC-3’); mouse SLC15A4 Mut2 R (5’-TCCTCTACTTTGTCTTCTG-3’);
human SLC15A3 Mut1F (5’-AAGACAGCCTgcggccCCCAGAGGAGCAAGGGGACC-3’); and human SLC15A3 Mut1R (5’-TCACCGGGAACTCTGGGT-3’).
Gel-based analysis of crosslinked HA tagged proteins in cells.
Probe labelled cell pellets were lysed in Dulbecco’s PBS (DPBS) buffer supplemented with 1% NP-40 (v/v) and 1× halt protease inhibitors cocktail (Thermo Fisher Scientific). Lysates were cleared by centrifugation at 14,000 ×g for 10 min at 4 °C. Protein concentration was normalized to 1.5–2.0 mg ml−1 in 1 ml volume with lysis buffer. Anti-HA magnetic beads (Thermo Fisher Scientific) was equilibrated with lysis buffer and added to normalized lysate at dose of 20 μL original slurry/1 mg lysate. HA-tagged proteins were enriched by rotating overnight at 4°C. Magnetic beads were washed with 500 μL lysis buffer for 6 times. HA-tagged proteins were eluted by adding 50 μl DPBS buffer supplemented with 1% NP-40 (v/v) and 2 mg ml−1 HA peptide and incubating in orbital shakers at 37 °C for 30 min. Deglycosylation of eluted protein was performed by incubating with 0.02 μg μl−1 of recombinant PNGase F at 37 °C for 30 mins (shown in the figure below). 3 μl of freshly prepared “click” reagent mixture containing 1.5 μl tris(benzyltriazolylmethyl)amine (TBTA) (1.7 mM in 1:4 DMSO:t-ButOH), 0.5 μl CuSO4 (50 mM in H2O), 0.5 μl Rhodamine-azide (1.25 mM in DMSO), and 0.5 μl tris(2-carboxyethyl)phosphine HCl (TCEP) (50 mM in H2O) was added to 25 μl deglycosylated sample and incubated for 1 hr at room temperature in the dark. 10 μl SDS sample buffer (4× stock) was added to quench the reaction. 10–20 μl sample was resolved by 10% SDS-PAGE and visualized for in-gel fluorescence on Bio-Rad ChemiDoc Imaging System. Proteins were transferred to a polyvinylidene fluoride (PVDF) membrane using Trans-Blot Turbo RTA Mini 0.45 μm PVDF transfer kit and blotted with anti-HA antibody in 5% (w/v) non-fat milk in TBST (50 mM Tris, 150 mM NaCl, 0.2% (v/v) Tween-20, pH 7.5) buffer. Comparison of HA tagged SLC15A4 labeling with or without PNGase are provided in Supplementary Fig. 2.
SLC15A4-mediated NOD2 activation (transport) reporter assay
To generate the SLC15A4 transport assay, the plasmid pGL4.32[luc2P NF-kB-RE Hygro] (Promega) was stably expressed in A549 cells. The various codon optimized mutants of SLC15A4 were cloned in frame with the mCherry protein, bridged by glycine-serine bridge into the pLPC retrovirus backbone using IDT gBlock fragments to enable expression within the reporter containing A549 cells. NOD2 was overexpressed in these cells using the pHAGE lentiviral backbone. Reporter cells were cultured in DMEM supplemented with 2 mM L-glutamine, 25 mM HEPES, 10% heat-inactivated FBS, 1× non-essential amino acids, and 55 μM β-Mercaptoethanol. Prior to use, 2.5 × 104 cells ml−1 cells were seeded in a 96 well flat-bottom dish and then treated with either compound or vehicle (DMSO). Following treatment, cells were stimulated with MDP (500 ng ml−1, unless otherwise indicated) and luminescence is read using the ClarioStar plate reader (BMG Labtech) after 24 h.
Cellular Thermal Shift Assay (CETSA)
For CETSA experiment, CAL-1 cells stably overexpressing HA-SLC15A4 were washed with cold DBPS for two times (400g, 4 min, 4 °C) and incubated with DMSO or indicated concentration of compounds in DPBS (2.0 × 106 cells ml−1) for 30 minutes at 37 °C. 50 μl aliquots were transferred to PCR tubes and heated at different temperatures on Bio-Rad C1000 Thermal Cycler for 3 mins, then incubated further for 3 minutes at 25°C. 2 μl 5% NP-40 and 0.5 μl 100× protease cocktail were added to each 50 μl aliquots and mixed by pipetting. Each PCR tube undergoes 3 snap freeze-thaw cycles in liquid nitrogen. Lysates were then transferred to 1.5 ml Eppendorf tubes and centrifuged at 20,000 ×g for 15 minutes at 4 °C. 36 μl Soluble protein was transferred to 12 μl 4× SDS sample buffer and stored in −80°C before western blot analysis. Remaining insoluble protein was resuspended in 50 μl 1× SDS sample buffer supplemented with 1× protease inhibitor cocktail, sonicated to break up DNA knots, and stored in −80°C before western blot analysis.
Flow Cytometry
Flow cytometry was performed in the Scripps Research Flow Core using either the NovoCyte Analyzers (ACEA Biosciences, Inc) or the LSRII Analyzers (BD Biosciences). Cells were resuspended in 1× PBS and stained using eBioscience Fixable Viability Dye eFluor 780 according to manufacturer’s instructions for 20 minutes. The Live/Dead cell stain was quenched, and cells were washed once using 1× FACS buffer. Surface marker staining was then performed in 1× FACS buffer for 30 minutes. Following the staining for surface markers, intracellular staining to assess phosphorylation status of signaling proteins was performed using BD Cytofix/Cytoperm Fixation/Permeabilization Solution Kit (BD Biosciences, 554714) following manufacturer’s instructions. The acidic compartment in B cells post TLR7 stimulation was evaluated using LysoSensor Green DND-189 (1 μM) (Thermo Fisher Scientific). All FACS data was analyzed using FlowJo version v10.
Quantitative PCR
The total RNA for quantitative real-time PCR was extracted using RNeasy Plus Mini (Qiagen) according to the protocol of the manufacturer. Genomic DNA was removed using Turbo DNA-free kit and cDNA was generated using M-MLV Reverse Transcriptase (200 U/μL) (Thermo Fisher Scientific), and RT-qPCR was performed with Fast SYBR™ Green Master Mix (Thermo Fisher Scientific) using QuantStudio 6 Pro Real-Time PCR System (Thermo Fisher Scientific). The following primers were used: IFNB F: 5’-tccaaattgctctcctgttgtgct-3’ and IFNB R: 5’-ccacaggagcttctgacactgaaaa-3’; ACTB F: 5’-aggtcatcaccattggcaatgag-3’; ACTB R: 5’-tctttgcggatgtccacgtca-3’. The results were normalized to the expression of ACTB and 2 h AJ2–18 treated group using the ΔΔCt method. Each sample was measured in technical duplicates, and each condition was tested with two biological replicates.
Lysosomal Isolation and vATPase Assay
Splenic mouse B cells were isolated using the EasySep Mouse B cell Isolation kit (StemCell Technologies) as described above. Once isolated, B cells were cultured in the following culture medium: RPMI 1640, 2 mM L-glutamine, 25 mM HEPES, 10% heat-inactivated FBS, 1× non-essential amino acids, and 55 μM β-Mercaptoethanol at a concentration of 2.0 × 106 cells ml−1. 20 million treated mouse B cells were resuspended in 1 ml of 1× PBS and then homogenized using a 2 ml homogenizer. The homogenate was incubated with rabbit anti-mouse LAMP2 antibody (Invitrogen) for 15 minutes and the lysosomal fraction was then isolated using Dynabeads M-280 Sheep anti-Rabbit IgG using the DynaMag magnet. The lysosomal fraction was gently washed 3× using PBS using the DynaMag magnet and then treated with following compounds: DMSO, AJ2–18 (5 μM), AJ2–30, (5 μM), or bafilomycin A1 (1 μM), respectively. vATPase activity of these treated lysosomal fractions was then assessed with EnzChek Phosphate Assay Kit (Thermo Fisher Scientific, E6646) according to manufacturer’s instructions.
THP1-Dual reporter assay
1 × 105 THP1-Dual cells (InvivoGen, #thpd-nfis) in 100 μl culturing media were plated per 96 well, incubated with varying concentration of compounds and stimulated with 5 μg ml−1 R848 for 18 h. Plates were then centrifuged at 300 ×g for 5 min and supernatants were collected for NF-κB and IRF reporter analysis. 20 μL of supernatants were first added to each well of a fresh white bottom 96 well plate, followed by 50 μL of Quanti-Luc gold (InvivoGen) reagent. Plates were gently tapped for around 20 times to ensure mixing, then luminescence was measured with ClarioStar plate reader (BMG Labtech). For NF-κB reporter analysis, 20 μl of supernatants were first added to each well of another clear bottom 96 well plate, followed by 180 μl of Quanti-Blue (InvivoGen, # rep-qbs) reagent. Plate was incubated at 37 °C for 5 to 10 min, optical density was measured at 650 nm using ClarioStar plate reader (BMG Labtech).
Immunofluorescence
To detect IRF5 and IRF7 localization, primary human pDC cells were seeded on 0.02% poly-L-lysine (Sigma, P1274) coated coverslips at RT for 15 min. Cells were then fixed in 2% PFA (Electron Microscopy Sciences) for 10 min, and permeabilized with ice-cold methanol at −20 °C for 10 min. After fixation/permeabilization, cells were washed for 5 min with PBS, and then blocked with 3% bovine serum albumin (BSA)/PBST (PBS with 0.05% TX-100) for 10 min. Primary antibodies were diluted at indicated ratio with blocking buffer and incubated with cells for 1 h. After washing three times in PBST, cells were incubated with Alexa Fluor-labeled secondary antibodies (1:500) for additional 1 h. Cells were then washed three times with PBST. For the rest of immunostaining experiments, CAL-1 cells or B cells were seeded on 0.02% poly-L-lysine coated coverslips at RT for 15 min. Cells were then fixed in 4% PFA for 10 min, permeabilized and blocked with BD Perm/Wash™ Buffer from BD Cytofix/Cytoperm kit for 20min, incubated with primary antibodies for 1h, and sequentially stained with secondary antibodies for additional 1 h.
Cells were stained with NucBlue Probe (Thermo Fisher Scientific, R37606) for 5 min to visualize nucleus, and then mounted onto slides with ProLong Diamond Antifade Mountant (Thermo Fisher Scientific, P36965). Confocal images were carried out on a Zeiss LSM 780 using a 100× objective (NA = 1.40). 1 Airy Unit was set as a pinhole for each channel. ImageJ and ZEN (Zeiss) software were utilized for image processing and analysis.
For colocalization quantification, around 25–50 cells were chosen randomly from each condition, and thresholds for each channel. Colocalization was reported as colocalization coefficient calculated using ZEN 2011 SP7 software (ver 14.0.5.201; displayed as #pixels of protein A colocalized with protein B: # pixels of protein A). For LC3B puncta counts and area analysis, images were quantified using the default ‘analyze particles’ plugin in ImageJ.
In vivo pharmacokinetic analysis of AJ2–30
3.96 mg of AJ2–30 was dissolved by sequential vortexing and sonication in 0.132 mL of DMA (N, N-dimethylacetamide), 0.132 mL of Solutol, and 1.056 mL of water to reach concentration of 3 mg ml−1. 30 mg kg−1 of AJ2–30 was dosed in male Balb-c mice through Intraperitoneal (IP) administration. Blood was drawn at different time points (0.25 h, 0.5 h, 1 h, 2 h, 4 h, 8 h, 24 h). The desired serial concentrations of working solutions were achieved by diluting stock solution of analyte with 50% acetonitrile in water solution. 10 μL of working solutions (0.5, 1, 2, 5, 10, 50, 100, 500, 1000 ng mL−1) were added to 10 μl of the blank mouse plasma to achieve calibration standards of 0.5~1000 ng/mL (0.5, 1, 2, 5, 10, 50, 100, 500, 1000 ng ml−1) in a total volume of 20 μl. Four quality control samples at 1 ng ml−1, 2 ng ml−1, 50 ng ml−1, and 800 ng mL−1 for plasma were prepared independently of those used for the calibration curves. These QC samples were prepared on the day of analysis in the same way as calibration standards. 20 μl of standards, 20 μl of QC samples and 20 μl of unknown samples (10 μl of mouse plasma with 10 μL of blank solution) were added to 200 μl acetonitrile and proteins were precipitated respectively. Then the samples were vortexed for 30 seconds. After centrifugation at 4 °C, 4000 rpm for 15 minutes (min), the supernatant was diluted 5 times with water, 3 μl of diluted supernatant was injected into the LC/MS/MS system for quantitative analysis. PK parameters were estimated by non-compartmental model using WinNonlin 8.3.
Cytotoxicity evaluation
To assess the cytotoxicity of the compounds, multiple concentrations of AJ2–18, AJ2–30, AJ2–32, and AJ2–90 were added to unstimulated PBMCs. PBMCs were plated at a concentration of 1× 106 cells ml−1 in culturing media. PBMCs cultured with the aforementioned compounds, necessary control compounds or DMSO were prescribed by the CytoTox 96 Non-Radioactive Cytotoxicity Assay kit, were incubated for 24 h at 37 °C and 5% CO2. Samples were then centrifuged (500g, 5 min) and supernatant was transferred to a new plate. CytoTox 96 Non-Radioactive Cytotoxicity Assay was used, following manufacturer’s instructions, to assess cytotoxicity of the samples. Cytotoxicity levels for AJ2–18, AJ2–30, AJ2–32, and AJ2–90 were compared to that of PMBCs treated with DMSO.
Immunoblotting and Immunoprecipitation
After treatment with compounds and immune agonists for designated time, cells were washed with ice-cold PBS and lysed in RIPA buffer (Thermo Fisher Scientific, 89900) supplemented with 1× Halt protease and phosphatase inhibitor (Thermo Fisher Scientific, 78841) for 20 min on ice. After centrifuging at 14,000 ×g for 10 min at 4 °C to remove cell debris, supernatants were taken and protein concentrations were determined by the BioRad DC protein assay (#5000112). Equal amounts of proteins were mixed with the 4×SDS sample buffer and resolved by SDS-PAGE. Proteins were transferred to PVDF membrane, blocked with 3% BSA or 5% non-fat milk in TBST, sequentially incubated with primary and secondary antibody, and visualized with a Bio-Rad ChemiDoc Imaging System.
For immunoprecipitation, cells were resuspended in E1C buffer (50 mM HEPES, 250 mM NaCl, 5 mM EDTA, 0.3% (w/v) CHAPS, 1× Halt protease inhibitor (Thermo Fisher Scientific,78438) and sonicated for three times (15 milliseconds (msec) on, 40 msec off, 15% amplitude, 1 second (sec) total on). Lysates were cleared by centrifuge and normalized to 1.5–2.0 mg ml−1 using E1C buffer. 20 μl HA magnetic beads were added to supernatants and rotated overnight at 4°C. The beads were washed three times with the E1C buffer, eluted by incubating in 2× sample buffer for 30min, and resolved by SDS-PAGE.
In vivo challenge of mice with CpG-A
Prior to challenge, C57BL/6J mice were treated by intraperitoneal injection with AJ2–30 (50 mg kg−1; 10% DMSO, 10% Tween-80, 80% 1× PBS by volume) or vehicle (10% DMSO, 10% Tween-80, 80% 1× PBS by volume). Mice were subsequently challenged with 2 μg of CpG-A complexed with 12 μl DOTAP Liposomal transfection reagent (Roche), administered by intravenous tail vein injection as described previously6. To assess the efficacy of AJ2–30 to suppress cytokine production in an in vivo model, blood was drawn by a retinal orbital bleed, and serum was isolated using centrifugation 6 h following CpG-A/DOTAP administration. Serum cytokine levels were quantitated by either Mouse IFN-Alpha ELISA Kit or the Bio-Plex Pro Mouse Cytokine 23-plex assay. To confirm that AJ2–30 was capable of suppressing mTOR activation in this in vivo model, mouse spleens were removed 2 hours after CpG-A/DOTAP administration, and the mTOR and 4EBP1 phosphorylation levels of splenic pDCs was determined by flow cytometry based on the following protocol 61.
Statistical analysis
Fluorescent gel scanning and western blots were quantified using Image Lab with a minimum of three biological replicates. Cytokine production, THP1-Dual reporter signals, B cell activation, and transport assays were performed with a minimum of three biological replicates. Statistical analysis for comparison of AJ2–30 treated samples to DMSO and AJ2–18 treated samples was done using an ANOVA followed by multiple comparisons with significance reported for P values of <0.05. Statistical significance for lupus patient samples was assessed using Wilcoxon matched-pairs signed-rank test with significance reported for P values of <0.05. Compound effects at different doses were normalized to vehicle treated cells and fitted with Sigmoidal 4PL curve. All statistical analysis was performed using Graphpad Prism 9. Proteomic analysis was performed with the processing software Proteome Discoverer 2.4 (Thermo Fisher Scientific). Peptide sequences were identified by matching theoretical spectra derived from proteome databases with experimental fragmentation patterns via the SEQUEST HT algorithm 62. Fragment tolerances were set to 0.6 Da, and precursor mass tolerances set to 10 ppm with one missed cleavage site allowed. Carbamidomethyl (C, +57.02146) and TMT-tag (K and N-terminal, +229.1629 for 10plex, +304.2071 for 16plex) were specified as static modifications while oxidation (M, +15.994915) was specified as variable. Spectra were searched against the Homo Sapiens proteome database (Uniprot, 2018, 42,358 sequences) or the Mus musculus proteome database (Uniprot, 2021; 25,314 sequences) using a false discovery rate of 1% (Percolator) 63,64. MS3 peptide quantitation was performed with a mass tolerance of 20 ppm. The final list of reported proteins was required to have at least two unique peptides. For enrichment experiments, abundances in each channel were normalized to the endogenously biotinylated protein PCCA. For unenriched proteomics, abundances in each channel were median normalized such that the median of each channel equals the average of all non-normalized channel medians. TMT ratios obtained by Proteome Discoverer were transformed with log2(x), and p-values were calculated via Student’s two-tailed t-tests with two biological replicates (significant if p <0.05). Quantitative data are listed in Supplementary Dataset.
Chemistry Methods
See Supplemental Information for complete synthetic methods and characterization
Extended Data
Extended Data Fig. 1. Fully functionalized fragment (FFF) probe library and cell models for SLC15A4 inhibitor characterization, Related to Figure 1.
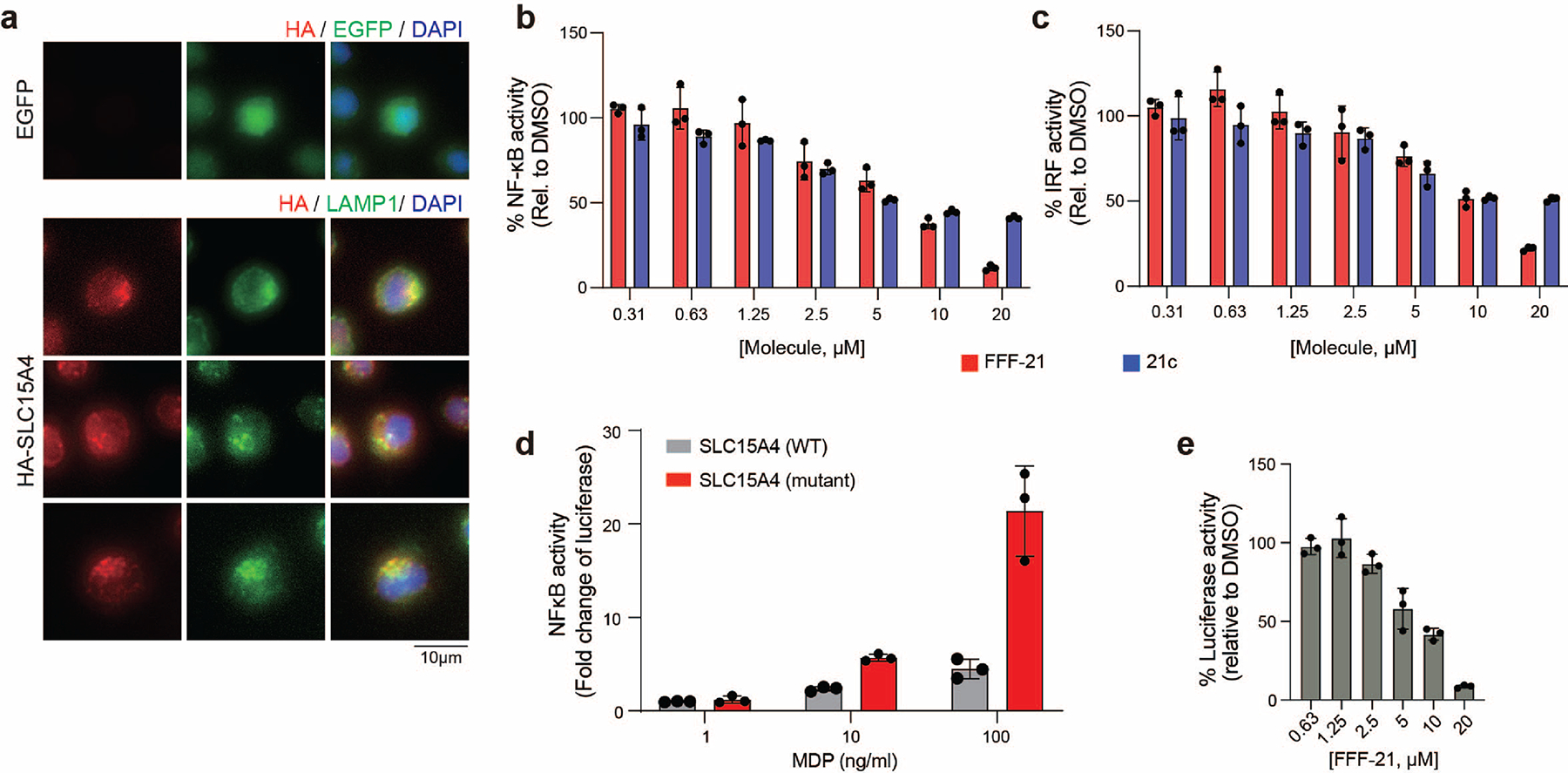
a, HA-SLC15A4 is localized to lysosomes in CAL-1 cells. Immunostaining of SLC15A4 in CAL-1 cells stably overexpressing EGFP or HA-tagged SLC15A4. Cells were co-stained with LAMP1 as a lysosome marker. Scale bar: 10 μm. Data are representative of two independent experiments.
b, c, FFF-21 and 21c inhibit TLR7/8 mediated signaling. THP1-DUAL reporter cells were co-incubated with R848 (5 μg/mL) and escalating doses of FFF-21 or 21c. IRF inhibition was monitored by measuring activity of secreted luciferase. NF-kB inhibition was monitored by measuring activity of secreted alkaline phosphatase. Data is plotted as the mean ± s.d. (n = 3 biological independent replicates).
d, Mutant human SLC15A4 (L14A, L15A, L318A, V319A) results in amplified MDP induced NOD activation relative to WT (24 h) as measured by luciferase activity in A549 cells expressing NOD2 and NF-κB luciferase reporter. Data is plotted as the mean ± s.d. (n = 3).
e, Concentration dependent inhibition of SLC15A4-dependent MDP transport by FFF-21. A549 cells were engineered to express an NF-κB-luciferase reporter, NOD2, and either membrane-localized SLC15A4. SLC15A4 transport cells were co-treated with FFF-21 and MDP (500 ng/mL) for 24 h. Data is plotted as the mean ± s.d. (n = 3 biological independent replicates).
Extended Data Fig. 2. Characterization of SLC15A4 chemical probes and control compounds, Related to Fig. 2.
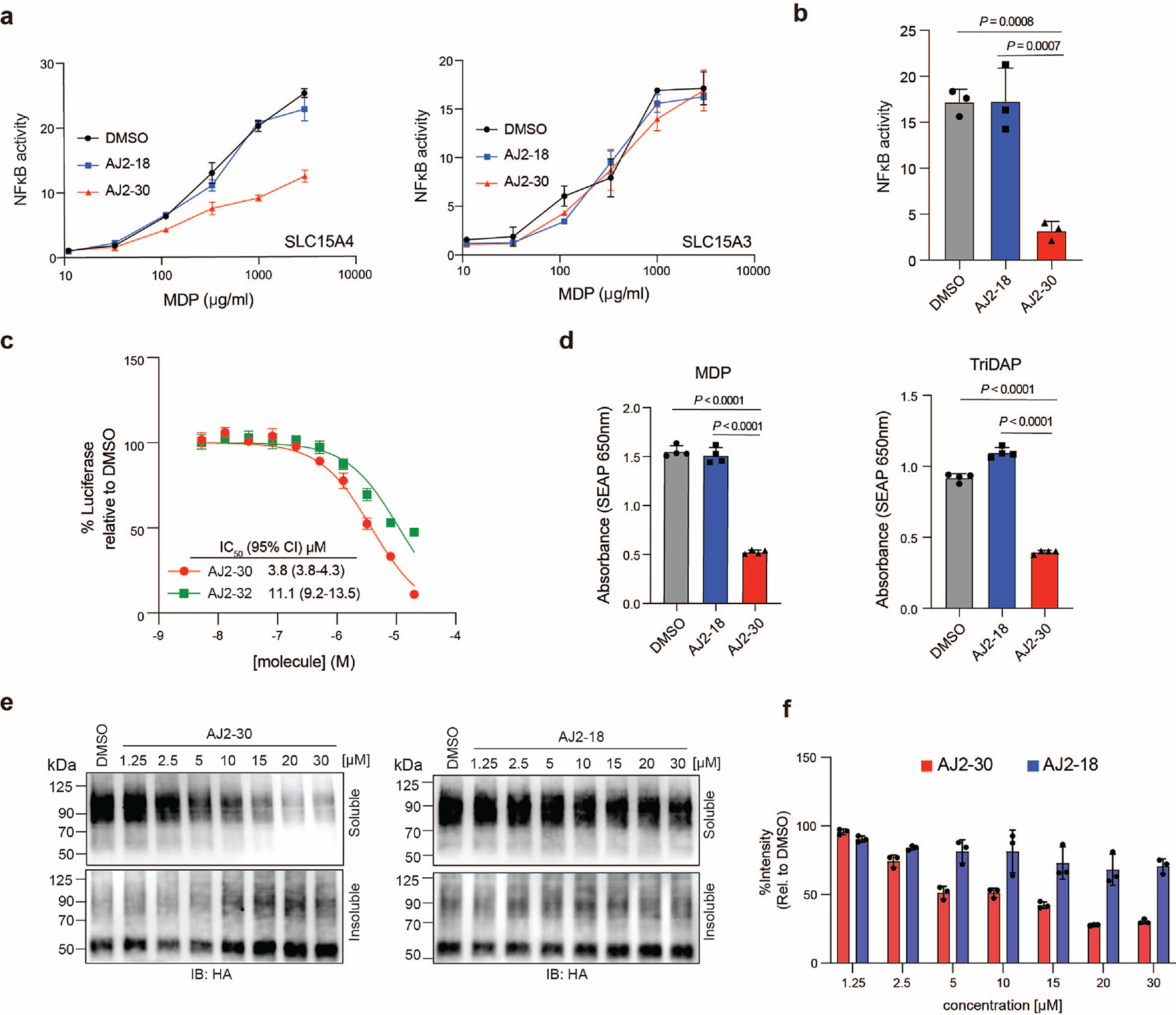
a, A549 cells engineered to express an NFκB luciferase reporter, human NOD2, and membrane-localized human SLC15A4 or SLC15A3. Cells were treated with AJ2–18 or AJ2–30 (5 μM) along with increasing quantities of MDP (24 h). Data is plotted as the mean ± s.d. (n = 3).
b, A549 cells transfected with membrane trafficked murine Slc15a4 as well as NOD2 were stimulated with MDP and treated with AJ2–18 or AJ2–30 (5 μM) for 24 h. Luciferase activity was measured in the cells at 24 h post-stimulation. Data is plotted as the mean ± s.d. (n = 3). P-values are shown.
c, THP1-DUAL reporter cells were co-incubated with 5 μg/ml R848 and escalating doses of AJ2–30 or AJ2–32 for 18 h. IRF inhibition was monitored by measuring activity of secreted luciferase. Data is plotted as the mean ± s.d. (n = 3)
d, THP1-DUAL reporter cells were stimulated with MDP and TriDAP in the presence of AJ2–30 or AJ2–18 and SEAP levels were assessed at 24 h. Data is plotted as the mean ± s.d. (n = 4 biological independent replicates). P-values are shown.
e, f, CETSA for AJ2–30 and AJ2–18 at varying doses. CAL-1 cells expressing HA-SLC15A4 were treated with increasing dosage of AJ2–30 or AJ2–18. Soluble and insoluble fractions were isolated by centrifugation and analyzed by immunoblot. Immunoblots were quantified and plotted as the mean ± s.d. of three independent replicates in (f).
Results are presented as mean ± s.d. of at least n = 3 independent experiments. Statistical analysis was performed using ANOVA analysis followed by multiple comparisons test. P-values are shown.
Extended Data Fig. 3. AJ2–30 inhibition of endo-lysosomal TLR signaling is selective and dependent on SLC15A4, Related to Fig. 4.
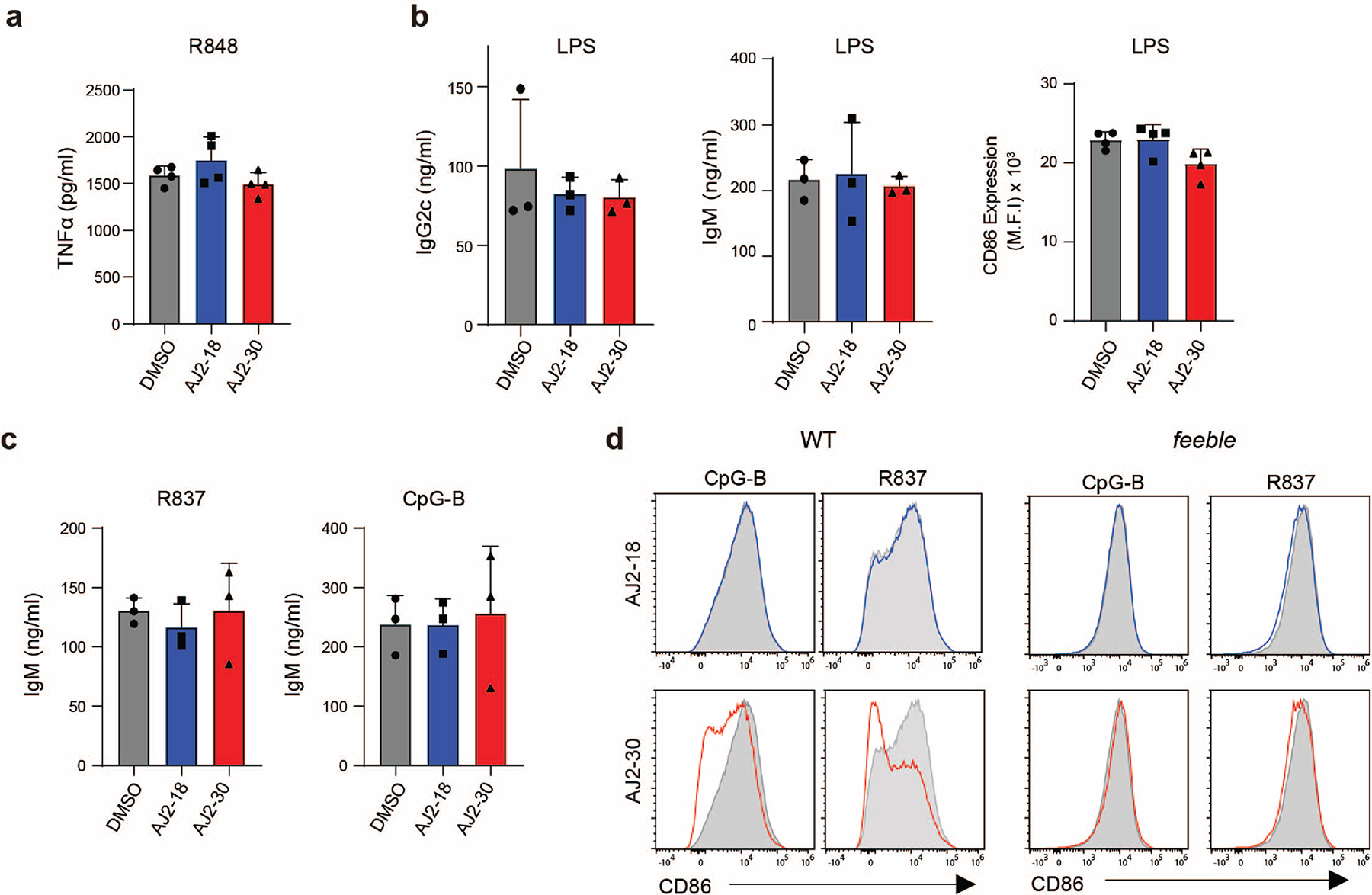
a, AJ2–30 does not inhibit TLR7/8 signaling in primary macrophages. Primary human derived macrophages were treated with AJ2–18 or AJ2–30 (5 μM), polarized with IFN-γ, and then stimulated with R848 (5 μg/mL) for 22 h.
b, AJ2–30 does not inhibit production of IgG2c, IgM, or CD86 expression in wild-type mouse B cells after LPS treatment.
c, AJ2–30 does not inhibit production of IgG2c, IgM in feeble mouse B cells after R837 and CpG-B treatment.
d, AJ2–30 suppresses CD86 expression in wild-type but not feeble B cells.
Results are presented as the mean ± s.d. of n = 3 (n = 4 for a) independent experiments. Statistical analysis was performed using ANOVA analysis followed by multiple comparisons test.
Extended Data Fig. 4. Mechanistic characterization of SLC15A4 inhibition by AJ2–30, Related to Fig. 5.
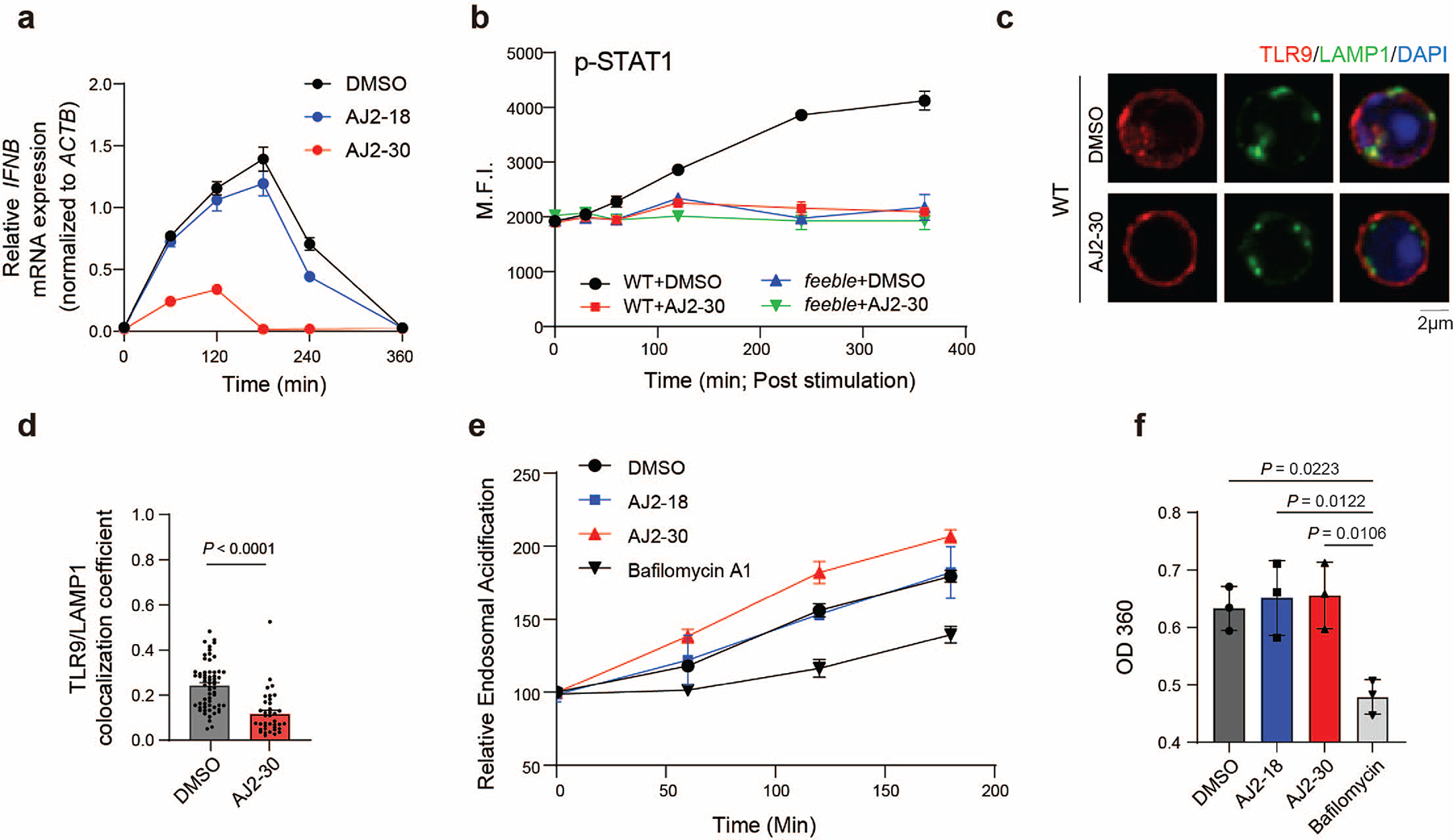
a, IFNB mRNA expression assessed by quantitative PCR in CAL-1 cells treated with compounds (5μM) or DMSO. Results were normalized to beta actin and values indicate mean ± s.d. (n = 2).
b, FACS analysis of STAT1 phosphorylation in WT and feeble mouse B cells with co-treatment of 1 μM CpG-B and 5 μM AJ2–30 (n = 3 biological independent replicates; mean ± s.d.).
c, Immunostaining of TLR9 in WT mouse B cells. Cells were co-treated with AJ2–30 (5 μM) orDMSO and stimuli 1 μM CpG-B for 15 min before fixation. Cells were co-stained with LAMP1 as a lysosome marker. Scale bar: 2 μm.
d, Quantitation of the colocalization coefficient of TLR9 with LAMP1 (n = 35 for DMSO versus n = 38 for AJ2–30 treatment; mean ± s.d.; unpaired two tailed t test). Images are representative of two independent experiments.
e, Acidification of lysosomes upon CpG-B stimulation in B cells. WT mouse B cells were treated with either DMSO, AJ2–18 (5 μM) AJ2–30 (5 μM), or Bafilomycin A (250 nM) and stimulated with 1 μM CpG-B for the indicated periods and labeled with LysoSensor DND-189. The fluorescence was analyzed by flow cytometry. Data is plotted as the mean ± s.d. (n = 3). f, Effect of AJ2–30 on ATPase activity of the LAMP1-enriched cellular fraction. Isolated lysosomal fraction was treated with DMSO, AJ2–18 (5 μM), AJ2–30 (5 μM), or bafilomycin A1 (1 μM). ATPase activity was measure using EnzChek™ Phosphate Assay Kit following manufacturer’s instructions. Statistical analysis was performed using ANOVA analysis followed by multiple comparisons test. Data is plotted as the mean ± s.d. (n = 3). P-values are shown.
Extended Data Fig. 5. Mechanistic characterization of SLC15A4 inhibition by AJ2–30, Related to Fig. 5.
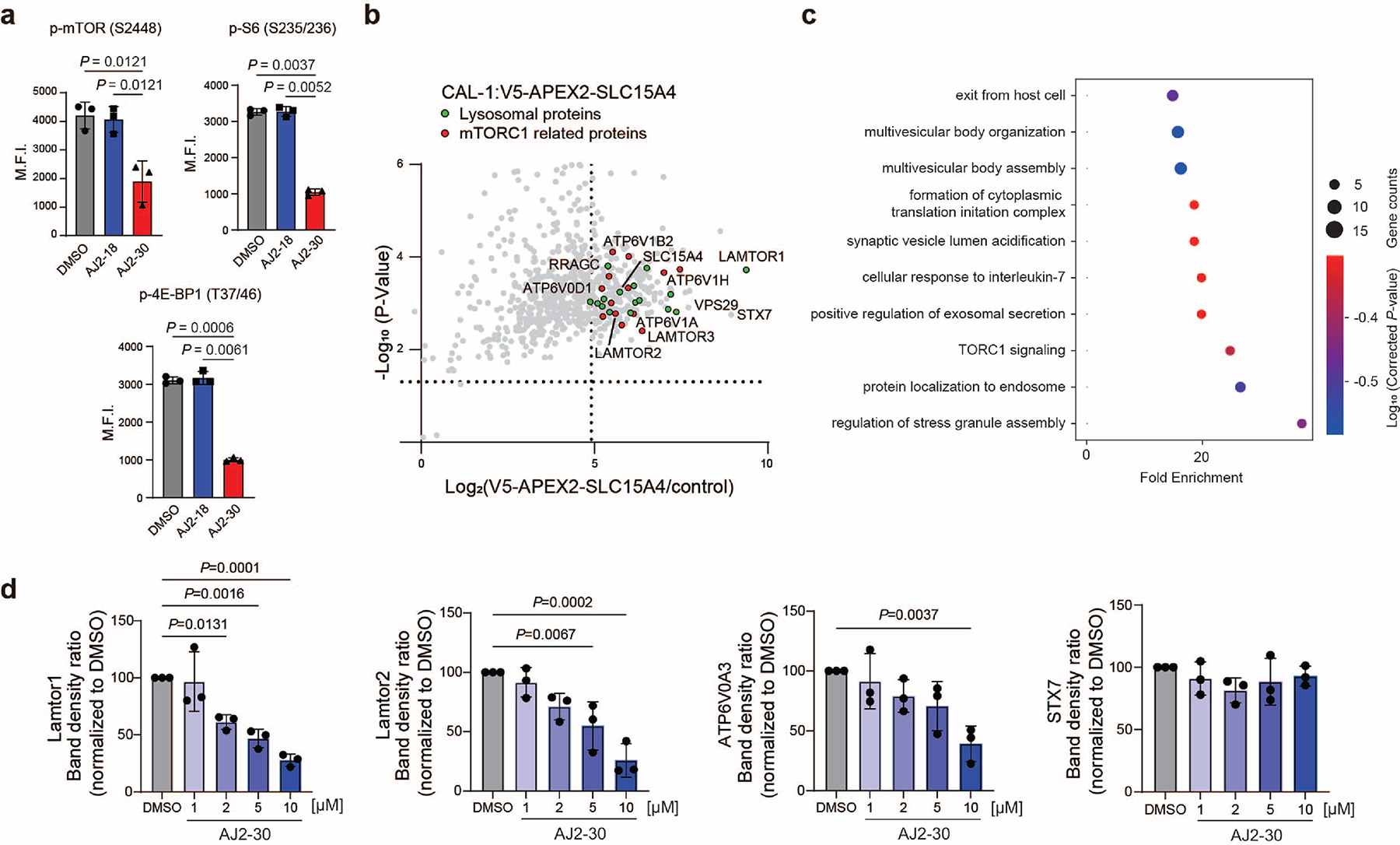
a, FACS analysis of mTORc1 activation by measuring the phosphorylation of mTOR (S2448), 4E-BP1 (T37/46), and S6 (S235/236) in human pDCs isolated from PBMCs. Primary human pDCs were treated with DMSO, AJ2–18 (5 μM), or AJ2–30 (5 μM) and then stimulated with CpG-A (1 μM) for 4 h. Statistical analysis was performed using ANOVA analysis followed by multiple comparisons test. Data is plotted as the mean ± s.d. (n = 3). P-values are shown.
b, Proximity labeling experiments of V5-APEX2-SLC15A4 in CAL-1 cells. Scatter plot displaying the fold change (FC) of proteins enriched in V5-APEX2 tagged SLC15A4 cells versus HA tagged SLC15A4 (negative control) cells. Proteins considered enriched if FC between V5A-PEX2-SLC15A4 and control >30, p < 0.05 (analyzed by unpaired two tailed t test). Data are presented as mean of n = 3 biologically independent samples. Enriched mTORC1 related proteins depicted as red circles, lysosomal associated proteins as green circles, and all other proteins as gray circles. Associated data set provided in Supplementary Dataset.
c, GO Enrichment Analysis of enriched proteins identified in proximity labeling enriched datasets include mTOR signaling among top 3 enriched pathways59.
d, Quantitation of indicated immunoprecipitated protein levels upon AJ2–30 treatment. Data presented as mean ± s.d. of n = 3 three biologically independent treatment samples and normalized to DMSO. Statistics were performed using ANOVA analysis. P-values are shown.
Extended Data Fig. 6. Mechanistic characterization of SLC15A4 inhibition by AJ2–30, Related to Fig. 5.
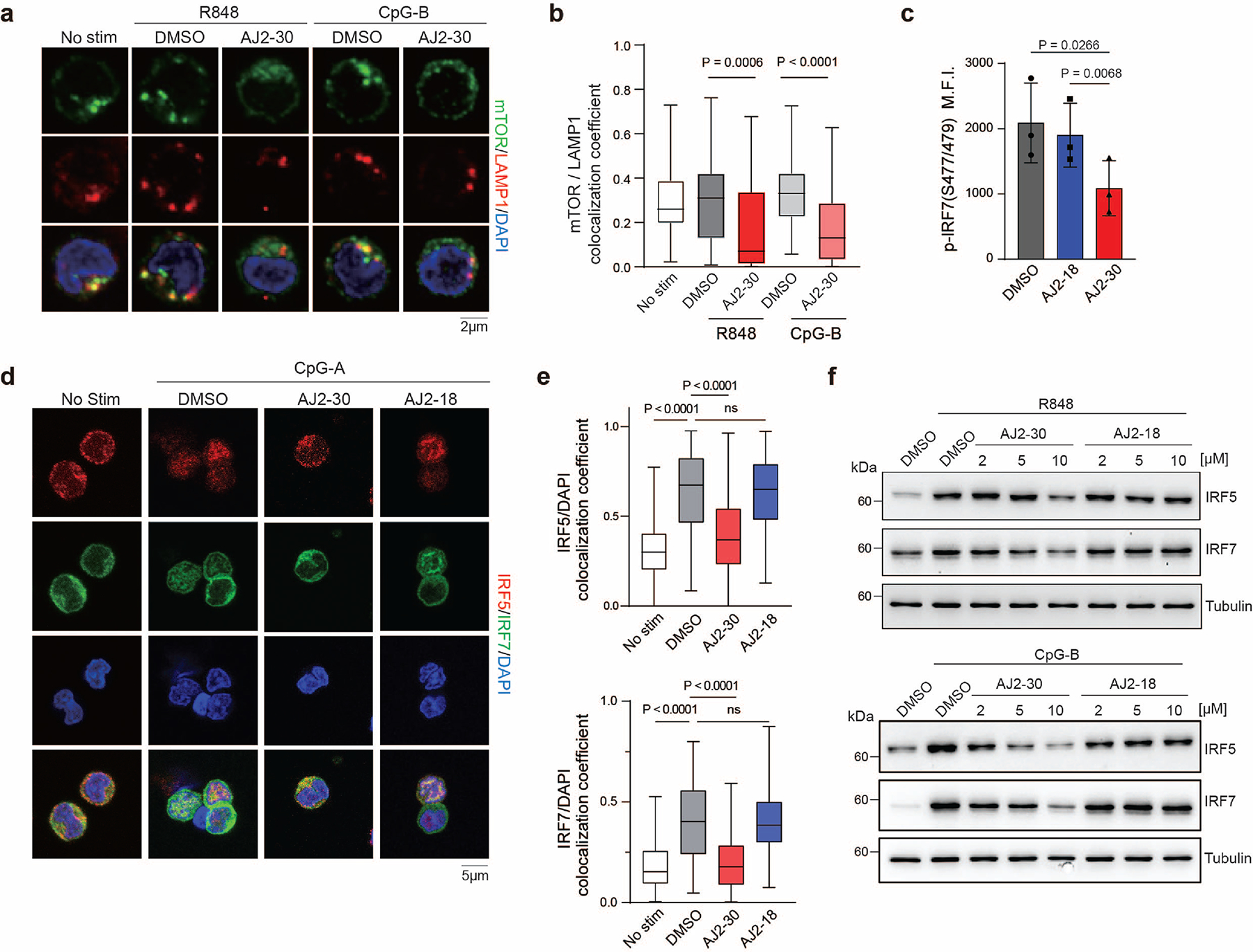
a, Immunostaining of mTOR in human B cells isolated from PBMCs. Cells were co-treated with AJ2–30 (5 μM) or DMSO and stimuli 5 μg/ml R848 or 1 μM CpG-B for 4h before fixation. Cells were co-stained with LAMP1 as a lysosome marker. Scale bar: 2 μm.
b, Quantitation of the colocalization coefficient of mTOR with LAMP1 (n = 64, 75, 47, 74, 76 cells from left to right). Images are representative of two independent experiments (two healthy donors). Data are presented as box plots. Centre line indicated the median, upper to lower bounds indicates 75th – 25th percentile with whiskers at maximum and minimum values. Statistics was performed using Student two-tail t-test analysis.
c, IRF7 activation was assessed by measuring the phosphorylation of IRF7 (S477/479) by flow cytometry in human pDCs isolated from PBMCs. Primary human pDCs were treated with DMSO, AJ2–18 (5 μM), or AJ2–30 (5 μM) and then stimulated with CpG-A (1 μM) for 4 h. Data is plotted as the mean ± s.d. (n = 3). Statistics were performed using ANOVA analysis. P-values are shown.
d, Immunostaining of IRF5 and IRF7 in primary human pDCs. Cells were co-treated with compounds (5 μM) or DMSO and stimuli 1 μM CpG-A for 4h before fixation. Cells were co-stained with DAPI as a nucleus marker. Scale bar: 5 μm.
e, Quantitation of the colocalization coefficient of IRF5 or IRF7 with DAPI (n = 121, 120, 153, 110 cells from left to right). Images are representative of three independent experiments. Data are presented as box plots and defined as Extended Figure 8B. Statistics were performed using ANOVA analysis. P-values are shown.
f, AJ2–30 suppresses TLR7/8 or TLR9-induced IRF5 and IRF7 translation in human B cells. Cells were co-treated with compounds (5 μM) or DMSO and stimuli (1 μM CpG-B or 5 μg/mL R848) for 8 h. Data are representative of two independent experiments.
Extended Data Fig. 7. AJ2–30 down-regulates SLC15A4 in CAL-1 and mouse B cells.
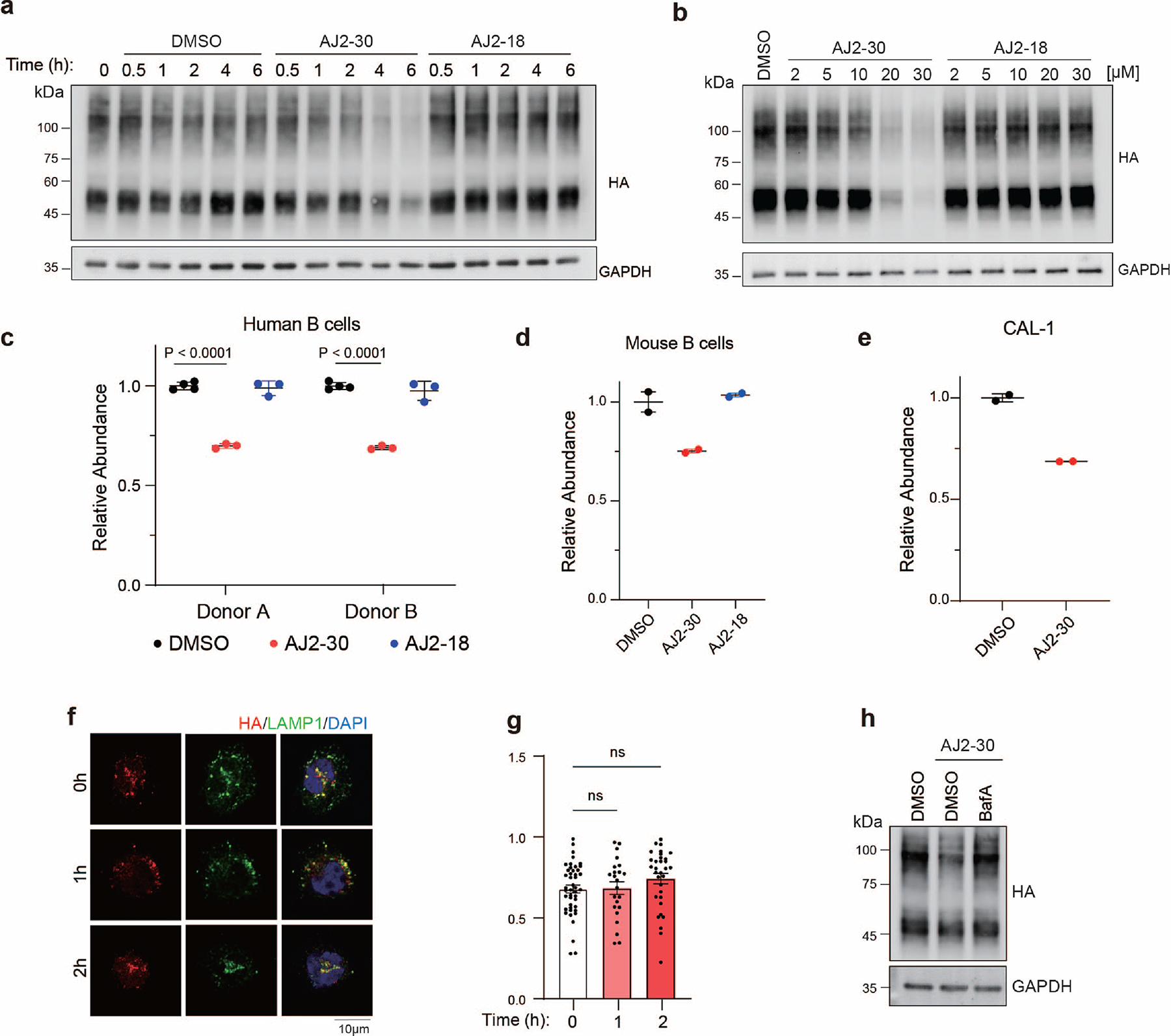
a, b, AJ2–30 down-regulates HA-SLC15A4. CAL-1 cells stably expressing HA-SLC15A4 were treated with compounds (20 μM), or DMSO for indicated time (a) or escalating doses at 4h (b). HA-SLC15A4 abundance was determined by immunoblot. Data are representative of three independent experiments.
c, Human B cells were treated with compounds (10 μM) or DMSO for 16 h. Abundance of SLC15A4 was analyzed by quantitative proteomics and normalized to DMSO treated samples. Data is plotted as the mean ± s.d. (n = 4). P-values are shown. Associated data set provided in Supplementary Dataset.
d, e, (d) mouse B cells were treatment with AJ2–30 (20 μM), AJ2–18 (20 μM), or DMSO for 4 h. (e) CAL-1 cells were treatment with AJ2–30 (10 μM) or DMSO for 16 h.
Abundance of SLC15A4 was analyzed by quantitative proteomics and normalized to DMSO treated samples (n = 2). Associated data set provided in Supplementary Dataset.
f, Localization of SLC15A4 in CAL-1 cells. CAL-1 cells stably expressing HA-SLC15A4 were co-treated with AJ2–30 (10 μM) or DMSO for different timepoints. 0h indicated cells were fixed before treatment. Cells were co-stained with LAMP1 as a lysosome marker. Scale bar: 10 μm.
g, Quantitation of the colocalization coefficient of HA-SLC15A4 with LAMP1 (n = 44, 23, 32 cells from left to right; mean ± s.d.). Statistics were performed using ANOVA analysis. Images are representative of two independent experiments.
h, Bafilomycin A blocks AJ2–30 degradation in CAL-1 cells. CAL-1 cells stably expressing HA-SLC15A4 were pre-treated with BafA for 1h and co-incubated with AJ2–30 (10 μM) or DMSO for 4h. HA-SLC15A4 abundance was determined by immunoblot. Images are representative of two independent experiments.
Extended Data Fig. 8. AJ2–30 suppresses costimulatory molecules expression of lupus patient B cells.
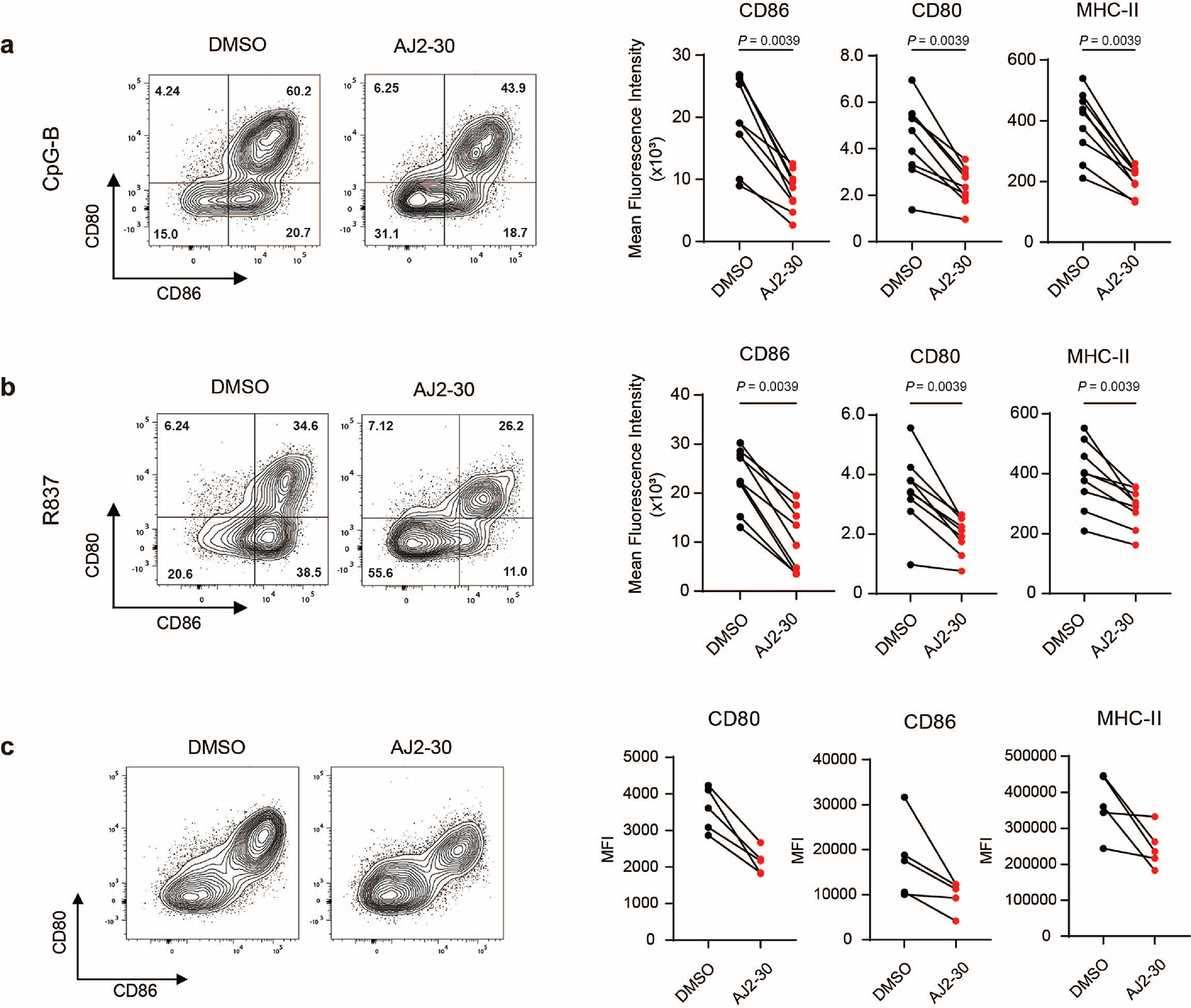
a, b, Expression of the costimulatory molecules CD80, CD86, and MHC-II on B cells following R837 or CpG-B stimulation when treated with either DMSO or AJ2–30 (5 μM) for 24h. Data is plotted (n = 8).
c, Expression of the costimulatory molecules CD80, CD86, and MHC-II on unstimulated B cells when treated with either DMSO or AJ2–30 (5 μM) for 24 h.
Data is plotted (n = 5). Statistical analysis was performed using Wilcoxon matched-pairs signed rank two-tailed test. P-values are shown.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Belharra Therapeutics for their assistance with in vivo and kinome profiling studies. We also thank B.F. Cravatt for his invaluable discussions and insights. This work was supported by the National Institute of Allergic and Infectious Diseases NIAID/ R01 AI156268 (C.G.P.) and T32 AI007244 (J.M.W.).
Footnotes
Competing Interests Statement
C.G.P., J.R.T., D.C.L., and A.M.J., are inventors on a patent application (WO2021174023) submitted by The Scripps Research Institute that covers small molecule inhibitors of SLC15A4. C.G.P. and J.R.T. are founders and scientific advisors to Belharra Therapeutics, a biotechnology company interested in using chemical proteomic methods to develop small molecule therapeutics. The remaining authors declare no competing interests.
Peer Review Information:
Nature Chemical Biology thanks Claire Steppan and the other, anonymous reviewers for their contribution to the peer review of this work.
Data availability
The data that support the findings in this work are available in the paper and Supplementary Information. Uncropped, full western blot images and gels are provided in Supplementary Fig 11. Gating strategies are described in Supplementary Fig 12. Processed proteomic data is provided in Supplementary Dataset. Homo Sapiens proteome database (Uniprot, 2018, 42,358 sequences) or the Mus musculus proteome database (Uniprot, 2021; 25,314 sequences) was used in proteomic searches. All raw proteomics data files have been deposited to the PRIDE repository and are available under the accession PXD045715 (for enrichment datasets; http://www.ebi.ac.uk/pride/archive/projects/PXD045715) and PXD045712 (for unenriched datasets; http://www.ebi.ac.uk/pride/archive/projects/PXD045712).
REFERENCES
- 1.Theofilopoulos AN, Kono DH & Baccala R The multiple pathways to autoimmunity. Nat Immunol 18, 716–724 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marshak-Rothstein A Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol 6, 823–835 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kono DH, Baccala R & Theofilopoulos AN TLRs and interferons: a central paradigm in autoimmunity. Curr Opin Immunol 25, 720–727 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lallana EC & Fadul CE Toxicities of immunosuppressive treatment of autoimmune neurologic diseases. Curr Neuropharmacol 9, 468–477 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Salliot C & van der Heijde D Long-term safety of methotrexate monotherapy in patients with rheumatoid arthritis: a systematic literature research. Ann Rheum Dis 68, 1100–1104 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blasius AL et al. Slc15a4, AP-3, and Hermansky-Pudlak syndrome proteins are required for Toll-like receptor signaling in plasmacytoid dendritic cells. Proc. Natl. Acad. Sci. U. S. A. 107, 19973–19978 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rimann I et al. The solute carrier SLC15A4 is required for optimal trafficking of nucleic acid-sensing TLRs and ligands to endolysosomes. Proc. Natl. Acad. Sci. U. S. A. 119, e2200544119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu Y, Song F, Jiang H, Nunez G & Smith DE SLC15A2 and SLC15A4 Mediate the Transport of Bacterially Derived Di/Tripeptides To Enhance the Nucleotide-Binding Oligomerization Domain-Dependent Immune Response in Mouse Bone Marrow-Derived Macrophages. J Immunol 201, 652–662 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakamura N et al. Endosomes are specialized platforms for bacterial sensing and NOD2 signalling. Nature 509, 240–244 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Sasawatari S et al. The solute carrier family 15A4 regulates TLR9 and NOD1 functions in the innate immune system and promotes colitis in mice. Gastroenterology 140, 1513–1525 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Heinz LX et al. TASL is the SLC15A4-associated adaptor for IRF5 activation by TLR7–9. Nature 581, 316–322 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kobayashi T et al. Human SLC15A4 is crucial for TLR-mediated type I interferon production and mitochondrial integrity. Int. Immunol. 33, 399–406 (2021). [DOI] [PubMed] [Google Scholar]
- 13.Kobayashi T et al. The histidine transporter SLC15A4 coordinates mTOR-dependent inflammatory responses and pathogenic antibody production. Immunity 41, 375–388 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Katewa A et al. The peptide symporter SLC15a4 is essential for the development of systemic lupus erythematosus in murine models. PLoS One 16, e0244439 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baccala R et al. Essential requirement for IRF8 and SLC15A4 implicates plasmacytoid dendritic cells in the pathogenesis of lupus. Proc Natl Acad Sci U S A 110, 2940–2945 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Griffith AD et al. A requirement for slc15a4 in imiquimod-induced systemic inflammation and psoriasiform inflammation in mice. Sci. Rep. 8, 1–10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zuo XB et al. Variants in TNFSF4, TNFAIP3, TNIP1, BLK, SLC15A4 and UBE2L3 interact to confer risk of systemic lupus erythematosus in Chinese population. Rheumatology International 34, 459–464 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Wang C et al. Genes identified in Asian SLE GWASs are also associated with SLE in Caucasian populations. European Journal of Human Genetics 21, 994–999 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Han JW et al. Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nature Genetics 41, 1234–U98 (2009). [DOI] [PubMed] [Google Scholar]
- 20.He CF et al. TNIP1, SLC15A4, ETS1, RasGRP3 and IKZF1 are associated with clinical features of systemic lupus erythematosus in a Chinese Han population. Lupus 19, 1181–1186 (2010). [DOI] [PubMed] [Google Scholar]
- 21.Zhang MW, Chen FR, Zhang DM, Zhai ZF & Hao F Association Study Between SLC15A4 Polymorphisms and Haplotypes and Systemic Lupus Erythematosus in a Han Chinese Population. Genetic Testing and Molecular Biomarkers 20, 451–458 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang WW, Gallo L, Jadhav A, Hawkins R & Parker CG The Druggability of Solute Carriers. J Med Chem 63, 3834–3867 (2020). [DOI] [PubMed] [Google Scholar]
- 23.Parker CG et al. Ligand and Target Discovery by Fragment-Based Screening in Human Cells. Cell 168, 527–541.e29 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y et al. Expedited mapping of the ligandable proteome using fully functionalized enantiomeric probe pairs. Nat. Chem. 11, 1113–1123 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang J et al. Synthesis of portimines reveals the basis of their anti-cancer activity. Nature 622, 507–513 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maeda T et al. A novel plasmacytoid dendritic cell line, CAL-1, established from a patient with blastic natural killer cell lymphoma. Int J Hematol 81, 148–154 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Caruso R, Warner N, Inohara N & Núñez G NOD1 and NOD2: signaling, host defense, and inflammatory disease. Immunity 41, 898–908 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee J et al. pH-dependent internalization of muramyl peptides from early endosomes enables Nod1 and Nod2 signaling. J. Biol. Chem. 284, 23818–23829 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Song F, Hu Y, Wang Y, Smith DE & Jiang H Functional characterization of human peptide/histidine transporter 1 in stably transfected MDCK cells. Mol. Pharm. 15, 385–393 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martinez Molina D et al. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 341, 84–87 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Ganguly D et al. Self-RNA-antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J Exp Med 206, 1983–1994 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kobayashi T et al. SLC15A4 mediates M1-prone metabolic shifts in macrophages and guards immune cells from metabolic stress. Proc. Natl. Acad. Sci. U. S. A. 118, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kobayashi T et al. Lysosome biogenesis regulated by the amino-acid transporter SLC15A4 is critical for functional integrity of mast cells. Int. Immunol. 29, 551–566 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mindell JA Lysosomal acidification mechanisms. Annu Rev Physiol 74, 69–86 (2012). [DOI] [PubMed] [Google Scholar]
- 35.Zoncu R et al. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 334, 678–683 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yamashita T et al. Cloning and functional expression of a brain peptide/histidine transporter. J. Biol. Chem. 272, 10205–10211 (1997). [DOI] [PubMed] [Google Scholar]
- 37.Lam SS et al. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat. Methods 12, 51–54 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim E, Goraksha-Hicks P, Li L, Neufeld TP & Guan K-L Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol. 10, 935–945 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sancak Y et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320, 1496–1501 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sancak Y et al. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141, 290–303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bar-Peled L, Schweitzer LD, Zoncu R & Sabatini DM Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 150, 1196–1208 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Perera RM & Zoncu R The lysosome as a regulatory hub. Annu. Rev. Cell Dev. Biol. 32, 223–253 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.de Araujo MEG et al. Crystal structure of the human lysosomal mTORC1 scaffold complex and its impact on signaling. Science 358, 377–381 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Saxton RA & Sabatini DM MTOR signaling in growth, metabolism, and disease. Cell 168, 960–976 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yonehara R et al. Structural basis for the assembly of the Ragulator-Rag GTPase complex. Nat. Commun. 8, 1–11 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lopez-Haber C et al. The phagosomal solute transporter SLC15A4 promotes inflammasome activity via mTORC1 signaling and autophagy restraint in dendritic cells. Embo Journal 41, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miyake K et al. Endolysosomal compartments as platforms for orchestrating innate immune and metabolic sensors. J. Leukoc. Biol. 106, 853–862 (2019). [DOI] [PubMed] [Google Scholar]
- 48.Cao W et al. Toll-like receptor-mediated induction of type I interferon in plasmacytoid dendritic cells requires the rapamycin-sensitive PI(3)K-mTOR-p70S6K pathway. Nat. Immunol. 9, 1157–1164 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schmitz F et al. Mammalian target of rapamycin (mTOR) orchestrates the defense program of innate immune cells. Eur. J. Immunol. 38, 2981–2992 (2008). [DOI] [PubMed] [Google Scholar]
- 50.Yoshimori T, Yamamoto A, Moriyama Y, Futai M & Tashiro Y Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J. Biol. Chem. 266, 17707–17712 (1991). [PubMed] [Google Scholar]
- 51.Crow MK Type I interferon in the pathogenesis of lupus. J Immunol 192, 5459–5468 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Banchereau J & Pascual V Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity 25, 383–392 (2006). [DOI] [PubMed] [Google Scholar]
- 53.Niewold TB, Hua J, Lehman TJ, Harley JB & Crow MK High serum IFN-alpha activity is a heritable risk factor for systemic lupus erythematosus. Genes Immun 8, 492–502 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Custódio TF et al. Molecular basis of TASL recruitment by the peptide/histidine transporter 1, PHT1. Nat. Commun. 14, 5696 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thomson AW, Turnquist HR & Raimondi G Immunoregulatory functions of mTOR inhibition. Nat. Rev. Immunol. 9, 324–337 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sardana R & Emr SD Membrane Protein Quality Control Mechanisms in the Endo-Lysosome System. Trends Cell Biol 31, 269–283 (2021). [DOI] [PubMed] [Google Scholar]
- 57.Lee C, Lamech L, Johns E & Overholtzer M Selective Lysosome Membrane Turnover Is Induced by Nutrient Starvation. Developmental Cell 55, 289–+ (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weichhart T, Hengstschläger M & Linke M Regulation of innate immune cell function by mTOR. Nat. Rev. Immunol. 15, 599–614 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Joeh E, Reeves AE, Parker CG & Huang ML Mapping Interactions between Glycans and Glycan-Binding Proteins by Live Cell Proximity Tagging. Curr Protoc 1, e104 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McAlister GC et al. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal Chem 86, 7150–7158 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Teijaro JR et al. S1PR1-mediated IFNAR1 degradation modulates plasmacytoid dendritic cell interferon-α autoamplification. Proc. Natl. Acad. Sci. U. S. A. 113, 1351–1356 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Eng JK, McCormack AL & Yates JR An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom 5, 976–989 (1994). [DOI] [PubMed] [Google Scholar]
- 63.Kall L, Canterbury JD, Weston J, Noble WS & MacCoss MJ Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat Methods 4, 923–925 (2007). [DOI] [PubMed] [Google Scholar]
- 64.Elias JE & Gygi SP Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods 4, 207–214 (2007) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings in this work are available in the paper and Supplementary Information. Uncropped, full western blot images and gels are provided in Supplementary Fig 11. Gating strategies are described in Supplementary Fig 12. Processed proteomic data is provided in Supplementary Dataset. Homo Sapiens proteome database (Uniprot, 2018, 42,358 sequences) or the Mus musculus proteome database (Uniprot, 2021; 25,314 sequences) was used in proteomic searches. All raw proteomics data files have been deposited to the PRIDE repository and are available under the accession PXD045715 (for enrichment datasets; http://www.ebi.ac.uk/pride/archive/projects/PXD045715) and PXD045712 (for unenriched datasets; http://www.ebi.ac.uk/pride/archive/projects/PXD045712).